

Abstract
BACKGROUND AND AIMS:
Nonalcoholic steatohepatitis (NASH) is associated with obesity and an increased risk for liver cirrhosis and cancer. Neutral ceramidase (NcDase), highly expressed in the intestinal brush border of the small intestine, plays a critical role in digesting dietary sphingolipids (ceramide) to regulate the balance of sphingosine and free fatty acids (FFAs). It remains unresolved whether obesity-associated alteration of NcDase contributes to the manifestation of NASH. Here we revealed that neutral ceramidase deficiency in murine models of NASH prevents hepatic inflammation and fibrosis, but not steatosis.
APPROACH AND RESULTS:
NcDase−/− mice display reduced SCD1 expression with a compositional decrease of monounsaturated fatty acids (MUFAs) under the different dietary conditions. We further found that neutral ceramidase is a functional regulator of intestinal B cells and influences the abundance and quality of the secretory IgA response toward commensal bacteria. Analysis of composition of the gut microbiota found that Clostridiales colonization was increased in NcDase−/− mice. The colonization of germ-free mice with gut microbiota from NcDase−/− mice resulted in a greater decrease in the expression of SCD1 and the level of MUFAs in the liver relative to gut microbiota from wild-type (WT) littermates, which are associated with the alternation of IgA bound bacteria including increase of Ruminococcaceae and reduction of Desulfovibrio.
CONCLUSIONS:
Mechanistically, NcDase is a crucial link that controls the expression of SCD1 and MUFAs‐mediated activation of the Wnt/β-catenin. Very importantly, our experiments further demonstrated that Wnt3a stimulation can enhance the activity of neutral ceramidase in hepatocytes. Thus, the NcDase-SCD1-Wnt feedback loop promotes the diet-induced steatohepatitis and fibrosis through the regulation of intestinal IgA+ immune cells.
Keywords: Sphingolipid, extracellular vesicle, gut microbiota, Wnt signaling, SCD1
Introduction
The progression of nonalcoholic steatohepatitis (NASH) is closely associated with dysregulated intracellular lipid accumulation and metabolism, including sphingolipid metabolism(1). High-fat diet (HFD)/palmitate alters expression and activity of the enzymes involved in ceramide synthesis (ceramide synthases [CerSs]), specifically CerS2 and CerS6 (1–3). Through catabolic pathways, ceramides are also derived from the hydrolysis of sphingomyelins by the action of sphingomyelinases or the catabolism of glycosphingolipids. In the salvage pathway, ceramides are synthesized from sphingosine and fatty acyl-CoA by CerSs. Ceramides can be hydrolyzed to produce sphingosine and free fatty acids (FAs) by the action of ceramidases encoded by five distinct genes (N-acylsphingosine amidohydrolase [ASAH] 1, ASAH2, ACER1, ACER2, and ACER3) (3, 4). Neutral ceramidase (NcDase), encoded by ASAH2, is highly expressed in the small intestine and colon along the brush border and facilitates the deacylation of ceramide (5). However, the specific role of NcDase in sphingolipid crosstalk with fatty acids (FAs) in the development/progression of NASH remains poorly defined.
FA composition of lipid species, especially the homeostasis of saturated fatty acids (SFAs) and monounsaturated fatty acids (MUFAs), is an important determinant of the development of hepatic insulin resistance and energy metabolism(6). Stearoyl-CoA desaturase (SCD) is a central lipogenic enzyme that catalyzes the synthesis of MUFAs, primarily oleate and palmitoleate (POA), from SFAs, stearate and palmitate, respectively(7). MUFAs serve as better substrates for the synthesis of hepatic neutral lipids, and thus play a role in increasing tissue lipid load and may initiate insulin resistance and NASH(8). The level of hepatic FA desaturation by SCD1 might depend on the gut microbial load(9, 10). Microbiota have emerged as a key regulator of metabolism within the mammalian host(11). Gut immune responses, especially B cells-derived molecule immunoglobulin A (IgA), are critical in regulating the composition of the microbiota(12). Overweight individuals and high fat diet (HFD)-fed obese mice evince some diminished immune responses, such as reduced levels of mucosal IgA, which was demonstrated as a critical regulator of HFD-induced intestinal inflammation and insulin resistance (13, 14). However, the relationship between gut IgA, MUFAs, and the development of NASH remains unknown.
Activation of the canonical Wnt pathway is a hallmark of both liver fibrogenesis and carcinogenesis(15). The MUFA, palmitoleate, plays a major structural role in mediating the interaction of Wnt with its receptor frizzled(16). Wnt fatty acylation is necessary in order to produce secreted, fully active Wnt protein(17). Thus, SCD1 is required for production of active palmitoleated Wnt proteins and plays a critical role in the development of liver fibrosis(18). Here, we found a deficiency of NcDase alters the pattern of MUFAs in the liver at least in part by regulating the activity of hepatic SCD1 and gut B cells IgA response, and disrupting the composition of commensal bacteria. Thus, the altered Wnt fatty acylation renders the NcDase−/− mice not susceptible to diet-induced steatohepatitis and fibrosis.
Methods
Animals and treatments
C57BL/6 mice were obtained from Jackson Laboratory. NcDase knockout mice (NcDase−/−) were from Dr. Yusuf A. Hannun (Stony Brook University) and had been backcrossed at least 8 generations to C57BL/6(5). NcDase−/− mice and WT littermates were placed on standard chow or a high-fat diet (HFD) containing 60% kcal % Fat (D12492; Research Diets, Inc.) at 5 weeks of age for 7 months (induction of steatosis by HFD) or 14 months (induction of NASH by HFD). For other NASH experiments, groups of mice were placed on a Hydrogenated Vegetable Oil-HVO, sucrose, palmitate, cholesterol (“HSPC”) diet (Teklad, TD. 160785) for four months or a methioninecholine- deficient (MCD) diet (Teklad, TD. 90262) for 8 weeks. Studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Louisville Institutional Animal Care and Use Committee.
Details of other methods used in this study are described in the Supplemental Materials and Methods.
Results
Deletion of NcDase suppresses liver inflammation and fibrosis in mouse models of NASH
We analyzed the expression of NcDase in liver and intestinal extracts from HFD mice with NASH versus steatosis and normal mice and observed a significant increase in NcDase expression in the NASH livers (Figures 1A and 1B). The expression and activity of NcDase was found at its highest level in the gut of mice with NASH (Figures 1A–1C, and Figure S1A). We further explored NcDase expression in another mouse model of NASH induced by a HSPC diet that was rich in HVO, Sucrose, Palmitate, Cholesterol (19, 20). Higher NcDase mRNA expression was also found in the gut of HSPC fed mice (Figure S1B). To test the role of NcDase on diet-induced NASH, we compared NcDase−/− mice with WT littermates maintained on a HFD for 7 or 14 months. HFD-fed NcDase−/− mice had both lower total body weight and a lower liver:body weight ratio than HFD-fed WT mice (Figure S1C). Liver sections showed marked reductions in fibrosis (Figure 1D) and inflammatory cell infiltration (Figure 1E) in the NcDase−/− mice, while steatosis lipid (hematoxylin and eosin and Oil Red O) was not affected (Figure 1E). The levels of hepatic fibrosis markers, including collagen type I alpha 1 chain (Col1a1), fibronectin, and alpha-smooth muscle actin (α-SMA) mRNAs and protein in NcDase−/− mice compared with WT mice (Figures 1F and 1G), and there was also a reduction in α-SMA+ cells (Figure 1H). Additionally, NcDase−/− mice showed significant decreases in alanine aminotransferase (ALT), aspartate aminotransferase (AST) and plasma insulin levels (Figure 1I). However, plasma choline, plasma TG, plasma VLDL, food intake, serum leptin, serum FFAs and liver FFAs were similar in the HFD-fed WT mice and NcDase−/− mice (Figures S1D–S1J). Low choline perturbs mitochondrial bioenergetics and fatty acid beta oxidation, which are critical mechanisms in the pathogenesis of NASH(21). More interestingly, liver choline was significantly increased in the livers of obese NcDase−/− mice, especially in the mice with NASH (Figure 1J). Real-time PCR results revealed that amounts of TNF-α, chemokine (C-X-C motif) ligand 1 (CXCL1), IL-6, TGFβ and S100A8 mRNA in the liver were decreased in HFD-fed NcDase−/− mice (Figure S2A). Furthermore, NcDase deficiency significantly reduced the infiltration of neutrophils and macrophages into the liver (Figures S2B and S2C). FACS analysis confirmed that HFD-fed NcDase−/− mice have decreased accumulation of F4/80-positive macrophages, CD11b+Gr-1+ immature myeloid cells (iMCs)/myeloid-derived suppressor cells (MDSCs) and CD8+ T cells, but not but not T helper (Th) 1 cells and Th17 cells (Figure S2D) in the liver compared with HFD-fed WT mice. Thus, deletion of NcDase improved or reversed key liver parameters related to inflammation and fibrosis.
Figure 1. NcDase deficiency reduces liver inflammation and fibrosis in HFD-fed mice.
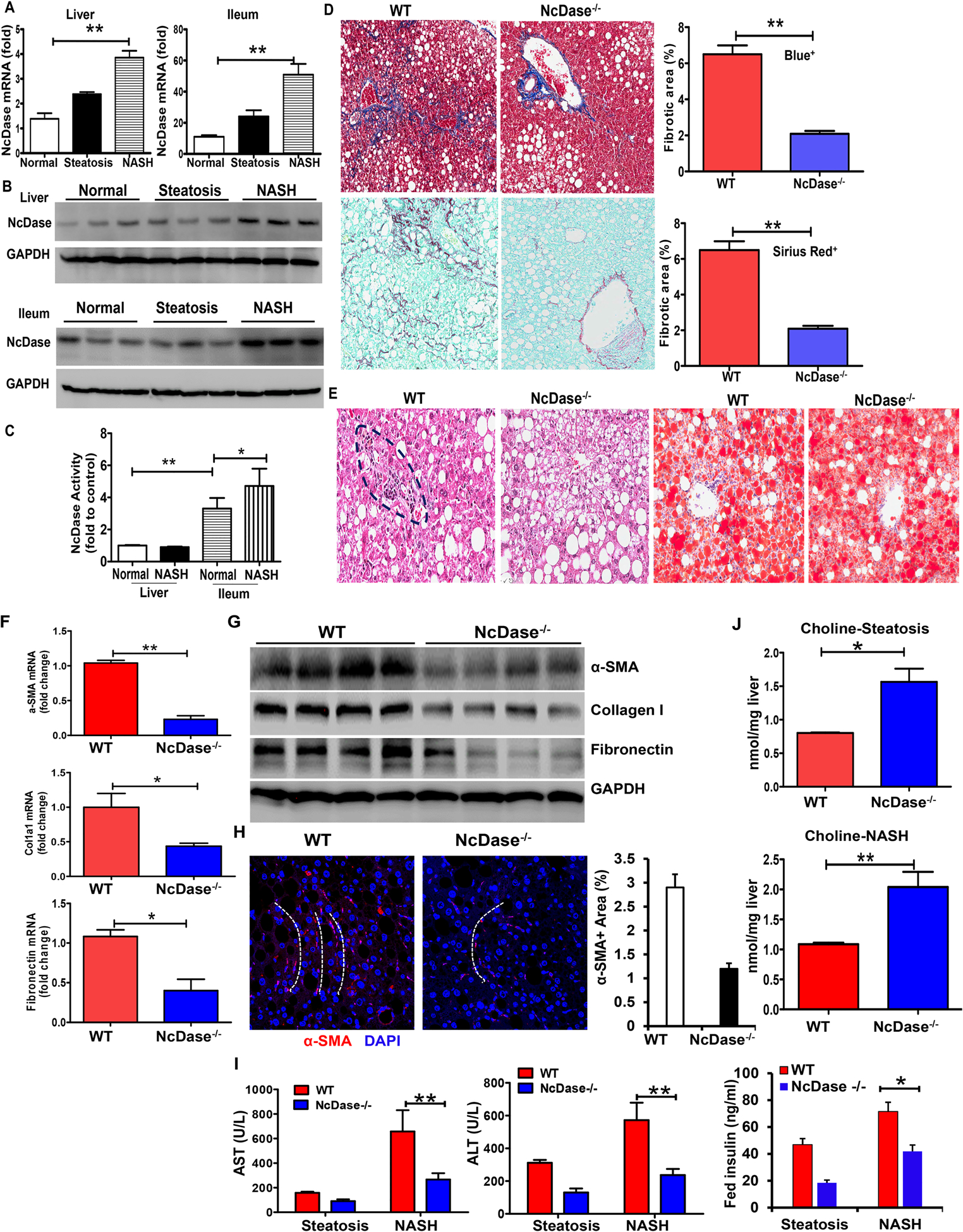
WT mice or NcDase−/− mice were fed the HFD diet for 7 months (inducing steatosis) or 14 months (inducing NASH).
(A) Quantification of NcDase mRNA in liver and ileum in normal, steatotic and NASH mice.
(B) Immunoblots of NcDase in liver and ileum in normal, steatotic and NASH mice.
(C) The activity of NcDase in liver and ileum in normal and NASH mice
(D-H) Liver section from mice with induction of NASH.
(D) Staining for Masson’s trichrome and Aniline blue-positive area (Top) and for Sirius red and Sirius red-positive area (Bottom).
(E) Staining for H&E and Oil red O.
(F) mRNA levels of the indicated genes related to fibrosis in liver.
(G) Immunoblot of the protein related to fibrosis in liver.
(H) α-SMA immunofluorescence (red) and DAPI counterstain (blue) and quantification. The white dot lines indicate the fibrotic pattern of α-SMA+(red) cells.
(I-J) Serum ALT and AST, and insulin levels (I) or liver choline levels (J) in mice with steatosis or NASH.
Mean ± SEM; n = 10, *p < 0.05, **p < 0.01. Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
To achieve NASH features within an experimentally acceptable time frame, we further studied the role of NcDase in the HSPC diet-induced NASH model. After 16 weeks of HSPC diet feeding, mouse body weight, liver:body weight ratio, fasting blood glucose, and fasting blood insulin were similar in the NcDase−/− mice and WT control groups (Figures S3A–3D). However, both inflammatory cell infiltration and fibrosis were reduced in the livers of HSPC-fed NcDase−/− mice as compared to WT mice (Figures 2A–2C). Plasma ALT was decreased in NcDase−/− mice (Figures 2D). Deficiency of NcDase also caused a robust reduction in the expression of mRNAs related to hepatic inflammation (CXCL1 and IL-6) and fibrosis, including α-SMA, tissue inhibitor of metalloproteinase 1 (Timp1), and Col1a1 (Figures 2E). These changes were accompanied by decreases in F4/80+ macrophages, but not in iMCs/MDSCs (Figure 2F). We further confirmed these results in the MCD diet-induced NASH model, in which deletion of NcDase showed decreased both hepatic injury and fibrosis (Figure 2G and Figure S4A), as well as the same steatosis and loss of body weight, liver:body weight ratio (Figure 2G and Figure S4B) and reduced inflammatory and fibrotic gene expression and α-SMA+ cells in the liver (Figures S4C–S4D).
Figure 2. NcDase deficiency reduces liver inflammation, fibrosis, and cell death in HSPC-or MCD-fed mice.
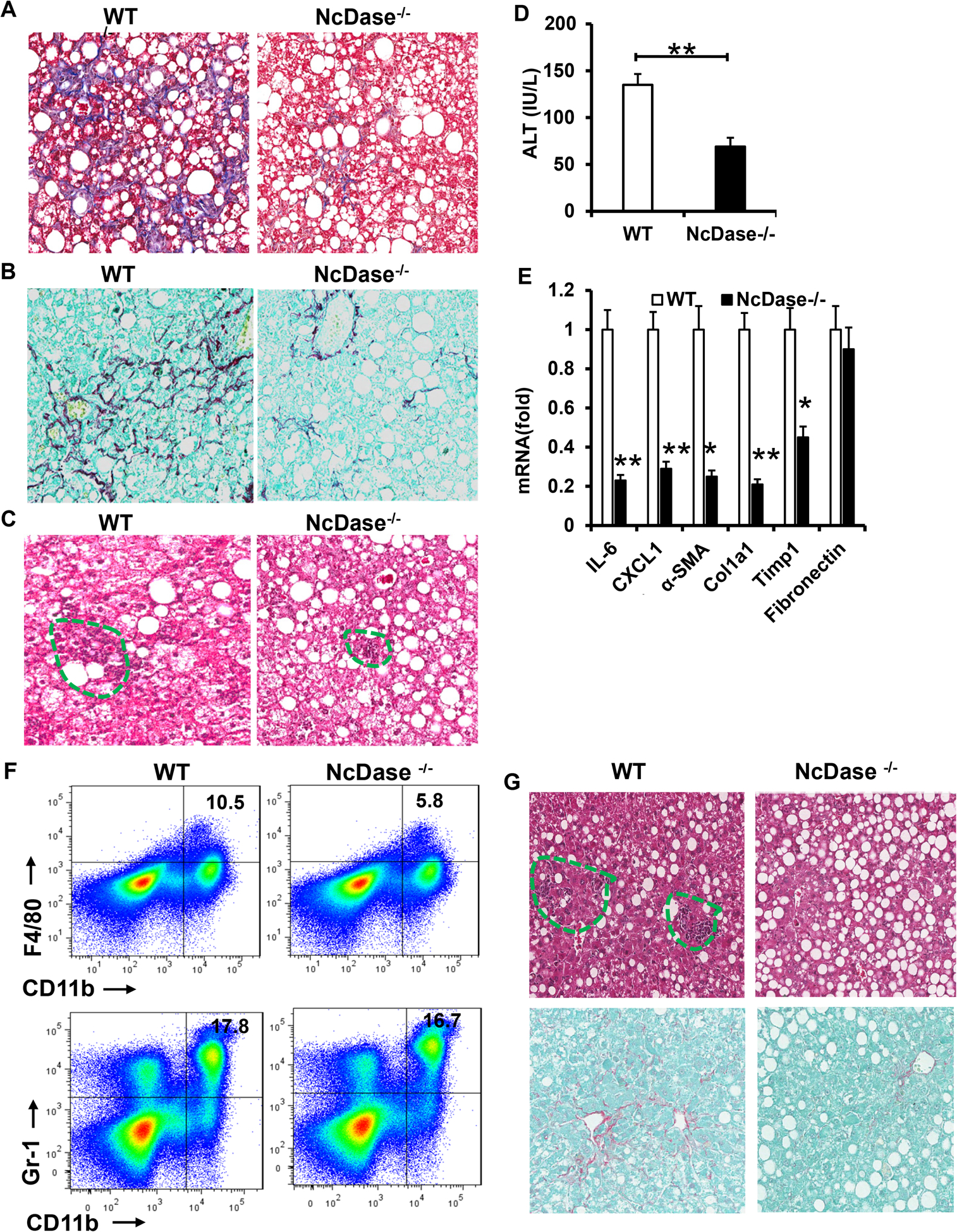
WT mice or NcDase−/− mice were fed the HSPC diet (A-F) or MCD diet (G).
(A-C) Masson’s trichrome staining (A), Sirius red staining (B) or H&E staining (C) of liver sections.
(D) Serum ALT levels.
(E) mRNA levels of the indicated genes related to inflammation and fibrosis in liver.
(F) FACS analysis of CD11b+F4/80+ macrophages and CD11b+Gr-1+ iMCs/MDSCs in the liver.
(G) H&E staining (top) or Sirius red (bottom) staining of liver sections in MCD-fed mice.
Mean ± SEM; n = 10, *p < 0.05, **p < 0.01.
HFD-fed NcDase−/− mice display decreased MUFA-enriched hepatic neutral lipids
To examine the HFD-induced biochemical changes, we analyzed liver ceramide metabolism and fatty acid composition including triglycerides (TGs), cholesterol esters (CE), and phospholipids (PLs) in NcDase−/− mice and their WT littermates after a 14-month-HFD feeding. Surprisingly, the level of total ceramide, acylation patterns of ceramide and sphingosine-1-phosphate (S1p) were not significantly different in WT versus NcDase−/− mice in serum, liver and intestine tissue (Figure 3A and Figures S5A–S5B), possibly resulting from a compensatory decrease in CerS5 and CerS6 transcripts in the livers and intestines of NcDase−/− mice (Figure S5C). However, we saw a striking decrease in NcDase metabolite sphingosine in the intestines of NcDase−/− mice (Figure S5A). Furthermore, NcDase−/− mice with NASH displayed a significant decrease in the hepatic neutral lipid CE, and an increase in PL compared to WT littermates (Figure 3B). Of note, NASH NcDase−/− mice had markedly reduced levels of the MUFAs palmitoleate (C16:1) and oleate (C18:1), but had similar levels of palmitic acid (C16:0) and stearic acid (C18:0) in total liver lipids (Figure 3C). The reduced oleate was mainly attributed to a decrease in C18:1 n-7 in the TG, CE and PL fractions and a significant decrease in C18:1 n-9 in the CE. (Figures 3D and 3E). Interestingly, palmitoleate (C16:1), a minor product of SCD1, was also significantly decreased in the TG and CE fractions in NcDase−/− mice as compared to WT, but was not statistically different in the PL fraction of NcDase−/− mice and WT littermates (Figure 3F). Furthermore, in the liver CE and PL, but not the TG fraction, C18:2 levels were significantly increased in NcDase−/− mice relative to WT littermates (Figure 3G).
Figure 3. Decreased MUFAs and hepatic neutral lipids in HFD NcDase−/− mice.
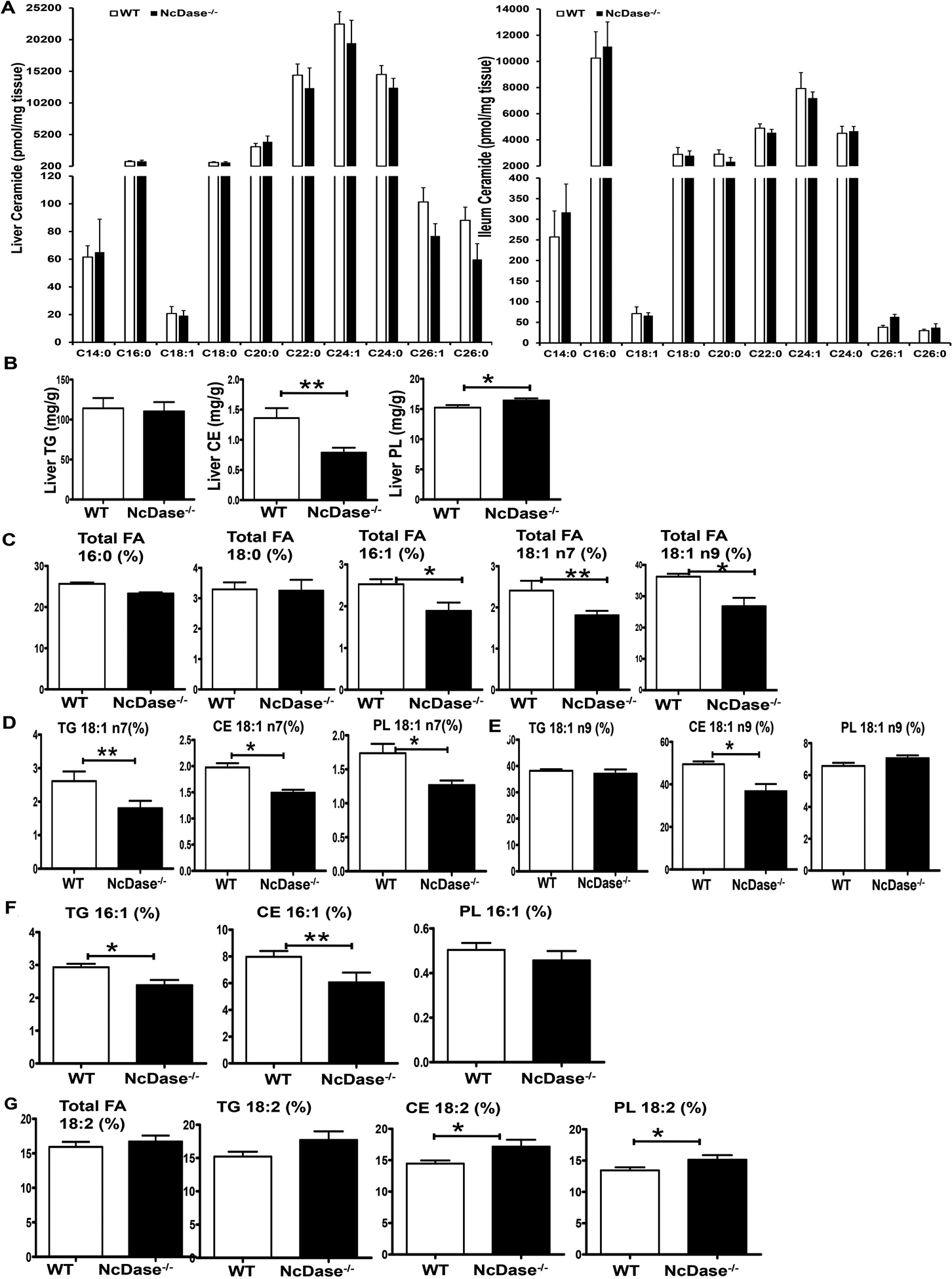
Age-matched NcDase−/− male mice and their WT littermates were fed HFD for 30–60 weeks.
(A) Levels of acyl-chain ceramides in the liver and ileum.
(B) Gas chromatography analysis of hepatic lipids TG, CEs and PLs.
(C-G) Gas chromatography analysis of fatty acid composition of hepatic lipid fractions, represented as (%). Fatty acid composition in total hepatic lipid (C); C18:1 n7 (D), C18:1 n9 (E) or C16:1(F) in TG, CEs and PLs; C18:2 in total hepatic lipid, TG, CEs and PLs (G)
Mean ± SEM; n=10, *p < 0.05, **p < 0.01.
SCD1 is a member of the fatty acid desaturase family and catalyzes mainly the conversion of saturated fatty acids (SFAs) to MUFAs (22). We hypothesized the changes in MUFAs profile may subsequently attribute to the inhibition of SCD1 gene expression in the liver of NcDase−/− mice. Indeed, the expression of SCD1 was reduced significantly in steatotic and NASH livers in the NcDase−/− mice (Figure 4A). In addition, the levels of SCD1 mRNA were downregulated about 20, 100, 300-fold in the livers of steatotic NcDase−/− mice with 3, 5 and 7 months of HFD feeding, respectively, compared to their WT littermates (Figure 4B). Moreover, the livers of NcDase−/− mice with NASH had the lowest levels of SCD1, about a 900-fold decrease, at 14 months of the HFD feeding (Figure 4B). SCD1 expression in the gut was also abolished in NcDase−/− mice with NASH (Figures 4C and 4D). The mRNAs for MUFA elongation, elongation of very long chain fatty acids (Elovl) 5 and Elovl6 elongase, also exhibited decreased expression in NcDase−/− mice (Figure 4E). No changes were observed in the level of lipogenesis genes sterol regulatory element binding protein 1c, acetyl-Coenzyme A carboxylase alpha, and fatty acid synthase mRNA (Figure S5D) in NcDase−/− mice, however, peroxisome proliferator-activated receptor gamma expression was found significantly downregulated (Figure 4E). We observed a significant decrease in fatty acid-binding protein (Fabp) 1 and Fabp4 expression and similar expression of CD36 and Fabp5 for FA uptake in HFD-fed NcDase−/− mice (Figure S5D). NcDase−/− mice exhibited higher 3-hydroxy-3-methyl-glutaryl- CoA [HMG-CoA] reductase and acyl CoA: cholesterol acyl transferase 2 mRNA expression for cholesterol synthesis and TG but lower aldehyde oxidase 1 mRNA expression for fatty oxidation and similar diacylglycerol acyl transferase 1 and 2 (Dgat1 and 2) mRNA expression compared with WT mice (Figure S5E). Thus, all of our observations suggest that a decrease of hepatic SCD1 might be the main cause of the altered balance between SFAs and MUFAs in HFD-fed NcDase−/− mice.
Figure 4. NcDase deficiency reduces the expression of SCD1.
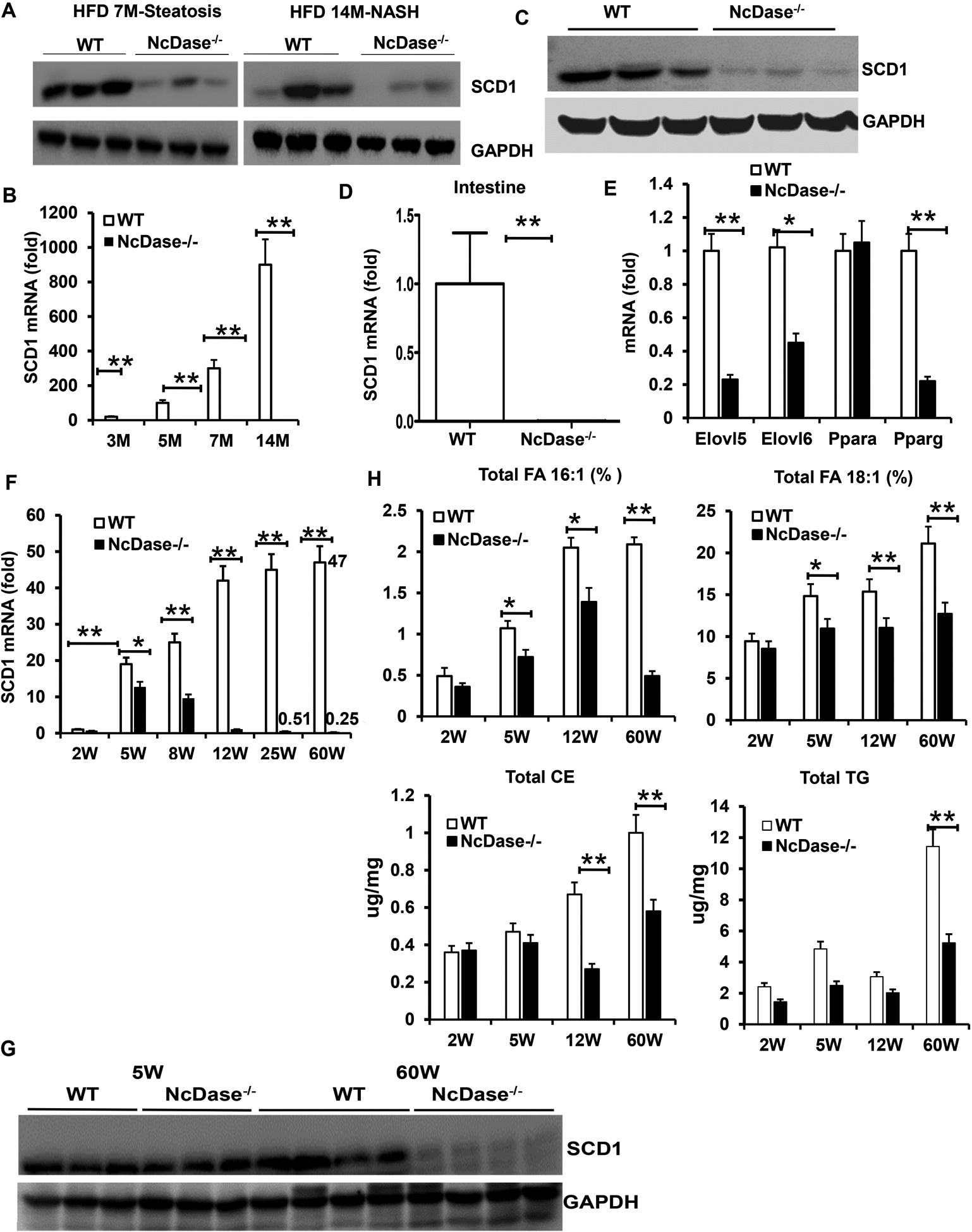
(A and C) Immunoblots of SCD1 in the liver (A) and gut (C) of HFD-fed WT and NcDase−/− mice.
(B and D) Fold changes of hepatic (B) and gut (D) SCD1 mRNAs in WT mice related to NcDase−/− mice during HFD feeding.
(E) mRNA levels of the indicated genes related to lipid metabolism in the liver in HFD-fed mice.
(F) Fold changes of hepatic SCD1 mRNAs in NCD-fed mice.
(G) Immunoblots of SCD1 in the liver of NCD-fed mice.
(H) Gas chromatography analysis of hepatic total TG and CE and fatty acid composition in total lipids in NCD-fed mice.
Mean ± SEM; n = 10, *p < 0.05, **p < 0.01.
Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PPAR, peroxisome proliferator-activated receptor.
Given that weaning is accompanied by substantial alterations in energy expenditure and maturation of the microbiota, and that liver SCD1 expression is microbiota dependent, we next asked whether the decreased SCD1 expression in the liver of NcDase−/− mice is present at birth or occurs after weaning onto the normal chow diet (NCD). At the time of suckling (2 weeks), WT and NcDase−/− mice displayed similar levels of SCD1 in the liver, which increased 2 weeks after weaning in both WT and NcDase−/− mice (Figure 4F). However, the fold-increase in SCD1 mRNAs in WT littermates 2 weeks after weaning was about 20-fold higher compared to the suckling stage, which was significantly higher than the fold-change in NcDase−/− mice (Figure 4F). Interestingly, after 12 weeks of age, the expression of SCD1 began to decrease in NcDase−/− mice, but not in WT mice. Notably, the levels of SCD1 downregulated about 90-fold and 180-fold compared to WT mice at the age of 25 and 60 weeks, respectively (Figure 4F). Similarly, the levels of liver SCD1 protein were also significantly downregulated in 60-week-old, but not in 5-week-old NcDase−/− mice, compared to their WT littermates (Figure 4G). Next, we asked whether the level of MUFAs in livers of NcDase−/− mice changed upon the suckling-to-weaning transition. As expected, both WT and NcDase−/− mice displayed similar levels of MUFAs in the liver at the time of suckling. Although levels of MUFAs increased in both WT and NcDase−/− mice after weaning (Figure 4H); the percent increase in NcDase−/− mice was significantly lower than in WT littermates. At 60 weeks of age, the levels of C18:1 and C16:1 FAs decreased strikingly in NcDase−/− mice when fed the NCD, which was similar to the HFD fed mice (Figure 4H). In addition to C18:1, liver total TG and CE levels decreased with age in NcDase−/− mice (Figure 4H). Thus, the level of SCD1 and MUFAs in the liver is dependent on NcDase.
Neutral ceramidase deficiency improves obesity-associated lipid abnormalities via gut microbiota
The intestinal microbiota are a known contributor to the metabolic function of SCD1 and MUFAs and have been linked with the development of human obesity(23, 24). To determine if the gut microbiota specifically modulate the SCD1 expression and lipogenesis in the liver of NcDase−/− mice, an antibiotic cocktail (Abx) was provided. Antibiotic treatment clearly downregulated the expression of SCD1 and ELOVL5 (Figure 5A), and reduced the differences in the mRNA levels of SCD1 and ELOVL5 between the NcDase−/− mice and WT littermates (Figure 5A). Antibiotic treatment also decreased C16:1 and C18:1 in total lipid and TG and CE fractions in both NcDase−/− mice and WT littermates and caused the levels of hepatic C16:1 and C18:1 in WT mice to approximate the levels in the NcDase−/− mice (Figure 5B). To test whether the gut microbiota of NcDase−/− mice fed the HFD could attenuate the production of MUFAs, we transplanted the fecal microbiota from 7-month-HFD fed NcDase−/− mice or WT mice into recipient germ-free (GF) mice and then fed both groups of recipient mice a normal control diet (NCD) for 5 weeks. Strikingly, GF mice that received microbiota from NcDase−/− mice gained less SCD1 and ELOVL5 expression than those that received microbiota from WT mice (Figures 5C and 5D). As shown in Figure 5E, C16:1 and C18:1 n9 levels in total lipid, TG, and CE in GF mice that received microbiota from NcDase−/− mice were significantly lower than GF mice that received microbiota from WT mice (Figure 5E). These results clearly support the notion that NcDase affects MUFA species generated by SCD1 via gut microbiota.
Figure 5. Decreased hepatic SCD1 expression and MUFAs levels are associated with gut microbiota in NcDase−/− mice.
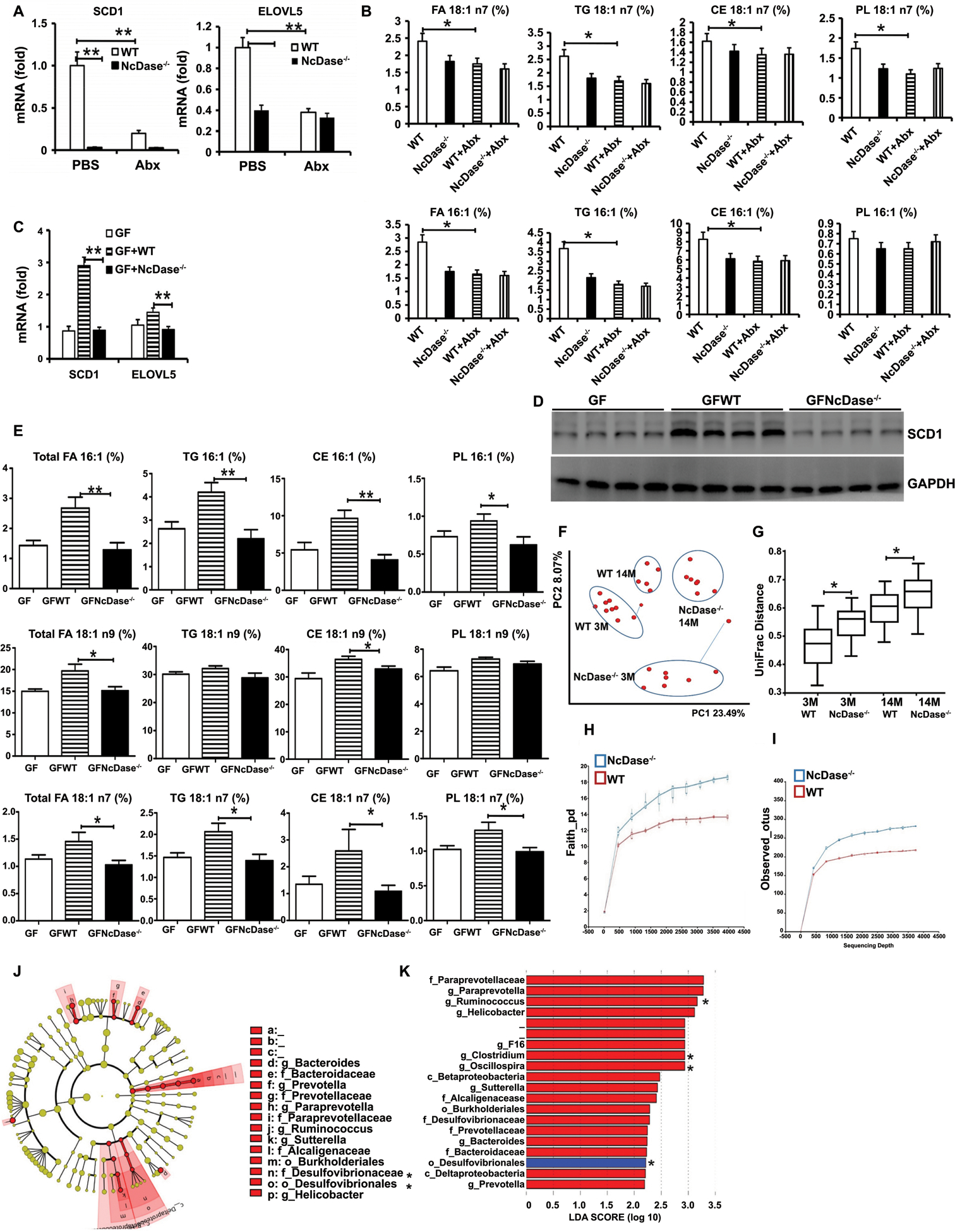
(A) mRNA levels of the SCD1 and Elovl5 in the liver after antibiotics (Abx) treatment.
(B) Hepatic C18:1 and C16:1 FA in total hepatic lipid, TG, CEs and PLs after antibiotics treatment.
(C-E) Eight-week-old male GF C57BL/6 WT mice were orally administered fecal microbiota from HFD-fed either WT (GFWT) or NcDase−/− (GFNcDase) mice and housed for 5 weeks.
(C) mRNA levels of the SCD1 and ELOVL5 in the liver.
(D) Immunoblots of SCD1 in the liver.
(E) Hepatic MUFAs in in total hepatic lipid, TG, CEs and PLs.
(F and G) Principal component (PC) analysis plot showing microbial unweighted UniFrac compositional differences (F), quantified by UniFrac distance (G) between WT and NcDase−/− mice on NCD at different ages (in months).
(H and I) Rarefaction curves of species richness and diversity between WT and NcDase−/− mice samples based on (H) Faith’s phylogenetic diversity (PD) measure and (I) observed OTUs.
(J) Cladogram generated from LEfSe analysis, showing the most differentially abundant taxa enriched in microbiota from WT and NcDase−/− mice.
(K) Linear discriminant analysis (LDA) scores of the differentially abundant taxa shown in (J). Taxa enriched in microbiota from NcDase−/− mice (red) are indicated with a positive LDA score related to that from WT mice (blue) (Kruskal-Qallis test, p<0.05 and LDA ≥ 2).
Mean ± SEM; n = 8–12, *p < 0.05, **p < 0.01. Abbreviation: PC1, Principal component 1.
In order to determine the features of the microbiota that affect MUFAs in NcDase−/− mice, we performed 16S ribosomal RNA (rRNA) gene sequencing on mice fed the NCD. There were significantly different communities in the fecal contents of NcDase−/− and WT mice, as shown by a principal component analysis (PCA) and quantified by UniFrac dissimilarity distance (Figures 5F and 5G). Additionally, there was a significantly increased species richness in the feces of NcDase−/− mice as observed in the Faith_RD and operational taxonomic units (OTU) results (Figures 5H–5I, and Figures S6A–S6B). Linear discriminant analysis effect size (LEfSe) indicated that Order_Bacteroidales (f_Paraprevotellaceae, f_Prevotellaceae), Order_Clostridiales (f_Lachnospiraceae, f_Ruminococcaceae and f_Mogibacteriaceae), Order_Burkholderiales (f_Alcaligenaceae) and Order_CW040 (F16) were increased significantly in NcDase−/− mice (Figures 5J–5K, and Figure S6C). Of note, when compared with WT mice, NcDase−/− mice showed broad increases in diversity and overall abundance of multiple Clostridiales taxa, including genus Ruminococcus, genus Clostridium and genus Oscillospira (Ruminococcaceae family) (Figure 5K). Thus, Clostridiales present in the NcDase−/− mice might have a reduced functional contribution to MUFAs.
Neutral ceramidase coordinates IgA responses against the microbiota
Alteration in sphingolipid S1p/Sphingosine-1-phosphate receptor 1 (S1PR1) signal during B cell differentiation in the Peyer’s patches (PPs) controls the emigration of IgA plasma cell precursors(25). Based on the observed diversity in gut microbiota in NcDase−/− mice, intestinal B cell function was examined. Percentages of IgM+B220+ B cells were increased significantly in the small intestine lamina propria (SI-LP) of NcDase−/− mice relative to WT littermate controls (Figure 6A). The percentages and the number of B220−IgA+ plasmablasts were significantly increased in the small intestine but not large intestine lamina propria (LI-LP) of NcDase−/− mice (Figures 6B and 6C). Furthermore, B220+IgA+ cells slightly, but not significantly, increased in the LI-LP of NcDase−/− mice compared to WT mice (Figure 6B). No differences were seen in the frequency of B220+IgM+ cells in the colon and PP or in B220+IgA+ cells in the PP of NcDase−/− and WT mice (Figures 6A and 6B). Using immunofluorescence microscopy, we confirmed IgA-committed plasma cells (or plasmablasts) exhibited an increase in the LP of NcDase−/− mice compared to WT mice (Figure 6D). These studies showed the physiological requirement for NcDase in establishing an adequate plasma cell distribution in the intestine.
Figure 6. NcDase regulates the secretory IgA immunity.
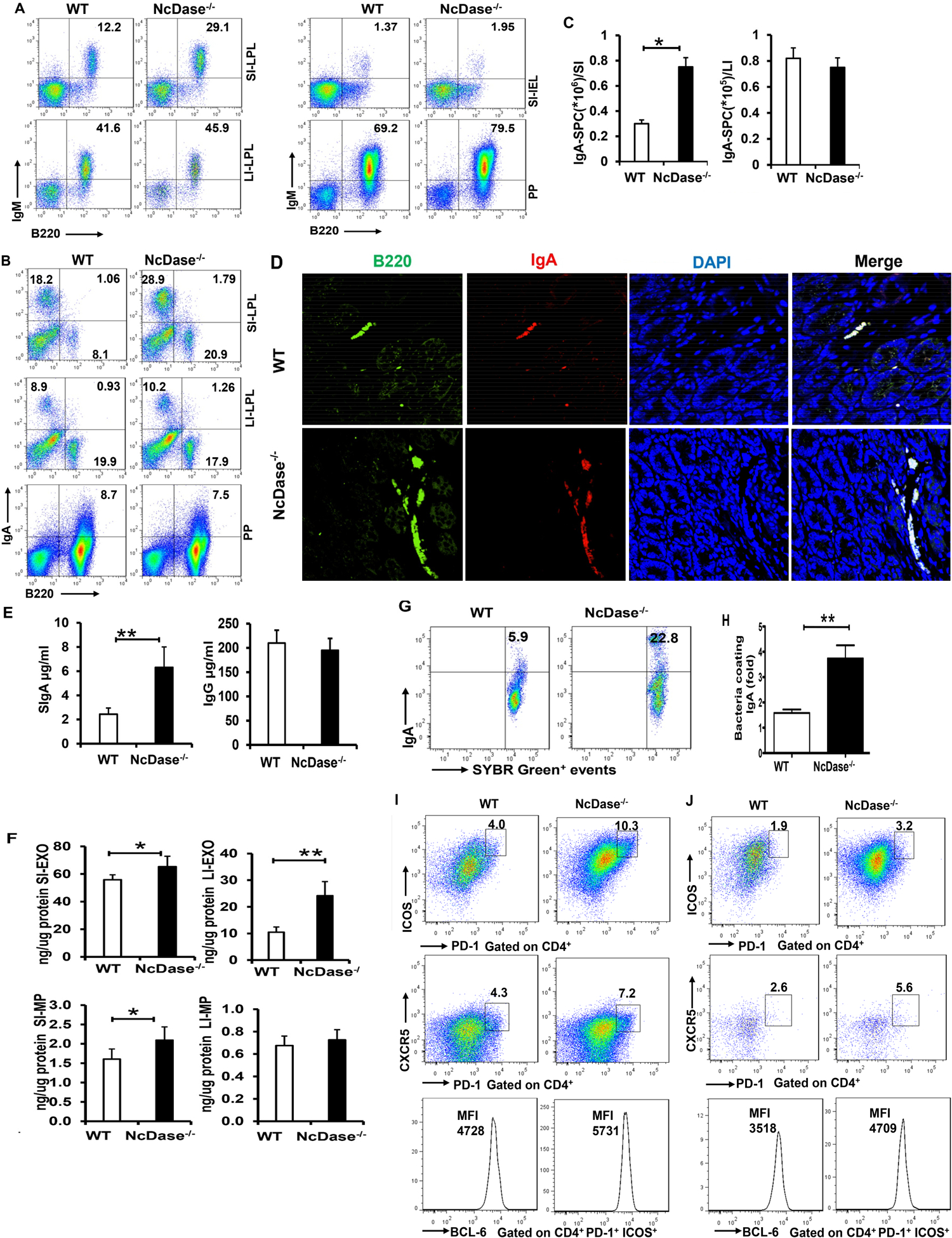
(A-B) FACS analysis of B220+IgM+ cells (A) and B220−IgA+ plasma cells (B) in the lamina propria lymphocytes (LPL) from large intestine (LI-LPL), small intestine (SI-LPL), intestinal epithelial lymphocytes (IEL) from small intestine (SI-IEL) or Peyer's Patch (PP).
(C) The number of IgA-secreting plasma cells (SPC) in lamina propria of small (SI) or large (LI) intestine.
(D) Immunofluorescence staining of IgA+ B cells (B220 with green, IgA with red) in the lamina propria.
(E) ELISA analysis of secretory IgA and IgG in the gut lumen.
(F) ELISA analysis of IgA in luminal particles including exosome-like particles (EXO) and microparticles (MP) in the small and large intestine.
(G) IgA-bound bacteria in the intestinal lumen was measured by flow cytometry. Representative plots were gated on SYBR green.
(H) Fold change of frequencies of IgA-bound bacteria (n=8 per group).
(I-J) FACS analysis of PD-1+ICOS+ Tfh cells, PD-1+CXCR5+ Tfh cells or geometric mean fluorescence intensity (MFI) of BCL6+ PD-1+ICOS+ Tfh cells in the Peyer's Patch (PP, I) and lamina propria lymphocytes (LPL) from small intestine (SI-LPL, J).
Mean ± SEM; n = 8, *p < 0.05, **p < 0.01. Abbreviation: BCL6, B cell lymphoma 6.
Next, we examined the functional consequences of NcDase on gut B cells by studying intestinal IgA secretion in NcDase−/− mice. We found that the production of secretory IgA (SIgA), but not IgG, was increased in NcDase−/− mice relative to WT controls (Figure 6E), illustrating the importance of neutral ceramidase for the generation of IgA. Our studies further revealed that the quantity of IgA correlated with the quantity of luminal extracellular vesicles (EVs), which appeared to be predominantly carried by luminal exosome-like particles rather than by luminal microparticles in WT mice (Figure 6F). Interestingly, IgA in the luminal exosomes from large and small intestine were increased in NcDase−/− mice (Figure 6F). However, there were no significant differences in the luminal microparticle-related IgA quantities observed from large intestine between NcDase−/− mice and WT littermate controls (Figure 6F). Next, we found that 3%-10% of microbiota were IgA+SYBR+ in naïve WT mice, while a 3-fold increase in IgA targeting of the microbiota was observed in naïve NcDase−/− mice (Figures 6G and 6H). Studies have identified CD4+CD25+ regulatory T cells as major helper cells for IgA responses to microbial antigens, presumably through their secretion of TGF-β(26). However, our results reveal similar frequency of CD4+CD25+Foxp3+ T cells in the colon of NcDase−/− mice relative to that observed in WT mice (data not shown). C-X-C chemokine receptor type 5+ (CXCR5+) T follicular helper (Tfh) cells in B cell zones of lymphoid tissues possess significant capacity for supporting IgA production by B cells (27, 28), we next examined the impact of NcDase deficiency on the frequency of Tfh cells in the intestine. Flow cytometric analysis revealed an enrichment of the PD-1+CXCR5+ (Programmed cell death protein [PD]) and PD-1+ICOS+ (Inducible T cell Costimulator [ICOS]) population of Tfh cells of PPs (Figure 6I) and SI-LPL (Figure 6J) in the NcDase−/− mice compared to their WT littermate controls. The expression of B cell lymphoma 6, a transcription factor required for Tfh differentiation, was also higher in the Tfh cells in NcDase−/− mice (Figure 6I and 6J). Thus, Tfh cells, which are known to be involved in B cell activation(29), and likely thus contribute to the ability of NcDase to regulate secretory intestinal immunity. Together, these data revealed that a NcDase deficiency leads to increases in both the quantity and activity of IgA+ plasma cells in intestinal tissues, and indicated that neutral ceramidase influences the abundance and quality of the SIgA response toward commensal bacteria.
As animals that lack NcDase have increased B cell responses within the gut, we asked whether the IgA-bound microbiota of NcDase−/− mice was sufficient to influence hepatic lipid MUFA levels. To do this, IgA-bound bacteria from HFD fed NcDase−/− or WT mice were sorted and transplanted into GF-WT mice. One month after transfer, GF mice administered WT IgA-bound bacteria showed a three-fold increase in the expression of SCD1 compared to GF mice. Strikingly, GF mice that received IgA-bound microbiota from NcDase−/− mice gained lower SCD1 expression in the liver than WT mice (Figure 7A). Analysis of fatty acid profiles indicated that C16:1, C18:1n9, and C18:1 n7 levels in total FA, TG, and CE, but not TG and CE C18:1n9 levels, were significantly lower in the liver of GF mice that received NcDase−/− IgA-bound microbiota than GF mice that received WT IgA-bound microbiota (Figures 7B). Mice receiving IgA-bound bacteria from NcDase−/− mice were ultimately colonized with significantly higher amounts of the Ruminococcaceae family (Figure 7C). Three taxa at the genus level were differentially targeted by IgA, including the Ruminococcus and Oscillospira genus of Clostridiales (Figure 7C). Interestingly, analysis of bacteria in GF mice transferred with IgA-bound bacteria from WT mice showed increased Desulfovibrio (Figure 7C), which antagonizes stable colonization of Clostridia and causes metabolic disease (13). Indeed, there was a significant negative correlation between the abundance of MUFA 16:1 in total FAs and Ruminococcaceae (Figure 7D). We further explored how IgA-bound bacteria regulates the expression of SCD1. Gnotobiotic mice colonized with the Ruminococcaceae alone had significant reductions in SCD1 expression in the liver and ileum when compared with GF mice (Figure 7E). Interestingly, the colonization of GF mice with Desulfovibrio significantly increased the expression of SCD1 in the liver (Figure 7F and 7G). The administration of probiotic Ruminococcaceae to GF mice colonized with the Desulfovibrioto led to a decrease in SCD1 expression in the liver (Figure 7F and 7G). Moreover, supernatants collected from the cultured Desulfovibrio significantly elevated SCD1 in cultured primary hepatocytes (Figure 7H). By contrast, supernatants collected from cultured Ruminococcaceae directly down-regulated the expression of SCD1 on hepatocytes (Figure 7H). Thus, Desulfovibrio-derived metabolites from WT mice can directly regulate the SCD1 expression in the liver. However, IgA-bound Clostridia species, likely Ruminococcaceae, from NcDase−/− mice have more of an ability to suppress the expression of hepatic SCD1.
Figure 7. NcDase modulates the expression of SCD1 and MUFAs levels in the liver by IgA-bound bacteria.
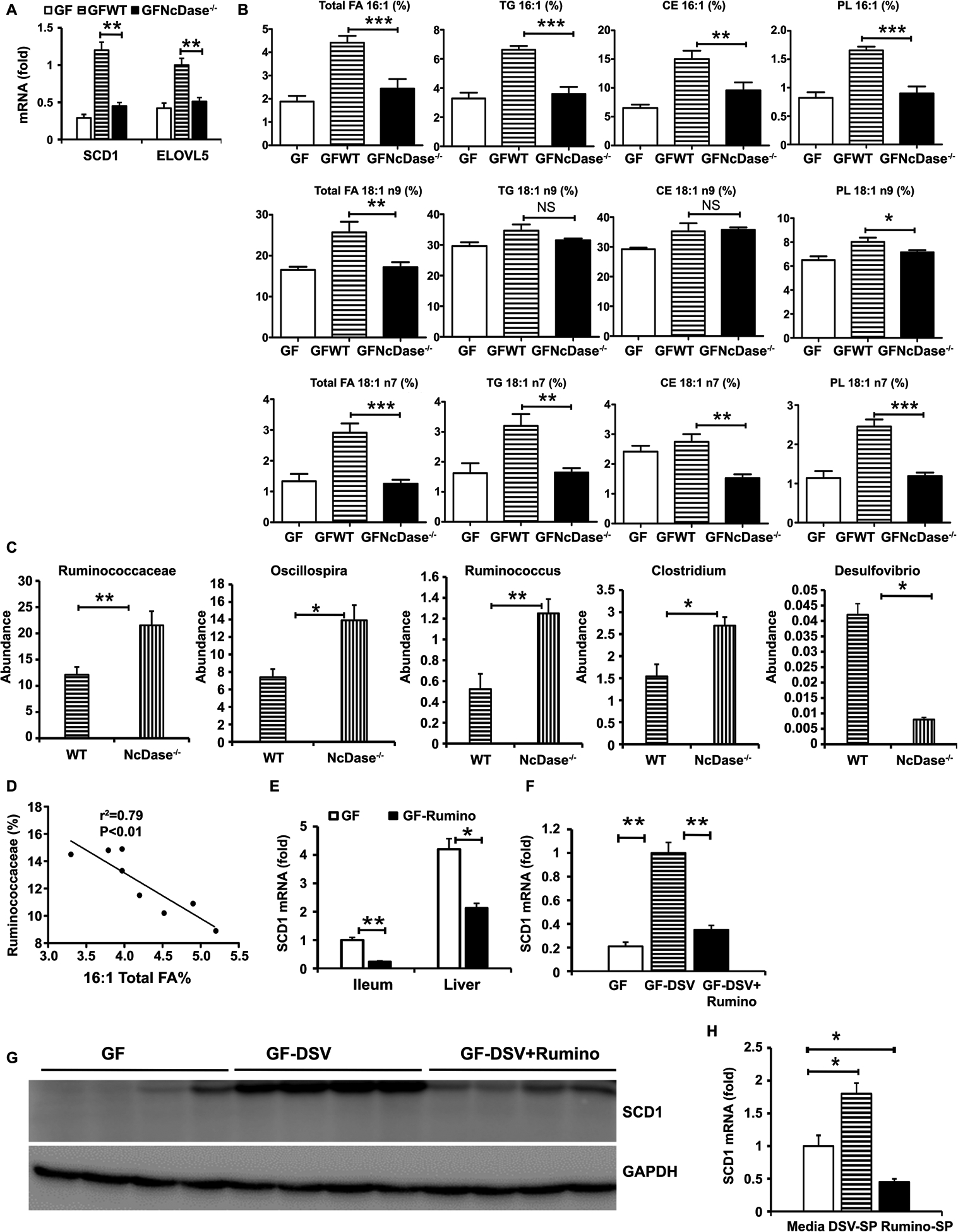
(A-D) Eight-week-old male GF C57BL/6 mice (n = 5) were orally administered IgA-bound microbiota from HFD-fed NcDase−/− (GFNcDase) or WT (GFWT) mice and housed for 3–5 weeks.
(A) mRNA levels of the SCD1 and ELOVL5 in the liver after microbiota transplantation.
(B) Hepatic MUFAs in in total hepatic lipid, TG, CEs and PLs.
(C) Relative abundance of indicated bacteria in GF mice colonized with IgA-bound microbiota.
(D) Correlation between MUFAs and Ruminococcaceae abundance in GF mice colonized with IgA-bound microbiota from WT mice. Mean ± SEM; n = 5, *P < 0.05, **P < 0.01, ***P < 0.001.
(E-G) GF mice with colonization of PBS, Ruminococcaceae (Rumino), Desulfovibrio. desulfuricans (DSV) or Both housed for 3 weeks. Mean ± SEM; n = 4, *p < 0.05, **p < 0.01,
(E) SCD1 mRNA expression in the liver and in the ileum.
(F) SCD1 mRNA expression in the liver.
(G) Immunoblots of SCD1 in the liver.
(H) SCD1 mRNA expression in primary hepatocytes incubated for 12 hours with media or bacterial supernatant (SP).
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
NcDase is Wnt target gene induced in hepatocytes and stabilizes β-catenin in a manner dependent on MUFAs.
We sought to explore possible mechanisms linking NcDase to SCD1-related fibrosis progression. Previous studies have demonstrated that SCD is implicated in obesity and fatty liver, and MUFAs produced by SCD provided a feedback loop to amplify Wnt signaling, contributing to liver fibrosis and tumor growth(18). We therefore considered the hypothesis that increased SCD1/MUFA levels lead to the activation of Wnt-β-catenin signaling, which promotes the expression of pro-fibrotic genes. We found the expression of Wnt ligands (Wnt3a, Wnt7a, Wnt7b) and receptor receptor frizzled homolog 6 (Drosophila) was downregulated by NcDase deletion (Figure 8A). The phosphorylation of β-catenin is also lower in the NcDase-null liver lysates independent of altered 5′ AMP-activated protein kinase phosphorylation (AMPK), indicative of inactivated β-catenin (Figure 8B). Decreased phosphorylation of low-density lipoprotein receptor-related protein 6 (LRP6), phosphorylation of GSK3β, and Wnt3a demonstrated lower activation of the Wnt-β-catenin signal had occurred in the NcDase-null livers (Figure 8B). We also found mRNA expression of Axin2, Myc, and Cyclin (Ccnd) 1, which are downstream targets of the β-catenin pathway, showed markedly lower expression in livers of NASH NcDase−/− mice than in the livers of control mice (Figure 8C). These results indicate a novel role for NcDase as a potential mediator for Wnt activation (Wnt3a and Wnt7b) in the development of NASH.
Figure 8. Wnt signaling is reduced in fibrotic livers of NcDase−/− mice.
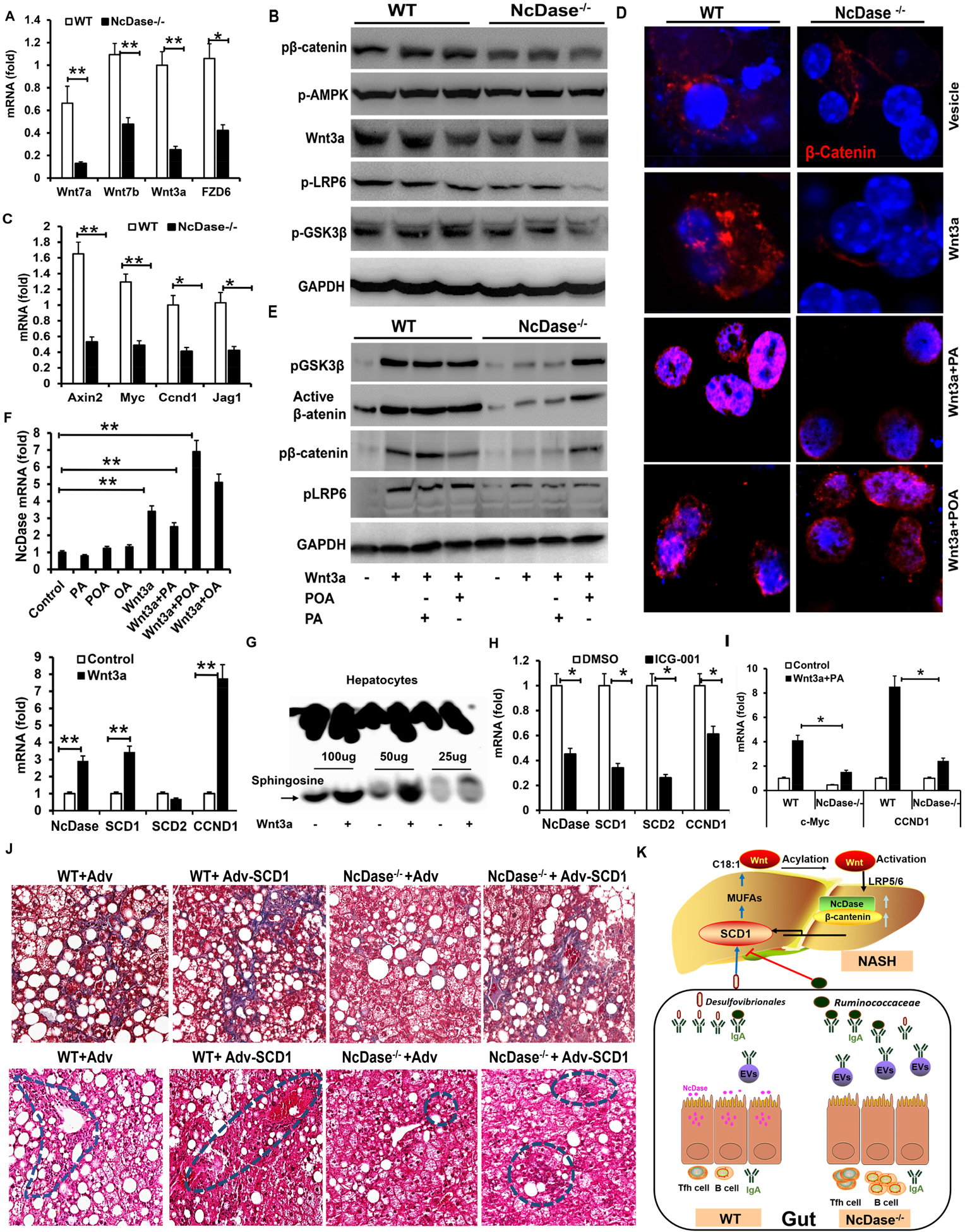
(A-D) Livers of HFD-induced NASH NcDase−/− mice and WT mice were examined.
(A) Real time PCR analysis for the expression of Wnt ligands and receptors in livers.
(B) Immunoblotting analysis of Wnt signal in the liver.
(C) Real-time PCR analysis for the expression of Wnt target genes.
(D-E) Primary hepatocytes were treated with control media, or media from L-Wnt3a with/without a final concentration of 200 μM BSA-conjugated fatty acids or vehicle control for 12h-48h.
(D) Immunofluorescence staining of β-catenin in hepatocytes.
(E) Immunoblotting analysis of Wnt signal in hepatocytes 24h after treatment.
(F) Real time PCR analysis for the mRNA levels of NcDase, SCD1, SCD2 and CCND1 12h-24h after treatment in primary hepatocytes from WT mice.
(G) The activity of NcDase in primary WT 24h after Wnt3a treatment.
(H) Primary hepatocytes were treated with DMSO or Wnt3a inhibitor ICG-001 for 24h. Real-time PCR analysis for the mRNA levels of indicated target genes.
(I) Primary hepatocytes from NcDase−/− or WT mice were treated with control or PA+Wnt3a for 24h. Real-time PCR analysis for the mRNA levels of indicated target genes.
Mean ± SEM; n = 7. *p < 0.05, **p < 0.01.
(J) Masson’s trichrome (top) and H&E (bottom) staining of liver sections. Four groups of mice, including WT mice injected with control adenovirus (WT-Adv), WT mice injected with SCD1 adenovirus (WT-Adv-SCD1), NcDase−/− mice injected with control adenovirus (NcDase−/−-Adv), NcDase−/− mice injected with SCD1 adenovirus (NcDase−/−-Adv-SCD1), were fed with HSPC for 16 weeks. n=5.
(K) Schematic diagrams of how NcDase regulates the SCD1-Wnt/β-catenin loop to protect from NASH via gut IgA associated microbiota. NcDase regulates IgA production, which appropriately affects the expansion of Desulfovibrio and the colonization of beneficial Ruminococcaceae family. Ruminococcaceae function to temper expression of SCD1 and MUFAs production. MUFAs promotes Wnt fatty acylation and β-catenin activation, which leads to NASH. Abbreviations: AXIN2, axis inhibition protein 2; EV, extracellular vesicle; FZD6, frizzled homolog 6 (Drosophila); GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Jag1, jagged canonical Notch ligand 1; Myc, MYC proto-oncogene; OA, Oleic acid; p-AMPK, AMP-activated protein kinase phosphorylation.
To explore the possibility of whether NcDase-induced Wnt activation is mediated by SCD1/MUFAs, we moved to an in vitro model using primary murine hepatocytes. Wnt3a treatment promoted the nuclear translocation of endogenous β-catenin (Figure 8D) in hepatocytes from WT mice, but not from NcDase−/− mice. Importantly, the decreased level and nuclear translocation of β-catenin are rescued by addition of MUFAs (palmitoleate, POA), but not with SFAs (palmitic acid, PA), in hepatocytes from NcDase−/− mice (Figure 8D). Deletion of NcDase abrogates Wnt activity in hepatocytes, as assessed by the phosphorylation of LRP6 and GSK3β at S9, and abrogates the level of the nonphosphorylated and active form of β-catenin (non-p-β-catenin); these effects are rescued by POA but not by PA (Figure 8E). Interestingly, addition of Wnt3a significantly induced the level of NcDase mRNA and enhanced ceramidase activity in primary hepatocytes (Figures 8F and 8G) and HepG2 cells (data not shown) cultured for 24 h. NcDase expression is induced at a >6-fold higher level by Wnt3a+ POA than Wnt3a (Figure 8F). We further demonstrated by real-time PCR analysis of cultured mouse hepatocytes treated with wnt3a canonical Wnt inhibitor ICG-001 that both NcDase and SCD1 are putative Wnt target genes (Figure 8H), indicating that the NcDase expression level is increased upon Wnt signaling. Consistent with our in vivo data, Wnt3a plus PA treatment resulted in lower expression of Wnt-dependent genes MYC proto-oncogene (cMyc) and Ccnd1 (cyclin D1) in the NcDase−/− hepatocytes compared with WT hepatocytes (Figure 8I), suggesting that NcDase may support the Wnt-β-catenin pathway and Wnt3a feedback upregulates NcDase activity. Collectively, these results demonstrate that MUFA, generated by neutral ceramidase-dependent SCD1, provides a positive feedback loop to enhance Wnt activity and stabilize β-catenin in hepatocytes.
NcDase-SCD1-Wnt loop promotes NASH
To demonstrate that PA-induced Wnt3a secretion from NcDase-null hepatocytes has less ability to activate hepatic stellate cells (HSC) due to a lower level of switched POA, we moved to an in vitro model using primary murine HSCs. PA-conditioned medium (PA-CM) and bovine serum albumin (BSA)-conditioned medium (BSA-CM) from NcDase−/− or WT hepatocytes, were added to primary murine HSCs. Compared with BSA-CM, hepatocyte PA-CM from WT hepatocytes markedly induced higher levels of osteopontin (Opn), Timp1 and Col1a1 mRNA, which are involved in HSC-induced fibrosis in NASH when compared to the results for PA-CM from NcDase−/− hepatocytes (Figure S7A). To make a more direct link between POA and NcDase-induced Wnt3a in the activation of HSCs, we restored Wnt3a in NcDase−/− hepatocytes by Wnt3a transfection. We found that restoration of Wnt3a in these NcDase−/− hepatocytes rescued PA-CM-induced HSC gene expression (Figure S7B). Together, these studies suggest that POA-activated Wnt3a promotes the expression of pro-fibrotic genes in HSCs. To validate the hypothesis that NcDase may play the critical role in NASH progression via the interaction of SCD1-Wnt, NcDase−/− mice were injected with SCD1 adenovirus or control virus and then fed the HSPC diet for 16 weeks. Compared to mice transfected with control adenovirus, there was a striking increase in hepatic SCD1 expression and inflammation or fibrosis (Figure 8J, and Figure S8A–S8C), but no appreciable steatosis in WT mice who received SCD1-Adv and fed the HSPC diet (Figure 8J). Of note, the livers of NcDase−/− mice who received SCD1-Adv showed marked increases at 16 weeks in fibrosis endpoints, inflammatory cells, inflammatory-related and fibrosis-related genes, F4/80+ macrophages, and α-SMA+ cells but not steatosis compared with those in NcDase−/− mice received control adenovirus (Figure 8J, and Figure S8A–S8C). Serum ALT was also increased by SCD1-adv (Figure S8D). These data suggest that the NcDase-SCD1 axis is particularly important in the critical processes that promote steatosis-to-NASH progression.
Discussion
Using several different animal models (HFD, HSPC, MCD and adenoviral overexpression), we clearly demonstrate that NcDase plays an important role in promoting NASH development. Deficiency for NcDase did not affect total ceramide levels, likely because of compensatory decreases in transcripts encoding CerSs (such as CerS5 and CerS6), which function to control metabolic levels of ceramide in cells. A very recent study showed that deficiency of alkaline ceramidase 3 (ACER3) alleviates the pathogenesis of NASH by regulating the hepatic levels of C18:1-ceramide (30). It is intriguing that NcDase activity influences the composition of MUFAs and the reduction of particularly FA18:1 n-9 and FA16:1 in the total lipid fraction in TG, cholesterol ester, and the phospholipid fractions in NcDase−/− mice (Figure 3). The protective effect of NcDase deficiency on NASH could be a result of a reduction in MUFAs, which might have effects on the inflammatory cascade, reactive oxygen species generation, kinase activity, and apoptosis. Experiments with a combination of NcDase deficiency and SCD1 overexpression by adenoviruses indicated that the subsequent SCD1‐mediated SFA desaturation plays an important role in steatosis- to-NASH conversion and provides new insight into NASH.
Understanding the mechanisms by which neutral ceramidase governs hepatic SCD1 activity will be an important endeavor. Recent studies provide strong evidence that the presence of microbes increases desaturation of palmitate by SCD1(9, 10). Notably, we identified that neutral ceramidase tends to target the expression of hepatic SCD1 in not only obese mice, but also in healthy animals. Importantly, our results showed that hepatic SCD1 level is substantially reduced in GF and microbiota-ablated mice, indicating that liver SCD1 is regulated in part via gut microbiota. Examination of mechanisms potentially responsible for increased total colonic fecal IgA in NcDase−/− mice revealed an increased T follicular helper (Tfh) cells required for the formation of long-lived plasma B cells and promoting secretory IgA immunity (29, 31). However, we cannot exclude that the increased B cell IgA response might be caused by migration/trafficking because S1p-S1PR1 signaling regulates the B cell trafficking/migration in the PPs for the intestinal IgA production (25). Therefore, disturbance of the sphingosine/S1p-S1PR1 signal in NcDase−/− mice might affect the migration of IgA committed B cells from the PPs to the lamina propria of intestine. It has been reported that individuals that are obese and have type 2 diabetes have lower mucosal IgA and decreased responses to immunizations(32, 33). Several studies using rodent models of NAFLD/NASH also showed altered intestinal IgA secretion (13, 14) (34, 35). We found that gut microbiota displays increased IgA coating in NcDase−/− mice, and GF mice have decreased expression of SCD1 and the levels of hepatic MUFAs after receiving IgA bound bacteria from HFD fed NcDase−/− mice compared to those from WT mice. Our data suggest that neutral ceramidase-dependent targeting of the microbiota is important for the maintenance of a healthy community. We found that NcDase−/− mice display decreased Desulfovibrio and increased numbers of several Clostridiales, including the Ruminococcaceae family, Ruminococcus and Oscillospira genus. A recent study showed that the inappropriate targeting of Clostridia by IgA may change their metabolic functions to influence development of obesity(13, 36). We further showed that Desulfovibrio-colonized GF animals upregulated SCD1, whereas the addition of Ruminococcaceae downregulated the expression of SCD1. Our results indicate that the IgA-bound Clostridia species, likely Ruminococcaceae, might contribute to decreased SCD1/MUFAs in NcDase−/− mice. The mechanism by which Desulfovibrio and Clostridia alter SCD1 remains unknown, and future studies will be needed to elucidate this interaction.
Aberrant activation of the Wnt pathway is common in NASH, and SCD1 upregulation is not limited to obesity, but also is evident in liver fibrosis and cancer(37, 38) via mechanisms possibly involving β-catenin signaling(18). Our current study establishes a novel role for steatosis in driving fibrosis by the recruitment of MUFAs that promote Wnt activation. Increased levels of MUFAs, particularly palmitoleic acid, have been reported in NASH patients(39). Wnt proteins contain palmitoleic acid, with an unusual lipid modification. Production of an active Wnt signal depends on the attachment of palmitoleic acid to Wnt. SCD1 generates palmitoleic acid that is then transferred by Porcupine to Wnt for activation(17, 40). Our findings indicate that NcDase may lead to Wnt activation through SCD1-regulated palmitoleic acid (Figures 8D and 8E). Very importantly, our in vitro experiments also demonstrated that Wnt3a stimulation can increase the expression of neutral ceramidase and enhance the activity of neutral ceramidase (Figures 8F and 8G), which triggers β-catenin-dependent gene activation. Thus, the Wnt-NcDase crosstalk is elegantly coupled to the previously described SCD-Wnt release pathway to maximize the positive Wnt-SCD1-NcDase feedback loop by promoting both release and signal transduction of Wnt (Figure 8K). Although NcDase is more highly expressed in the ileum of steatotic and NASH mice, more in-depth mechanistic approaches including studies in mice with liver/gut-specific deletion of NcDase will be required to fully understand the mechanism of MUFA-mediated NASH.
In conclusion, we demonstrate that expression of neutral ceramidase is a regulator of gut B cells that produce IgA to control gut microbiota homeostasis. Gut microbiota regulates the activity of SCD1 that produces MUFA to activate the canonical WNT pathway. Understanding the differential effects of specific FAs on the downstream pathway is critical for understanding NASH development and progression. Our findings suggest that NcDase might be an attractive therapeutic target for NASH and associated complications such as cirrhosis and hepatocellular carcinoma. Future investigations should be focused on restoring gut IgA in WT mice or identifying the metabolites from IgA-bound bacteria in NcDase−/− mice, and studying the interaction between the gut IgA immune response and MUFAs in individuals with NASH, which may hold promise for obesity-related NASH.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH R21AA025724, R21AI128206 and R01 DK115406 (Z.D.). We thank Dr. J. Ainsworth and Marion McClain for editorial assistance.
Footnotes
Conflicts of Interest
The authors disclose no conflicts of interest.
The authors declare that they have no competing financial interests
Reference:
- 1.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 2014;20:678–686. [DOI] [PubMed] [Google Scholar]
- 2.Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, et al. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metabolism 2015;22:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raichur S, Wang ST, Chan PW, Li Y, Ching JH, Chaurasia B, Dogra S, et al. CerS2 Haploinsufficiency Inhibits beta-Oxidation and Confers Susceptibility to Diet-Induced Steatohepatitis and Insulin Resistance. Cell Metabolism 2014;20:687–695. [DOI] [PubMed] [Google Scholar]
- 4.Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta 2008;1781:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, Bielawski J, et al. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. Journal of Biological Chemistry 2006;281:7324–7331. [DOI] [PubMed] [Google Scholar]
- 6.Hodson L, Fielding BA. Stearoyl-CoA desaturase: rogue or innocent bystander. Progress in Lipid Research 2013;52:15–42. [DOI] [PubMed] [Google Scholar]
- 7.Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, Ntambi JM. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metabolism 2007;6:484–496. [DOI] [PubMed] [Google Scholar]
- 8.Matsuzaka T, Atsumi A, Matsumori R, Nie T, Shinozaki H, Suzuki-Kemuriyama N, Kuba M, et al. Elovl6 promotes nonalcoholic steatohepatitis. Hepatology 2012;56:2199–2208. [DOI] [PubMed] [Google Scholar]
- 9.Kindt A, Liebisch G, Clavel T, Haller D, Hormannsperger G, Yoon H, Kolmeder D, et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nature Communications 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh V, Chassaing B, Zhang LM, Yeoh BS, Xiao X, Kumar M, Baker MT, et al. Microbiota-Dependent Hepatic Lipogenesis Mediated by Stearoyl CoA Desaturase 1 (SCD1) Promotes Metabolic Syndrome in TLR5-Deficient Mice. Cell Metabolism 2015;22:983–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–1031. [DOI] [PubMed] [Google Scholar]
- 12.Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O'Connell RM, Round JL. MyD88 Signaling in T Cells Directs IgA-Mediated Control of the Microbiota to Promote Health. Cell Host & Microbe 2015;17:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen C, Bell R, Klag KA, Lee SH, Soto R, Ghazaryan A, Buhrke K, et al. T cell-mediated regulation of the microbiota protects against obesity. Science 2019;365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, Chakraborty M, et al. Gut-associated IgA(+) immune cells regulate obesity-related insulin resistance. Nat Commun 2019;10:3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monga SP. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015;148:1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science 2012;337:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rios-Esteves J, Resh MD. Stearoyl CoA Desaturase Is Required to Produce Active, Lipid-Modified Wnt Proteins. Cell Reports 2013;4:1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai KKY, Kweon SM, Chi F, Hwang E, Kabe Y, Higashiyama R, Qin L, et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology 2017;152:1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, et al. High-Fructose, Medium Chain Trans Fat Diet Induces Liver Fibrosis and Elevates Plasma Coenzyme Q9 in a Novel Murine Model of Obesity and Nonalcoholic Steatohepatitis. Hepatology 2010;52:934–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XB, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai BS, Masia R, et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metabolism 2016;24:848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teodoro JS, Rolo AP, Duarte FV, Simoes AM, Palmeira CM. Differential alterations in mitochondrial function induced by a choline-deficient diet: understanding fatty liver disease progression. Mitochondrion 2008;8:367–376. [DOI] [PubMed] [Google Scholar]
- 22.Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, Ishibashi S, et al. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003;38:1529–1539. [DOI] [PubMed] [Google Scholar]
- 23.Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng LX, et al. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metabolism 2015;22:516–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500:541–546. [DOI] [PubMed] [Google Scholar]
- 25.Gohda M, Kunisawa J, Miura F, Kagiyama Y, Kurashima Y, Higuchi M, Ishikawa I, et al. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer's patches for intestinal IgA responses. J Immunol 2008;180:5335–5343. [DOI] [PubMed] [Google Scholar]
- 26.Chu VT, Beller A, Rausch S, Strandmark J, Zanker M, Arbach O, Kruglov A, et al. Eosinophils Promote Generation and Maintenance of Immunoglobulin-A-Expressing Plasma Cells and Contribute to Gut Immune Homeostasis. Immunity 2014;40:582–593. [DOI] [PubMed] [Google Scholar]
- 27.Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular Helper T Cells: Lineage and Location. Immunity 2009;30:324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science 2012;336:485–489. [DOI] [PubMed] [Google Scholar]
- 29.Good-Jacobson KL, Szumilas CG, Chen LP, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nature Immunology 2010;11:535–U107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Li CJ, Lin XX, Sun H, Xu RJ, Li QP, Wei YR, et al. Targeting alkaline ceramidase 3 alleviates the severity of nonalcoholic steatohepatitis by reducing oxidative stress (vol 11, 28, 2020). Cell Death & Disease 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 2000;13:443–451. [DOI] [PubMed] [Google Scholar]
- 32.Pallaro A, Barbeito S, Taberner P, Marino P, Franchello A, Strasnoy I, Ramos O, et al. Total salivary IgA, serum C3c and IgA in obese school children. Journal of Nutritional Biochemistry 2002;13:539–542. [DOI] [PubMed] [Google Scholar]
- 33.Inamine T, Schnabl B. Immunoglobulin A and liver diseases. Journal of Gastroenterology 2018;53:691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okazaki Y, Tomotake H, Tsujimoto K, Sasaki M, Kato N. Consumption of a Resistant Protein, Sericin, Elevates Fecal Immunoglobulin A, Mucins, and Cecal Organic Acids in Rats Fed a High-Fat Diet. Journal of Nutrition 2011;141:1975–1981. [DOI] [PubMed] [Google Scholar]
- 35.Taira T, Yamaguchi S, Takahashi A, Okazaki Y, Yamaguchi A, Sakaguchi H, Chiji H. Dietary polyphenols increase fecal mucin and immunoglobulin A and ameliorate the disturbance in gut microbiota caused by a high fat diet. Journal of Clinical Biochemistry and Nutrition 2015;57:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin JJ, Li YR, Cai ZM, Li SH, Zhu JF, Zhang F, Liang SS, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55–60. [DOI] [PubMed] [Google Scholar]
- 37.Muir K, Hazim A, He Y, Peyressatre M, Kim DY, Song XL, Beretta L. Proteomic and Lipidomic Signatures of Lipid Metabolism in NASH-Associated Hepatocellular Carcinoma. Cancer Research 2013;73:4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roongta UV, Pabalan JG, Wang XY, Ryseck RP, Fargnoli J, Henley BJ, Yang WP, et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Molecular Cancer Research 2011;9:1551–1561. [DOI] [PubMed] [Google Scholar]
- 39.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081–1090. [DOI] [PubMed] [Google Scholar]
- 40.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, et al. Monounsaturated fatty acid modification of Wnt protein: Its role in Wnt secretion. Developmental Cell 2006;11:791–801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.