

Abstract
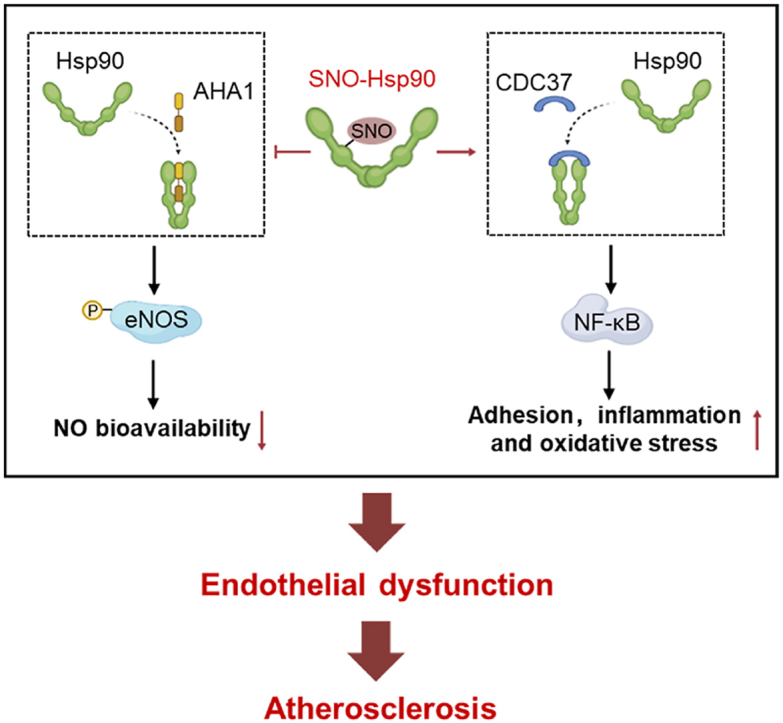
Endothelial dysfunction is the initial process of atherosclerosis. Heat shock protein 90 (Hsp90), as a molecular chaperone, plays a crucial role in various cardiovascular diseases. Hsp90 function is regulated by S-nitrosylation (SNO). However, the precise role of SNO-Hsp90 in endothelial dysfunction during atherosclerosis remains unclear. We here identified Hsp90 as a highly S-nitrosylated target in endothelial cells (ECs) by biotin switch assay combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS). The elevation of SNO-Hsp90 was observed in atherosclerotic human and rodent aortas as well as in oxidized LDL (oxLDL)-treated ECs. Inhibition of inducible nitric oxide synthase (iNOS) or transfection with Hsp90 cysteine 521 (Cys521) mutation plasmid decreased the level of SNO-Hsp90 in oxLDL-cultured ECs. Coimmunoprecipitation and proximity ligation assay demonstrated that SNO-Hsp90 at Cys521 suppressed the interaction between Hsp90 and activator of Hsp90 ATPase activity 1 (AHA1), but promoted the association of Hsp90 and cell division cycle 37 (CDC37). Hsp90 Cys521 mutation increased endothelial nitric oxide synthase (eNOS) activity and inhibited nuclear factor kappa-B (NF-κB) signaling, thereby increasing nitric oxide (NO) bioavailability and alleviating endothelial adhesion, inflammation and oxidative stress in oxLDL-treated ECs. Also, administration of endothelial-specific adeno-associated viruses of Cys521-mutated Hsp90 significantly mitigated vascular oxidative stress, macrophage infiltration and atherosclerosis lesion areas in high fat diet-fed ApoE-/- mice. In conclusion, SNO-Hsp90 at Cys521, that serves as a conformational switch, disrupts Hsp90/AHA1 interaction but promotes recruitment of CDC37 to exacerbate atherosclerosis.
Keywords: Heat shock protein 90, S-nitrosylation, Atherosclerosis, Oxidative stress, Endothelial dysfunction
Graphical abstract
Highlights
-
•
Hsp90 S-nitrosylation at Cys521 acts as a conformational switch to modulate Hsp90/AHA1 and Hsp90/CDC37 interaction.
-
•
SNO-Hsp90 induces endothelial adhesion, inflammation and oxidative stress.
-
•
SNO-Hsp90 mediates endothelial dysfunction to exacerbate atherosclerosis.
1. Introduction
Atherosclerosis is a chronic inflammatory disease and initially caused by endothelial dysfunction. Endothelial cells (ECs), continuously locating in the lumen of blood vessels, act as a pivotal regulatory node for homeostatic vascular network. Endothelial dysfunction involves upregulation of chemokines and adhesion molecules, the focal permeation, recruitment of circulating monocytes into the intima to become foam cells, ultimately leads to formation of atherosclerotic lesion [1,2].
Nitric oxide (NO), as a gasotransmitter, is essential for maintaining the vascular homeostasis [3]. Except for the conventional function in mediating vasodilation, it is acknowledged that NO can react with reactive cysteine thiols of target proteins to form nitrosothiols, termed as S-nitrosylation (SNO)-a covalent posttranslational modification [4]. It is well verified that SNO mediated by NO is crucial for cardiovascular system homeostasis via regulating enzymatic activity, protein-protein interaction (PPI) or signal transduction of targeted proteins [[5], [6], [7]]. For all that, the precise role of SNO in the endothelial dysfunction and atherosclerotic cardiovascular disease requires in-depth investigation.
Heat shock protein 90 (Hsp90), as a molecular chaperone, functions as promoting the maturation, structural maintenance and proper regulation of specific client proteins involved for instance enzymatic activity and signal transduction to further participate in diverse cellular processes in cardiovascular system [8,9]. The interaction of Hsp90 with distinct client proteins is regulated by posttranslational modifications, including phosphorylation, acetylation, SUMOylation and also S-nitrosylation [10,11]. Our previous studies have demonstrated that S-nitrosylation of Hsp90 (SNO-Hsp90) modulates its interaction with downstream chaperones to aggravate cardiac hypertrophy and fibrosis [12,13]. Although SNO-Hsp90 induces the inhibition of its ATPase and chaperone activity [14,15], the precise role and mechanism of SNO-Hsp90 in endothelial dysfunction and atherosclerosis remain unclear.
In this study, using biotin switch assay combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS) for detection of S-nitrosylated proteins and determination of S-nitrosylated sites, we identify that Hsp90 is also a S-nitrosylated target in endothelium. By selectively site-directed mutagenesis of S-nitrosylated cysteine residue in Hsp90 protein, we illustrate a mechanism by which the S-nitrosylation at Cys521 breaks Hsp90 conformational cycle equilibrium to decrease NO bioavailability and facilitate adhesion, inflammation and oxidative stress in endothelium to exacerbate atherosclerosis.
2. Materials and methods
2.1. Experimental animals
Male apolipoprotein E knockout (ApoE-/-) mice of 4 or 8 weeks old were purchased from the Changzhou Cavens Laboratory Animal Co. Ltd. (Changzhou, China), and housed at a constant temperature (22 ± 1 °C) and 55–60% relative humidity under a 12-h light/dark cycle with free access to food and water.
Endothelial-enhanced adeno-associated virus (AAVendo) packaging was achieved according to a protocol from Yu Huang (Chinese University of Hong Kong, China) [16]. The shuttle RGDLRVS-AAV9-cap plasmid was a gift from O. J. Müller (Universität Heidelberg, Germany). The AAVendo driving the expression of wild-type Hsp90 (AAVendo-Hsp90-WT) or Cys521 S-nitrosylation site-mutated Hsp90 (AAVendo-Hsp90-C521A, Cys521 mutated to alanine) was constructed by Kunshan Renyuan Biotechnology Company (Suzhou, China). Male ApoE-/- mice of 4 weeks old were injected with AAVendo-Hsp90-WT or AAVendo-Hsp90-C521A via tail veins at a dose of 5 × 1011 GC/mouse. Four weeks after injection, mice were fed a normal chow (NC) or a high fat diet (HFD) for eight weeks. All animal experiments were approved by the Committee on Animal Care of Nanjing Medical University and were conducted according to the NIH Guidelines for the Care and Use of Laboratory Animals. All studies involving animals are reported in accordance with the ARRIVE guidelines.
2.2. Human artery specimens
Human atherosclerotic artery samples were obtained from patients undertaking coronary artery bypass grafting or carotid endarterectomy. Non-atherosclerotic artery samples were obtained from patients undergoing aortic valve replacement. Informed consent was obtained from each patient prior to surgery. All studies involving human samples were approved by the Ethics Committee of the Affiliated Drum Tower Hospital of Nanjing University Medical School.
2.3. Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords according to a previously described method [17] and were cultured in complete endothelial cell medium (ECM) (ScienCell, Carlsbad, USA) supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth medium supplement mix (ECGS) and 1% penicillin/streptomycin. HUVECs within seven passages were used in vitro studies. EA.hy926 endothelial cell line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and maintained in DMEM (Gibco, Carlsbad, USA) supplemented with 10% (v/v) FBS (Gibco, Carlsbad, USA). Primary mouse aortic endothelial cells (MAECs) were isolated from the aorta of wild-type or iNOS-/- mice. In brief, aortas were quickly collected and transferred into HBSS (Gibco). Then, the aortas were longitudinally opened to expose the endothelium and cut into small segments, which were placed onto gelatin-coated plates. After five days, the aortic segments were removed, and endothelial cells were resuspended. MAECs were cultured in ECM and used between passages two and five. Confluent cells (80–85%) were incubated with oxidized LDL (oxLDL; 100 μg/mL; Yiyuan Biotechnology, China).
2.4. Plasmid transfection
The plasmid containing HA-tagged Hsp90-WT was purchased from Addgene (Cambridge, MA). Single mutation at Cys521 to Ala (Hsp90-C521A, Haibio, Shanghai, China) was confirmed by DNA sequencing. Plasmids were transfected into 75% confluent HUVECs and EA.hy926 cells with Lipofectamine 3000 reagent (Invitrogen, Carlsbad, USA). After 4–6 h of transfection, medium was changed to fresh ECM, and cells were maintained for an additional 24 h before use in experiments.
2.5. Biotin switch assay
S-nitrosylation of Hsp90 (SNO-Hsp90) was determined by biotin switch assay with S-nitrosylation Protein Detection Assay Kit (Cayman Chemical; item No. 10006518) as described previously [7]. Briefly, cell lysates were incubated with blocking buffer for 30 min to block free thiols and proteins were precipitated with cold acetone at −20 °C for 1 h. Then, the S-nitrosothiols were reduced to free thiols with reducing buffer (Vitamin C, Vc) and labelled with biotin. Samples without reducing buffer incubation were used as negative controls (-Vc). The biotinylated proteins were purified by incubating with avidin-coupled agarose beads (Thermo Scientific) overnight at 4 °C. The SNO-Hsp90 level was detected by western blotting with anti-Hsp90 or anti-HA antibody.
2.6. Mass spectrometry for S-nitrosylated proteins identification
Cell lysates from endothelial cells incubated with oxLDL (100 μg/mL, 24 h) were blocked with N-ethylmaleimide and labelled with biotin-maleimide. Then, the biotinylated proteins were immunoprecipitated with avidin-coupled agarose beads and trypsinized (using a 1:50 ratio of protein:trypsin) for liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis performed by the Analysis Center, Nanjing Medical University.
2.7. RNA-sequence analysis
HUVECs transfected with Hsp90-WT or Hsp90-C521A, followed by oxLDL treatment (100 μg/mL, 24 h) were extracted for RNA isolation by TRIzol™ Reagent (Invitrogen). The concentration and integrity of RNA were assessed using Qubit Fluorometer (Invitrogen) and Bioanalyzer 2100 system (Agilent), respectively. The 23S and 16S rRNA was depleted using a MICROB Express kit (Ambion). Genomic DNA was removed with two digestions using amplification-grade DNase 1 (Invitrogen). cDNA libraries were generated using NEB Next Ultra Directional RNA Library Prep Kit (NEB) following the manufacturer's instructions. Then, the library was used for sequencing with the Illumina sequencing technology on an Illumina Hiseq2500.
2.8. Proximity Ligation Assay (PLA)
The interactions of Hsp90 with AHA1 or CDC37 in HUVECs were detected by Duolink® Proximity Ligation Assay Kit (Sigma). Endothelial cells were transfected with Hsp90-WT or Hsp90-C521A, followed by oxLDL treatment (100 μg/mL, 24 h), and then fixed, permeabilized and incubated with primary antibodies such as in immunofluorescence assays. The following steps were performed as recommended by the manufacturer of PLA Kit. Quantification of fluorescence intensity was performed by using ImageJ software.
2.9. En face analysis of atherosclerosis and plaque histology
Mouse aortas were collected from the base ascending aorta to the iliac bifurcation. Atherosclerosis lesion in whole aortas was detected by Oil Red O (ORO) staining. The sections from the aortic roots of each mouse were stained with ORO to show the atherosclerotic lesion. The plaque areas were quantified with ImageJ software.
2.10. Assessment of endothelium-dependent relaxation
Endothelium-dependent relaxation was detected by wire myograph (DMT-620 M, Denmark). Briefly, isolated mouse aortic ring segments were incubated in organ baths containing warmed (37 °C), oxygenated Kreb's solution and stretched to a resting tension of 9.8 mN for 1 h. After equilibration for 60 min, aortic ring segments were precontracted by adding 80 mM KCl for 15 min, and washed by Krebs' solution. Then, norepinephrine (NE, 0.1 μM) was added for vasoconstriction. Once NE-induced steady tension, acetylcholine (Ach, 10−9-10−5 M) was added cumulatively to induce endothelium-dependent relaxation. Relaxation at each concentration was calculated as the percentage of force generated in response to NE.
2.11. Monocyte adhesion assay
THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS. For adhesion assay, THP-1 cells were first labelled with CM-Dil (2.5 μg/mL) for 10 min and then incubated with HUVECs with different treatments for 60 min at 37 °C. Nonadherent THP-1 cells were washed away with PBS for 3 times. Fluorescence images of adherent THP-1 cells were captured and analyzed using ImageJ software to estimate the number of adherent monocytes.
2.12. Monocyte transmigration assay
HUVECs were cultured in 3-μm-pore-size Transwell chambers (Millipore) to form a confluent monolayer with complete ECM containing 5% FBS by different treatments. THP-1 cells were labelled with CM-Dil (2.5 μg/mL) for 10 min and then added onto upper chamber and allowed to migrate over for 4 h. The migrated THP-1 cells were collected and counted from the lower chamber with fluorescence intensity measured by a plate reader (Spectramax M2, Molecular Devices, CA, USA).
2.13. Measurement of cyclic GMP (cGMP) level
cGMP levels in HUVECs were measured using the Cyclic GMP Complete ELISA Kit (Abcam), following the manufacturer's instructions and the OD absorbance was read by a plate reader (Spectramax M2, Molecular Devices).
2.14. Immunofluorescence staining
Sections or cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, blocked with 5% BSA, and then incubated with indicated antibodies against HA (Santa Cruz, California, USA), AHA1 (Abcam, Cambridge, UK), CDC37 (Abcam) or CD68 (CST, MA, USA) overnight at 4 °C. Alexa Fluor® 488- or 594-conjugated fluorescent secondary antibodies (Life Technologies, Thermo Fisher Scientific) were incubated at 37 °C for 1 h, and nuclei were counterstained with DAPI (Santa Cruz). Images were captured using a confocal microscope (Zeiss LSM 410, Oberkochen, Germany).
2.15. Measurement of ROS formation
Superoxide production in endothelial cells or aorta sections was detected with dihydroethidium (DHE) (Beyotime Biotechnology, China). Briefly, cells or tissue samples were incubated with DHE (1 μM) for 30 min. Fluorescence was detected with an OLYMPUS BX53 fluorescence microscope.
2.16. Detection of superoxide dismutase (SOD) and NADPH oxidase (NOX) activity
The endothelial SOD and NOX activity were measured to assess intercellular oxidative stress with commercial kits (Jiancheng Bioengineering Ltd, China) according to the manufacturer's instructions.
2.17. Detection of malondialdehyde (MDA) level
The serum MDA level in HFD-fed ApoE-/- mice with AAVendo-Hsp90-WT or AAVendo-Hsp90-C521A injection was measured with commercial kits (Beyotime Biotechnology) according to the manufacturer's instructions.
2.18. Western blotting
Equal amounts of protein were resolved in 8% or 10% SDS-PAGE and western blotting analysis was performed as previously described [5]. Primary antibodies included anti-Hsp90 antibody (Abcam), anti-iNOS antibody (CST), anti-eNOS antibody (CST), anti-p-eNOS antibody (CST), anti-GSNOR antibody (Abcam), anti-Trx antibody (Abcam), anti-HA antibody (Santa Cruz), anti-AHA1 antibody (Abcam), anti-CDC37 antibody (Abcam), anti-IκB antibody (Abcam), anti-p-IκB antibody (Abcam), anti-p65 antibody (Abcam), anti-p-p65 antibody (Abcam) and anti-GAPDH (CST). Band intensities were analyzed by ImageJ software.
2.19. Coimmunoprecipitation
Proteins of mouse aortic tissue or endothelial cells were extracted in lysis buffer containing 40 mM HEPES (pH 7.4), 2 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 0.5% Triton and 1% protease inhibitor cocktail. Supernatants were harvested by centrifugation at 12000 g for 10 min. Whole-cell lysates (50 μL) were loaded as input sample. The remaining cell lysates were incubated with indicated antibody overnight at 4 °C, followed by precipitation with protein A/G-agarose beads for 4 h at 4 °C. All samples were subjected to SDS-PAGE separation and western blotting with the indicated antibodies.
2.20. Real-time quantitative PCR (qPCR) analysis
Total RNA was extracted with Trizol (Takara Bio, Shiga, Japan) and converted to cDNA using HiScript II Q RT SuperMix for qPCR Kit (Vazyme, Nanjing, China). The purity of total RNA was evaluated based on the absorbance ratio at 260 and 280 nm. Real-time quantitative PCR reactions was performed by SYBR Green (Vazyme) on a QuantStudio qPCR System (Thermo Fisher Scientific Inc). The primer sequences used for qPCR are listed in Table 1.
Table 1.
The primer sequences used in this study.
| mouse Icam1 | Forward: GTGATGCTCAGGTATCCATCCA |
| Reverse: CACAGTTCTCAAAGCACAGCG | |
| mouse Vcam1 | Forward: AGTTGGGGATTCGGTTGTTCT |
| Reverse: CCCCTCATTCCTTACCACCC | |
| mouse Sele | Forward: ATGCCTCGCGCTTTCTCTC |
| Reverse: GTAGTCCCGCTGACAGTATGC | |
| mouse Ccl2 | Forward: TTAAAAACCTGGATCGGAACCAA |
| Reverse: GCATTAGCTTCAGATTTACGGGT | |
| mouse Cxcl2 | Forward: GGCGGTCAAAAAGTTTGCCT |
| Reverse: TTCTTCCGTTGAGGGACAGC | |
| mouse Gapdh | Forward: AGGTCGGTGTGAACGGATTTG |
| Reverse: TGTAGACCATGTAGTTGAGGTCA | |
| human ICAM1 | Forward: TGACCGTGAATGTGCTCTCC |
| Reverse: TCCCTTTTTGGGCCTGTTGT | |
| human VCAM1 | Forward: AATGCCTGGGAAGATGGTCG |
| Reverse: GATGTGGTCCCCTCATTCGT | |
| human SELE | Forward: TGATCTTTCCCGGAACTGCC |
| Reverse: TGGTGAGGTGTGCTCATTCC | |
| human CCL2 | Forward: TTTGCTTGTCCAGGTGGTCC |
| Reverse: GATCTCAGTGCAGAGGCTCG | |
| human CXCL2 | Forward: TCTCTGCTCTAACACAGAGGGA |
| Reverse: AGATCAATGTGACGGCAGGG | |
| human CXCL8 | Forward: TTCTCAGCCCTCTTCAAAAACT |
| Reverse: ACTCCAAACCTTTCCACCCC | |
| human GAPDH | Forward: GGAGCGAGATCCCTCCAAAAT |
| Reverse: GGCTGTTGTCATACTTCTCATGG |
2.21. Statistical analysis
All values are presented in the figures as mean ± standard error of the mean (SEM), with *P < 0.05, **P < 0.01, ***P < 0.001. Unpaired two-tailed Student's t-test was used for comparisons between two groups when data passed normality and equal variance test, otherwise Mann–Whitney U test was used. Differences among groups were evaluated using one-way ANOVA followed by Turkey's post-hoc test. All graphs and statistical analyses were performed by GraphPad Prism 8.
3. Results
3.1. SNO-Hsp90 is elevated in atherosclerotic aortas and oxLDL-treated ECs
To discover the S-nitrosylated protein targets that may participate in atherosclerosis, first, we purified the S-nitrosylated proteins from ECs exposed to oxLDL through biotin switch and then scanned the S-nitrosylated proteins by LC-MS/MS analysis. The result showed that Hsp90 was identified as a highly S-nitrosylated protein upon oxLDL treatment (Fig. 1A). For further verification, we assessed the SNO-Hsp90 in arteries from patients with atherosclerosis and found that the level of SNO-Hsp90 was significantly increased in atherosclerotic samples (Fig. 1B). Then, we fed ApoE-/- mice with HFD for eight weeks to induce atherosclerosis and detected the level of SNO-Hsp90 in mouse aortas. The result showed that, compared with control groups, the SNO-Hsp90 was obviously elevated in atherosclerotic ApoE-/- mice (Fig. 1C). Meanwhile, the elevation of SNO-Hsp90 was also observed in oxLDL-treated HUVECs (Fig. 1D). These results reveal that the endothelial S-nitrosylation of Hsp90 is increased in atherosclerosis.
Fig. 1.

SNO-Hsp90 levels are significantly increased in atherosclerotic aortas and oxLDL-treated endothelial cells. A: Liquid chromatography-tandem mass spectrometry (LC–MS/MS) scans of S-nitrosylated proteins in endothelial cells were performed. Representative LC–MS/MS spectra showed a target modification site (Cys521) in the peptide fragmentation of Hsp90. B: The SNO-Hsp90 levels in non-atherosclerotic (Non-AS) or atherosclerotic (AS) arteries from patients were detected by biotin switch. (n = 3 for Non-AS, n = 5 for AS, ***P < 0.001). C: Eight-week-old ApoE-/- mice were fed with high fat diet (HFD) for eight weeks. The SNO-Hsp90 levels in mice aortas were detected by biotin switch. (n = 6, ***P < 0.001). D: HUVECs were treated by oxidized LDL (oxLDL; 100 μg/mL) for 24 h. The SNO-Hsp90 levels were detected by biotin switch. (n = 6, ***P < 0.001).
3.2. iNOS contributes to the S-nitrosylation of Hsp90 at Cys521
The cellular S-nitrosylation of proteins is strictly regulated by several enzymes. Three isoforms of nitric oxide synthase (NOS) - neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS) derived NO promotes the protein S-nitrosylation [18]. Besides, thioredoxin (Trx) and S-nitrosoglutathione reductase (GSNOR) are two main enzymes known to mediate the de-nitrosylation through converting nitrosothiol (-SNO) to sulfydryl (-SH) by reduction reactions [19,20]. To explore the upstream enzyme that is responsible for the elevation of SNO-Hsp90 in atherosclerosis, we detected the expression of these enzymes in oxLDL-treated ECs. Compared with control groups, iNOS expression was significantly increased and eNOS activity (showed by phosphorylated eNOS) was reduced upon oxLDL treatment, whereas Trx and GSNOR protein levels were unchanged (Fig. 2A). Moreover, we found that both 1400W (an iNOS inhibitor) treatment and iNOS knockout reversed the elevated SNO-Hsp90 induced by oxLDL in ECs (Fig. 2B and C), suggesting the enhanced SNO-Hsp90 in endothelium is triggered by iNOS under pathological condition. The peptide of Hsp90 identified by mass spectrometry showed that Hsp90 S-nitrosylation occurs at the Cys521 (Fig. 1A). For further verification, we constructed mutagenesis plasmids by substituting Cys521 of Hsp90 with the non-nitrosylable alanine (C521A) (Fig. 2D). ECs were transfected with Hsp90-WT and Hsp90-C521A, followed by oxLDL treatment. The biotin switch assay showed that Cys521 mutation totally inhibited the increased SNO-Hsp90 induced by oxLDL (Fig. 2E), confirming that Cys521 is the SNO site of Hsp90.
Fig. 2.

iNOS mediates the S-nitrosylation of Hsp90 at Cys521. A: HUVECs were treated by oxLDL (100 μg/mL) for 24 h. The levels of iNOS, p-eNOS, eNOS, GSNOR and TRX were detected by Western blot. (n = 5, ***P < 0.001). B: HUVECs were pre-incubated with 1400W (an iNOS inhibitor, 10 μM) for 1 h followed by treatment with oxLDL for 24 h. The SNO-Hsp90 levels were detected by biotin switch. (n = 5, ***P < 0.001). C: Primary mouse aortic endothelial cells (MAECs) were isolated from WT and iNOS-/- mice, followed by oxLDL treatment for 24 h. The SNO-Hsp90 levels were detected by biotin switch. (n = 3, **P < 0.01). D: Construction of mutagenesis Hsp90 plasmids by substituting the Cys521 with the non-nitrosylable alanine. E: EA.hy926 endothelial cells were transfected with Hsp90-WT or Hsp90-C521A plasmids for 24 h, followed by exposure to oxLDL for 24 h. The SNO-Hsp90 levels were detected by biotin switch. (n = 6, ***P < 0.001).
3.3. SNO-Hsp90 at Cys521 reduces endothelial NO bioavailability via Hsp90/AHA1/eNOS pathway
The defective phosphorylation of eNOS is a hallmark of endothelial dysfunction [21], while the activation of eNOS in ECs is dependent on the association between Hsp90 and its primary chaperone-AHA1 [22]. Co-IP assay showed the decreased interaction of Hsp90 and AHA1 was reversed by Cys521 mutation in oxLDL-cultured ECs (Fig. 3A). Hsp90 contains an N-terminal ATP-binding domain, a C-terminal dimerization domain and a middle domain that is known to bind the different client proteins [23]. AHA1 binds to the middle domain of Hsp90 [24] and the S-nitrosylation occurs at Cys521, that locates in the middle domain of Hsp90, which explains the repressive binding of AHA1 to Hsp90 after S-nitrosylation. The results of proximity ligation assay and immunofluorescence also showed that, compared with Hsp90-WT transfected ECs, Cys521 mutation enhanced the association of Hsp90 and AHA1 upon oxLDL stimulation (Fig. 3B and Fig. S1). Meanwhile, oxLDL treatment suppressed the phosphorylation of eNOS in Hsp90-WT transfected ECs and this effect was abolished by Cys521 mutation (Fig. 3C). NO/soluble guanylate cyclase (sGC)/cyclic guanosine monophosphate (cGMP) signaling is essential for endothelial cell function. Our data showed that Hsp90 Cys521 mutation could prevent the decrease of cGMP content under oxLDL treatment (Fig. 3D). Taken together, these results demonstrate that SNO-Hsp90 at Cys521 inhibits the interaction of Hsp90 and AHA1, thereby suppressing the eNOS phosphorylation to decrease NO bioavailability in ECs (Fig. 3E).
Fig. 3.

Cys521 S-nitrosylation restrains Hsp90 and AHA1 interaction to inhibit eNOS activity. Endothelial cells were transfected with Hsp90-WT or Hsp90-C521A plasmids for 24 h, followed by exposure to oxLDL for 24 h. A: Lysates from EA.hy926 endothelial cells were immunoprecipitated with an anti-AHA1 antibody and blotted with anti-HA(Hsp90) and anti-AHA1 antibody. An aliquot of total lysate was analyzed for HA(Hsp90) and AHA1 expression. (n = 5, *P < 0.05). B: Specific interactions (red spots) of Hsp90 with AHA1 in HUVECs were determined by proximity ligation assay (PLA). (Blue: DAPI, Scale bars = 20 μm, n = 3, *P < 0.05, **P < 0.01). C: The expression of p-eNOS and eNOS in HUVECs was detected by Western blot. (n = 5, **P < 0.01). D: The cGMP levels in HUVECs were measured by Cyclic GMP Complete ELISA Kit. (n = 5, *P < 0.05, ***P < 0.001). E: Schematic diagram showed that SNO-Hsp90 inhibits the Hsp90/AHA1 interaction to decrease eNOS phosphorylation.
3.4. SNO-Hsp90 at Cys521 induces endothelial adhesion, inflammation and oxidative stress via Hsp90/CDC37/NF-κB signaling
To further investigate the effect and mechanism of SNO-Hsp90 on atherosclerotic endothelial dysfunction, we performed RNA-sequencing (RNA-seq) analysis in oxLDL-treated ECs with Hsp90-WT or Hsp90-C521A transfection to scan the differentially expressed genes. There were 121 genes with significantly differential expression among WT, WT + oxLDL and C521A + oxLDL groups (Fig. 4A). GO biological processes analysis showed that these genes mainly related to cytokine-mediated signaling, cell-cell adhesion, IκB kinase/NF-κB signaling, inflammatory and immune response (Fig. 4B). KEGG pathway analysis further revealed that NF-κB signaling was the most dominating pathway regulated by SNO-Hsp90 (Fig. 4C).
Fig. 4.

Cys521 S-nitrosylation promotes Hsp90 and CDC37 interaction to activate NF-κB signaling. Endothelial cells were transfected with Hsp90-WT or Hsp90-C521A plasmids for 24 h, followed by exposure to oxLDL for 24 h. A: RNA-sequence analysis identified the 121 genes with significantly differential expression between WT, WT + oxLDL and C521A + oxLDL groups. B: The GO analysis for these 121 genes in biological processes. C: The KEGG analysis for these 121 genes in signaling pathway. D: Lysates from EA.hy926 endothelial cells were immunoprecipitated with an anti-CDC37 antibody and blotted with anti-HA(Hsp90) and anti-CDC37 antibody. An aliquot of total lysate was analyzed for HA(Hsp90) and CDC37 expression. (n = 5, ***P < 0.001). E: Specific interactions (red spots) of Hsp90 with CDC37 in HUVECs were determined by proximity ligation assay (PLA). (Blue: DAPI, Scale bars = 20 μm, n = 3, *P < 0.05). F: The expression of p-IκB, IκB, p-p65 and p65 in HUVECs was detected by Western blot. (n = 5, *P < 0.05). G: Nuclear extracts prepared from HUVECs were subjected to western blotting analysis for detecting the nuclear localization of p65. (n = 3, *P < 0.05, **P < 0.01). H: Schematic diagram showed that SNO-Hsp90 promotes the interaction of Hsp90 and CDC37 to activate NF-κB signaling pathway.
The function of Hsp90 depends on proceeding through a cycle of conformational changes by recruiting either AHA1 or CDC37 circularly [25]. CDC37, as another crucial co-chaperone of Hsp90, is essential for assembly of Hsp90/CDC37/IKK complex to further mediate NF-κB signaling activation [26,27]. Our result showed that oxLDL increased the interaction of Hsp90 with CDC37 in Hsp90-WT-transfected but not Hsp90-C521A-transfected ECs (Fig. 4D). The proximity ligation assay showed that, compared with Hsp90-WT transfected ECs, Cys521 mutation inhibited the association of Hsp90 and CDC37 upon oxLDL stimulation (Fig. 4E). The similar phenomenon was also observed by immunofluorescence (Fig. S2). Meanwhile, the phosphorylated IκB and p65 (subunit of NF-κB) levels (Fig. 4F), as well as the nuclear p65 level (Fig. 4 G) were increased after oxLDL stimulation in Hsp90-WT-transfected but not in Hsp90-C521A-transfected ECs, demonstrating the SNO-Hsp90 at Cys521 promotes the interaction of Hsp90 and CDC37 to activate NF-κB signaling (Fig. 4H). The expressions of cellular adhesion molecules and inflammatory factors modulated by NF-κB play pivotal roles in inflammation response and oxidative stress [28,29]. We found that Hsp90 Cys521 mutation abolished the upregulation of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), selectin-E (SELE), C–C motif chemokine ligand 2 (CCL2), C-X-C motif chemokine ligand 2 (CXCL2) and C-X-C motif chemokine ligand 8 (CXCL8) induced by oxLDL in ECs (Fig. 5A). OxLDL treatment significantly increased the adhesion and transmigration of monocytes to ECs, which were attenuated by Cys521 mutation (Fig. 5B and C). Cys521 mutation also increased endothelial SOD activity (Fig. 5D), mitigated NOX activity (Fig. 5E) and decreased intracellular MDA level (Fig. 5F) under oxLDL treatment. Meanwhile, the DHE staining and electron paramagnetic resonance (EPR) analysis revealed that Cys521 mutation decreased the endothelial ROS level upon oxLDL stimulation (Fig. 5G and Fig. S3). These results demonstrate that the S-nitrosylation of Hsp90 at Cys521 promotes the interaction of Hsp90 and CDC37 to activate NF-κB signaling, further mediating the endothelial adhesion, inflammatory response and oxidative stress.
Fig. 5.

Cys521 mutation attenuates the endothelial adhesion, inflammation and oxidative stress upon oxLDL treatment. Endothelial cells transfected with Hsp90-WT or Hsp90-C521A plasmids for 24 h, followed by exposure to oxLDL for 24 h. A: The mRNA levels of ICAM1, VACM1, SELE, CCL2, CXCL2 and CXCL8 in HUVECs were detected by qPCR. (n = 5, *P < 0.05, **P < 0.01, ***P < 0.001). B: The representative images and quantification of THP-1 monocytes adhesion to HUVECs. (Scale bars = 200 μm n = 5, ***P < 0.001). C: The quantification of THP-1 monocytes transmigration to HUVECs. (n = 5, **P < 0.01, **P < 0.001). D: The SOD activity in HUVECs was detected by SOD assay kit. (n = 5, ***P < 0.001). E: The NOX activity in HUVECs was detected by NOX assay kit. (n = 5, *P < 0.05, ***P < 0.001). F: The MDA level in HUVECs was detected by MDA assay kit. (n = 3, **P < 0.01). G: The ROS level in HUVECs was detected by DHE staining. Recombinant SOD (2000 U/mL, 50 μL) was incubated for 3 h as a control. (Scale bars = 200 μm n = 3, **P < 0.01, ***P < 0.001).
3.5. Inhibition of SNO-Hsp90 by Cys521 mutation alleviates atherosclerosis in ApoE-/- mice
To verify the effect of SNO-Hsp90 on atherosclerosis in vivo, ApoE-/- mice were injected with endothelial cell-enhanced AAV vector (AAVendo) containing Hsp90-WT or Hsp90-C521A and subjected to normal chow (NC) or HFD (Fig. 6A). Biotin switch assay showed delivering AAVendo-Hsp90-C521A significantly inhibited the aortic SNO-Hsp90 after HFD treatment (Fig. 6B). Accordingly, Hsp90 Cys521 mutation reversed the suppression of endothelial Hsp90/AHA1 interaction and the promotion of Hsp90/CDC37 complex formation in atherosclerotic mice (Fig. S4 and Fig. S5). Compared to AAVendo-Hsp90-WT, Hsp90 Cys521 mutation alleviated the impairment in endothelium-dependent relaxation in HFD-fed ApoE-/- mice (Fig. 6C). Meanwhile, AAVendo-Hsp90-C521A-delivered mice exhibited less atherosclerotic lesions than AAVendo-Hsp90-WT, as showed by Oil Red O staining of whole aortas and aortic roots (Fig. 6D and E). Delivery of AAVendo-Hsp90-C521A also reduced macrophage infiltration in aorta roots from HFD-fed ApoE-/- mice (Fig. 6F). Hsp90 Cys521 mutation attenuated ROS level both in aortic rings (Fig. 6G and Fig. S6) and in plaques of aortic roots (Fig. S7), decreased serum MDA level (Fig. 6H) and reduced the expressions of adhesion molecules and inflammatory cytokines in aortas (Fig. 6I). In addition, Cys521 mutation had no effect on lipid metabolism in atherosclerotic ApoE-/- mice (Table 2). These data verify that inhibition of SNO-Hsp90 by Cys521 mutation significantly improves endothelial function to protect against atherosclerosis.
Fig. 6.

Hsp90 Cys521 mutation improves atherosclerosis in ApoE-/-mice. A: Four-week-old ApoE-/- mice were intravenously injected with AAVendo-Hsp90-WT or AAVendo-Hsp90-C521A. Four weeks later, these mice were fed with normal chow (NC) or high fat diet (HFD) for eight weeks. B: The SNO-Hsp90 levels in mice aortas were detected by biotin switch. (n = 5, ***P < 0.001). C: Endothelium-dependent vasorelaxation to acetylcholine (Ach) of precontracted aorta was assessed. (n = 5, *p < 0.05). D: The Oil Red O (ORO) staining of mice whole aortas. (Scale bars = 5 mm, n = 5, ***P < 0.001). E: ORO staining of mice aortic roots. (Scale bars = 500 μm, n = 5, ***P < 0.001). F: The macrophage infiltration was detected by immunofluorescence of CD68 (red) in plaque (dotted outline) of aorta roots. (Blue: DAPI, Scale bars = 100 μm, n = 5, ***P < 0.001). G: The ROS level in aortas was detected by DHE staining. (Scale bars = 200 μm, n = 3, ***P < 0.001). H: The MDA level in serum was detected by MDA assay kit. (n = 5, **P < 0.01, ***P < 0.001). I: The mRNA levels of Icam1, Vcam1, Sele, Ccl2, and Cxcl2 in aortas were detected by qPCR. (n = 5, *P < 0.05, **P < 0.01, ***P < 0.001).
Table 2.
Lipid profile of serum in AAVendo-Hsp90-WT or AAVendo-Hsp90-C521A delivered ApoE−/- mice fed with HFD for 8 weeks.
| Hsp90-WT +NC |
Hsp90-WT +HFD |
Hsp90-C521A +NC |
Hsp90-C521A +HFD |
|
|---|---|---|---|---|
| T-CHO (mmol/L) | 17.46 ± 1.630 | 24.79 ± 2.404* | 18.30 ± 1.319 | 23.92 ± 1.372* |
| TG (mmol/L) | 1.420 ± 0.254 | 1.726 ± 0.159 | 1.382 ± 0.236 | 1.788 ± 0.162 |
| LDL-C (mmol/L) | 3.120 ± 1.449 | 8.562 ± 1.030* | 3.306 ± 0.925 | 8.900 ± 1.672* |
| HDL-C (mmol/L) | 0.456 ± 0.092 | 0.346 ± 0.022 | 0.442 ± 0.075 | 0.380 ± 0.080 |
Values are shown as means ± SD (n = 5 mice per group, *P < 0.05 vs NC group). T-CHO: total cholesterol; TG: triglycerides; LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol.
4. Discussion
Physiologically, NO maintains endothelial homeostasis, while by pathological stimulus, excessive NO production from iNOS and higher reactive oxide species lead to the high-level ONOO−, which could nitrosylate target proteins to cause nitrative stress and aggravate cellular stress. Our and other groups have proved several S-nitrosylated proteins in endothelium that contribute to endothelial dysfunction [7,[30], [31], [32]]. Here, we identified Hsp90 as another S-nitrosylated target in endothelial cells by LC-MS/MS. Our previous study demonstrated the precise role of SNO-Hsp90 at Cys589 in cardiac remodeling [12,13]. Interestingly, we found that the S-nitrosylation occurs at Cys521 in endothelium, suggesting the distinguishing effect of S-nitrosylation in different cellular environments, like phosphorylation occurring at different amino acids of Hsp90 by disparate manners to mediate diverse cellular processes [10]. Analogously, the S-nitrosylation of Akt at Cys296/344 in endothelium damages cardiac function after myocardial infarction [33]. While S-nitrosylation occurring at Cys224 of Akt in skeletal muscle is involved in insulin resistance during diabetes [34]. However, the underlying regulatory mechanisms for different S-nitrosylated sites in proteins under disparate conditions are waiting for further explorations. We here investigated the pathophysiological effect of SNO-Hsp90 at Cys521 on endothelial function and atherosclerosis.
Hsp90, a molecular chaperone, is in charge of the structural maturation, stability and activation of its client proteins, including enzymes, signaling kinases and transcription factors [35]. The biological function of Hsp90 is ATP-dependent and relies on its co-chaperones [36]. AHA1, one of co-chaperones of Hsp90, acts as an ATPase activator, that plays a crucial role in Hsp90 chaperone machinery [37]. Post-translational modifications in Hsp90 are able to influence its binding affinity to AHA1 by allosteric regulation [11]. It is reported that SUMOylation of Hsp90 at Lys178 facilitates recruitment of AHA1 [38]. The phosphorylation of Hsp90 at different threonines or tyrosines is proved to enhance Hsp90/AHA1 interaction [24,39,40], while S-nitrosylation at cysteines in Hsp90 has been shown to reduce the binding affinity of AHA1 to Hsp90 [14,15]. Our findings identify a novel S-nitrosylated site-Cys521 of Hsp90, that serves as a conformational switch. Cys521 locates at the middle domain of Hsp90, which is also the binding domain to combine AHA1 directly [41]. We confirm that substituting the Cys521 with the non-nitrosylable alanine abolished the effect of SNO-Hsp90 on inhibiting Hsp90/AHA1 interaction. In consideration of the essentiality of association between Hsp90 and AHA1 in eNOS activation [22,42], the SNO-Hsp90 at Cys521 is responsible for the impairment in eNOS activation upon pathological stimulus. The Hsp90 chaperone machinery relies on the conformational cycle of sequential association and dissociation of AHA1 and CDC37 [43]. On account of the inhibition of Hsp90 ATPase activity by CDC37, AHA1 and CDC37 do not bind to Hsp90 simultaneously [44,45]. There is evidence that phosphorylation at tyrosines of Hsp90 disrupts the Hsp90/CDC37 complex and promotes AHA1 recruitment [25]. Interestingly, our data reveals that S-nitrosylation of Cys521 suppresses Hsp90 and AHA1 interaction, but promotes CDC37 recruitment. Usually, CDC37 binds to Hsp90 to regulate the activity of client kinases [46,47]. The RNA-seq data showed that SNO-Hsp90 mainly contributes to the activation of NF-κB signaling. The formation of Hsp90/CDC37 complex is important for the recruitment of IKK and the subsequent activation of NF-κB signaling [26,48], and our results are consistent with the hypothesis that S-nitrosylation at Cys521 facilitates the formation of Hsp90/CDC37 complex to activate NF-κB signaling, further aggravating endothelial adhesion and inflammation response.
Taken together, our results show that the SNO-Hsp90 levels are increased in atherosclerotic human and murine aortas, as well as in oxLDL-treated ECs. The elevated SNO-Hsp90 levels in endothelium are triggered by upregulated iNOS expression under pathological conditions. SNO-Hsp90 at Cys521 inhibits the association of AHA1 and Hsp90, leading to the inactivation of eNOS and impairment of vasodilation. Meanwhile, SNO-Hsp90 at Cys521 promotes the interaction of CDC37 and Hsp90, further activating NF-κB signaling pathway and inducing endothelial adhesion, inflammation response and oxidative stress, ultimately leading to endothelial dysfunction and the development of atherosclerosis. The inhibition of SNO-Hsp90 levels by Cys521 mutation could increase NO bioavailability, attenuate endothelial adhesion, inflammation and oxidative stress, thereby mitigating endothelial dysfunction and protecting against atherosclerosis. Our findings reveal a novel regulatory mechanism for Hsp90 function and provide a therapeutic strategy for atherosclerosis.
Author contributions
YJ, HW and ZRH designed the research; SZ, XT, ZAM, YRC, JWC, TYS, DTY, YQZ, ZL, DW and ZGS performed the experiments; DJW and XLT collected the clinical samples; SLC, LSW, AHG, FC, HW, LPX and YJ analyzed data; SZ and LPX wrote the paper.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgements
We thank Qian Jiang (Institute of Soil Science, Chinese Academy of Sciences, Nanjing) for the electron paramagnetic resonance (EPR) analysis. This work was supported by grants from the National Key Research and Development Program of China (2019YFA0802704), National Natural Science Foundation of China (grant NOs. 82121001, 82030013, 81870183, 81820108002, 82000231), China Postdoctoral Science Foundation (2020M681667, 2021T140337), Jiangsu Province Postdoctoral Research Funding (2021Z141), Natural Science Foundation of Jiangsu Province (BK20200673) and Natural Science Foundation of Jiangsu Higher Education Institutions of China (20KJB310016).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102290.
Contributor Information
Zhengrong Huang, Email: huangzhengrong@xmu.edu.cn.
Hong Wang, Email: hongw@temple.edu.
Yong Ji, Email: yongji@njmu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Souilhol C., Serbanovic-Canic J., Fragiadaki M., Chico T.J., Ridger V., Roddie H., Evans P.C. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat. Rev. Cardiol. 2020;17:52–63. doi: 10.1038/s41569-019-0239-5. [DOI] [PubMed] [Google Scholar]
- 2.Gimbrone M.A., Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tousoulis D., Kampoli A.M., Tentolouris C., Papageorgiou N., Stefanadis C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012;10:4–18. doi: 10.2174/157016112798829760. [DOI] [PubMed] [Google Scholar]
- 4.Hess D.T., Matsumoto A., Kim S.O., Marshall H.E., Stamler J.S. Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 5.Tang X., Pan L., Zhao S., Dai F., Chao M., Jiang H., Li X., Lin Z., Huang Z., Meng G., Wang C., Chen C., Liu J., Wang X., Ferro A., Wang H., Chen H., Gao Y., Lu Q., Xie L., Han Y., Ji Y. SNO-MLP (S-nitrosylation of muscle LIM protein) facilitates myocardial hypertrophy through TLR3 (Toll-Like receptor 3)-mediated RIP3 (Receptor-Interacting protein kinase 3) and NLRP3 (NOD-Like receptor pyrin domain containing 3) inflammasome activation. Circulation. 2020;141:984–1000. doi: 10.1161/CIRCULATIONAHA.119.042336. [DOI] [PubMed] [Google Scholar]
- 6.Lima B., Forrester M.T., Hess D.T., Stamler J.S. S-nitrosylation in cardiovascular signaling. Circ. Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W., Wang D., Kong C., Li S., Xie L., Lin Z., Zheng Y., Zhou J., Han Y., Ji Y. eNOS S-nitrosylation mediated OxLDL-induced endothelial dysfunction via increasing the interaction of eNOS with betacatenin. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2019;1865:1793–1801. doi: 10.1016/j.bbadis.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Genest O., Wickner S., Doyle S.M. Hsp90 and Hsp70 chaperones: collaborators in protein remodeling. J. Biol. Chem. 2019;294:2109–2120. doi: 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latchman D.S. Heat shock proteins and cardiac protection. Cardiovasc. Res. 2001;51:637–646. doi: 10.1016/s0008-6363(01)00354-6. [DOI] [PubMed] [Google Scholar]
- 10.Mollapour M., Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta. 2012;1823:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backe S.J., Sager R.A., Woodford M.R., Makedon A.M., Mollapour M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020;295:11099–11117. doi: 10.1074/jbc.REV120.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao S., Song T.Y., Wang Z.Y., Gao J., Cao J.W., Hu L.L., Huang Z.R., Xie L.P., Ji Y. S-nitrosylation of Hsp90 promotes cardiac hypertrophy in mice through GSK3beta signaling. Acta Pharmacol. Sin. 2021 doi: 10.1038/s41401-021-00828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X., Zhang Y., Miao Q., Shi Z., Hu L., Liu S., Gao J., Zhao S., Chen H., Huang Z., Han Y., Ji Y., Xie L. Inhibition of HSP90 S-nitrosylation alleviates cardiac fibrosis via TGFbeta/SMAD3 signalling pathway. Br. J. Pharmacol. 2021;178:4608–4625. doi: 10.1111/bph.15626. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Ruiz A., Villanueva L., Gonzalez de Orduna C., Lopez-Ferrer D., Higueras M.A., Tarin C., Rodriguez-Crespo I., Vazquez J., Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. U. S. A. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Retzlaff M., Stahl M., Eberl H.C., Lagleder S., Beck J., Kessler H., Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009;10:1147–1153. doi: 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L., Luo J.Y., Li B., Tian X.Y., Chen L.J., Huang Y., Liu J., Deng D., Lau C.W., Wan S., Ai D., Mak K.K., Tong K.K., Kwan K.M., Wang N., Chiu J.J., Zhu Y., Huang Y. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540:579–582. doi: 10.1038/nature20602. [DOI] [PubMed] [Google Scholar]
- 17.Xie L., Gu Y., Wen M., Zhao S., Wang W., Ma Y., Meng G., Han Y., Wang Y., Liu G., Moore P.K., Wang X., Wang H., Zhang Z., Yu Y., Ferro A., Huang Z., Ji Y. Hydrogen sulfide induces keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65:3171–3184. doi: 10.2337/db16-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farah C., Michel L.Y.M., Balligand J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018;15:292–316. doi: 10.1038/nrcardio.2017.224. [DOI] [PubMed] [Google Scholar]
- 19.Barnett S.D., Buxton I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017;52:340–354. doi: 10.1080/10409238.2017.1304353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sengupta R., Holmgren A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxidants Redox Signal. 2013;18:259–269. doi: 10.1089/ars.2012.4716. [DOI] [PubMed] [Google Scholar]
- 21.Forstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017;120:713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 22.Desjardins F., Delisle C., Gratton J.P. Modulation of the cochaperone AHA1 regulates heat-shock protein 90 and endothelial NO synthase activation by vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2012;32:2484–2492. doi: 10.1161/ATVBAHA.112.256008. [DOI] [PubMed] [Google Scholar]
- 23.Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., Buchner J. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell. 2010;37:344–354. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Xu W., Beebe K., Chavez J.D., Boysen M., Lu Y., Zuehlke A.D., Keramisanou D., Trepel J.B., Prodromou C., Mayer M.P., Bruce J.E., Gelis I., Neckers L. Hsp90 middle domain phosphorylation initiates a complex conformational program to recruit the ATPase-stimulating cochaperone Aha1. Nat. Commun. 2019;10:2574. doi: 10.1038/s41467-019-10463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W., Mollapour M., Prodromou C., Wang S., Scroggins B.T., Palchick Z., Beebe K., Siderius M., Lee M.J., Couvillon A., Trepel J.B., Miyata Y., Matts R., Neckers L. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine. Mol. Cell. 2012;47:434–443. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G., Cao P., Goeddel D.V. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell. 2002;9:401–410. doi: 10.1016/s1097-2765(02)00450-1. [DOI] [PubMed] [Google Scholar]
- 27.Hinz M., Broemer M., Arslan S.C., Otto A., Mueller E.C., Dettmer R., Scheidereit C. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007;282:32311–32319. doi: 10.1074/jbc.M705785200. [DOI] [PubMed] [Google Scholar]
- 28.Zhong L., Simard M.J., Huot J. Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation. Faseb. J. 2018;32:4070–4084. doi: 10.1096/fj.201701536R. [DOI] [PubMed] [Google Scholar]
- 29.El Assar M., Angulo J., Rodriguez-Manas L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013;65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Pan L., Lin Z., Tang X., Tian J., Zheng Q., Jing J., Xie L., Chen H., Lu Q., Wang H., Li Q., Han Y., Ji Y. S-nitrosylation of plastin-3 exacerbates thoracic aortic dissection formation via endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2020;40:175–188. doi: 10.1161/ATVBAHA.119.313440. [DOI] [PubMed] [Google Scholar]
- 31.Xu X., Qiu H., Shi F., Wang Z., Wang X., Jin L., Chi L., Zhang Q. The protein S-nitrosylation of splicing and translational machinery in vascular endothelial cells is susceptible to oxidative stress induced by oxidized low-density lipoprotein. J. Proteonomics. 2019;195:11–22. doi: 10.1016/j.jprot.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Thibeault S., Rautureau Y., Oubaha M., Faubert D., Wilkes B.C., Delisle C., Gratton J.P. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell. 2010;39:468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Li X.Y., Zhang H.M., An G.P., Liu M.Y., Han S.F., Jin Q., Song Y., Lin Y.M., Dong B., Wang S.X., Meng L.B. S-Nitrosylation of Akt by organic nitrate delays revascularization and the recovery of cardiac function in mice following myocardial infarction. J. Cell Mol. Med. 2021;25:27–36. doi: 10.1111/jcmm.15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasukawa T., Tokunaga E., Ota H., Sugita H., Martyn J.A., Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J. Biol. Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 35.McClellan A.J., Xia Y., Deutschbauer A.M., Davis R.W., Gerstein M., Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 36.Schopf F.H., Biebl M.M., Buchner J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 37.Schulze A., Beliu G., Helmerich D.A., Schubert J., Pearl L.H., Prodromou C., Neuweiler H. Cooperation of local motions in the Hsp90 molecular chaperone ATPase mechanism. Nat. Chem. Biol. 2016;12:628–635. doi: 10.1038/nchembio.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mollapour M., Bourboulia D., Beebe K., Woodford M.R., Polier S., Hoang A., Chelluri R., Li Y., Guo A., Lee M.J., Fotooh-Abadi E., Khan S., Prince T., Miyajima N., Yoshida S., Tsutsumi S., Xu W., Panaretou B., Stetler-Stevenson W.G., Bratslavsky G., Trepel J.B., Prodromou C., Neckers L. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell. 2014;53:317–329. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mollapour M., Tsutsumi S., Donnelly A.C., Beebe K., Tokita M.J., Lee M.J., Lee S., Morra G., Bourboulia D., Scroggins B.T., Colombo G., Blagg B.S., Panaretou B., Stetler-Stevenson W.G., Trepel J.B., Piper P.W., Prodromou C., Pearl L.H., Neckers L. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol. Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mollapour M., Tsutsumi S., Truman A.W., Xu W., Vaughan C.K., Beebe K., Konstantinova A., Vourganti S., Panaretou B., Piper P.W., Trepel J.B., Prodromou C., Pearl L.H., Neckers L. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell. 2011;41:672–681. doi: 10.1016/j.molcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koulov A.V., LaPointe P., Lu B., Razvi A., Coppinger J., Dong M.Q., Matteson J., Laister R., Arrowsmith C., Yates J.R., 3rd, Balch W.E. Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell. 2010;21:871–884. doi: 10.1091/mbc.E09-12-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swick L., Kapatos G. A yeast 2-hybrid analysis of human GTP cyclohydrolase I protein interactions. J. Neurochem. 2006;97:1447–1455. doi: 10.1111/j.1471-4159.2006.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taipale M., Jarosz D.F., Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 44.Siligardi G., Panaretou B., Meyer P., Singh S., Woolfson D.N., Piper P.W., Pearl L.H., Prodromou C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002;277:20151–20159. doi: 10.1074/jbc.M201287200. [DOI] [PubMed] [Google Scholar]
- 45.Harst A., Lin H., Obermann W.M. Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 2005;387:789–796. doi: 10.1042/BJ20041283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyata Y., Nishida E. Analysis of the CK2-dependent phosphorylation of serine 13 in Cdc37 using a phospho-specific antibody and phospho-affinity gel electrophoresis. FEBS J. 2007;274:5690–5703. doi: 10.1111/j.1742-4658.2007.06090.x. [DOI] [PubMed] [Google Scholar]
- 47.Shao J., Prince T., Hartson S.D., Matts R.L. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J. Biol. Chem. 2003;278:38117–38120. doi: 10.1074/jbc.C300330200. [DOI] [PubMed] [Google Scholar]
- 48.Li J., Soroka J., Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.