

Abstract
Background & Aims:
Achieving HBsAg loss is an important landmark in the natural history of chronic hepatitis B. A more personalized approach to prediction of HBsAg loss is relevant in couseling patients. This study sought to develop and validate a prediction model for HBsAg loss based on quantitative HBsAg levels and other baseline characteristics.
Methods:
Hepatitis B Research Network (HBRN) is a prospective cohort including 1240 untreated HBeAg-negative patients (1150 adults, 90 children) with median follow-up of 5.5 years. Incidence rates of HBsAg loss and anti-HBs acquisition were determined and a predictor score of HBsAg loss using readily available variables was developed and externally validated.
Results:
Crude incidence rates of HBsAg loss and anti-HBs acquisition were 1.6 and 1.1 per 100 person-years (PY); 67 achieved sustained HBsAg loss for an incidence rate of 1.2 per 100 PY. Increased HBsAg loss was significantly associated with older age, non-Asian race, HBV phenotype (inactive carrier vs others), HBV genotype A, lower HBV DNA levels and lower and greater change in quantitative HBsAg (ΔqHBsAg). The HBRN-SQuARe (sex,ΔquantHBsAg, age, race) score predicted HBsAg loss over time with AUROC (95% confidence intervals) at 1 and 3 years of 0.99 (95% CI: 0.987–1.00) and 0.95 (95% CI 0.91–1.00), respectively. Validation in another cohort of 1253 HBeAg-negative patients with median follow-up of 3.1 years, HBRN-SQuARe predicted HBsAg loss at 1 and 3 years with AUROC values of 0.99 [0.98–1.00] and 0.88 [0.77–0.99], respectively.
Conclusion:
HBsAg loss in predominantly untreated patients with HBeAg-negative chronic hepatitis B can be accurately predicted over a 3-year horizon using a simple validated score (HBRN-SQuARe). This prognostication tool can be used to support patient care and counseling.
Keywords: functional cure, HBeAg, anti-HBe, HBV genotype, seroreversion
Introduction
Functional cure of chronic hepatitis B (CHB) is defined as a sustained loss of hepatitis B surface antigen (HBsAg) and hepatitis B virus DNA (HBV DNA) from serum, with or without hepatitis B surface antibody (anti-HBs) seroconversion1. The importance of achieving functional cure is emphasized by the lower rates of liver complications, including hepatocellular carcinoma (HCC), in patients who have lost HBsAg versus those with sustained HBV DNA suppression but persistence of HBsAg2. Patients who achieve a functional cure remain at risk for spontaneous or immunosuppressive therapy-induced-HBV reactivation3. Complete cure, with eradication of integrated HBV DNA and covalently closed circular DNA (cccDNA), is the more desirable objective, but largely beyond reach with currently approved therapies1. Thus, functional cure is generally regarded as the optimal outcome for patients with CHB and is the primary endpoint for many of the novel therapies under development.
Recent systematic reviews highlighted the relative rarity of HBsAg loss among persons with CHB estimating the pooled annual incidence of spontaneous HBsAg loss at ~1% per year4,5. Comparison of treated cohorts suggests peg-interferon alone or in combination with NA may enhance HBsAg loss to 3–8% at end of study 6–8 but overall, the rates of HBsAg loss are low. These data emphasize both the need for better therapies to enhance rates of HBsAg loss as well as the need for prognostic models to better estimate the trajectory of HBsAg loss. The factor most frequently associated with loss of HBsAg is quantitative levels of HBsAg, but the specific threshold and factors influence rate of HBsAg loss are not well established 9.
Much of what is known about the natural history of CHB has emerged from Asian or European cohorts, with much more limited data from North American CHB patients. While CHB in North America is primarily found among immigrant populations10–12 the existent data from other countries on CHB outcomes may not be applicable. Differing epidemiologic and environmental factors, liver disease comorbidities, access to treatment and monitoring may influence the natural history of CHB and likelihood of achieving HBsAg loss, thereby stimulating the need to study these key questions in a representative North American population with CHB.
As HBeAg loss precedes HBsAg loss, patients with HBeAg-negative CHB are the prime group to evaluate predictors of HBsAg loss or functional cure. The Hepatitis B Research Network (HBRN), a North American, prospective, multiethnic, multicenter cohort of children and adults provided opportunity to determine the incidence of key events in the natural history of HBeAg-negative CHB and to develop and validate a prognostic score for HBsAg loss over a several year time span.
Methods:
Study Population:
The HBRN is a collaborative network including 28 clinical sites (21 adult, 7 pediatric) from diverse regions in the U.S. and Canada that prospectively followed participants with CHB from January 2011 to December 2019 funded by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, NCT01263587) 13,14. At enrollment, all participants were HBsAg-positive and not currently receiving treatment. Those with hepatic decompensation, liver cancer, solid organ or bone marrow transplantation, human immunodeficiency virus infection or active hepatitis C or D coinfections were excluded. Participants with less than 24 weeks of follow-up from baseline were also excluded. Treatment after enrollment occurred as per standard of care at individual sites or through an HBRN clinical trial. Follow-up visits and laboratory testing were completed at week 12 after enrollment and then every 24 weeks thereafter in adults, and at 24 weeks and 48 weeks and annually thereafter in children. For this analysis, all HBsAg-positive adult and pediatric participants whose first available HBeAg test showed a negative result, irrespective of the presence of anti-HBe, were included. Historical data on HBeAg status (prior to enrollment) was not captured. HBRN phenotypes were defined as previously published (15).
The HBRN Steering Committee approved all study protocols. Institutional review board or ethics committee approval was obtained from all clinical sites. All participants or their parents/guardians provided written informed consent.
Definitions and Outcomes
The primary outcome for this analysis was HBsAg loss. HBsAg loss was categorized as transient or sustained, with the latter defined as having persistently negative results for 6 months or more. Additional outcomes included hepatitis B surface antibody (anti-HBs) seroconversion, HBeAg seroreversion, alanine aminotransferase (ALT) flares, and new onset of cirrhosis. If there were missing data between two timepoints, we assumed that the outcome had not changed; in other words, the outcome at the first timepoint was carried forward to the second timepoint with an outcome.
Anti-HBs seroconversion was categorized as transient and sustained, with the latter defined as having persistently detectable anti-HBs positivity for 6 months or more. HBeAg seroreversion was defined as transient or sustained, with the latter defined by detectable HBeAg measured at least 6 months apart without any intervening negative tests. ALT flares were defined as ALT level ≥10 × the upper limit of the normal range (ULN). The ULN for adult men and women were 30 U/L and 20 U/L15, respectively, and for children were 33 U/L for male and female infants <1 year old; 25 U/L for male and female children 1 year to <13 years old; and 25 U/L for males and 22 U/L for female between 13 years and <18 years old16. New onset cirrhosis was based on clinical, radiologic (splenomegaly or a nodular liver), laboratory (platelet count <120,000 mm3) and histologic data and a cirrhosis diagnosis within 6 months of cohort entry was considered to be present at baseline. All outcomes were reviewed and confirmed by an adjudication committee17.
Laboratory testing
HBsAg and HBeAg quantitation, qualitative anti-HBs and HBV DNA level were measured centrally at an HBRN-funded virology laboratory (University of Washington, Seattle, WA). Additional qualitative anti-HBs was performed at the University of Texas Southwestern. Local laboratory results were used to supplement missing values. HBV DNA was tested using a real-time polymerase chain reaction assay (ROCHE COBAS Ampliprep/COBAS Taqman HBV Test, v2.0) with lower limit of detection of 10 IU/mL. Quantitative HBsAg and HBeAg levels were measured by Elecsys HBsAg II Quant and Elecsys HBeAg II Quant assay (Roche Molecular Systems, Inc) with lower limits of detection of 0.05 and 0.3 IU/mL, respectively. Central testing of qualitative anti-HBs used the FDA approved EIA assay (ETI-AB-AUK PLUS, DiaSorin, Italy) with values ≥10 mIU/mL being considered positive. Qualitative assays for HBsAg, anti-HBs, HBeAg and hepatitis B e antibody (anti-HBe) were performed locally using commercially available ELISA assays. Genotyping of HBV was performed by the Centers for Disease Control and Prevention using mass spectrometry18. Precore (PC) and basal core promoter (BCP) variants were determined by Sanger sequencing19. All other laboratory results were obtained locally. Hyperlipidemia and diabetes were self-reported clinical diagnoses or use of diagnosis-specific medications. AST-to-platelet ratio index (APRI) was calculated as: (AST/upper limit of normal)/platelet count (expressed as platelets × 109/L) × 10020.
Statistical Analyses
The baseline categorical variables of participants were summarized using frequencies and percentages, and continuous variables summarized with medians and interquartile ranges. Duration of follow-up for each individual was determined by the time between baseline and the last follow-up visit. Rates of clinical outcomes were calculated and expressed per 100-person-years (PY). The confidence intervals for PY rates were calculated using Poisson regression (log-linear regression for rates with log follow-up time as offset).
Poisson regression with log of the follow-up time as offset was used to assess the association between baseline characteristics and incidence rates of HBsAg loss with results expressed as rate ratios (IRR) and corresponding 95% confidence intervals or adjusted rate ratios (ARR) when adjusted for other covariates in the model. The Poisson regression was preferred to Cox regression as follow-up were not as frequent. Each variable was evaluated in univariable and then multivariable Poisson regression models. Since genotype and race were highly correlated, multivariable models only included race. The receipt of ≥24 weeks of antiviral therapy during follow-up was evaluated as a time-varying covariate. Sensitivity analysis was conducted with censoring of participants at the initiation of treatment. A similar approach was used for the analysis of anti-HBs seroconversion rates.
Prediction Model Development and Validation
A parametric Weibull regression model with time to HBsAg loss as the outcome was fit using age, sex, race, qHBsAg, qHBsAg change in a year (at max 3 measurements within 48 weeks), and their interaction. A parametric regression model over semi-parametric Cox proportional hazard model was chosen considering the small number of events (HBsAg loss) over time. The variables selected were pre-specified with a focus on qHBsAg and its change over a period of one year as predictive markers in addition to age, sex, and race (surrogate for genotype) which are other known predictors of HBsAg loss. The pre-specification of covariates were done to prevent model over-fitting due to small number of events and to mostly evaluate the predictive ability of qHBsAg and its change. For the same reasons, no model calibration was considered. The model including qHBsAg change per year as a predictor (hereafter referred to as HBRN-SQuARe) required participants to have more than one year of follow-up with at least two measurements of qHBsAg during the first year. For each participant, the qHBsAg change in the first year was calculated as the slope of a simple linear regression model of qHBsAg on time during the first year, meaning that if a participant had only two measurements during this period the resulting change could be calculated by dividing their difference by the time between the two measurements. For this model formulation, “time origin” to calculate time to HBsAg loss was taken to be the time of last qHBsAg measurement within the first year. Age was treated as a categorical variable to provide predictions for specific age groups. Additional models evaluated HBV genotype (B/C versus others) in lieu of race and a single baseline HBsAg. For models that did not include qHBsAg change, the time origin was the baseline visit.
Missing covariate data were treated as missing completely at random whereas the censoring was assumed independent of observed data. This implied that the likelihood-based estimation in models such as that in the Poisson and Weibull regression were consistent. The goodness of fit for the models was investigated using the time-dependent area-under-the-receiver-operating-characteristics curve (AUROC) values as well as the sensitivity and specificity values. We specifically evaluated the AUROCs at years 1, 2, and 3 since the time origin. AUROC values from various models were compared using Wald-type tests. An additional sensitivity analysis, excluded the 131 participants who had any prior history of HBV treatment. Finally, the HBRN-SQuARe score was externally validated on an HBeAg-negative cohort of 1253 patients (51 of whom lost HBsAg) from the Toronto Centre for Liver Disease with similar inclusion criteria to the HBRN cohort.
The model HBRN-SQuARe was applied to the validation cohort to calculate the predicted probability of HBsAg loss at years 1, 2, and 3. Model performance was assessed using the time-dependent AUROC, similar to the development process.
All analyses were conducted using SAS 9.4 (SAS Institute Inc., Cary, NC.) and R version 3.5.3 (R Core Team, 2013). All tests reported were two-sided and p-values less than 0.05 were considered statistically significant. All authors had access to the study data and reviewed and approved the final manuscript.
Results
Cohort characteristics
Of the 2458 HBsAg-positive participants at entry, 1408 met initial eligibility criteria for this study. Following exclusion of those HBeAg-negative participants with less than 24 weeks of follow-up from baseline, a total of 1240 were eligible for the longitudinal analytic sample - 1150 adults and 90 children (sFigure 1). All participants had at least 2 and at maximum 18 follow-up visits, with the majority of participants (79.4%) had 5 or more follow-up visits. The median duration of follow-up was 5.5 years and 17.1% of participants had 8 years or more of follow-up. Of the 1057 participants who reached one year of follow-up, 7.9% had initiated HBV therapy within the prior 24 weeks, and at 5 years of follow-up, 21.1% (125/588) had initiated antiviral therapy.
The baseline characteristics of the study cohort are summarized in Table 1. The mean age of children was 12 years and of adults was 43 years. Children and young adults (<30 years of age) represented 17.7% of the participants, while 11.9% were older than age 60. The majority (69.9%) of participants were of Asian ethnicity with near equal representation of males and females. With respect to virological and laboratory characteristics, the median HBV DNA was 3.2 log10 IU/mL, 96.6% were anti-HBe positive (96.4% of these were consistently positive), and 34.2% had normal ALT levels. With respect to the phase of HBV infection, 24.7% met the HBRN criteria for immune active CHB, 31.1% were inactive carriers and 44% of indeterminant phenotype15,16. Genotypes B (40.2%) and C (27.5%) were most prevalent followed by genotype A (18.6%); 71.7% of those tested (n=628) had evidence of PC/BCP variants. The median HBsAg level at baseline was 3.2 log10 IU/mL (interquartile range [IQR] 2.8 to 3.8). Only 3% of the population had evidence of advanced fibrosis or cirrhosis at entry with APRI scores >1.5.
Table 1.
Baseline characteristics of eligible adult and children with HBeAg-negative CHB
| Baseline Characteristics | N, Value |
|---|---|
|
| |
| Age (yrs) | n=1240 |
| <18 | 90 (7.3%) |
| 18-<30 | 130 (10.5%) |
| 30-<50 | 619 (49.9%) |
| 50-<60 | 254 (20.5%) |
| >=60 | 147 (11.9%) |
|
| |
| Female | 610 (49.2%) |
|
| |
| Race | n=1236 |
| White | 154 (12.5%) |
| Black | 185 (15.0%) |
| Asian | 864 (69.9%) |
| Other/Mixed | 33 (2.7%) |
|
| |
| Phenotype * | n=1176 |
| HBeAg- CHB | 290 (24.7%) |
| Inactive carrier | 366 (31.1%) |
| Indeterminant (IND) | 520 (44.2%) |
| IND B | 465 (89.4%) |
| IND C | 55 (10.6%) |
|
| |
| HBV Genotype | n=1096 |
| A | 204 (18.6%) |
| B | 441 (40.2%) |
| C | 301 (27.5%) |
| D | 109 (9.9%) |
| Other (E, F, and multiple) | 41 (3.8%) |
|
| |
| Quantitative HBsAg (log10 IU/mL) | n=1110 |
| Median(25th:75th) | 3.2 (2.5: 3.8) |
| < 2 | 159 (14.3%) |
| 2 – 3 | 299 (26.9%) |
| > 3 | 652 (58.7%) |
|
| |
| Anti-HBe | n=1036 |
| Positive | 1001 (96.6%) |
|
| |
| HBV DNA (log10 IU/mL) | n=1237 |
| Median(25th:75th) | 3.2 (2.4: 4.3) |
| <3 | 549 (44.4%) |
| 3 - < 4 | 329 (26.6%) |
| 4 - < 5 | 187 (15.1%) |
| ≥ 5 | 172 (13.9%) |
|
| |
| ALTxULN | n=1216 |
| Median(25th:75th) | 1.3 (0.9: 1.8) |
| ≤1 ULN | 416 (34.2%) |
| 1< - ≤2 ULN | 558 (45.9%) |
| > 2 ULN | 242 (19.9%) |
|
| |
| Platelets (x103/mm3) | n=1044 |
| Median(25th:75th) | 222.0 (182.5: 262.0) |
|
| |
| APRI (AST-platelet-ratio index) | n=1035 |
| Median(25th:75th) | 0.3 (0.2: 0.4) |
| ≤0.50 | 831 (80.3%) |
| >0.50–1.50 | 173 (16.7%) |
| >1.50 | 31 (3.0%) |
|
| |
| BCP/PC Mutation (A1762T, G1764A, G1896A) | n=628 |
| Wild type | 178 (28.3%) |
| BCP only | 152 (24.2%) |
| PC only | 174 (27.7%) |
| BCP & PC | 124 (19.7%) |
|
| |
| Prior HBV treatment | n=131 (10.6%) |
| Median(25th:75th) year-to-entry | 5.0 (1.3, 9.0) |
| Interferon | 29 (17.9%) |
| Entecavir | 23 (14.2%) |
| Telbivudine | 6 (3.7%) |
| Lamivudine | 48 (29.6%) |
| Adefovir | 13 (8.0%) |
| Peg-interferon | 19 (11.7%) |
| Tenofovir | 10 (6.2%) |
| Unknown | 14 (8.6%) |
as per HBRN phenotype definitions (15). Inactive CHB defined by HBeAg-negative, normal ALT and HBV DNA ≤10,000 IU/mL, IND-B by HBeAg-negative, elevated ALT and HBV DNA ≤10,000 IU/mL and IND-C by HBeAg-negative, normal ALT and HBV DNA >10,000 IU/mL. Normal ALT was <30 U/L for males and <20 U/L for females.
Incidence of Clinical Outcomes
Clinical outcomes, including key serologic events are summarized in Table 2. Among the 1240 HBeAg-negative participants, 92 ever became HBsAg negative, for a crude incidence rate of 1.6 per 100 person-years (PY), of whom 71 met criteria for sustained HBsAg loss for an incidence rate of 1.2 per 100 PY. The time course for HBsAg loss among those who ever lost HBsAg is shown in Figure 1. Baseline characteristics of participants with and without sustained HBsAg loss is shown in sTable 1.
Table 2.
Summary of clinical outcomes
| Outcomes1 | Post baseline | |||
|---|---|---|---|---|
| Number of events | Number of events within 24 weeks of baseline | Median Follow-up Time in Weeks (min, max) | Incidence (per 100 PY, 95% CI)2 | |
| Ever HBsAg negative 1 | 92 | 0 | 211 (25, 384) | 1.6 (1.3, 2.0) |
| Sustained HBsAg negative 3 | 71 | 0 | 194 (25, 345) | 1.2 (1.0, 1.6) |
| Ever HBeAg seroreversion | 25 | 10 | 26 (0.4, 362) | 0.4 (0.3, 0.6) |
| Sustained HBeAg seroreversion 3 | 8 | 4 | 25 (20, 360) | 0.1 (0.1, 0.3) |
| Ever anti-HBs positive 4 | 63 | 1 | 193 (23, 399) | 1.4 (1.1, 1.8) |
| Sustained anti-HBs positive 3,4 | 33 | 0 | 148 (40, 338) | 0.7 (0.5, 1.0) |
| Initiation of any HBV treatment 5 | 291 | 63 | 92 (0.1, 376) | 5.9 (5.2, 6.6) |
| New onset of ALT flare 6 | 44 | 8 | 52 (1, 313) | 0.8 (0.6, 1.0) |
| New onset of Cirrhosis | 23 | 10 | 29 (1, 167) | 0.4 (0.3, 0.6) |
Includes transient and sustained HBsAg loss.
PY= person-year. Incidence estimate and 95% CI were obtained from Poisson regression.
Defined as confirmed first HBsAg(−)/HBeAg(+)/anti-HBs(+) measurement ≥24 weeks apart without any HBsAg(+)/HBeAg(−)/anti-HBs(−) measurement in between.
Among the 1240 HBeAg negative participants, 926 (74.7%) had negative anti-HBs at study baseline and are eligible for anti-HBs analysis.
Only NA therapies were given. None were treated with peg-IFN.
ALT flare is defined as ≥10 × ULN
Figure 1: Evolution of HBsAg status amongst participants with HBsAg loss during study years.

Each participant’s follow-up is shown as a row, with the time of follow-up on the x-axis. Each time interval is represented by a box: Pink boxes indicate HBsAg is positive, Light green boxes indicate HBsAg is negative and Grey boxes indicate missing test results. Further, the participants are grouped into 3 broad categories indicated by the colored bar on the far left: with those with sustained HBsAg loss (BLACK bar), transient HBsAg loss (BLUE bar) and insufficient follow-up (ORANGE bar). The majority of (67/92) participants had sustained HBsAg loss.
A total of 926 (74.7%) were tested and negative for anti-HBs at study baseline and eligible for an evaluation on anti-HBs development. The remainder were not tested within the window used to define baseline. Of the 926 with testing at baseline, 594 (64.1%) had at least one anti-HBs test in follow-up (median number of tests =1, IQR 0 to 5). Sixty-three ever became anti-HBs positive for a crude incidence rate of 1.4 per 100 PY, and it was sustained in 33, for an incidence rate of 0.7 per 100 PY. The time course for anti-HBs seroconversion among those who ever lost HBsAg is shown in sFigure 2. Of the 92 participants with HBsAg loss, 24 (26.1%) participants had anti-HBs detected at the same (n=19) or an earlier time point (n=5), 25 (27.1%) following HBsAg loss (11 within one year and 14 beyond one year of HBsAg loss) and the remainder were without anti-HBs (n=37) or testing (n=6) at last follow-up. In a time to event analysis restricted to patients with anti-HBs testing at least annually after HBsAg loss, the estimated median time between HBsAg loss and anti-HBs detection was approximately 1.5 years (95% CI: 0.7 – 2.2).
Cirrhosis developed in 23 participants for an incidence rate of 0.4 per 100 PY. As 10 occurred within the first 24 weeks of cohort entry and thus were likely present at baseline, excluding these resulted in an incidence rate of de novo cirrhosis of 0.2 per 100 PY (95% CI 0.1–0.4). No participant with HBsAg loss progressed to cirrhosis. ALT flares, defined as a serum ALT ≥10X ULN, were observed in 44 participants for an incidence rate of 0.8 per 100 PY. Only one participant had an ALT flare prior to HBsAg loss (11 months before). Baseline characteristics associated with ALT flares were HBeAg-negative CHB and indeterminant phenotypes (versus inactive CHB) (p<0.001), higher HBV DNA levels (p<0.001) and lower platelet count (p=0.04). Ever experiencing HBeAg seroreversion was observed in 25 participants for an incidence rate of 0.4 per 100 PY and was sustained in only 6 (0.1 per 100 PY); 8 (32%) were anti-HBe negative at baseline. Seroreversion was not associated with receipt of immunosuppressive therapy. Two hundred and ninety-one HBeAg negative participants started treatment over the duration of follow-up (5.9 per 100 PY).
Baseline and Time-Varying Factors Associated with HBsAg Loss and Seroconversion
The crude incidence rate of ever HBsAg loss varied by baseline characteristics (sTable 2 and Figure 2). In Poisson regression analysis, the incidence rate ratios for HBsAg loss were significantly associated with increasing age, non-Asian race, baseline HBV phenotype (inactive carrier versus others), HBV genotype, and lower quantitative HBsAg and HBV DNA (Table 3). Those older than 60 years had an almost 80% higher rate of HBsAg loss compared to those younger than 30 years (p=0.001). Participants of White and Black race had a 37% higher rate of HBsAg loss compared to Asians (p=0.03). Participants with an inactive CHB phenotype had the highest rate of HBsAg loss compared to participants with other HBV phenotypes (p<0.001). Rate ratios of HBsAg loss were significantly lower in those with high qHBsAg and high HBV DNA levels with the rate declining by 75% for each log10 increase in qHBsAg level (p<0.001) and by 65% for each log10 increase in HBV DNA level (p<0.001). Treatment for a period longer than 24 weeks as time-varying covariate was associated with a 66% lower rate of HBsAg loss compared to not starting treatment (p=0.02).
Figure 2: Crude incidence rate (per 100 person-years) for ever HBsAg loss by selected baseline characteristics.
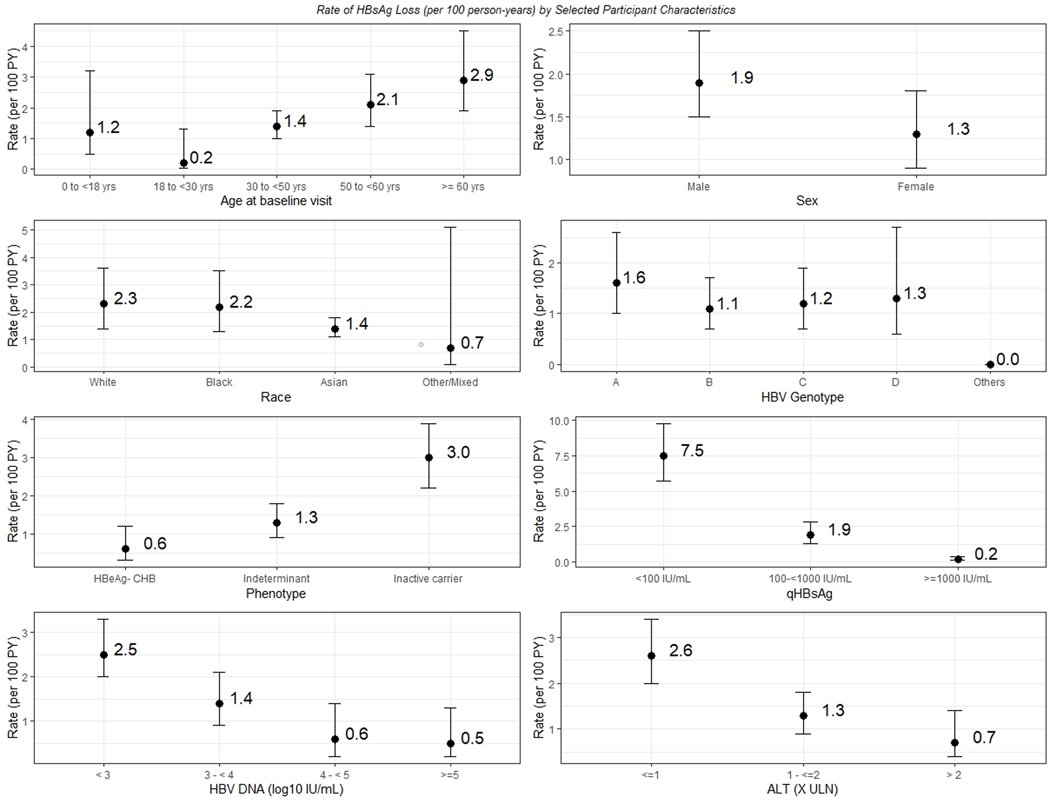
Participants with ever HBsAg loss differed by baseline characteristics. Crude incidence rates by key baseline factors are shown: Age, Sex, Race, HBV genotype, CHB phenotype, ALT, HBV DNA and HBsAg quantitation are shown.
Table 3.
Association between patient characteristics and incidence of first HBsAg loss expressed as rate ratios (RRs)1
| N | Unadjusted RR (95% CI) | P Value | N | Adjusted RR (95% CI)2 | P value | |
|---|---|---|---|---|---|---|
| Baseline Characteristics | ||||||
| Age, years (ref>=60) | 1240 | 0.01 | ||||
| <18 | 0.41 (0.14, 1.16) | |||||
| 18 to <30 | 0.06 (0.01, 0.45) | |||||
| 30-<50 | 0.47 (0.28, 0.79) | |||||
| 50-<60 | 0.71 (0.40, 1.24) | |||||
| Female sex | 1240 | 0.67 (0.45, 1.02) | 0.06 | |||
| Asian (Ref: Non-Asian) 3 | 1203 | 0.63 (0.41, 0.95) | 0.03 | |||
| Phenotype (Ref: Inactive carrier) | 1176 | <0.001 | 1143 | <0.001 | ||
| HBeAg- CHB | 0.22 (0.11, 0.44) | 0.24 (0.12, 0.49) | ||||
| Indeterminant | 0.43 (0.27, 0.66) | 0.44 (0.28, 0.69) | ||||
| Non-A Genotype 4 | 1055 | 0.72 (0.41, 1.27) | 0.26 | 1055 | 0.77 (0.43, 1.35) | 0.36 |
| BCP/PC mutation (Ref: wild type) | 628 | 0.07 | 609 | 0.11 | ||
| BCP only | 0.41 (0.15, 1.11) | 0.42 (0.15, 1.16) | ||||
| PC only | 0.27 (0.09, 0.81) | 0.29 (0.10, 0.87) | ||||
| BCP & PC | 0.72 (0.30, 1.76) | 0.63 (0.23, 1.74) | ||||
| Time-Varying Variables | ||||||
| HBV treatment (≥24 wks in past 36 wks) | 1240 | 0.34 (0.14, 0.84) | 0.02 | |||
| Quantitative HBsAg per 1 log10 IU/mL increase | 1219 | 0.25 (0.18, 0.35) | 0.001 | 1182 | 0.24 (0.18, 0.32) | <0.001 |
| HBV DNA per 1 log10 IU/mL increase | 1240 | 0.35 (0.28, 0.43) | <0.001 | 1203 | 0.22 (0.16, 0.30) | <0.001 |
| ALT per 1x ULN increase | 1234 | 0.56 (0.31, 1.02) | 0.06 | 1197 | 0.63 (0.36, 1.11) | 0.11 |
| Platelet per 103/mm3 increase | 1185 | 1.002 (0.998, 1.01) | 0.03 | 1150 | 1.003 (0.999, 1.01) | 0.11 |
The ratio of the rate at which participants experienced HBsAg loss where a lower rate suggests a longer time to HBsAg negative. Thus, a value <1 indicates longer time to HBsAg loss.
Each estimate is from a unique model adjusting for age group, sex, race, and treatment with one exception; genotype is not adjusted for race. For adjusted model, age was regrouped to combine all <30 in one group because of lack of HBsAg loss in lower age groups [Supplemental sTable 1].
Does not include “Other/Mixed” race (n=33).
Does not include “Other (E, F, and multiple)” genotype (n=41).
The relationships between HBsAg loss and age, race, HBV phenotype, qHBsAg and HBV DNA, were robust and remained significant in adjusted models (Table 3). Male sex, which was not associated with HBsAg loss in unadjusted analysis, became statistically significant after controlling for age, race and treatment. Conversely, AST and APRI were no longer significantly associated with HBsAg loss after controlling for age, race, sex and treatment. HBsAg loss was not significantly associated with HBV genotype, presence of BCP/PC mutation, serum ALT and platelet count in adjusted models.
The crude incidence rate of ever developing anti-HBs varied by baseline characteristics is shown in sTable 3. In Poisson regression analysis, the incidence rate ratios for anti-HBs acquisition were significantly higher among those older than 60 years, with low baseline qHBsAg and low HBV DNA level (sTable 4).
Prediction of HBsAg Loss
Two models were evaluated using age, sex, race, and either a single qHBsAg value or rate of qHBsAg change (minimum 2 and maximum 3 measurements) over a year. The median (IQR) time between qHBsAg measurements was 48.9 (46.6,51.7) weeks. Based on 1077 HBeAg-negative participants, a model using age, sex, race, and a single qHBsAg value at baseline was developed to predict HBsAg loss over time (Table 4, Model 1). The AUROC for predicting loss of HBsAg at 1 year was 0.95 (95% CI: 0.91– 0.99), at 2 years was 0.94 (95% CI: 0.91–0.97), and at 3 years was 0.94 (95% CI: 0.92 – 0.97). The second model with the rate of qHBsAg change (minimum 2 and maximum 3 measurements) over a year as well as the baseline age, sex, race and baseline qHBsAg was based on total of 739 individuals with 56 achieving HBsAg loss (Table 4, Model 2). The AUROC for predicting loss of HBsAg at 1 year was 0.99 (95% CI: 0.987–1.00), at 2 years was 0.96 (95% 0.89–1.00) and at 3 years was 0.95 (95% CI 0.91–1.00) and was significantly better than model 1 in the first year (p = 0.01). We refer to this as the HBRN-SQuARe (Sex, ΔQuantitave HBsAg, Age, Race) model. The ROC curves for models 1 and 2 for years 1–3 are given in Figure 3. Inclusion of an interaction term for baseline qHBsAg and change in HBsAg did not improve model prediction. Graphical depiction of the predicted probability of HBsAg loss in representative patient groups are shown in Figure 4.
Table 4.
Model for predicting log time to HBsAg loss based on one-time measure of quantitative HBsAg level at baseline, age, race, and sex [Model 1; n = 1077, event=80] and one-year change in qHBsAg level, age, race, and sex [Model 2; n = 739, event=56].
| Model 1 | Model 2 (HBRN SQuARe) | |||
|---|---|---|---|---|
| Parameter | Estimate (SE) | p-value | Estimate (SE) | p-value |
| Intercept | 1.11(0.17) | <.0001 | 1.94(0.24) | <.0001 |
| Age (years) [Ref: >=60 years] | ||||
| < 30 years | 1.11(0.58) | 0.05 | 0.65(0.44) | 0.14 |
| 30 - < 50 | −0.03(0.16) | 0.86 | −0.18(0.17) | 0.29 |
| 50 - <60 | −0.08(0.17) | 0.64 | −0.47(0.17) | 0.01 |
| Sex (Female vs. Male) | 0.41(0.14) | 0.004 | 0.33(0.13) | 0.004 |
| Race (Asian vs. Others) | 0.65(0.14) | <.0001 | 0.12(0.15) | <.0001 |
| qHBsAg (log 10 IU/ml) baseline | 0.55(0.06) | <.0001 | 0.37(0.07) | <.0001 |
| Rate of change in qHBsAg (log 10 IU/mL/year) | 1.26(0.18) | <.0001 | ||
| Interaction (qHBsAg * rate of change in qHBsAg) | −0.25(0.08) | <.0001 | ||
| Akaike Information Criterion (AIC) | 462.19 | 258.71 | ||
Estimates refer to mean change in log time to HBsAg loss with respect to one unit change in continuous variable, and the mean difference in log time to HBsAg loss between comparison and reference groups. Thus, a positive coefficient indicates longer time to HBsAg loss, or reduced likelihood of HBsAg loss.
Figure 3: ROC curves for predicting HBsAg loss using HBRN SQuAre Models.
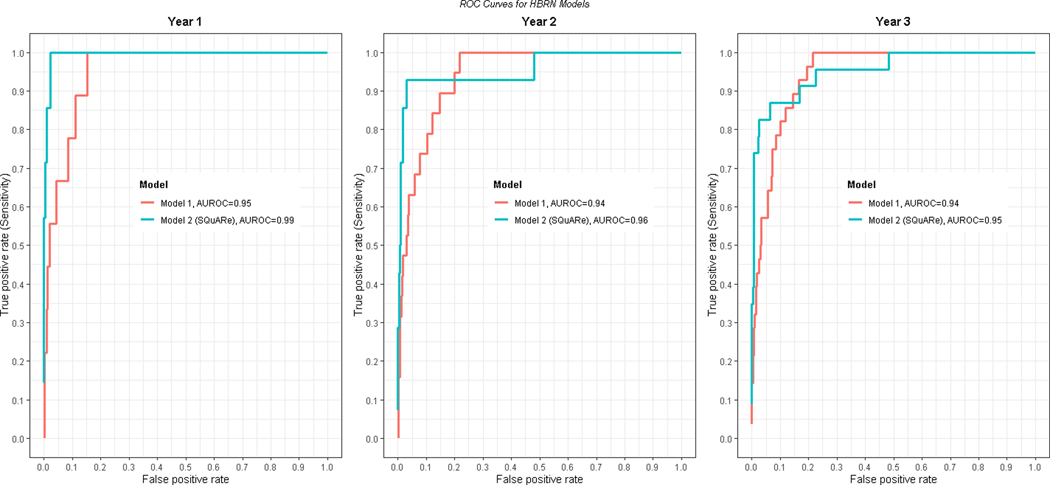
The SQuARe model can estimate predicted probability of HBsAg loss over time (https://abduswahed.shinyapps.io/RShiny/ ).
Figure 4: Examples of predicting probability of HBsAg loss using HBRN SQuARe Model.
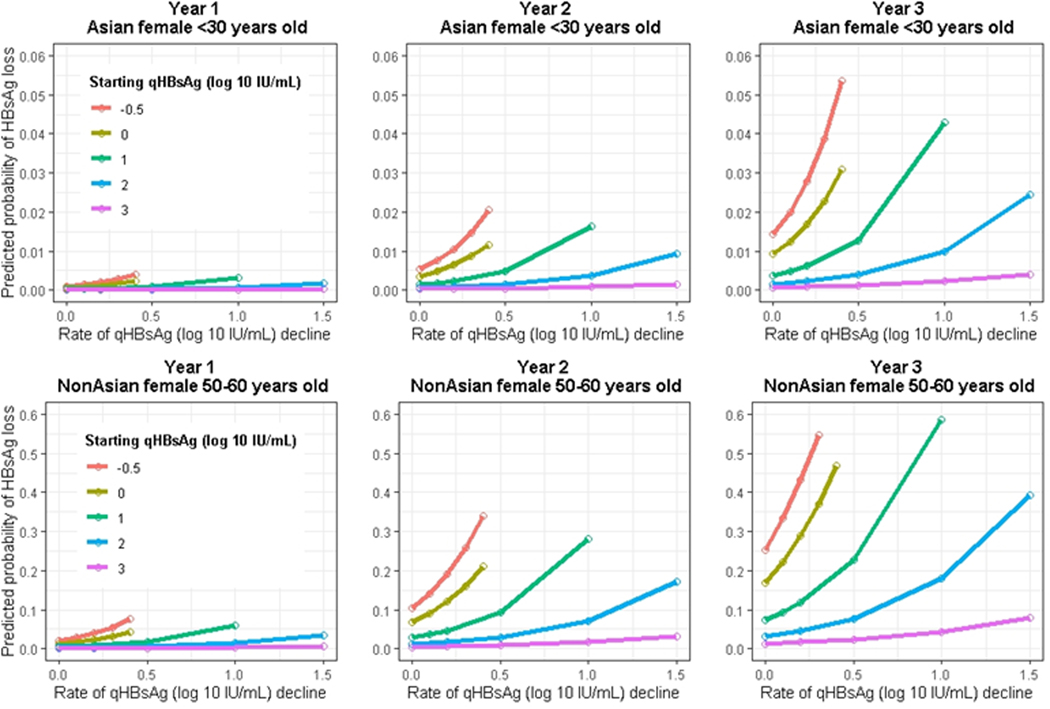
Predicted probability of HBsAg loss as a function of baseline quantitative HBsAg levels and change over a one-year period for selected participants of varying race, sex, and age. Upper row for a <30 year old Asian female participant and the lower panel for a non-Asian female in their 50’s.
Other models evaluating prediction of HBsAg loss evaluated genotype instead of race and genotype plus race in the model but were not superior to the HBRN-SQuARe model (model 2) (sTable 5). HBRN-SQuARe also yielded very similar results after excluding 131 participants who had any prior history of HBV treatment, except that the race was no longer a significant predictor (results not shown). Finally, the model was re-run by replacing the continuous qHBsAg with baseline HBsAg categories (<100, 100 to <1000, and >1000) with very similar AIC statistic compared of the HBRN-SQuARe model. A web application for the HBRN-SQuARe model is provided https://abduswahed.shinyapps.io/HBRN-SQuARe/.
External Validation of HBRN-SQuARe model
For external validation of the predictive model, HBRN SQuARe Models 1 and 2 were validated retrospectively in a separate single center (Toronto Centre for Liver Disease). Baseline characteristics of the validation cohort (sTable 7) included: 71% (n=893) Asian, 45% (n=558) female, with young adults (<30 years of age but no children) comprising only 6% of the cohort; in contrast to the HBRN which included children and 18% of participants were under age 30. Further, the proportion of patients in the Toronto cohort over the age of 60 was 28% versus 18% of the HBRN cohort. Baseline qHBsAg, proportion female and proportion Asian were similar. The mean follow up duration for patients was 3.1 (SD: 1.9) years with 10% of patients having 6 years or more of follow up. A total of 69 (5.5%) patients achieved ever HBsAg loss for an incidence rate of 1.77 per 100 PY, very similar to our cohort.
Validation of models 1 and 2 was performed by assessing performance of predicting HBsAg loss at year 1, 2, and 3 by estimating the AUROC. AUROC estimates with 95% CI for model 1 at Year 1, 2, and 3, were 0.70 [0.54–0.86], 0.75 [0.65–0.85], and 0.77 [0.69–0.85], respectively. AUROC for model 2 (HBRN SQuARe) estimates with 95% CI at Years 1, 2, and 3 were 0.99 [0.98–1.00], 0.90 [0.80–1], and 0.88 [0.77–0.99], respectively. A total of 728 individuals with 22 (3%) achieving HBsAg loss met criteria for inclusion in this latter model. The ROC curves for the validation models are shown in sFigure 3.
Discussion
HBsAg loss is an infrequent but critically important event in the natural history of chronic HBV infection. In this large, multiethnic North American cohort with median follow-up of 5.5 years (nearly 20% with 8 years or more), we found that HBsAg loss occurred at a rate of 1.6 per 100 PY, similar to what has been reported in other cohorts and systematic reviews4,5. However, the rates of HBsAg loss differed markedly by baseline characteristics, ranging from <0.5 per 100 PY to over 3 per 100 PY. Using sex, age, race and quantitative HBsAg, the HBRN SQuARe model was developed and internally and externally validated to accurately predict HBsAg loss over a 3-year period. The information provided by the HBRN SQuARe score can be used to convey likelihood of HBsAg loss with a 3-year horizon for untreated HBeAg-negative CHB patients under medical care. Identification of patients who are more or less likely to spontaneously clear HBsAg allows for prognostication, which is clearly important for patients, but also may help prioritize individuals for novel therapies. For example, those who are unlikely to clear HBsAg spontaneously may be more inclined to opt for new therapies, while patients predicted to have higher spontaneous clearance rates may be informed that need for additional therapies to enhance HBsAg loss is less urgent.
Understanding the predictors of HBsAg loss is important for many reasons. Long-term follow-up data consistently confirm the importance of HBsAg loss, with markedly lower rates of progressive liver disease and HCC, particularly when HBsAg loss occurs in patients younger than 50 years without cirrhosis21,22. The clear clinical benefits and durability of HBsAg loss have made it the preferred therapeutic endpoint for novel HBV therapies in development. Yet, given the heterogeneity in patients with CHB, models that can help to stratify likelihood of HBsAg loss in the absence of treatment may be especially helpful in the design of randomized trials to ensure balance across treatment arms and potentially to allow for improved matching of patients with specific therapeutic approaches that are more likely to be effective based on a given clinical profile.
We found the strongest predictors of HBsAg loss were older age, non-Asian ethnicity, inactive disease and low HBsAg and HBV DNA levels. The association of very low or undetectable levels of HBsAg and HBV DNA with HBsAg loss, suggests that clearance may occur when declining cccDNA levels and/or transcriptional activity fall below a threshold, not necessarily invoking a specific viral or immunological event that triggers clearance. Importantly, we were not able to distinguish HBsAg derived from cccDNA from HBsAg that is transcribed from HBV DNA integrated into the host genome. Tools to distinguish the source of HBsAg would be very helpful, particularly if the prognostic implications of the HBsAg from integrated versus cccDNA differ. Understanding the processes by which HBsAg clearance occurs could have important implications for the development of treatments that lead to functional cure.
Few studies have examined the durability of spontaneous HBsAg loss and -seroreversion. We found that HBsAg loss was durable in the majority of participants (73%), with anti-HBs developing concurrent with or in the subsequent year in approximately half the participants. As anti-HBs testing was less frequent than HBsAg testing in the cohort, the timing of anti-HBs acquisition is less precise than the trajectory of HBsAg loss and seroreversion. However, similar rates of durable spontaneous HBsAg loss were reported other large cohort studies23–25. HBsAg seroreversion was rare in our study with an incidence of 0.4% per year, which was lower than the 4.1% reported in the Hong Kong cohort25. Differences may reflect differences in the baseline characteristics of the population, with our study more limited to those that entered the cohort having already achieved HBeAg negativity. Similar to other studies, we observed a variable interval between HBsAg loss and anti-HBs seroconversion. Thus, current definition of HBV functional cure does not mandate detection of anti-HBs.
Clinical events among the cohort with HBeAg-negative CHB were infrequent, as previously highlighted in the outcomes among all adult HBRN participantsand also in the pediatric cohort 17,26. ALT flares defined by ALT values greater than 10 times normal occurred in 44 patients at a median of 52 weeks of follow-up, for a rate of 0.8% per year, a rate lower than reported in the entire HBRN cohort (incidence = 1.51 per 100 PY) but reflective of differences in ALT flare rates between HBeAg-positive versus HBeAg-negative CHB. Treatment initiation occurred at a rate of 5.9% in this cohort and was associated with a lower rate of HBsAg loss compared to those who remained untreated. It is likely that treatment was a surrogate for active disease rather than truly negatively influencing the likelihood of HBsAg clearance. Treatment decisions were made by individual health care providers (not protocolized) and final models adjusted for treatment initiation as a time-varying covariate. In post-hoc analysis, factors associated with treatment initiation were elevated ALT and HBV DNA levels (data not shown), supporting our hypothesis that treatment initiation was a surrogate for active CHB. Finally, progression to cirrhosis, as determined by an adjudication committee, occurred in 23 patients (rate of 0.4% per year) but nearly half (10/23) were diagnosed within first 24 weeks, suggesting it was likely present at baseline and none of the participants with HBsAg loss progressed to cirrhosis. This low rate of progression to cirrhosis aligns with our previously reported low rate of clinically relevant outcomes in the HBRN cohort and likely reflects the benefits of active management of chronic HBV infection17.
This study has some limitations. HBsAg loss was an infrequent event, limiting the number of covariates that could be included in the prediction models. However, the cohort represents a large multiethnic population from North America with long-term and comprehensive prospective follow-up and the risk prediction score developed from our cohort was externally validated with good accuracy. HBV genotype representation is dominated by genotype B and C, though genotypes A-G are also represented. This limited our ability to perform genotype-specific models for HBsAg loss. Regarding our prediction models, internal cross-validation was not possible due to the modest number of HBsAg loss events but external validation in another North American cohort strengthens this work. As expected, the accuracy of HBRN SQuARe in predicting HBsAg in the validation cohort was less than in the original cohort, possibly related to the difference in the percentage of patients at the extremes of age, with the Toronto cohort having a smaller proportion under age 30 (6% versus 18% in HBRN). Nonetheless, the AUROC value of 0.77 at 3 years for HBRN SQuARe in the externally validated cohort indicates good to excellent discrimination. We noted that use of serial qHBsAg data through week 48 performed better in the Toronto cohort (AUROC=0.88 at 3 years), but this was not true in the HBRN development cohort. Fewer patients were eligible for inclusion in the model using sequential qHBsAg and this reduced the overall size of the cohort for modeling. Therefore the preferred final model was HBRN SQuARe, based on a single qHBsAg value. However these data suggest serial qHBsAg levels may provide important incremental value in clinical practice.
In conclusion, we found that HBsAg loss occurred infrequently in a large cohort of predominantly untreated North American patients with HBeAg-negative CHB. HBsAg loss was associated with older age, non-Asian ethnicity and inactive hepatitis B and can be predicted over 3 years using a simple validated HBRN SQuARe score model. This information is novel and important to disease prognostication and prioritization of future therapeutic approaches in CHB.
Supplementary Material
Acknowledgements:
In addition to the authors, the HBRN would like to acknowledge the contributions of the following: Harvard Consortium: Jianghe Niu, PhD, Asad Javaid, MBBS, Bilal Nasir, MBBS, Ammu Susheela, MBBS, Imad Nasser, MD (Beth Israel Deaconess Medical Center, Boston, MA), Arley Donovan, Nifasha Rusibamayila, Cara Foley (Massachusetts General Hospital, Boston, MA). Minnesota Alliance for Research in Chronic Hepatitis B: Alisha C. Stahler, Linda Stadheim, RN (Mayo Clinic Rochester, Rochester, MN), John Lake, MD, Philip Lacher (University of Minnesota, Minneapolis, MN), Shannon M. Riggs, LPN, AS (Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN). Midwest Hepatitis B Consortium: Kathryn Rushing, RN, Rosemary A. Nagy, RDN, LD, MBA, Jacki Cerkoski, RN, MSN (Saint Louis University School of Medicine, St Louis, MO), Debra DeMarco Shaw, RN, BSN, Lisa Kessels, RN, Michael K. Klebert, PhD, RN, ANP-BC (Washington University School of Medicine, St. Louis, MO). University of Toronto Consortium: Seham Noureldin, PhD, Danie La, RN, Lucie Liu, MSc, CCRP, Diana Kaznowski, RN, Jiayun Chen, Fengfei Huang, Doinita Vladutu, Orlando Cerocchi (Toronto General Hospital, Toronto, Ontario), Athena Hau, BSc (Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario). HBV CRN North Texas Consortium: Debra Rowan, LVN (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Sheila Bass (University of Texas Southwestern, Dallas, TX), Barbara Lilly, BS (Baylor University Medical Center, Dallas, TX), Laurie A. Rodgers-Augustyniak, RN, Shirley Montanye, RN (Department of Pediatrics, UTSW, Dallas, TX). Los Angeles Hepatitis B Consortium: Samuel French, MD, Velma Peacock, RN (David Geffen School of Med, UCLA, Los Angeles, CA). San Francisco Hepatitis B Research Group Consortium: Marion Peters, MD, Ashley Shobe, MS, Rayshawnda Davis, Romuald Kuras, Claudia Ayala, MS, Ivy Lau, BS (University of California-San Francisco, San Francisco, CA), Veronika Podolskaya, BS, NCPT, Anna von Bakonyi, LVN, CCRC, Nata DeVole, RN (California Pacific Medical Center Research Institute, San Francisco, CA), Natasha Feier, MS, Joel Feier, BS, Camille Langlois, MS (Department of Pediatrics, UCSF, San Francisco, CA). Michigan Hawaii Consortium: Barbara McKenna, MD, Karen Choi, MD, Kelly Oberhelman, PAC, Sravanthi Kaza, Bpharm, Isabel Moran (University of Michigan, Ann Arbor, MI), Leslie Huddleston, NP, Richmond Wong (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: A. Sidney Barritt, M.D., Tiffany Marsh, BA, Vikki Metheny, ANP, Danielle Cardona, PA-C (University of North Carolina at Chapel Hill, Chapel Hill, NC). Virginia Commonwealth University Medical Center: Paula G. Smith, RN, BSN, Charlotte Hofmann, RN (Virginia Commonwealth University Health System, Richmond, VA). PNW/Alaska Clinical Center Consortium: Alycia Wolfstone, RN, MN (University of Washington Medical Center, Seattle, WA) Jody Mooney, Lupita Cardona-Gonzalez (Virginia Mason Medical Center, Seattle, WA), Kara L. Cooper (Center for Clinical and Translational Research, Seattle Children’s Institute, Seattle, WA). Johns Hopkins University: Hongxia Li, MBBS, MS, Douglas Mogul, MD, Robert Anders, MD, PhD, Hejab Imteyaz, Peter Lee, MD, Kiyoko Oshima, MD, Kim Kafka, RN, Naureen Islam, BS (Department of Pediatrics, Johns Hopkins Medical Institutions, Baltimore, MD). Liver Diseases Branch, NIDDK, NIH: Nancy Fryzek, RN, BSN, Elenita Rivera, BSN, Nevitt Morris, Vanessa Haynes-Williams, Amy Huang, RN, Catherine Nadal, RN, MS, Jaha Norman-Wheeler, RN, BA (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK, NIH: Averell H. Sherker, MD, Rebecca J. Torrance, RN, MS, Sherry R. Hall, MS (National Institutes of Health, Bethesda, MD). Immunology Center: Mary E. Valiga, RN, Keith Torrey, BS, Danielle Levine, BS, James Keith, BS, Michael Betts, PhD (University of Pennsylvania, Philadelphia, PA), Luis J. Montaner, DVM, DPhil (Wistar Institute, Philadelphia, PA). Data Coordinating Center: Frani Averbach, MPH, Tamara Haller, Regina Hardison, MS, Stephanie Kelley, MS, Christina M. Lalama, MS, Sharon Lawlor, MBA, Hsing-Hua S. Lin, MS, PhD, Manuel Lombardero, MS, Andrew Pelesko, BS, Donna Stoliker, Melissa Weiner, MPH, Ella Zadorozny, MS, Qian Zhao, PhD (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA).
Grant support: The HBRN was funded as a Cooperative Agreement between the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the following investigators: Anna Suk-Fong Lok, MD (U01-DK082863), Steven H. Belle, PhD, MScHyg (U01-DK082864), Adrian M. Di Bisceglie, MD (U01-DK082871), William M. Lee, MD (U01-DK082872), Harry L. A. Janssen, MD, PhD (U01-DK082874), Daryl T-Y Lau, MD, MPH (U01-DK082919), Richard K. Sterling, MD, MSc (U01-DK082923), Mandana Khalili, MD (U01-DK082944), an interagency agreement with NIDDK: Lilia M. Ganova-Raeva, PhD (A-DK-3002-001) and support from the intramural program, NIDDK, NIH: Marc G. Ghany, MD, Intramural Research Program, NIDDK, NIH. Additional funding to support this study was provided to NIH Public Health Service Research Grant M01-RR00040), Richard K. Sterling, MD, MSc (UL1TR000058, NCATS (National Center for Advancing Translational Sciences, NIH), Norah A. Terrault, MD, MPH (CTSA Grant Number UL1TR000004), and Anna Suk-Fong Lok (CTSA Grant Number UL1RR024986, U54TR001959.) Additional support was provided by Gilead Sciences, Inc. and Roche Molecular Systems via a CRADA through the NIDDK.
Terrault: Institutional grant support from GSK, Roche-Genentech, Gilead Sciences
Wahed AS: Nothing to disclose
Feld JJ: Institutional grant support from Abbvie, Eiger, Enanta, Gilead, Janssen and consulting honoraria from Abbvie, Arbutus, Gilead, GSK, Roche
Cooper SL: Nothing to disclose
Ghany MG: Nothing to disclose
Lisker-Melman M: Speaker Bureau: Gilead, AbbVie
Perrillo R: Nothing to disclose
Sterling RK: Grant support from Abbott, AbbVie, Roche, and Gilead and serves on a DSMB for Pfizer and AskBio.
Khalili M: : Institutional grant support from Gilead Sciences Inc and Intercept Pharmaceutical, consultant Gilead Sciences Inc.
Chung RT: Institutional grant support from Gilead, BMS, Janssen, Roche, GSK
Rosenthal P: Grant support: Gilead, Abbvie, Merck, Travere Therapeutics, Albireo, Mirum, Arrowhead. Consulting: Gilead, Abbvie, Travere Therapeutics, Albireo, Mirum, Audentes, Dicerna, Encoded, Vertex.
Fontana RJ: Institutional grant support from Gilead, BMS, and Abbvie; consulting for Sanofi.
Sarowar A: Nothing to Disclose
Lau D: Research support: Gilead, Abbott Diagnostic, Janssen Pharmaceutica. Consultant: Abbott Diagnostics
Wang, J: Nothing to disclose
Lok AS: Institutional grant support from Bristol-Myers Squibb and Gilead Sciences
Janssen HLA: Received grants from: AbbVie, Arbutus, Bristol Myers Squibb, Gilead Sciences, Janssen, Medimmune, Merck, Roche./ Consultant for: Arbutus, Arena, Enyo, Gilead Sciences, GlaxoSmithKline, Janssen, Medimmune, Merck, Roche, Vir Biotechnology Inc., Viroclinics.
The HBRN: Minnesota Alliance for Research in Chronic Hepatitis B Consortium: Lewis R. Roberts, MB, ChB, PhD (Mayo Clinic Rochester, Rochester, MN), Mohamed A. Hassan, MD (University of Minnesota, Minneapolis, MN), Sarah Jane Schwarzenberg, MD (Department of Pediatrics, University of Minnesota Masonic Children’s Hospital, Minneapolis, MN). Midwest Hepatitis B Consortium: Adrian M. Di Bisceglie, MD, (Saint Louis University School of Medicine, St Louis, MO), Jeffrey Teckman, MD (Department of Pediatrics, Cardinal Glennon Children’s Medical Center, Saint Louis University, St. Louis, MO). University of Toronto Consortium: David K. Wong, MD (Toronto General Hospital, Toronto, Ontario), Joshua Juan, MD (Toronto General Hospital, Toronto, Ontario), Colina Yim, NP, MN (Toronto General Hospital, Toronto, Ontario), Keyur Patel, MD (Toronto General Hospital, Toronto, Ontario), Simon C. Ling, MBChB (Department of Paediatrics, The Hospital for Sick Children, University of Toronto, Toronto, Ontario). HBV CRN North Texas Consortium: William M. Lee, MD (Division of Digestive and Liver Diseases, University of Texas Southwestern Medical Center at Dallas, Dallas, TX), Son Do, MD (University of Texas Southwestern, Dallas, TX), Norberto Rodriguez-Baez, MD (Department of Pediatrics, University of Texas Southwestern, Dallas, TX). Los Angeles Hepatitis B Consortium: Steven-Huy B. Han, MD (David Geffen School of Medicine, UCLA, Los Angeles, CA), Tram T. Tran, MD (Cedars Sinai Medical Center, Los Angeles, CA). Michigan Hawaii Consortium: Naoky Tsai, MD (The Queen’s Medical Center, University of Hawaii, Honolulu, HI), Barak Younoszai, DO (The Queen’s Medical Center, University of Hawaii, Honolulu, HI). Chapel Hill, NC Consortium: Michael W. Fried, MD, (University of North Carolina at Chapel Hill, Chapel Hill, NC), Andrew Muir, M.D. (Duke University Medical Center, Durham, NC), Donna Evon, Ph.D. (University of North Carolina at Chapel Hill, Chapel Hill, NC), Jama M. Darling, MD (University of North Carolina at Chapel Hill, NC). PNW/Alaska Clinical Center Consortium: Robert C. Carithers, MD (University of Washington Medical Center, Seattle WA), Margaret Shuhart, M.D. (Harborview Medical Center, Seattle WA), Kris V. Kowdley, MD (Virginia Mason Medical Center, Seattle WA), Chia C. Wang, MD (Virginia Mason Medical Center, Seattle WA), Karen F. Murray, MD (Department of Pediatrics, University of Washington, Seattle, WA). Virginia Commonwealth University Medical Center: Velimir A. Luketic, MD (Virginia Commonwealth University Health System, Richmond, VA). Johns Hopkins University: Kathleen B. Schwarz, MD (Department of Pediatrics, Johns Hopkins Medical Institutions, Baltimore, MD). Liver Diseases Branch, NIDDK: T. Jake Liang, MD (National Institutes of Health, Bethesda, MD). Liver Disease Research Branch, NIDDK: Jay H. Hoofnagle, MD (National Institutes of Health, Bethesda, MD), Edward Doo, MD (National Institutes of Health, Bethesda, MD). Immunology Center: Kyong-Mi Chang, MD, (University of Pennsylvania Perelman School of Medicine, Philadelphia, PA), Jang-June Park, PhD (University of Pennsylvania Perelman School of Medicine, Philadelphia, PA). Data Coordinating Center: Steven H. Belle, PhD, MScHyg (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA), Wendy C. King, PhD (Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA). Central Pathology: David Kleiner, MD, PhD. (Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD).
Footnotes
Conflicts of Interest:
References:
- 1.Cornberg M, Lok AS, Terrault NA, Zoulim F, Faculty E-AHTEC. Guidance for design and endpoints of clinical trials in chronic hepatitis B - Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference(double dagger). J Hepatol 2020;72:539–57. [DOI] [PubMed] [Google Scholar]
- 2.Yip TC, Wong GL, Chan HL, et al. HBsAg seroclearance further reduces hepatocellular carcinoma risk after complete viral suppression with nucleos(t)ide analogues. J Hepatol 2019;70:361–70. [DOI] [PubMed] [Google Scholar]
- 3.Su YC, Lin PC, Yu HC, Wu CC. Hepatitis B virus reactivation in patients with resolved hepatitis B virus infection receiving chemotherapy or immunosuppressive therapy. Eur J Gastroenterol Hepatol 2018;30:925–9. [DOI] [PubMed] [Google Scholar]
- 4.Zhou K, Contag C, Whitaker E, Terrault N. Spontaneous loss of surface antigen among adults living with chronic hepatitis B virus infection: a systematic review and pooled meta-analyses. Lancet Gastroenterol Hepatol 2019;4:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeo YH, Ho HJ, Yang HI, et al. Factors Associated With Rates of HBsAg Seroclearance in Adults With Chronic HBV Infection: A Systematic Review and Meta-analysis. Gastroenterology 2019;156:635–46 e9. [DOI] [PubMed] [Google Scholar]
- 6.Buster EH, Flink HJ, Cakaloglu Y, et al. Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon alpha-2b. Gastroenterology 2008;135:459–67. [DOI] [PubMed] [Google Scholar]
- 7.Marcellin P, Bonino F, Lau GK, et al. Sustained response of hepatitis B e antigen-negative patients 3 years after treatment with peginterferon alpha-2a. Gastroenterology 2009;136:2169–79 e1–4. [DOI] [PubMed] [Google Scholar]
- 8.Bourliere M, Rabiega P, Ganne-Carrie N, et al. Effect on HBs antigen clearance of addition of pegylated interferon alfa-2a to nucleos(t)ide analogue therapy versus nucleos(t)ide analogue therapy alone in patients with HBe antigen-negative chronic hepatitis B and sustained undetectable plasma hepatitis B virus DNA: a randomised, controlled, open-label trial. Lancet Gastroenterol Hepatol 2017;2:177–88. [DOI] [PubMed] [Google Scholar]
- 9.Liaw YF. Clinical utility of HBV surface antigen quantification in HBV e antigen-negative chronic HBV infection. Nat Rev Gastroenterol Hepatol 2019;16:631–41. [DOI] [PubMed] [Google Scholar]
- 10.Rein DB, Lesesne SB, O’Fallon A, Weinbaum CM. Prevalence of hepatitis B surface antigen among refugees entering the United States between 2006 and 2008. Hepatology 2010;51:431–4. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell T, Armstrong GL, Hu DJ, Wasley A, Painter JA. The increasing burden of imported chronic hepatitis B--United States, 1974–2008. PloS one 2011;6:e27717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coffin CS, Ramji A, Cooper CL, et al. Epidemiologic and clinical features of chronic hepatitis B virus infection in 8 Canadian provinces: a descriptive study by the Canadian HBV Network. CMAJ Open 2019;7:E610–E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghany MG, Perrillo R, Li R, et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin Gastroenterol Hepatol 2015;13:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarz KB, Cloonan YK, Ling SC, et al. Children with Chronic Hepatitis B in the United States and Canada. J Pediatr 2015;167:1287–94 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Bisceglie AM, Lombardero M, Teckman J, et al. Determination of hepatitis B phenotype using biochemical and serological markers. J Viral Hepat 2017;24:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz KB, Lombardero M, Di Bisceglie AM, et al. Phenotypes of Chronic Hepatitis B in Children From a Large North American Cohort. J Pediatr Gastroenterol Nutr 2019;69:588–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lok AS, Perrillo R, Lalama CM, et al. medLow Incidence of Adverse Outcomes in Adults with Chronic Hepatitis B Virus Infection in the Era of Antiviral Therapy. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganova-Raeva L, Ramachandran S, Honisch C, Forbi JC, Zhai X, Khudyakov Y. Robust hepatitis B virus genotyping by mass spectrometry. J Clin Microbiol 2010;48:4161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau DTY, Ganova-Raeva L, Wang J, et al. Precore and basal core promoter hepatitis B virus (HBV) variants are present from a young age and differ across HBV genotypes. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao G, Yang J, Yan L. Comparison of diagnostic accuracy of aspartate aminotransferase to platelet ratio index and fibrosis-4 index for detecting liver fibrosis in adult patients with chronic hepatitis B virus infection: a systemic review and meta-analysis. Hepatology 2015;61:292–302. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Yang HI, Lee MH, et al. Serum Levels of Hepatitis B Surface Antigen and DNA Can Predict Inactive Carriers With Low Risk of Disease Progression. Hepatology 2016;64:381–9. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Yang HI, Lee MH, et al. Spontaneous seroclearance of hepatitis B seromarkers and subsequent risk of hepatocellular carcinoma. Gut 2014;63:1648–57. [DOI] [PubMed] [Google Scholar]
- 23.Lok AS, Zoulim F, Dusheiko G, et al. Durability of Hepatitis B Surface Antigen Loss With Nucleotide Analogue and Peginterferon Therapy in Patients With Chronic Hepatitis B. Hepatol Commun 2020;4:8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alawad AS, Auh S, Suarez D, Ghany MG. Durability of Spontaneous and Treatment-Related Loss of Hepatitis B s Antigen. Clin Gastroenterol Hepatol 2020;18:700–9 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yip TC, Wong GL, Wong VW, et al. Durability of hepatitis B surface antigen seroclearance in untreated and nucleos(t)ide analogue-treated patients. J Hepatol 2017. [DOI] [PubMed] [Google Scholar]
- 26.Ling SC. A Longitudinal Study of Clinical, Virological, Laboratory Characteristics and Health Outcomes of Children with Chronic Hepatitis B Infection in the USA and Canada. 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.