

Abstract
Introduction
The U.S. Food and Drug Administration (FDA)'s guidances help describe the agency's current thinking on regulatory issues and serve as a means of informal policymaking that is non‐binding. This study examines the impact of two guidance documents for Alzheimer's disease (AD) trials. The first guidance in 2013 encouraged the use of cognitive/functional endpoints, while the second in 2018 modified such recommendation.
Methods
Using pivotal trial data, we applied a regression discontinuity in time (RDiT) framework to examine trialist response to these guidance documents. Results were stratified by disease‐modifying therapy (DMT) status, and controlled for disease staging, FDA registration status, and trial phase.
Results
Among AD DMT trials, annual use of cognitive/functional composite endpoints significantly increased after the 2013 guidance (+12.9%, P < .001), and significantly decreased after the 2018 guidance (–19.9%, P = .022).
Discussion
Although guidance documents do not set new legal standards or impose binding requirements, our findings indicate they are broadly followed by AD trialists.
Keywords: Alzheimer's disease, Clinical Dementia Rating Sum of Boxes, clinical trials, composite endpoints, Food and Drug Administration, guidance documents, legal, mild cognitive impairment, primary endpoints, prodromal, regulatory
1. BACKGROUND
In clinical trials, primary endpoint selection governs the nature of the evidence produced and serves as an important input for the review of the drug by regulatory authorities like the US Food and Drug Administration (FDA) 1 . Endpoint choices depend on several factors, including the primary objective of the clinical trial, the endpoint's meaningfulness to end‐users, ease of measurement, interpretability, and the likelihood of meeting the endpoint 2 , 3 . Selection of endpoints may also be influenced in part by regulatory authorities through formal as well as informal policymaking, which is the focus of this study.
In the United States, the FDA, a federal agency within the US Department of Health and Human Services, oversees clinical trials for drug, biologic, and medical device products. It has several regulatory tools at its disposal 4 . Among these are guidance documents that it issues to help describe its current thinking on regulatory issues 5 , 6 , 7 . Guidance documents are not legally binding on the public or the FDA itself 5 , 6 , 7 . They are intended to provide insight on the agency's current thinking and offer advice related to trial elements as diverse as recruitment, the conduct and monitoring of trials, ethics, and trial design—including primary endpoint selection 8 . Trialists may use alternative approaches to these non‐binding guidances, as long as they satisfy the requirements of the applicable statutes and regulations 9 .
To formalize ongoing deliberations for clinical trial design in early Alzheimer's disease (AD), the FDA released draft guidance in 2013 that offered non‐binding recommendations for early‐stage AD trials 10 . The 2013 draft guidance recommended that drugs demonstrate efficacy on both a cognitive and a functional or global assessment scale. It acknowledged the challenges to measuring cognitive improvements in early disease (i.e., mild cognitive impairment [MCI]), and suggested the use of a composite cognitive and functional score as a suitable tool for assessment in early disease. The guidance then specifically suggested Clinical Dementia Rating–Sum of Boxes (CDR‐SB) as an example of such an endpoint for early disease. A follow‐on draft guidance released in 2018 for early AD trials shifted away from such blanket recommendations, and suggested a broader array of endpoints including biomarkers, coprimary endpoints, and integrated scales (i.e., cognitive, functional), to establish efficacy depending on the type of early AD that a trial is assessing 11 . This study seeks to examine and quantify trialist response to language in non‐binding FDA guidances, and use these two guidance documents in AD as a case study.
As guidance documents are not enforceable through administrative actions or through courts, their effectiveness is not immediately obvious 4 . Many guidances remain perpetually in draft form—including the guidances in our study—and until finalized they do not technically reflect the current thinking of the FDA, which leaves guidances as “doubly noncommittal” tools 9 , 12 , 13 . Furthermore, the multi‐stakeholder nature of the FDA approval process clouds the potential effect of a non‐binding opinion offered by the agency. For example, in the case of the FDA's approval of Aduhelm, an external panel of experts opposed the drug's authorization and held a viewpoint counter to that of the FDA 14 . This external panel's recommendation historically has held great influence in the FDA's decision‐making for approvals. Thus, given the multi‐stakeholder nature, compliance with FDA guidances is not immediately obvious. On top of this, trialists adhering to earlier guidance that is later replaced with conflicting recommendations cannot hold the FDA accountable to its earlier expressed views—a form of non‐estoppel that could hinder adherence 15 . Last, we have identified a meaningful number of investigators that elected not to follow draft guidances of the FDA. As such, non‐binding guidances are an imperfect tool whose efficacy needs to be established. At present the FDA issues guidance without knowing the extent to which that advice will impact trialist behavior, and no evidence in the literature yet supports the view that non‐binding guidances have their intended effects.
It also remains unclear whether the FDA is passively responding to trends in clinical research by issuing reactive guidance documents, or rather trying to advance the design of trials by issuing proactive guidances. Our analysis takes advantage of two shifts in guidance that help to distinguish between these two hypotheses for guidances related to AD. To the best of our knowledge, this is the first empirical study of trialist response to FDA guidances, and it applies a framework that could be used in the future to gauge the impact of other guidance changes more generally.
In this study, we analyze historical AD primary endpoint use over time and by clinical domain, by examining differences in how they evolve immediately after these two guidance documents of interest are issued. We also examine the responsiveness of private versus public sponsors to these guidance documents.
2. METHODS
To understand the impact of the 2013 and 2018 FDA non‐binding draft guidances, we compiled a chronology of pivotal AD trials, assigned primary endpoints to outcome domains of interest, and conducted regression discontinuity in time (RDiT) analyses.
2.1. Trial selection
Pivotal AD trials were identified in the Citeline TrialTrove database, which is a global registry of clinical trials. This dataset was extracted in June 2020, and AD trials were identified based on the following International Classification of Diseases, 10th revision codes: G30.0, G30.1, G30.8, G30.9 16 . We define pivotal trials as later phase trials, and included phase II/III, III, III/IV trials in our dataset. The dataset was then compared to prior AD pipeline reviews 17 , 18 , 19 , 20 , 21 , to confirm that all pivotal trials identified in those publications were present in the dataset.
Trials with a start date of 1997 and later were then kept, as trial reporting from this point forward became more comprehensive (applicable statutes include the FDA Modernization Act [FDAMA] enacted in 1997 and the FDA Amendments Act [FDAA] enacted in 2007) 22 . Such acts created the ClinicalTrials.Gov registry among other reforms, and thus ensured that trial data from this year forward would be more comprehensive. In cases for which trial start dates were unavailable, an imputation of the trial start date was conducted by subtracting trial length as predicted by trial sample size and individual trial site regions, from trial end dates. Sensitivity analyses were conducted to confirm study findings were robust even when excluding imputed trials.
2.2. Primary endpoints
Primary endpoint data were sourced from the TrialTrove dataset. In cases for which primary endpoint information had not been extracted, a manual review was completed using trial registry websites, as well as published manuscripts and abstracts, to identify primary endpoints. Because some primary endpoints varied simply due to version (e.g., Alzheimer's Disease Assessment Scale [ADAS]‐Cog 12, ADAS‐Cog Korean), we collapsed endpoints into broader categories (e.g., ADAS‐Cog) to better summarize trends in endpoint use. Trials that were solely focused on safety and/or tolerability were excluded as our aim was to study trials focused on efficacy. Finally, trials without any primary endpoint data were dropped as the goal of this study was to examine primary endpoint trends.
2.3. Domain and disease‐modifying therapy assignment
All primary endpoints were assigned to domains, which included cognitive, functional, behavioral, and biomarker. Domain assignment was performed based on a review of instrument documentation, published literature, and the medical expert opinion of a neurologist. Trials were then classified as either disease‐modifying therapy (DMT) therapies or non‐DMT therapies, based on prior published classifications 17 , 18 , 19 , 20 , 21 as well as medical expert opinion.
2.4. Regression specification
This study applies a RDiT framework, which is a type of time‐series analysis, to estimate the impact of the 2013 and 2018 FDA draft guidance documents on primary endpoint selection. The RDiT design relies on time‐series variation to identify the effects of policy changes on outcomes, and has been used empirically in a number of economic studies 23 , 24 , 25 , 26 . In a RDiT regression, time is used as a continuous longitudinal measure to determine treatment assignment, and treatment begins at a particular threshold in time. RDiT is engineered to overcome some of the limitations of a simple pre/post or Chi‐squared analysis by targeting a very specific date on which a change occurs, and allows us to evaluate outcomes in a continuous longitudinal manner.
The RDiT design was specified as a piecewise linear regression. We regressed the use of a cognitive/functional composite endpoint (or use of CDR‐SB) on trial start date, and placed a knot on trial start date at March 1, 2013, as well as March 1, 2018, to study the effects of the 2013 and 2018 FDA draft guidances (Appendix Figure A1). Because trials examining DMT therapies are thought to be more likely to respond to these two sets of guidance documents, as they seek to demonstrate cognitive and functional/global improvements, we stratified models by DMT status. The models controlled for AD staging, FDA registration status, and trial phase. Cognitive/functional composite endpoints were defined as endpoints that measured both cognition and function, regardless of whether they measured other additional domains (such as behavior). AD staging was defined based on the 2011 National Institute on Aging–Alzheimer's Association diagnostic guidelines 27 : (1) asymptomatic (i.e., presence of AD biomarkers without cognitive changes); (2) symptomatic predementia (also known as prodromal AD or MCI; i.e., cognitive impairment with preserved functional independence); and (3) dementia due to AD, including mild, moderate, or severe dementia. Model robustness was tested by evaluating model fit and knot P‐values under alternative model specifications in which the policy‐relevant knot was varied.
Analyses were then conducted using Stata Version 13.1 (StataCorp).
3. RESULTS
3.1. Trial selection
A total of 3227 unique global AD trials were identified in the TrialTrove dataset, of which 487 were phase II/III, III, or III/IV trials (Figure 1). Thirteen trials with a trial start date predating 1997 were excluded, as well as 62 trials that focused solely on safety and/or tolerability. Thirty‐nine trials without a trial start date were excluded, and 59 trials without any identifiable primary endpoint information were also excluded. After excluding all these trials, 314 trials remained eligible for this study.
FIGURE 1.
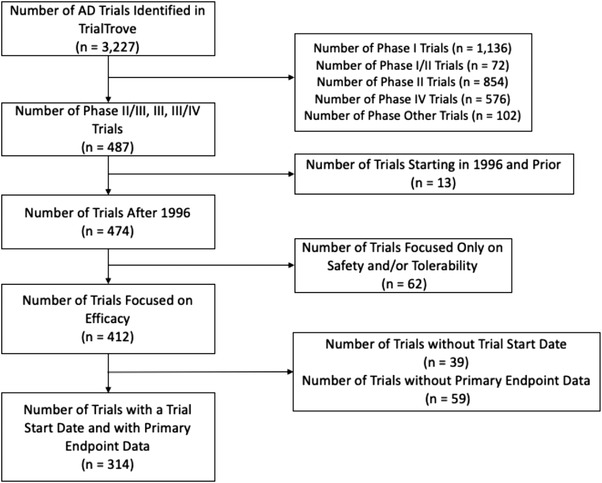
Diagram summarizing the selection of pivotal Alzheimer's disease clinical trials included for the study analysis
3.2. Characteristics of the selected trials
Of these trials, 34 (10.8%) were phase II/III trials, 279 (88.9%) were phase III trials, and 1 (0.3%) was a phase III/IV trial (Appendix Table A1). AD trial initiation in these trials reached its peak during the period from 2001 to 2005. Among the trials studied, 124 (39.5%) investigated DMTs. In terms of endpoint frequency, 159 (50.6%) trials had just one primary endpoint, and 155 (49.4%) trials had two or more primary endpoints. One hundred eighty‐two (58.0%) trials had at least one site in North America, 150 (47.8%) had at least one site in Europe, and 100 (31.9%) had at least one site in Asia.
3.3. Primary endpoint use
A total of 128 unique primary endpoints were identified from the sample of trials (Table 1 and Appendix Table A2). Over the course of the study period, the five most frequently used primary endpoints were ADAS‐Cog (n = 145), Mini‐Mental State Examination (MMSE; n = 45), Clinician's Interview‐Based Impression of Change–Plus Caregiver Input (CIBIC+; n = 36), Neuropsychiatric Inventory (NPI; n = 35), and the Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS‐ADL) (n = 33). In terms of cognitive‐only endpoints, ADAS‐Cog was by far the most frequently selected. Regarding multi‐domain endpoint use, the three most frequently used endpoints were the CIBIC+ (n = 36), CDR‐SB (n = 30), and Clinical Global Impression of Change (CGIC; n = 21).
TABLE 1.
List of primary endpoints used in pivotal Alzheimer's disease trials, grouped by domain classification
| Domain classification | Frequency |
|---|---|
| Cognitive only | |
| Alzheimer's Disease Assessment Scale–Cognitive Subscale | 145 |
| Mini‐Mental State Examination | 45 |
| Severe Impairment Battery | 24 |
| Global Deterioration Scale | 6 |
| Neuropsychological Test Battery | 3 |
| Trail Making Test | 3 |
| Behavioral only | |
| Neuropsychiatric Inventory | 35 |
| Cohen‐Mansfield Agitation Inventory | 19 |
| Brief Psychiatric Rating Scale | 4 |
| Geriatric Depression Scale | 4 |
| Behavioral Pathology in Alzheimer's Disease | 3 |
| Functional only | |
| Alzheimer's Disease Cooperative Study–Activities of Daily Living | 33 |
| Activities of Daily Living | 9 |
| Disability Assessment for Dementia | 9 |
| Instrumental Activities of Daily Living | 7 |
| Physical Self‐Maintenance Scale | 2 |
| PROs only | |
| Nighttime Total Sleep Time | 4 |
| Progressive Deterioration Scale | 3 |
| Caregiver Stress Scale | 2 |
| Quality of Life–Alzheimer's Disease | 2 |
| Biomarkers only | |
| Magnetic Resonance Imaging | 3 |
| Positron Emission Tomography | 2 |
| Total Antioxidant Capacity | 2 |
| Multi‐domain | |
| Clinician's Interview‐Based Impression of Change–Plus Caregiver Input | 36 |
| Clinical Dementia Rating Scale–Sum of Boxes | 30 |
| Clinical Global Impression of Change | 21 |
| Clinical Global Impression | 16 |
| Clinical Dementia Rating Scale | 7 |
Note : For brevity, only the five most frequently used primary endpoints are shown for each domain grouping. A complete list of primary endpoints, including labeled cognitive/functional composite endpoints, is available in Appendix Table A2.
3.4. Longitudinal trends in primary endpoint use
In Figure 2, the 10 most commonly used primary endpoints are presented across time. Proportions are presented relative to March 2013, which is when the 2013 FDA guidance for AD trials was released. Use of ADAS‐Cog has remained relatively consistent, between 37.9% and 46.3% of trials, across the evaluated time periods (Figure 2). However, the use of the CDR‐SB tool rose sharply, from a low of 1.9% in the period before March 2013, to a high of 16.7% in the period after. A similar rise was also seen for the Cohen‐Mansfield Agitation Inventory (CMAI), which may reflect the increase in novel non‐DMT assets targeting agitation during this period. The use of global endpoints such as CIBIC+, CGIC, and Clinical Global Impression (CGI) fell steadily across the study period, reflecting a shift toward alternatives such as CDR‐SB.
FIGURE 2.
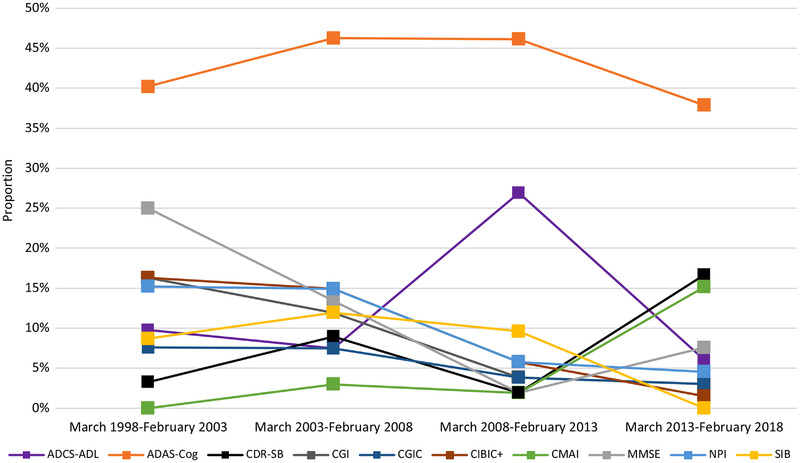
Primary endpoint use in pivotal Alzheimer's disease clinical trials across time. ADAS‐Cog, Alzheimer's Disease Assessment Scale–Cognitive Subscale; ADCS‐ADL, Alzheimer's Disease Cooperative Study – Activities of Daily Living; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; CGI, Clinical Global Impression CGIC, Clinical Global Impression of Change; CIBIC+, Clinician's Interview‐Based Impression of Change–Plus Caregiver Input; CMAI, Cohen‐Mansfield Agitation Inventory; MMSE, Mini‐Mental State Examination; NPI, Neuropsychiatric Inventory; SIB, Severe Impairent Battery
Broader trends in AD endpoint selection are more apparent in the types of endpoint domains used over time. Figure 3 shows changes in proportions relative to March 2013 for the various domains. Most trials used a primary endpoint that featured only cognition, at rates between 56.1% to 77.6% across the evaluated time periods—this reflects a majority. And while only 15.4% of trials initiated between March 2008 and March 2013 used a primary endpoint that measured more than one domain, this percentage rose to 27.3% in the 5 years after.
FIGURE 3.
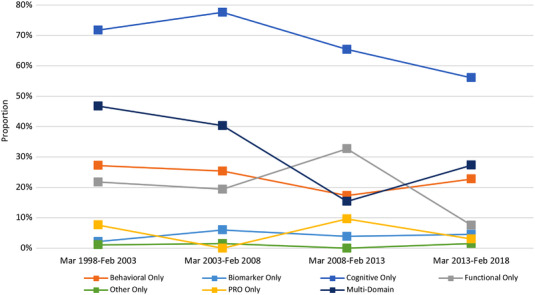
Primary endpoint domain use in pivotal Alzheimer's disease clinical trials across time
3.5. Impact of the FDA 2013 and 2018 non‐binding guidances
We first descriptively analyze trends in the use of the recommended primary endpoints over time. In Figure 4, LOWESS (locally weighted scatterplot smoothing) plots of the proportion of trials using a cognitive/functional composite endpoint (and CDR‐SB) are presented among both DMT and non‐DMT trials. Across time, the use of these two types of endpoints steadily declined among non‐DMT trials. Among DMT trials, a decline in cognitive/functional composite endpoint use was also seen prior to the March 2013 guidance; however, their use, and the use of CDR‐SB, increased sharply in the subsequent period. The LOWESS plots show decreased use of both types of endpoints in the period after March 2018, which reflects the March 2018 guidance that offered alternative options for early AD trials.
FIGURE 4.
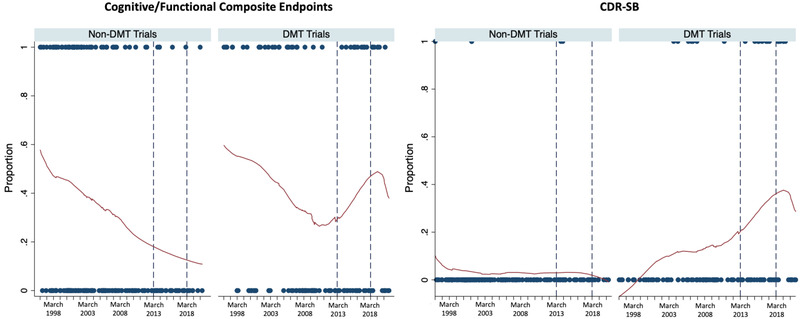
Locally weighted scatterplot smoothing (LOWESS) plots of the use of cognitive/functional composite endpoints and CDR‐SB. CDR‐SB, Clinical Dementia Rating–Sum of Boxes; DMT, disease‐modifying therapy
A model based on a RDiT framework was then used to investigate empirically the effects of the 2013 and 2018 draft guidance documents (Table 2). Among DMT trials, the proportion of trials using a cognitive/functional composite endpoint significantly increased yearly (+12.9%; P < .001) from March 2013 to March 2018, and significantly decreased yearly after March 2018 (–19.9%; P = .022). Similarly, the proportion of trials using CDR‐SB significantly increased yearly from March 2013 to March 2018 (+11.5%; P < .001), and significantly decreased yearly after March 2018 (–14.8%; P = .017). Among non‐DMT trials, we did not identify the same type of effects for either of these endpoints. Notably, use of CDR‐SB is significantly higher (+25.2%; P < .001) among DMT trials that were FDA registered.
TABLE 2.
Regression discontinuity in time (RDiT) linear model to investigate the impact of the 2013 and 2018 FDA draft guidances on the selection of primary endpoints in AD DMT trials
| Cognitive/functional composite endpoint | CDR‐SB | |||||||
|---|---|---|---|---|---|---|---|---|
| DMT | Non‐DMT | DMT | Non‐DMT | |||||
| Independent variables | Coefficient | P‐value | Coefficient | P‐value | Coefficient | P‐value | Coefficient | P‐value |
| Intercept | 2.015 *** | .000 | 1.292 *** | .001 | 0.278 | .518 | −0.057 | .686 |
| Years prior to March 1, 2013 | −0.037 *** | .002 | −0.022 ** | .012 | −0.013 ** | .042 | 0.001 | .730 |
| Years between March 1, 2013, and March 1, 2018 | 0.129 *** | .000 | −0.021 | .418 | 0.115 *** | .000 | −0.005 | .509 |
| Years after March 1, 2018 | −0.199 ** | .022 | 0.105 | .459 | ‐0.148 ** | .017 | −0.006 | .451 |
| AD stage (Reference = overt AD) | ||||||||
| Presymptomatic | −0.016 | .933 | — | — | −0.240 *** | .002 | — | — |
| Prodromal/MCI | 0.075 | .506 | 0.240 | .124 | 0.145 *** | .098 | 0.367 ** | .033 |
| FDA registered trial | 0.065 | .557 | 0.074 | .329 | 0.252 *** | .000 | 0.041 * | .092 |
| Phase (reference = phase III) | ||||||||
| Phase II/III | −0.124 | .226 | −0.077 | .513 | −0.203 *** | .005 | −0.032 | .101 |
| Phase III/IV | 0.739 *** | .000 | — | — | ‐0.239 *** | .000 | — | — |
| Number of observations | 124 | 190 | 124 | 190 | ||||
| R‐squared | 0.141 | 0.087 | 0.293 | 0.228 | ||||
Abbreviations: AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; CI, confidence interval; DMT, disease‐modifying therapy; FDA, Food and Drug Administration; MCI, mild cognitive impairment.
* P < .10, ** P < .05, *** P < .01.
The robustness of the model was tested by evaluating model fit and spline P‐values under alternative model specifications in which the policy‐relevant spline was varied where applicable (Appendix Table A3A and A3B). The base‐case specification offered the highest R2 and lowest spline P‐value, suggesting a robust goodness of fit.
Among DMT trials with private sponsors, the use of cognitive/functional composite endpoints significantly increased yearly in the period from March 2013 to March 2018 (+17.0%; P = .001; Appendix Table A4), as did the use of CDR‐SB (+13.3%; P = .005). Among DMT trials with only public sponsors, no such significant yearly response was seen for cognitive/functional composite endpoints (–12.4%; P = .137) or CDR‐SB (–5.8%; P = .337) after the 2013 guidance. Evidence for the effects after the 2018 draft guidance was non‐significant for DMT trials with either type of sponsor.
We also find that trials with a cognitive/functional composite endpoint (+140.1; P = .020) or CDR‐SB (+227.9; P = .005) had significantly larger sample sizes than trials without these types of endpoints (Appendix Tables A5A & A5B).
4. DISCUSSION
It seems clear from this work that these two guidance documents in the field of AD may have had a strong influence on trial design. After the 2013 draft guidance recommended the use of cognitive/functional composite endpoints, their use increased significantly. Furthermore, after the release of the 2018 draft guidance which changed this recommendation, these trends reversed. Although we show this degree of compliance to guidances, we also identify a number of investigators that elected to use other endpoints (i.e., a single cognitive‐only endpoint or a co‐primary endpoint) rather than those suggested by the guidances. This observation suggests that non‐binding guidance imperfectly influences trialist behavior. These observations are important because they provide quantitative evidence rather than anecdotal observation to demonstrate the effects of regulatory guidance on industry practice.
Our research also shows that suggestions offered in guidances may be interpreted as regulatory requirements. Concerns had in fact been expressed that the FDA's specific suggestion of CDR‐SB would drive the adoption of CDR‐SB as a standard of efficacy in early AD. Because research practices in early AD have yet to be fully established, FDA suggestions might impact the measurement of clinical benefit and even innovation itself if the standard for trial success is constrained 28 . Thus, how the FDA decides to operationalize the guidances to describe trial design is crucial. The guidances also did not clarify whether composite endpoints should report effects by component, which could unwittingly lead to findings that are hard to interpret. For instance, it may be hard to tell whether results are driven solely by a cognitive or functional component, or driven by small effects from several components 28 , 29 ; however, it is understood that when combined, composite endpoints can increase statistical power in trials 30 , 31 .
Unlike laws and associated regulations that undergo a rigorous and time‐consuming “notice and comment” period for stakeholder input before they are enacted formally, guidance documents are developed to provide insight on the current thinking of regulators, intended as advice rather than legally binding instruments 32 . Guidance documents from the FDA are becoming increasingly common, because they can be changed relatively quickly as new scientific information becomes known. The potential “recipes” that they provide for best practice have been viewed as a form of “soft law,” because trialists seeking FDA authorization for a marketed product are incentivized to follow guidance, to increase the likelihood that their efficacy data will be considered acceptable during regulatory reviews. It was therefore unsurprising that the advice from guidances studied here appears most strongly correlated with changes in practice among privately sponsored trials because these types of trials typically are more likely to support marketing applications for AD drugs. These strong effects occurred even though the guidances were still in draft form. The trials supported by public sponsors may also explore questions of mechanisms of action or biological pathways, so they have more freedom to accept endpoints based on first principles rather than perceived instruction from guidance documents. Lower rates of adherence among private sponsors may be due to the relative inexperience of smaller/newer firms, a lack of scientific consensus on the merits of different endpoints, and the expected likelihood of meeting different endpoints.
To our understanding, the responses of clinical trial stakeholders to FDA guidances have not been studied previously with quantitative methods in any disease area. This study adds a new dimension to prior work that examined compliance with traditional standards for primary endpoint selection in AD trials (i.e., a co‐primary approach involving one cognitive endpoint and one functional/global endpoint) 33 . Our work also adds on to past research on guidance documents, which has shown their effects on future innovation 34 , and updates previous work on the landscape of outcome measures in AD 35 . In addition, this study provides greater context regarding the first approved DMT treatment in AD, whose trial initiated after the 2013 draft guidance and used CDR‐SB as its single primary endpoint 36 , 37 . Although this work specifically studies the impact of the 2013 and 2018 draft guidances for AD, it also provides a framework to evaluate the impact of changing regulatory guidance in other disease indications.
We acknowledge that our results come with certain limitations. First, trial start dates were imputed for 17.8% of trials for which data were unavailable in the TrialTrove dataset. However, we conducted sensitivity analyses in which we excluded trials with imputed dates, and found our findings to hold. Second, it is possible that the FDA guidance documents may have simply mirrored changes in endpoint selection by AD trialists or regulatory advice that would have been given without such documents. However, our robustness checks suggest this to be unlikely. Third, the effect sizes measured after the first guidance may have been augmented by the release of the European Medicines Agency's (EMA's) January 2016 draft guidance for AD trials, which reiterated the FDA's March 2013 draft guidance on composite endpoints for prodromal AD or MCI trials 38 . However, the effects found in our analysis emerged well before 2016, and it is possible the EMA release only augmented this effect. Given the limited sample size, we are unable to test EMA‐specific trials to identify effects within this subgroup. Fourth, our policy effect models did not stratify by AD staging, but specified AD staging in the models instead, to optimize sample size and capture disease‐wide effects. Finally, we did not restrict our dataset by trials registered with the FDA, so our findings may have been attenuated by the possibly lower responsiveness of non–US‐centered trials—regulatory spillovers should be tested in further work.
The study also finds that trials with more comprehensive endpoints such as those evaluated require larger trials. This may suggest the 2013 and 2018 FDA draft guidances affected clinical trial costs, as larger clinical trials tend to correlate with higher costs; however, more research on this is needed 39 .
Unlike acts such as the Federal Food, Drug, and Cosmetic Act (FD&C Act), and FDA regulations, guidance documents put forth by the FDA are non‐binding in nature. While these guidance documents do not set new legal standards or impose new requirements, they may be taken as regulatory expectations by trialists and impact clinical trial design, as shown in our RDiT findings—especially by private‐sector trialists. Future research may expand the scope of our analysis to other disease areas or study the effect of FDA guidances on other trial design choices.
CONFLICTS OF INTEREST
JCY received funding through a University of Southern California (USC) School of Pharmacy Travel Award to attend academic conferences. In the past 3 years, JPH has received speaker fees from FTI Consulting. EJ has received pilot funding from her home institution (USC), which supports her work as a primary investigator. EJ has received salary support from the NIA Alzheimer's Disease Research Centers (ADRCs; Award P30‐AG066530) that was paid to USC and supports her work as a co‐investigator. EJ has also received an honoraria payment that was paid to USC to attend the Science of Alzheimer's Disease and Related Dementias for Social Scientists workshop (P30AG043073‐07S1). FJR received funding from the European Commission to serve as an expert reviewer for the Horizon 2020 program (which was called H2020‐SC1‐2020‐Single‐Stage‐RTD) and has served as the Chair of the Conflicts of Interest Committee at USC. In the past 3 years, Darius N. Lakdawalla has received speaker fees or consulting income from the following sources: Amgen, Genentech, GRAIL, Mylan, Novartis, Otsuka, Perrigo, and Pfizer. Dr. Lakdawalla owns equity in Precision Medicine Group, and previously served as a consultant for them. Dr. Lakdawalla also owns equity in EntityRisk, Inc., and serves as its Chief Scientific Officer. The Leonard D. Schaeffer Center for Health Policy & Economics has received an unrestricted grant from Biogen to support research at the center. This work was supported by the National Institutes of Health (NIH; Award R01AG062277).
ACKNOWLEDGMENTS
The authors are grateful to Bryan Tysinger and others who have provided advice to the research team. This work was supported by the National Institutes of Health (Award R01AG062277), as well as the Alzheimer's Disease Research Center (ADRC; Award P30‐AG066530).
APPENDIX A.
A.1.
We summarize the characteristics of the AD pivotal clinical trials in our dataset in Table A1.
TABLE A1.
Characteristics of Alzheimer's disease pivotal clinical trials
| Trial characteristic | Estimate |
|---|---|
| N | 314 |
| Trial phase | |
| II/III | 34 (10.8%) |
| III | 279 (88.9%) |
| III/IV | 1 (0.3%) |
| Trial start year | |
| 1997–2000 | 62 (19.1%) |
| 2001–2005 | 76 (24.2%) |
| 2006–2010 | 68 (21.7%) |
| 2011–2015 | 56 (17.8%) |
| 2016–2020 | 52 (16.6%) |
| DMT status | |
| Yes | 124 (39.5%) |
| Number of unique primary endpoints | |
| 1 | 159 (50.6%) |
| 2 | 112 (35.7%) |
| 3 | 23 (7.3%) |
| 4 | 12 (3.8%) |
| 5 | 3 (1.0%) |
| 6 | 2 (0.6%) |
| 7 | 1 (0.3%) |
| 8 | 2 (0.6%) |
| Trial region (not mutually exclusive) | |
| Americas | 187 (59.6%) |
| North America | 182 (58.0%) |
| South America | 43 (13.7%) |
| Caribbean/Central America | 13 (4.1%) |
| Europe | 150 (47.8%) |
| Eastern Europe | 75 (23.9%) |
| Asia | 100 (31.9%) |
| Australia/Oceania | 60 (19.1%) |
| Western Asia/Middle East | 25 (8.0%) |
| Africa | 0 (0.0%) |
| Not Available | 2 (0.6%) |
Abbreviation: DMT, disease‐modifying therapy.
In Figure A1, we present in equation format the linear model specification for our RDiT analysis.
FIGURE A1.

Regression discontinuity in time (RDiT) linear model specification
The primary endpoints used in our clinical trials are summarized in Table A2 by domain classification.
TABLE A2.
List of primary endpoints used in phase III‐type trials, as grouped by domain classification
| Domain classification |
|---|
| Cognitive only |
| AKT (Alters‐Konzentrations‐Test) |
| Alzheimer's Disease Cooperative Study ‐ Preclinical Alzheimer Cognitive Composite (ADCS‐PACC) |
| Alzheimer's Prevention Initiative Composite Cognitive (APCC) Test Score |
| Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS‐Cog) |
| Brief Praxis Test |
| Buschke Selective Reminding Test |
| Cambridge Examination for Mental Disorders in the Elderly–Cognitive Section (CAMCOG) |
| Category/Letter Fluency |
| Change on a composite score of immediate and delayed recall of verbal and visual memory |
| Choice Reaction Time (CRT) |
| Clock Drawing Test |
| Composite Score of a Broad Cognitive Test Battery |
| Composite Z Score (combining 4 different cognitive tests including 10 Mini‐Mental State Examination [MMSE] questions) |
| Consortium to Establish a Registry for Alzheimer's Disease–Neuropsychological (CERAD‐NP) |
| Delayed Recall Test |
| Dementia Rating Scale |
| DemTect |
| Free and Cued Selective Reminding Test |
| Global Deterioration Scale |
| Gruber & Buscke Test |
| Hasegawa's Dementia Scale‐Revised (HDS‐R) |
| Mini‐Mental State Examination |
| National Institute of Neurological Disorders and Stroke (NINDS)‐initiated EXecutive Abilities: Measures and Instruments for Neurobehavioral Evaluation and Research or (EXAMINER) Tool Box |
| Neurospsychological Test Battery |
| New York University (NYU) Paragraph Delayed Recall Test |
| Preclinical Alzheimer's Cognitive Composite‐5 (PACC‐5) |
| Rey Auditory Verbal Learning Test |
| Seoul Neuropsychologic Screening Battery |
| Seven Minute Test |
| Severe Impairment Battery |
| Stroop Interference Condition ‐ Completion Time and Errors |
| Syndrom Kurz Test (SKT) |
| Trail Making Test |
| Vascular Dementia Assessment Scale ‐ Cognitive Subscale (VaDAS‐cog) |
| Verbal Learning Task Scores |
| Visual Continuous Performance Test (CPT) |
| Wechsler Memory Scale‐Revised Logical Memory (WMS‐R LM) for Immediate and Delayed Recall |
| Behavioral only |
| Alzheimer's Disease Assessment Scale (ADAS) ‐ Behavioral Subscale |
| Apathy Evaluation Scale (AES) |
| Assessment of the Clinical Significance of Behavioral Change Rated by the Study Clinician |
| Behavioral Pathology in Alzheimer's Disease (BEHAVE‐AD) |
| Brief Psychiatric Rating Scale |
| Cohen‐Mansfield Agitation Inventory (CMAI) |
| Columbia Suicide Severity Rating Scale (C‐SSRS) |
| Cornell Scale for Depression in Dementia |
| Emotional Lability Scale |
| Frontal Systems Behavior Scale |
| Geriatric Depression Scale |
| Global Assessment of Functioning (GAF) |
| Hamilton Depression Scale |
| Italian “Scala per la valutazione del benessere emotivo dell'anziano (SVEBA)” |
| NeuroBehavioral Rating Scale‐ Agitation (NBRS‐A) |
| Neuropsychiatric Inventory (NPI) |
| Pittsburgh Agitation Scale |
| Positive and Negative Syndrome Scale (PANSS) |
| Profile of Mood States (POMS) |
| Relapse (Hallucintions, Delusions from Dementia‐Related Psychosis) |
| Sandoz Clinical Assessment‐Geriatric |
| Sheehan Suicidality Tracking Scale (S‐STS) Score |
| Social Dysfunction and Aggression Scale ‐ 9 (SDAS‐9) |
| Functional only |
| Activities of Daily Living (ADL) |
| Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS‐ADL) |
| Alzheimer's Disease Cooperative Study–Instrumental Activities of Daily Living (ADCS‐IADL) |
| Barthel Index |
| Bristol Activities of Daily Living Scale (BrADL) |
| Caregiver Activity Survey |
| Dependence Scale |
| Disability Assessment for Dementia (DAD) |
| Functional Assessment Staging Tool (FAST) |
| Gait Speed |
| Instrumental Activities of Daily Living (IADL) |
| Minimum Data Set Activities of Daily Living Test (MDS‐ADL) |
| Nuremberg Gerontopsychological Rating Scale for Activities of Daily Living (NAI‐NAA) |
| Physical Self‐Maintenance Scale (PSMS) |
| Timed Up and Go (TUG) Test Score |
| PROs only |
| Abbey Pain Scale |
| Caregiver Stress Scale |
| Epworth Sleepiness Scale (ESS) Score |
| Goal Attainment Scaling (GAS) Score |
| Intensity Rating Scale |
| Mean Total Sleep Time |
| Nighttime Total Sleep Time |
| Pain Assessment Checklist for Seniors with Limited Ability to Communicate (PACSLAC) |
| Pain Assessment in Advanced Dementia (PAINAD) |
| Progressive Deterioration Scale |
| Quality of Life–Alzheimer's Disease (QoL‐AD) |
| Screen for Caregiver Burden |
| Short‐Form Health Survey (SF‐36) |
| Visual Analog Scale |
| Biomarkers only |
| Amyloid PET scan |
| APOE4 |
| Brain Volume Change (as measured by Magnetic Resonance Imaging [MRI]) |
| Cerebrospinal Fluid (CSF) |
| Ferric Reducing Ability of Plasma |
| PMN (Polymorphonuclear Leukocytes) Production |
| Glutathione |
| Interleuken‐1‐Beta |
| Interleukin‐6 |
| Magnetic Resonance Imaging (MRI) |
| Malondialdehyde |
| Modified Hachinski Ischemia Score |
| Oxygenated Hemoglobin Concentration |
| Positron Emission Tomography |
| Regional cerebral blood flow using arterial spin‐labeling Magnetic Resonance Imaging (MRI) |
| Serum Brain‐Derived Neurotrophic Factor (BDNF) Levels |
| Serum Levels of Malondialdehyde |
| Superoxid Dismutase |
| TNF‐Alpha |
| Total Antioxidant Capacity |
| Weight change in kilograms |
| Multi‐domain |
| Alzheimer's Disease Assessment Scale (ADAS) Total |
| Alzheimer's Disease Rating Scale* |
| Alzheimer's Disease Cooperative Study – Clinical Global Impression (ADCS‐CGI)* |
| Blessed‐Roth Scale |
| Clinical Dementia Rating – Sum of Boxes (CDR‐SB)* |
| Clinical Dementia Rating (CDR)* |
| Clinical Global Impression of Change (CGIC)* |
| Clinician's Interview‐Based Impression of Change–Plus Caregiver Input (CIBIC+)* |
| Clinician's Interview‐Based Impression of Change (CIBIC)* |
| Global Improvement Scale (GIS)* |
| Gottfries‐Brane‐Steen Scale* |
| Incidence of Alzheimer's Disease (AD) (based on NICDS [National Institute For Communicable Diseases], ADRD [Alzheimer's Disease and Related Dementias], and DSM‐IV [Diagnostic and Statistical Manual of Mental Disorders] Criteria)* |
| mADCS‐CGI* |
| Nuremberg Activities Inventory* |
| Patient Diary |
| Polysomnography |
| Time to clinical diagnosis of AD by NINCDS‐ADRDA (National Institute of Neurological and Communicative Disorders and Stroke ‐ Alzheimer's Disease and Related Disorders Association) and DSM‐IV Criteria* |
| Time to diagnosis of Mild Cognitive Impairment (MCI) due to Alzheimer's Disease (AD) or dementia due to AD* |
Note: *Cognitive/functional composite endpoints which measure cognition/function ± behavior.
Through Appendix Table A3A, we show how the RDiT model's R2 (also known as the coefficient of determination) changes when we vary the discontinuity years. Findings indicate that the base case offers the highest R2, which implies robust model fit.
TABLE A3A.
Regression discontinuity in time (RDiT) design linear model – model R2 when varying discontinuity year
| Spline for 2013 FDA guidance | Model R2 |
|---|---|
| March 2007 | 0.0576 |
| March 2008 | 0.0788 |
| March 2009 | 0.0969 |
| March 2010 | 0.1099 |
| March 2011 | 0.1213 |
| March 2012 | 0.1343 |
| March 2013 (Base Case) | 0.1407 |
| March 2014 | 0.1378 |
| March 2015 | 0.1182 |
| March 2016 | 0.0958 |
| March 2017 | 0.0761 |
| Spline for 2018 FDA guidance | Model R2 |
| March 2014 | 0.0999 |
| March 2015 | 0.1163 |
| March 2016 | 0.1245 |
| March 2017 | 0.1350 |
| March 2018 (base case) | 0.1407 |
Abbreviation: FDA, Food and Drug Administration.
Through Appendix Table A3B, we show the RDiT model's spline P‐values when we vary the discontinuity years. Findings indicate that the base case offers the lowest P‐value, which implies model robustness.
TABLE A3B.
Regression discontinuity in time (RDiT) design linear model – Spline P‐values when varying discontinuity year
| Spline for 2013 FDA guidance | Spline P‐value |
|---|---|
| March 2007 | .2789 |
| March 2008 | .0716 |
| March 2009 | .0189 |
| March 2010 | .0062 |
| March 2011 | .0024 |
| March 2012 | .0006 |
| March 2013 (base case) | .0003 |
| March 2014 | .0004 |
| March 2015 | .0016 |
| March 2016 | .0094 |
| March 2017 | .0313 |
| Spline for 2018 FDA guidance | Spline P |
| March 2014 | .7516 |
| March 2015 | .6839 |
| March 2016 | .3498 |
| March 2017 | .0951 |
| March 2018 (Base Case) | .0220 |
Abbreviation: FDA, Food and Drug Administration.
In Appendix Table A4, we developed an additional RDiT model, which suggests that DMT trials with private sponsors may be more responsive to guidance documents than DMT trials with public sponsors.
TABLE A4.
Regression discontinuity in time (RDiT) design linear model to investigate the impact of the 2013 and 2018 FDA guidances on the selection of primary endpoints in Alzheimer's disease DMT clinical trials, by sponsor type
| Cognitive/functional composite endpoint | CDR‐SB | |||||||
|---|---|---|---|---|---|---|---|---|
| Trials with private sponsors | Trials with public sponsors | Trials with private sponsors | Trials with public sponsors | |||||
| Independent variables | Coefficient | P‐value | Coefficient | P‐value | Coefficient | P‐value | Coefficient | P‐value |
| Intercept | 3.483 *** | .000 | −0.978 | .490 | 1.130 ** | .041 | −0.627 | .353 |
| Years prior to March 1, 2013 | −0.068 *** | .000 | 0.030 | .366 | −0.028 ** | .030 | 0.017 | .344 |
| Years between March 1, 2013, and March 1, 2018 | 0.170 *** | .001 | −0.124 | .137 | 0.133 *** | .005 | −0.058 | .337 |
| Years after March 1, 2018 | −0.091 | .733 | 0.105 | .538 | 0.037 | .878 | 0.109 | .341 |
| AD stage (reference = overt AD) | ||||||||
| Presymptomatic | 0.514 ** | .028 | 0.310 | .244 | −0.386 * | .070 | 0.040 | .383 |
| Prodromal/MCI | 0.072 | .674 | −0.203 | .458 | 0.175 | .191 | −0.127 | .356 |
| FDA registered trial | 0.156 | .282 | −0.139 | .462 | 0.406 *** | .001 | −0.101 | .342 |
| Phase (Reference = phase III) | ||||||||
| Phase II/III | −0.133 | .290 | 0.122 | .541 | −0.150 | .209 | 0.003 | .880 |
| Phase III/IV | 0.765 *** | .000 | — | — | −0.236 *** | .001 | — | — |
| Number of observations | 84 | 34 | 84 | 34 | ||||
| R‐Squared | 0.337 | 0.1014 | 0.409 | 0.145 | ||||
= Abbreviations: AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating–Sum of Boxes; CI, confidence interval; DMT, disease‐modifying therapy; MCI, mild cognitive impairment.
* P < .10, ** P < .05, *** P < .01.
In Appendix Table A5A, we use a linear model to show that DMT trials with cognitive/functional composite primary endpoints have larger sample sizes than DMT trials without.
TABLE A5A.
Linear regression model to investigate whether trials with cognitive/functional composite primary endpoints have larger sample sizes than trials without, among Alzheimer's disease DMT clinical trials
| Sample size | ||
|---|---|---|
| Independent variables | Coefficient | P |
| Intercept | 1075.760 | .251 |
| Has a cognitive/functional composite endpoint | 140.110 ** | .020 |
| AD stage (reference = overt AD) | ||
| Presymptomatic | −822.074 | .390 |
| Prodromal/MCI | −918.275 | .331 |
| FDA registered trial | 288.643 *** | .000 |
| Phase (reference = phase III) | ||
| Phase II/III | −286.396 ** | .032 |
| Phase III/IV | −316.799 *** | .000 |
| DMT status | 240.565 *** | .000 |
| Number of observations | 305 | |
| R‐squared | 0.213 | |
Abbreviations: AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating; DMT, disease‐modifying therapy; FDA, Food and Drug Administration; MCI, mild cognitive impairment.
** P < .05, ***P < .01.
In Appendix Table A5B, we use a linear model to show that DMT trials with CDR‐SB as their primary endpoint have larger sample sizes than DMT trials without.
TABLE A5B.
Linear regression model to investigate whether trials with CDR‐SB as a primary endpoint have larger sample sizes than trials without, among Alzheimer's disease DMT clinical trials
| Sample size | ||
|---|---|---|
| Independent variables | Coefficient | P |
| Intercept | 1210.540 | .210 |
| Has CDR‐SB | 277.909 *** | .005 |
| AD stage (reference = overt AD) | ||
| Presymptomatic | −961.732 | .322 |
| Prodromal/MCI | −996.694 | .301 |
| FDA registered trial | 254.405 *** | .000 |
| Phase (reference = phase III) | ||
| Phase II/III | −283.080 ** | .039 |
| Phase III/IV | −176.911 ** | .010 |
| DMT status | 218.662 *** | .000 |
| Number of observations | 305 | |
| R‐squared | 0.215 | |
Abbreviations: AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating; DMT, disease‐modifying therapy; FDA, Food and Drug Administration; MCI, mild cognitive impairment.
** P < .05, ***P < .01.
Yu JC, Hlávka JP, Joe E, Richmond FJ, Lakdawalla DN. Impact of non‐binding FDA guidances on primary endpoint selection in Alzheimer's disease trials. Alzheimer's Dement. 2022;8:e12280. 10.1002/trc2.12280
REFERENCES
- 1. International Conference on Harmonisation. ICH Harmonised Tripartite Guideline. Statistical principles for clinical trials. International Conference on Harmonisation E9 Expert Working Group. Stat Med. 1999;18(15):1905‐1942. [PubMed] [Google Scholar]
- 2. McLeod C, Norman R, Litton E, Saville BR, Webb S, Snelling TL. Choosing primary endpoints for clinical trials of health care interventions. Contemp Clin Trials Commun. 2019;16:100486. 10.1016/j.conctc.2019.100486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Evans S, McRae‐McKee K, Wong MM, Hadjichrysanthou C, De Wolf F, Anderson R. The importance of endpoint selection: how effective does a drug need to be for success in a clinical trial of a possible Alzheimer's disease treatment?. Eur J Epidemiol. 2018;33(7):635‐644. 10.1007/s10654-018-0381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.FDA Related Laws, Regulations, and Guidances. U.S. Food and Drug Administration. Accessed 2021. https://www.fda.gov/drugs/cder‐small‐business‐industry‐assistance‐sbia/fda‐related‐laws‐regulations‐and‐guidances
- 5.What is the difference between the Federal Food, Drug, and Cosmetic (FD&C Act), FDA regulations, and FDA guidance? U.S. Food and Drug Administration. Accessed 2020. https://www.fda.gov/about‐fda/fda‐basics/what‐difference‐between‐federal‐food‐drug‐and‐cosmetic‐act‐fdc‐act‐fda‐regulations‐and‐fda‐guidance#:∼:text=FDA%20develops%20regulations%20based%20on,law%2C%20to%20issue%20FDA%20regulations.&text=FDA%20guidance%20describes%20the%20agency's%20current%20thinking%20on%20a%20regulatory%20issue
- 6. Noah L. Governance by the Backdoor: Administrative Law(lessness?) at the FDA. Nebraska Law Review. 2014;93(1):89‐138. [Google Scholar]
- 7. Noah L. Law, Medicine, and Medical Technology, Cases and Materials. 5 ed. University Casebook Series. Foundation Press; 2022. [Google Scholar]
- 8.Search for FDA Guidance Documents. U.S. Food and Drug Administration. Accessed 2021. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents#guidancesearch
- 9. Lewis MS. Halted Innovation: The Expansion of Federal Jurisdiction over Medicine and the Human Body. Utah Law Review. 2018;2018(5). [Google Scholar]
- 10.Early Alzheimer's Disease: Developing Drugs for Treatment – Guidance for Industry. U.S. Food and Drug Administration. 2013. Accessed 2020. https://isctm.org/public_access/FDAGuidance_AD_Developing_Drugs_Early_Stage_Treatment.pdf
- 11.Early Alzheimer's Disease: Developing Drugs for Treatment – Guidance for Industry. U.S. Food and Drug Administration. 2018. Accessed 2020. https://www.fda.gov/media/110903/download
- 12.Alexander L, Burr R, Isakson J, Hatch OG. United States Committee on Health, Education, Labor, and Pensions. 2016. Accessed 2022. https://www.help.senate.gov/imo/media/doc/5‐6‐2016%20FDA%20Guidance%20Letter.pdf
- 13.Noah L. Guidance Gone Wild?: FDA's Regrettable Retreat from Legislative Rulemaking. Washington Legal Foundation. 2015. Accessed 2022. https://s3.us‐east‐2.amazonaws.com/washlegal‐uploads/upload/legalstudies/legalbackgrounder/100915LB_Noah.pdf
- 14. Joseph A. Member of FDA's expert panel resigns over controversial Alzheimer's therapy approval. Stat News. 2021. https://www.statnews.com/2021/06/08/fda‐expert‐panel‐resigns‐alzheimers‐approval/. [Google Scholar]
- 15. Lars N. The FDA's New Policy on Guidelines: Having Your Cake and Eating It Too. Catholic University Law Review. 1997;47(1). [Google Scholar]
- 16. Taylor C, Greenlund S, McGuire L, Lu H, Croft J. Deaths from Alzheimer's Disease – United States, 1994‐2014. MMWR Morb Mortal Wkly Rep. 2017;66(20):521‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cummings J, Morstorf T, Lee G. Alzheimer's drug‐development pipeline: 2016. Alzheimers Dement (N Y). 2016;2(4):222‐232. 10.1016/j.trci.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement (N Y). 2017;3(3):367‐384. 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer's disease drug development pipeline: 2018. Alzheimers Dement (N Y). 2018;4:195‐214. 10.1016/j.trci.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272‐293. 10.1016/j.trci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimers Dement (N Y). 2020;6(1):e12050. 10.1002/trc2.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.ClinicalTrials.Gov Background. U.S. Food and Drug Administration. Accessed 2021. https://clinicaltrials.gov/ct2/about‐site/background
- 23. Hausman C, Rapson D. Regression Discontinuity in Time: Considerations for Empirical Applications. Annual Review of Resource Economics. 2018;10:533‐552. [Google Scholar]
- 24. Busse M, Jorge S, Zettelmeyer F. $1,000 Cash Back: The Pass‐Through of Auto‐Manufacturer Promotions. Am Econ Rev. 2006;96(4):1253‐1270. [Google Scholar]
- 25. Burger N, Daniel T, Bo Y. Did California's Hand‐Held Cell Phone Ban Reduce Accidents?. Trans Res Part A. 2014;66(1):162‐172. [Google Scholar]
- 26. M Anderson. Subways, Strikes, and Slowdowns: The Impacts of Public Transit on Traffic Congestion. Am Econ Rev. 2014;104(9):2763‐2796. [Google Scholar]
- 27. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280‐292. 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schneider LS. Rethinking the Food and Drug Administration's 2013 guidance on developing drugs for early‐stage Alzheimer's disease. Alzheimers Dement. 2014;10(2):247‐250. 10.1016/j.jalz.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy CE. Understanding the Use of Composite Endpoints in Clinical Trials. West J Emerg Med. 2018;19(4):631‐634. 10.5811/westjem.2018.4.38383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Insel PS, Donohue MC, Mackin RS, et al. Cognitive and functional changes associated with Aβ pathology and the progression to mild cognitive impairment. Neurobiol Aging. 2016;48:172‐181. 10.1016/j.neurobiolaging.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 31. Wang J, Logovinsky V, Hendrix SB, et al. ADCOMS: a composite clinical outcome for prodromal Alzheimer's disease trials. J Neurol Neurosurg Psychiatry. 2016;87(9):993‐999. 10.1136/jnnp-2015-312383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strauss P. Domesticating Guidance. Lewis Clark Environ Law Rev. 2019;49(3):765‐792. [Google Scholar]
- 33. Takeshima N, Ishiwata K, Sozu T, Furukawa TA. Primary Endpoints in Current Phase II/III Trials for Alzheimer Disease: a Systematic Survey of Trials Registered at ClinicalTrials.gov. Alzheimer Dis Assoc Disord. 2020;34(1):97‐100. 10.1097/WAD.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 34. Weitzman RE, Stern AD, Kramer DB. Food and Drug Administration Guidance Documents and New Medical Devices: the Case of Breast Prostheses. Am J Ther. 2021;28(1):e127‐e130. 10.1097/MJT.0000000000000888. [DOI] [PubMed] [Google Scholar]
- 35. Harrison JK, Noel‐Storr AH, Demeyere N, Reynish EL, Quinn TJ. Outcomes measures in a decade of dementia and mild cognitive impairment trials. Alzheimers Res Ther. 2016;8(1):48. 10.1186/s13195-016-0216-8. 11 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease (EMERGE). ClinicalTrials.gov. Accessed 2021. https://clinicaltrials.gov/ct2/show/NCT02484547
- 37.221AD301 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer's Disease (ENGAGE). ClinicalTrials.gov. Accessed 2021. https://clinicaltrials.gov/ct2/show/NCT02477800
- 38. European Medicines Agency . Draft guideline on the clinical investigation on medicines for the treatment of Alzheimer's disease and other dementias. European Medicines Agency. Accessed 2021. https://www.ema.europa.eu/en/documents/scientific‐guideline/draft‐guideline‐clinicalinvestigation‐medicines‐treatment‐alzheimers‐disease‐other‐dementias_en.pdf
- 39. Cox Grad, S . Trial Design. 2009. New Jersey: John Wiley. [Google Scholar]