

SUMMARY
Telomere length control is critical for cellular lifespan and tumor suppression. Telomerase is transiently activated in the inner cell mass of the developing blastocyst to reset telomere reserves. Its silencing upon differentiation leads to gradual telomere shortening in somatic cells. Here, we report that transcriptional regulation through cis-regulatory elements only partially accounts for telomerase activation in pluripotent cells. Instead, developmental control of telomerase is primarily driven by an alternative splicing event, centered around hTERT exon 2. Skipping of exon 2 triggers hTERT mRNA decay in differentiated cells, and conversely, its retention promotes telomerase accumulation in pluripotent cells. We identify SON as a regulator of exon 2 alternative splicing and report a patient carrying a SON mutation and suffering from insufficient telomerase and short telomeres. In summary, our study highlights a critical role for hTERT alternative splicing in the developmental regulation of telomerase and implicates defective splicing in telomere biology disorders.
Graphical Abstract
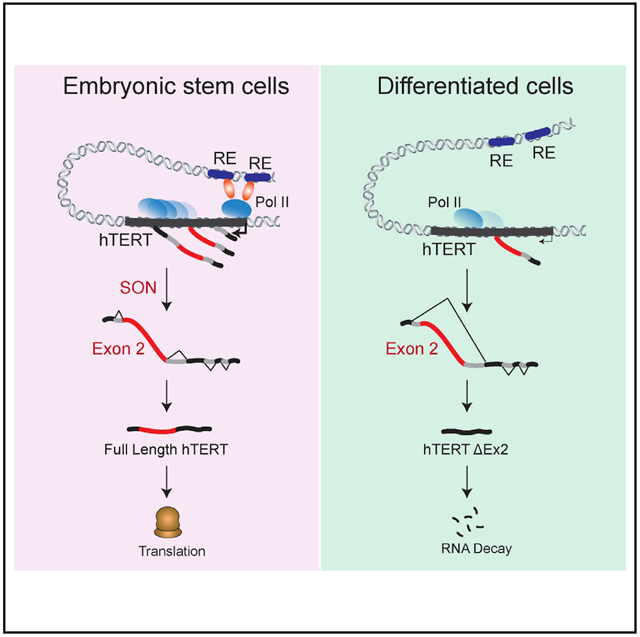
In brief
Human telomerase is transiently activated in the inner cell mass of the developing blastocyst to reset telomere length. Penev et al. identify a critical role for hTERT alternative splicing in ensuring robust telomerase activation in pluripotent cells and its repression in somatic cells.
INTRODUCTION
Mammalian telomeres consist of tracts of TTAGGG repeats that are replenished by telomerase, a specialized ribonucleoprotein composed of telomerase reverse transcriptase (TERT) and an integral RNA component. An active telomerase also contains two sets of H/ACA proteins (dyskerin, NHP2, NOP10, and GAR1) and the RNA chaperone TCAB1 (Roake and Artandi, 2020). Human telomerase RNA (hTR) is ubiquitously expressed and highly abundant (Feng et al., 1995; Venteicher et al., 2008). In contrast, expression of human TERT (hTERT) is tightly regulated and often a limiting factor during telomerase complex assembly (Meyerson et al., 1997). Embryonic stem cells (ESCs) are an exception; they accumulate high levels of hTERT, and as a result, other subunits, including hTR, become rate limiting (Chiba et al., 2015).
During embryogenesis, telomerase is activated in the inner cell mass (ICM) of the blastocyst, where it resets telomere length. As embryonic development progresses, the reverse transcriptase is repressed as a function of cellular differentiation (Shay and Wright, 2011; Wright et al., 1996). In effect, telomerase silencing in somatic cells leads to gradual shortening of telomeres, which limits proliferative capacity and exerts a tumor-suppressive function (Feldser and Greider, 2007; Greenberg et al., 1999; Kim et al., 1994; McNally et al., 2019). Reactivation of hTERT also takes place during nuclear reprograming of somatic cells to induced pluripotent stem cells (iPSCs) (Takahashi et al., 2007), and high telomerase activity is essential to sustain telomeres in pluripotent cells (Batista et al., 2011). Telomerase insufficiency in stem cells leads to telomere attrition and dysfunction that result in premature exhaustion of stem cell pools (Hao et al., 2005) and manifest in bone marrow failure, lung fibrosis, and other short-telomere syndromes. Short-telomere disorders, including idiopathic pulmonary fibrosis and dyskeratosis congenita (DC) are driven primarily by mutations in hTERT, hTR, and other telomerase-pathway genes (Armanios and Blackburn, 2012). Despite the importance of developmental regulation of hTERT, its activation in pluripotent cells and repression in differentiated cells remain poorly understood.
hTERT is reactivated in ~90% of all cancers, and its upregulation is critical to prevent telomere erosion and allow tumor cells to proliferate indefinitely (Kim et al., 1994). The core hTERT promoter contains binding sites for several growth-related transcription factors, including Myc, Klf4, and Sp1, that act as transcriptional regulators of TERT (Greenberg et al., 1999; Oh et al., 2001; Wong et al., 2010; Wu et al., 1999). However, expression of these factors in somatic cells does not reverse TERT silencing and fails to induce cellular immortalization. Recently, whole-genome sequencing (WGS) of human cancers demonstrated that highly recurrent promoter mutations drive strong monoallelic re-expression of hTERT. Common promoter mutations include −124C > T and −146C > T, whereby a single base pair mutation creates a novel binding site for the ETS family of transcription factors and boosts hTERT transcription (Horn et al., 2013; Huang et al., 2013; Stern et al., 2015). hTERT promoter mutations are currently the most common non-coding somatic mutations in cancer and are present in melanoma (67%), liposarcoma (79%), hepatocellular carcinoma (44%), and many other types of cancers (Roake and Artandi, 2020).
In addition to transcriptional regulation, studies conducted primarily in cancer cells indicated that hTERT is subject to post-transcriptional processing, most notably by alternative splicing. The reverse transcriptase comprises 16 exons that generate >20 splice isoforms (Cong et al., 1999; Hrdlicková et al., 2012; Kilian et al., 1997; Ludlow et al., 2018; Withers et al., 2012; Zhu et al., 2014). Well-characterized variants include hTERT α and β that code for a catalytically inactive reverse transcriptase (Cong et al., 1999; Kilian et al., 1997; Ludlow et al., 2018; Sayed et al., 2019; Wick et al., 1999). hTERT α and β isoforms were also detected in the developing human heart, liver, and kidney (Ulaner et al., 1998). Another common splice isoform is hTERT-ΔEx2, generated by skipping hTERT exons 2, that was first detected in cancer cell lines and noted in other primates (Withers et al., 2012). The potential implication of the various hTERT splice isoforms in the developmental regulation of telomerase remains unknown.
Here, we uncover the molecular basis of telomerase regulation as a function of pluripotency. We map and functionally interrogate hTERT enhancer elements and conclude that transcriptional regulation does not fully account for developmental regulation of hTERT. Instead, we show that hTERT mRNA levels are determined largely by a SON-mediated alternative splicing event involving exon 2. Specifically, skipping of hTERT exon 2 in differentiated cells generates a transcript that is subject to nonsense-mediated decay. Conversely, inclusion of exon 2 in pluripotent cells promotes telomerase accumulation. Last, we report on a patient carrying a germline mutation in the splicing co-factor SON (Kim et al., 2016) and displaying short telomeres and low telomerase activity. Altogether, our study highlights alternative splicing as a molecular gatekeeper that ensures strict telomerase repression in somatic cells in which a few telomerase molecules can trigger cellular immortality and facilitate tumorigenesis. Our study also implicates dysregulated hTERT splicing in telomerase insufficiency and human disease.
RESULTS
Identification of hTERT cis-acting regulatory elements
To interrogate transcriptional control of hTERT, we performed a comprehensive analysis of cis-regulatory elements (REs) that could account for ~100-fold increase in hTERT mRNA levels in pluripotent cells relative to somatic cells (Figure S1A). We applied circular chromosomal conformation capture (4C) (Simonis et al., 2006) to H7 human ESCs and retinal pigment epithelial (ARPE) cells. Specifically, we used a 1.1 kb fragment spanning the telomerase promoter as a bait and uncovered several genomic regions that are in close spatial proximity to the hTERT locus. Analysis of Illumina sequencing reads with 4C-ker (Raviram et al., 2016; van de Werken et al., 2012) revealed that most promoter interactions detected by 4C-seq occurred within the topologically associated domain (TAD) boundary surrounding hTERT and were common to both cell types (Figures S1B and S1C). In addition, our analysis identified a number of genomic regions that interacted differentially with the hTERT promoter in H7 versus ARPE cells (Figures 1A and 1B; Figure S1C). In an independent set of experiments, we performed assay for transposase-accessible chromatin using sequencing (ATAC-seq) and profiled genome-wide chromatin accessibility (Buenrostro et al., 2015). As predicted, the hTERT promoter was marked by open chromatin in H7 cells but displayed closed chromatin configuration in ARPE cells (Figure S1D). We then superimposed 4C interactions onto chromatin accessibility maps and identified five putative enhancers elements, termed REs 1–5 (RE1–RE5), that had increased contact with the hTERT promoter and displayed open chromatin in H7 cells (Figures 1A and 1B). With the exception of RE4, which was shown to enhance telomerase activity in cancer cell lines (Akıncılar et al., 2016), the remaining candidate enhancers were unknown. Our analysis also recognized three putative hTERT transcriptional silencer elements (RE6–RE8) that showed interaction with the promoter and increased chromatin accessibility in differentiated cells (Figures 1A and 1B).
Figure 1. Identification of novel cis-regulatory elements (REs) that modulate hTERT expression as a function of pluripotency.
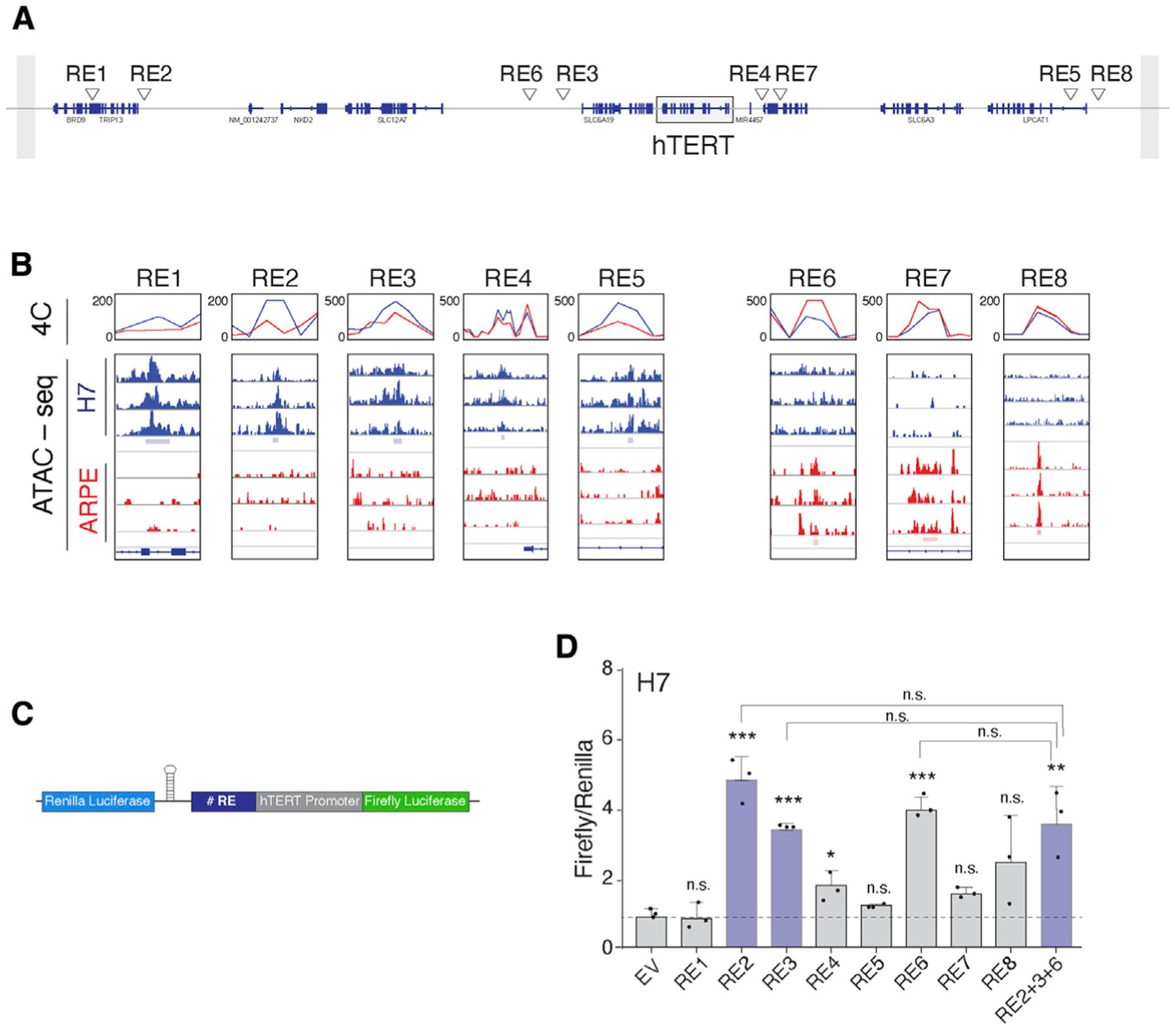
(A) Refseq of annotated genes on chr5, depicting candidate RE elements within the TAD boundary surrounding the hTERT locus (box).
(B) Overlay of data obtained from ATAC-seq and 4C-seq to identify candidate hTERT REs. Candidate enhancers (left, RE1–RE5) preferentially interact with hTERT promoter and display open chromatin in embryonic stem cells (ESCs). Silencer elements (right, RE6–RE8) preferentially interact with hTERT promoter in differentiated cells. 4Cker software was used to determine interaction score (line graphs) and aligned to ATAC-seq results from three independent replicates.
(C) Schematic of the dual-luciferase reporter used to interrogate the function of individual REs. hTERT core promoter: 500 bp sequence upstream of transcription start site (TSS) and 100 bp downstream of start codon. Individual REs cloned upstream of hTERT promoter. Expression of Renilla luciferase driven by a constitutive promoter for normalization. A hairpin downstream of the Renilla gene prevents transcriptional read-through.
(D) Bar graph represents the ratio of Firefly to Renilla luciferase in ESCs expressing the reporter plasmid with the indicated RE. n = 3 independent biological replicates. *p < 0.05, **p < 0.01, and ***p < 0.001, statistics calculated using ANOVA with multiple comparisons and comparing each value with empty vector (EV) control; similarly, ANOVA performed comparing RE2, RE3, and RE6 with RE2+3+6.
To validate putative REs, we inserted RE1–RE8 upstream of the hTERT promoter in a dual-luciferase reporter plasmid and assayed for luciferase activity in H7 and ARPE cells (Figures 1C and 1D; Figures S1E and S1F). Our data revealed that RE2 and RE3, both located downstream of the hTERT locus (Figure 1A), significantly enhanced hTERT promoter activity in ESCs but not in somatic cells (Figure 1D; Figure S1F), thus implicating ESC-specific transcription factors in hTERT activation. Paradoxically, the putative repressor element, RE6, resulted in enhanced promoter activity in H7 cells. This observation can be explained in the event that RE6 contained a transcription factor binding site that was rendered accessible when removed from its genomic context. We introduced RE2, RE3, and RE6 in tandem upstream of the hTERT promoter and noted no additive effect on luciferase activity (Figure 1D), potentially ruling out cooperativity in enhancer function during hTERT transcription. To further characterize these putative hTERT REs, we mined publicly available ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data for histone modifications characteristic of enhancers (Davis et al., 2018). Consistent with RE2 and RE3 acting as active enhancers, both were enriched for H3K4me1, whereas no enrichment of H3K4me1 was evident at the RE6 locus (Figures S1G and S1H).
Minimal impact of enhancer regulation in developmental control of hTERT
To investigate the enhancer function of RE2 and RE3 in vivo, we used CRISPR-Cas9 gene editing in ESCs and replaced the enhancers with donor sequences containing a puromycin resistance cassette and flanked by two LoxP sites (Figure 2A). We derived independent ESC clones for each enhancer and confirmed homozygous targeting by genotyping PCR and Sanger sequencing (Figure 2B and data not shown). Targeted cells were transduced with a lentiviral Cre-recombinase to excise puromycin and yield RE2Δ/Δ and RE3V/V cells (Figure 2B). qRT-PCR detected significant reduction in hTERT mRNA levels in enhancer-deleted cells (Figure 2C). In addition, we generated RE2Δ/Δ; RE3 V/V double-knockout ESCs and showed that loss of both enhancers did not further inhibit hTERT transcription (Figure 2C; Figure S2A). These results corroborate that RE2 and RE3 are activating enhancers but do not act cooperatively during hTERT transcriptional activation. Despite the significant reduction in hTERT mRNA levels, Telomere Repeat Amplification Protocol (TRAP) assay revealed similar telomerase activity in cells lacking RE2 and RE3 compared with non-targeted cells (Figure 2D). Given that the reverse transcriptase is not a limiting factor for telomerase complex assembly in human ESCs (Chiba et al., 2015), a 4-fold reduction in hTERT mRNA is not predicted to affect the overall activity of telomerase in H7 cells.
Figure 2. Reduced telomerase in ESCs upon deletion of hTERT enhancers.
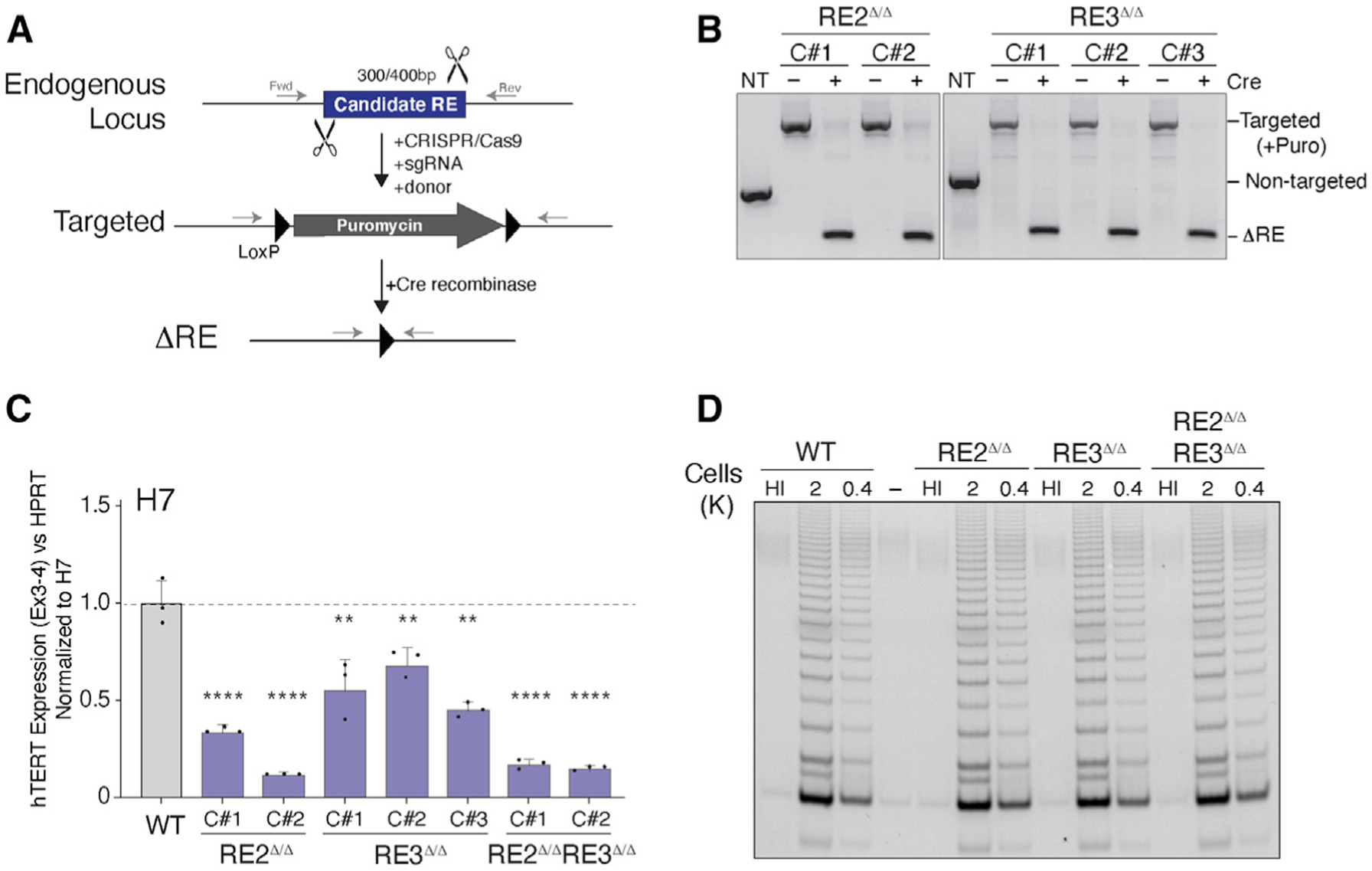
(A) Schematic for CRISPR-Cas9-mediated gene editing of RE2 (300 bp) and RE3 (400 bp) in ESCs.
(B) Genotyping PCR on cells with the indicated genotype and Cre treatment using primers highlighted in (A). PCR products (RE2/RE3): wild-type, 560/700 bp; targeted, 1,500/1,700 bp; null, 250/300 bp.
(C) qRT-PCR for hTERT mRNA in cells with the indicated genotype and Cre treatment. n = 3 independent biological replicates. RE3Δ/Δ, p < 0.01; RE2 Δ/Δ and DKO, p < 0.0001. Expression values normalized to non-targeted ESC. p values calculated using ANOVA with multiple comparisons with non-targeted ESC.
(D) Telomere Repeat Amplification Protocol (TRAP) assay to measure telomerase activity in clonally derived ESCs lacking the indicated RE. HI, heat inactivated. Two dilutions used for each sample.
In summary, our data highlighted RE2 and RE3 as putative enhancer elements that modulate the levels of hTERT mRNA as a function of pluripotency. However, the overall reduction in hTERT expression in RE2Δ/Δ and RE3V/V ESCs was significantly less than the 100-fold decrease observed following differentiation (Figure S1A). Although we cannot rule out the presence of additional enhancer elements and transcriptional regulators, we postulated that transcriptional regulation does not fully account for the robust activation of hTERT in ESCs and its tight repression in differentiated cells.
Skipping of hTERT exon 2 in differentiated cells
We next focused on alternative splicing of pre-mRNA as a regulatory pathway that has been previously shown to control developmentally regulated genes and several pluripotency-associated factors (Das et al., 2011; Gabut et al., 2011; Han et al., 2013; Venables et al., 2013; Yamazaki et al., 2018a). We performed targeted sequencing using RNA CaptureSeq, in which tiling arrays of biotinylated oligos are used to capture and enrich for low-abundance transcripts prior to RNA sequencing (RNA-seq) analysis (Mercer et al., 2014). Using hTERT CaptureSeq, we identified several mRNA isoforms, including previously known splice variants (Kilian et al., 1997; Ludlow et al., 2018; Withers et al., 2012; Yi et al., 2001) that we detected at similar levels in H7 and ARPE cells. We also identified reads spanning the junction of exon 1 to exon 3, indicative of hTERT transcripts lacking exon 2 (hTERT-ΔEx2) (Withers et al., 2012). Notably, the hTERT-ΔEx2 variant was abundant in differentiated cells but undetectable in ESCs (Figures 3A and 3B). To validate these data, we designed a junction-specific qRT-PCR assay, in which cDNA was generated using a gene-specific primer in hTERT exon 4 and followed by exon-spanning qPCR to determine the ratio of hTERT-ΔEx2 (143 bp amplicon) relative to full-length hTERT mRNA (91 bp amplicon) (Figures 3C and 3D). Our data showed that hTERT-ΔEx2 was enriched in cells lacking telomerase activity, including epithelial cells (ARPE) and fibroblasts (BJ). In contrast, iPSCs and ESCs had diminished levels of hTERT-ΔEx2. As a control, we showed that the relative abundance of hTERT-ΔEx2 was also reduced upon overexpression of full-length hTERT cDNA in differentiated cells (Figure 3D). In conclusion, we observed a positive correlation between inclusion of exon 2 and high telomerase activity, and this was recapitulated in human tissues. Specifically, hTERT-ΔEx2 was abundant in brain and skeletal muscle that lack telomerase activity. In contrast, tissues with stem cell pools (liver, colon, and lung) and those previously shown to possess high telomerase activity (bone marrow, peripheral blood mononuclear cells [PBMCs], and testis) had diminished hTERT-ΔEx2 levels (Figure 3E).
Figure 3. Inclusion exon 2 correlates with hTERT mRNA abundance.
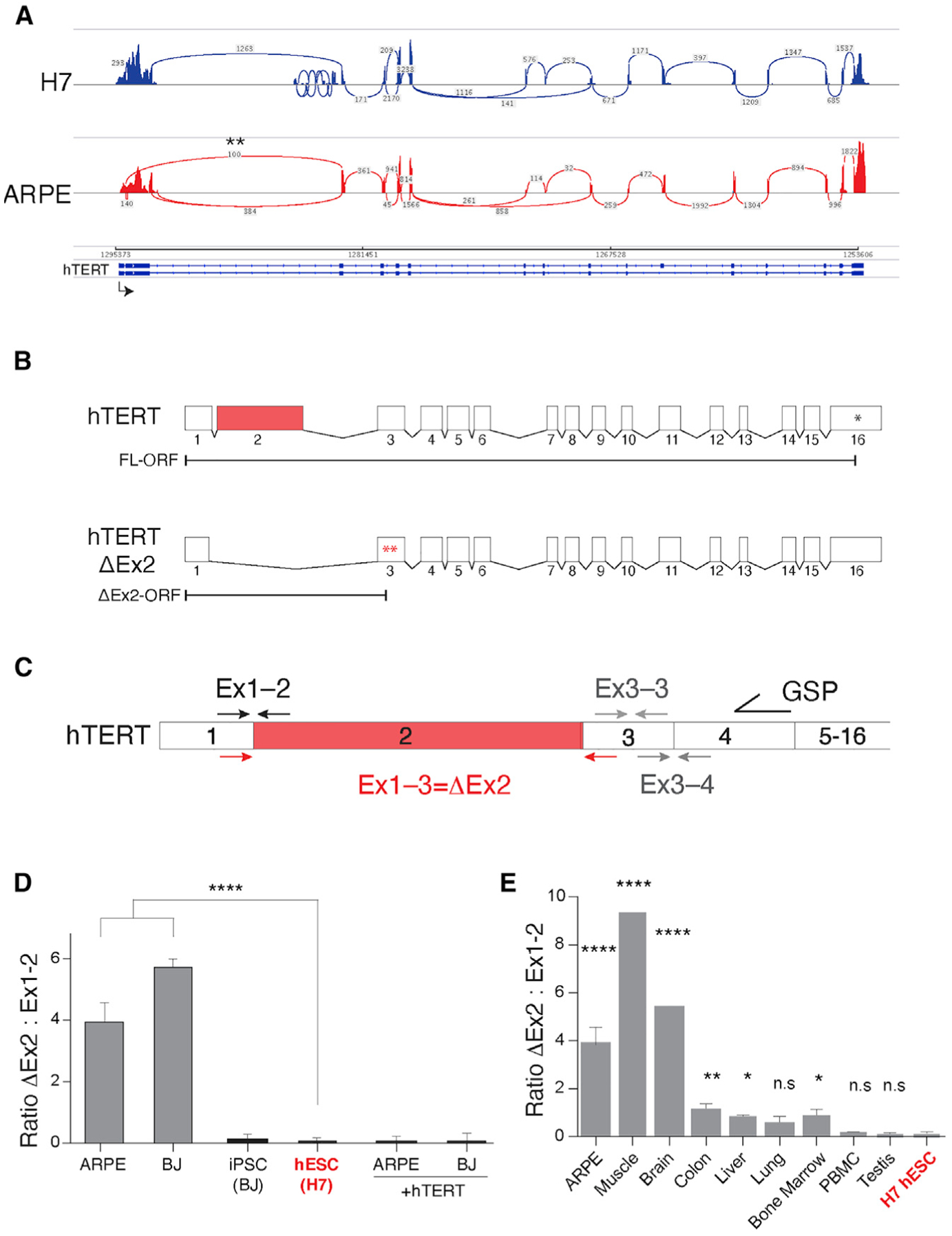
(A) Sashimi plot representing hTERT RNA CaptureSeq in H7 and ARPE cells. Transcription is from left to right. Reads from exons are depicted as pileups and exon-exon junctions denoted with arcs. Asterisks mark hTERT-ΔEx2 splice variant.
(B) Schematic of full-length and hTERT-ΔEx2 transcripts, demonstrating the position of the premature termination codons (asterisks) upon exon 2 skipping.
(C) Schematic illustration of junction-spanning PCR strategy used to assay hTERT-ΔEx2 abundance by qRT-PCR. RNA was reverse-transcribed using an hTERT gene-specific primer (GSP), and cDNA was purified and equalized between samples prior to PCR amplification with the indicated primers (91 bp for the exon1-2 amplicon and 143 bp for the exon1-3 amplicon).
(D) Quantification of the ratio of hTERT-ΔEx2 relative to full-length as determined using qRT-PCR in mortal cell lines (ARPE and BJ), cell lines overexpressing hTERT, iPSCs derived from BJ cells, and human ESCs (n = 3, p < 0.0001). Expression levels of exon1-2 and exon1-3 we first normalized to exon3-4, a non-alternatively spliced junction. We then determined the relative expression of each normalized hTERT junction to that of HeLa cells that express equivalent levels of exon1-2 and exon1-3. The relative expression values for exon1-3, indicative of ΔEx2, were divided by the relative expression of exon1-2, to produce the ratio of ΔEx2 to full-length hTERT. Statistics were computed using ANOVA with multiple comparisons, comparing the ratio in each cell line to the ratio in H7 ESCs (in red).
(E) Quantification of hTERT-ΔEx2 abundance relative to full-length by qRT-PCR in whole human tissue RNA (Thermo). Human testis and PBMCs express high levels of hTERT (Wright et al., 1996) and display low hTERT-ΔEx2 to full-length ratio (n = 3 biological replicates, p < 0.01). Colon, liver, lung, and bone marrow are highly regenerative tissues with adult stem cell populations that have some telomerase activity. n = 3 biological replicates.*p < 0.05 and **p < 0.01. Brain and skeletal muscle are telomerase negative (Wright et al., 1996) and display high hTERT-ΔEx2 to full-length ratio (n = 1 biological replicates). Ratios and statistics computed as in (D).
Rapid decay of hTERT transcripts lacking exon 2
Exclusion of hTERT exon 2 creates a frameshift that generates two tandem premature stop codons in exon 3, which are predicted to trigger nonsense-mediated RNA decay (Figure 3B; Withers et al., 2012; Hug et al., 2016). Using junction-specific qRT-PCR, we noted increased accumulation of hTERT-ΔEx2 transcripts in HeLa cells transiently depleted of the nonsense-mediated decay factor, UPF1, and the RNA exosome adapters SKIV2L2 and ZFC3H1 (Ogami et al., 2017; Figure S3A). We obtained similar results in cells treated with small molecule inhibitors of the RNA decay machinery (Figure S3B), indicating that exon 2 skipping generates hTERT transcripts that are rapidly degraded. In an independent approach, we treated HeLa cells with splice-blocking anti-sense morpholinos designed to prevent the inclusion of hTERT exon 2. Using junction-specific qRT-PCR, we observed an increase in the levels of hTERT-ΔEx2 variant and a concomitant reduction in exon 2-containing transcripts (Figure S3C). In agreement with impaired telomerase function as a result of exon 2 skipping, we noted a significant reduction in TRAP activity in HeLa cells treated with splice-blocking morpholinos against exon 2 (Figures S3D–S3F). Taken together, our results highlight a critical role for exon 2 alternative splicing in telomerase regulation, where hTERT transcripts lacking exon 2 are processed by the nonsense-mediated decay machinery.
Forced inclusion of exon 2 prevents hTERT silencing upon differentiation
So far, our data have underscored a positive correlation between exon 2 inclusion and increased abundance of hTERT transcripts in pluripotent cells. To establish causality, we tested whether constitutive retention of hTERT exon 2 would prevent telomerase silencing in differentiated cells. To do so, we deleted the first intron of hTERT in human ESCs using CRISPR-Cas9 gene targeting. Deletion of hTERT intron 1 is predicted to fuse exon 1 and exon 2 and prevent exon 2 skipping (Figure 4A). We performed two separate rounds of gene editing and isolated six independent hTERTΔin1/Δin1 clones that we validated using genotyping PCR and Sanger sequencing (Figure 4B; Figure S4A). hTERTΔin1/Δin1 ESCs maintained proper morphology and expressed pluripotency-associated cell surface markers (Takahashi et al., 2007; Figures S4B and S4C). Consistent with the reverse transcriptase not being limiting for telomerase assembly in human ESCs, telomerase activity was similar in hTERTΔin1/Δin1 and hTERT+/+ ESCs (Figure S4D). Differentiation of hTERTΔin1/Δin1 and hTERT+/+ ESCs resulted in the anticipated downregulation of key pluripotency genes and a concomitant upregulation of fibroblast-specific genes (Figures S4B and S4C). As expected, we observed complete silencing of telomerase in fibroblasts derived upon differentiation of hTERT+/+ ESCs (Figure 4C). In contrast, hTERTΔin1/Δin1 fibroblasts retained significant hTERT mRNA by qRT-PCR (Figure 4C). We quantified hTERT mRNA directly using custom NanoString-designed probes and confirmed the accumulation of several hTERT exons in hTERTΔin1/Δin1 fibroblasts (Figure 4D). In a complementary approach, we examined hTERT mRNA abundance and localization using single-molecule RNA-FISH (fluorescence in situ hybridization) (sm-FISH) (Tsanov et al., 2016) and noted a strong enrichment of telomerase transcripts in hTERTΔin1/Δin1 cells, whereas no FISH signal was detected in hTERT+/+ fibroblasts (Figure S4E). To rule out that intron 1 contained cis-REs, we generated ESCs in which we scrambled intron 1 sequence while retaining splice junctions (Figure S5A) and showed that hTERTScr1/Scr1 ESCs had similar hTERT mRNA levels compared with control cells (Figure S5B). The failure to silence telomerase in hTERTΔin1/Δin1 cells was not limited to fibroblasts, as we obtained similar results when ESCs were differentiated to hepatocyte-like cells (Mallanna and Duncan, 2013; Figures S5C and S5D). Notably, TRAP assay revealed an increase in telomerase activity in hTERTΔin1/Δin1 fibroblasts relative to differentiated hTERT+/+ cells (Figure 4E; Figure S5E). It is worth noting that despite accumulating high levels of hTERT mRNA, hTERTΔin1/Δin1 fibroblasts had significantly less telomerase activity than ESCs. It is well known that splicing is linked to nuclear export of mRNA and often coupled to post-transcriptional modifications such as m6-adenosine deposition, both of which could be necessary for efficient hTERT translation (Barbieri et al., 2017). Consistent with this idea, sm-FISH analysis revealed that pre-spliced hTERT transcripts expressing from cDNA are more efficiently exported than transcripts generated from the endogenous hTERT locus (Figure S4E).
Figure 4. Forced retention of exon 2 prevents silencing of hTERT upon differentiation.
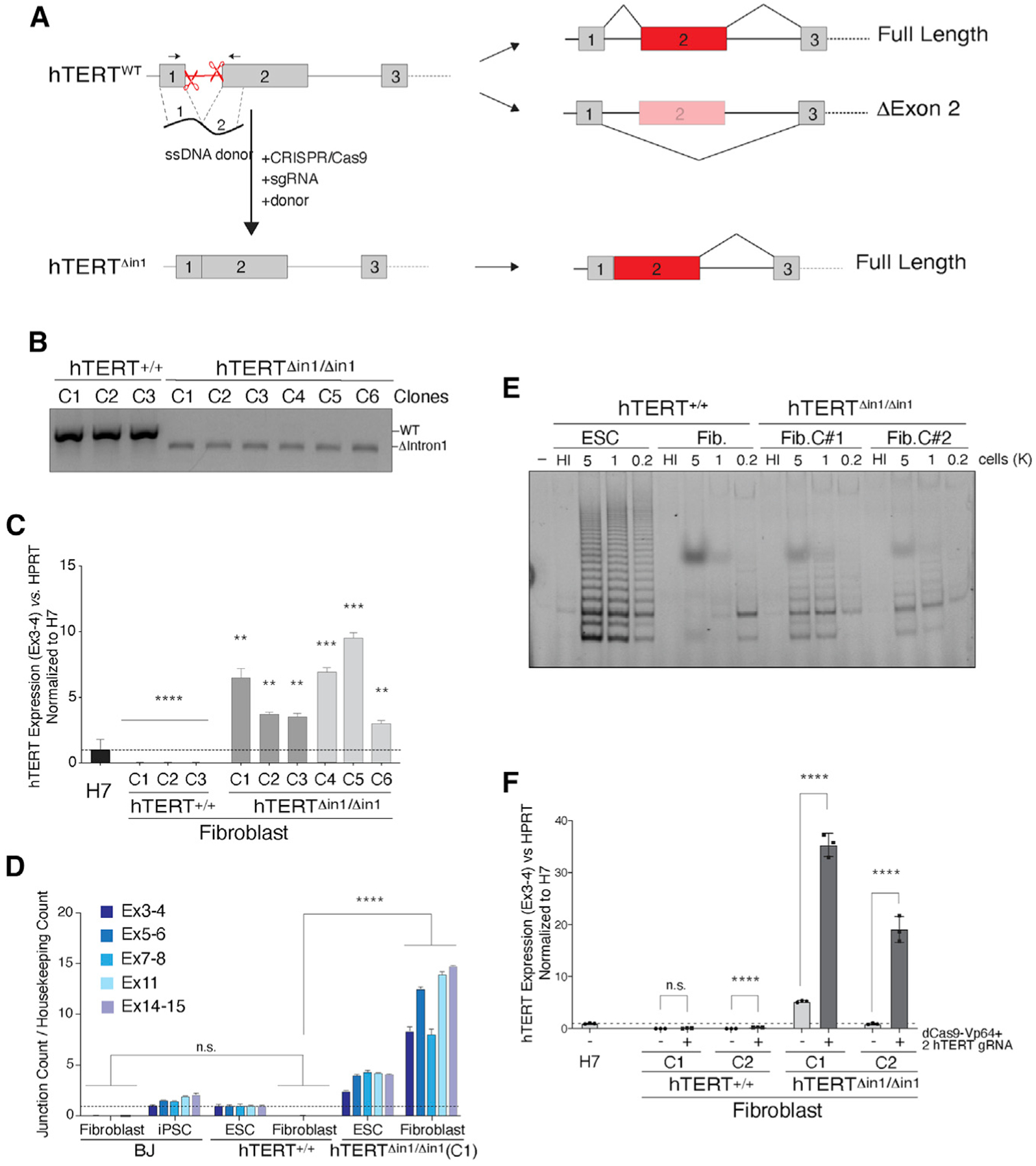
(A) Schematic illustration of intron 1 deletion by CRISPR-Cas9 gene editing and the predicted splicing pattern. Cells were co-transfected with two single guide RNAs (sgRNAs) that cleave within hTERT intron 1 and a 200 bp single-stranded (ss) DNA donor containing 100 bp sequence from exon 1 and exon 2 directly concatenated.
(B) Genotyping PCR from cells with the indicated genotype. PCR products: wild-type, 584 bp; Δintron1, 480 bp.
(C) qRT-PCR for hTERT mRNA in cells with the indicated genotype. Dark gray and light gray bars represent two independent CRISPR-Cas9 targeting experiments, and three independently derived clonal cell lines were generated from each. Values are normalized to hTERT+/+ H7 ESC. n = 3 biological replicates. **p < 0.01, ***p < 0.001, and ****p < 0.0001; statistics were computed using ANOVA with multiple comparisons, comparing hTERT expression with that of H7 ESCs.
(D) Absolute quantification of multiple hTERT exon-exon junctions using direct NanoString quantification. Data normalized to hTERT+/+ ESCs. n = 3 biological replicates. p < 0.001; statistics computed using ANOVA.
(E) TRAP assay for telomerase activity in cells with the indicated genotype shows that hTERTΔin1/ Δin1 fibroblasts retain telomerase activity compared with wild-type control cells. HI, heat inactivated.
(F) qRT-PCR for hTERT mRNA in differentiated fibroblasts following Vp64-Cas9 transcriptional activation of hTERT. Two hTERT+/+ and hTERTΔin1/Δin1 clones were transduced with lenti-virus particles expressing Vp64-dCas9 and two guide RNAs (gRNAs) targeting the hTERT promoter. Following fluorescence-activated cell sorting (FACS) selection of Vp64-dCas9-expressing cells, qRT-PCR was performed on non-transduced parental clonal lines and Vp64-dCas9-expressing cells. Values normalized to H7 cells. n = 3 biological replicates. p < 0.001, Student’s t test.
Transcriptional and post-transcriptional processes synergize to regulate hTERT mRNA levels
Having examined hTERT transcriptional activation and alternative splicing separately, our next goal was to investigate the interplay between these two processes. To that end, we transduced hTERTΔin1/Δin1 and hTERT+/+ fibroblasts with Vp64-dCas9 fusion protein and two gRNAs that target the hTERT promoter (Konermann et al., 2015). As a control, we showed that Vp64-dCas9-driven transcriptional activation resulted in an increase in hTERT transcripts in HeLa cells. Interestingly, hTERT mRNA abundance was not overtly increased in somatic cells transduced with Vp64-dCas9 targeting the hTERT promoter (Figures S5F–S5H; Figure 4F). However, we observed a synergistic effect on hTERT mRNA levels when the Vp64-dCas9 system was transduced in hTERTΔin1/ Δin1 cells (Figure 4F). Taken together, our data are consistent with hTERT transcriptional activation being coupled with exon 2 retention to mount high levels of hTERT in human ESCs.
A genetic screen using splicing reporters uncovers regulators of hTERT splicing
We next aimed to uncover the underlying mechanism that regulate alternative splicing of hTERT exon 2 and identify factors that influence exon choice. To do so, we carried out a small-scale RNAi screen using luciferase reporter genes tailored to quantify different hTERT splice isoforms. We generated two synthetic doxycycline-inducible “minigenes” comprising hTERT exons 1–3 and the spanning introns (Figure 5A). In the first minigene, Firefly luciferase was expressed upon inclusion of exon 2. Conversely, Nano luciferase was transcribed upon exclusion of exon 2 in the second minigene. Both minigenes were assembled in yeast by homologous recombination (Mitchell et al., 2015) and contained FRT sites to drive their genomic integration in T-REx-HeLa cells (Figures S6A and S6B) that express similar levels of hTERT-ΔEx2 and full-length hTERT (Figure S6C). We established a clonally derived cell line with heterozygous integration of both minigenes in the same locus. We then measured Firefly and Nano luciferase activity 48 h after doxycycline treatment and calculated the ratio of hTERT-ΔEx2 transcripts relative to full-length mRNA (Figure S6D).
Figure 5. SON is a key regulator of hTERT alternative splicing.
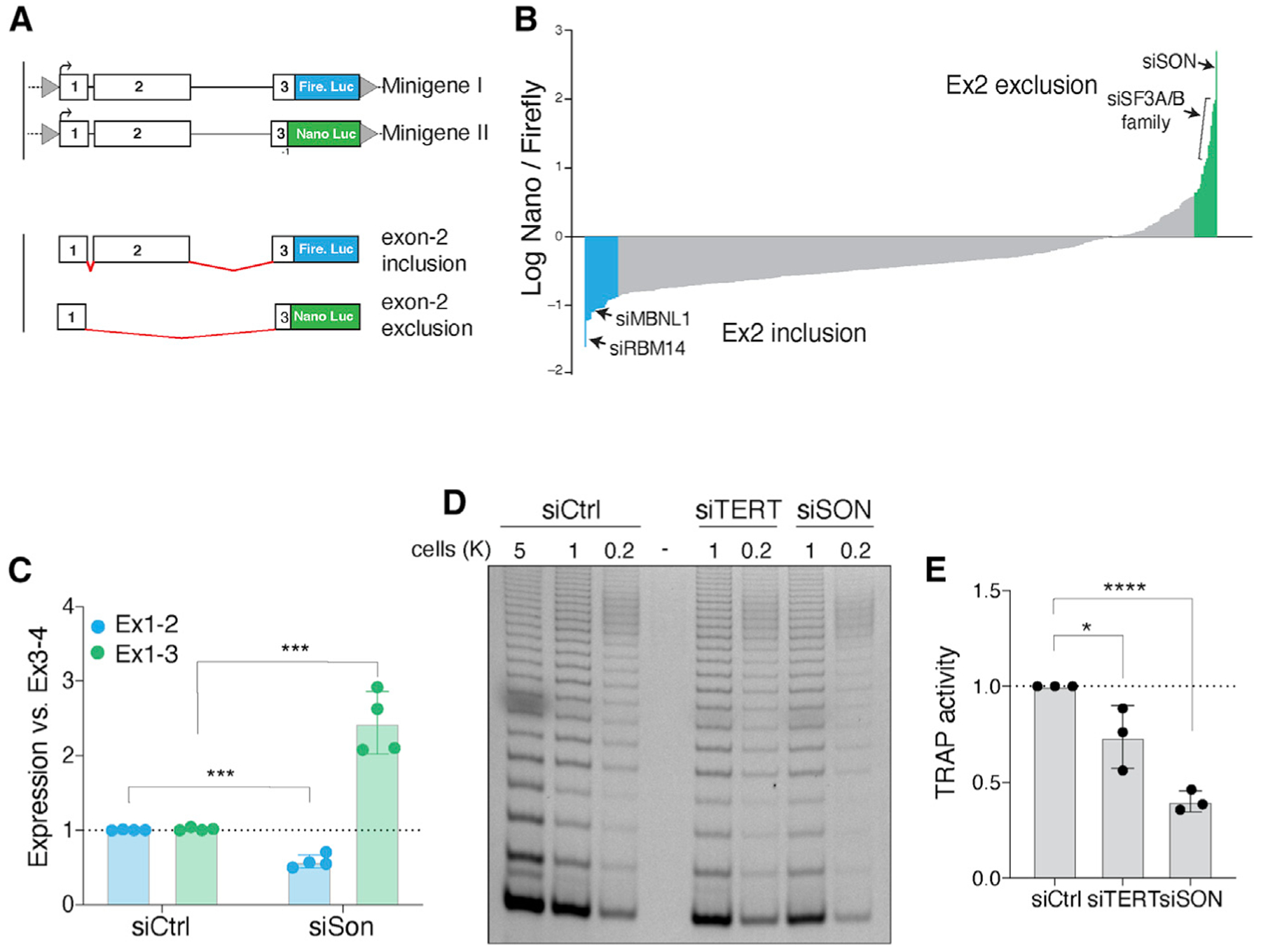
(A) Schematic of minigenes designed to measure the efficiency of hTERT mRNA splicing with Nano and Firefly luciferase. Retention of exon 2 leads to expression of Firefly luciferase. Conversely, exclusion of exon 2 leads to a (−1) frameshift in exon 3 prompting the expression of Nano luciferase while shifting Firefly luciferase out of frame.
(B) hTERT minigenes were integrated into HeLa cells, and a small-scale RNAi screen using a curated list of splicing factors and RNA-binding proteins was performed. Graph depicts the average ratio of Nano to Firefly luciferase for 442 genes and from three biological replicates. Data are presented as log ratios. We established confidence intervals representing a = 0.05 and a = 0.01 by calculating Z scores for each gene tested, using a stringent cutoff of 99% confidence on each end. Colors highlight genes with p values < 0.05. Genes highlighted in green are putative positive regulators of hTERT, and genes in blue are negative regulators of hTERT expression.
(C) Quantification of hTERT exon1-2 and exon1-3 splice junctions in ESCs following knockdown of SON. n = 4 biological replicates. p < 0.001, Student’s t test.
(D) Representative TRAP assay to detect telomerase activity in ESCs 48 h after SON depletion with siRNA.
(E) Quantification of the TRAP assay as in (D). n = 3 biological replicates. *p < 0.05 and ****p < 0.0001, Student’s t test.
Upon validating the minigene reporters, we assembled an RNAi mini-library targeting 442 genes annotated as RNA modifying enzymes, RNA-binding proteins, and splice factors (Table S1) and performed a small-scale genetic screen. We monitored dual-luciferase activity and found that knockdown of 77 genes significantly altered the Nano/Firefly ratio (p < 0.05) (Figure 5B). Candidates that enhanced exon 2 inclusion included RBM14, a paraspeckle protein involved in nuclear RNA sequestration (Yamazaki et al., 2018b) and Mbnl1, a splicing regulator that represses pluripotency-associated exon inclusion (Han et al., 2013). On the other hand, the splicing factor SRSF2 and the testis specific factor, BRDT, promoted hTERT exon 2 exclusion (Figures S6E and S6F).
SON, a nuclear speckle protein that regulates hTERT exon 2 splicing
The strongest hit from our small-scale RNAi screen was SON, a nuclear speckle protein and alternative splicing co-factor that is enriched in ESCs and promote splicing of pluripotency genes by facilitating the inclusion of exons with weak consensus splicing sites (Lu et al., 2013; Sharma et al., 2011). Knockdown of SON resulted in a significant increase in the ratio of Nano to Firefly luciferase, indicative of enhanced hTERT exon 2 exclusion (Figure 5B). Given that long-term SON inhibition impairs pluripotency (Lu et al., 2013), we transiently depleted the splicing co-factor by treating H7 and HeLa cells with small interfering RNA (siRNA) (Figures S7A and S7B) and noted a significant increase in hTERT-ΔEx2 (Figure 5C; Figure S7C). In agreement with exon 2 skipping leading to destabilization of hTERT mRNA, SON depletion resulted in a significant reduction in telomerase activity in ESCs (Figures 5D and 5E) and HeLa cells (Figures S7D and S7E). In conclusion, SON-mediated regulation of exon 2 splicing is a key determinant of telomerase activity in pluripotent cells.
Germline mutations in SON are associated with telomerase insufficiency and short telomeres
Germline de novo mutations in SON have been identified in patients with Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome (Kim et al., 2016). ZTTK syndrome is a rare disease marked by severe intellectual disability and associated with cerebral malformations, epilepsy, vision abnormalities, and dysmorphology (Tokita et al., 2016). SON alterations characterized to date are predominantly heterozygous loss-of-function mutations resulting in haploinsufficiency. A 3-year-old ZTTK syndrome patient (NCI-550-1) with a de novo SON mutation (p.Q96X) (Figure 6A) presented with a history of recurrent infection, unexplained fevers, and IgA deficiency and underwent telomere length testing as part of an immunodeficiency evaluation by her referring physician (Kim et al., 2016). Her total lymphocyte telomeres were close to the 1st percentile for age (Figure 6B). Her medical history was remarkable for birth at 33 weeks’ gestation with intrauterine growth restriction (IUGR) and fetal distress. Brain MRI revealed an abnormal corpus callosum, nodularity in the frontal horns, and white matter loss. She also has a submucous cleft palate, laryngeal cleft, clinodactyly, webbing of fingers, a sacral dimple, severe hypotonia that developed into spasticity, and significant developmental delay. We evaluated telomerase activity with TRAP assay on lysates obtained from PBMCs and found lower than expected telomerase activity. Notably, telomerase activity in PBMCs from NCI-550-1 was comparable with that derived from a patient with biallelic mutations in the gene encoding poly(A)-specific ribonuclease (PARN) (Figures 6C and 6D; Moon et al., 2015).
Figure 6. Patient-derived SON mutation is associated with short telomeres and telomerase insufficiency.
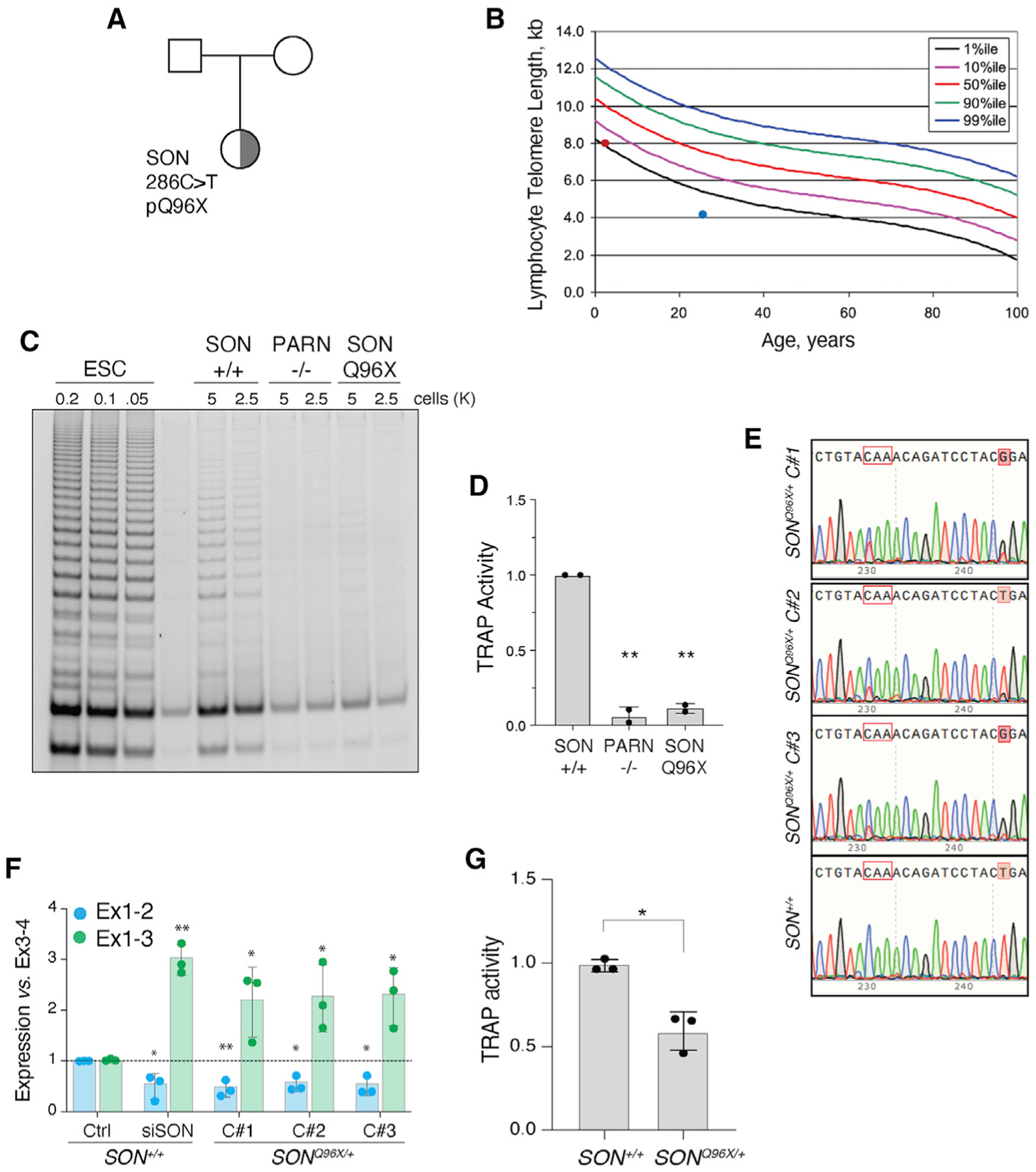
(A) Pedigree highlighting the proband, a female child of unaffected parents, carrying a de novo SON Q96X heterozygous mutation identified by exome sequencing.
(B) Telomere length measure by flow cytometry for total lymphocytes. Percentile curves are derived from healthy donors. Red dot represents lymph telomere length of SON carrier (NCI-550-1 with 8.1 kb telomeres). Blue dot represents a patient with PARN p.N7H mutation (NCI-382-1). Patient with lymph telomeres ~4.1 kb in length.
(C) Representative TRAP assay for telomerase activity in PBMC from a healthy donor (WT) and the proband (SON). PBMC from a PARN patient as a control.
(D) Quantification of the TRAP assay in (C) (p < 0.01). WT (TA 4646 0523), PARN p.N7H (c.19A > C) and deletion chr16:14,037,911 – 15,319,123 (NCI-382-1 – TA 2812 0860; patient published in Moon et al., 2015), SON mutation c.286C > T exon 3 p.Q96X (NCI-550-1 – TA 5330 0534).
(E) Sanger sequencing of three SONQ96X/+ independently derived clones and one SON+/+ clone. Tracks aligned to reference genome sequence. Codon in red outline encodes Q96. C > T mutation affects first nucleotide in codon. Shaded red boxes indicates an engineered silent T > G mutation to disturb gRNA binding site upon successful allele targeting.
(F) Quantification of the abundance of hTERT exon1-2 and exon1-3 splice junctions in SONQ96X/+ and SON+/+ ESCs. n = 3 biological replicates. *p < 0.05 and **p < 0.01, statistics calculated using ANOVA.
(G) Quantification of the TRAP assay in Figure S7F (p < 0.05).
To elucidate the effect that the patient-associated SON mutation could have on hTERT expression and telomerase activity in a developmental context, we recapitulated the patient mutation in human ESCs. We used CRISPR-Cas9 gene editing with single-stranded DNA (ssDNA) donors to introduce the SON Q96X point mutation into a single allele in H7 ESCs cells and generated three independently derived SONQ96X/+ clones (Figure 6E). In all clones, we observed an increase in hTERT-ΔEx2 transcripts coupled with significant loss in hTERT transcripts containing exon 2 (Figure 6F). Furthermore, in SONQ96X/+ ESCs displayed a reduction in telomerase activity compared with SON+/+ cells (Figures 6G and S7F), and this is consistent with the reduced TRAP activity observed upon SON depletion by RNAi and in a SON patient. As SON Q96X introduces a premature stop codon, the mutation effectively acts as a non-functional allele in ESCs, indicating that haploinsufficiency of SON affects hTERT exon 2 alternative splicing and subsequently reduces telomerase activity.
DISCUSSION
Increased telomerase activity in the early stages of embryonic development resets telomere reserves, which is critical for tissue renewal, prevention of degenerative disorders, and possibly determining longevity. So far, great strides have been made in our understanding of telomerase composition, biogenesis, trafficking through the nucleoplasm, and regulation in cancer cells (Roake and Artandi, 2020). However, the fundamental question of how hTERT gets transiently activated in the blastocyst stage of human embryonic development remains outstanding. On the basis of our data, we propose a model in which hTERT is dually regulated by transcriptional as well as post-transcriptional processes (Figure 7). We show that transcriptional regulation driven by cis-REs can influence hTERT promoter activity, although it does not fully account for the high levels of telomerase in pluripotent cells. An important aspect of our findings is the observation that hTERT exon 2 alternative splicing acts as a molecular gatekeeper to establish hTERT levels. This regulatory paradigm is reminiscent of alternative splicing-based regulation of tightly controlled pluripotency genes such as Oct4, Nanog (Das et al., 2011; Toh et al., 2016), and other factors that control transcriptional networks in ESCs (Gabut et al., 2011).
Figure 7. Dual regulation of hTERT mRNA as a function of pluripotency.
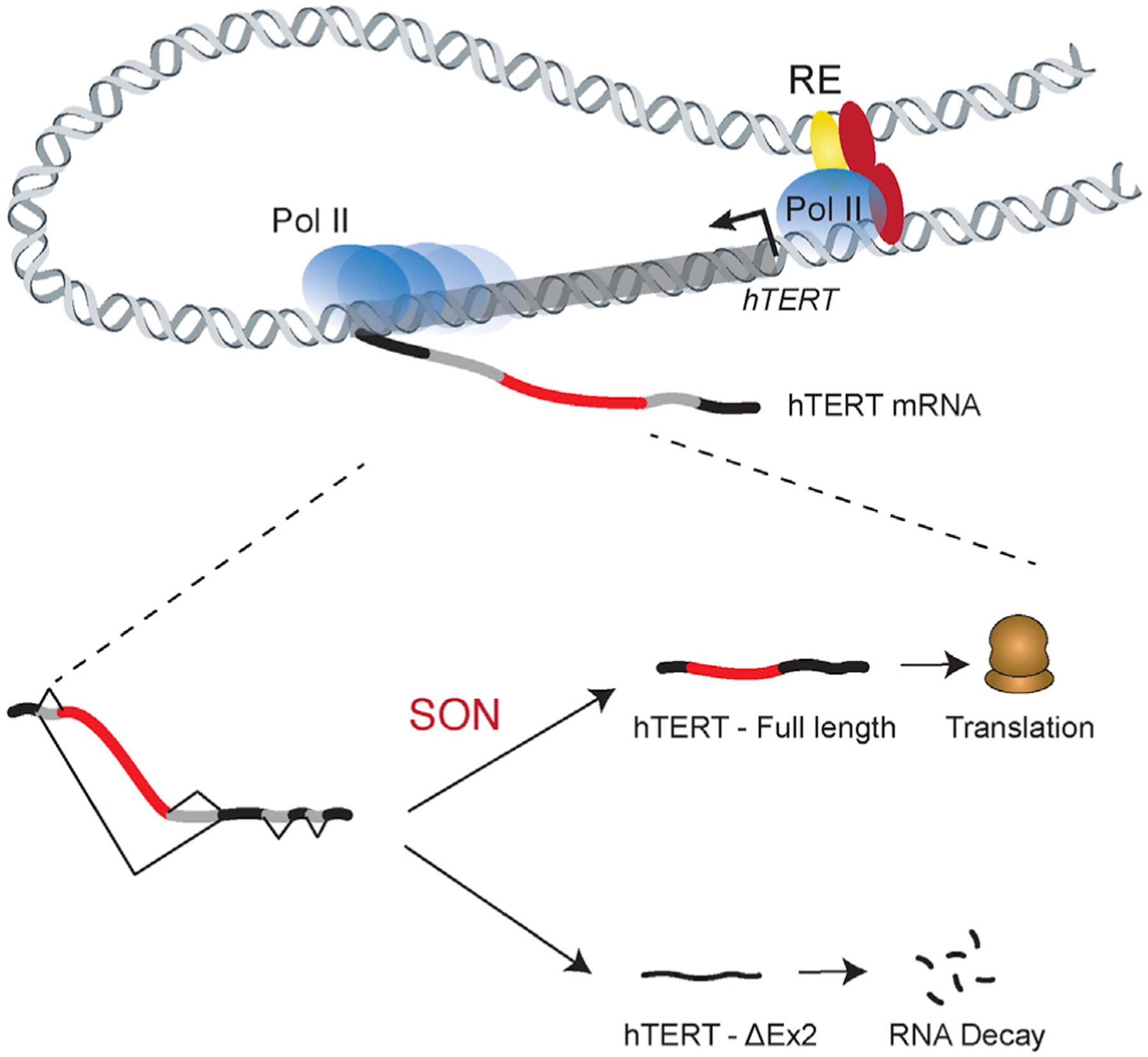
Expression of hTERT mRNA is regulated by transcriptional and post-transcriptional processes. At the hTERT promoter, transcription initiation is governed by the recruitment of distal enhancer elements coupled with transcription factor binding. A second and critical regulatory step is exerted post-transcriptionally by alternative splicing of exon 2. SON, and possibly additional splicing co-factors, regulate the inclusion of hTERT exon 2 to produce a stable transcript that is translated into a functional telomerase reverse transcriptase.
hTERT transcriptional regulation in pluripotent cells
hTERT promoter regulation, mainly as a result of activating mutations, is key to the regulation of telomerase activity in cancer cells (Okamoto and Seimiya, 2019). In this study, we superimposed datasets generated by 4C-seq, ATAC-seq, and ChIP-seq and ultimately identified two pluripotency-associated enhancers downstream of the hTERT locus. Genetic deletion of RE2 and RE3 confirmed their enhancer function (Figure 2C). However, the maximal reduction of hTERT transcripts in enhancer-deleted ESCs was significantly less than what is observed in differentiated cells (Figure S1A). At this stage, we cannot rule out the presence of additional distant hTERT enhancer elements. However, given the role of exon 2 alternative splicing in controlling mRNA accumulation (Figure 4), we propose that transcriptional control is insufficient to induce robust telomerase in pluripotent cells. RE2 and RE3 contain Myc binding motifs, suggesting that the recruitment of these enhancers to the vicinity of the hTERT promoter promotes its transcription. These findings are consistent with previous reports indicating that transcriptional activation of hTERT is driven by Myc (Greenberg et al., 1999; Oh et al., 2001; Wong et al., 2010; Wu et al., 1999). However, ectopic expression of Myc was reported to be insufficient to induce hTERT transcription (Wu et al., 1999), thus further implicating post-transcriptional processes in regulating telomerase in pluripotent cells.
Alternative splicing as a molecular gatekeeper for hTERT
hTERT alternative splicing has been predominantly surveyed in cancer cells with >20 depicted splice variants (Hrdlicková et al., 2012; Kilian et al., 1997). Notable isoforms include hTERT α/β variants that generate a dominant-negative telomerase allele (Cong et al., 1999; Hrdlicková et al., 2012; Kilian et al., 1997; Wick et al., 1999). CaptureSeq of hTERT transcripts unveiled the full spectrum of splice variants, most notable being hTERT-ΔEx2 that was enriched in somatic cells but diminished in ESCs (Figure 2A). Our findings are in line with previous RNA-seq meta-analysis that highlighted hTERT-ΔEx2 in somatic cells as well as cancer cell lines but failed to detect it in human ESCs (Withers et al., 2012). Importantly, we provide evidence to functionally implicate this specific splice variant in hTERT regulation as a function of pluripotency. Consistent with what has been previously proposed, we show that exon 2 skipping results in a frameshift mutation and two tandem stop codons in exon 3 that elicit mRNA decay and degrade hTERT transcripts (Withers et al., 2012). It is also possible that remnant hTERT-ΔEx2 transcript generate a small peptide that is yet to be characterized. Importantly, we show that forced inclusion of exon 2 abrogates hTERT silencing in differentiated cells (Figures 4C and 4D), thus underscoring the key role of alternative splicing during telomerase regulation. Taken together, we conclude that hTERT exon 2 alternative splicing is a regulated process that controls hTERT mRNA abundance via pre-programmed degradation and restricts hTERT expression to particular developmental stages.
Our study raises the obvious question of why is it necessary for splicing-mediated hTERT regulation. It is estimated that the number of catalytically active telomerase in a cancer cell is <250 molecules (Cohen et al., 2007; Xi and Cech, 2014) that are produced from ~20 mRNA (Yi et al., 2001). Accordingly, very few hTERT transcripts are sufficient for cellular immortalization that would then facilitate tumorigenesis. As such, alternative splicing and generation of non-functional hTERT transcripts could serve as a failsafe mechanism to ensure complete telomerase silencing in differentiated cells. Our limited analysis of exon 2 alternative splicing in HeLa and HT1080 cells (Figure S6C) suggested that reprogramming of hTERT splicing also takes place in some cancer cell lines. Furthermore, many splicing co-factors, including SON, are abnormally expressed in the context of human cancers (Sharma et al., 2011). Future experiments will help uncover a potential role for dysregulated hTERT mRNA splicing in tumorigenesis.
Transcript variants missing segments of hTERT exon 2 were previously reported in murine and avian models, but their function remains unknown (Hrdlicková et al., 2012; Rousseau et al., 2016). Similar TERT alternative splice variants were also identified in species as phylogenetically distant as Planarians (Tan et al., 2012) that express robust telomerase activity at the site of blastema formation and facilitate rapid tissue neogenesis. During regeneration, planarian TERT is alternatively spliced to include a cluster of exons that are equivalent to exon 2 (Tan et al., 2012). Therefore, hTERT “exon 2-like” alternative splicing is likely an evolutionarily conserved mechanism to support rapid proliferation in specific developmental time frames while protecting somatic cells.
Nuclear speckle protein and splicing co-factor SON regulates hTERT splicing
Our study identifies SON as a regulator of hTERT exon 2 splicing. SON is a nuclear speckle protein that is highly expressed in pluripotent cells and multiple cancer cell lines and promotes the inclusion of cassette exons with weak splice sites (Sharma et al., 2011). Interestingly, the reported DNA consensus sequence for SON binding (GA[GT]AN[CG][AG]CC) is adjacent to hTERT exon 2. Although the RNA-binding motif of SON has not been determined, future studies will help explore its direct binding to hTERT mRNA and provide insight into the mechanism by which it controls hTERT splicing. In addition, our study identifies a patient that suffers from insufficient telomerase activity and borderline short telomeres and harbors a heterozygous SON mutation but no other variants in genes linked to telomere biology disorders (Figure 6). SON heterozygote mutations leading to significant reduction in SON mRNA (up to 80%) were identified in a cohort of pediatric patients with mental disability and ZTTK syndrome (Kim et al., 2016). It would be interesting to examine telomerase levels and telomere dysfunction in the context ZTTK syndrome.
In summary, we uncover a previously unappreciated role for alternative splicing in the developmental regulation of hTERT expression and provide further insight into the understanding of replicative senescence and cellular immortalization. In addition, our results underscore a potential therapeutic benefit for targeting alternative splicing to promote hTERT exon 2 inclusion and increase telomerase activity in telomere biology disorders as well as regenerative medicine therapies.
Limitations of study
We show that SON depletion reduces telomerase activity in ESCs and in a cancer cell line. Analysis of clinical exome sequencing identified a SON mutation in a patient with telomerase insufficiency and short telomeres. A caveat in the patient analysis is that exome sequencing does not identity mutations in REs that might affect other genes involved in telomere maintenance. Furthermore, SON is a critical splicing regulator for many human genes (Lu et al., 2013; Sharma et al., 2011), and it is therefore possible that SON dysfunction affects hTERT splicing indirectly. It is worth noting that using a splicing reporter and analyzing SONQ96X/+ ESCs, we provide evidence for a direct role for SON in preventing skipping of hTERT exon 2, prompting us to conclude that SON is regulator of hTERT splicing. Last, our study highlights a single patient carrying a SON mutation and suffering from short telomeres. Future studies are necessary to identify additional patients and firmly establish the link between SON and telomere syndromes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Agnel Sfeir (SfeirA@mskcc.org).
Materials availability
Cell lines generated in this study are available from Dr. Agnel Sfeir. Plasmids generated in this study have been deposited to Addgene (www.addgene.org)
Data and code availability
The 4C and ATAC-seq datasets generated during this study are available at GEO: GSE168193. Raw data available at Mendeley: https://dx.doi.org/10.17632/jfmsvxnpbg.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All cells were grown in an incubator with O2 and CO2 maintained at 5%, and the temperature was maintained at 37°C. ARPE-19 cells (ATCC CRL-2302), HeLa (ATCC® CCL-2), T-REx-HeLa (Invitrogen) and BJ cells (ATCC® CRL-2522) in Dulbecco’s Modified Eagle Medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS, GIBCO), 2 mM L-glutamine (GIBCO), 100 U/ml Penicillin-Streptomycin (GIBCO), and 0.1 mM MEM non-essential amino acids (GIBCO). ARPE-19 and BJ cell lines were immortalized with hTERT and cultured in the same media conditions. Cells were passaged every 48–72 hours and maintained mycoplasma free by using Plasmocin (Invivogen) per manufacturer indication. H7 human embryonic stem cells were a kind gift from Lei Bu and cultured in mTeSR Plus (STEMCELL) media supplemented with 100 U/ml Penicillin-Streptomycin (GIBCO). ESCs were passaged every 2–3 days using 500 μM EDTA in PBS and recovered for one day in Y-27632 2HCl (5–10 μM, SelleckChem). 293T cells used for lentiviral packaging were cultured in DMEM supplemented with 10% bovine calf serum (BCS, Gemini), 2 mM L-glutamine (GIBCO), 100 U/ml Penicillin-Streptomycin (GIBCO), 0.1 mM MEM non-essential amino acids (GIBCO).
The patient (5 years old) was a participant the IRB-approved longitudinal cohort study titled Etiologic Investigation of Cancer Susceptibility in Inherited Bone Marrow Failure Syndromes (NCT-00027274). Informed consent was signed by her parents and data were collected through questionnaires and medical record review (Alter et al., 2018). Patient DNA underwent standard clinical exome sequencing at Baylor Miraca Genetics Laboratories, Houston, TX, with appropriate consent. The variant in SON was the only variant of clinical consequence identified. Blood samples are collected from patients at their clinic visits and processed at a large, centralized NIH facility using robust standard operating procedures and quality control methods (per IRB approved protocol, ClinicalTrials.gov Identifier NCT00027274). De-identified patient-derived cells were shipped from NIH to NYU at the same time, they were lysed at the same time, PCR/TRAP reactions were set up at the same time and run on the same gel.
METHOD DETAILS
Cell culture procedures and treatments
iPSC reprogramming of BJ fibroblasts was performed using CytoTune™- iPS Sendai Reprogramming kit (ThermoFisher) as per manufacturer instructions. Fibroblast differentiation from hESCs was performed in differentiation media containing 15% fetal bovine serum (FBS, GIBCO), 2 mM L-glutamine (GIBCO), 100 U/ml Penicillin-Streptomycin (GIBCO), and 0.1 mM MEM non-essential amino acids (GIBCO). Media was changed without passaging every two days for 21 days when fibroblast differentiation was complete. Cells were subsequently passaged every 4–5 days. Hepatocyte differentiation was performed as described in Mallanna and Duncan (2013) with media changes every 2 days. Briefly, ESCs were plated in Geltrex™ (GIBCO) coated 6-well plates and treated with RPMI 1640 (Corning) media containing 2% B27 supplement (w/o insulin, GIBCO), and additional recombinant factors including Activin A (100ng/ml, Thermo), BMP4 (10ng/ml, Thermo), FGF2 (20ng/ml, Thermo) for two days. Day 3–5 media contained only Activin A in B27-RPMI 1640 media. Day 6–10 media contained 2% B27 supplement with insulin (GIBCO) and BMP4 and FGF2 at previous concentrations. Days 11–15 media contained recombinant HGF (20ng/ml, Thermo) in B27+insulin RPMI 1640 media. Days 16–24 media contained recombinant Oncostatin M (20ng/ml, Thermo) in HCM media lacking EGF (Lonza). For cell treatments, the following compounds were used: 5-fluoro-uracil (50 μM, Sigma-Aldrich), Isoginkgetin (20 μM, Cayman Chemicals). hTERT minigene expression was induced with 2 μM doxycycline (Sigma-Aldrich) for 48 hours following siRNA transfections.
4C and ATAC-seq genome-wide sequencing and bioinformatic analysis
Library preparation for Circular Chromosome Conformation Capture (4C) was performed as described (van de Werken et al., 2012) with the exception of initial fixation and lysis procedure, which was adapted from (Miele and Dekker, 2009). 1×107 ARPE or H7 ESCs were dissociated and resuspended in 5ml PBS containing 10% BCS. Cells were crosslinked by adding 5ml of 4% formaldehyde in PBS and incubating at room temperature (RT) for 10 minutes with gentle rocking. Reaction was quenched by adding 2.5ml 2.5M glycine, mixing well and incubating at RT for 5 minutes, then on ice for 15 minutes. Cells were pelleted at 800×g for 10 minutes, resuspended in 1ml of cold Lysis Buffer 1 (10mM Tris pH 8.0, 10mM NaCl, 0.2% IGE-PAL, Protease inhibitor (Roche)) and incubated on ice for 15 minutes. Cells were then dounce-homogenized on ice with a B-type pestle twice for 15 minutes with a minute rest on ice in between. Sample was pelleted at 2500×g for 5 minutes and the pellet was washed with 1x Cutsmart buffer (NEB) and pelleted again. Samples were further processed as described in van de Werken et al. (2012) using DpnII primary digestion and Csp6I secondary digestion. Sequencing library was prepared as described using Expand Long Template polymerase (Roche) and Illumina adaptor sequence-containing primers unique to the hTERT fragend listed in Table S2. Sequencing was performed by the NYU Genome Technology Core on Illumina HiSeq 4000 sequencer. Sequencing data was analyzed using the 4Cker software package described in Raviram et al. (2016) using the recommended parameters.
ATAC-seq was performed on 5×105 ARPE and H7 cells in triplicate as described in Buenrostro et al. (2015). Sequencing of libraries was performed by NYU Genome Technology on Illumina HiSeq 2500 sequencer. Alignment was performed as in Buenrostro et al. (2013) to the hg19 human reference genome and peak calling was done using macs2 using parameters described in Corces et al. (2017). IGV was used to visualize sequencing data tracks and bedtools was used for find regions of overlap between ATAC-seq and 4C data.
Dual-Luciferase assay for hTERT promoter activity
Dual luciferase reporter plasmid pRF-HCV was a kind gift from Maria Barna. HCV promoter was replaced with hTERT core promoter sequence amplified from ESC genomic DNA and cloned into pRF using NcoI restriction enzyme sites. Candidate sequences were similarly amplified from genomic DNA and cloned into BbvCI restriction enzyme site upstream of hTERT promoter. Plasmid DNA was purified using Plasmid Plus Midi Kit (QIAGEN) or NucleoBond Xtra Midi Kit (Machery Nagel). Reporter plasmid was introduced to H7 and ARPE cells via transient transfection. Cells were lysed 48 hours after transfection and assayed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions using a Flexstation 3 Multi-mode Microplate Reader (Molecular Devices). Each sample was assayed in triplicate.
Real-Time RT-qPCR
Total RNA was purified with RNAeasy Mini Kit (QIAGEN) or NucleoSpin RNA Clean-up (Macherey-Nagel) following manufacturer instructions. Genomic DNA was eliminated by on-column digestion with DNaseI. A total of 1 μg of RNA was reverse-transcribed using iScript Reverse Transcription Supermix (Biorad) and qPCR (45 cycles) was performed on a Roche LightCycler480. Reactions were run in triplicates with ssoAdvanced SYBR green Supermix (Biorad) in a total volume of 10μl with standard cycling conditions. Relative gene expression was normalized using HPRT or TBP as housekeeping genes and all calculations were performed in Excel. A list of primers is available in Table S2.
RT-qPCR for hTERT exon-exon junctions
Total RNA was purified with RNAeasy Mini Kit (QIAGEN) or NucleoSpin RNA Clean-up (Macherey-Nagel) following manufacturer instructions. Genomic DNA was eliminated by on-column digestion with DNaseI. A total of 10 μg of RNA was reverse-transcribed using Superscript IV Reverse-Transcription kit (Invitrogen) using an hTERT specific probe in exon-4 (CCTGACCTCTGCTTCCGACAG). Primer was annealed to hTERT RNA at 65C for 5 minutes and then incubated at 0C for 5 minutes before addition of other kit reagents. Reaction was then incubated at 60C for 50 minutes and heat inactivated for 10 minutes at 80C. Each reaction was RNaseH (NEB) treated according to manufacturer instructions and cDNA was purified using MinElute Reaction Cleanup Kit (QIAGEN). cDNA concentration was determined using Qubit ssDNA Assay Kit (Invitrogen) and then equalized between samples with ddH2O. qPCR (50 cycles) was performed on a Roche LightCycler480. Reactions were run in triplicates with PrimeTime Gene Expression Master Mix (IDT) in a total volume of 10μl with standard cycling conditions. hTERT exon1-2 and exon1-3 expression was normalized using hTERT exon3-4 junctions and all calculations were performed in Excel. A list of primers is available in Table S2. To determine the hTERT exon-2 alternative-splicing ratios, we calculated the relative expression of each hTERT junction to that of the HeLa cell line (which had equivalent expression of exon1-2 and exon1-3 in our assays) and normalized the values to the non-alternatively spliced junction hTERT exon3-4. The relative expression values for exon1-3, indicating ΔEx2, were then divided by the relative expression of exon1-2, to produce the ratio of ΔEx2:full-length hTERT. Statistics were computed using ANOVA analysis with multiple comparisons, comparing the ratio in each cell line to the ratio in H7 ESCs.
CRISPR/Cas9 targeting
H7 RE2−/−, RE3−/− and RE2−/− RE3−/− double knockouts polyclonal populations were generated. Briefly H7 cells were transfected with a total of 2 gRNAs targeting the 500bp region for RE2 (TTCCCTTGCCCGCTAGAGGG and CCCCCAAGGGAATGAAAAAG) or RE3 (GTGTCTGGATGGACCAGCAG and GCAATGGTAACTCAGTGACT) cloned in a modified version of vector pX458, a kind gift from Feng Zhang (Addgene plasmid # 48138), and a plasmid DNA donor containing 500bp of upstream and downstream homology sequence for either RE2 or RE3 flanking a hPGK-driven Puromycin resistance gene surrounded by two LoxP sites. Five days following transfection, transfected cells were treated with puromycin (250ng/ml) and surviving clones were subjected to genotyping PCR using primers included in Table S2. Homozygous targeted clones were expanded and infected using lentivirally encoded Cre-recombinase with a Hygromycin resistance gene. Following infection, cells were treated with hygromycin (100ug/ml) and genotyping PCR was performed to confirm excision of the puromycin cassette by fragment size using primers in Table S2. H7 hTERTΔin/Δin ESCs were obtained by delivering a single-stranded 200nt template oligo containing abutting 100nt sequences from hTERT exon-1 and exon-2 and either an RNP complex of a single gRNA targeting hTERT intron-1 (CGGGGGGAACCAGCGACATG), tcrRNA and wild-type Cas9 protein (IDT) or 2 gRNAs targeting hTERT intron-1 (CGCATGTCGCTGGTTCCCCC and CGGGGGGAACCAGCGA CATG) cloned into a modified pX458 plasmid described above. Transfected cells were plated at clonal density and individual clones were picked for genotyping approximately one week later. Genotyping was performed by PCR looking for loss of 104bp in PCR amplicon spanning hTERT exons 1-2 (Table S2). Genomic DNA was extracted using the Quick-DNA Miniprep Kit (Zymo). Genotyping PCR was performed using Failsafe PCR 2x PreMix H (Lucigen) and Taq polymerase (NEB).
Lentiviral delivery
Cre-recombinase and Vp64 Cas9-Activation constructs (Konermann et al., 2015)were purchased in the pLenti backbone (Addgene 73795, 61425, 61426) and were introduced by 4 lentiviral infections at 12hr intervals in presence of 8 μg/ml polybrene (Sigma-Aldrich) using supernatant from transfected 293T cells. For targeting Vp64-dCas9 to hTERT promoter, two guide RNA sequences were used in combination (CCAGCTCCGCCTCCTCCGCG and CCAGGACCGCGCTTCCCACG). Vp64 antibiotic resistance genes were replaced with fluorescent protein (mCherry, sfGFP, and tagBFP) genes via several cloning strategies to allow for FACS selection of triple-positive cells containing all Cas9-activation components.
TRAP Assay for telomerase activity
Telomerase Repeat Addition Processivity (TRAP) was performed as described (Mender and Shay, 2015). In brief, cells were dissociated and counted on a manual hemocytometer (Fisher Scientific) using Trypan Blue (Corning) to count viable cells. Cells were pelleted and resuspended in CHAPS lysis buffer (Millipore) with Halt Protease+Phosphatase inhibitor (Thermo) and 3μM β-mercaptoethanol at a concentration of 5×103 cells per μl. Lysates were incubated on ice for 30 minutes, vortexed twice during incubation, and then clarified at 12,000 g for 20 minutes. Serial dilutions and heat inactivated samples were prepared and 2μl of lysate or dilution was used in each PCR reaction as described in Mender and Shay (2015). Cycling conditions were as follows: incubate 30 minutes at 30°C, boil 5 minutes at 95°C, then melt 30 s at 95°C, anneal 30 s at 59°C, extend 1 minute at 72°C; 25 cycles were used for ESC samples, 26 cycles for HeLa, and 30 cycles for fibroblast and PBMC samples, and in all cases reaction concluded with a final extension for 10 minutes at 72°C. Reactions were run on a 10% acrylamide gel (19:1, Fisher Scientific) and imaged on a ChemiDoc MP apparatus (Biorad).
RNA-capture sequencing for hTERT transcripts
Total RNA was purified with RNAeasy Mini Kit (QIAGEN) following manufacturer instructions. Genomic DNA was eliminated by on-column digestion with DNaseI. A total of 25 μg of RNA was reverse-transcribed using Superscript IV Reverse-Transcription kit (Invitrogen) in multiple PCR-tubes using a mix of hTERT specific primers in exon-4, exon-9, exon-12, and exon-16 (Table S2). Primers were annealed to hTERT RNA at 65°C for 5 minutes and then incubated at 0C for 5 minutes before addition of other kit reagents. Reaction was then incubated at 60°C for 50 minutes and heat inactivated for 10 minutes at 80°C. Each reaction was RNaseH (NEB) treated according to manufacturer instructions and second-strand cDNA was synthesized using Second Strand DNA Synthesis kit (NEB) per manufacturer’s instructions. NYU Genome Technology core designed custom probe-library tiling hTERT exons and flanking intragenic sequences using X-Gen probe design software proceeded with Illumina sequencing library preparation of double-stranded cDNA and hybridization and purification of target sequences. Libraries were sequenced using the Illumina MiSeq and data was aligned to hg19 reference genome using Bowtie2 and alternative splicing analysis performed using TopHat.
Transient transfection of plasmid DNA and siRNA
Purified plasmid DNA (2–3 μg) was introduced to ARPE cells via transient transfection using Lipofectamine 3000 transfection reagent (ThermoFisher) or to H7 ESCs using Genejuice transfection reagent (MilliporeSigma) according to manufacturer’s instructions. For candidate validation experiments, 2–10 pmol of 4-oligo ON-TARGETplus siRNA pools (Horizon, Dharmacon) or non-targeting control pools (Horizon, Dharmacon) were introduced to H7 ESCs and HeLa cells using 4D-Nucleofector (Lonza) electroporation as per manufacturer instructions for each cell line (ESC: CA-137 in P3 solution, HeLa: CN-113 in SE solution). Splice-blocking morpholino (ASO) for hTERT exon-2 5′ splice site (AGGACACCTGCGGGGGAAGCG) was ordered from Gene Tools, LLC according to their design recommendations with a 3′-Carboxyfluorescein residue to assess transfection efficiency. ASO was delivered to cells by nucleofection as described above.
hTERT exon 1-3 splicing minigene assembly by homologous recombination
DNA inserts were PCR amplified with oligos containing the corresponding VEGAS adapters and gel purified while the VEGAS backbone36 was digested with BsaI and gel purified. ~100 ng of each fragment along with the linearized VEGAS backbone were transformed using the standard lithium acetate method into Saccharomyces cerevisiae strain BY4741 (Brachmann et al., 1998) and plated onto SC–Ura plates. After 48 hours colonies were replica plated onto SC–Ura plates containing G418. After an additional two days of growth colonies were screened using PCR to verify the presence of each fragment-fragment junction as well as the initial and terminal backbone-fragment junctions. Colonies containing all junctions were grown overnight in liquid SC–Ura media containing G418. Plasmids were extracted from yeast as follows. 1.5 mL of overnight culture was spun down and resuspended in 250 μL P1 buffer with RNase (QIAGEN) and 200 μL of glass beads (Sigma) in an eppendorf tube and shaken for 10 minutes to break the cells. 250 μL of P2 buffer (QIAGEN) was added, mixed by inversion, and incubated for 5 minutes at room temperature. 350 μL of P3 buffer (QIAGEN) was added and mixed by inversion. This mixture was spun at 10000×g for 10 minutes in a tabletop centrifuge. The supernatant was transferred to a Zyppy miniprep column (Zymo Research) and spun at 10000×g for one minute; the flowthrough was discarded. The column was washed with 200 μL endo wash buffer, spun at 10000×g for one minute, then washed with 400 μL Zyppy wash buffer and spun again for one minute with flow through discarded. The column was spun one more time to remove residual wash buffer. DNA was eluted with 10 μL of elution buffer. 3 μL of eluted DNA was transformed into E. coli. Plasmid DNA recovered from E. coli was digested with AflIII and NotI to release minigene fragment from shuttle vector and clone into pcDNA5/FRT/TO (Thermo) mammalian expression vector for integration into HeLa cells by co-transfection with pOG44-Flpase (Thermo). Hygromycin (150ug/ml) was used to select for cells with integration and Nano-Glo Dual-luciferase assay kit (Promega) was used to confirm expression of both Firefly and Nano-luciferase in clonally isolated cells.
RNAi luciferase screen for hTERT alternative splicing factors
4.5 pmol of Ambion® Silencer® Select siRNA pools (ThermoFisher) was spotted in a 96-well plate format and transfection complexes were formed in OptiMEM (GIBCO) using Lipofectamine RNAiMAX (Invitrogen™) according to manufacturer’s instructions. We then introduced the T-REx-HeLa cell line with heterozygous integration of two minigene constructs suspended in doxycycline-containing DMEM media and incubated for 48 hours at 37°C. Cells were lysed and assayed for luciferase activity using the Nano-Glo Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions using a EnVision® Multilabel Plate Reader (PerkinElmer). Each plate was assayed in triplicate.
Absolute quantification of hTERT mRNA
Total RNA was purified with RNAeasy Mini Kit (QIAGEN) following manufacturer instructions. Genomic DNA was eliminated by on-column digestion with DNaseI. A total of 1 μg of RNA was hybridized for 24 hours with a custom library of multiply-labeled fluorescent oligos per manufacturer’s instructions to detect specific hTERT exon-exon junctions to derive and absolute quantification of mature spliced transcripts. Predesigned and validated probes against housekeeping genes, TBP and HPRT, were used for subsequent normalization. Probe sequences are listed in Table S2. Hybridized RNA:probe mixture was then purified and immobilized on nCounter chips using nCounter Prep Station (NanoString) and then data was acquired using the nCounter Digital Analyzer (nCounter FLEX Analysis System, NanoString). All instruments were run and maintained at the NYU Genome Technology Core. Data was analyzed using nSolver™ software package (v4.0, NanoString).
Immunofluorescence (IF) and microscopy
Cells were plated on 12 mm circular glass coverslips (Fisher Scientific) and analyzed for IF with standard techniques. Briefly, cells were fixed with 4% (v/v) paraformaldehyde in PBS (Santa Cruz Biotechnology, Inc.) for 5 minutes at room temperature. Cells were washed with PBS, permeabilized with 0.5% (v/v) Triton X for 10 minutes and blocked for 30 minutes with PBS containing 3% goat serum (Sigma-Aldrich), 1 mg/ml bovine serum albumin (BSA, Sigma-Aldrich), 0.1% Triton X-100 and 1mM EDTA. Cells were incubated with the same buffer containing primary antibodies for 2 hours at room temperature followed by secondary antibodies incubations for 1 hour at room temperature. Cells were mounted with ProLong Gold Antifade (Thermo Fisher Scientific), imaged on a Nikon Eclipse 55i upright fluorescence microscope at 20X and analyzed with Nikon software. Additional contrast/brightness enhancement and export were performed with Fiji-ImageJ software (Schindelin et al., 2012; Schneider et al., 2012). DNA was counterstained with 5 μg/mL DAPI as needed. A complete list of antibodies used in the study and relative dilutions is available in Table S3.
Single-molecule hTERT mRNA FISH
Cells were plated on 12 mm circular glass coverslips (Fisher Scientific) and analyzed for smiRNA-FISH using techniques described by Tsanov et al. (2016). In brief, cells were fixed with 4% (v/v) EM-grade paraformaldehyde (Electron Microscopy Sciences) for 20 minutes at room temperature. Cells were washed with RNase-free PBS, and permeabilized in 70% ethanol for 1 hour at 4°C. Cells were rehydrated in RNase-free 1x SSC buffer containing 15% (v/v) formamide (Sigma-Aldrich) for 15 minutes and then hybridization solution was applied to coverslips overnight, containing 1x SSC, 15% (v/v) formamide, BSA (2mg/ml, NEB), dextran sulfate (10%, Sigma-Aldrich), VRC (2mM, NEB), tRNA (0.5mg/ml) and 8pmol of flap-annealed probe oligos for hTERT (IDT) designed using Oligostan (Tsanov et al., 2016) software, listed in Table S2. Flap-annealing of probe oligos was done as in Tsanov et al. (2016). After hybridization, cells were washed twice in 1X SSC w/ 15% formamide, and twice with 1x PBS, before DNA was counterstained with 5 μg/mL DAPI. Cells were mounted with ProLong Gold Antifade (Thermo Fisher Scientific), imaged on a Nikon Eclipse Ti2 spinning-disc confocal microscope at 60X and analyzed with Nikon software. Additional contrast/brightness enhancement, quantification of foci and export were performed with Fiji-ImageJ software.
Western blot analysis
Cells were harvested by trypsinization, lysed in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% SDS, 1% NP-40, 1% sodium deoxycholate) at about 104 cell/μl. After 2 cycles of water bath sonication at medium settings lysates were incubated at 4°C on a rotator for additional 30 minutes. Lysates were clarified by spinning 30 minutes at 14800 rpm, 4°C and supernatant protein concentration was quantified with Enhanced BCA protocol (Thermo Fisher Scientific, Pierce). Equivalent amounts of proteins were separated on an SDS-page (approximately 30 μg) and transferred to a nitrocellulose membrane. Membranes were blocked in 5% milk in TBST (137 mM NaCl, 2.7 mM KCl, 19 mM Tris Base and 0.1% Tween-20). Incubation with primary antibodies was performed overnight at 4°C. Membranes were washed and incubated with HRP conjugated secondary antibodies at 1:5000 dilution, developed with Clarity ECL (Biorad) and acquired with a ChemiDoc MP apparatus (Biorad). Antibodies against GAPDH were used as loading control. A full list of antibodies used in the study and relative dilutions is available in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of experiments was performed using Graphpad Prism software. The details of the statistical analysis used for each experiment can be found in the figure legends. Significance was defined as a p value < 0.05. Samtools (Li et al., 2009) and Bowtie2 (Langmead and Salzberg, 2012) algorithms were used.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Anti-SON | Novus Biologicals | NBP1-88706 |
| Mouse monoclonal GAPDH (0411) | Santa Cruz Biotech. | sc-47724 |
| Mouse monoclonal SSEA-4 (MC-813–70) | R&D Systems | MAB1435 |
| Mouse monoclonal COL1A1 (3G3) | Santa Cruz Biotech. | sc-293182 |
| Bacterial and virus strains | ||
| Cytotune iPS Sendai Reprogramming kit | ThermoFisher | A16517 |
| One Shot Stbl3 Chemically Competent E. coli | ThermoFisher | C737303 |
| Biological samples | ||
| Patient derived PBMCs | Dr. Savage, NIH | NCT-00027274 |
| Chemicals, peptides, and recombinant proteins | ||
| Y-27632 2HCl | SelleckChem | S1049 |
| Isoginkgetin | Tocris | 6483 |
| Critical commercial assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | E1910 |
| Nano-Glo® Dual-Luciferase® Reporter Assay System | Promega | N1610 |
| DNA Prep Kit, Tagmentation | Illumina | 20018704 |
| Deposited data | ||
| Raw and analyzed sequencing data | This paper | GEO accession: GSE168193 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Raw data at Mendeley | This paper | https://dx.doi.org/10.17632/jfmsvxnpbg.1 |
| Experimental models: cell lines | ||
| Human: BJ Fibroblast | ATCC | CRL-2522 |
| Human: ARPE-19 | ATCC | CRL-2302 |
| Human: HeLa | ATCC | CCL-2 |
| Human: TREx-HeLa | Invitrogen | R71407 |
| Human: H7 ESC | Dr. Lei Bu | WA07 |
| Oligonucleotides | ||
| See Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pRF-HCV | Maria Barna | n/a |
| pNL1.1 | Promega | N1001 |
| pcDNA5/FRT/TO | ThermoFisher | V652020 |
| Software and algorithms | ||
| 4C-ker | Raviram et al., 2016 | https://github.com/rr1859/R.4Cker |
| Oligostan | Tsanov et al., 2016 | https://bitbucket.org/muellerflorian/fish_quant |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
Highlights.
Enhancers have a minor role in regulating telomerase during development
Alternative splicing of exon 2 is a major determinant of hTERT levels
Exon 2 skipping triggers hTERT mRNA decay in somatic cells
SON regulates hTERT splicing, and its mutation manifests in telomerase insufficiency
ACKNOWLEDGMENTS
We thank Ashley S. Thompson for assistance with gene and variant curation and Marion Pouillard and Mike Al-Kareh for technical support. We acknowledge Eros-Lazzerini Denchi and members of the Sfeir lab for comments on the manuscript. We thank Luis Batista for sharing methods for hepatocyte differentiation. We are grateful to the patients, their families, and the referring clinicians for their valuable contributions. Lei Bu, Jerry Shay, and Maria Barna are thanked for reagents. We acknowledge the genome technology core (GTC), high throughput biology (HTB) core, Michael Cammer, and the microscopy core at the NYU School of Medicine. This work was supported in part by a grant from the NYSTEM and Irma T. Hirschl Foundation to A.S., an NIH fellowship to A.P., and an NIH grant (RM1HG009491) to J.D.B. The work in the lab of S.A.S. is supported by the intramural program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH. The authors would like to dedicate this study to the memory of Woodring E. Wright.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.03.033.
DECLARATION OF INTERESTS
A.S. is a co-founder, consultant, and shareholder in Repare Therapeutics. J.D.B. is a founder and director of CDI Labs, Inc., a founder of Neochromosome, Inc., a founder and scientific advisory board (SAB) member of ReOpen Diagnostics, and serves or served on the SABs of Sangamo, Inc., Modern Meadow, Inc., Sample6, Inc., and the Wyss Institute.
REFERENCES
- Akıncılar SC, Khattar E, Boon PL, Unal B, Fullwood MJ, and Tergaonkar V (2016). Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 6, 1276–1291. [DOI] [PubMed] [Google Scholar]
- Alter BP, Giri N, Savage SA, and Rosenberg PS (2018). Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M, and Blackburn EH (2012). The telomere syndromes. Nat. Rev. Genet 13, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 552, 126–131, 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474, 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, and Boeke JD (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, and Hockemeyer D (2015). Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife 4, e07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, and Reddel RR (2007). Protein composition of catalytically active human telomerase from immortal cells. Science 315, 1850–1853. [DOI] [PubMed] [Google Scholar]
- Cong YS, Wen J, and Bacchetti S (1999). The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum. Mol. Genet 8, 137–142. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Jena S, and Levasseur DN (2011). Alternative splicing produces Nanog protein variants with different capacities for self-renewal and pluripotency in embryonic stem cells. J. Biol. Chem 286, 42690–42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018). The Encyclopedia of DNA Elements (ENCODE): data portal update. Nucleic Acids Res. 46 (D1), D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldser DM, and Greider CW (2007). Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, Yu J, et al. (1995). The RNA component of human telomerase. Science 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O’Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, et al. (2011). An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147, 132–146. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, and DePinho RA (1999). Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 97, 515–525. [DOI] [PubMed] [Google Scholar]
- Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, et al. (2013). MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao LY, Armanios M, Strong MA, Karim B, Feldser DM, Huso D, and Greider CW (2005). Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 123, 1121–1131. [DOI] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. (2013). TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961. [DOI] [PubMed] [Google Scholar]
- Hrdlicková R, Nehyba J, and Bose HR Jr. (2012). Alternatively spliced telomerase reverse transcriptase variants lacking telomerase activity stimulate cell proliferation. Mol. Cell. Biol 32, 4283–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, and Garraway LA (2013). Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Longman D, and Cáceres JF (2016). Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, and Jefferson RA (1997). Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum. Mol. Genet 6, 2011–2019. [DOI] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, and Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, Bosch DGM, Bubulya PA, Shashi V, Petrovski S, et al. ; University of Washington Center for Mendelian Genomics; Deciphering Developmental Disorders Study (2016). De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet 99, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Göke J, Sachs F, Jacques PE, Liang H, Feng B, Bourque G, Bubulya PA, and Ng HH (2013). SON connects the splicing-regulatory network with pluripotency in human embryonic stem cells. Nat. Cell Biol 15, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow AT, Wong MS, Robin JD, Batten K, Yuan L, Lai TP, Dahlson N, Zhang L, Mender I, Tedone E, et al. (2018). NOVA1 regulates hTERT splicing and cell growth in non-small cell lung cancer. Nat. Commun 9, 3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallanna SK, and Duncan SA (2013). Differentiation of hepatocytes from pluripotent stem cells. Curr. Protoc. Stem Cell. Biol 26, 1G.4.1–1G.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EJ, Luncsford PJ, and Armanios M (2019). Long telomeres and cancer risk: the price of cellular immortality. J. Clin. Invest 129, 3474–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, and Shay JW (2015). Telomerase repeated amplification protocol (TRAP). Biol. Protoc 5, e1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Crawford J, Brunck ME, Gerhardt DJ, Taft RJ, Nielsen LK, Dinger ME, and Mattick JS (2014). Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat. Protoc 9, 989–1009. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, et al. (1997). hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90, 785–795. [DOI] [PubMed] [Google Scholar]
- Miele A, and Dekker J (2009). Mapping cis- and trans- chromatin interaction networks using chromosome conformation capture (3C). Methods Mol. Biol 464, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LA, Chuang J, Agmon N, Khunsriraksakul C, Phillips NA, Cai Y, Truong DM, Veerakumar A, Wang Y, Mayorga M, et al. (2015). Versatile genetic assembly system (VEGAS) to assemble pathways for expression in S. cerevisiae. Nucleic Acids Res. 43, 6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, and Agarwal S (2015). Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat. Genet 47, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogami K, Richard P, Chen Y, Hoque M, Li W, Moresco JJ, Yates JR 3rd, Tian B, and Manley JL (2017). An Mtr4/ZFC3H1 complex facilitates turnover of unstable nuclear RNAs to prevent their cytoplasmic transport and global translational repression. Genes Dev. 31, 1257–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ST, Kyo S, and Laimins LA (2001). Telomerase activation by human papillomavirus type 16 E6 protein: induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol 75, 5559–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, and Seimiya H (2019). Revisiting telomere shortening in cancer. Cells 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviram R, Rocha PP, Müller CL, Miraldi ER, Badri S, Fu Y, Swanzey E, Proudhon C, Snetkova V, Bonneau R, and Skok JA (2016). 4C-ker: a method to reproducibly identify genome-wide interactions captured by 4C-Seq experiments. PLoS Comput. Biol 12, e1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake CM, and Artandi SE (2020). Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol 21, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau P, Khondaker S, Zhu S, Lauzon C, Mai S, and Autexier C (2016). An intact putative mouse telomerase essential N-terminal domain is necessary for proper telomere maintenance. Biol. Cell 108, 96–112. [DOI] [PubMed] [Google Scholar]
- Sayed ME, Yuan L, Robin JD, Tedone E, Batten K, Dahlson N, Wright WE, Shay JW, and Ludlow AT (2019). NOVA1 directs PTBP1 to hTERT pre-mRNA and promotes telomerase activity in cancer cells. Oncogene 38, 2937–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Markey M, Torres-Muñoz K, Varia S, Kadakia M, Bubulya A, and Bubulya PA (2011). Son maintains accurate splicing for a subset of human pre-mRNAs. J. Cell Sci 124, 4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, and Wright WE (2011). Role of telomeres and telomerase in cancer. Semin. Cancer Biol 21, 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, and de Laat W (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet 38, 1348–1354. [DOI] [PubMed] [Google Scholar]
- Stern JL, Theodorescu D, Vogelstein B, Papadopoulos N, and Cech TR (2015). Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 29, 2219–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Tan TC, Rahman R, Jaber-Hijazi F, Felix DA, Chen C, Louis EJ, and Aboobaker A (2012). Telomere maintenance and telomerase activity are differentially regulated in asexual and sexual worms. Proc. Natl. Acad. Sci. U S A 109, 4209–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh CX, Chan JW, Chong ZS, Wang HF, Guo HC, Satapathy S, Ma D, Goh GY, Khattar E, Yang L, et al. (2016). RNAi reveals phase-specific global regulators of human somatic cell reprogramming. Cell Rep. 15, 2597–2607. [DOI] [PubMed] [Google Scholar]
- Tokita MJ, Braxton AA, Shao Y, Lewis AM, Vincent M, Küry S, Besnard T, Isidor B, Latypova X, Bézieau S, et al. (2016). De novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. Am. J. Hum. Genet 99, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M, et al. (2016). smiFISH and FISH-quant—a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res. 44, e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulaner GA, Hu JF, Vu TH, Giudice LC, and Hoffman AR (1998). Telomerase activity in human development is regulated by human telomerase reverse transcriptase (hTERT) transcription and by alternate splicing of hTERT transcripts. Cancer Res. 58, 4168–4172. [PubMed] [Google Scholar]
- van de Werken HJ, de Vree PJ, Splinter E, Holwerda SJ, Klous P, de Wit E, and de Laat W (2012). 4C technology: protocols and data analysis. Methods Enzymol. 513, 89–112. [DOI] [PubMed] [Google Scholar]
- Venables JP, Lapasset L, Gadea G, Fort P, Klinck R, Irimia M, Vignal E, Thibault P, Prinos P, Chabot B, et al. (2013). MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun 4, 2480. [DOI] [PubMed] [Google Scholar]
- Venteicher AS, Meng Z, Mason PJ, Veenstra TD, and Artandi SE (2008). Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132, 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick M, Zubov D, and Hagen G (1999). Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT). Gene 232, 97–106. [DOI] [PubMed] [Google Scholar]
- Withers JB, Ashvetiya T, and Beemon KL (2012). Exclusion of exon 2 is a common mRNA splice variant of primate telomerase reverse transcriptases. PLoS ONE 7, e48016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CW, Hou PS, Tseng SF, Chien CL, Wu KJ, Chen HF, Ho HN, Kyo S, and Teng SC (2010). Krüppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 28, 1510–1517. [DOI] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, and Shay JW (1996). Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet 18, 173–179. [DOI] [PubMed] [Google Scholar]
- Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J, and Dalla-Favera R (1999). Direct activation of TERT transcription by c-MYC. Nat. Genet 21, 220–224. [DOI] [PubMed] [Google Scholar]
- Xi L, and Cech TR (2014). Inventory of telomerase components in human cells reveals multiple subpopulations of hTR and hTERT. Nucleic Acids Res. 42, 8565–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Liu L, Lazarev D, Al-Zain A, Fomin V, Yeung PL, Chambers SM, Lu CW, Studer L, and Manley JL (2018a). TCF3 alternative splicing controlled by hnRNP H/F regulates E-cadherin expression and hESC pluripotency. Genes Dev. 32, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, and Hirose T (2018b). Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell 70, 1038–1053.e7. [DOI] [PubMed] [Google Scholar]
- Yi X, Shay JW, and Wright WE (2001). Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 29, 4818–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Rousseau P, Lauzon C, Gandin V, Topisirovic I, and Autexier C (2014). Inactive C-terminal telomerase reverse transcriptase insertion splicing variants are dominant-negative inhibitors of telomerase. Biochimie 101, 93–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 4C and ATAC-seq datasets generated during this study are available at GEO: GSE168193. Raw data available at Mendeley: https://dx.doi.org/10.17632/jfmsvxnpbg.1