

Abstract
Duchenne muscular dystrophy involves an absence of dystrophin, a cytoskeletal protein which supports cell structural integrity and scaffolding for signalling molecules in myocytes. Affected individuals experience progressive muscle degeneration that leads to irreversible loss of ambulation and respiratory diaphragm function. Although clinical management has greatly advanced, heart failure due to myocardial cell loss and fibrosis remains the major cause of death. We examined cardiac morphology and function in D2.B10-Dmdmdx/J (D2-mdx) mice, a relatively new mouse model of muscular dystrophy, which we compared to their wild-type background DBA/2J mice (DBA/2). We also tested whether drug treatment with a specific blocker of mitochondrial permeability transition pore opening (Debio-025), or ACE inhibition (Perindopril), had any effect on dystrophy-related cardiomyopathy. D2-mdx mice were treated for six weeks with Vehicle control, Debio-025 (20 mg/kg/day), Perindopril (2 mg/kg/day), or a combination (n = 8/group). At 18 weeks, compared to DBA/2, D2-mdx hearts displayed greater ventricular collagen, lower cell density, greater cell diameter, and greater protein expression levels of IL-6, TLR4, BAX/Bcl2, caspase-3, PGC-1α, and notably monoamine oxidases A and B. Remarkably, these adaptations in D2-mdx mice were associated with preserved resting left ventricular function similar to DBA/2 mice. Compared to vehicle, although Perindopril partly attenuated the increase in heart weight and collagen at 18 weeks, the drug treatments had no marked impact on dystrophic cardiomyopathy.
1. Introduction
Duchenne muscular dystrophy (DMD) is an X chromosome-linked muscle-wasting disease that affects approximately 1 in 3600-6000 boys [1, 2]. A mutation in the dystrophin gene prevents synthesis of functional dystrophin protein resulting in mechanical and molecular signalling-related cell injury and mitochondria-related cell death in skeletal and cardiac muscle [3–8]. Gene and cell-based therapies for directly restoring dystrophin are clinically unavailable due to current technological limitations. Notably, clinical management with corticosteroids, manually assisted coughing devices, and mechanical ventilation has greatly extended patient survival to adulthood [9, 10]. Cardiomyopathic heart failure now prevails for these patients, with most presenting cardiac-related complications by the age of 20 [11–13]. The current standard of care recommended by the DMD Care Considerations Working Group to delay the onset of cardiomyopathy includes the use of angiotensin-converting enzyme (ACE) inhibitors (i.e., Perindopril) by 10 years of age [2]. Early ACE inhibitor therapy has been shown to preserve cardiac function in patients with DMD and “mdx” mouse models [14, 15].
Myocardial fibrosis is one of the earliest clinically detectable cardiac features of DMD that contributes to the maladaptive remodelling [16]. Cardiomyocytes demonstrate membrane instability and dysregulated Ca2+ handling which augments Ca2+ influx, leading to the formation of fibrosis and cellular necrosis [17, 18]. Aside from increasing the stiffness of the myocardium and impeding its ability to contract and relax, myocardial fibrosis promotes compensatory hypertrophy of neighbouring cardiomyocytes that disrupts the electrical conduction and coupling system in the heart, thus increasing the risk of serious arrhythmias [19–21]. As the onset of fibrosis begins early in DMD, slowing the progression of myocardial fibrosis is an important therapeutic target to delay the onset of cardiomyopathy.
Mitochondrial dysfunction is another potential therapeutic target in DMD. Diminished mitochondrial energy production, Ca2+ dysregulation, oxidative stress, and mitochondrial-mediated cell death underlie muscle degeneration associated with DMD cardiomyopathy [6, 7, 17, 22–24]. Impairment of mitochondrial respiration and prolonged increases in cytosolic Ca2+ contribute to opening of the mitochondrial permeability transition pore (mPTP) and subsequent mitochondrial-dependent cell death signalling [25]. The mPTP is a large, voltage-dependent channel in the inner mitochondrial membrane that opens with inner membrane depolarization [ 26, 27]. Prolonged, irreversible openings of the mPTP have been linked to ischemic cell death associated with stroke, myocardial infarction, and cardiomyopathy associated with DMD [28]. An overload of Ca2+ in the mitochondrial matrix and excessive production of ROS contribute to the persistent opening of the mPTP, inner mitochondrial membrane depolarization and loss of oxidative phosphorylation-dependent ATP production [26, 29–32], mitochondrial swelling, outer mitochondrial membrane rupture, oxidation of cardiolipin, release of cytochrome c, and proapoptotic protein activity [30, 33, 34]. Loss of mitochondria beyond a critical threshold ultimately causes cell death and exacerbation of myocardial complications in DMD [35]. Thus, a drug that prevents irreversible opening of mPTP may delay myocyte death in DMD. Debio-025 is a prolyl isomerase inhibitor that inhibits the mPTP-sensitizing protein, cyclophilin D, to prevent mitochondrial swelling and apoptosis [36]. Preliminary studies conducted with mouse models of DMD have shown that Debio-025 protects skeletal and diaphragm muscles from cell death, improves myofibril organisation, reduces mitochondrial swelling and fibrosis in skeletal muscle, and improves overall muscle function [24, 36, 37]. It has also been shown to improve skeletal muscle fibre attachment to the myotendinous junction, normalise the organisation and ultrastructure of cristae, and prevent mPTP opening in isolated mitochondria from DMD patients [38]. However, the effect of Debio-025 on heart function has not been examined in patients with DMD.
Mice featuring a mutation in the dystrophin gene (i.e., “mdx”) have been crossed with different genetic backgrounds to create a disease model that mimics the human condition. Complicating model development, the genetic background of wild-type mice has been shown to significantly affect the phenotype of a given single gene mutation due to the presence of modifier genes [11, 39]. Mounting evidence has shown that, compared to the most commonly used C57BL/10-Dmdmdx (BL10-mdx) on a C57BL/10ScSn background, the recently described D2.B10-Dmdmdx/J (D2-mdx) on a DBA2/J background displays a more pronounced dystrophic phenotype that is characterized by muscular atrophy, increased inflammation and fibrosis, and reduced skeletal muscle function as early as seven weeks of age [3, 40–43]. Using the γ-sarcoglycan- (Sgcg-) null mouse model of muscular dystrophy and cardiomyopathy on a DBA2/J background, a polymorphism in the coding region of the latent TGF-β-binding protein 4 gene (Ltbp4) resulting in a 12-amino-acid deletion in LTBP4 protein was found to be responsible for increased proteolysis, fibrosis, and atrophy in these mice [44]. As LTBP4 usually sequesters TGF-β to limiting signalling, importantly, polymorphism of the human Ltbp4 locus has been shown to be a genetic modifier of disease severity in DMD patients [45, 46]. D2-mdx mice have also been shown to have a dysfunctional annexin A6 (anxa6) gene which is responsible for decreased self-renewal capacity of satellite cells [47]. D2-mdx mice also carry the dystrophic calcinosis 1 (dyscalc1) gene locus which contributes susceptibility to calcinosis in the heart and skeletal muscles [48]. Recently, study of the D2-mdx muscle phenotype has confirmed early presence of impaired mitochondrial function in the heart [25]; however, whether this has early impact on in vivo cardiac function that can be targeted by therapeutic intervention requires investigation. Thus, the aims of the current study were to characterize the extent of cardiac pathology and dysfunction in D2-mdx mice, compared to wild-type DBA/2J background strain, and to test the effects of ACE inhibition with Perindopril and/or mPTP blockade with Debio-025.
2. Materials and Methods
2.1. Animal Handling and Care
The study was approved by the Murdoch Children's Research Institute (MCRI) Animal Ethics Committee (A859) and was performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (8th Edition, 2013). DBA/2J (referred to as DBA/2) male mice (n = 4) were purchased from the Animal Resources Centre (Western Australia, Australia), and D2.B10-Dmdmdx/J (D2-mdx) male mice (JAX stock #013141; n = 32) were purchased from Jackson Laboratory (Bar Harbor, ME). D2-mdx mice were bred from C57BL/10-mdx mice that carry a spontaneous mutation resulting in a stop codon at position 3185 within exon 23 of the dystrophin gene on the X chromosome and were backcrossed with DBA/2J inbred mice [3, 40, 49]. Mice were housed at a density of up to five males in individually ventilated cages (Techniplast Blue Line) maintained at 65-75 air changes per hour. Rooms were maintained at 18-24°C with dawn/dusk timed lighting. Mice had access to irradiated rodent diet (18% protein, Teklad) and water ad libitum. Animals were monitored and weighed daily.
2.2. Drug Treatment
D2-mdx mice were randomised into four treatment groups (n = 8/group) as follows: (1) Vehicle control (phosphate-buffered saline (PBS)+Debio-025 carrier), (2) Perindopril (2 mg/kg/day; P0094, Sigma-Aldrich, St. Louis, MO, USA), (3) Debio-025 (20 mg/kg/day; Alisporivir; Debiopharm Research & Manufacturing SA, Martigny, Switzerland), and (4) Perindopril (2 mg/kg/day)+Debio-025 (20 mg/kg/day). Treatments were delivered via oral gavage as a volume of 0.1 mL. Treatment began at 12 weeks of age for six weeks.
2.3. Echocardiography
Ultrasound imaging at 10 and 18 weeks (before and after treatment) was performed with a Vevo 3100 Ultrasound Machine (FUJIFILM VisualSonics, Canada) to obtain LV functional and structural measurements. Mice were anaesthetized with 4% isoflurane and maintained with 2% isoflurane/oxygen flow. Hair was removed from the chest with depilatory hair removal cream to facilitate ultrasound transmission gel (Aquasonic100, Parker Laboratories Inc., Fairfield, NJ) and ultrasound probe placement. Heart rate and rectal body temperature were monitored throughout the procedure. Two-dimensional brightness mode (B-mode) and motion mode (M-mode) images were acquired from a parasternal long axis and parasternal short axis LV view and used to obtain LV end-diastolic (LVED) dimension, LV end-systolic (LVES) dimension, interventricular septum (IVS) diameter during systole and diastole, and posterior wall thickness (PWT) during systole and diastole. LV mass, LV fractional shortening (FS), and LV ejection fraction (EF) (measures of systolic function) were calculated based on LV chamber diameter and volume measurements using VevoLAB Version 3.1.0 software package (FS = ((LVED diameter − LVES diameter)/LVED diameter) × 100; EF = ((LVED volume − LVES volume)/LVED volume) × 100).
2.4. Forelimb Grip Strength Test
As the skeletal muscle phenotype of this mouse model has been previously characterized and the current study focus was the heart, a single skeletal muscle function test was performed. Forelimb grip strength was measured in vivo at 10 and 18 weeks (before and after treatment) for DBA/2 and D2-mdx mice, using a grip strength meter (Columbus Instruments, Columbus, OH, USA) according to TREAT-NMD SOP DMD_M.2.2.001 protocol. Using a bar attached to an isometric force transducer, forelimb grip strength was measured as the amount of horizontal force produced to break the grip of a mouse, while suspended by its tail [50]. With a minute of rest between each pull, five consecutive measurements were recorded for each mouse and then averaged and normalised to body weight.
2.5. Tissue Collection
At 18 weeks of age, mice were anaesthetized with 4% isoflurane/oxygen flow. Once unconscious and assessments were complete, a cardiac puncture was performed to collect blood directly from the heart. Mice were then killed by cervical dislocation and dissected to obtain the heart, diaphragm, and tibia. All tissues were individually weighed, snap frozen in liquid N2, and stored at -80°C until required. Diaphragms and transverse heart slices were also fixed in 10% buffered formalin, then embedded in paraffin wax, sectioned at 3 μm, and stained using Masson's trichrome to detect collagen as a measure of fibrosis [51].
Whole heart transverse sections were digitally imaged using a Mirax Digital Slide Scanner (3DHISTECH Ltd., Budapest, Hungary). Whole diaphragms were imaged using a Leica DM 1000 Light Microscope (Leica Microsystems, Wetzlar, Germany). Quantification of fibrosis was performed using ImagePro (Version 9.2). The percentage of fibrotic tissue was determined by dividing the total of the blue collagen areas by the total area. For each mouse, the RV, LV, and septum were also imaged (Leica DM1000 Light Microscope, Wetzlar, Germany) to determine cell density (cell count/area) and cell diameter. For each heart region, a total of 5 areas of size 150 × 130 μm (that excluded regions of dystrophic calcinosis) were assessed for a minimum of 100 cells using ImageJ (https://imagej.nih.gov/ij/). Cell diameter measurements were performed using Feret's diameter in ImageJ.
2.6. Electron Microscopy
Electron microscopy was conducted on LV heart sections of D2-mdx mice from each treatment group to visualise differences in cellular and intracellular structure and organisation, particularly focusing on mitochondria. Excised myocardial samples were cut into ~1 mm cubed blocks and fixed in PBS with 2.5% cacodylate-buffered glutaraldehyde, 2% paraformaldehyde, and 1% osmium tetroxide for 24 hours. Samples were then rinsed in fresh PBS, dehydrated in an alcohol series, and embedded in Epon 812. Sections were examined with a Zeiss Libra 120 PLUS Energy-Filtered Transmission Electron Microscope (Zeiss, Oberkochen, Germany).
2.7. Protein Analysis
Western immunoblotting was performed using heart tissue frozen at necropsy to determine expression of key proteins as per Table 1. Protein was extracted from 50 mg of frozen heart tissue powder per mouse following addition of modified radioimmunoprecipitation assay (RIPA) buffer (0.5 mL) containing 50 mM Tris-HCl at pH 7.4 (15567-027, Thermo Fisher), 1% Nonyl Phenoxylpolethoxylethanol (Tergitol; NP40S, Sigma-Aldrich), 150 mM sodium chloride (NaCl; 1.06404.5000, Merck-Millipore, Burlington, MA), and 500 mM ethylenediaminetetraacetic acid (EDTA; T9285, Sigma-Aldrich). One tablet of protease inhibitor cocktail (05892970001, Roche, Basel, Switzerland) and one tablet of phosphatase cocktail (04906837001, Roche) were added per 10 mL of RIPA buffer. The mixture was vortexed for 60 seconds and placed on ice for 45 minutes. Samples were further homogenised via TissueRuptor (Qiagen, Hilden, Germany) in two bursts of 30 seconds' duration. Samples were centrifuged at 2000 × g for 10 minutes at 4°C, and supernatants were collected and transferred into a fresh tube. A bicinchoninic acid (BCA) assay kit (B9643, Sigma-Aldrich) was used to determine total protein concentration as per manufacturer's protocol (A2153, Sigma-Aldrich). All samples were diluted 1 : 25 with nuclease free water (9937, Ambion, Austin, TX), and 10 μL aliquots were pipetted in triplicate into a 96-well spectrophotometric plate, with 80 μL of BCA. The plate was then incubated (23°C, 30 minutes), and then, optical density was determined at 560 nm with a Fluostar Optima spectrophotometric plate reader (BMG LabTech, Victoria, Australia).
Table 1.
Antibody details for western immunoblotting.
| Marker | Catalogue number | Company | Molecular weight (kDa) | Source | Dilution |
|---|---|---|---|---|---|
| Bax | 2772 | Cell Signalling, USA | 20 | Rabbit | 1/1000 |
| Bcl-2 | 2876 | Cell Signalling, USA | 28 | Rabbit | 1/1000 |
| Beclin-1 | 3495 | Cell Signalling, USA | 60 | Rabbit | 1/500 |
| Caspase-3 | 9665 | Cell Signalling, USA | 35 | Rabbit | 1/500 |
| GAPDH | 5174 | Cell Signalling, USA | 37 | Rabbit | 1/50,000 |
| LC3B | 3868 | Cell Signalling, USA | 16 | Rabbit | 1/1000 |
| MAO-A | ab126751 | Abcam, UK | 60 | Rabbit | 1/1000 |
| MAO-B | ab137778 | Abcam, UK | 60 | Rabbit | 1/1500 |
| NF-κB | 12540 | Cell Signalling, USA | 50 | Rabbit | 1/500 |
| p62 | 5114 | Cell Signalling, USA | 62 | Rabbit | 1/1000 |
| p-Akt | 4060 | Cell Signalling, USA | 60 | Rabbit | 1/1000 |
| p-GSK3β | 5558 | Cell Signalling, USA | 46 | Rabbit | 1/1000 |
| t-GSK3β | 9315 | Cell Signalling, USA | 46 | Rabbit | 1/1000 |
| t-Akt | 4691 | Cell Signalling, USA | 60 | Rabbit | 1/1000 |
| TNF-α | 11948 | Cell Signalling, USA | 28 | Rabbit | 1/500 |
Bax = Bcl-2-associated X; Bcl-2 = B cell lymphoma 2; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; LC3B = light chain 3B; MAO = monoamine oxidase A and B; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B; p-Akt = phosphorylated protein kinase B; p-GSK3β = phosphorylated glycogen synthase kinase 3β; t-GSK3β = total glycogen synthase kinase 3β; t-Akt = total protein kinase B; TNF-α = tumour necrosis factor α.
Protein samples were separated on 10% or 15% polyacrylamide gels of 1.5 mm thickness. Stacking gel contained 0.5 M Trizma base (T1504, Sigma-Aldrich) at pH 6.8, 10% sodium dodecyl sulfate (SDS; L3771, Sigma-Aldrich), 30% acrylamide (161-0156, Bio-Rad, Hercules, CA), 10% ammonium persulfate solution (APS; A3678, Sigma-Aldrich), tetramethylenediamine (TEMED; T9281, Sigma-Aldrich), and dH2O. Separating gel contained 1.5 M Trizma base at pH 8.8, 10% SDS, 30% acrylamide, 10% APS, TEMED, and distilled H2O.
Samples were diluted in SDS buffer (10% w/v SDS, 10 mM dithiothreitol, 20% v/v glycerol, 0.2 M Tris-HCl at pH 6.8, 0.05% w/v Bromophenol blue (all Sigma-Aldrich)) and Milli-Q H2O. Protein samples were denatured at 60°C for 20 minutes. 20 μg of protein was loaded per lane, and 5 μL of precision plus western C (161-0376, Bio-Rad) was loaded into the first lane of each gel to use as a molecular ladder. The gels were run for 30 minutes at 60 V in SDS running buffer containing 25 mM Tris-HCl, 200 mM glycine (Sigma-Aldrich), and 0.1% w/v SDS. When the dye front had moved through the stacking gel, gels were run for a further 1-1.5 hours at 110 V. Gels were electrotransferred onto polyscreen polyvinylidene difluoride (PVDF) membranes (NEF100200, Perkin-Elmer, Waltham, MA) for 1.5 hours at 100 V at 4°C using transfer buffer containing 0.058% Trizma base, 10% SDS, 20% methanol, and 0.029% glycine. Following transfer, PVDF membranes were blocked with 30 mL of 0.5% skim milk solution containing Tris-buffered saline (TBST, Trizma base, NaCl at pH 7.5 and Tween20 (Sigma-Aldrich)) using the SNAP i.d. system (Merck Millipore). PVDF membranes were incubated with primary antibodies (Table 1) diluted in 0.5% skim milk/TBST overnight at 4°C on a blood tube roller.
The following day, membranes were washed with one litre of phosphate-buffered saline/Tween20 (PBST). Secondary antibody, goat anti-rabbit IgG (H+L)-conjugate (170-6515, Bio-Rad) was diluted in 0.5% skim milk/TBST solution, added to each membrane, and incubated at room temperature for 10 minutes. Membranes were then washed with one litre of PBST. Enhanced chemiluminescence (Merck Millipore) was added to the membranes and incubated at room temperature for one minute. Membranes were exposed to medical imaging film (28906839, GE Healthcare Life Sciences, Marlborough, MA). Film slides were scanned, and densitometry analysis was performed using ImageJ (https://imagej.nih.gov/ij/).
Few studies have reported western blot analyses using the D2-mdx mouse model. GAPDH is a standard western assay protein loading control that has also commonly been used in mdx mouse models [52]. Although mRNA expression did not vary (see below), protein expression of GAPDH was found to vary greatly between age-matched D2-mdx mice. Consequently, gels were run for Coomassie blue staining and normalisation to total protein content. Highly variable GAPDH expression in the D2-mdx mouse model was also recently reported by Kennedy and colleagues [53]. A recent study by Hildyard and others [54] in mdx mice showed that GAPDH gene expression exhibited a twofold decrease in dystrophic tissue highlighting GAPDH gene expression instability and marked muscle and mdx-specific differences.
After electrophoresis, separated proteins on acrylamide gels were placed in a clean container and rinsed three times for five minutes each with deionized water to remove excess SDS. 40 mL of EZBlue™ gel staining reagent (Coomassie blue G1041, Sigma-Aldrich) was added to each gel, and the gel was placed on a shaker and left to stain for one hour at room temperature. Following this, the gel was washed with deionized water every 10-15 minutes for four hours. The gel was then left on the shaker in deionized water overnight to destain. Images of the gel were taken the following day and analysed by densitometry.
2.8. RNA Analysis
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis was performed using left ventricular myocardium (50 mg) from DBA/2 and D2-mdx mice frozen at necropsy to determine messenger RNA (mRNA) expression of several key genes. Total RNA extraction from TRI Reagent-treated tissue homogenates was performed as per manufacturer's instructions (Applied Biosystems, USA). RNA concentration and quality (Abs260/280 ratio between 1.8 and 2) of each sample were measured using the NanoDrop ND-1000 Spectrophotometer (Biolab, Canada). Extracted RNA samples were diluted to 100 ng/μL and reverse transcribed to produce complementary deoxyribonucleic acid (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and TaqMan assays listed in Table 2 were performed according to the manufacturer's protocol (Applied Biosystems). All reactions were run in triplicate with a 7900HT Fast Real-Timer PCR System (Applied Biosystems) with the thermocycler conditions: stage 1: 50°C for two minutes, 1 cycle; stage 2: 95°C for 10 min, 95°C for 15 seconds and 60°C for one minute, 40 cycles.
Table 2.
Primers for RT-qPCR.
| Primer | Catalogue number | Target species | Company |
|---|---|---|---|
| GAPDH | 4352932E | Mouse | Applied Biosystems |
| PGC-1α | Mm01208835_m1 | Mouse | Applied Biosystems |
| IL-6 | Mm00446190_m1 | Mouse | Applied Biosystems |
| NF-κB | Mm00482418_m1 | Mouse | Applied Biosystems |
| TNF-α | Mm00443258_m1 | Mouse | Applied Biosystems |
| Col1a1 | Mm00801666_g1 | Mouse | Applied Biosystems |
| TLR4 | Mm00445273_m1 | Mouse | Applied Biosystems |
GAPDH = glyceraldehyde 3-phosphate dehydrogenase; PGC-1α = peroxisome proliferator-activated receptor-γ coactivator 1; IL-6 = interleukin 6; NF-κB=nuclear factor ĸ-light-chain-enhancer of activated B cells; TNF-α = tumour necrosis factor α; Col1a1 = collagen type I α 1 chain; TLR4 = toll-like receptor 4.
Expression of mRNA was evaluated from the threshold cycle (CT) values. The CT value is the fractional PCR cycle at which the fluorescence signal passes the fixed threshold and reaches a level of detection that can be used to indicate the level of mRNA of the target gene. CT levels were analysed using the ∆CT method of relative quantification whereby ∆CT values are obtained by normalising each mRNA of interest to a reference mRNA (i.e., GAPDH) [55]. Relative fold change (FC) in gene expression was calculated as 2-∆∆CT, where FC = 1 means no change; FC > 1 = increase; FC < 1 = decrease in gene expression.
2.9. Statistical Analyses
All graphs and statistical analyses were completed using GraphPad Prism Software Version 9 (GraphPad Software Inc., La Jolla, CA). The Kolmogorov-Smirnov test was used to determine whether data was normally distributed, and statistical tests to contrast potential group differences were selected accordingly. For normally distributed data, one-way ANOVA, repeated measures two-way ANOVA, and Dunnett's multiple comparisons were used, comparing each group to the D2-mdx Vehicle group. Data are presented as mean ± standard deviation (SD). Where required, a Mann–Whitney U test was used, with data presented as individual values and group median.
3. Results
3.1. Skeletal and Respiratory Muscle in the D2-mdx Mouse
Male D2-mdx mice weighed less than age-matched DBA/2 mice at 15, 16, 17, and 18 weeks of age (p < 0.05; Table 3), and quadriceps weight was significantly lighter at 18 weeks (p = 0.0058; Table 4). Forelimb grip strength of the D2-mdx mice was weaker at 10 and 18 weeks compared to that of the DBA/2 mice (p = 0.004, p < 0.0001, respectively; Table 5). Diaphragms of the D2-mdx had a greater percentage of collagen compared to those of the DBA/2 mice at 18 weeks (p < 0.0001; Figure 1). No differences in any of these parameters were observed in the Perindopril, Debio-025, and Debio-025+Perindopril groups compared to the D2-mdx Vehicle group (Tables 3–5).
Table 3.
Body weights of DBA/2 and D2-mdx mice from 10 to 18 weeks.
| Body weight (g) | |||||
|---|---|---|---|---|---|
| DBA/2 | D2-mdx | ||||
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | ||
| 10 weeks | 25.6 ± 2.6 | 21.8 ± 1.3 | 21.8 ± 1.3 | 21.7 ± 2.6 | 20.7 ± 2.4 |
| 11 weeks | 24.8 ± 1.6 | 22.7 ± 1.7 | 22.7 ± 1.4 | 22.6 ± 2.2 | 21.4 ± 2.4 |
| 12 weeks | 25.3 ± 1.8 | 22.9 ± 2.0 | 23.0 ± 1.2 | 22.8 ± 2.3 | 21.6 ± 2.1 |
| 13 weeks | 25.4 ± 1.9 | 23.5 ± 1.8 | 23.4 ± 1.5 | 23.3 ± 2.2 | 21.9 ± 2.4 |
| 14 weeks | 25.8 ± 1.5 | 23.9 ± 1.8 | 23.7 ± 1.0 | 23.9 ± 2.4 | 22.0 ± 2.7 |
| 15 weeks | 27.2 ± 1.7∗ | 23.7 ± 1.5 | 23.6 ± 1.2 | 23.5 ± 2.4 | 21.9 ± 2.2 |
| 16 weeks | 28.2 ± 2.1∗ | 23.9 ± 2.1 | 24.1 ± 1.3 | 23.9 ± 2.4 | 22.2 ± 2.2 |
| 17 weeks | 28.3 ± 1.3∗ | 24.7 ± 1.7 | 24.7 ± 1.2 | 24.3 ± 2.6 | 23.1 ± 1.5 |
| 18 weeks | 29.0 ± 1.7∗ | 24.9 ± 2.1 | 24.6 ± 1.3 | 23.8 ± 2.2 | 23.1 ± 1.2 |
Table 4.
Tibialis anterior and quadriceps weights of DBA/2 and D2-mdx mice at 18 weeks.
| DBA/2 | D2-mdx | ||||
|---|---|---|---|---|---|
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | ||
| TA (mg/cm) | 19.8 ± 1.4 | 17.3 ± 2.9 | 16.5 ± 3.7 | 18.4 ± 2.7 | 13.7 ± 1.9 |
| Qu (mg/cm) | 56.1 ± 12.0∗∗ | 42.1 ± 5.0 | 38.5 ± 7.9 | 41.1 ± 4.4 | 38.9 ± 5.0 |
Table 5.
Forelimb grip strength of DBA/2 and D2-mdx mice at 10 and 18 weeks.
| Forelimb grip strength normalised to body weight (N/g) | |||||
|---|---|---|---|---|---|
| DBA/2 | D2-mdx | ||||
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | ||
| 10 weeks | 0.03 ± 0.01∗∗ | 0.01 ± 0.003 | 0.01 ± 0.004 | 0.01 ± 0.003 | 0.01 ± 0.004 |
| 18 weeks | 0.04 ± 0.004∗∗∗ | 0.02 ± 0.008 | 0.02 ± 0.005 | 0.025 ± 0.007 | 0.03 ± 0.005 |
Figure 1.
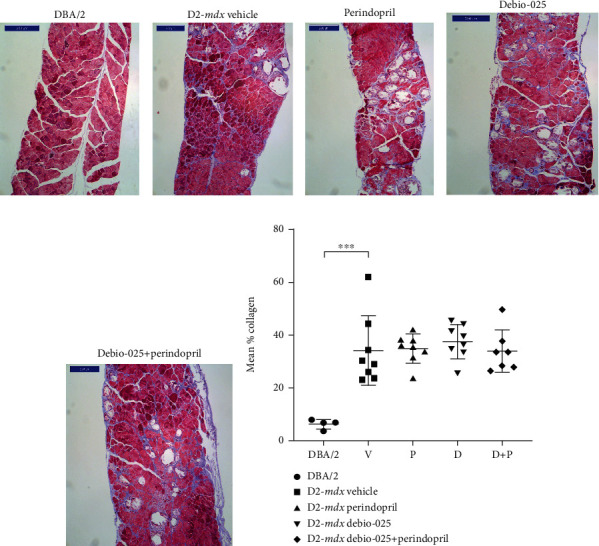
Fibrosis in diaphragms of DBA/2 and D2-mdx mice at 18 weeks. Representative transverse sections of diaphragms (Masson's trichrome stain, scale bar = 200 μm) from (a) DBA/2 and D2-mdx treated to (b) Vehicle, (c) Perindopril, (d) Debio-025, and (e) Debio-025+Perindopril. Mean % collagen for each group (f). Data analysed by one-way ANOVA and Dunnett's multiple comparisons and presented as mean ± SD and individual values; ∗∗∗p < 0.001.
Data analysed by repeated measures two-way ANOVA and Dunnett's multiple comparison tests and presented as mean ± SD; ∗p < 0.05 vs. Vehicle D2-mdx.
Tibialis anterior (TA) and quadriceps (Qu) weights were normalised to tibia lengths. Tibial bone lengths were similar in all animals and did not differ between groups. Data analysed by one-way ANOVA and Dunnett's multiple comparisons and presented as mean ± SD; ∗∗p < 0.01 vs. Vehicle D2-mdx.
Forelimb grip strength is normalised to body weight. Data were analysed by repeated measures two-way ANOVA and Dunnett's multiple comparisons tests and presented as mean ± SD; ∗∗p < 0.01 and ∗∗∗p < 0.0001 vs. Vehicle D2-mdx.
3.2. Echocardiography
The structural and functional parameters of LV were measured at 10 and 18 weeks of age using echocardiography (Tables 6 and 7). No differences in fractional shortening and ejection fraction were observed between the DBA/2 and D2-mdx mice. Heart rate was greater in the DBA/2 mice compared to the D2-mdx mice at 18 weeks (p < 0.05). However, heart rates in all groups remained above 300 bpm and other functional parameters were not different between DBA/2 and D2-mdx. No differences were observed in LV structural or functional parameters in the treatment groups compared to the D2-mdx Vehicle group.
Table 6.
LV functional parameters in DBA/2 and D2-mdx mice at 10 and 18 weeks.
| DBA/2 | D2-mdx | |||||
|---|---|---|---|---|---|---|
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | |||
| FS (%) | 10 weeks | 38 ± 9.50 | 39.5 ± 7.4 | 41.3 ± 6.7 | 33.4 ± 7.8 | 38.5 ± 5.9 |
| 18 weeks | 46.8 ± 9.4 | 40.8 ± 6.6 | 37.6 ± 9.2 | 39.9 ± 7.2 | 31.4 ± 8.4 | |
| EF (%) | 10 weeks | 68.6 ± 12.0 | 70.6 ± 8.8 | 72.7 ± 8.4 | 62.6 ± 11.4 | 69.6 ± 7.6 |
| 18 weeks | 78.2 ± 9.0 | 72.1 ± 8.3 | 67.4 ± 11.5 | 71.1 ± 9.0 | 59.0 ± 11.9 | |
| HR (bpm) | 10 weeks | 415.3 ± 50.3 | 363.5 ± 42.2 | 378.3 ± 53.3 | 342.3 ± 41.4 | 315.0 ± 43.6 |
| 18 weeks | 442.8 ± 58.5∗ | 348.0 ± 61.7 | 317.8 ± 56.5 | 309.8 ± 27.4 | 341.0 ± 63.1 | |
Table 7.
LV structural parameters in DBA/2 and D2-mdx mice at 10 and 18 weeks.
| DBA/2 | D2-mdx | |||||
|---|---|---|---|---|---|---|
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | |||
| LVEDd (mm) | 10 weeks | 3.7 ± 0.2 | 3.3 ± 0.3 | 3.4 ± 0.5 | 3.5 ± 0.3 | 3.3 ± 0.4 |
| 18 weeks | 3.5 ± 0.2 | 3.5 ± 0.5 | 3.6 ± 0.4 | 3.4 ± 0.4 | 3.7 ± 0.4 | |
| LVESd (mm) | 10 weeks | 2.3 ± 0.4 | 2.0 ± 0.4 | 2.0 ± 0.4 | 2.3 ± 0.4 | 2.1 ± 0.4 |
| 18 weeks | 1.9 ± 0.4 | 2.1 ± 0.5 | 2.4 ± 0.7 | 2.1 ± 0.3 | 2.6 ± 0.5 | |
| IVSd (diastole) (mm) | 10 weeks | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 |
| 18 weeks | 0.7 ± 0.1 | 0.7 ± 0.1 | 0.7 ± 0.2 | 0.8 ± 0.2 | 0.8 ± 0.2 | |
| IVSd (systole) (mm) | 10 weeks | 0.8 ± 0.2 | 1.0 ± 0.3 | 1.1 ± 0.1 | 0.9 ± 0.2 | 1.0 ± 0.2 |
| 18 weeks | 1.2 ± 0.1 | 1.2 ± 0.2 | 1.2 ± 0.2 | 1.2 ± 0.2 | 1.2 ± 0.2 | |
| PWT (diastole) (mm) | 10 weeks | 0.5 ± 0.1 | 0.7 ± 0.2 | 0.6 ± 0.1 | 0.6 ± 0.2 | 0.7 ± 0.1 |
| 18 weeks | 0.8 ± 0.2 | 0.8 ± 0.2 | 0.8 ± 0.1 | 0.7 ± 0.2 | 0.8 ± 0.2 | |
| PWT (systole) (mm) | 10 weeks | 1.0 ± 0.2 | 1.0 ± 0.3 | 0.9 ± 0.2 | 0.9 ± 0.2 | 0.9 ± 0.2 |
| 18 weeks | 1.1 ± 0.1 | 1.1 ± 0.3 | 1.2 ± 0.1 | 1.0 ± 0.2 | 1.0 ± 0.3 | |
| LV mass (mg) | 10 weeks | 59.0 ± 11.6 | 63.6 ± 13.9 | 63.3 ± 15.2 | 69.0 ± 21.4 | 67.2 ± 15.3 |
| 18 weeks | 82.4 ± 21.2 | 85.3 ± 20.6 | 90.2 ± 10.9 | 85.4 ± 25.4 | 100.1 ± 28.1 | |
FS = fractional shortening; EF = ejection fraction; HR = heart rate; bpm = beats per minute. Data analysed by repeated measures two-way ANOVA and Dunnett's multiple comparisons tests and presented as mean ± SD; ∗p < 0.05 vs. D2-mdx Vehicle.
LV = left ventricle; LVED = LV end-diastolic; LVES = LV end-systolic; IVS = interventricular septum; PWT = posterior wall thickness. Data analysed by repeated measures two-way ANOVA and presented as mean ± SD.
3.3. Cardiac Mass
No differences were observed in heart weights of the DBA/2 mice compared to those of the D2-mdx Vehicle mice at 18 weeks (Figure 2). Whole hearts from the Perindopril and Debio-025+Perindopril groups were lighter than those from the D2-mdx Vehicle mice (p = 0.03 and p = 0.009, respectively).
Figure 2.
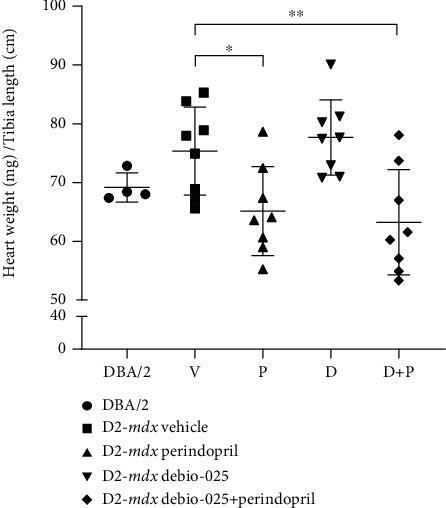
Heart weights (normalised to tibial length) from DBA/2 and D2-mdx mice at 18 weeks. Tibial bone lengths were similar in all animals and did not differ between groups. Data analysed by one-way ANOVA and Dunnett's multiple comparisons tests and presented as mean ± SD and individual values; ∗p < 0.05 and ∗∗p < 0.01.
3.4. Cardiac Fibrosis
Transverse heart sections were assessed at 18 weeks of age using Masson's trichrome staining (Figures 3(a)–(e) All D2-mdx hearts featured an epicardial fibrotic calcinosis layer surrounding the right ventricle and extending beyond the edges of the LV. In contrast, while all the DBA/2 also had some RV epicardial calcinosis, this was very limited and restricted to the RV. The percentage of total area with fibrotic tissue was quantified using a digital segmentation method. Hearts of the D2-mdx mice had a greater percentage of collagen stain compared to those of the DBA/2 mice at 18 weeks (p = 0.004; Figure 3(f)), and Col1a1 mRNA expression measured by RT-qPCR was greater in the D2-mdx hearts compared to the DBA/2 (FC = 1.89, p = 0.0008). Hearts from the D2-mdx Perindopril group had a lower percentage of collagen stain compared to those from the D2-mdx Vehicle group (p = 0.006; Figure 3(f)).
Figure 3.
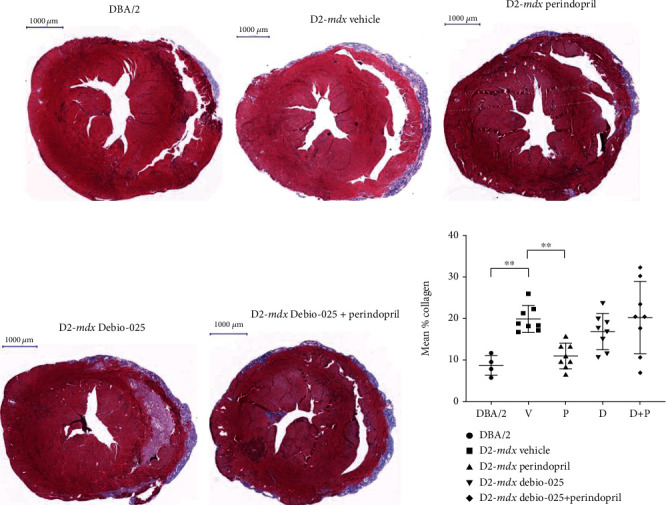
Cardiac fibrosis in DBA/2 and D2-mdx mice at 18 weeks. Representative sections of (a) DBA/2, (b) D2-mdx Vehicle, (c) D2-mdx Perindopril, (d) D2-mdx Debio-025, and (e) D2-mdx Debio-025+Perindopril mouse hearts stained with Masson's trichrome stain (scale = 1000 μm). (f) Mean % collagen for DBA/2 and D2-mdx hearts. Data analysed by one-way ANOVA and Dunnett's multiple comparisons tests and presented as mean ± SD and individual values; ∗∗p < 0.01.
In order to determine potential changes in cardiomyocyte content and cell size, average cell density and cell diameter were measured in the RV, LV, and septum of DBA/2 and D2-mdx mice at 18 weeks (Figures 4(a)–4(e) Greater RV cell density was observed in the DBA/2-, Debio-025-, and Debio-025+Perindopril-treated groups compared to the D2-mdx Vehicle group (p = 0.0002, p = 0.045, and p = 0.048, respectively, Figure 5). Greater LV and septum cell density was observed in the DBA/2 mice compared to the D2-mdx mouse model (p = 0.006 and p = 0.002, respectively). Septum cell density was greater in the Perindopril and Debio-025+Perindopril groups compared to Vehicle (p = 0.024 and p = 0.02, respectively).
Figure 4.
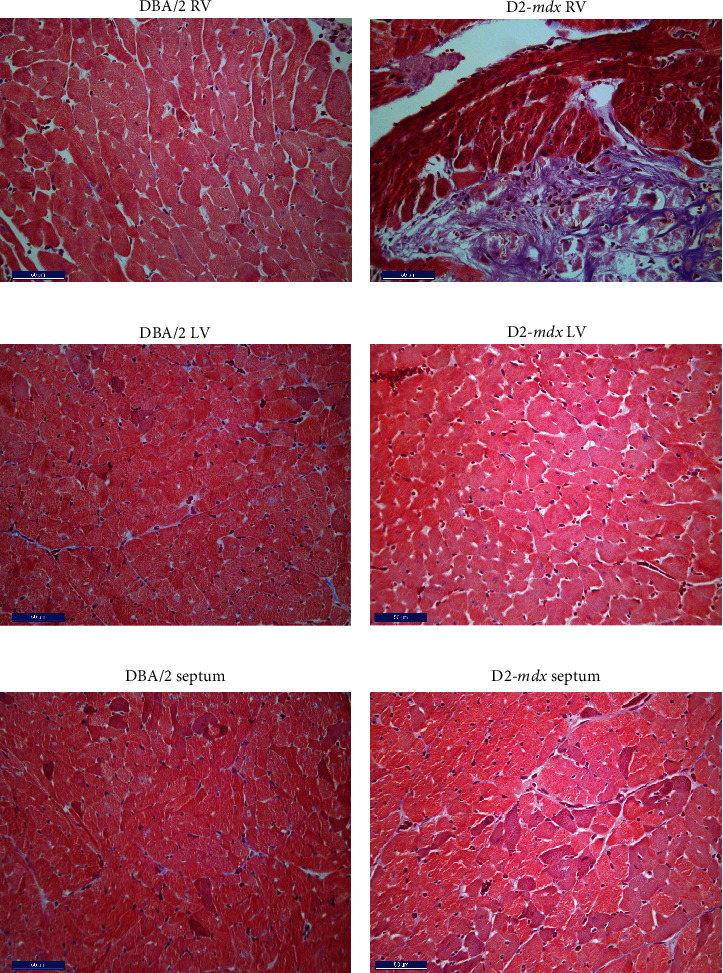
Cross-sectional images from the RV, LV, and septum of DBA/2 and D2-mdx hearts at 18 weeks. Representative sections of (a) DBA/2 RV, (b) D2-mdx RV, (c) DBA/2 LV, (d) D2-mdx LV, (e) DBA/2 septum, and (f) D2-mdx septum stained with Masson's trichrome (scale bar = 50 μm).
Figure 5.
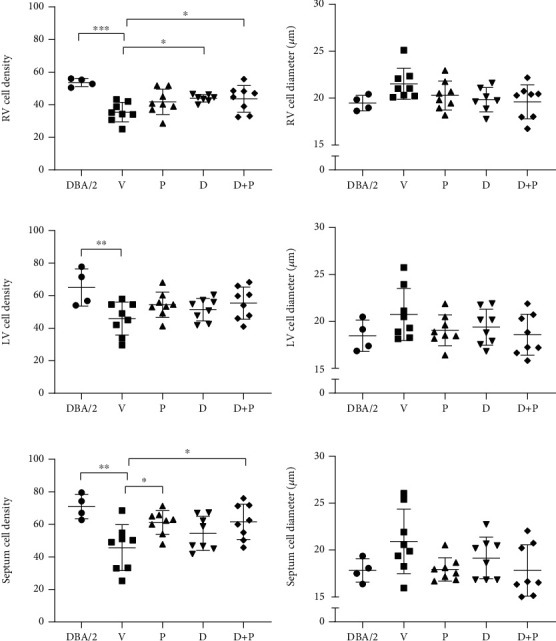
RV, LV, and septum cell density (number of cells/area) and diameter in DBA/2 and treated D2-mdx mice at 18 weeks. The number of cells within an area of 19,500 μm2 (150 × 130 μm) for (a) RV, (c) LV, and (e) septum and average cell diameter for (b) RV, (d) LV, and (f) septum. Values represent the average of five areas of size 150 × 130 μm assessed for each tissue region. Data were analysed by nested one-way ANOVA and presented as mean ± SD and individual values; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
3.5. Inflammation and Proapoptotic Activity
IL-6 and TLR4 mRNA expression levels were greater in the D2-mdx hearts compared to the DBA/2 (FC = 4.03, p = 0.006; FC = 2.13, p = 0.004, respectively; Figures 6(a) and 6(b) No significant differences were observed for NF-κB (FC = 1.0) and TNF-α (FC = 0.607) mRNA. Expression of Bax (proapoptotic marker) and Bcl-2 (antiapoptotic marker) proteins was lower in the D2-mdx hearts compared to the DBA/2 (p = 0.008 and p = 0.004, respectively; Figures 6(c) and 6(d)). Expression of the Bax/Bcl-2 ratio and caspase-3 was greater in the D2-mdx hearts compared to the DBA/2 (p = 0.049 and p = 0.049, respectively; Figures 6(e) and 6(f)). Lower levels of LC3B protein expression were observed in the D2-mdx hearts compared to the DBA/2 (p = 0.008; Figure 6(h)), but there was no difference in Beclin-1 expression. Compared to Vehicle, treatment with Debio-025, Perindopril, or their combination had no significant effect on relative expression of these inflammatory and apoptotic proteins (Table 8). See Supplement 1 for western assay blots.
Figure 6.
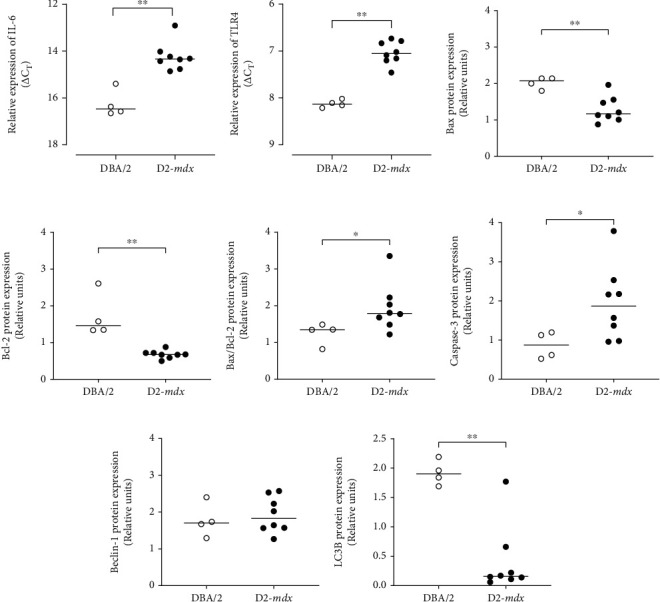
Relative expression of mRNA and proteins involved in inflammation, apoptosis, and autophagy in DBA/2 and D2-mdx hearts at 18 weeks. Expression of (a) IL-6 mRNA and (b) TLR4 mRNA obtained from RT-qPCR and presented as individual ΔCT values, where a lower ΔCT denotes greater mRNA expression. Relative protein expression of (c) Bax, (d) Bcl-2, (e) Bax/Bcl-2 ratio, (f) caspase-3, (g) Beclin-1, and (h) LC3B obtained from western immunoblots. Intact western blots are shown in Supplement 1 Data analysed by Mann–Whitney U test and presented as median and individual values; ∗p < 0.05 and ∗∗p < 0.01.
Table 8.
Summary of relative protein expression from western blot assays in treated D2-mdx hearts.
| Vehicle | Perindopril | Debio-025 | Debio-025+Perindopril | |
|---|---|---|---|---|
| Bax | 2.23 ± 0.52 | 2.03 ± 0.47 | 2.56 ± 1.03 | 2.67 ± 0.63 |
| Bcl-2 | 1.19 ± 0.26 | 1.34 ± 0.33 | 1.12 ± 0.30 | 1.18 ± 0.31 |
| Bax/Bcl-2 | 1.95 ± 0.65 | 1.61 ± 0.54 | 2.33 ± 0.68 | 2.10 ± 0.75 |
| Caspase-3 | 3.35 ± 1.58 | 3.0 ± 1.59 | 3.52 ± 0.97 | 4.10 ± 1.11 |
| Beclin-1 | 3.37 ± 0.87 | 3.0 ± 0.54 | 3.10 ± 0.69 | 2.97 ± 0.79 |
| MAO-A | 4.18 ± 0.54 | 4.36 ± 1.06 | 4.07 ± 0.90 | 4.18 ± 1.44 |
| MAO-B | 7.09 ± 0.70 | 7.26 ± 0.76 | 6.65 ± 0.49 | 6.48 ± 0.51 |
| NF-κB | 0.38 ± 0.19 | 0.44 ± 0.34 | 0.63 ± 0.62 | 0.63 ± 0.66 |
| TNF-α | 1.16 ± 0.84 | 0.47 ± 0.54 | 3.07 ± 1.78 | 0.72 ± 0.61 |
Data presented as mean ± SD. Western blots are shown in Supplement 1.
3.6. Mitochondrial Perturbations
Relative expression of the PGC-1α gene and monoamine oxidase A and monoamine oxidase B proteins (MAO-A, MAO-B) was greater in the hearts of the D2-mdx mice compared to the DBA/2 mice at 18 weeks (FC = 1.19, p = 0.028; p = 0.004; p = 0.016, respectively; Figure 7). No effect of treatment with Debio-025 (FC = 0.999, p = 0.999), Perindopril (FC = 1.19, p = 0.079), or their combination (FC = 1.19, p = 0.145) was evident for PGC-1α or MAO-A and MAO-B proteins (Table 8). Representative electron micrographs of the LV from the D2-mdx Vehicle group were comparable to those from the Perindopril, Debio-025, and Perindopril+Debio-025 treatment groups (Figure 8). Despite heterogeneous areas of disorganisation and ultrastructural variation, all treatment groups also displayed discrete areas of evenly distributed interfibrillar mitochondria between organised myofibrils. All D2-mdx group mitochondria also featured distinct areas of mitochondrial proliferation, mitochondrial swelling, loss of cristae, and mitophagy. Although the electron micrograph assessment did not permit quantitative analysis, the mitochondrial and myofibril morphology indicates that these adaptive perturbations within anaesthetized D2-mdx mice were adequate to sustain resting LV function.
Figure 7.
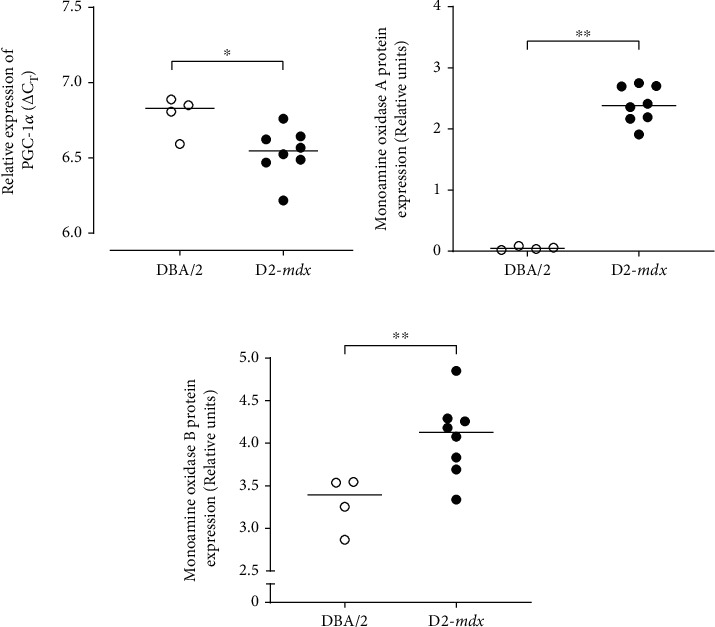
Relative expression pattern of PGC-1α gene and protein expression of markers of mitochondrial ROS signalling from monoamine oxidase in DBA/2 and D2-mdx hearts at 18 weeks. (a) Relative expression of PGC-1α obtained from RT-qPCR and presented as individual ΔCT values, where a lower ΔCT denotes greater mRNA expression. (b, c) Relative protein expression of monoamine oxidase A and monoamine oxidase B obtained from western immunoblots. Data analysed by Mann–Whitney U test and presented as median and individual values; ∗p < 0.05 and ∗∗p < 0.01.
Figure 8.

Representative electron micrographs of LV myocardial tissue from D2-mdx mice at 18 weeks. (a) An example from the D2-mdx Vehicle group that shows areas of randomly distributed mitochondria between poorly organised mitochondria, mitochondrial proliferation, and also some areas that are preserved (3000x mag). (b) Highly preserved regions with evenly sized and distributed mitochondria in the D2-mdx Debio-025+Perindopril group (4000x mag). (c) Examples of mitophagy and mitochondrial swelling in the D2-mdx Vehicle group (10,000x mag). (d) Examples of enlarged mitochondria with thick membranes and sparse cristae in the D2-mdx Debio-025 group (10,000x mag).
4. Discussion
In the current era of improved respiratory management in DMD patients, cardiomyopathy progressing to heart failure is the main cause of death [11]. In this study, the D2-mdx mouse model was characterized and tested to determine whether the ACE inhibitor Perindopril, already in clinical use, and Debio-025, a specific mPTP blocker with a human safety profile, either alone or in combination could have an impact on the extent of cardiomyopathy. The main findings were as follows: (1) at 18 weeks of age, compared to DBA/2 mice, D2-mdx mice had a severe cardiac pathology characterized by fewer myocytes (less cell density), larger myocytes (hypertrophy), and intense epicardial fibrosis, most prominently in the RV; (2) resting LV function was comparable to age-matched DBA/2 mice at 18 weeks; (3) following a six-week drug treatment period from 12 to 18 weeks, Perindopril marginally reduced total heart weight and collagen content at 18 weeks of age in D2-mdx mice. However, Perindopril, Debio-025, and their combination had no significant impact on LV function, and treatment with both drugs did not further limit the extent of fibrosis. Notably, the indirect echocardiographic measure of LV mass was not altered by Perindopril treatment.
The lower forelimb force, body, and muscle weights in comparison to the DBA/2 animals are consistent with other reports of an atrophic phenotype in D2-mdx mice [3, 25, 40–43]. Despite the overt epicardial pathology in D2-mdx mice at 18 weeks, it appears that by this age these mice maintain sufficient adaption to sustain normal resting LV function. Thus, not surprisingly, Debio-025, Perindopril, and their combination had no discernible impact on LV performance. However, the decreased heart rate in the D2-mdx mice compared to the DBA/2 at 18 weeks linked with relatively similar LV dimensions and ejection fraction, but lower heart weight suggests potential for lower cardiac output. If the LV length in D2-mdx mice is shorter than that in DBA/2 mice, then a more globular LV geometric shape may lead to overestimated ejection fraction because the present cardiac ultrasound analysis is based on m-mode assumptions of similar LV diameter and length.
An early study reported marked cardiac dysfunction in the D2-mdx mice compared to DBA/2 [40]. In contrast, our study and other studies to date have not been able to successfully reproduce these findings. Kennedy and colleagues [53] assessed cardiac function at 21 weeks using cardiac MRI and found that, compared to DBA/2 mice, D2-mdx mice showed elevated LV and RV EF. Yet, at 28 weeks, these differences were no longer apparent. Vohra and colleagues [41] used cardiac MRI and were not able to detect LV dysfunction compared to DBA/2 mice at 28 weeks. Hughes and colleagues [25] used echocardiography and detected no overt cardiac dysfunction at four weeks, and Hakim and colleagues [43] used a cardiac catheter assay and also found no differences between the D2-mdx and DBA/2 mice at six months, also reporting high EF values. However, they did detect characteristic ECG changes such as increased QRS duration and QT interval in the D2-mdx mice compared to the DBA/2. Our findings at 10 and 18 weeks indicate that there are no major differences in LV function.
4.1. Fibrosis and Calcinosis
Dystrophic myocardial calcinosis has been reported for several mouse strains including DBA/2 mice [56], which is consequent to inflammation, necrosis, fibrosis, and calcium mineralisation and is also linked to dyscalc1 expression in D2-mdx mice [48]. The severe epicardial fibrosis measured in the D2-mdx mice prominently features calcinosis in a thick fibrotic outer layer surrounding the RV. This result is consistent with Kennedy and colleagues and van Putten and colleagues who also reported an increase in myocardial fibrosis at similar ages (10 and 21 weeks, respectively), as well as prominent necrosis and calcification [42, 53]. Hughes and colleagues [25] reported no differences in the degree of fibrosis between the D2-mdx and DBA/2 mice at four weeks of age, indicating severe myocardial fibrosis is likely to occur between four and 10 weeks of age. The present study is the first to examine average myocyte density and diameter in the RV, LV, and septum of DBA/2 and D2-mdx mice. Although Hughes et al. [25] did assess LV cardiomyocyte size, no myofibril atrophy or hypertrophy was detected in D2-mdx mice at four weeks.
In support of our trichrome stain histology results, we also observed elevated expression of Col1a1 in the hearts of D2-mdx mice compared to DBA/2 mice at 18 weeks. This result supports the findings of van Putten et al. [42] who reported similar elevated expression at 10 weeks of age. Col1a1 is a gene involved in collagen synthesis and is likely to be elevated in these animals due to the identified polymorphism in the Ltpb4 gene found in their DBA/2 background [44]. This polymorphism results in upregulated signalling of the profibrotic TGF-β pathway, increasing Col1a1 gene expression, a major component of the extracellular matrix [56]. TGF-β is a potent profibrogenic factor that, in conjunction with downstream SMAD signalling, stimulates fibroblasts to produce extracellular matrix proteins such as collagen and fibronectin and is activated in response to stress or injury [57]. Contrary to our findings and previous literature comparing the D2-mdx and DBA/2 mice, Hakim and colleagues reported pathological changes present in the DBA/2 wild-type mice as well as the D2-mdx43. Specifically, they observed epicardial fibrotic and calcified areas that were located on the surface of the RV, as well as regions of fibrosis throughout the septum and LV, comparable to the pathology detected in the D2-mdx mice [43]. In our study, we observed only a small degree of fibrosis in the DBA/2 hearts. Variation in these results may be explained by the fact that previous reports have found that only approximately half the mice on the DBA background show cardiac calcification [56]. It is also possible that the polymorphism found in the background DBA/2 of these animals contributed to a greater propensity for fibrosis in the D2-mdx mice. However, a limitation of our study was that, due to division of the same hearts for other measures, histological assessment was conducted only at a single cut plane, thus preventing detailed quantitative analyses through each entire heart. Given the limitations on available animals and the unique epicardial RV-focused fibrosis and calcinosis in this study that is not evident in patients with DMD, the present data and animal model are limited with regard to direct experimental support of the current clinical use of Perindopril where it is primarily used to reduce myocardial fibrosis to delay the onset of cardiomyopathy in DMD patients [58].
4.2. Adaptive Cell Signalling Processes
The expression of genes and proteins involved in apoptosis, autophagy, inflammation, and mitochondrial ROS signalling in the myocardium of D2-mdx animals has not previously been studied in detail. We found that expression of Bax/Bcl-2 and caspase-3 was significantly increased in the D2-mdx mouse hearts compared to the DBA/2, suggesting greater likelihood of apoptosis in D2-mdx high ratios of Bax (proapoptotic factor) to Bcl-2 (antiapoptotic factor) are indicative of propensity for apoptosis in mitochondrial-mediated apoptotic pathways that involve sustained opening of the mPTP and therefore release cytochrome c from the mitochondria [59]. The release of mitochondrial cytochrome c associates with apoptotic protease-activating factor and procaspase-9 to form the apoptosome, which then activates caspase-9 and caspase-3 [59, 60].
Contrary to previous reports, we found no differences in Akt or GSK3β between D2-mdx and DBA/2 hearts. Recent studies have suggested a role for these two proteins in the development of cardiac hypertrophy through Akt activation and GSK3β inhibition [61]. GSK3β inhibition can occur via stimuli that activate phosphatidylinositol 3-kinase (PI3K), which acts via Akt, an upstream regulator of GSK3β, and by activation of the Wnt signalling pathway [62, 63]. We were however unable to confirm Kennedy and colleagues' report of increased phosphorylated GSK3β relative to total GSK3β expression in the D2-mdx hearts compared to DBA/2 at 21 weeks [53].
As a measure of cardiac hypertrophy, heart weight was normalised to tibia length, and although there was a trend for greater heart weight in the D2-mdx mice, there was no significant difference compared to the DBA/2. Although we detected larger, but fewer cardiomyocytes, these histological measures were limited to a single plane and not a systematic series of sections through the entire heart; thus, a more detailed quantitative histological study is required.
We also assessed markers of autophagy, which is an important protective mechanism that promotes degradation and recycling of cellular components and occurs in normal heart maintenance [64]. A recent study by Fiacco and colleagues [65] showed that activation of autophagy was impaired during the late stages of DMD progression and that this correlated with a decline in muscle regeneration and the accumulation of fibrotic tissue in mdx mice. Our current finding suggests that at least LC3B-related activity may be impeded in D2-mdx hearts. In addition, the previous reports of chronic inflammation associated with DMD and various mdx models [66, 67] were supported by our findings of greater cardiac TLR4 and IL-6 gene expression in D2-mdx compared to DBA/2 mice. However, these inflammatory and autophagy signalling markers were not responsive to our current drug treatment regimes.
4.3. Mitochondrial Adaptations
We have identified unique and important evidence for mitochondrial adaptations and increased ROS in the hearts of the D2-mdx mice, as indicated by elevated PGC-1α, MAO-A, and MAO-B. MAO-A and MAO-B are two isoenzymes located on the outer mitochondrial membrane and play a role in oxidative deamination of monoamines (from neurotransmitters and diet) producing aldehydes, ammonia, and H2O2, with potential to contribute to ROS and hydroxyl radical accumulation in the heart [68, 69]. Increased MAO results in production of H2O2 that can contribute to important adaptations via intracellular signalling of ERK1/2, NFAT3/4, and matrix metalloproteases, which in turn promote cell hypertrophy, fibroblast proliferation, and extracellular matrix remodelling [70]. Subsequent maladaptation causes mitochondrial perturbation, mPTP opening, apoptosis, and ultimately cardiac dysfunction and further remodelling [70]. Increased expression of PGC-1α in the D2-mdx mice suggests that mitochondrial biogenesis and mitochondrial proliferation may be upregulated as an adaptation to maintain viable mitochondria and energy production. Recent studies have found that upregulating PGC-1α in mdx mice improves myofibril recovery following injury and reduces degeneration [71]. Our results extend the findings of Hughes and colleagues who identified mitochondrial dysfunction at four weeks of age in D2-mdx mice [25]. Their work identified complex I-supported H2O2 emission and impaired mitochondrial respiration as preceding steps before any signs of cardiac dysfunction contrast to the BL10-mdx mouse model where early mitochondrial dysfunction in dystrophic hearts remains undetected due to the extensive muscle regeneration experienced by these animals [72]. Hughes and colleagues [25] have proposed that early mitochondrial dysfunction is easier to detect because the D2-mdx mice have abnormal regenerative capacity in skeletal muscle.
Representative electron micrographs of LV from the D2-mdx showed extensive compensatory mitochondrial proliferation and resultant disrupted myofibril organisation. Within the zones of mitochondrial proliferation, there was also evidence of varying stages of mitochondrial swelling and mitophagy. Although a systematic quantitative electron micrograph study was not possible, compared to Vehicle controls, drug-treated hearts featured extensive discrete areas of mitochondrial proliferation and dense cristae and areas of mitochondrial abundance amongst ordered myofibrils. Generally, all D2-mdx showed heterogeneous areas of disorganisation in contrast to discrete areas showing evenly distributed interfibrillar mitochondria between organised myofibrils. These organised areas are congruent with capacity for the normal resting LV function measured at 18 weeks.
4.4. Study Limitations
A limitation of our study is that cardiac ultrasound measures were made in anaesthetized animals with low resting heart rates. Increasing demand for cardiac output by workload due to exercise metabolism and increased heart rate may have evoked functional limitations in the LV. The present study was also limited due to the inability to obtain RV ultrasound imaging of sufficient quality for quantitative assessment of function in D2-mdx mice. Imaging of the RV in mice in situ is challenging due to interference from the sternum which affects accurate positioning of the ultrasound probe. Sternum artefact can also obscure portions of the heart, particularly the endocardial borders, making it difficult to assess RV function. Yet from our morphology sections, the extensive RV fibrosis in these mice suggests potential for RV dysfunction; thus, future studies require direct RV investigation with magnetic resonance imaging or pressure catheters in D2-mdx mice.
Another limitation of our study is that we were not able to initiate treatments at a younger age (i.e., at weaning) due to the commercial purchase and overseas shipment of the mice. Although no LV dysfunction was evident at 10 weeks of age, early progression of fibrosis may have limited potential for drug treatment impact on fibrosis. Debio-025 has been shown to be most effective when used in a preventative manner, prior to myofibril loss and replacement with fibrotic tissue [24, 37]. Thus, future study designs need to include capacity for treatment before the onset of fibrosis, i.e., in utero, at birth, or postweaning, and at later more advanced phases of adverse muscle remodelling. Another important study limitation was the use of DBA/2 mice as the comparator control group, due to the variability and propensity for early onset fibrosis and calcinosis in these mice; future studies of D2-mdx should concurrently include additional comparator control groups with other backgrounds.
5. Conclusions
D2-mdx mice displayed a severe diaphragm myopathy characterized by large regions devoid of myocytes and collagen replacement, consistent with other studies [40, 42] and DMD patients [73]. Treatment with Perindopril, Debio-025, or in combination did not reduce diaphragm collagen content compared to the Vehicle group. Similar to DMD patients [74, 75], we confirm that D2-mdx mice display severe fibrosis in advance of LV dysfunction, though predominantly in the RV. In contrast to DMD patients with early subendocardial and inferobasal LV fibrosis [75], D2-mdx mice had severe fibrosis predominantly in the RV, and there was no overt LV dysfunction in D2-mdx mice compared to age-matched DBA/2. Given the extensive respiratory diaphragm and RV fibrosis, it is likely that D2-mdx mice may have respiratory and RV dysfunction, which should be directly investigated in future studies.
We have identified novel characteristics of the D2-mdx mice that have not previously been reported, including reduced myocyte number, myocyte hypertrophy, augmented mitochondrial MAO and PGC1α expression, and increased myocardial fibrosis, most prominently in the RV. Thus, at 18 weeks of age, the D2-mdx mice appear sufficiently adapted to maintain resting LV function. Chronic Perindopril and Debio-025 treatment had no impact on fibrosis and myocyte disruption. Our results suggest that the adaptations evident in the D2-mdx mouse model have cardiomyopathy features which are only partly in common with those seen in patients with DMD, and thus, further detailed longitudinal and molecular investigations of this mouse model are required.
Acknowledgments
We thank Debiopharm Research & Manufacturing SA, Martigny, Switzerland, for their gift of Debio-025 (Alisporivir). Funding of the work was provided in part by The Royal Children's Hospital 1000 Program and the Victorian Government's Operational Infrastructure Support Program to the Murdoch Children's Research Institute.
Data Availability
Data can be found in the manuscript and supplementary data file.
Disclosure
Debiopharm provided no financial funding support and had no role in the design of the study, data analysis, and conclusions.
Conflicts of Interest
All authors declare that they have no conflicts of financial interest.
Authors' Contributions
The authors contributed to the manuscript content as follows: conception and design: HH, SP, AP, JW, and MC; data acquisition: HH, SP, JA, JK, MT, JM, AP, and JW; data analysis and interpretation: HH, SP, AP, JW, and MC; manuscript draft and revisions: HH, SP, AP, JW, and MC. All authors read and approved the final manuscript.
Supplementary Materials
Western blot protein expression assays (Supplement 1).
References
- 1.Zimmermann J. F., Butler C. R. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurology . 2018;17(6):p. 251. doi: 10.1016/S1474-4422(18)30024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birnkrant D. J., Bushby K., Bann C. M., et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurology . 2018;17(4):347–361. doi: 10.1016/S1474-4422(18)30025-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukada S., Morikawa D., Yamamoto Y., et al. Genetic background affects properties of satellite cells and _mdx_ phenotypes. The American Journal of Pathology . 2010;176(5):2414–2424. doi: 10.2353/ajpath.2010.090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fairclough R. J., Wood M. J., Davies K. E. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nature Reviews Genetics . 2013;14(6):373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 5.Whitehead N. P., Yeung E. W., Allen D. G. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clinical and Experimental Pharmacology & Physiology . 2006;33(7):657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- 6.Allen D. G., Whitehead N. P., Froehner S. C. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiological Reviews . 2016;96(1):253–305. doi: 10.1152/physrev.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rybalka E., Timpani C. A., Cooke M. B., Williams A. D., Hayes A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS One . 2014;9(12, article e115763) doi: 10.1371/journal.pone.0115763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Percival J. M., Siegel M. P., Knowels G., Marcinek D. J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Human Molecular Genetics . 2013;22(1):153–167. doi: 10.1093/hmg/dds415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inkley S. R., Oldenburg F. C., Vignos P. J. Pulmonary function in Duchenne muscular dystrophy related to stage of disease. American Journal of Medicine. . 1974;56(3):297–306. doi: 10.1016/0002-9343(74)90611-1. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa Y., Miura T., Ishikawa Y., et al. Duchenne muscular dystrophy: survival by cardio-respiratory interventions. Neuromuscular Disorders . 2011;21(1):47–51. doi: 10.1016/j.nmd.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 11.McNally E. M. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annual Review of Medicine . 2007;58(1):75–88. doi: 10.1146/annurev.med.58.011706.144703. [DOI] [PubMed] [Google Scholar]
- 12.Politano L., Nigro G. Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myologica . 2012;31:24–30. [PMC free article] [PubMed] [Google Scholar]
- 13.Ballmann C., Denney T. S., Beyers R. J., et al. Lifelong quercetin enrichment and cardioprotection in Mdx/Utrn+/-mice. American Journal of Physiology-Heart and Circulatory Physiology. . 2017;312(1):H128–H140. doi: 10.1152/ajpheart.00552.2016. [DOI] [PubMed] [Google Scholar]
- 14.Duboc D., Meune C., Pierre B., et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow-up. American Heart Journal . 2007;154(3):596–602. doi: 10.1016/j.ahj.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Rafael-Fortney J. A., Chimanji N. S., Schill K. E., et al. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation . 2011;124(5):582–588. doi: 10.1161/CIRCULATIONAHA.111.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verhaert D., Richards K., Rafael-Fortney J. A., Raman S. V. Cardiac involvement in patients with muscular dystrophies magnetic resonance imaging phenotype and genotypic considerations. Circulation-Cardiovascular Imaging . 2011;4(1):67–76. doi: 10.1161/CIRCIMAGING.110.960740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viola H. M., Davies S. M. K., Filipovska A., Hool L. C. L-type Ca2+channel contributes to alterations in mitochondrial calcium handling in themdxventricular myocyte. American Journal of Physiology-Heart and Circulatory Physiology . 2013;304(6):H767–H775. doi: 10.1152/ajpheart.00700.2012. [DOI] [PubMed] [Google Scholar]
- 18.Johnstone V. P., Viola H. M., Hool L. C. Dystrophic cardiomyopathy-potential role of calcium in pathogenesis, treatment and novel therapies. Genes . 2017;8(4) doi: 10.3390/genes8040108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Westering T. L. E., Betts C. A., Wood M. J. A. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules . 2015;20(5):8823–8855. doi: 10.3390/molecules20058823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Jong S., van Veen T. A. B., van Rijen H. V. M., de Bakker J. M. T. Fibrosis and cardiac arrhythmias. Journal of Cardiovascular Pharmacology . 2011;57(6):630–638. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 21.Corrado G., Lissoni A., Beretta S., et al. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. American Journal of Cardiology . 2002;89(7):838–841. doi: 10.1016/S0002-9149(02)02195-1. [DOI] [PubMed] [Google Scholar]
- 22.Zabłocka B., Górecki D. C., Zabłocki K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. International Journal of Molecular Sciences . 2021;22(20):p. 11040. doi: 10.3390/ijms222011040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duelen R., Costamagna D., Gilbert G., et al. Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem Cell Reports . 2022;17(2):352–368. doi: 10.1016/j.stemcr.2021.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Millay D. P., Sargent M. A., Osinska H., et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nature Medicine . 2008;14(4):442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes M. C., Ramos S. V., Turnbull P. C., et al. Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. The Journal of Physiology . 2020;598(7):1377–1392. doi: 10.1113/JP277306. [DOI] [PubMed] [Google Scholar]
- 26.Bernardi P., Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. Journal of Molecular and Cellular Cardiology . 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haworth R. A., Hunter D. R. The Ca2+-induced membrane transition in mitochondria. Archives of Biochemistry and Biophysics . 1979;195(2):460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 28.Halestrap A. P., Clarke S. J., Javadov S. A. Mitochondrial permeability transition pore opening during myocardial reperfusion - a target for cardioprotection. Cardiovascular Research . 2004;61(3):372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 29.Kroemer G., Galluzzi L., Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews . 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 30.Petronilli V., Penzo D., Scorrano L., Bernardi P., Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Journal of Biological Chemistry . 2001;276(15):12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- 31.Elustondo P. A., Nichols M., Negoda A., et al. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discovery . 2016;2(1):p. 16070. doi: 10.1038/cddiscovery.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonora M., Morganti C., Morciano G., et al. Mitochondrial permeability transition involves dissociation of F1FOATPsynthase dimers and C-ring conformation. Embo Reports . 2017;18(7):1077–1089. doi: 10.15252/embr.201643602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernardi P., Krauskopf A., Basso E., et al. The mitochondrial permeability transition from in vitro artifact to disease target. Febs Journal . 2006;273(10):2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 34.Zorov D. B., Juhaszova M., Sollott S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological Reviews . 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baines C. P., Kaiser R. A., Purcell N. H., et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature . 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 36.Reutenauer J., Dorchies O. M., Patthey-Vuadens O., Vuagniaux G., Ruegg U. T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. British Journal of Pharmacology . 2008;155(4):574–584. doi: 10.1038/bjp.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wissing E. R., Millay D. P., Vuagniaux G., Molkentin J. D. Debio-025 is more effective than prednisone in reducing muscular pathology in _mdx_ mice. Neuromuscular Disorders . 2010;20(11):753–760. doi: 10.1016/j.nmd.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schiavone M., Zulian A., Menazza S., et al. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacological Research . 2017;125(Part B):122–131. doi: 10.1016/j.phrs.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 39.McNally E. M., Passos-Bueno M. R., Bönnemann C. G., et al. Mild and severe muscular dystrophy caused by a single gamma-sarcoglycan mutation. American Journal of Human Genetics . 1996;59(5):1040–1047. [PMC free article] [PubMed] [Google Scholar]
- 40.Coley W. D., Bogdanik L., Vila M. C., et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Human Molecular Genetics . 2016;25(1):130–145. doi: 10.1093/hmg/ddv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vohra R., Batra A., Forbes S. C., Vandenborne K., Walter G. A. Magnetic resonance monitoring of disease progression in _mdx_ mice on different genetic backgrounds. The American Journal of Pathology . 2017;187(9):2060–2070. doi: 10.1016/j.ajpath.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Putten M., Putker K., Overzier M., et al. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. The FASEB Journal . 2019;33(7):8110–8124. doi: 10.1096/fj.201802488R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hakim C. H., Wasala N. B., Pan X., et al. A five-repeat micro-Dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Molecular Therapy-Methods & Clinical Development . 2017;6:216–230. doi: 10.1016/j.omtm.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heydemann A., Ceco E., Lim J. E., et al. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. The Journal of Clinical Investigation . 2009;119(12):3703–3712. doi: 10.1172/JCI39845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van den Bergen J. C., Hiller M., Bohringer S., et al. Validation of genetic modifiers for Duchenne muscular dystrophy: a multicentre study assessingSPP1andLTBP4variants. Journal of Neurology, Neurosurgery, and Psychiatry . 2015;86(10):1060–1065. doi: 10.1136/jnnp-2014-308409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flanigan K. M., Ceco E., Lamar K. M., et al. LTBP4genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Annals of Neurology . 2013;73(4):481–488. doi: 10.1002/ana.23819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodrigues M., Echigoya Y., Maruyama R., Lim K. R., Fukada S. I., Yokota T. Impaired regenerative capacity and lower revertant fibre expansion in dystrophin-deficient _mdx_ muscles on DBA/2 background. Scientific Reports . 2016;6(1, article 38371) doi: 10.1038/srep38371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts N. W., Holley-Cuthrell J., Gonzalez-Vega M., Mull A. J., Heydemann A. Biochemical and functional comparisons of mdx and Sgcg−/− muscular dystrophy mouse models. BioMed Research International . 2015;2015 doi: 10.1155/2015/131436.131436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito T., Ogawa R., Uezumi A., et al. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-marker expression in muscle mesenchymal progenitors. Neuromuscular Disorders . 2013;23(4):349–356. doi: 10.1016/j.nmd.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 50.Takeshita H., Yamamoto K., Nozato S., et al. Modified forelimb grip strength test detects aging-associated physiological decline in skeletal muscle function in male mice. Scientific Reports . 2017;7(1, article 42323) doi: 10.1038/srep42323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomori G. A rapid one-step trichrome stain. American Journal of Clinical Pathology . 1950;20(7):661–664. doi: 10.1093/ajcp/20.7_ts.661. [DOI] [PubMed] [Google Scholar]
- 52.Camerino G. M., Cannone M., Giustino A., et al. Gene expression in mdx mouse muscle in relation to age and exercise: aberrant mechanical-metabolic coupling and implications for pre-clinical studies in Duchenne muscular dystrophy. Human Molecular Genetics . 2014;23(21):5720–5732. doi: 10.1093/hmg/ddu287. [DOI] [PubMed] [Google Scholar]
- 53.Kennedy T. L., Guiraud S., Edwards B., et al. Micro-utrophin improves cardiac and skeletal muscle function of severely affected D2/ mdx mice. Molecular Therapy-Methods & Clinical Development . 2018;11:92–105. doi: 10.1016/j.omtm.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildyard J. C. W., Finch A. M., Wells D. J. Identification of qPCR reference genes suitable for normalizing gene expression in the mdx mouse model of Duchenne muscular dystrophy. PLoS One . 2019;14(1, article e0211384) doi: 10.1371/journal.pone.0211384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Livak K. J., Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔ CT method. Methods . 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 56.Eaton G. J., Custer R. P., Johnson F. N., Stabenow K. T. Dystrophic cardiac calcinosis in mice genetic, hormonal, and dietary influences. The American Journal of Pathology . 1978;90(1):173–186. [PMC free article] [PubMed] [Google Scholar]
- 57.Pan X. D., Chen Z. P., Huang R., Yao Y. Y., Ma G. S. Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoSOne . 2013;8(4, article e60335) doi: 10.1371/journal.pone.0060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Russo V., Papa A. A., Williams E. A., et al. ACE inhibition to slow progression of myocardial fibrosis in muscular dystrophies. Trends in Cardiovascular Medicine . 2018;28(5):330–337. doi: 10.1016/j.tcm.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Kwak H. B. Effects of aging and exercise training on apoptosis in the heart. Journal of Exercise Rehabilitation . 2013;9(2):212–219. doi: 10.12965/jer.130002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Narula J., Pandey P., Arbustini E., et al. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America . 1999;96(14):8144–8149. doi: 10.1073/pnas.96.14.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desai M. S., Shabier Z., Taylor M., et al. Hypertrophic cardiomyopathy and dysregulation of cardiac energetics in a mouse model of biliary fibrosis. Hepatology . 2010;51(6):2097–2107. doi: 10.1002/hep.23585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hardt S. E., Sadoshima J. Glycogen synthase kinase-3β. Circulation Research . 2002;90(10):1055–1063. doi: 10.1161/01.RES.0000018952.70505.F1. [DOI] [PubMed] [Google Scholar]
- 63.Sugden P. H., Fuller S. J., Weiss S. C., Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. British Journal of Pharmacology . 2008;153(Supplement 1):S137–S153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li B., Chi R. F., Qin F. Z., Guo X. F. Distinct changes of myocyte autophagy during myocardial hypertrophy and heart failure: association with oxidative stress. Experimental Physiology . 2016;101(8):1050–1063. doi: 10.1113/EP085586. [DOI] [PubMed] [Google Scholar]
- 65.Fiacco E., Castagnetti F., Bianconi V., et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death and Differentiation . 2016;23(11):1839–1849. doi: 10.1038/cdd.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giordano C., Mojumdar K., Liang F., et al. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Human Molecular Genetics . 2015;24(8):2147–2162. doi: 10.1093/hmg/ddu735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurek J. B., Nouri S., Kannourakis G., Murphy M., Austin L. Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle & Nerve . 1996;19(10):1291–1301. doi: 10.1002/(SICI)1097-4598(199610)19:10<1291::AID-MUS6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 68.Menazza S., Blaauw B., Tiepolo T., et al. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Human Molecular Genetics. . 2010;19(21):4207–4215. doi: 10.1093/hmg/ddq339. [DOI] [PubMed] [Google Scholar]
- 69.Mialet-Perez J., Bianchi P., Kunduzova O., Parini A. New insights on receptor-dependent and monoamine oxidase-dependent effects of serotonin in the heart. Journal of Neural Transmission . 2007;114(6):823–827. doi: 10.1007/s00702-007-0695-7. [DOI] [PubMed] [Google Scholar]
- 70.Kaludercic N., Carpi A., Menabo R., Di Lisa F., Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochimica et Biophysica Acta . 2011;1813(7):1323–1332. doi: 10.1016/j.bbamcr.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan M. C., Rowe G. C., Raghuram S., Patten I. S., Farrell C., Arany Z. Post-natal induction of PGC-1α protects against severe muscle dystrophy independently of utrophin. Skeletal Muscle . 2014;4(1):p. 2. doi: 10.1186/2044-5040-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang A. C. Y., Ong S. G., LaGory E. L., et al. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America . 2016;113(46):13120–13125. doi: 10.1073/pnas.1615340113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mead A. F., Petrov M., Malik A. S., et al. Diaphragm remodeling and compensatory respiratory mechanics in a canine model of Duchenne muscular dystrophy. Journal of Applied Physiology . 2014;116(7):807–815. doi: 10.1152/japplphysiol.00833.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kharraz Y., Guerra J., Pessina P., Serrano A. L., Munoz-Canoves P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Research International . 2014;2014 doi: 10.1155/2014/965631.965631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tandon A., Villa C. R., Hor K. N., et al. Myocardial fibrosis burden predicts left ventricular ejection fraction and is associated with age and steroid treatment duration in Duchenne muscular dystrophy. Journal of the American Heart Association . 2015;4(4, article e001338) doi: 10.1161/JAHA.114.001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blot protein expression assays (Supplement 1).
Data Availability Statement
Data can be found in the manuscript and supplementary data file.