

Abstract
Human (h) carbonic anhydrase (CAs, EC 4.2.1.1) isoforms IX and XII were recently confirmed as anticancer targets against solid hypoxic tumours. The “three-tails approach” has been proposed as an extension of the forerunner “tail” and “dual-tail approach” to fully exploit the amino acid differences at the medium/outer active site rims among different hCAs and to obtain more isoform-selective inhibitors. Many three-tailed inhibitors (TTIs) showed higher selectivity against the tumour-associated isoforms hCA IX and XII with respect to the off-targets hCA I and II. X-ray crystallography studies were performed to investigate the binding mode of four TTIs in complex with a hCA IX mimic. The ability of the most potent and selective TTIs to reduce in vitro the viability of colon cancer (HT29), prostate adenocarcinoma (PC3), and breast cancer (ZR75-1) cell lines was evaluated in normoxic (21% O2) and hypoxic (3% O2) conditions demonstrating relevant anti-proliferative effects.
Keywords: Tail approach, carbonic anhydrase, inhibitor, X-ray crystallography, hypoxic tumour
1. Introduction
Tumour growth, malignant progression, and resistance to chemotherapy and radiotherapy appear to be strongly associated with tumour hypoxia1–5. Hypoxia is the main cause responsible for the overexpression of the hypoxia-inducible factor (HIF-1) and the Warburg effect in tumours, an indispensable metabolic reprogramming of cancer cells from glycolytic metabolism to fermentation1–8. In order to survive in hypoxic conditions and acidosis due to fermentative metabolism, HIF-1 triggers a signalling cascade, that upregulates the expression of several genes, coding for the lactate-proton symporters (MCT4), other proton-transporters, and the tumour-associated isoforms IX and XII of carbonic anhydrases (CAs, EC 4.2.1.1), that catalyse the reversible hydration of carbon dioxide (CO2) into a proton (H+) and bicarbonate (HCO3−)1,2,6–11. These proton export mechanisms, in concert with poor vascular drainage, are responsible to maintain an intracellular pH of 7.2–7.4, acidifying the extracellular pH to 6.2–6.8, which is strongly associated with the propagation, malignant progression, and resistance to chemotherapy and radiotherapy of tumours1,2,6–11. In detail, the CA IX and XII expression is strongly increased in many types of tumours9,12–21 and is downregulated by the wild-type von Hippel–Lindau tumour suppressor protein (pVHL)2,22,23. In some cancer cells, the VHL gene is mutated leading to the strong upregulation of tumour-associated CA isoforms as a consequence of constitutive HIF activation24,25. Recent studies have shown that isoform hCA IV, prevalently anchored on the membrane of the astrocytes, are responsible for regulating interstitial pH26 and for regulating transmembrane lactate transport, interacting with the chaperones of the monocarboxylate transporters in the brain cells27–30. Whereas targeting of hCA IV with inhibitors does not yet have clear antitumor therapeutic applications31, in the last decades, several studies corroborated CA IX and XII as targets for the development of carbonic anhydrases inhibitors (CAIs) as novel anticancer drugs and, to date, the sulphonamide inhibitor SLC-0111 is in phase two clinical trials as an antitumor agent32–34.
Among the large number of CAI chemotypes, the zinc binder sulphonamides led to many potent and fruitful inhibitory molecules34,35. However, their lack of selectivity and inability to discern among the 15 human (h) CA isoforms, prevents their wider use as therapeutic agents, at least for the first and second generation of such inhibitors28,29. In fact, the inhibition of ubiquitous and cytosolic isoforms hCA I and II is responsible for the side effect in the treatment with CAIs34,35. To overcome their promiscuous inhibition, the “three-tails approach” was applied as an extension of the previously proposed “tail” and “dual tails approach” (Figure 1(A))36–39. A careful 3D analysis has shown different dimensions of the 15 hCAs active site that together with diverse architecture and extension of the hydrophilic and lipophilic areas, line disparate pockets that could be targeted by specific tails35. The three-tails approach consists of appending three pendants of various nature on a CA inhibitory (CAI) scaffold (e.g. benzenesulfonamide) in order to interact with the most variable residues among the fifteen hCAs in the middle/outer rim of the active site, conferring to the sulphonamide inhibitors some important properties, such as water solubility37 and subsequently membrane (im)permeability40, improving the interactions with the hydrophilic and hydrophobic halves of the active sites and increasing the matching and fitting of the ligand-target contacts to attain the proper hCA selective inhibition35.
Figure 1.

Schematic representation of the (A) “three-tails” approach for the design of zinc-binding CAIs. (B) Binding mode of a Three-Tailed Inhibitor (TTIs).
2. Material and methods
2.1. Chemistry
The synthesis and characterisation of sulphonamides 1–50 was reported earlier by our group36.
2.2. Carbonic anhydrase inhibition
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity41 as reported earlier42. Enzyme concentrations were in the range 5–18 nM. All CA isoforms were recombinant ones obtained in-house as reported earlier43.
2.3. X-ray crystallography
2.3.1. Protein expression and purification
Competent BL21 Escherichia coli cells were transformed with plasmid DNA containing the hCA IX-mimic gene using standard protocols as described earlier44,45.
2.3.2. Crystallisation
Inhibitors were successfully cocrystalised with CAII and CAIX-mimic via the hanging-drop vapour diffusion method. 0.5 mL of mother liquor consisting of 1.6 M sodium citrate and 50 mM Tris at pH 7.8 was used in the wells for setting up crystal trays. Each cover slip contained a 1:1 ratio of 10 mg/mL protein to mother liquor. DMSO was used to dissolve inhibitors to 1 mM, with the drops final concentration ∼100 µM. Cocrystals of CAII and CAIX formed within a week.
2.3.3. Data collection and processing
Diffraction data were collected via the F1 beamline at Cornell High Energy Synchrotron Source (CHESS, Ithaca, NY) at 0.977 Å wavelength and at Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA). A Pilatus 6 M detector collected data sets with a crystal-to-detector distance of 270 mm, 1° oscillation, and 4 s image exposure, for a total of 180 images. Diffraction data were indexed and integrated with XDS46. Data were scaled in space group P21 via AIMLESS47 from the CCP4 program suite48. Phases were determined via molecular replacement using PDB: 4ZAO49 as a search model. Modifications to the model, such as addition of inhibitor, ligand (glycerol), zinc, and water to the active site of CA were executed in Coot50 along with ligand PDB file modifications. Refinements were completed and ligand restraint files were created in Phenix51. Figures were generated with PyMol (Schrödinger). Protein-ligand bond lengths and active site interactions were observed with (LigPlot Plus, Hinxton, Cambridgeshire, UK)52 .
2.4. Antiproliferative assays
2.4.1. Cell culture and treatments
Human prostate cancer cell line PC3, human breast cancer cell line ZR75-1, and human colon cancer cell line HT-29 were obtained from American Type Culture Collection (Rockville, MD). PC3, ZR75-1, and HT-29 were cultured in DMEM high glucose with 10% FBS in 5% CO2 atmosphere at 37 °C. Media contained 2 mM L-glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Sigma, Milan, Italy). Cells were plated in 96-well cell culture (1.104/well) and, 24 h after, treated with the tested compounds (0 − 200 μM) for 48 h. Low oxygen conditions were acquired in a hypoxic workstation (Concept 400 anaerobic incubator, Ruskinn Technology Ltd., Bridgend, UK). The atmosphere in the chamber consisted of 1% O2 (hypoxia), 5% CO2, and residual N2. In parallel, normoxic (20% O2) dishes were incubated in air with 5% CO2.
2.4.2. Cell viability assay
PC3, ZR75-1, and HT-29 cell viability were evaluated by the reduction of 3–(4,5-dimethylthiozol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as an index of mitochondrial compartment functionality. Cells were plated and treated as described. Post-treatment, after extensive washing, 1 mg/mL MTT was added into each well and incubated for 30 min at 37 °C. After washing, the formazan crystals were dissolved in 150 μL of DMSO. The absorbance was measured at 550 nm. Experiments were performed in quadruplicate on at least three different cell batches.
2.4.3. Statistical analysis
Results were expressed as mean ± SEM, and the analysis of variance was performed by one-way ANOVA. A Bonferroni’s significant difference procedure was used as post-hoc comparison. p Values of less than .05 were considered as significant. Data were analysed using (Origin version 9.1 software, Hinxton, Cambridgeshire, UK).
3. Results and discussion
Several drug design studies of CAIs adopted the p-substituted benzenesulfonamide as a main scaffold against heteroaromatic sulphonamides to notably simplify the synthetic procedures and allow to focus on the attachment of variable pendants on the inhibitor structure38,53. A p-substituted benzenesulfonamide was adopted by us as a CAI scaffold to converge efforts and attention on studying the three-tailing effects on CA inhibition36. The structure of the three-tailed inhibitors (TTIs) was selected to merge an easy and versatile chemistry with the opportunity to expand it to many different chemical groups, which is significant for generating a range of tail combinations (Figure 1(B)). The synthetic strategies adopted to yield the TTI derivatives here discussed were previously reported by us36.
3.1. Carbonic anhydrase inhibition
In this first screening, mono-tailed (1–7) and three-tailed (18–50) compounds were analysed by a stopped-flow kinetic assay41 with: the tumour-associated isoforms CA IV, IX, and XII and the main off-target isoforms CA I and II, if considered the anticancer application (Table 1)1,9–11. The selectivity index of mono-tailed and three-tailed compounds vs. the off-target isoforms CA I and II are reported in Table S1 (Supplementary Information).
Table 1.
Inhibition data of human CA isoforms I, II, IV, IX, and XII with sulphonamides 19–50 reported here and the standard sulphonamide inhibitor acetazolamide (AAZ) by a stopped-flow CO2 hydrase assay41.
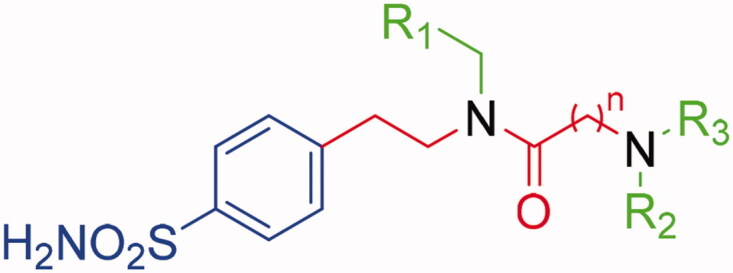
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | n | R1 | R2 | R3 | KIa |
||||
| hCA Ib | hCA IIb | hCA IVb | hCA IX | hCA XIIb | |||||
| 1 | – | C6H5 | – | – | 95.3 | 98.4 | 2854.4 | 78.1 | 65.4 |
| 2 | – | 4-NO2-C6H4 | – | – | 224.3 | 120.9 | 1685.3 | 89.2 | 77.4 |
| 3 | – | 4-F-C6H4 | – | – | 112.8 | 78.5 | 1196.7 | 56.5 | 60.1 |
| 4 | – | 2-Naph | – | – | 458.1 | 87.1 | 6248.1 | 71.2 | 78.6 |
| 5 | – | Fu | – | – | 68.4 | 62.8 | 1584.5 | 64.6 | 55.4 |
| 6 | – | CH2CN | – | – | 105.3 | 153.7 | 5547.2 | 104.7 | 113.2 |
| 7 | – | CH2C6H5 | – | – | 278.4 | 89.1 | 3587.4 | 101.9 | 104.3 |
| 18 | 1 | C6H5 | CH2CH3 | CH2CH3 | 786.6 | 8.3 | 4147.5 | 30.6 | 43.9 |
| 19 | 1 | C6H5 | CH2CH3 | CH2C6H5 | 4210.4 | 391.6 | >10,000 | 118.8 | 82.6 |
| 20 | 1 | C6H5 | CH2C6H5 | CH2C6H5 | 865.9 | 412.3 | >10,000 | 234.3 | 98.8 |
| 21 | 1 | C6H5 | (CH2)4CH3 | (CH2)4CH3 | 506.1 | 124.5 | >10,000 | 48.5 | 69.4 |
| 22 | 1 | C6H5 | (CH2)5CH3 | (CH2)5CH3 | 878.7 | 237.0 | >10,000 | 123.4 | 92.8 |
| 23 | 1 | C6H5 | (CH2)7CH3 | (CH2)7CH3 | 946.7 | 843.8 | >10,000 | 271.2 | 99.4 |
| 24 | 2 | C6H5 | CH2CH3 | CH2CH3 | 184.7 | 8.9 | 3928.8 | 31.1 | 61.1 |
| 25 | 2 | C6H5 | CH2CH3 | CH2C6H5 | 544.3 | 79.6 | >10,000 | 165.7 | 90.4 |
| 26 | 2 | C6H5 | CH2C6H5 | CH2C6H5 | 692.3 | 559.2 | 4640.8 | 106.4 | 302.5 |
| 27 | 2 | C6H5 | (CH2)4CH3 | (CH2)4CH3 | 563.6 | 522.6 | 3244.8 | 133.8 | 100.3 |
| 28 | 2 | C6H5 | (CH2)5CH3 | (CH2)5CH3 | 308.2 | 578.4 | 3455.4 | 94.2 | 77.8 |
| 29 | 2 | C6H5 | (CH2)7CH3 | (CH2)7CH3 | 209.3 | 778.8 | >10,000 | 146.5 | 280.0 |
| 30 | 2 | CH2C6H5 | (CH2)5CH3 | (CH2)5CH3 | 518.4 | 780.8 | 3413.2 | 42.9 | 62.5 |
| 31 | 2 | Fu | (CH2)5CH3 | (CH2)5CH3 | 220.1 | 60.4 | 3153.7 | 47.1 | 9.7 |
| 32 | 1 | 2-Naph | (CH2)5CH3 | (CH2)5CH3 | 541.4 | 4562.9 | >10,000 | 295.5 | 61.7 |
| 33 | 1 | CH2CN | (CH2)5CH3 | (CH2)5CH3 | 395.9 | 52.5 | 3478.3 | 154.5 | 8.6 |
| 34 | 1 | CH2C6H5 | (CH2)2C6H5 | (CH2)2CN | 777.3 | 368.5 | >10,000 | 32.3 | 75.5 |
| 35 | 1 | Fu | (CH2)2C6H5 | (CH2)2CN | 300.8 | 73.2 | 457.4 | 95.1 | 8.7 |
| 36 | 1 | 4-F-C6H5 | (CH2)2C6H5 | (CH2)2CN | 676.4 | 133.0 | 4133.8 | 3.0 | 9.8 |
| 37 | 1 | 2-Naph | (CH2)2C6H5 | (CH2)2CN | 685.0 | 247.5 | 3812.9 | 151.3 | 64.9 |
| 38 | 1 | 4-NO2-C6H5 | (CH2)2C6H5 | (CH2)2CN | 407.5 | 264.2 | 2421.5 | 201.5 | 89.5 |
| 39 | 1 | CH2CN | (CH2)2C6H5 | (CH2)2CN | 61.6 | 0.7 | 726.6 | 19.3 | 8.9 |
| 40 | 1 | CH2C6H5 | (CH2)2C6H5 | (CH2)3NH2 | 242.4 | 367.3 | 2149.2 | 4.0 | 83.7 |
| 41 | 1 | Fu | (CH2)2C6H5 | (CH2)3NH2 | 246.7 | 57.0 | 374.1 | 11.1 | 42.7 |
| 42 | 1 | 4-F-C6H5 | (CH2)2C6H5 | (CH2)3NH2 | 451.4 | 30.4 | 365.3 | 4.8 | 0.6 |
| 43 | 1 | 2-Naph | (CH2)2C6H5 | (CH2)3NH2 | 506.7 | 5.6 | 819.2 | 34.3 | 10.5 |
| 44 | 1 | (CH2)2NH2 | (CH2)5CH3 | (CH2)5CH3 | 435.8 | 2924.8 | 913.9 | 115.6 | 32.5 |
| 45 | 1 | CH2C6H5 | (CH2)2C6H5 | (CH2)2COOH | 203.5 | 72.0 | 2330.5 | 7.5 | 29.7 |
| 46 | 1 | Fu | (CH2)2C6H5 | (CH2)2COOH | 79.5 | 2.4 | 335.5 | 1.2 | 7.1 |
| 47 | 1 | 4-F-C6H5 | (CH2)2C6H5 | (CH2)2COOH | 95.8 | 23.5 | 419.3 | 11.1 | 8.8 |
| 48 | 1 | 2-Naph | (CH2)2C6H5 | (CH2)2COOH | 197.0 | 72.5 | 680.6 | 22.1 | 6.8 |
| 49 | 1 | CH2COOH | (CH2)5CH3 | (CH2)5CH3 | 285.5 | 585.7 | 45.8 | 68.3 | 9.9 |
| 50 | 1 | Fu | (CH2)2C6H5 | (CH2)2CONHoleyl | 737.9 | 132.0 | 1807.1 | 61.1 | 5.5 |
| AAZ | – | – | – | – | 250 | 12 | 74 | 25 | 5.7 |
aMean from three different assays, by a stopped-flow technique (errors were in the range of ±5 − 10% of the reported values).
bData reported in Bonardi et al. [36].
While the structure–activity relationship (SAR) against CA I, II, and IV have already been discussed in depth36, only the inhibitory action of mono-tailed inhibitors and TTIs against the tumour-associated isoforms CA IX and XII was here investigated and compared.
Generally, the inhibition data reported in Table 1 highlighted that mono-tailed compounds 1–7 were medium to high nanomolar inhibitors of CA I (KI = 68.4 − 458.1 nM), II (KI = 62.8 − 153.7 nM), IX (KI = 56.5 − 108.7 nM) and XII (KI = 55.4–113.2 nM), and weak inhibitors of CA IV with inhibition constant (KI) values in the low micromolar range (1.1 − 6.2 μM).
In detail, the tumour-associated isoforms CAs IX and XII were inhibited almost similarly by the single-tail compounds 1–7. Nonetheless, derivatives 3 (R1= 4-F-C6H4) and 5 (R1= Fu) stood out as the best inhibitors of CA IX (KI = 56.5 nM) and XII (KI = 55.4 nM), respectively, whereas the cyanoalkyl- and phenethyl-tailed compounds 6 and 7 exhibit KIs above 100 nM against both isoforms.
As observed by the data in Table 1, the development of 1–7 upon inclusion of two other tails to synthesise compounds 18–50 significantly impacted the inhibition profiles against the panel of CA isoforms, as well as the selectivity indexes against CA I and II were improved (Table S1, Supplementary Information). In fact, TTIs showed lightly decreased or markedly improved inhibition of CA IX (KIs = 1.2 − 295.5 nM) and XII (KIs = 0.6 − 302.5 nM). CA IV remained the less inhibited isozyme, though inhibition improvement of one or two orders of magnitude was testified for some compounds (KIs = 45.8 –> 10,000 nM). On the whole, no significant improvement of the ubiquitous CA I inhibition was detected with TTIs (KIs = 79.5 − 4210.4 nM). The off-target CA II showed the inhibition profiles most affected, both positively and negatively, upon inclusion of additional tails on the scaffold of 1–7 (KIs = 0.7 − 4562.9 nM).
To better discuss TTIs’ SAR from Table 1, compounds and related data were distinguished in five subsets: (i) 18–29 (with R1 = C6H5), (ii) 30–33, 44, 49 (with R2 = R3 = (CH2)5CH3), (iii) 34–39 (R2 = (CH2)2C6H5 and R3 = (CH2)2CN), (iv) 40–43 (R2 = (CH2)2C6H5 and R3 = (CH2)3NH2), and (v) 45–48 (R2 = (CH2)2C6H5 and R3 = (CH2)2COOH).
(i) Compounds 18, 21, and 24 resulted in the best CA IX inhibitors of the first subset (KI = 30.6, 48.5, and 31.1 nM, respectively) while 20 and 21 were the worst (KI = 234.3 and 271.2 nM). Instead, all derivatives potently inhibited the tumour-associated isoform CA XII with KI values below 100 nM, except for compounds 26 and 29 that were also the worst inhibitors among all the synthesised compounds against this isoform (KI = 280.0 and 302.5 nM), whereas 18 showed the best inhibitory profile of this series (KI = 43.9 nM). In this subset, compound 19 resulted in the fourth most selective CA XII inhibitor vs. CA I (CA I/CA XII = 51.0).
The importance of the linker length (n = 1, 2) is pointed out from the activity analysis of this first subset. In fact, the elongation of the chain between R1 and R2/R3 increased the activity against CA I, II, and IV which possess the smallest binding cavities, as a longer linker (n = 2) can shift the tails R2/R3 towards the rim of the active site removing the ligand-target steric encumbrance. On the other hand, the roomier active sites of CA IX and XII are able to host bulky substituents and the introduction of the linker n = 2, which drives out the tails R2/R3 from the active site, may decrease the activity weakening the ligand-target interactions.
(ii) Comparing the second subset (30–33, 44, 49 with R2 = R3 = (CH2)5CH3) compounds with the first subset R2/R3-analogues 22 and 28, it was highlighted that the introduction of CH2C6H5 and Fu in R1 increased the activity against both the tumour-associated isoforms, such as observed in compounds 30 (CA IX KI = 42.9 nM; CA XII KI = 62.5 nM) and 31 (CA IX KI = 47.1 nM; CA XII KI = 9.7 nM), while the presence of 2-Naph (32) and CH2CN (33) increase the activity only against CA XII (KI = 61.7 and 8.6 nM, respectively). Furthermore, the tail R1 = CH2CN reduction of compounds 33 into amine 44 or its hydrolysis into carboxylic acid 49 worsened or did not affect the activity against CA XII (KI = 32.5 and 9.9 nM, respectively) but increased the inhibition profile vs. CA IX (KI = 115.6 and 68.3 nM, respectively).
In particular, compounds 30, 31, and 49 were the best CA IX inhibitors of this second subset with KI values below 100 nM (42.9, 47.1, and 68.3 nM, respectively), while derivatives 32, 33, and 44 were high nanomolar inhibitors of this isozyme (295.5, 154.5, and 115.6 nM, respectively). Compounds 44, 30, and 32 resulted, respectively, in the third (CA II/CA IX = 25.3), fourth (CA II/CA IX = 18.2), and fifth (CA II/CA IX = 15.4) most selective derivatives against CA IX with respect to the ubiquitous CA II.
In the case of CA XII all compounds showed a good activity: compounds 31 (KI = 9.7 nM), 33 (KI = 8.6 nM), and 49 (KI = 9.9 nM) inhibited this isoform with KI in the low nanomolar range while 30, 32, and 44 acted as medium nanomolar inhibitors (KI = 32.5–62.5 nM). TTIs 44, 32, and 49 resulted, respectively, in the first (CA II/CA XII = 90.0), second (CA II/CA XII = 74.0), and third (CA II/CA XII = 59.2) most selective compounds against CA XII vs. the off-target CA II.
Generally, it was observed for this subset that the concomitant presence of R2 = R3 = (CH2)5CH3 with substituent 2-Naph in R1 worsened the activity by 19 times against CA II (KI = 4.5 μM) but only 2.5 times against CA IX (KI = 295.5 nM) with respect to the analogue 22 (R1 = C6H5), increasing the CA II/CA IX selective index.
(iii) The third subset (34–39) is characterised by the introduction of a hydrophobic tail R2 = (CH2)2C6H5, a polar one R3 = (CH2)2CN, and a variable pendant R1. Only compounds 37 and 38 inhibited CA IX with KI values in the high nanomolar range (151.3 and 201.5 nM, respectively), derivatives 34, 35, and 39 were medium nanomolar inhibitors (KI = 32.3, 95.1, and 19.3 nM, respectively), while 36 (R1 = 4-F-C6H5) resulted in the first and second most selective inhibitor vs. the off-targets CA I (CA I/CA IX = 225.5) and CA II (CA II/CA IX = 44.3), respectively, among all the synthesised compounds with KI = 3.0 nM.
The target CA XII was strongly inhibited by all compounds of the subset with compounds 35, 36, and 39 acting in a low nanomolar range (KI = 8.7, 9.8, and 8.9 nM, respectively), while 34, 37, and 38 were medium nanomolar inhibitors (KI = 75.5, 64.9, and 89.5 nM, respectively). In this case, derivative 36 resulted also in the third most selective inhibitor against CA XII (CA I/CA XII = 69.8).
The comparison of compounds 37 and 39 from subset iii with the second subset analogues 32 and 33 (R2 = R3 = (CH2)5CH3) pointed out that the substitution of R2 and R3 with the tails (CH2)2C6H5 and (CH2)2CN, respectively, generally increased the activity against CA II, IV, and IX, with opposite effect against CA I and no significant effect against CA XII.
(iv) The fourth series (40–43) was obtained by reducing R3 = (CH2)2CN to amine tails in the aforesaid derivatives 34–37, introducing a potentially positively charged pendant. This structural modification led to a general increment of the activity against CA I, II, IV, IX, and XII, suggesting that a strong polar interaction is favourable for the binding and might take place in all five active sites.
In detail, CA IX was potently inhibited in the low nanomolar range (KI = 4.0–34.3 nM) and inhibitors 42 (KI = 4.8 nM) and 40 (KI = 4.0 nM) resulted in the second and fourth most selective among all the synthesised compounds against this isozyme with respect to CA I (CA I/CA IX = 94.0 and 60.6, respectively), while derivatives 41 and 43 were medium nanomolar inhibitors with KI of 11.1 and 34.3 nM, respectively. Again, derivative 40 is the most selective inhibitor against CA IX vs. the off-target CA II (CA II/CA IX = 91.8).
The tumour-associated CA XII was strongly inhibited by 42 with a subnanomolar KI = 0.6 nM that makes it the most potent and selective compounds against this isoform (CA I/CA XII = 752.3; CA II/CA XII = 50.7), whereas 40 (KI = 83.7 nM), 41 (KI = 42.7 nM), and 43 (KI = 10.5 nM) acted with a KI in the medium nanomolar range.
(v) The fifth subset (45–48) obtained by the introduction of a potentially negatively charged tail in R3 showed a general increment of the inhibition activity against CA I, II, IV, IX, and XII compared to their analogues 34, 35, 37, and 38. Against CA IX compound 45 and 46 acted in the low nanomolar range (KI = 7.5 and 1.2 nM, respectively), where the last one resulted to be the most potent and the third most selective inhibitor vs. CA I (CA I/CA IX = 66), while 47 (KI = 11.1 nM) and 48 (KI = 22.1 nM) inhibited this isoform with KI in the medium nanomolar range.
Moreover, derivatives 46–48 were low nanomolar inhibitors of CA XII (KI = 7.1, 8.8, and 6.8 nM, respectively), whereas 48 and 46 resulted to be the second and third most potent inhibitors of this glaucoma-associated isoform, while compound 45 acted with a KI of 29.7 nM.
Comparing the fourth (40–43) and the fifth subset (45–48) it was detected that the presence of R3 = (CH2)2COOH in place of amine tails shifted the activity against CA I.
Finally, the loss of the hydrophilic tail R3 in 50 decreased the activity against CA I (KI = 737.9 nM), II (KI = 132.0 nM), IV (KI = 1.8 μM), and IX (KI = 61.1 nM) without effects against CA XII (KI = 5.5 nM), obtaining the second most potent and selective compound against this isoform (CA I/CA XII = 134.2).
3.2. X-ray crystallography
Co-crystallisation of hCA II36 and hCA IX-mimic with some of the new inhibitors resulted in solved crystal structures with resolutions between 1.39 and 1.56 Å (Figure 2 and Table S2, Supplementary Information). All inhibitors contain a conserved benzenesulfonamide in the same orientation that acts as a zinc-binding group that displaces zinc bound water (ZBW) to form a hydrogen bond between the amide backbone of Thr199 and oxygen of sulphonamide (2.9–3.0 Å). With these similarities in the inhibitors, any difference in observed binding affinity results from modifications to the tail regions.
Figure 2.

Cartoon representation of A) hCA II36 and B) hCA IX-mimic overlayed on a surface view with zinc (magenta sphere) and His94, His96, and His119 (sticks) shown in the active site with inhibitors bound. 34 (purple), 41 (yellow), 42 (cyan), 46 (green), and 48 (orange) are shown in both panels.
In complex with hCA IX-mimic, compound 41 showed an observed omit map electron density lacking the whole entirety of T2 and T3 tails (PDB: 7SUW, Figure 3(A)). The T1 furyl moiety accommodated within the lipophilic pocket constituted by residues Val135, Leu198, Pro202, and Ala204, as it did within the hCA II active site (Figure 4(B)).
Figure 3.

Electron density of A) 41, B) 42, C) 46, and D) 48 in hCA IX-mimic active site with a sigma of 1.0.
Figure 4.

X-ray crystallography: active site view of hCA IX-mimic in adduct with A) no inhibitor, B) 41, C) 42, D) 46, and E) 48. H-bonds are depicted as black dashed lines. Water molecules involved in water-bridged H-bonds are shown as red spheres. Amino acids are labelled with one letter symbols: D, Asp; E, Glu; F, Phe; H, His; I, Ile; L, Leu; N, Asn; P, Pro; Q, Gln; T, Thr; V, Val; W, Trp.
Compound 42 reported a strong observed omit map electron density, which may indicate stronger binding and/or a high binding occupancy (PDB: 7SUY, Figure 3(B)). Again, the swap from a furyl (41) to a 4-F-benzyl (42) in T1 induced the halo aromatic ring to lie over the lipophilic cleft made by Val131, Val135, Pro202, and Ala204. In contrast, the phenethyl portion in T2 interacts with the opposite side of the hCA IX-mimic hydrophobic half, namely with Trp5, Phe20, and Pro201 (Figure 4(C)). The protonated aminopropyl tail was exposed to bulk solvent.
Compound 46 had a weak observed omit map electron density around the benzene ring and carboxylic acid tail (PDB: 7SV8, Figure 3(C)). The furylmethyl tail flipped almost 180° with respect to that of derivative 41, being in hydrophobic contact with Pro201 and Pro202. The phenethyl tail exhibited lipophilic interactions with Gly132 and Val135. The COO- in T3 was involved in a water-mediated H-bond network with Gln92 that included the amide carbonyl group, while the T3 alkyl tail showed interactions with Leu91 (Figure 4(D)).
Compound 48 showed a weak observed omit map electron density around the benzene ring in T2 (PDB: 7SV1, Figure 3(D)). Expectedly, not being able to occupy the lipophilic pocket nearby Leu198 for steric hindrance reasons, the naphthyl ring in T1 lied over the region lined by Trp5, Phe20, Pro201, and Pro202. The benzene ring in T3 was interestingly found to interact with the outer portion of the α-helix including residues 130–136 and is partially exposed to bulk solvent, whereas the carboxyethyl tail folded back towards the cavity, making an intramolecular water bridge with the protonated amine group branching T2 and T3. However, it held water-bridged H-bonds to Gln92 together with the amide carbonyl group (Figure 4(E)).
Again, the binding mode exhibited by 46 was related to the best hCA IX inhibition measured in vitro (KI of 1.2 nM), though a minor difference was detected among the co-crystallised ligands with respect to hCA II. Likewise, the binding mode of 42, that mostly deviated from that of the other ligands and also within hCA IX-mimic, led to an efficient hCA IX inhibition of 4.8 nM. The 20-fold drop of efficacy passing from 46 to 48 (KI from 1.2 to 22.1 nM) might be related again to the furyl/napthyl switch that provoked a significant loss of favourable contacts within the active site.
Figure 5 depicts the superimposed binding orientations of compounds 41, 42, 46, and 48 within the active site of hCA II36 and hCA IX-mimic. Although partially missing electron density was observed for compound 41 in the hCA IX-mimic active site, a similar orientation to the ligand binding mode in hCA II was observed for T1 and for the linker up to the T2/T3 branching junction. Hence, very similar ligand orientations exist for inhibitors 41 (Figure 5(A)) and 42 (Figure 5(B)) bound to hCA II and hCA IX-mimic. In contrary, greater differences were detected when ligands 46 and 48 bound to the active site of the two isoforms. Nonetheless, it should be stressed that all four compounds showed a significantly greater efficacy as inhibitors of hCA IX than hCA II, with selectivity that spans between 2- and 6-fold. In fact, hCA II had an average KI of 41 nM (for compounds 41, 42, 46, and 48), whereas hCA IX had an average KI of 10 nM for these same inhibitors. In this context, Val131 in hCA IX compared to Phe131 in hCA II is the major difference in active sites between the two isoforms. Inhibitors more easily enter the active site in hCA IX due to the smaller amino acid at residue 131. What is more, Phe131 of hCA II can produce less favourable positioning and conformational geometry of the inhibitors with respect to hCA IX-mimic.
Figure 5.

Superimposition of the crystallographic binding orientations adopted in the active site of hCA II (grey) [36] and hCA IX-mimic (light blue) for A) 41, B) 42, C) 46, and D) 48. Colours are as in Figure 6 of Bonardi et al. [36] and Figure 4.
Comparing compound 46 between hCA II and hCA IX-mimic shows that in the latter, the amide linker shifted towards the hydrophobic region most likely as a result of less steric hindrance from Val131. As a result, the two cyclic tails rotate in towards the active site, preventing clashes with the enzymes surface residues (Figure 5(C)). As for compound 48, there was also less steric hindrance from Val131 which allowed the phenyl tails to rotate about the linker (Figure 5(D)). The clearer case was that of compound 42; although a very similar orientation was observed when bound within the two active sites, the inhibitor tails slightly moved towards the hydrophobic half of hCA IX-mimic accordingly to the less steric hindrance from Val131 (Figure 5(B)) just enough to improve the binding and thus inhibition efficacy.
3.3. Antiproliferative studies
Hypoxic tumours are a heterogeneous mass of cells with different degrees of oxygen supply1,6–8,54. The cells in the internal part of the mass grow under hypoxic conditions while the external ones have a more physiological supply of oxygen. In normoxic cells (where CA IX and XII are normally expressed on the membranes), CAIs could predominantly act on the cytosolic isoforms, blocking the ability of cells to maintain the intracellular and extracellular pH values compliant for their survival. Instead, in the hypoxic ones, CAIs also inhibit the tumour-membrane-associated isoforms of CA IX and XII that result overexpressed in these conditions. Obviously, achieving selective inhibition of CA IX and XII, expressed in both cell types, is preferable to avoid the side effects related to the inhibition of the off-target isoform also present in the healthy cells with CAIs.
To evaluate in vitro the effects on the viability of colon (HT29), prostate adenocarcinoma (PC3) and breast cancer (ZR75-1) cell lines, TTIs 28, 34, 36, and 50 were selected among all the synthesised derivatives for their selectivity against CA IX and XII and also considering the nature of the tails. Cells were incubated for 48 h with three different concentration of inhibitors (10, 30, and 100 μM) in normoxic (21% O2) and hypoxic (3% O2) conditions.
All compounds act in a dose-dependent manner similarly in normoxia and hypoxia against HT29 and PC3 cancer cell lines, resulting in a possession of a slightly most potent effect in hypoxic conditions vs. the cell line ZR75-1. Moreover, inhibitors 28 and 36 were the most active against all the used cell lines while derivative 50 reduced the ZR75-1 cell viability.
In detail, inhibitor 28 was effective at the lowest concentration (10 µM) only against HT-29 cells, decreasing viability by about 55% in normoxia and 40% in hypoxia. The other concentrations (30–100 µM) further reduced viability by more than 90% in HT-29 and PC3 cell lines and were slightly less effective in ZR75-1 where the reduction was around 80%.
Inhibitors 34, 36, and 50, on the other hand, have no effect at a concentration of 10 µM, except for compound 36 on PC3 cells in normoxia and on ZR75-1 cells in both conditions (15% of decrease). Interestingly, compound 50 at 10 µM on ZR75-1 was effective in hypoxic condition compared to normoxia, inducing a 15% of reduction (Figure 6).
Figure 6.

In vitro cell viability assay of colon adenocarcinoma (HT-29), prostate adenocarcinoma (PC3), and breast cancer (ZR75-1) cell lines after 48 h of treatment with three different concentrations (10, 30, and 100 µM) of three-tailed inhibitors 28 (orange), 34 (yellow), 36 (blue), and 50 (green) in normoxic (21% O2) and hypoxic (3% O2) conditions. Control cells are arbitrarly set at 100% and results are expressed as the mean ± SEM of three experiments. One-way ANOVA was performed followed by a Bonferroni’s significant difference procedure. *p < .05, **p < .01, and ***p < .001 vs. control; ∧p < .05 and ∧∧p < .01 vs. normoxia.
Inhibitor 34 at a concentration of 30 µM affected the viability under normoxic condition up to 40% in HT-29 cells and 15–20% in PC3 and ZR75-1 cells. In hypoxic condition, it was less effective on HT-29 and PC3 cell lines, while ZR75-1 cell viability decreased by 30% compared to control and by10–15% respect to normoxic condition. Even the concentration 100 µM of inhibitor 34 was able to induce cell death in a similar percentage for the three cell lines (50–60%) under normoxic condition, and in hypoxia ZR75-1 cells were confirmed as the most sensitive compared to the other cell lines.
Derivative 36 was more effective against PC3 and ZR75-1 at 30 µM and 100 µM in both conditions, with a reduction of cell viability by around 80–90%, except for the 30 µM in hypoxia on PC3 cells where the viability decreased only by 50%. On HT-29 cells inhibitor 36 induced a 40% and 90% reduction at 30 and 100 µM, respectively, in both conditions.
Finally, inhibitor 50 had no effect at 30 µM on HT-29 cells, and at 100 µM a 40% reduction was observed in both conditions; on PC3 cells under hypoxic condition the concentration 30 µM of derivative 50 induced a reduction by 30%, while at 100 µM a more effective action under hypoxic condition was determined (almost 50% compared to normoxia); also ZR75-1 cells were sensitive to inhibitor 50, which was active at 30 µM (respectively, 20% and 30% of reduction in normoxia and hypoxia) and at 100 µM was able to induce 65% of death in normoxia and a significant further decrease in hypoxia (5–10% less)55–58.
4. Conclusion
In recent decades the human (h) CAs (EC 4.2.1.1) isoforms IX and XII were validated as anticancer targets against solid hypoxic tumours. The lack of selectivity of sulphonamide CAIs prevents their wider use as therapeutic agents owing to the side effects onset, mainly due to the inhibition of the ubiquitous human (h) CA I and II. To overcome this issue the “three-tails approach” is here proposed as an extension of the forerunner “tail” and “dual-tail approach” to fully exploit the amino acid differences at the medium/outer active sites rim among the different hCA active sites. The majority of the thirty-three synthesised TTIs resulted in a higher selectivity against the tumour-associated isoforms hCA IX and XII with respect to the off-targets hCA I and II than the mono-tailed compounds (CA I/CA IX = 1.8–225.5; CA II/CA IX = 1.3–91.8; CA I/CA XII = 2.3–752.3; CA II/CA XII = 1.3–90.0). X-ray crystallography studies were performed to investigate the binding mode of four TTIs (41, 42, 46, and 48) in complex with hCA IX mimic. Moreover, the ability of the most potent and selective TTIs (28, 34, 36, and 50) to reduce in vitro the viability of colon (HT-29), prostate adenocarcinoma (PC3), and breast cancer (ZR75-1) cell lines was evaluated in normoxic (21% O2) and hypoxic (3% O2) conditions. In particular, all tested compounds act in a concentration-dependent manner similarly in normoxia and hypoxia against HT-29 and PC3 cancer cell lines, and with a slightly most potent effect in hypoxic conditions against ZR75-1 cell line. Moreover, inhibitors 28 and 36 resulted in the most active derivatives against all the cell lines used while derivative 50 was able to strongly affect PC3 and ZR75-1 cell viability under hypoxic condition compared to normoxia.
Supplementary Material
Funding Statement
This project was funded by the Researchers Supporting Project number [RSP-2021/405], King Saud University, Riyadh, Saudi Arabia (SMO) and by the Italian Ministry for University and Research (Ministero dell'Istruzione, dell'Università e della Ricerca [MIUR]), grant PRIN: prot. 2017XYBP2R (CTS).
Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this article. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Neri D, Supuran CT.. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. [DOI] [PubMed] [Google Scholar]
- 2.Thiry A, Dogné JM, Masereel B, et al. Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol Sci 2006;27:566–73. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev 2007;26:223–4. [DOI] [PubMed] [Google Scholar]
- 4.Brahimi-Horn MC, Pouyssegur J.. Oxygen, a source of life and stress. FEBS Lett 2007;581:3582–91. [DOI] [PubMed] [Google Scholar]
- 5.Hon WC, Wilson MI, Harlos K, et al. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002;417:975–8. [DOI] [PubMed] [Google Scholar]
- 6.Vaupel P, Multhoff G.. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol 2021;599:1745–57. [DOI] [PubMed] [Google Scholar]
- 7.Vaupel P, Schmidberger H, Mayer A.. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol 2019;95:912–9. [DOI] [PubMed] [Google Scholar]
- 8.Riemann A, Schneider B, Gündel D, et al. Acidosis promotes metastasis formation by enhancing tumor cell motility. Adv Exp Med Biol 2016;876:215–20. [DOI] [PubMed] [Google Scholar]
- 9.Supuran CT. Carbonic anhydrase inhibitors: an update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs 2021;30:1197–208. [DOI] [PubMed] [Google Scholar]
- 10.Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whittington DA, Waheed A, Ulmasov B, et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA 2001;98:9545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chafe SC, Vizeacoumar FS, Venkateswaran G, et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci Adv 2021;7:eabj0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Hypoxia-activated tumor pathways of angiogenesis and pH regulation independent of anemia in head-and-neck cancer. Int J Radiat Oncol Biol Phys 2004;59:67–71. [DOI] [PubMed] [Google Scholar]
- 14.Angeli A, Carta F, Nocentini A, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites 2020;10:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potter CP, Harris AL.. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br J Cancer 2003;89:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald PC, Chafe SC, Brown WS, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology 2019;157:823–37. [DOI] [PubMed] [Google Scholar]
- 17.Swinson DE, Jones JL, Richardson D, et al. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J Clin Oncol 2003;21:473–82. [DOI] [PubMed] [Google Scholar]
- 18.Swayampakula M, McDonald PC, Vallejo M, et al. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 2017;36:6244–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchison GJ, Valentine HR, Loncaster JA, et al. West hypoxia-inducible factor 1alpha expression as an intrinsic marker of hypoxia: correlation with tumor oxygen, pimonidazole measurements, and outcome in locally advanced carcinoma of the cervix. Clin Cancer Res 2004;10:8405–12. [DOI] [PubMed] [Google Scholar]
- 21.Dorai T, Sawczuk I, Pastorek J, et al. Role of carbonic anhydrases in the progression of renal cell carcinoma subtypes: proposal of a unified hypothesis. Cancer Invest 2006;24:754–79. [DOI] [PubMed] [Google Scholar]
- 22.Cazzamalli S, Ziffels B, Widmayer F, et al. D. Enhanced therapeutic activity of non-internalizing small-molecule-drug conjugates targeting carbonic anhydrase IX in combination with targeted interleukin-2. Clin Cancer Res 2018;24:3656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Testa C, Papini AM, Zeidler R, et al. First studies on tumor associated carbonic anhydrases IX and XII monoclonal antibodies conjugated to small molecule inhibitors. J Enzyme Inhib Med Chem 2022;37:592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maxwell PH, Wiesener MS, Chang CW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271–5. [DOI] [PubMed] [Google Scholar]
- 25.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292:468–72. [DOI] [PubMed] [Google Scholar]
- 26.Tong CK, Brion LP, Suarez C, et al. Interstitial carbonic anhydrase (CA) activity in brain is attributable to membrane-bound CA type IV. J Neurosci 2000;20:8247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svichar N, Chesler M.. Surface carbonic anhydrase activity on astrocytes and neurons facilitates lactate transport. Glia 2003;41:415–9. [DOI] [PubMed] [Google Scholar]
- 28.Svichar N, Esquenazi S, Waheed A, et al. Functional demonstration of surface carbonic anhydrase IV activity on rat astrocytes. Glia 2006;53:241–7. [DOI] [PubMed] [Google Scholar]
- 29.Stridh MH, Alt MD, Wittmann S, et al. Lactate flux in astrocytes is enhanced by a non-catalytic action of carbonic anhydrase II. J Physiol 2012;590:2333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forero-Quintero LS, Ames S, Schneider H-P, et al. Membrane-anchored carbonic anhydrase IV interacts with monocarboxylate transporters via their chaperones CD147 and GP70. J Biol Chem 2019;294:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 2011;12:238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pacchiano F, Carta F, McDonald PC, et al. Supuran. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902. [DOI] [PubMed] [Google Scholar]
- 33.Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3. [DOI] [PubMed] [Google Scholar]
- 34.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. [DOI] [PubMed] [Google Scholar]
- 35.Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. [DOI] [PubMed] [Google Scholar]
- 36.Bonardi A, Nocentini A, Bua S, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem 2020;63:7422–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring. J Med Chem 1999;42:2641–50. [DOI] [PubMed] [Google Scholar]
- 38.Tanpure RP, Ren B, Peat TS, et al. Carbonic anhydrase inhibitors with dual-tail moieties to match the hydrophobic and hydrophilic halves of the carbonic anhydrase active site. J Med Chem 2015;58:1494–501. [DOI] [PubMed] [Google Scholar]
- 39.Fares M, Eldehna WM, Bua S, et al. Discovery of potent dual-tailed benzenesulfonamide inhibitors of human carbonic anhydrases implicated in glaucoma and in vivo profiling of their intraocular pressure-lowering action. J Med Chem 2020;63:3317–26. [DOI] [PubMed] [Google Scholar]
- 40.Wilkinson BL, Bornaghi LF, Houston TA, et al. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing”. J Med Chem 2006;49:6539–48. [DOI] [PubMed] [Google Scholar]
- 41.Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73. [PubMed] [Google Scholar]
- 42.Petreni A, Bonardi A, Lomelino C, et al. Inclusion of a 5-fluorouracil moiety in nitrogenous bases derivatives as human carbonic anhydrase IX and XII inhibitors produced a targeted action against MDA-MB-231 and T47D breast cancer cells. Eur J Med Chem 2020;190:112112. [DOI] [PubMed] [Google Scholar]
- 43.a) Scozzafava A, Briganti F, Mincione G, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, aminoacyl/dipeptidyl sulfonamides possessing long-lasting intraocular pressure-lowering properties via the topical route. J Med Chem 2021;42:3669–700; b) Berrino E, Michelet B, Martin-Mingot A, et al. Modulating the efficacy of carbonic anhydrase inhibitors through fluorine substitution. Angew Chem Int Ed Engl 60: 23068–82. [DOI] [PubMed] [Google Scholar]
- 44.Pinard MA, Boone CD, Rife BD, et al. Structural study of interaction between brinzolamide and dorzolamide inhibition of human carbonic anhydrases. Bioorg Med Chem 2013;21:7210–5. [DOI] [PubMed] [Google Scholar]
- 45.Tanhauser SM, Jewell DA, Tu CK, et al. A T7 expression vector optimized for site-directed mutagenesis using oligodeoxyribonucleotide cassettes. Gene 1992;117:113–7. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 2010;66:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans PR, Murshudov GN.. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 2013;69:1204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011;67:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Avvaru BS, Kim CU, Sippel KH, et al. A short, strong hydrogen bond in the active site of human carbonic anhydrase II. Biochemistry 2010;49:249–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004;D60:2126–32. [DOI] [PubMed] [Google Scholar]
- 51.Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010;66:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laskowski RA, Swindells MB.. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011;51:2778–86. [DOI] [PubMed] [Google Scholar]
- 53.a) Nocentini A, Supuran CT.. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discovery 14:1175–97; b) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci (Lond) 2021;135:1233–49. [Google Scholar]
- 54.Terry S, Buart S, Chouaib S.. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front Immunol 2017;8:1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836. [DOI] [PubMed] [Google Scholar]
- 56.Bua S, Lucarini L, Micheli L, et al. Bioisosteric development of multitarget nonsteroidal anti-inflammatory drug-carbonic anhydrases inhibitor hybrids for the management of rheumatoid arthritis. J Med Chem 2020;63:2325–42. [DOI] [PubMed] [Google Scholar]
- 57.a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. [DOI] [PubMed] [Google Scholar]; b) Nocentini A, Angeli A, Carta F, et al. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem 2021;36:561–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bua S, Supuran CT.. Diagnostic markers for glaucoma: a patent and literature review (2013-2019). Expert Opin Ther Pat 2019;29:829–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.