

Abstract
Hydrogen [11C]cyanide ([11C]HCN) is a versatile 11C-labelling agent for the production of 11C-labelled compounds used for positron emission tomography (PET). However, the traditional method for [11C]HCN production requires a dedicated infrastructure, limiting accessibility to [11C]HCN. Herein, we report a simple and efficient [11C]HCN production method that can be easily implemented in 11C production facilities. The immediate production of [11C]HCN was achieved by passing gaseous [11C]methyl iodide ([11C]CH3I) through a small two-layered reaction column. The first layer contained an N-oxide and a sulfoxide for conversion of [11C]CH3I to [11C]formaldehyde ([11C]CH2O). The [11C]CH2O produced was subsequently converted to [11C]HCN in a second layer containing hydroxylamine-O-sulfonic acid. The yield of [11C]HCN produced by the current method was comparable to that of [11C]HCN produced by the traditional method. The use of oxymatrine and diphenyl sulfoxide for [11C]CH2O production prevented deterioration of the molar activity of [11C]HCN. Using this method, compounds labelled with [11C]HCN are now made easily accessible for PET synthesis applications using readily available labware, without the need for the ‘traditional’ dedicated cyanide synthesis infrastructure.
In a reaction column, gaseous [11C]methyl iodide was converted to [11C]formaldehyde in a first layer containing N-oxide and then transformed into hydrogen [11C]cyanide in a second layer containing hydroxylamine-O-sulfonic acid within 2 minutes.
Introduction
Positron emission tomography using compounds labelled with positron emitters is now a standard imaging modality used for diagnosis and clinical investigations. This has also been applied to investigation of the in vivo kinetics of drug candidates and biomolecules in animals and even plants.1 The use of 11C-labelled isotopologues of organic compounds of interest is vital to trace their in vivo behaviour. Therefore, expansion of the synthetic toolbox for 11C-labelling is greatly needed.2
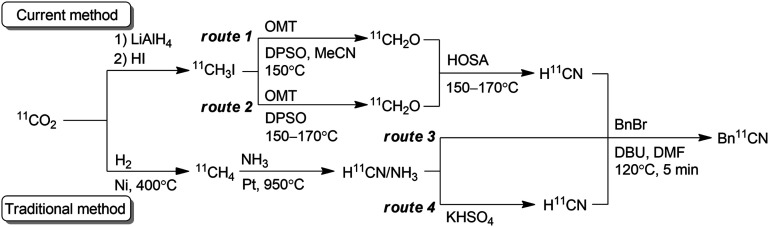
11C must be incorporated into a compound with a short synthesis time at the late stage of synthesis owing to its short half-life (20.4 min). Hydrogen [11C]cyanide ([11C]HCN) is a useful and versatile 11C-labelling agent prepared from the cyclotron-produced primary starting material [11C]carbon dioxide ([11C]CO2). [11C]HCN is used to prepare labelled nitriles by 11C-cyanation. The 11C-nitrile group can be readily converted to other 11C-labelled functional groups, such as amines, amides, and carboxylic acids, by reduction or hydrolysis.3 Furthermore, [11C]HCN can be converted to further 11C-labelling agents, such as [11C]cyanogen bromide and [11C]thiocyanate.4 As [11C]lactic acid was prepared with [11C]cyanide in 1941,511C-cyanation with [11C]HCN has been used to prepare a diverse array of 11C-labelled compounds, such as neurotransmitters, fatty acids, amino acids, sugars, receptor ligands, and a plant hormone.3 However, only a few facilities produce [11C]HCN, and the compounds labelled using [11C]HCN remain under-utilised, despite its usefulness. This is mainly attributed to the method for [11C]HCN production. For half a century, [11C]HCN has generally been prepared based on the BMA process,6 in which [11C]methane is passed over platinum at 900–1000 °C under ammonia gas flow (Scheme 1).7 The starting material, [11C]methane, is generally obtained by heating [11C]CO2 over nickel under hydrogen gas flow at 400 °C, or sometimes by proton irradiation of the N2/H2 gas target. Therefore, [11C]HCN production using the traditional method requires a dedicated infrastructure, including H2 and NH3 gas lines, and furnaces for high-temperature heating in the limited space of a shielded radiation containment chamber. Furthermore, optimising the reaction conditions to obtain reliable production of [11C]HCN for routine use is difficult.3b,7c,7d
Scheme 1. Traditional method for the production of H11CN and on-column conversion of 11CH3I to 11C-labelling agents. HOSA, hydroxylamine-O-sulfonic acid.

To make [11C]HCN widely accessible, we sought a practical and efficient method for the production of [11C]HCN without any special equipment and reagents. Herein, we propose a route for the preparation of [11C]HCN from [11C]methyl iodide ([11C]CH3I), which is established as the most widely used 11C-labelling agent. We employed a simple on-line system using a reaction column, in which [11C]CH3I was first converted to [11C]formaldehyde ([11C]CH2O) and then transformed into [11C]HCN in a short period (Scheme 1). The conversion of [11C]CH3I to further 11C-labelling agents using a reaction column has been commonly performed. Useful 11C-labelling agents [11C]methyl triflate and [11C]nitromethane have been frequently prepared by passing [11C]CH3I through a heated reaction column filled with silver triflate and silver nitrite, respectively.8 Therefore, the production of [11C]HCN from [11C]CH3I using a reaction column can be easily implemented in 11C production facilities.
Results and discussion
Production of [11C]CH2O in solution
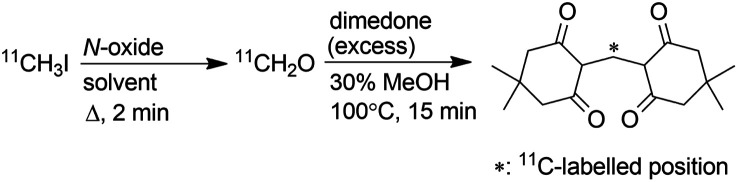
First, an efficient on-column method to produce gaseous [11C]CH2O was required. There are several methods to prepare [11C]CH2O.9 They are classified into three groups on the basis of the starting materials ([11C]methanol, [11C]CO2 and [11C]CH2O). Among them, the methods using [11C]methanol would not be suitable for the current purpose because the production of [11C]methanol requires incorporation of an additional synthesis system in the 11C production facilities that are used to produce [11C]CH3I from [11C]methane. For [11C]CO2, the reduction of [11C]CO2 to yield [11C]CH2O requires a low temperature to prevent over-reduction and the reducing agent is quenched by adding an acid before [11C]CH2O distillation at high temperature. These procedures are not easily applicable to the on-column reaction. In contrast to these methods, the reaction of [11C]CH3I with an N-oxide was considered suitable for the on-column production of gaseous [11C]CH2O, because [11C]CH3I is available in most 11C production facilities and the reaction is simple and efficient.9j Passing [11C]CH3I through the reaction column containing an N-oxide solution was expected to afford gaseous [11C]CH2O. However, the solvent used in this reaction caused some concern. Alkyl halides are known to react with trimethylamine N-oxide (TMAO) to afford the corresponding aldehydes,10 and dimethyl sulfoxide (DMSO) has been reported as a good solvent for this reaction.11 However, using DMSO as solvent in the reaction of [11C]CH3I with TMAO gave a low yield of [11C]CH2O, whilst using N,N-dimethylformamide (DMF) as solvent afforded [11C]CH2O in high yield.9j In our strategy, gaseous [11C]CH2O was extracted from the reaction mixture by distillation. Therefore, DMSO was considered more favourable than DMF because of its higher boiling point.
According to these considerations, the efficiency of [11C]CH2O production by the reaction of [11C]CH3I with TMAO in DMF and DMSO was re-evaluated. A DMF or DMSO solution of [11C]CH3I (300 μL) was added to a reaction vial containing fine crystals of TMAO (4 mg, 53 μmol) and the suspended solution was allowed to stand for 2 min at 70 °C. These reaction conditions were the same as those used in a previous study.9j The radiochemical yields of [11C]CH2O were determined using the dimedone precipitation method9g to directly compare the values in the previous study, measured using this method, with those in this study. In DMF, [11C]CH3I was converted to [11C]CH2O in moderate yield (Table 1, entry 7), as reported previously.9j,12 Efficient conversion of [11C]CH3I to [11C]CH2O was observed in the reaction with DMSO (80 ± 1.9%, entry 1) despite the previous study reporting a much lower value (23 ± 7%).9j The reason for this discrepancy remains unclear. As anhydrous TMAO is sparingly soluble in DMSO, sonication and slight warming were necessary to dissolve TMAO. The low yield of [11C]CH2O reported in the previous study was initially anticipated to be due to the poor solubility of TMAO in DMSO. However, a high [11C]CH2O yield was observed even in the reaction with TMAO suspended in DMSO.
Effects of solvents and N-oxides on the yield of [11C]CH2Oa.
| |||||
|---|---|---|---|---|---|
| Entry | Solvent | N-Oxide (mg) | T (°C) | Yieldsc (%) | n d |
| 1b | DMSO | TMAO (4 mg, 53 μmol) | 70 | 80 ± 1.9 | 4 |
| 2 | DMSO | TMAO (4) | 70 | 90 ± 1.9 | 4 |
| 3 | DMSO | TMAO 2H2O (4) | 70 | 87 ± 3.5 | 3 |
| 4 | DMSO | NMO (6 mg, 51 μmol) | 70 | 83 ± 2.2 | 3 |
| 5 | DMSO | OMT (15 mg, 57 μmol) | 90 | 71 ± 6.3 | 3 |
| 6 | TMSO | TMAO (4) | 70 | 66 ± 1.0 | 3 |
| 7b | DMF | TMAO (4) | 70 | 73 ± 6.1 | 7 |
| 8 | DMF | TMAO (4) | 70 | 48 ± 1.7 | 5 |
| 9b | MeCN | TMAO (4) | 70 | 76 ± 1.7 | 4 |
| 10 | MeCN | TMAO (4) | 70 | 50 ± 1.4 | 4 |
Reaction conditions: For reactions in N-oxide suspended solution (entries 1, 7, and 9), a solution of [11C]CH3I (37–74 MBq, 300 μL) was added to a reaction vial containing a given amount of N-oxide, then allowed to stand for 2 min at the designated temperature. For reactions with N-oxide completely dissolved in the solution, a solution of 11CH3I (37–74 MBq, 100 μL) was added to N-oxide solution (200 μL) and reacted for 2 min at the designated temperature.
Reactions were performed in the N-oxide suspension.
Yields of [11C]CH2O were measured by dimedone precipitation assay.9g Values are presented as mean ± sd.
Number of experiments.
We further investigated the efficiency of [11C]CH2O production using TMAO in solution. A solution of [11C]CH3I (100 μL) was added to a solution (200 μL) in which TMAO was completely dissolved, and the mixture was reacted for 2 min. [11C]CH3I was completely consumed within 2 min, even at ambient temperature using TMAO (4 mg) dissolved in DMSO. In the reaction in DMSO at 70 °C, using fully dissolved TMAO increased the [11C]CH2O yield (Table 1, entry 2) compared with the TMAO suspension (entry 1). The use of the dihydrate of TMAO did not affect the excellent yield of [11C]CH2O (entry 3).
In contrast, using TMAO fully dissolved in DMF solution significantly decreased the yield of [11C]CH2O (entry 8) compared with the TMAO suspension (entry 7). These results indicated that DMSO was a favourable solvent for the preparation of [11C]CH2O by reacting [11C]CH3I with TMAO in terms of reaction efficiency.
In terms of molar activity,13 which is the activity of a radiolabelled compound divided by the total amount of the compound in moles, DMSO might not be a suitable solvent for the preparation of [11C]CH2O, because DMSO degradation generates nonradioactive CH2O.14 The generation of nonradioactive CH2O reduces the molar activity of [11C]CH2O, resulting in deterioration of the molar activity of [11C]HCN and compounds labelled with it. A radiolabelled compound with high molar activity is often required to avoid toxicological effects, and visualise and quantify biological functions, especially in a study using radiolabelled xenobiotics.1c Tetramethylene sulfoxide (TMSO), which has no methyl group, was not expected to generate CH2O by thermal degradation (Chart 1). When TMSO was applied as the solvent instead of DMSO, TMAO converted [11C]CH3I to [11C]CH2O in moderate yield (Table 1, entry 6). This result suggested that sulfoxides with no S-methyl group were preferable solvents for these reactions.
Chart 1. N-Oxides and sulfoxides used in this study. TMAO, trimethylammonium N-oxide; NMO, N-methylmorpholine N-oxide; OMT, oxymatrine; DMSO, dimethyl sulfoxide; TMSO, tetramethylene sulfoxide; DPSO, diphenyl sulfoxide.

In a previous report, the reaction of [11C]CH3I with TMAO in acetonitrile (MeCN) was suggested to afford [11C]CH2O with relatively high molar activity.12 Furthermore, in MeCN, we observed that TMAO converted [11C]CH3I to [11C]CH2O in moderate yield (Table 1, entries 9 and 10) despite a previous study reporting a much lower value (27 ± 7%).9j Although MeCN was not a suitable solvent for the present study owing to its volatile nature, it might be a good solvent for in situ generation of [11C]CH2O in the one-pot 11C-labelling with reductive N-methylation and a Pictet–Spengler reaction.
The use of N-methylmorpholine N-oxide (NMO) dissolved in DMSO afforded [11C]CH2O in moderate yield (Table 1, entry 4). However, TMAO and NMO, which possess an N-methyl group, underwent thermal degradation to form CH2O.15 The generation of CH2O would significantly deteriorate the molar activity of [11C]CH2O, even if only 0.1% of TMAO (4 mg, 53 μmol) was decomposed to form CH2O (53 nmol). Therefore, using an N-oxide with no N-methyl group was preferable to prevent reducing the molar activity of [11C]CH2O. Triethylamine N-oxide and quinuclidine N-oxide have been reported as effective for this reaction, whilst pyridine N-oxide was ineffective.9j,12,16 However, triethylamine N-oxide is a thermolabile compound that decomposes via Cope elimination and Meisenheimer rearrangement.17 Quinuclidine N-oxide is also not suitable for routine use because it is highly hygroscopic and deliquescent when synthesised, as previously reported, and not commercially available.18 Furthermore, the use of quinuclidine N-oxide instead of TMAO did not improve the molar activity of [11C]CH2O.12 Next, oxymatrine (OMT), a natural N-oxide quinolizidine derivative with no N-methyl group (Chart 1), was applied instead of TMAO and NMO for the production of [11C]CH2O from [11C]CH3I in DMSO. Using OMT as oxidant afforded a moderate yield of [11C]CH2O, although a higher reaction temperature was required (entry 5).
Production of gaseous [11C]CH2O
Gaseous [11C]CH2O was produced by passing [11C]CH3I through quartz wool wetted with N-oxide solution. Quartz wool was compressed to 25 mm in a small glass column (syringe barrel type, approx. 8.5 mm ID × 70 mm). The quartz wool was soaked with N-oxide solution and then excess solution was purged with nitrogen gas flow (100 mL min−1) at 50 °C for 10 min. [11C]CH3I in the carrier gas was introduced into the reaction column, which was heated during the reaction with a silicone rubber heater,8c at a flow rate of 50 mL min−1. The flow rate was the same value as used in the routine 11C-methylation procedure with [11C]CH3I to minimise changes in the sequential program for automated synthesis.
As a preliminary study, [11C]CH3I was introduced into a heated reaction column (120 °C), which was prepared with a solution of TMAO (10 mg) dissolved in only MeCN (1 mL). The eluted gas containing the radioactive product from the reaction column was passed through an 80% aqueous DMSO solution (cooled with an ice bath) to trap the [11C]CH2O and unreacted [11C]CH3I. A considerable amount of unreacted [11C]CH3I was recovered in the trapped solution (Fig. S1 in ESI†). This result suggested that a less volatile solvent would be needed for an efficient reaction in the reaction column, because almost all MeCN was evaporated and removed from the reaction column during preparation. However, when a reaction column prepared with a solution of TMAO dissolved in only DMSO was used, unreacted [11C]CH3I was not observed, but a considerable amount of radioactivity was trapped in the column (48%). Next, a mixture of MeCN (750 μL) and DMSO (250 μL) was used as solvent for the TMAO solution in reaction column preparation to reduce the amount of DMSO retained in the quartz wool. By passing [11C]CH3I through a reaction column prepared with this solution at 120 °C for 3 min, [11C]CH3I was almost completely captured in the column and the radioactivity in the column was efficiently distilled.
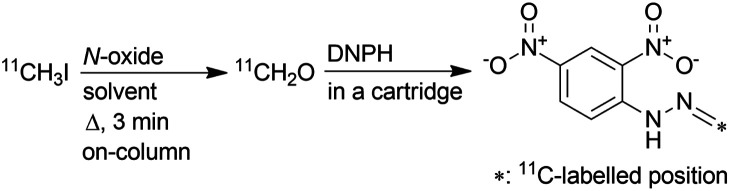
Subsequently, eluted gas from the reaction column was introduced into a 2,4-dinitrophenylhydrazine (DNPH) cartridge to detect the production of [11C]CH2O and measure its molar activity. The remaining radioactive substances, which were not trapped in the DNPH cartridge, were collected in a sampling bag. The activity trapped in the DNPH cartridge was found to be more than 83% of the total activity introduced into the reaction column. When [11C]CH3I was passed through a heated reaction column (120 °C), which was prepared with a solution of TMAO in a mixture of MeCN and DMSO, [11C]CH2O-2,4-dinitrophenylhydrazone ([11C]CH2O-DNPH) was formed in the DNPH cartridge in moderate yield (Table 2, entry 1). However, the molar activity of [11C]CH2O-DNPH was much lower than that of [11C]CH3I (345 ± 70 GBq μmol−1) at the start of [11C]CH3I transfer, as expected (entry 1). The use of NMO instead of TMAO ameliorated the molar activity of [11C]CH2O-DNPH (entry 2), but the value was lower than that of [11C]CH3I. Unexpectedly, the molar activity was not improved, even when using OMT, which has no methyl group (entry 3). The mechanism of this phenomenon was unclear, but it was also observed in experiments in which NMO and OMT were heated with or without DMSO. Although NMO generated CH2O by heating without DMSO, whilst OMT did not, heating their DMSO solutions released comparable amounts of CH2O (Fig. S2 in ESI†). Diphenyl sulfoxide (DPSO), which has no methyl group, was then used instead of DMSO to ameliorate the molar activity of [11C]CH2O. With the reaction column prepared using a solution of OMT (25 mg), DPSO (250 mg), and MeCN (750 μL), [11C]CH2O-DNPH was obtained in moderate yield with high molar activity comparable to that of [11C]CH3I (entry 4). When using DPSO, the column temperature was increased to 150 °C because [11C]CH3I breakthrough increased by reacting at 120 °C. Using DMF as solvent resulted in a low yield of [11C]CH2O-DNPH (entry 5). In this case, 56% of the total activity introduced into the reaction column was retained in the column after the reaction. This might be due to the relatively low efficiency of the conversion of [11C]CH3I to [11C]CH2O in DMF, and the reaction of [11C]CH2O with amines generated by the decomposition of DMF under heating conditions.
Yield and molar activity of [11C]CH2O-DNPHa.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Solventb | N-Oxide | T c (°C) | Yield (%) | A m d (GBq μmol−1) | n g | |
| EOBe | EOTf | EOTf | |||||
| 1 | DMSO | TMAO | 120 | 55 ± 7.0 | 37 ± 4.3 | 90 ± 36 | 3 |
| 2 | DMSO | NMO | 120 | 57 ± 9.3 | 38 ± 6.2 | 190 ± 29 | 5 |
| 3 | DMSO | OMT | 120 | 55 ± 9.5 | 37 ± 6.2 | 158 ± 41 | 6 |
| 4 | DPSO | OMT | 150 | 57 ± 1.5 | 39 ± 0.8 | 335 ± 52 | 4 |
| 5 | DMF | TMAO | 120 | 36 | 23 | 123 | 1 |
Values are presented as mean ± sd. [11C]CH2O eluted from the reaction column was introduced into a cartridge containing 2,4-dinitrophenylhydrazine (DNPH) to yield [11C]CH2O-2,4-dinitrophenylhydrazone ([11C]CH2O-DNPH).
The designated solvents (DMSO and DMF, 250 μL; DPSO, 250 mg) were mixed with MeCN (750 μL) and N-oxide (TMAO and NMO, 10 mg; OMT, 25 mg) and used to prepare the reaction column.
Temperature of reaction column.
Molar activity.
At the end of bombardment.
At the end of [11C]CH2O transfer.
Number of experiments.
Production of gaseous [11C]HCN
Although the radiochemical yield of [11C]CH2O was not directly determined, the yield should be higher than that of [11C]CH2O-DNPH (around 55% at the end of bombardment, Table 2). This encouraged us to investigate the conversion of [11C]CH2O to [11C]HCN using hydroxylamine-O-sulfonic acid (HOSA). HOSA has been used to convert aldehydes to nitriles,19 but its applicability to the conversion of CH2O to HCN was unclear. [11C]CH2O was then reacted with HOSA in DMSO or water at 120 or 100 °C, respectively, for 3 min. In both solvents, [11C]HCN was predominantly formed, whilst [11C]CH2O was almost completely consumed (Fig. S3 in ESI†). This result suggested that DMSO vapor and moisture would not interfere with the conversion of [11C]CH2O to [11C]HCN by HOSA.
Subsequently, the reaction of [11C]CH2O with HOSA under solvent-free conditions was studied. Gaseous [11C]CH2O, which was generated by passing [11C]CH3I through a solution of NMO (40 mg) in DMSO (1 mL) at 120 °C, was introduced into the reaction column, which included a mixture of HOSA (40 mg) and quartz sand (2.3 g), and the eluted radioactive substances were collected in ice-cold DMF and a sampling bag sequentially. When heating the reaction column at 120 °C, [11C]HCN (96%) and [11C]CH3I (4%) were observed in DMF. This indicated that HOSA efficiently captured [11C]CH2O and converted it to [11C]HCN without solvent. However, 40% of the total activity introduced was retained in the column after the reaction. When the reaction temperature of the column was increased to 150 °C, the activity retained in the column was reduced to 19%, with 74% of the total activity introduced being recovered in DMF. The radiochemical purity of [11C]HCN collected in DMF was 97% and the major impurity was found to be [11C]CH3I (2.5%).
Finally, [11C]HCN was produced by passing [11C]CH3I through a reaction column in which a mixture of HOSA (40 mg) and quartz sand (2.3 g) was placed on the quartz wool layer containing OMT and DPSO (Fig. 1). The reaction temperature was set at 150 °C based on the preliminary study described above. The synthesis time from the end of bombardment to the end of collection of [11C]HCN in ice-cold DMF was 10 ± 0.3 min (n = 3). The radioactivity in DMF reached a plateau within 2 min of starting the [11C]CH3I transfer. The [11C]HCN yield at the end of collection in DMF was comparable to that of the traditional method (Table 3, entries 1 and 3). The radiochemical purity of [11C]HCN trapped in DMF was sufficiently high (94%, entry 1). The [11C]CH3I content observed in DMF was 1.3 ± 1.1% (Fig. S4 in ESI†). The molar activity of [11C]HCN was determined by derivatisation to benzyl [11C]cyanide ([11C]BnCN), achieved by reacting benzyl bromide with [11C]HCN in DMF. Although the total synthesis time of [11C]BnCN after the end of bombardment in the current method and traditional method was comparable, the molar activity of [11C]BnCN produced with the current method was much higher than that produced with the traditional method (entries 1 and 3). The presence of ammonia (5%) in the carrier gas of [11C]HCN was suspected to be the cause of the relatively low molar activity of [11C]BnCN produced by the traditional method, because ammonia in DMF potentially generates cyanide.20 However, decreasing the ammonia content (<0.2 ppm) in the carrier gas introduced into the reaction solution did not affect the molar activity of [11C]BnCN (entry 4). This indicated that the low molar activity of [11C]BnCN was attributed to that of [11C]HCN produced by the traditional method. The relatively low molar activity of [11C]HCN produced by the traditional method has been occasionally mentioned in literature.3b,21
Fig. 1. External view of reaction column. The video showing the method for the preparation of the reaction column is available in ESI.†.

Yield, purity, and molar activity of [11C]HCN.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Method | Routea | [11C]HCN | [11C]BnCNf | ||||
| Yield (%) | Purity (%) | Yield (%) | A m g (GBq μmol−1) | |||||
| EOBh | EOTi | EOSj | EOTi | EOSj | ||||
| 1 | Current | Route 1b | 60 ± 2.5 | 42 ± 1.6 | 94 ± 0.8 | 11 ± 1.4 | 349 ± 29 | 198 ± 18 |
| 2 | Current | Route 2c | 52 ± 4.1 | 37 ± 2.7 | 88 ± 1.6 | 9.5 ± 0.8 | 340 ± 62 | 193 ± 38 |
| 3 | Traditional | Route 3d | 51 ± 5.6 | 41 ± 4.8 | >99% | 11 ± 1.9 | 103 ± 69 | 53 ± 36 |
| 4 | Traditional | Route 4e | — | — | — | 7.8 ± 1.2 | 109 ± 35 | 55 ± 17 |
Values are presented as mean ± sd (n = 3). Synthetic route for the preparation of [11C]HCN.
The N-oxide layer of reaction column was prepared with the OMT solution.
A mixture of powdered OMT, DPSO, and SiO2 granules was used as the N-oxide layer.
Ordinary synthetic route for [11C]HCN preparation with NH3 included in the carrier gas of [11C]HCN (5% in N2).
NH3 in the carrier gas of [11C]HCN was removed (<0.2 ppm).
Reaction conditions: [11C]HCN was transferred to a reaction vessel containing a DMF solution (300 μL) of benzyl bromide (BnBr, 1 μL) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 1 μL) at −20 °C until the radioactivity in the vessel reached a plateau. [11C]HCN was then reacted at 120 °C for 5 min.
Molar activity.
At the end of bombardment.
At the end of [11C]HCN transfer.
At the end of synthesis (purification).
With decay correction, the molar activity of [11C]HCN produced with the current method was estimated to be 349 ± 29 GBq μmol−1 at the end of collection in the reaction solution (entry 1). This value was comparable to that of [11C]CH3I (345 ± 70 GBq μmol−1) at the start of [11C]CH3I transfer. This indicated that nonradioactive cyanide was not generated or present in the current method. As the molar activity of [11C]HCN produced by the current method depended on that of [11C]CH3I, [11C]HCN with even higher molar activity would be obtained using [11C]CH3I with higher molar activity.
The current method allowed [11C]HCN to be obtained easily in sufficient yield and with high molar activity. Furthermore, [11C]HCN was directly applied to a nucleophilic 11C-cyanation reaction. However, preparation of the reaction column for routine use was somewhat inconvenient, because it required a syringe barrel-type glass tube, the removal of excess solution, the use of HCN-free MeCN, and some operator experience to achieve adequate compression of the quartz wool. These disadvantages were attributed to the method for preparation of the N-oxide layer in the reaction column. To eliminate these problems, the reagents, namely, OMT (25 mg), DPSO (75 mg), and SiO2 granules (1.6 g), were simply mixed and used as the N-oxide layer instead of quartz wool wetted with the MeCN solution of these reagents. DPSO with a melting point of around 70 °C was expected to act as a solvent during heating of the reaction column. Furthermore, OMT was expected to convert [11C]CH3I to [11C]CH2O efficiently, even if not completely dissolved in DPSO, because efficient conversion was observed in the suspension of TMAO in DMSO, as described above (Table 1, entry 1). Along with using the reaction column prepared by this simplified procedure, the reaction conditions were slightly modified to decrease the breakthrough of [11C]CH3I. The reaction temperature further increased to 170 °C from introducing [11C]CH3I at 150–160 °C and the flow rate was reduced from 50 to 40 mL min−1. Although the reaction conditions were not fully optimised, the yield and the molar activity of [11C]HCN produced with the dry reaction column were comparable to those of [11C]HCN produced with the wetted reaction column (Table 3, entries 1 and 2). The radiochemical purity of [11C]HCN in the collected solution was slightly decreased (88%, entry 2) compared with that obtained using the wetted quartz wool (94%, entry 1), whilst [11C]CH3I breakthrough was increased (6.1 ± 3.1% in the collection solution, Fig. S4 in ESI†). These results indicated that the reaction column prepared simply with the reagent mixture was applicable to [11C]HCN production. Furthermore, [11C]HCN produced with the dry reaction column was directly applied to the preparation of [11C]BnCN. The radioactive impurities including [11C]CH3I would not have a significant impact on the 11C-cyanation reaction. However, any impurities or radioactive substances derived from them would potentially make the purification of the desired 11C-labelled compound difficult. In this case, further optimisation or an additional purification process to reduce radioactive impurities is required.
Conclusions
In this study, a simple and efficient [11C]HCN production method was developed. The method is immediately implementable in every 11C production facility. The current method does not require any specialised equipment. By passing [11C]CH3I through the heated reaction column, [11C]HCN was obtained within 2 min after [11C]CH3I transfer into the column, and the yield was comparable to that of the traditional method. In addition to facilitating access to [11C]HCN, this method has further advantages over the traditional method. When OMT and DPSO were used for [11C]CH2O production, this method afforded [11C]HCN with high molar activity corresponding to that of [11C]CH3I used. When [11C]HCN with high molar activity is not required, NMO, which is cheaper and widely available, can be applied instead of OMT. For example, [11C]HCN with a high molar activity might not be necessary for 11C-labelling of biomolecules, such as glucose, amino acids, and fatty acids, which are present in the blood. The other advantage of this method is that the carrier gas of [11C]HCN does not include ammonia, which can potentially interfere with the 11C-cyanation reaction. Furthermore, gaseous [11C]CH2O can be produced within the reaction column including only an N-oxide layer. However, as a disadvantage, whilst the traditional method does not require special advance preparation, the current method requires preparation of the reaction column prior to each [11C]HCN production. Therefore, the reaction column preparation method was simplified as much as possible to reduce this burden. Although [11C]CH3I is produced by two different routes (Scheme 1), [11C]CH3I produced by either route would be applicable to this method. This method allows compounds to be labelled with [11C]HCN for broad application to studies with positron emission tomography.
Data availability
Data supporting this article are available in the ESI.†
Author contributions
Conceptualisation: T. K., M. O., and A. D. G.; funding acquisition: T. K. and M.-R. Z.; investigation: T. K., M. O., and T. O.; methodology: T. K.; project administration: T. K. and M.-R. Z.; validation: T. K., M. O., and T. O.; visualisation: T. K., writing-original draft: T. K.; writing-review and editing: T. K., M. O., T. O., A. D. G., and M.-R. Z.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We would like to thank the technical team of the Cyclotron Section of our institute for their support during cyclotron operation and radioisotope production. We also thank our technical staff members, especially Yusuke Kurihara and Sayaka Shibata, for their assistance in the production of 11C-labelled compounds. This work was supported in part by JSPS KAKENHI Grant Number JP19K08189. We thank Simon Partridge, PhD, from Edanz (https://jp.edanz.com/ac), for editing a draft of this manuscript.
Electronic supplementary information (ESI) available: Experimental details and supplementary figure. See DOI: 10.1039/d1sc07033a
Notes and references
- For selected reviews, see: ; (a) Phelps M. E. Proc. Natl. Acad. Sci. 2000;97:9226–9233. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Willmann J. K. van Bruggen N. Dinkelborg L. M. Gambhir S. S. Nat. Rev. Drug Discovery. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]; (c) Piel M. Vernaleken I. Rösch F. J. Med. Chem. 2014;57:9232–9258. doi: 10.1021/jm5001858. [DOI] [PubMed] [Google Scholar]; (d) Neumann K. Flavell R. Wilson D. M. Semin. Nucl. Med. 2017;47:461–473. doi: 10.1053/j.semnuclmed.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schmidt M. P. Mamet S. D. Ferrieri R. A. Peak D. Siciliano S. D. Mol. Imaging. 2020;19:1–31. doi: 10.1177/1536012120966405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews, see: ; (a) Miller P. W. Long N. J. Vilar R. Gee A. D. Angew. Chem., Int. Ed. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]; (b) Deng X. Rong J. Wang L. Vasdev N. Zhang L. Josephson L. Liang S. H. Angew. Chem., Int. Ed. 2019;58:2580–2605. doi: 10.1002/anie.201805501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews, see: ; (a) Xu Y. Qu W. Eur. J. Org. Chem. 2021;2021:4653–4682. doi: 10.1002/ejoc.202100651. [DOI] [Google Scholar]; (b) Zhou Y. P. Makaravage K. J. Brugarolas P. Nucl. Med. Biol. 2021;102–103:56–86. doi: 10.1016/j.nucmedbio.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Westerberg G. Långström B. Appl. Radiat. Isot. 1997;48:459–461. doi: 10.1016/S0969-8043(96)00303-X. [DOI] [PubMed] [Google Scholar]; (b) Stone-Elander S. Roland P. Halldin C. Hassan M. Seitz R. Int. J. Radiat. Appl. Instrum., Part B. 1989;16:741–746. doi: 10.1016/0883-2897(89)90148-7. [DOI] [PubMed] [Google Scholar]; (c) Johnström P. Bergman L. Varnäs K. Malmquist J. Halldin C. Farde L. Nucl. Med. Biol. 2015;42:555–560. doi: 10.1016/j.nucmedbio.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Cramer R. D. Kistiakowsky G. B. J. Biol. Chem. 1941;137:549–555. doi: 10.1016/S0021-9258(19)56160-9. [DOI] [Google Scholar]
- Endter F. Chem. Ing. Tech. 1958;30:305–310. doi: 10.1002/cite.330300506. [DOI] [Google Scholar]
- (a) Christman D. R. Finn R. D. Karlstrom K. I. Wolf A. P. J. Nucl. Med. 1973;14:864–866. [PubMed] [Google Scholar]; (b) Christman D. R. Finn R. D. Karlstrom K. Wolf A. P. Int. J. Appl. Radiat. Isot. 1975;26:435–442. doi: 10.1016/0020-708X(75)90057-5. [DOI] [Google Scholar]; (c) Iwata R. Ido T. Takahashi T. Nakanishi H. Iida S. Int. J. Rad. Appl. Instrum. A. 1987;38:97–102. doi: 10.1016/0883-2889(87)90003-7. [DOI] [PubMed] [Google Scholar]; (d) Ellison P. A. Jedele A. M. Barnhart T. E. Nickles R. J. Murali D. DeJesus O. T. Proceedings of the 15th International Workshop on Targetry and Target Chemistry. 2015:159–163. [Google Scholar]
- (a) Jewett D. M. Int. J. Rad. Appl. Instrum. A. 1992;43:1383–1385. doi: 10.1016/0883-2889(92)90012-4. [DOI] [PubMed] [Google Scholar]; (b) Schoeps K. O. Stone-Elander S. Halldin C. Int. J. Rad. Appl. Instrum. A. 1989;40:261–262. doi: 10.1016/0883-2889(89)90159-7. [DOI] [PubMed] [Google Scholar]; (c) Kato K. Zhang M. R. Minegishi K. Nengaki N. Takei M. Suzuki K. J. Labelled Compd. Radiopharm. 2011;54:140–144. doi: 10.1002/jlcr.1833. [DOI] [Google Scholar]
- (a) Christman D. R. Crawford E. J. Friedkin M. Wolf A. P. Proc. Natl. Acad. Sci. U. S. A. 1972;69:988–992. doi: 10.1073/pnas.69.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marazano C. Maziére M. Berger G. Comar D. Int. J. Appl. Radiat. Isot. 1977;28:49–52. doi: 10.1016/0020-708X(77)90159-4. [DOI] [PubMed] [Google Scholar]; (c) Halldin C. Långström B. Acta Chem. Scand., Ser. B. 1984;38:1–4. doi: 10.3891/acta.chem.scand.38b-0001. [DOI] [Google Scholar]; (d) Slegers G. Lambrecht R. H. Vandewalle T. Meulewaeter L. Vandecasteele C. J. Nucl. Med. 1984;25:338–342. [PubMed] [Google Scholar]; (e) Hughes J. A. Jay M. Nucl. Med. Biol. 1995;22:105–109. doi: 10.1016/0969-8051(94)00073-S. [DOI] [PubMed] [Google Scholar]; (f) Nader M. W. Zeisler S. K. Theobald A. Oberdorfer F. Appl. Radiat. Isot. 1998;48:1599–1603. doi: 10.1016/S0969-8043(97)10156-7. [DOI] [Google Scholar]; (g) Roeda D. Crouzel C. Appl. Radiat. Isot. 2001;54:935–939. doi: 10.1016/S0969-8043(00)00352-3. [DOI] [PubMed] [Google Scholar]; (h) Roeda D. Dollé F. J. Labelled Compd. Radiopharm. 2003;46:449–458. doi: 10.1002/jlcr.686. [DOI] [Google Scholar]; (i) Van der Mey M. Windhorst A. D. Klok R. P. Herscheid J. D. M. Kennis L. E. Bischoff F. Bakker M. Langlois X. Heylen L. Jurzak M. Leysen J. E. Bioorg. Med. Chem. 2006;14:4526–4534. doi: 10.1016/j.bmc.2006.02.029. [DOI] [PubMed] [Google Scholar]; (j) Hooker J. M. Schönberger M. Schieferstein H. Fowler J. S. Angew. Chem., Int. Ed. 2008;47:5989–5992. doi: 10.1002/anie.200800991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Nader M. Oberdorfer F. Herrmann K. Appl. Radiat. Isot. 2019;148:178–183. doi: 10.1016/j.apradiso.2019.03.032. [DOI] [PubMed] [Google Scholar]
- (a) Franzen V. Otto S. Chem. Ber. 1961;94:1360–1363. doi: 10.1002/cber.19610940530. [DOI] [Google Scholar]; (b) Franzen V. Org. Synth. 1967;47:96. doi: 10.15227/orgsyn.047.0096. [DOI] [Google Scholar]
- Godfrey A. G. Ganem B. Tetrahedron Lett. 1990;31:4825–4826. doi: 10.1016/S0040-4039(00)97742-6. [DOI] [Google Scholar]
- Maurer A. Leonov A. Ryazanov S. Herfert K. Kuebler L. Buss S. Schmidt F. Weckbecker D. Linder R. Bender D. Giese A. Pichler B. J. Griesinger C. ChemMedChem. 2020;15:411–415. doi: 10.1002/cmdc.201900689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen H. H. Gee A. D. Adam M. Antoni G. Cutler C. S. Fujibayashi Y. Jeong J. M. Mach R. H. Mindt T. L. Pike V. W. Windhorst A. D. Nucl. Med. Biol. 2017;55:v–xi. doi: 10.1016/j.nucmedbio.2017.09.004. [DOI] [PubMed] [Google Scholar]
- (a) Traynelis V. J. Hergenrother W. L. J. Org. Chem. 1964;29:221–222. doi: 10.1021/jo01024a505. [DOI] [Google Scholar]; (b) Yang X. Zhang X. Guo Z. Bai W. Hao L. Wei H. Thermochim. Acta. 2013;559:76–78. doi: 10.1016/j.tca.2013.02.027. [DOI] [Google Scholar]; (c) Lin J. Zhou Q. Zhu W. Li Q. Xu L. Chen W. Wang J. Zhang X. Hu W. Li M. J. Pharm. Biomed. Anal. 2020;188:113361. doi: 10.1016/j.jpba.2020.113361. [DOI] [PubMed] [Google Scholar]
- (a) Lindegård B. Mathiasson L. Jönsson J. Å. Åkesson B. J. Chromatogr. 1990;514:293–304. doi: 10.1016/S0021-9673(01)89401-2. [DOI] [PubMed] [Google Scholar]; (b) Potthast A. Rosenau T. Kosma P. Schelosky N. Baldinger T. Holzforschung. 2000;54:641–646. [Google Scholar]
- Neelamegam R. Hellenbrand T. Schroeder F. A. Wang C. Hooker J. M. J. Med. Chem. 2014;57:1488–1494. doi: 10.1021/jm401802f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Meisenheimer J. Chem. Ber. 1919;52:1667–1677. doi: 10.1002/cber.19190520830. [DOI] [Google Scholar]; (b) Hattori Y. Yakugaku Zasshi. 1940;60:24–45. doi: 10.1248/yakushi1881.60.1_24. [DOI] [Google Scholar]; (c) Cope A. C. Foster T. T. Towle P. H. J. Am. Chem. Soc. 1949;71:3929–3935. doi: 10.1021/ja01180a014. [DOI] [Google Scholar]
- (a) Naumann K. Zon G. Mislow K. J. Am. Chem. Soc. 1969;91:7012–7023. doi: 10.1021/ja01053a021. [DOI] [Google Scholar]; (b) Kubas G. J. Larson A. C. Ryan R. R. J. Org. Chem. 1979;44:3867–3871. doi: 10.1021/jo01336a026. [DOI] [Google Scholar]; (c) Kolb H. C. Andersson P. G. Sharpless K. B. J. Am. Chem. Soc. 1994;116:1278–1291. doi: 10.1021/ja00083a014. [DOI] [Google Scholar]
- (a) Kasuga K. Hirobe M. Okamoto T. Yakugaku Zasshi. 1974;94:945–951. doi: 10.1248/yakushi1947.94.8_945. [DOI] [PubMed] [Google Scholar]; (b) Fizet C. Streith J. Tetrahedron Lett. 1974;15:3187–3188. doi: 10.1016/S0040-4039(01)91857-X. [DOI] [Google Scholar]; (c) Streith J. Fizet C. Fritz H. Helv. Chim. Acta. 1976;59:2786–2792. doi: 10.1002/hlca.19760590817. [DOI] [Google Scholar]; (d) Quinn D. J. Haun G. J. Moura-Letts G. Tetrahedron Lett. 2016;57:3844–3847. doi: 10.1016/j.tetlet.2016.07.047. [DOI] [Google Scholar]
- (a) Kim J. Chang S. J. Am. Chem. Soc. 2010;132:10272–10274. doi: 10.1021/ja104917t. [DOI] [PubMed] [Google Scholar]; (b) Kim J. Kim H. Chang S. Org. Lett. 2012;14:3924–3927. doi: 10.1021/ol301674m. [DOI] [PubMed] [Google Scholar]; (c) Pawar A. B. Chang S. Chem. Commun. 2014;50:448–450. doi: 10.1039/C3CC47926A. [DOI] [PubMed] [Google Scholar]
- (a) Pike V. W. Curr. Med. Chem. 2016;23:1818–1869. doi: 10.2174/0929867323666160418114826. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lohith T. G. Tsujikawa T. Siméon F. G. Veronese M. Zoghbi S. S. Lyoo C. H. Kimura Y. Morse C. L. Pike V. W. Fujita M. Innis R. B. J. Cereb. Blood Flow Metab. 2017;37:2458–2470. doi: 10.1177/0271678X16668891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting this article are available in the ESI.†