

Abstract

This investigation is focused on the synthesis of two halo-functionalized crystalline Schiff base (imine) compounds: (E)-2-methoxy-6-(((3-(trifluoromethyl)phenyl)imino)methyl)phenol (MFIP) and (E)-1-(((2-fluorophenyl)imino)methyl)naphthalen-2-ol (FPIN) by the condensation reaction of substituted benzaldehydes and substituted aniline. The crystal structures of MFIP and FPIN were determined unambiguously by single-crystal X-ray diffraction (SC-XRD) studies. Intermolecular interactions and the role of fluorine atoms in the stabilization of the crystal packing are explored for both compounds using Hirshfeld surface analysis. Accompanied with experimental studies, quantum chemical calculations were also performed for comprehensive structure elucidation at the M06/6-311G(d,p) level of theory. A comparison of experimental and density functional theory results for geometrical parameters exhibited excellent agreement. Interestingly, Frontier molecular orbitals and natural bond orbital (NBO) findings revealed that intramolecular charge transfer and hyper-conjugation interactions had played a significant role to stabilize the molecules. Both compounds exhibited a relatively larger value of hardness with a smaller global softness, which, as proposed by the SC-XRD and NBO study, shows a higher stability. Nonlinear optical (NLO) findings showed that FPIN manifested a larger value of linear polarizability (<a> = 293.06 a.u.) and second-order hyperpolarizability (<γ> = 3.31 × 105 a.u.) than MFIP (<a> = 252.42 and <γ> = 2.08 × 105 a.u.) due to an extended conjugation. The above-mentioned findings of the entitled compounds may play a crucial role in NLO applications.
1. Introduction
Synthesis of new crystalline organic compounds and their computational exploration are a significant and a well-recognized research area in modern times. Schiff bases/imines are a substantial class of organic compounds achieved via condensation of carbonyl compounds (aldehydes or ketones) and primary amines to generate an azomethine (−C=N−) functionality.1 Compounds of this class have shown significant applications in the field of analytical, coordination, and pharmaceutical chemistry. The compounds having imine/azomethine (−C=N−) functionality have been recognized as valuable ligands in the field of coordination chemistry in order to prepare various metal complexes of valuable interest.2 Compounds having a Schiff base feature have been explored for their potential applications in the field of medicinal chemistry, and various members of this class are found to possess valuable biological activities such as anticancer,3 analgesic and anti-inflammatory,4 anti-tubercular,5 antimicrobial,6 anthelmintic,7 antioxidant,8 anticonvulsant,9 and so forth. Schiff bases/imines may also exist in the form of zwitterion or inner salt that play a key role to contribute to the physical properties such as enhancing permeability and solubility of these compounds that are highly demanding characteristics in the field of drug designing.10 In the asymmetric organic synthesis (where chiral secondary amines are used as organic catalysts) and multi-component reaction chemistry such as the Ugi four-component reaction, the reaction began with the formation of iminium ion/imines/Schiff bases and is considered as the key intermediate for useful transformations, and they play a vital role in the production of alkaloids and other nitrogen-based heterocycles.11 Recently, overlapping of diverse research fields for the exploration, investigation, and justification of the key electronic features of the newly manufactured compounds has been getting valuable recognition. In this scenario, synthetic organic chemistry is well supported by the DFT calculations in order to find the kinetic and structural properties. DFT calculations are highly acknowledged as an influential contrivance for the exploration of key electronic features of the newly synthesized organic compounds such as nonlinear optical properties, non-covalent interactions, and so forth.12 The non-covalent interactive capacity of organic compounds predicted by DFT calculations plays a significant role in the pharmacological potential of these chemical building blocks.13
Imines are also a class of organic chemical architecture that has been reconnoitered by DFT analysis in order to investigate their crucial electronic properties such as non-covalent interaction, nonlinear optical properties, and so forth.14 The computational tool has also been applied to the imine-based zwitterion compounds in order to explore its stability (supported by NBO investigations) accompanied with reactivity in the redox process.15
Herein, we are presenting our findings toward the synthesis, DFT study, and crystal packing of the crystalline imine/Schiff base compounds: (E)-2-methoxy-6-(((3-(trifluoromethyl) phenyl)imino)methyl)phenol (MFIP) and (E)-1-(((2-fluorophenyl)imino)methyl)naphthalen-2-ol (FPIN).
2. Results and Discussion
2.1. Exploration of the Crystal Structure of MFIP and FPIN
SC-XRD characterization was performed for both compounds and the Cambridge Structural Database confirmed the novelty of the crystal structure of both compounds. Inspection related to the molecular configuration and how the molecules are packed in both the crystals was carried out thoroughly. Table S1 provides the experimental details related to SC-XRD, whereas Table 1 specifies the comparison of selected bond lengths and bond angles determined by SC-XRD and DFT studies of both compounds. In the literature, the effect of phenyl and naphthyl groups on the conformational properties of nitrogenated compounds is found, which is kept in view while describing the crystal structure of FPIN and MFIP.16
Table 1. Comparison of Selected Bond Lengths (Å) and Bond Angles (°) of MFIP and FPIN by SC-XRD and DFT.
| bond (selected) lengths in MFIP | bond (selected) lengths in FPIN | ||||
|---|---|---|---|---|---|
| SC-XRD | DFT | SC-XRD | DFT | ||
| N1–C8 | 1.275 (3) | 1.283 | N1–C11 | 1.286 (4) | 1.287 |
| N1–C9 | 1.416 (3) | 1.399 | N1–C12 | 1.412 (4) | 1.393 |
| O1–C2 | 1.353 (3) | 1.326 | O1–C1 | 1.325 (4) | 1.321 |
| O2–C3 | 1.371 (3) | 1.348 | F1–C13 | 1.353 (4) | 1.333 |
| O2–C7 | 1.420 (4) | 1.404 | C1–C10 | 1.400 (4) | 1.404 |
| C15–F1A | 1.336 (2) | 1.333 | |||
| C15–F2A | 1.340 (2) | 1.338 | |||
| C15–F3A | 1.352 (2) | 1.338 | |||
| C1–C8–N1 | 122.7 (2) | 123.3 | C10–C11–N1 | 121.6 (3) | 122.7 |
| C8–N1–C9 | 119.7 (2) | 120.3 | C11–N1–C12 | 122.0 (3) | 120.9 |
| C1–C2–O1 | 122.6 (2) | 123.4 | C10–C1–O1 | 122.7 (3) | 123.5 |
| O1–C2–C3 | 117.9 (2) | 117.7 | O1–C1–C2 | 117.2 (3) | 116.1 |
| C2–C3–O2 | 115.1 (2) | 114.9 | C11–C10–C1 | 119.0 (3) | 119.4 |
| C3–O2–C7 | 117.2 (3) | 117.7 | C12–C13–F1 | 117.7 (3) | 118.7 |
The title Schiff base compound MFIP (Figure 1a, Table S1) has a phenol tautomeric form as evident from the bond lengths O1–C2 and N1–C8 of 1.353 (3) and 1.275 (3) Å. O–H···N intramolecular H-bonding stabilizes the molecular configuration to form the pseudo-six-membered ring as found in a closely related crystal structure.17 The planarity of the o-cresol group A (C1–C6/C8/O1) and p-toluidine group B (C9–C15/N1) is clear by the root-mean-square (rms) deviation of 0.0049 of group A and 0.0110 Å of group B. The dihedral angle between group A and group B is 39.5 (9)°. The C-atom and O-atom of the methoxy group C (C7/O2) are deviated by 0.2472 (5) and 0.0141 (3) Å from the plane formed by group A, respectively. The atoms of the trifluoro group are disordered over three sets of sites with the first or a major part of almost 45%, second or middle part of 41%, and third or minor part of 14%. The major part of the disordered trifluoro group D (F1A/F2A/F3A) forms the dihedral angle of 5.1 (2)° with the middle part E (F1B/F2B/F3B) and 5.7 (3)° with the minor part F (F1C/F2C/F3C). The dihedral angles formed by groups D, E, and F with the parent group B are 88.7 (6)°, 86.2 (8)°, and 85.6 (9)°, which indicates that the planes of the disordered parts are almost perpendicular to the plane formed by atoms of group B. The molecules of the title compounds are inter-connected through weak non-classical H-bonding named as C–H···O to make the C9 chain that runs along the c crystallographic axis (Table 2). The fluorine atoms play an important role in the stabilization of the crystal packing as these atoms are involved in C–H···F bonding and interlink the molecules to make the C8 zigzag chain that runs along the b crystallographic axis (Figure 2). Aamer Saeed et al.18 efficiently explored the role of C–H···F bonding in the stabilization of the crystal packing of the crystal structures containing fluorine-substituted phenyl rings. The chains of molecules along the c crystallographic axis are further strengthened by off-set π···π stacking interactions (Figure 3). The phenyl ring substituted by methoxy and hydroxyl groups of a molecule located at the symmetric position is involved in off-set π···π stacking with similar phenyl rings of a neighboring molecule with symmetry 1/2–x, y, −1/2 + z. This is weak interaction as the inter-centroid distance is 4.44 Å and 2.671 to 3.049 Å is the range of the ring off-set. The crystal packing of MFIP is compared with very closely related crystal structures found in the literature. The Cambridge structural database search for Schiff-bases with one phenyl ring substituted by the hydroxyl group on one end and, on the other end, a phenyl ring substituted by the trifluoromethyl group is performed. This search gives numerous crystal structures that are relevant to MFIP. Out of the relevant structures, three most closely related crystal structures are compared with the crystal structure of MFIP. Reference codes are DUJNUS (containing a 4-methoxy-substituted phenyl ring),19FUGWEK (containing a 4-hydroxy-substituted phenyl ring),20 and YIFLEF (containing a 4-methyl-substituted phenyl ring).21 Like in MFIP, the molecular configuration of the relevant structures is stabilized by intra-molecular H-bonding (O–H···N). The crystal packing of MFIP is different from the crystal packing in DUJNUS, FUGWEK, and YIFLEF. In DUJNUS, C–H···O and C–H···π interactions are the main aspects of the crystal packing. In the crystal packing of FUGWEK, strong H-bonding of type O–H···O bonding is present. In the crystal packing of FUGWEK, no significant intermolecular H-bonding is present, but in MFIP, C–H···O and the off-set π···π stacking interactions are the main aspects of the crystal packing.
Figure 1.

ORTEP diagrams of (a) MFIP and (b) FPIN that are drawn at a 30% probability level. Small circles of arbitrary radii represent H-atoms.
Table 2. Hydrogen-Bond Geometry (Å, °) for MFIP and FPINa.
| MFIP | D—H···A | D—H | H···A | D···A | <(D—H···A)° |
| O1–H1···N1 | 0.82 | 1.89 | 2.618 (3) | 147 | |
| C10–H10···O2i | 0.93 | 2.52 | 3.441 (4) | 170 | |
| C8–H8···F1Cii | 0.93 | 2.48 | 3.39 (3) | 166 | |
| FPIN | D—H···A | D—H | H···A | D···A | <(D—H···A)° |
| O1–H1···N1 | 0.82 | 1.79 | 2.510 (4) | 146 | |
| C17–H17···O1iii | 0.93 | 2.66 | 3.442 (4) | 142 |
Symmetry codes: Symmetry code: (i) −x+1/2, y, z+1/2; (ii) x, y–1/2, z+1/2; (iii) 1 + x, y, z.
Figure 2.

Packing diagram of MFIP. Only H-atoms that are involved in H-bonding are displayed.
Figure 3.

Graphical representation of the C9 zigzag chain and the off-set π···π stacking interaction in the crystal packing of MFIP. Distances shown are measured in Å.
The title Schiff base compound FPIN (Figure 1b, Table S1) also has a phenol tautomeric form as evident from the bond length O1–C1 and N1–C11 of values 1.325 (4) and 1.286 (4) Å, respectively. As in MFIP, the intramolecular H-bonding (O–H···N) is responsible for the stabilization of the molecular configuration for a tilted compound, but the intramolecular H-bonding in FPIN is stronger as compared to the intramolecular H-bonding in MFIP (Table 2). This may be due to the strain produced by the trifluoromethyl group attached to the phenyl ring in MFIP. The planarity of 3-methylnaphthalen-2-ol group A (C1–C11/O1) and 2-(iminomethyl)-6-methoxyphenol group B (C12–C17/N1/F1) is clear by the rms deviation of 0.0156 Å for group A and 0.0083 Å for group B. The dihedral angle between groups A and B is 38.3°.4 As in MFIP, C–H···O bonding connects the molecules of FPIN. However, in contrast to the C9 chains along the c-axis in MFIP, infinite C8 chains are formed in FPIN that run along the a crystallographic axis (Figure 4). In MFIP, the hydroxyl group does not form any intermolecular H-bond, whereas in FPIN, the hydroxyl group participates not only in intramolecular but also in intermolecular H-bonding. The crystal packing is further strengthened by a weak interaction of type off-set π···π. The (C1–C4/C9/C10) aromatic part of the naphthalene ring of a molecule that is located at a symmetric position is involved in off-set π···π stacking with a similar aromatic part of the naphthalene ring of a neighboring molecule connected by symmetry −1–x, −1/2 + y, −z (Figure S1). This interaction is very weak as the inter-centroid distance is 4.42 Å with ring off-set ranges from 2.7135 to 4.3048 Å. The Cambridge structural database search for Schiff-bases with a naphthalen-2-ol ring at one end and a substituted phenyl ring on the other end is performed. This search gives a lot of crystal structures that are relevant to FPIN. Out of the relevant structures, three most closely related crystal structures are compared with the crystal structure of FPIN. Reference codes are NUQSAU (containing a 2-fluoro-substituted phenyl ring, a polymorphous of FPIN solved in the P212121 space group),22FEQWEE (containing a 3-fluoro-substituted phenyl ring),23 and MUDGAT (containing a 4-fluoro-substituted phenyl ring).24NUQSAU is polymorphous of FPIN that has both enol and keto forms with occupancies of 57 and 43%, respectively, whereas FPIN is different from NUQSAU as it has an enol form. No intermolecular H-bonding is present in NUQSAU, whereas H-bonding of type C–H···O and C–H···F is found in the crystal packing of FPIN. As in FPIN, FEQWEE and MUDGAT also adopt an enol tautomeric form, and the same type of intramolecular H-bonding is found, but the crystal packing is not explored in FEQWEE and MUDGAT.
Figure 4.

Packing diagram of FPIN showing C8 chains formed by molecules that run along the a crystallographic axis. Only H-atoms that are involved in H-bonding are displayed.
2.2. Hirshfeld Surface Analysis
As far as the crystal packing is concerned, the various non-covalent interactions are the main aspect to be explored because they provide us the unique information about the arrangement of molecules throughout the crystalline material. For the exploration of the non-covalent interactions, the Hirshfeld surface (HS) inspection is accomplished using Crystal Explorer (17.5).25 The HS is popped up by a strive to specify the space inhabited by a molecule for the sake of dividing the density of an electron of the whole crystal into the electronic density of molecular fragments.26 The HS can be made by using different features such as the shape index, dnorm (normalized distance), electrostatic potential, and so forth. Each surface presents exceptional information regarding the non-covalent interactions. The HS of a molecule plotted over property dnorm contains three colors, red, blue, and white. Red and white colors on the HS indicate the inter-atomic contacts where the distance among the atoms is less than or equal to the summation of van der Waals radii of the involved atoms, respectively. Blue regions on the HS show that the space among the atoms is more than the summation of van der Waals radii of the involved atoms. We can say that red colored, white colored, and blue colored spots on the HS indicate strong, comparatively weak, and negligibly weak intermolecular interactions, respectively. Around the hydroxyl group of both compounds, one of the CH of the (trifluoromethyl)phenyl group in MFIP and one of the CH of the fluorophenyl group in FPIN, the HS has red spots, which indicates that these specific atoms are promising in the H-bonding interaction. C–H···F bonding is present in MFIP (Table 2), but it involves a minor part of the disordered trifluoro group, and we made the HS by taking mainly the disordered group portion. Therefore, around an F-atom of the disordered group (Figure 5a), the HS has no red spot; however, around an F-atom of FPIN (Figure 5b), the HS containing a red spot indicates that the F-atom is engaged in an H-bonding interaction. Not only H-bonding interactions can be visualized by plotting HS, but we can explore a weak interaction of type π···π stacking interaction. In order to visualize this interaction, the HS is plotted on a shape index. A π···π stacking interaction is indicated on the HS by the presence of consecutive triangular regions of red and blue around the aromatic rings.27,28 For both compounds, the consecutive triangular regions of red and blue are present on the HS around the aromatic rings, which indicates that the π···π stacking interaction is present (Figure 5c,d). In H-bonding interactions, hydrogen bond acceptors and donors can be codified or separately recognized by plotting the HS over the property named as the electrostatic potential. Blue and red spots on this HS around the atoms specify H-bond donors and H-bond acceptors, respectively (Figure 5e,f).
Figure 5.

HS plotted over a property named as dnorm for (a) MFIP in the range −0.175 to 1.341 a.u. (b) FPIN in the range −0.047 to 1.308 a.u. HS plotted over a property named as shape index in the range −1 to 1 a.u. for (c) MFIP and (d) FPIN. HS plotted over the electrostatic potential for (e) MFIP in the range −0.083 to 0.052 a.u. and (f) FPIN in the range −0.069 to 0.034 a.u.
We are going to express the non-covalent interactions in a quantitative manner by finding out the role that the interatomic contacts play in the stabilization of the crystal packing.29 We make a comparative study of some important 2D plots of MFIP and FPIN with some nitrogen–oxygen–halogen-rich compounds found from the literature. We choose compounds with the CSD reference code EQIDAL (CCDC number1410273),30ILUHIJ (CCDC number 2018498),31 and JIPZUG (CCDC number 1884126)32 for the 2D plots’ comparison with MFIP and FPIN. The 2D plots provide us the endowment of the inter-atomic contacts in percentage. The distance from the HS to the nearest atom outside and inside the HS is denoted by de and di, respectively. Computation of each interatomic contact includes the contribution of its reciprocal contact. Figure 6a,e,i,m,q shows the 2D plot for all the possible interactions in the crystal packing of MFIP, FPIN, EQIDAL, ILUHIJ, and JIPZUG, respectively. A π···π stacking interaction in all the above-mentioned compounds is indicated by the central region of sky blue color. For MFIP, the interatomic contact of the greatest contributor in the crystal packing is F···H, with a percentage contribution of 33.3% (Figure 6d). For FPIN, the contact of the greatest contributor is H···H (36.1%) (Figure 6f). The contributions of the H···H contact for MFIP, EQIDAL, ILUHIJ, and JIPZUG are 25.1% (Figure 6b), 72% (Figure 6j), 66.5% (Figure 6n), and 33.9% (Figure 6r), respectively. The contribution of the C···H contact in the crystal packing is the greatest for FPIN (35.8%, Figure 6g) as compared to the contribution of the C···H contact in other compounds. The contributions of the C···H contact for MFIP, EQIDAL, ILUHIJ, and JIPZUG are 20% (Figure 6c), 10.4% (Figure 6k), 22% (Figure 6o), and 26.6% (Figure 6s), respectively. The contribution of the F···H contact is the greatest for MFIP as it contains a trifluoro group, which is absent in the other compounds. The other interatomic contacts for MFIP and FPIN are compared in Figure S2. O···H, C···C, O···C, and N···C contacts have contributions of 11.1, 4.6, 1.8, and 1.5% (Figure S2a–d), respectively, for MFIP. For FPIN, O···H, C···C, O···C, and N···C contacts have contributions of 7.5, 3.5, 0.6, and 0.4% (Figure S2e–h), respectively. The enrichment ratio33 is computed for all the possible pairs of chemical species (X, X) and (X, Y) in MFIP and FPIN in order to determine the tendency of a pair of chemical species to form crystal packing interactions. The results are summarized in Table S2 for MFIP and Table S3 for FPIN. The results infer that the F···H/H···F contact has the highest propensity to form crystal packing interactions in MFIP with an enrichment ratio of 1.67, whereas the O···H/H···O contact has the highest propensity to form crystal packing interactions in FPIN with an enrichment ratio of 1.45.
Figure 6.

Important 2D finger print plots of MFIP (a–d) and FPIN (e–h) that are compared with important 2D finger print plots of some nitrogen–oxygen–halogen-rich compounds found from the literature with CSD reference codes EQIDAL (i–l), ILUHIJ (m–p), and JIPZUG (q–t).
As we explore the quantitative analysis of the interatomic contact, now we are going to explore the crystal packing in a unique way by the determination of the interaction of an atom in the HS with all the atoms of the neighboring molecules (ALL).34 For both compounds, the strongest interaction of that kind is H-ALL, having a 56.1% percentage contribution for MFIP (Figure S3a) and 60.4% for FPIN (Figure S3b). The other interactions of such a kind are C-ALL, F-ALL, O-ALL, and N-ALL for both compounds with percentage contributions of 17.7, 17.6, 7.1, and 1.5%, respectively, for MFIP and 26.9, 6.8, 4.3, and 1.6%, respectively, for FPIN. All the atoms present inside the HS (ALL) interact with an atom of the neighboring molecules, so we find ALL-atom interactions. The ALL-H interaction is the strongest interaction for both compounds with percentage contributions of 59.8% for MFIP (Figure S3c) and 67.4% for FPIN (Figure S3d). The other interactions of such a nature for MFIP are ALL-F, ALL-C, ALL-O, and ALL-N with percentage contributions of 16.9, 15.4, 6.5, and 1.3%, respectively. Similarly, for FPIN, such interactions other than the strongest contributor are ALL-C, ALL-F, ALL-O, and ALL-N, having percentage contributions of 20.7, 6.8, 3.8, and 1.2%, respectively.
By the application of an external force to a single crystal, a strain is produced in it. If a single crystal is not mechanically stable, then it cannot bear the external force of significant magnitude and it will break. In order to find out whether the single crystals of MFIP and FPIN are mechanically stabilized or not, we find out the voids for both compounds. The void analysis we perform is based on an assumption, that is, all the atoms of a molecule are spherically symmetric and then the electron density of each atom is added up.35 The void volumes are 412.25 Å3 for MFIP and 79.19 Å3 for FPIN (Figure 7). The percentage spaces occupied by the molecules in the crystal packing are almost 85% for MFIP and 88% for FPIN. This analysis shows that both compounds are mechanically stable. Both compounds contain no large cavity in the crystal packing and the strength of the crystal packing is almost equal in both compounds.
Figure 7.

Void analysis using an isosurface value of 0.0002 a.u. for (a) MFIP, a view along the c-axis, and for (b) FPIN, a view along the b-axis.
3. Computational Procedure
By utilizing the Gaussian 09 package program,36 quantum chemical analyses for synthesized compounds were accomplished. The primary structures of FPIN and MFIP were attained by the SC-XRD. Then, a DFT37 study was approached utilizing M06/6-311G(d,p) as the level of theory for optimizing geometries.38 The absence of negative frequencies represented optimization at true minima. Additionally, the natural bond orbital (NBO), the Frontier molecular orbital (FMO), and the nonlinear optical (NLO) investigations were also performed by utilizing optimized structures of the title compounds at the aforesaid functional. All results were interpreted by utilizing GaussSum,39 ChemCraft,40 Avogadro,41 and GaussView 5.042 computational programs.
3.1. NBO Analysis
The NBO approach is an outstanding method that shows useful aspects for understanding interactions between atoms and provides a suitable foundation for representing charge transfer between vacant and filled atomic orbitals.43 Further, NBO analysis is also beneficial to explain the proper and valid picture of the charge densities and intramolecular delocalization from the donor to acceptor moiety. In NBO analysis, second-order perturbation-based stabilization energy of compound structures can be computed by using eq 1.
| 1 |
In the above equation, the donor is signified as (i), the acceptor is signified as (j), the stabilization energy is represented by E(2), and the diagonal, off-diagonal, orbital occupancy, and NBO Fock matrix elements are represented by εi, εj, qi, and Fi,j , respectively. NBO analysis of the title compounds has been performed and their characteristic interactions are tabulated in Table 3, while the remaining data are collected in Tables S4 and S5.
Table 3. NBO Analysis of the Studied Compounds.
| comp | donor (i) | type | acceptor (j) | type | E(2)a[kcal/mol] | E(J)E(i)b(a.u) | F(I,j)c(a.u |
|---|---|---|---|---|---|---|---|
| MFIP | C26–C28 | π | C30–C31 | π* | 23.1 | 0.30 | 0.074 |
| C8–C9 | π | C8–C9 | π* | 1.82 | 0.29 | 0.021 | |
| C30–C31 | σ | C28–C30 | σ* | 5.96 | 1.30 | 0.079 | |
| F2–C33 | σ | F3–C33 | σ* | 0.68 | 1.33 | 0.027 | |
| O4 | LP(2) | C8–C9 | π* | 39.52 | 0.36 | 0.113 | |
| F1 | LP(1) | C30–C33 | σ* | 0.52 | 1.49 | 0.025 | |
| FPIN | C5–C20 | π | N4–C21 | π* | 26.35 | 0.29 | 0.08 |
| C5–C20 | π | C5–C20 | π* | 3.48 | 0.29 | 0.029 | |
| C11–H12 | σ | C10–C19 | σ* | 5.20 | 1.08 | 0.067 | |
| C15–H16 | σ | C13–C15 | σ* | 0.52 | 1.09 | 0.021 | |
| O2 | LP(2) | C5–C20 | π* | 44.37 | 0.36 | 0.118 | |
| O2 | LP(1) | C5–C6 | σ* | 0.73 | 1.14 | 0.026 |
Some dominant π → π* transitions are examined such as π(C26–C28) → π*(C30–C31) and π(C5–C20) → π*(N4–C21) having greater stabilization energies: 23.1 and 26.35 kcal/mol in MFIP and FPIN, respectively (Table 3). Furthermore, some other significant π → π* electronic transitions of the title compounds are presented in Tables S4 and S5. Nevertheless, π(C8–C9) → π*(C8–C9) and π(C5–C20) → π*(C5–C20) transitions are noted with the least values of stabilization energy of 1.82 and 3.48 kcal/mol in MFIP and FPIN, respectively.
Due to the resonance process, the massive stabilization energy values: 39.52 and 44.37 kcal/mol, respectively, are found for the transition LP2(O4) → π*(C8–C9) in MFIP and LP2(O2) → π*(C5–C20)) in FPIN. Similarly, LP1(F1) → σ*(C30–C33) and LP1(O2) → σ*(C5–C6) transitions are examined with least values (0.52 and 0.73 kcal/mol) of stabilization energy in MFIP and FPIN, respectively. The electronic transitions associated with σ → σ* are known to be weak transitions aroused as a result of a weak interaction between donor and acceptor moieties. Among all σ → σ* transitions in both MFIP and FPIN, σ(C30–C31) → σ*(C28–C30) and σ(C11–H12) → σ*(C10–C19) are found having the highest stabilization energies of 5.96 and 5.20 kcal/mol, respectively. However, σ(F2–C33) → σ*(F3–C33) and σ(C15–H16) → σ*(C13–C15) transitions exhibiting the lowest energy values as 0.68 and 0.52 kcal/mol are observed in MFIP and FPIN, respectively. The above discussion revealed that hyper-conjugative interactions and extended conjugation are responsible for the stability of the title compounds.
3.2. FMO Analysis
An FMO investigation is considered to be a very effective parameter to elaborate the charge transfer and chemical stability of compounds.44,45 As a consequence of having an electron-rich nature, the electron-donating ability is found good in higher occupied molecular orbitals (HOMOs), while the electron-accepting capability is observed in lower unoccupied molecular orbitals (LUMOs).46 Moreover, the band gap significantly demonstrates the NLO properties of the studied compounds, and the energy difference between the HOMOs and LUMOs is known by the formula Egap = ELUMO – EHOMO. It is conjectured that the chromophores with a smaller band gap are more polarized and can be considered as good NLO materials.47
The energies for HOMO are noted to be −6.32 and −6.24 eV while those for LUMO are found to be −1.93 and −2.07 eV for MFIP and FPIN, respectively. Further, Table 4 shows that the HOMO/LUMO band gap calculated for MFIP is found higher (4.39 eV) than for FPIN (4.17 eV).
Table 4. Computed Orbital Energies and Their Band Gapsa.
| MOs | MFIP | ΔE | FPINΔE | |
|---|---|---|---|---|
| LUMO | –1.93 | 4.39 | –2.07 | 4.17 |
| HOMO | –6.32 | –6.24 | ||
| LUMO + 1 | –0.86 | 6.02 | –0.89 | 6.12 |
| HOMO – 1 | –6.87 | –7.01 | ||
| LUMO + 2 | –0.27 | 7.36 | –0.39 | 6.77 |
| HOMO – 2 | –7.64 | –7.16 | ||
MO = molecular orbital, ΔE = EL – EH, units in eV.
However, in UV–vis spectra (Figures S4 and S5), the maximum wavelength (λmax) for MFIP is found to be 312 nm at a maximum oscillator strength (0.572) with MO contributions: H-1 → L (88%), H-4 → L (3%), and H-2 → L (7%), smaller than λmax = 371 nm of FPIN at a maximum oscillator strength (0.578) with MO contributions: H → L (98%) (Tables S5 and S6). This might be due to the extended conjugation owing to the presence of the benzene ring in FPIN, which stabilized the molecule by lowering its Eg.48 Accompanied with the energies, the charge transfer between the molecules can also be explained by FMOs.49 Therefore, we calculated the intramolecular charge transfer (ICT) between the orbitals and their pictographs, which are displayed in Figures 8 and S6. In MFIP, for HOMO, the electronic cloud is concentrated over the methoxy group and benzene ring, while the LUMO is located over the entire crystals except for the trifluoromethyl group. In FPIN, the charge density regarding HOMO/LUMO is intensely located all over the molecule, as shown in Figure 8.
Figure 8.

HOMOs and LUMOs of the studied compounds.
3.3. Global Reactivity Parameters
The HOMO/LUMO band gap (ΔE) is a crucial factor to elaborate the global reactivity parameters (GRPs) such as chemical hardness (η), chemical softness (σ), global electrophilicity (ω), electronegativity (X), ionization energy (IP), and electron affinity (EA) of the compounds.50 With the help of these parameters, the kinetic stability and reactivity of compounds can be explained. The ionization potential of MFIP and FPIN can be calculated to express their electron-donating and -accepting ability, which is the amount of energy required to pull out an electron from HOMO. The ionization potential is found significant to measure the amount of energy needed to pull an electron out from HOMO, which is endowed to be useful to manifest their capability to donate and accept electrons. Being a significant chemical property, electronegativity can determine the capability of compounds to gain electrons coming toward them. The stability of compounds can be analyzed by the chemical hardness (η) and chemical potential (μ). These parameters can be calculated using the following equations for the title compounds, and the results are listed in Table 5
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
Table 5. Global Reactivity Descriptors of the Entitled Compoundsa.
| comp | I | EA | X | η | ω | σ | μ |
|---|---|---|---|---|---|---|---|
| MFIP | 6.319 | 1.932 | 4.125 | 2.193 | 3.879 | 0.227 | –4.125 |
| FPIN | 6.237 | 2.069 | 4.153 | 2.084 | 4.138 | 0.239 | –4.153 |
Units are in eV.
From Table 5, it is found that a higher value of ionization energy is found for MFIP (6.319 eV) than for FPIN (6.237 eV). Interestingly, a greater value of the electron affinity (EA = 2.069 eV) is found in FPIN with a larger electronegativity (X=4.153 eV) than in MFIP (EA = 1.932 eV and X = 4.125 eV). However, both parameters are comparable in MFIP and FPIN, which indicated their greater ability to attract the electron toward themselves due to the fluoro groups. The energy gap and hardness of a compound are directly proportional to each other. For this, the compound having a more dominant band gap is observed to be harder, less reactive, more kinetically stable, and more resistant against the change in electronic configuration. Contrarily, the compounds with a smaller energy band gap are considered to be soft and having a lower kinetic stability and more chemical reactivity.51 Considering the above-mentioned information, MFIP has a higher value of global hardness (η = 2.193 eV) with a small value of softness (σ = 0.227 eV) than that of FPIN (η = 2.084 eV and σ = 0.239 eV). The aforesaid facts revealed that FPIN is somehow less stable and a more reactive specie than MFIP. This is may be due to the presence of more resonance effect due to the presence of two benzene rings in the donor unit of FPIN, which also reduces the energy band gap of the orbitals.
3.4. NLO Properties
Organic crystalline structures with extended conjugation and significant ICT exhibited remarkable NLO properties.52 Organic NLO materials have low dielectric constants with rapid electronic responses, which increase their demand and use in various optoelectronic applications. Organic materials are also used in optical fiber telecommunications due to their delocalized electronic systems and the ability to transmit intramolecular charge.53 Therefore, we calculated the linear polarizability, dipole moment, and second-order hyperpolarizability of our compounds, and the results are tabulated in Table S6. The investigations presented in Table S6 show that MFIP exhibited the higher value of dipole moment (μtotal = 2.35 a.u.) than FPIN (μtotal = 1.52 a.u.). This might be due to the presence of the trifluoromethyl (−CF3) group in MFIP, which is a strong electron-withdrawing group.54Table S6 also holds major contributing tensors along x, y, and z directions of μtotal, <a> and <γ>. Both MFIP and FPIN compounds showed a higher value of dipole moment along the y-axis (μy = 1.73 and −1.48 a.u., respectively). Between both compounds, interestingly, FPIN manifested a larger value of linear polarizability (<a> = 293.06 a.u.) and second-order hyperpolarizability (<γ> = 3.31 × 105 a.u.) than MFIP(<a> = 252.42 and <γ> = 2.08 × 105 a.u.), which might be due to an extended conjugation in FPIN. In MFIP and FPIN, a higher value of the <a> and <γ> tensors along the x-direction (αxx) is studied [(αxx = 392.63 and 457.07 a.u., respectively), (1.90 × 105 and 2.95 × 105 a.u., respectively)]. A higher value of <γ> of the title compounds designated them as the remarkable NLO material.
4. Conclusions
The synthesis of two halo-functionalized crystalline organic compounds (MFIP and FPIN), SC-XRD, the HS, and DFT studies have been reported. SC-XRD infers that intramolecular H-bonding of type O–H···N stabilizes the molecular configuration of both compounds to form a pseudo-six-membered ring. Weak H-bonding and off-set π···π stacking interaction stabilize the crystal packing of both compounds. The non-covalent interactions are explored for both compounds by HS analysis, and 2D fingerprint plots of both compounds are compared with some nitrogen–oxygen–halogen-rich compounds found from the literature. The comparative study of DFT and SC-XRD findings showed a good agreement for geometrical parameters. Additionally, the NBO investigation uncovered the hyper-conjugative interactions in both compounds. The energies of the HOMO/LUMO for MFIP and FPIN are found to be −6.32/–1.93 with a band gap of 4.39 eV and −6.24/–2.07 eV with a band gap of 4.17 eV, respectively. The values of hardness for MFIP and FPIN (η = 2.193 eV and 2.084 eV) were reported larger than their values of global softness (σ = 0.239 eV and 277 eV). Consequently, the GRP disclosed that both MFIP and FPIN have a greater stability as suggested by SC-XRD and NBO investigations, and both compounds unveiled noteworthy NLO responses.
5. Experimental Section
5.1. Synthesis and Crystallization
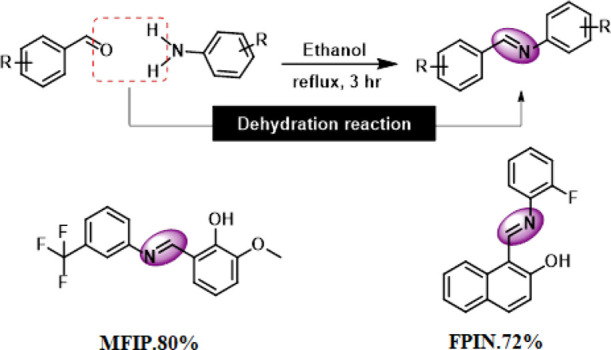
The imine/Schiff base compounds MFIP and FPIN were synthesized by the condensation reaction of 3-(trifluoromethyl) aniline and 2-hydroxy-3-methoxybenzaldehyde in the case of MFIP and 2-hydroxy-1-naphthaldehyde and 2-fluoroaniline in the case of FPIN.
5.1.1. Synthesis of MFIP(55,56)
In a 50 mL round bottom flask, 2-hydroxy-3-methoxybenzaldehyde (1 mmol, 1 equiv) was dissolved in 30 mL of dry ethanol and then 3-(trifluoromethyl) aniline (1.2 mmol, 1.2 equiv) was added. The stirring reaction mixture was refluxed for 3 h. After completion (indicated by TLC), the mixture was allowed to cool at room temperature and transferred to a 50 mL beaker. The beaker was covered by an aluminum foil leaving small holes at the surface and left overnight to get pure crystals of the final product that were analyzed by XRD analysis.
5.1.2. Synthesis of FPIN
The compound FPIN was also synthesized by employing the above same procedure except for using 2-hydroxy-1-naphthaldehyde and 2-fluoroaniline as aldehyde and aniline components, respectively. Scheme 1.
Scheme 1. Synthetic Scheme of Crystalline Imines (Schiff Bases) MFIP and FPIN.
5.2. Materials and Methods
The fluoro-functionalized imines were prepared using the substituted benzaldehyde and substituted anilines of highest purity. Simple distillation was employed for solvent purification. The pre-coated silica gel aluminum sheet (Merck company) was used for thin-layer chromatography to check the reaction progress. The Bruker Kappa Apex-II diffractometer characterized by Mo Kα radiation was used for X-ray data collection. X-ray data collection was performed by using software Apex-II,57 whereas a software named SAINT was used for the purpose of integration of data.58 A software called SHEXS-97 was used59 for the structure solution of the raw data, whereas SHEXL 2018/360 was employed for data refinement. Anisotropic displacement parameters were allotted to all atoms other than H atoms, whereas H-atoms were located in ideal sites and were refined as riding atoms with relative isotropic displacement parameters. For graphical illustration of SC-XRD results, PLATON,61 Mercury 4.0,62 and ORTEP-363 were employed.
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no. (D-298-130-1443). The authors, therefore, gratefully acknowledge DSR technical and financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00288.
Crystallographic data for MPIF (CIF)
Crystallographic data for FPIN (CIF)
SC-XRD; experimental details of MFIP and FPIN; enrichment ratio for pairs of chemical species (X, X) and (X, Y) in compound MFIP and compound FPIN; NBO analysis; dipole polarizability, polarizability, second-order hyperpolarizability and their major contributing tensor (a.u), wavelengths (λmax), energies, and oscillator strengths; off-set π···π stacking interaction in the crystal packing of FPIN; comparison of the remaining 2D plots of MFIP (a–d, i–h) with FPIN (e–h, m,n); summary of interactions of an atom present inside the HS with the neighboring molecules; simulated absorption spectra of MFIP and FPIN; FMOs of the entitled compounds; and bond lengths in Å and bond angles in ° for MFIP and FPIN (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cimerman Z.; Miljanić S.; Galić N. Schiff bases derived from aminopyridines as spectrofluorimetric analytical reagents. Croat. Chem. Acta 2000, 73, 81–95. [Google Scholar]
- Abu-Dief A. M.; Mohamed I. M. A. A review on versatile applications of transition metal complexes incorporating Schiff bases. Beni-Seuf Univ. J. Appl. Sci. 2015, 4, 119–133. 10.1016/j.bjbas.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S. M. M.; Azad M. A. K.; Jesmin M.; Ahsan S.; Rahman M. M.; Khanam J. A.; Islam M. N.; Shahriar S. M. S. In vivo anticancer activity of vanillin semicarbazone. Asian Pac. J. Trop. Biomed. 2012, 2, 438–442. 10.1016/s2221-1691(12)60072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi S. M.; Singh N.; Kumar A.; Lozach O.; Meijer L. Synthesis, anti-inflammatory, analgesic and kinase (CDK-1, CDK-5 and GSK-3) inhibition activity evaluation of benzimidazole/benzoxazole derivatives and some Schiff’s bases. Bioorg. Med. Chem. 2006, 14, 3758–3765. 10.1016/j.bmc.2006.01.054. [DOI] [PubMed] [Google Scholar]
- Aboul-Fadl T.; Mohammed F. A.-H.; Hassan E. A.-S. Synthesis, antitubercular activity and pharmacokinetic studies of some schiff bases derived from 1- alkylisatin and isonicotinic acid hydrazide (inh). Arch Pharm. Res. 2003, 26, 778–784. 10.1007/bf02980020. [DOI] [PubMed] [Google Scholar]
- Mounika K.; Pragathi A.; Gyanakumari C. Synthesis, Characterization and Biological Activity of a Schiff Base Derived from 3-Ethoxy Salicylaldehyde and 2-Amino Benzoic acid and its Transition Metal Complexes. J. Sci. Res. 2010, 2, 513. 10.3329/jsr.v2i3.4899. [DOI] [Google Scholar]
- Avaji P. G.; Vinod Kumar C. H.; Patil S. A.; Shivananda K. N.; Nagaraju C. Synthesis, spectral characterization, in-vitro microbiological evaluation and cytotoxic activities of novel macrocyclic bis hydrazone. Eur. J. Med. Chem. 2009, 44, 3552–3559. 10.1016/j.ejmech.2009.03.032. [DOI] [PubMed] [Google Scholar]
- Wei D.; Li N.; Lu G.; Yao K. Synthesis, catalytic and biological activity of novel dinuclear copper complex with Schiff base. Sci. China, Ser. B: Chem. 2006, 49, 225–229. 10.1007/s11426-006-0225-8. [DOI] [Google Scholar]
- Kumar C. A.; Pandeya S. N. Synthesis & anticonvulsant activity (Chemo Shock) of Schiff and Mannich bases of Isatin derivatives with 2-Amino pyridine (mechanism of action). Int. J. PharmTech Res. 2012, 4, 590–598. [Google Scholar]
- Bighley L. D.; Berge S. M.; Monkhouse D. C. Salt forms of drugs and absorption. Encycl. Pharm. Technol. 1996, 13, 453–499. [Google Scholar]
- a Martin S. F. Recent applications of imines as key intermediates in the synthesis of alkaloids and novel nitrogen heterocycles. Pure Appl. Chem. 2009, 81, 195–204. 10.1351/pac-con-08-07-03. [DOI] [PMC free article] [PubMed] [Google Scholar]; b de la Torre1 A. F.; Ali A.; Galetto F. Z.; Braga A. L.; Delgado J. A. C.; Paixão M. W. One-pot organocatalytic/multicomponent approach for the preparation of novel enantioenriched non-natural selenium-based peptoids and peptide–peptoid conjugates. Mol. Divers. 2020, 24, 1–10. 10.1007/s11030-019-09923-w. [DOI] [PubMed] [Google Scholar]; c de la Torre A. F.; Ali A.; Concepcion O.; Montero-Alejo A. L.; Muñiz F. M.; Jiménez C. A.; Belmar J.; Velázquez-Libera J. L.; Hernández-Rodríguez E. W.; Caballero J. A study of the cis-trans isomerization preference of N-alkylated peptides containing phosphorus in the side chain and backbone. New J. Chem. 2019, 43, 12804–12813. 10.1039/c9nj02234a. [DOI] [Google Scholar]
- a Ali A.; Khalid M.; Rehman M. F. U.; Haq S.; Ali A.; Tahir M. N.; Ashfaq M.; Rasool F.; Braga A. A. C. Efficient Synthesis, SC-XRD, and Theoretical Studies of O-Benzenesulfonylated Pyrimidines: Role of Noncovalent Interaction Influence in Their Supramolecular Network. ACS Omega 2020, 5, 15115–15128. 10.1021/acsomega.0c00975. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Khalid M.; Ali A.; Abid S.; Tahir M. N.; Khan M. U.; Ashfaq M.; Imran M.; Ahmad A. Facile Ultrasound-Based Synthesis, SC-XRD, DFT Exploration of the Substituted Acyl-Hydrazones: An Experimental and Theoretical Slant towards Supramolecular Chemistry. ChemistrySelect 2020, 5, 14844–14856. 10.1002/slct.202003589. [DOI] [Google Scholar]; c Khalid M.; Ali A.; De la Torre A. F.; Marrugo K. P.; Concepcion O.; Kamal G. M.; Muhammad S.; Al-Sehemi A. G. Facile Synthesis, Spectral (IR, Mass, UV–Vis, NMR), Linear and Nonlinear Investigation of the Novel Phosphonate Compounds: A Combined Experimental and Simulation Study. ChemistrySelect 2020, 5, 2994–3006. 10.1002/slct.201904224. [DOI] [Google Scholar]
- Maharramov A. M.; Mahmudov K. T.; Kopylovich M. N.; Pombeiro A. J.. Non-Covalent Interactions in the Synthesis and Design of New Compounds; Wiley Online Library, 2016. [Google Scholar]
- Ali A.; Khalid M.; Marrugo K. P.; Kamal G. M.; Saleem M.; Khan M. U.; Concepción O.; de la Torre A. F. Spectroscopic and DFT/TDDFT insights of the novel phosphonate imine compounds. J. Mol. Struct. 2020, 1207, 127838. 10.1016/j.molstruc.2020.127838. [DOI] [Google Scholar]
- Malik A. N.; Kuznetsov A.; Ali A.; Ashfaq M.; Tahir M. N.; Siddique A. Imine-Based Zwitterion: Synthesis, Single-Crystal Characterization, and Computational Investigation. J. Mol. Struct. 2022, 1253, 132237. 10.1016/j.molstruc.2021.132237. [DOI] [Google Scholar]
- Saeed A.; Ashraf S.; White J. M.; Soria D. B.; Franca C. A.; Erben M. F. Synthesis, X-ray crystal structure, thermal behavior and spectroscopic analysis of 1-(1-naphthoyl)-3-(halo-phenyl)-thioureas complemented with quantum chemical calculations. Spectrochim. Acta, Part A 2015, 150, 409–418. 10.1016/j.saa.2015.05.068. [DOI] [PubMed] [Google Scholar]
- González D. L. N.; Saeed A.; Shabir G.; Flörke U.; Erben M. F. Conformational and crystal structure of acyl thiourea compounds: the case of the simple (2, 2-dimethyl-propionyl) thiourea derivative. J. Mol. Struct. 2020, 1215, 128227. 10.1016/j.molstruc.2020.128227. [DOI] [Google Scholar]
- Saeed A.; Erben M. F.; Flörke U. Effect of fluorine substitution on the crystal structures and vibrational properties of phenylthiourea isomers. J. Mol. Struct. 2010, 982, 91–99. 10.1016/j.molstruc.2010.08.012. [DOI] [Google Scholar]
- Keleşoğlu Z.; Büyükgüngör O.; Albayrak Ç.; Odabaşoğlu M. (E)-4-Meth-oxy-2-[3-(trifluoro-meth-yl)phenyl-imino-meth-yl]phenol. Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, 65, o3245–6. 10.1107/S160053680905034X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şahin Z. S.; Gümüş S.; Macit M.; Işık Ş. (E)-2-[3-(Trifluoromethyl) phenyliminomethyl] benzene-1, 4-diol. Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, 65, o2754. 10.1107/s1600536809041610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şahin Z. S.; Ağar E.; Işık Ş. (E)-4-Methyl-2-[3-(trifluoromethyl) phenyliminomethyl] phenol. Acta Crystallogr., Sect. E: Struct. Rep. Online 2007, 63, O2854. 10.1107/S1600536807017655. [DOI] [Google Scholar]
- Alpaslan G.; Macit M.; Büyükgüngör O.; Erdönmez A. (E)-1-[(2-Fluorophenyl)iminomethyl]-2-naphthol-(Z)-1-[(2-fluorophenyl)aminomethylidene]naphthalen-2(1H)-one (0.57/0.43). Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, o1178. 10.1107/s1600536810014777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ünver H.; Karakaş A.; Elmali A.; Durlu T. N. The investigation of nonlinear optical properties of N-(3-fluorophenyl) naphthaldimine. J. Mol. Struct. 2005, 737, 131–137. 10.1016/j.molstruc.2004.10.016. [DOI] [Google Scholar]
- Ünver H.; Zengi D. M.; Durlu T. N. Crystal structure of 1-[N-(4-fluorophenyl)] naphthaldimine. Anal. Sci. 2001, 17, 1021–1022. 10.2116/analsci.17.1021. [DOI] [PubMed] [Google Scholar]
- Wolff M. A.; Grimwood S. K.; McKinnon D. J.; Turner J. J.; Jayatilaka M. J.; Spackman D.. Crystal Explorer 17.5; University of Western Australia, 2012. [Google Scholar]
- a Spackman M. A.; Jayatilaka D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. 10.1039/b818330a. [DOI] [Google Scholar]; b Tahir M. N.; Ashfaq M.; De la Torre A. F.; Caballero J.; Hernández-Rodríguez E. W.; Ali A. Rationalizing the stability and interactions of 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 2-hydroxy-3,5-dinitrobenzoate salt. J. Mol. Struct. 2019, 1193, 185–194. 10.1016/j.molstruc.2019.05.003. [DOI] [Google Scholar]; c Khalid M.; Ali A.; Tariq J.; Tahir M. N.; Aliabad H. A. R.; Hussain I.; Ashfaq M.; Khan M. U. Stabilization of Supramolecular Assembly of N-Substituted Benzylidene Acetohydrazide Analogs by Non-Covalent Interactions: A Concise Experimental and Theoretical Approach. ChemistrySelect 2020, 5, 10618–10631. 10.1002/slct.202002653. [DOI] [Google Scholar]; d Ashfaq M.; Tahir M. N.; Kuznetsov A.; Mirza S. H.; Khalid M.; Ali A. DFT and single crystal analysis of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chloro-phenyl)-6-ethylpyrimidin-1-ium:4-hydroxybenzoate:methanol:hydrate (1:1:1:1) (DEHMH). J. Mol. Struct. 2020, 1199, 127041. 10.1016/j.molstruc.2019.127041. [DOI] [Google Scholar]; e Madni M.; Ahmed M. N.; Hafeez M.; Ashfaq M.; Tahir M. N.; Gil D. M.; Galmés B.; Hameed S.; Frontera A. Recurrent π-π stacking motifs in three new 4,5-dihydropyrazolyl-thiazole-coumarin hybrids: X-ray characterization, Hirshfeld surface analysis and DFT calculations. New J. Chem. 2020, 44, 14592–14603. 10.1039/d0nj02931a. [DOI] [Google Scholar]
- a Saeed A.; Ashraf S.; Flörke U.; Delgado Espinoza Z. Y.; Erben M. F.; Pérez H. Supramolecular self-assembly of a coumarine-based acylthiourea synthon directed by π-stacking interactions: Crystal structure and Hirshfeld surface analysis. J. Mol. Struct. 2016, 1111, 76–83. 10.1016/j.molstruc.2016.01.074. [DOI] [Google Scholar]
- Ketata I.; Bejaoui L.; Brahmia A.; Ben Hassen R. Synthesis, crystal structure, Hirshfeld surface analysis, spectroscopic characterizations and electrical properties of the new complex of cobalt (III) and the Schiff-base of dihydroxycoumarin. J. Mol. Struct. 2021, 1246, 131161. 10.1016/j.molstruc.2021.131161. [DOI] [Google Scholar]
- a McKinnon J. J.; Jayatilaka D.; Spackman M. A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]; b Ali A.; Khalid M.; Abid S.; Tahir M.; Iqbal J.; Ashfaq M.; Kanwal F.; Lu C.; Rehman M. Green synthesis, SC-XRD, non-covalent interactive potential and electronic communication via DFT exploration of pyridine-based hydrazone. Crystals 2020, 10, 778. 10.3390/cryst10090778. [DOI] [Google Scholar]; c Khalid M.; Ali A.; Haq S.; Tahir M. N.; Iqbal J.; Braga A. A. C.; Ashfaq M.; Akhtar S. U. H. O-4-Acetylamino-benzenesulfonylated pyrimidine derivatives: synthesis, SC-XRD, DFT analysis and electronic behaviour investigation. J. Mol. Struct. 2021, 1224, 129308. 10.1016/j.molstruc.2020.129308. [DOI] [Google Scholar]; d Ashfaq M.; Bogdanov G.; Glebov V.; Ali A.; Tahir M. N.; Abdullah S. Single crystal investigation, Hirshfeld surface analysis and DFT exploration of the pyrimethamine-based novel organic salt: 2, 4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 3-carboxybenzoate hydrate (1: 1: 1). J. Mol. Struct. 2021, 1224, 129309. 10.1016/j.molstruc.2020.129309. [DOI] [Google Scholar]
- Saeed A.; Bolte M.; Erben M. F.; Pérez H. Intermolecular interactions in crystalline 1-(adamantane-1-carbonyl)-3-substituted thioureas with Hirshfeld surface analysis. CrystEngComm 2015, 17, 7551–7563. 10.1039/c5ce01373a. [DOI] [Google Scholar]
- Nasaruddin N. H.; Ahmad S. N.; Sirat S. S.; Wai T. K.; Zakaria N. A.; Bahron H. Structural Characterization, DFT, Hirshfeld Surface Analysis and Antibacterial Activity of a Schiff Base Derived from Cyclohexanediamine. J. Mol. Struct. 2021, 1232, 130066. 10.1016/j.molstruc.2021.130066. [DOI] [Google Scholar]
- Saeed A.; Khurshid A.; Flörke U.; Echeverría G. A.; Piro O. E.; Gil D. M.; Rocha M.; Frontera A.; El-Seedi H. R.; Mumtaz A.; Erben M. F. Intermolecular interactions in antipyrine-like derivatives 2-halo-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)benzamides: X-ray structure, Hirshfeld surface analysis and DFT calculations. New J. Chem. 2020, 44, 19541–19554. 10.1039/d0nj03958f. [DOI] [Google Scholar]
- Jelsch C.; Ejsmont K.; Huder L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. 10.1107/s2052252514003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ali A.; Kuznetsov A.; Khan M. U.; Tahir M. N.; Ashfaq M.; Raza A. R.; Muhammad S. 2-Amino-6-methylpyridine based co-crystal salt formation using succinic acid: Single-crystal analysis and computational exploration. J. Mol. Struct. 2021, 1230, 129893. 10.1016/j.molstruc.2021.129893. [DOI] [Google Scholar]; b Ali A.; Khalid M.; Tahir M. N.; Imran M.; Ashfaq M.; Hussain R.; Assiri M. A.; Khan I. Synthesis of Diaminopyrimidine Sulfonate Derivatives and Exploration of Their Structural and Quantum Chemical Insights via SC-XRD and the DFT Approach. ACS Omega 2021, 6, 7047–7057. 10.1021/acsomega.0c06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Turner M. J.; McKinnon J. J.; Jayatilaka D.; Spackman M. A. Visualisation and characterisation of voids in crystalline materials. CrystEngComm 2011, 13, 1804–1813. 10.1039/c0ce00683a. [DOI] [Google Scholar]; b Ashfaq M.; Tahir M. N.; Muhammad S.; Munawar K. S.; Ali A.; Bogdanov G.; Alarfaji S. S. Single-Crystal Investigation, Hirshfeld Surface Analysis, and DFT Study of Third-Order NLO Properties of Unsymmetrical Acyl Thiourea Derivatives. ACS Omega 2021, 6, 31211–31225. 10.1021/acsomega.1c04884. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ashfaq M.; Munawar K. S.; Tahir M. N.; Dege N.; Yaman M.; Muhammad S.; Alarfaji S. S.; Kargar H.; Arshad M. U. Synthesis, crystal structure, Hirshfeld surface analysis, and computational study of a novel organic salt obtained from benzylamine and an acidic component. ACS Omega 2021, 6, 22357–22366. 10.1021/acsomega.1c03078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.. Gaussian 09, Revision D. 01; Gaussian, Inc Wallingford CT, 2009.
- a Braga A. A. C.; Ujaque G.; Maseras F. A DFT Study of the Full Catalytic Cycle of the Suzuki–Miyaura Cross-Coupling on a Model System. Organometallics 2006, 25, 3647–3658. 10.1021/om060380i. [DOI] [Google Scholar]; b Braga A. A. C.; Morgon N. H.; Ujaque G.; Maseras F. Computational Characterization of the Role of the Base in the Suzuki–Miyaura Cross-Coupling Reaction. J. Am. Chem. Soc. 2005, 127, 9298–9307. 10.1021/ja050583i. [DOI] [PubMed] [Google Scholar]; c Braga A. A. C.; Morgon N. H.; Ujaque G.; Lledós A.; Maseras F. Computational study of the transmetalation process in the Suzuki-Miyaura cross-coupling of aryls. J. Organomet. Chem. 2006, 691, 4459–4466. 10.1016/j.jorganchem.2006.02.015. [DOI] [Google Scholar]
- Huang C.-C.; Huang Y.-C.; Hsieh W.-C.; Chen Y.-J.; Jiang S.-K.; Chen B.-H.; Hsu I.-J.; Lee J.-J. The effects of molecular structure and functional group of a rodlike Schiff base mesogen on blue phase stabilization in a chiral system. New J. Chem. 2019, 43, 1743–1756. 10.1039/c8nj04493g. [DOI] [Google Scholar]
- GaussSum Version 3.0, (n.d.), 2021. http://gausssum.sourceforge.net/DocBook/index.html. accessed June 10, 2021.
- Zhurko G. A.. Chemcraft, 2014. http://www.chemcraftprog.com, Receiv. Oct. 22, 2014.
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennington R. D.; Keith T. A.; Millam J. M.. GaussView 5.0. 8; Gaussian Inc., 2008, Google Scholar There Is No Corresponding Record for This Reference. (n.d.). - Google Search, (n.d.).
- a James C.; Raj A. A.; Reghunathan R.; Jayakumar V. S.; Joe I. H. Structural conformation and vibrational spectroscopic studies of 2,6-bis(p-N,N-dimethyl benzylidene)cyclohexanone using density functional theory. J. Raman Spectrosc. 2006, 37, 1381–1392. 10.1002/jrs.1554. [DOI] [Google Scholar]; b Szafran M.; Komasa A.; Bartoszak-Adamska E. Crystal and molecular structure of 4-carboxypiperidinium chloride (4-piperidinecarboxylic acid hydrochloride). J. Mol. Struct. 2007, 827, 101–107. 10.1016/j.molstruc.2006.05.012. [DOI] [Google Scholar]
- Srnec M.; Solomon E. I. Frontier molecular orbital contributions to chlorination versus hydroxylation selectivity in the non-heme iron halogenase SyrB2. J. Am. Chem. Soc. 2017, 139, 2396–2407. 10.1021/jacs.6b11995. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kandemirli F.; Sagdinc S. Theoretical study of corrosion inhibition of amides and thiosemicarbazones. Corros. Sci. 2007, 49, 2118–2130. 10.1016/j.corsci.2006.10.026. [DOI] [Google Scholar]
- Parvarinezhad S.; Salehi M. Synthesis, characterization, crystal structures, Hirshfeld surface analysis and DFT computational studies of new Schiff Bases derived from Phenylhydrazine. J. Mol. Struct. 2020, 1222, 128780. 10.1016/j.molstruc.2020.128780. [DOI] [Google Scholar]
- Soleimani Amiri S.; Makarem S.; Ahmar H.; Ashenagar S. Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1,3,4-oxadiazol-2-yl)thio)benzene-1,2-diol. J. Mol. Struct. 2016, 1119, 18–24. 10.1016/j.molstruc.2016.04.053. [DOI] [Google Scholar]
- a Parr R. G.; Szentpály L. v.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]; b Chattaraj P. K.; Roy D. R. Update 1 of: electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. 10.1021/cr078014b. [DOI] [Google Scholar]
- Activating and Deactivating Groups In Electrophilic Aromatic Substitution; Master Org. Chem., 2017. https://www.masterorganicchemistry.com/2017/09/26/activating-and-deactivating-groups-in-electrophilic-aromatic-substitution/(accessed June 9, 2021).
- Khan M. U.; Khalid M.; Ibrahim M.; Braga A. A. C.; Safdar M.; Al-Saadi A. A.; Janjua M. R. S. A. First Theoretical Framework of Triphenylamine-Dicyanovinylene-Based Nonlinear Optical Dyes: Structural Modification of π-Linkers. J. Phys. Chem. C 2018, 122, 4009–4018. 10.1021/acs.jpcc.7b12293. [DOI] [Google Scholar]
- Khalid M.; Ali M.; Aslam M.; Sumrra S. H.; Khan M. U.; Raza N.; Kumar N.; Imran M. Frontier molecular, Natural bond orbital, UV-Vis spectral study, Solvent influence on geometric parameters, Vibrational frequencies and solvation energies of 8-Hydroxyquinoline. Int. J. Pharma Sci. Res. 2017, 8, 457. 10.13040/IJPSR.0975-82328(2).457-69. [DOI] [Google Scholar]
- Ibrahim M.; Abbad M.; Khaild M. Phytochemical, crystal structure, spectroscopic, DFT based non covalent interactions and non-linear optical studies of neurada procumbens. J. Chem. Soc. Pak. 2018, 40, 749–760. [Google Scholar]
- a Medishetty R.; Zaręba J. K.; Mayer D.; Samoć M.; Fischer R. A. Nonlinear optical properties, upconversion and lasing in metal-organic frameworks. Chem. Soc. Rev. 2017, 46, 4976–5004. 10.1039/c7cs00162b. [DOI] [PubMed] [Google Scholar]; b Manjunatha K. B.; Rajarao R.; Umesh G.; Ramachandra Bhat B.; Poornesh P. Optical nonlinearity, limiting and switching characteristics of novel ruthenium metal-organic complex. Opt. Mater. 2017, 72, 513–517. 10.1016/j.optmat.2017.06.051. [DOI] [Google Scholar]; c Khan I.; Khalid M.; Adeel M.; Niaz S. I.; Shafiq I.; Muhammad S.; Braga A. A. C. Palladium-catalyzed synthesis of 5-(arylated) pyrimidines, their characterization, electronic communication, and non-linear optical evaluations. J. Mol. Struct. 2021, 1237, 130408. 10.1016/j.molstruc.2021.130408. [DOI] [Google Scholar]
- Khalid M.; Arshad M. N.; Tahir M. N.; Asiri A. M.; Naseer M. M.; Ishaq M.; Khan M. U.; Shafiq Z. An efficient synthesis, structural (SC-XRD) and spectroscopic (FTIR, 1HNMR, MS spectroscopic) characterization of novel benzofuran-based hydrazones: An experimental and theoretical studies. J. Mol. Struct. 2020, 1216, 128318. 10.1016/j.molstruc.2020.128318. [DOI] [Google Scholar]
- Lash T. D.Organic Chemistry, Carey Francis A., Ed.; ACS Publications, 1996. [Google Scholar]
- Hameed K.; Rubina S.; Raheel A. Design, synthesis, docking and in vitro antibacterial study of fluoro substituted schiff bases. J. Chem. Soc. Pak. 2018, 40, 782–791. [Google Scholar]
- Guzel B.; Avsar G. Fluorus Schiff base ligands and their nickel(II) complexes: synthesis, characterization and solubility properties in supercritical CO2. Inorg. Chem.: Indian J. 2008, 3, 1–4. [Google Scholar]
- Mohandas T.; Inbaseelan C. R. D.; Saravanan S.; Sakthivel P. Glycine-D-tartaric acid (1/1). Acta Crystallogr., Sect. E: Struct. Rep. Online 2013, 69, o236. 10.1107/s1600536813000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz R.; Martínez F.; Orcajo G.; Wojtas L.; Briones D. Synthesis of a honeycomb-like Cu-based metal-organic framework and its carbon dioxide adsorption behaviour. Dalton Trans. 2013, 42, 2392–2398. 10.1039/c2dt32138f. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. A short history ofSHELX. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 2008, 64, 112–122. 10.1107/s0108767307043930. [DOI] [PubMed] [Google Scholar]
- Kratzert D.; Holstein J. J.; Krossing I. DSR: enhanced modelling and refinement of disordered structures withSHELXL. J. Appl. Crystallogr. 2015, 48, 933–938. 10.1107/s1600576715005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuchuryukin A. V.; Huang R.; Lutz M.; Chadwick J. C.; Spek A. L.; Van Koten G. NCN-Pincer Metal Complexes (Ti, Cr, V, Zr, Hf, and Nb) of the Phebox Ligand (S,S)-2,6-Bis(4′-isopropyl-2′-oxazolinyl)phenyl. Organometallics 2011, 30, 2819–2830. 10.1021/om200170b. [DOI] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. 10.1107/s1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGXandORTEP for Windows: an update. J. Appl. Crystallogr. 2012, 45, 849–854. 10.1107/s0021889812029111. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.