

Abstract
Background
The incidence of coronary heart disease (CHD) in premenopausal women is significantly lower than that of men of the same age, suggesting protective roles of estrogen for the cardiovascular system against CHD. This study aimed to confirm the protective effect of estrogen on myocardium during myocardial ischemia/reperfusion (MI/R) injury and explore the underlying mechanisms.
Methods
Neonatal rat cardiomyocytes and Sprague–Dawley rats were used in this study. Different groups were treated by bilateral ovariectomy, 17β-estradiol (E2), adenoviral infection, or siRNA transfection. The expression of sarcoplasmic reticulum Ca2+ ATPase pump (SERCA2a) and endoplasmic reticulum (ER) stress-related proteins were measured in each group to examine the effect of different E2 levels and determine the relationship between SERCA2a and ER stress. The cell apoptosis, myocardial infarction size, levels of apoptosis and serum cardiac troponin I, ejection fraction, calcium transient, and morphology changes of the myocardium and ER were examined to verify the effects of E2 on the myocardium.
Results
Bilateral ovariectomy resulted in reduced SERCA2a levels and more severe MI/R injury. E2 treatment increased SERCA2a expression. Both E2 treatment and exogenous SERCA2a overexpression decreased levels of ER stress-related proteins and alleviated myocardial damage. In contrast, SERCA2a knockdown exacerbated ER stress and myocardial damage. Addition of E2 after SERCA2a knockdown did not effectively inhibit ER stress or reduce myocardial injury.
Conclusions
Our data demonstrate that estrogen inhibits ER stress and attenuates MI/R injury by upregulating SERCA2a. These results provide a new potential target for therapeutic intervention and drug discovery in CHD.
Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s12964-022-00842-2.
Keywords: Myocardial ischemia/reperfusion injury, Estrogen, Endoplasmic reticulum stress, SERCA2a
Background
Myocardial ischemia/reperfusion (MI/R) injury refers to the increased damage to cardiomyocytes and their resultant functional decrease after the recovery of blood supply, which caused no-reflow, severe arrhythmia disorders, and other myocardial defects [1]. Thus, coronary revascularization induces MI/R injury, which significantly influences the prognosis of coronary heart disease (CHD) patients. Therefore, we need to fully understand the mechanisms of MI/R injury to develop preventative measures.
There are dramatic gender differences in the incidence of CHD [2]; the prevalence of CHD in premenopausal women is low and symptoms usually occur a full decade later than in men [3], suggesting that female hormones, especially estrogen, may play an essential role in prevention of CHD. In the past 20 years, research into cardiovascular disease in women has increased our understanding of the factors that drive female CHD. Estrogen has several effects on cardiovascular function and disease: it modulates vascular function, the inflammatory response, metabolism, insulin sensitivity, cardiac myocytes, stem cell survival, and the development of hypertrophy [4]. It was observed that the female myocardium has stronger resistance to I/R injury in mice, rats, rabbits and, dogs [5, 6]. Young adult female rats exhibit better recovery of contractile function and fewer arrhythmias during reperfusion than age-matched males [7, 8]. Estrogen pretreatment was indicated to exert protective effects against I/R injury on the myocardium [9–12]. In addition, estrogen derivatives 7 has a cardioprotective effect on calcium channels activation and decreasing myocardial necrosis after I/R [13]. Besides, estrogen was also demonstrated to protect against I/R injury of the brain [14], kidney [15], intestine [16], and liver [17].
The mechanisms of MI/R injury primarily include endoplasmic reticulum (ER) stress, oxidative stress, calcium overload, mitochondrial dysfunction, apoptosis, autophagy activation, and epigenetic changes [18]. Among them, ER stress plays an important role. ER is where many proteins are translated and folded. Approximately 33% of transcripts are synthesized in ER, where they are folded and assembled with the assistance of molecular chaperones and oxidoreductases [19]. Various stimuli, such as oxidative stress, ischemia, and Ca2+ disorders, can cause unfolded or misfolded proteins to accumulate in the ER. This is termed ER stress, and ER stress signaling triggers the unfolded protein response (UPR) to maintain ER stability. The main pathways of the UPR are activated when GRP78 dissociates from the ER transmembrane sensors proteins. These sensors are inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activating transcription factor (ATF) 6.
Nevertheless, under persistent or severe ER stress, the UPR will induce apoptosis [20]. The UPR pathway mediated by ATF6 and PERK, which increase expression of the transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP). CHOP inhibits expression of the anti-apoptotic proteins Bcl-2 and Bnip3 in cardiomyocytes. ER stress can also activate caspase 12, which activates caspase 9/3 to mediate non-mitochondrial apoptosis [21].
Estrogen can inhibit ER stress-induced apoptosis in human endometrial cells, pancreatic β cells, and osteoblasts [22–24]. Many studies have investigated the effects of estrogen on ER stress in tumors and neurological diseases [25–27], but few have studied its impacts on cardiovascular diseases.
Sarcoplasmic reticulum Ca2+ ATPase pump (SERCA2a) is the primary protein involved in the excitation–contraction coupling of cardiomyocytes. SERCA2a pumps Ca2+ from cytoplasm into sarcoplasmic reticulum (SR) in an ATP-dependent manner against the concentration gradient, promoting myocardial relaxation and ensuring enough Ca2+ is present in the SR for release. This allows the myocardium to contract normally. Decreased expression or dysfunction of SERCA2a weakens the ability of the SR to uptake Ca2+, altering intracellular Ca2+ homeostasis [28], which is one of the leading causes of ER stress. Knocking out SERCA2a from mouse cardiomyocytes leads to ER stress and apoptosis [29]; conversely expressing exogenous SERCA2a in porcine myocardial ischemia reduces ER stress [30]. Our group has previously published that SERCA2a expression is significantly reduced in MI/R injury and the increase of SERCA2a could effectively relieve the myocardium damage [31].
Estrogen acts on a variety of tissues and cell, and also has a significant impact on SERCA2a expression in cardiomyocytes. A previous study showed that SERCA2a expression was significantly reduced in cardiomyocytes after rats underwent bilateral ovariectomy (OVX) [32] but increased in H9c2 cells cultured with 17β-estradiol (E2) [33].
Based on these findings, we propose the following hypothesis: (1) estrogen can inhibit ER stress during MI/R injury and reduce myocardial damage; (2) part of this effect of estrogen on ER stress in cardiomyocytes is achieved by increasing SERCA2a levels to maintain intracellular Ca2+ homeostasis. In this study, we will test this hypothesis by detecting SERCA2a expression, ER stress levels, and the degree of MI/R injury.
Methods
Cells and culture conditions
Neonatal rat cardiomyocytes (NRCMs) were isolated from the ventricular myocardium of 1-to-3-day-old Sprague–Dawley (SD) rats by enzymatic dissociation. The Animal Center of Xuzhou Medical University (Xuzhou, China) provided neonatal SD rats. The experimental procedures were approved by the Xuzhou Medical University Animal Ethics Committee (permit number: CMCACUC 2018-04-161).
Animals
Female SD rats (8–10 weeks old) weighing 200–220 g were purchased from the Animal Center of Xuzhou Medical University. The Animal Ethics Committee of Xuzhou Medical University approved all experimental protocols. Rats were housed four per cage under controlled temperature (24 ± 1 °C) and humidity (55% ± 5%) conditions, with luminosity cycles of 12-h light/12-h dark and free access to food and water.
In vitro assays
Cell culture, E2 administration, adenoviral infection, and siRNA transfection
The ventricular myocardium of 1-to-3-day-old SD rats was cut into pieces on ice. The myocardium tissue was then shredded and incubated in 0.08% type I collagenase and 0.08% trypsin for digestion. The digestion product was filtered and collected after 5–6 repetitions. Suspension cells were then gathered and centrifuged at 1000×g for 5 min. The cells were then resuspended in DMEM (Dulbecco's modified eagle medium) containing 20% charcoal-stripped FBS (fetal bovine serum) and 100 μg/mL penicillin/streptomycin and cultured in 5% CO2 for 90 min at 37 °C. Unwalled myocardial cell suspension were then transferred to 3-cm cell dishes at a density of 5 × 105 cells/mL and cultured in 5% CO2 at 37 °C.
After 48 h, Adenovirus-mediated gene transfer was implemented as previously described [34]. Briefly, NRCMs were infected with adenoviral vectors containing the Atp2a2 gene or EGFP (Ad-SERCA2a and Ad-EGFP, respectively, Hanbio Biotechnology Co., Ltd, Shanghai, China). Ad-EGFP was used as a control, and both constructs were used at a multiplicity of infection (MOI) of 200 pfu. Then replace the culture medium 6 h later.
The siRNA transfection and adenoviral infection were performed simultaneously. SERCA2 siRNA and scramble RNA were purchased from GenePharma (Shanghai, China) and transfected into NRCMs using X-tremeGENE™ siRNA Transfection Reagent (Sigma-Aldrich) following the manufacturer's protocol. The SERCA2 siRNA sequence is as follows: sense, 5′-GACUUACUAGUUAGAAUUUTT-3′, antisense, 5′-AAAUUCUAACUAGUAAGUCUU-3′. Transfected cells were cultured for 48 h before performing in vitro hypoxia/reoxygenation (H/R) assays.
After 12 h of siRNA transfection and adenoviral infection, E2 (Sigma-Aldrich, St. Louis, MO, USA) dissolved in DMSO (dimethyl sulfoxide) was added to the culture medium of the E2 intervention groups at a final concentration of 10 nM. The total acting time of E2 was approximately 36 h.
The H/R protocol
Hypoxia conditions were produced by culturing cells with D-Hanks solution (5.37 mM KCl, 136.89 mM NaCl, 0.44 mM KH2PO4, 0.338 mM Na2HPO4, 4.166 mM NaHCO3, and 5 mM D-glucose, pH 7.3–7.4) at 37 °C, saturated with 95% N2 and 5% CO2. The pH was maintained at 6.8 with lactate to mimic an ischemic solution. The cells were cultured for 4 h in a hypoxic incubator equilibrated with 1% O2, 5% CO2, and 94% N2. After hypoxia treatment, the culture medium was replaced with fresh DMEM containing 10% FBS for another 4 h to initiate reoxygenation.
Flow cytometry
The Annexin V-APC Apoptosis Detection Kit (KeyGen Biotech, Nanjing, China) was used to detect apoptosis. Briefly, 5 × 105 cells were collected, washed twice with phosphate buffered saline (PBS), resuspended in 500 μL binding buffer and incubated with 5 μL Annexin V-APC and 5 μL propidium iodide (PI) for 10 min before analysis using a FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Measurements and analysis of calcium transient
NRCMs were plated on coverslips in 3-cm dishes for routine cultivation. After carrying out all intervention protocols, NRCMs were eluted with Hanks Balanced Salt Solution (HBSS), and then incubated with 5 μM/mL Fura 2-AM (Dojindo Laboratories, Kumamoto Japan) in DMSO and diluted in HBSS for 30 min at 37 °C followed by another HBSS elution. NRCMs on coverslips were then sent for calcium transient recording using a dual-excitation fluorescence photomultiplier system (IonOptix, Milton, MA, USA). Briefly, the NRCMs were continuously covered with warm oxygenated Tyrode's solution (140 mM NaCl, 1 mM MgCl2, 6 mM KCl, 10 mM glucose, 2 mM CaCl2, and 5 mM HEPES, pH 7.4) [35]. Fluorescence intensity at 510 nm was detected with alternate scanning by fluorescence emissions at 340 nm and 380 nm. Intracellular Ca2+ levels and the intracellular Ca2+ transient decay time constant were recorded and analyzed by the SoftEdge MyoCam® system (IonOptix).
In vivo assays
OVX protocol and E2 administration
First, 8–10-week-old rats underwent OVX as previously described [36]. Briefly, rats were anesthetized with isoflurane and fixed in the prone position. The hair between the ribs and pelvis at the bottom of the back was shaved and the area disinfected. An approximately 1 cm incision was made parallel to the spine on the right side of the disinfected area. Then the muscle was separated, and small pink spherical ovaries appeared behind the kidney. The ovarian artery and fallopian tube under the ovary were then ligated, the ovaries were removed and the muscle and skin was sutured after resetting the abdominal organs. These steps were then repeated to remove the left ovary. In the sham operation group, only a small piece of adipose tissue from both sides of the incision was removed. Rats were given E2 (dissolved in olive oil) by subcutaneous injection (40 μg/kg/day) for 31 consecutive days starting 2 weeks after the sham operation or bilateral OVX.
Adenoviral infection and MI/R protocol
The Atp2a2 gene or EGFP (Hanbio Biotechnology Co., Ltd) were transferred into the heart by intramuscular injections. First, the left thorax was opened to expose the heart. There were three injection sites in the left ventricle free wall with a total volume of 15 uL of Ad-SERCA2a or Ad-EGFP (1 × 1010 pfu). Three days after transfection, the hearts underwent MI/R, which was induced by left anterior descending artery (LAD) ligation for 30 min followed by 24 h reperfusion.
Terminal deoxynucleotidyl transferase dUTP-nick end labeling (TUNEL)
TUNEL staining was performed according to the manufacturer's protocol (Beyotime Institute of Biotechnology, Shanghai, China). Briefly, after fixation in a 4% paraformaldehyde solution for 15 min, tissue slides were washed twice with PBS, covered with 20 µg/mL proteinase K without DNase for 25 min, and then with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein isothiocyanate-dUTP. The samples were analyzed in the range of 520 ± 20 nm (green) and 460 nm (blue) under a fluorescent and UV light microscope (Tokyo, Japan). For each group, five random fields were examined.
Evans blue and 2,3,5-triphenyltetrazolium chloride (TTC) staining
Evans blue and TTC (Sigma-Aldrich) staining were used to show the myocardial infarct size. After 24 h of reperfusion, the LAD was ligated again in the position of the previous ligation. Then the aorta was clamped, and 1.5 mL of 2% Evans blue was injected into the left atrium to stain the left ventricle myocardium. After 30 s, 10% KCl was injected to cause cardiac arrest. The heart was quickly removed and frozen for 10 min at − 80 °C, and then cut into five 2-mm thick slices. The myocardial slices were immersed in 1% TTC solution (preheated to 37 °C) for 30 min. The color of the non-ischemic area was blue, the white area was the infarct area, and the white/red area was the area at risk (AAR).
Enzyme-linked immunosorbent assay (ELISA)
To measure cardiac troponin‑I (cTn I) levels in the serum of each group, ELISA kits for cTn I were purchased from KeyGEN Bio tech. A multi-detection microplate reader (Synergy™HT, BioTek, Winooski, VT USA) was used for detection. All protocols were performed following the kit's manufacturing recommendations.
Echocardiography
Transthoracic echocardiography was used to measure the ejection fraction (EF%) of the rat heart and to observe the movement of left ventricular wall. The rats were anesthetized after 24 h of reperfusion, and the hair on their left front chest wall was removed. A Vevo 770 (Visual Sonics, Toronto, Canada) with a 13-MHz probe was used to performed this study.
Hematoxylin–eosin (HE) staining
HE staining was used to observe pathological changes to myocardial tissue. Paraffin sections of myocardial tissue were deparaffinized and then placed in a hematoxylin solution for staining. The sections were rinsed with running water for 1 h after color separation. Then the sections that turned blue after dehydration were stained in an eosin staining solution for 5 min. The samples were dehydrated again and soaked in xylene. The sections were then covered with cover glass and observed under an optical microscope.
Masson staining
Masson staining was used to identify fibrosis within the myocardial tissue. After dewaxing, paraffin sections of myocardial tissue were stained with Weigert's hematoxylin solution for 5–10 min, and then stained with Masson blue and ponceau magenta staining solutions for 5 min. The tissues were then placed into aniline blue for 2 min after washing with a phosphomolybdic acid solution. Finally, the sections were dehydrated, made transparent with xylene, and covered. Nuclei and collagen fibers were blue, while the red area was the cytoplasm, muscles, and erythrocytes.
Transmission electron microscopy (TEM)
Left ventricular tissues were fixed in 2.5% glutaraldehyde, and then dehydrated with graded ethanol and acetone solution. The tissues were then embedded and solidified, cut into 50–60 nm slices with an ultramicrotome (Leica EM UC7rt A-1170, Wetzlar, Germany), and then double-stained with 3% uranyl acetate-lead citrate. Finally, the samples were scanned under a TEM (Tecnai™ G2 Spirit TWIN, FEI Co., Hillsboro, Oregon, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from isolated myocardial tissue using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Then, 2 μg RNA samples from each group were assayed by qRT-PCR using the SuperReal PreMix Color kit (TIANGEN Biotech Co. Ltd., Beijing, China) and an ABI7500 real-time DNA detection system (Applied Biosystems, Waltham, MA, USA). Raw data were normalized to GAPDH, and relative expression was calculated using the 2−ΔΔCT. The primers used for qRT-PCR were as follows:
| SERCA2a | Forward: 5′-GTCTGGGTTGTCCTCCTTA-3′ |
| Reverse: 5′-TTTTCCGTTACCTGGCTAT-3′ | |
| GRP78 | Forward: 5′-GTGCGGGCGGAGGAGGAG-3′ |
| Reverse: 5′-ATACGACGGTGTGATGCGGTTG-3′ | |
| CHOP | Forward: 5′-GTCACAAGCACCTCCCAAAGCC-3′ |
| Reverse: 5′-CGCACTGACCACTCTGTTTCCG-3′ | |
| Calreticulin (CRT) | Forward : 5′-CGCACTGACCACTCTGTTTCCG-3′ |
| Reverse: 5′-AATCGGGCATCTTGGCTTGTCTG-3′ | |
| GAPDH | Forward: 5′-GAGAGGGAAATCGTGCGT-3′ |
| Reverse: 5′-GGAGGAAGAGGATGCGG-3′ |
Western blotting
Myocardial tissues were lysed to extract protein, which was subjected to SDS–polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membrane (Merck Millipore, Billerica, MA, USA). Western blotting was performed using primary antibodies against SERCA2a (1:20,000; Abcam, Cambridge, UK), GRP78 (1:10,000), CRT (1:1000), CHOP (1:500), GAPDH (1:10,000; all from Proteintech, Wuhan, China), Bcl-2 (1:1000), Bax (1:1000; both from Cell Signaling Technology, Danvers, MA, USA), and caspase 12 (1:1000; Affinity Biosciences, Inc., Cincinnati, OH, USA). Anti-mouse- and anti-rabbit-horseradish peroxidase-conjugated (HRP) secondary antibodies were purchased from Proteintech. Immunoreactive bands were visualized with ECL chemiluminescent HRP substrate (Bioworld Technology, Inc., Bloomington, MN, USA) using a chemiluminescence detection system (Tanon Imaging System, Tanon, Shanghai, China).
Statistical analysis
Data are expressed as mean ± SEM. All statistical analyses were conducted with GraphPad Prism version 6.0 (GraphPad Software, Inc., San Diego, CA, USA). For statistical analyses, one-way ANOVA was used for intergroup comparison, followed by Bonferroni's multiple comparisons exact probability test. P < 0.05 was considered statistically significant.
Results
E2 increased SERCA2a expression
First, we confirmed that transfecting Ad-SERCA2a and Ad-EGFP at an MOI of 200 pfu was safe and efficient (Fig. 1a). In vitro, SERCA2a protein levels were significantly increased after incubation with 10 nM E2 compared with the control (con) group (P < 0.001) (Fig. 1b). In rats that received intramyocardial injections with Ad-SERCA2a or Ad-EGFP, SERCA2a mRNA and protein expression in cardiomyocytes increased in the Ad-SERCA2a group (P < 0.001), but was unchanged in the Ad-EGFP group (Fig. 1c).
Fig. 1.

The efficiency of SERCA2a overexpression. E2 increased SERCA2a expression in NRCMs. a MOI of Ad-SERCA2a and Ad-EGFP in NRCMs. b The effects of different concentrations of E2 solution on SERCA2a expression in vitro (n = 3; ***P < 0.001 vs. con; ##P < 0.01, ### P < 0.001 vs. 10 nM). c The mRNA and protein expression of SERCA2a after Ad-SERCA2a or Ad-EGFP infection (n = 5; ***P < 0.001 vs. Sham; ###P < 0.001 vs. Ad-EGFP). d SERCA2a protein expression in vivo (n = 3; *P < 0.05, ***P < 0.001 vs. sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. OVX; $P < 0.05, $$$P < 0.001 vs. OVX + E2; %%P < 0.01, %%%P < 0.001 vs. I/R; &&P < 0.01, &&&P < 0.001 vs. OVX + I/R; @@@P < 0.001 vs. OVX + E2 + I/R; ^^^P < 0.001 vs. Ad-EGFP + I/R). e SERCA2a protein expression in vitro (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. con; #P < 0.05, ##P < 0.01, vs. H/R; $P < 0.05, $$P < 0.01 vs. E2 + H/R; %%P < 0.01 vs. Ad-EGFP + H/R; &&P < 0.01, &&&P < 0.001 vs. Ad-SERCA2a + H/R; @@P < 0.01 vs. si-NC + H/R; ^^^P < 0.001 vs. si-SERCA2a + H/R; aaaP < 0.001 vs. Ad-SERCA2a + E2 + H/R)
In vivo, SERCA2a protein levels were significantly decreased in the OVX and I/R groups compared with the sham group (both P < 0.001; Fig. 1d). In contrast, SERCA2a protein levels were increased in the Ad-SERCA2a + I/R group (P < 0.05). Compared with the I/R group, SERCA2a expression was lower in the OVX + I/R group (P < 0.01). SERCA2a expression in the OVX + E2 + I/R and Ad-EGFP + I/R groups were not statistically distinct from the I/R group.
In vitro, SERCA2a expression was decreased in both the H/R and si-SERCA2a + H/R groups compared with the con group (both P < 0.001; Fig. 1e). Nevertheless, the E2 + H/R group had increased SERCA2a expression compared with the H/R group (P < 0.05). SERCA2a was also significantly upregulated in the Ad-SERCA2a + E2 + H/R group, while there was no difference in its protein levels between the si-SERCA2a + E2 + H/R group and the si-SERCA2a + H/R group.
E2 treatment relieved ER stress during MI/R injury by upregulating SERCA2a
Increased expression of CHOP, CRT, caspase 12, and GRP78, which are ER stress-related proteins, is commonly used to detect ER stress [37–39]. In vivo, all the groups that underwent MI/R showed increased CHOP, CRT, caspase 12, and GRP78 expression (Fig. 2a). The OVX + I/R group had the most significant increase in the expression of these proteins. Meanwhile, these proteins showed decreased expression in the OVX + E2 + I/R and Ad-SERCA2a groups compared with the OVX + I/R group.
Fig. 2.

E2 reduced ER stress-related proteins by upregulating SERCA2a. a Protein levels of CHOP, CRT, caspase 12, and GRP78 in vivo (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. OVX; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs. OVX + E2; %P < 0.05, %%P < 0.01, %%%P < 0.001 vs. I/R; &P < 0.05, &&P < 0.01, &&&P < 0.001 vs. OVX + I/R; @P < 0.05, @@P < 0.01 vs. OVX + E2 + I/R; ^P < 0.05 vs. Ad-EGFP + I/R). b Protein levels of CHOP, CRT, caspase 12, and GRP78 in vitro (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. con; #P < 0.05, ##P < 0.01 vs. H/R; $P < 0.05, $$P < 0.01 vs. E2 + H/R; %P < 0.05, %%P < 0.01 vs. Ad-EGFP + H/R; &P < 0.05, &&P < 0.01 vs. Ad-SERCA2a + H/R; @P < 0.05, @@P < 0.01 vs. si-NC + H/R; ^^^P < 0.001 vs. si-SERCA2a + H/R; aaaP < 0.001 vs. Ad-SERCA2a + E2 + H/R)
In vitro, H/R caused increased expression of the ER stress-related proteins, especially in the si-SERCA2a + H/R group (Fig. 2b). In the E2 + H/R and Ad-SERCA2a groups, these proteins showed decreased expression compared with the H/R group. However, there were no significant differences between the si-SERCA2a + H/R group and the si-SERCA2a + E2 + H/R group.
E2 reduced cardiomyocyte apoptosis during MI/R by upregulating SERCA2a
In vivo, a higher percentage of TUNEL-positive cells means that there was more apoptosis in the tissue (Fig. 3a). Apoptotic cells were apparent in every group that underwent the MI/R protocol. Apoptosis levels were highest in the OVX + I/R group; however, the OVX + E2 + I/R and Ad-SERCA2a + I/R groups had less apoptosis than the OVX + I/R group (P < 0.01 and P < 0.001, respectively). Meanwhile, each group showed different levels of the apoptosis-related proteins Bcl-2 and Bax (Fig. 3c). The group with the lowest Bcl-2/Bax ratio had the greatest levels of apoptosis. The ratio in every I/R group was lower than that in the sham group, especially for the OVX + I/R group. However, the Bcl-2/Bax ratio was higher in the OVX + E2 + I/R and Ad-SERCA2a + I/R groups than in the OVX + I/R group (both P < 0.01).
Fig. 3.

E2 relieved apoptosis by upregulating SERCA2a. a Representative images of TUNEL-stained cardiomyocytes of each group in vivo (× 400) and the percentage of TUNEL-positive cells (n = 4). Blue: DAPI-stained nuclei; green: fragmented DNA. b Flow cytometry by staining with Annexin V-APC/PI and the apoptosis rate of each group in vitro (n = 4). c Bcl-2/Bax ratio in myocardial tissue of rats (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. OVX; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs. OVX + E2; &P < 0.05, &&P < 0.01, vs. I/R; @@P < 0.01 vs. OVX + I/R; %P < 0.05 vs. OVX + E2 + I/R; ^P < 0.05 vs. Ad-EGFP + I/R). d Bcl-2/Bax ratio in NRCMs (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. con; #P < 0.05, ##P < 0.01 vs. H/R; $P < 0.05, $$P < 0.01 vs. E2 + H/R; %P < 0.05, %%P < 0.01 vs. Ad-EGFP + H/R; &P < 0.05, &&P < 0.01 vs. Ad-SERCA2a + H/R; @P < 0.05, @@P < 0.01 vs. si-NC + H/R; ^^^P < 0.001 vs. si-SERCA2a + H/R; aaaP < 0.001 vs. Ad-SERCA2a + E2 + H/R)
In vitro, apoptosis rates were determined by apoptosis level of NRCMs (Fig. 3b), and the Bcl-2/Bax ratio (Fig. 3d). H/R increased the apoptosis rate of NRCMs. The si-SERCA2a + H/R group showed more apoptotic cells than the H/R group (P < 0.05). Moreover, the apoptosis rate was decreased in the E2 + H/R and Ad-SERCA2a + H/R groups compared with the H/R group (both P < 0.01). However, the si-SERCA2a + E2 + H/R group had a similar apoptosis rate to the si-SERCA2a + H/R group. The Bcl-2/Bax ratios of each group showed similar trends as the apoptosis rates.
E2 alleviated myocardial infarction and cTn I level by upregulating SERCA2a
In Fig. 4a, successful ligation of the LAD was confirmed by electrocardiography evidence. The ST elevation following LAD ligation returned during reperfusion. As shown in Fig. 4c–e, the percentages of AAR in each group were similar. I/R resulted in myocardial infarction, and the infarct size was increased in the OVX + I/R group compared with the I/R group (P < 0.05). Moreover, the infarct size was decreased in the OVX + E2 + I/R and Ad-SERCA2a + I/R groups compared with the OVX + I/R group (P < 0.01, P < 0.001, respectively).
Fig. 4.

E2 alleviated myocardial infarction and reduced cTn I level by upregulating SERCA2a. a The electrocardiography of rats during the establishment of the MI/R model. b CTn I levels of each group (n = 3). c Representative photographs of Evans blue/TTC-stained heart sections in each group. d Area at risk of each group. e Percentage of infarction size of each group (n = 3). d *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. OVX; $P < 0.05, $$P < 0.01, $$$P < 0.001 vs. OVX + E2; %P < 0.05 vs. I/R; @P < 0.05, @@P < 0.01, @@@P < 0.001 vs. OVX + I/R; &P < 0.05, &&P < 0.01 vs. OVX + E2 + I/R; ^P < 0.01, ^^P < 0.01 vs. Ad-EGFP + I/R
Serum cTn I levels in each group were consistent with the infarct size (Fig. 4b).
E2 increased SERCA2a expression and improved the EF% of the rat heart
I/R injury caused a decrease in the EF% of the heart (Fig. 5a, b). The OVX + I/R group had a lower EF% than the I/R group (P < 0.05). However, the OVX + E2 + I/R group showed no difference in EF% from the I/R group. The EF% of the Ad-SERCA2a + I/R group was higher than that of the I/R group (P < 0.05).
Fig. 5.

E2 ameliorated EF% and Ca2+ transient by increasing SERCA2a expression. a M-mode of echocardiography of each group. b EF% of the rats in each group (n = 4; **P < 0.01, ***P < 0.001 vs. sham; ##P < 0.01, ###P < 0.001 vs. OVX; $$P < 0.01, $$$P < 0.001 vs. OVX + E2; %P < 0.05 vs. I/R; &P < 0.05, &&P < 0.01 vs. OVX + I/R; @P < 0.05 vs. OVX + E2 + I/R; ^P < 0.01 vs. Ad-EGFP + I/R). c Wave of the ratio 340/380 nm in Ca2+ transient of each group in vitro. d–f The trend of the resting intracellular Ca2+ levels (F0), maximal value of intracellular Ca2+ transient (Peak), and the diastolic intracellular Ca2+ transient decay time constant (Tau) (n = 5; *P < 0.05, **P < 0.01, ***P < 0.001 vs. con; #P < 0.05, ##P < 0.01 vs. H/R; $P < 0.05, $$P < 0.01 vs. E2 + H/R; %P < 0.05, %%P < 0.01 vs. Ad-EGFP + H/R; &P < 0.05, &&P < 0.01 vs. Ad-SERCA2a + H/R; @P < 0.05, @@P < 0.01 vs. si-NC + H/R; ^^^P < 0.001 vs. si-SERCA2a + H/R; aaaP < 0.001 vs. Ad-SERCA2a + E2 + H/R)
E2 ameliorated Ca2+ transient by increasing SERCA2a expression
In cardiomyocytes, a transient rise in cytosolic Ca2+ concentration initiates contraction [40]. Ca2+ transient can reflect the contraction capacity of cardiomyocytes (Fig. 5c). There were no differences in resting intracellular Ca2+ levels (F0) among all groups (Fig. 5d). H/R caused a decrease in the maximum intracellular Ca2+ transient (peak) (Fig. 5e). Compared with the H/R group, the peak value of the si-SERCA2a + H/R group was lower (P < 0.05), while the E2 + H/R and Ad-SERCA2a + H/R groups were higher (P < 0.05 and P < 0.01, respectively). Moreover, the si-SERCA2a + E2 + H/R group was not significantly different from the si-SERCA2a + H/R group. The diastolic intracellular Ca2+ transient decay time constant (Tau) was prolonged in the H/R group compared with that in the con group (P < 0.001; Fig. 5f). Compared with the H/R group, Tau was increased in the si-SERCA2a + H/R group (P < 0.05). Meanwhile, Tau was decreased in the E2 + H/R and Ad-SERCA2a + H/R groups compared with the H/R group (both P < 0.01). Nevertheless, the si-SERCA2a + E2 + H/R group was not distinct from the si-SERCA2a + H/R group.
E2 relieved pathological myocardial damage, fibrosis, and ER swelling via increasing SERCA2a expression
As shown in Fig. 6a, cardiomyocytes in the Sham, OVX, and OVX + E2 groups were neatly arranged with normal morphologies and uniform staining. Nuclei were located in the center of myocardial fibers and the nucleolus was apparent. Cardiomyocytes in the I/R group were irregularly and disorderly arranged. Some cases showed edema with fuzzy and pyknotic nuclei. Additionally, myocardial interstitial edema, inflammatory cell infiltration, and local degradation of myocardial tissue was observed. This damage was more severe in the OVX + I/R group than in the I/R group. Some cells were dissolved, necrotic, and fused, with obvious edema, and had reduced nuclei. E2 treatment relieved this damage compared with the OVX + I/R group. Myocardial damage was less severe in the Ad-SERCA2a + I/R group than in the I/R group.
Fig. 6.
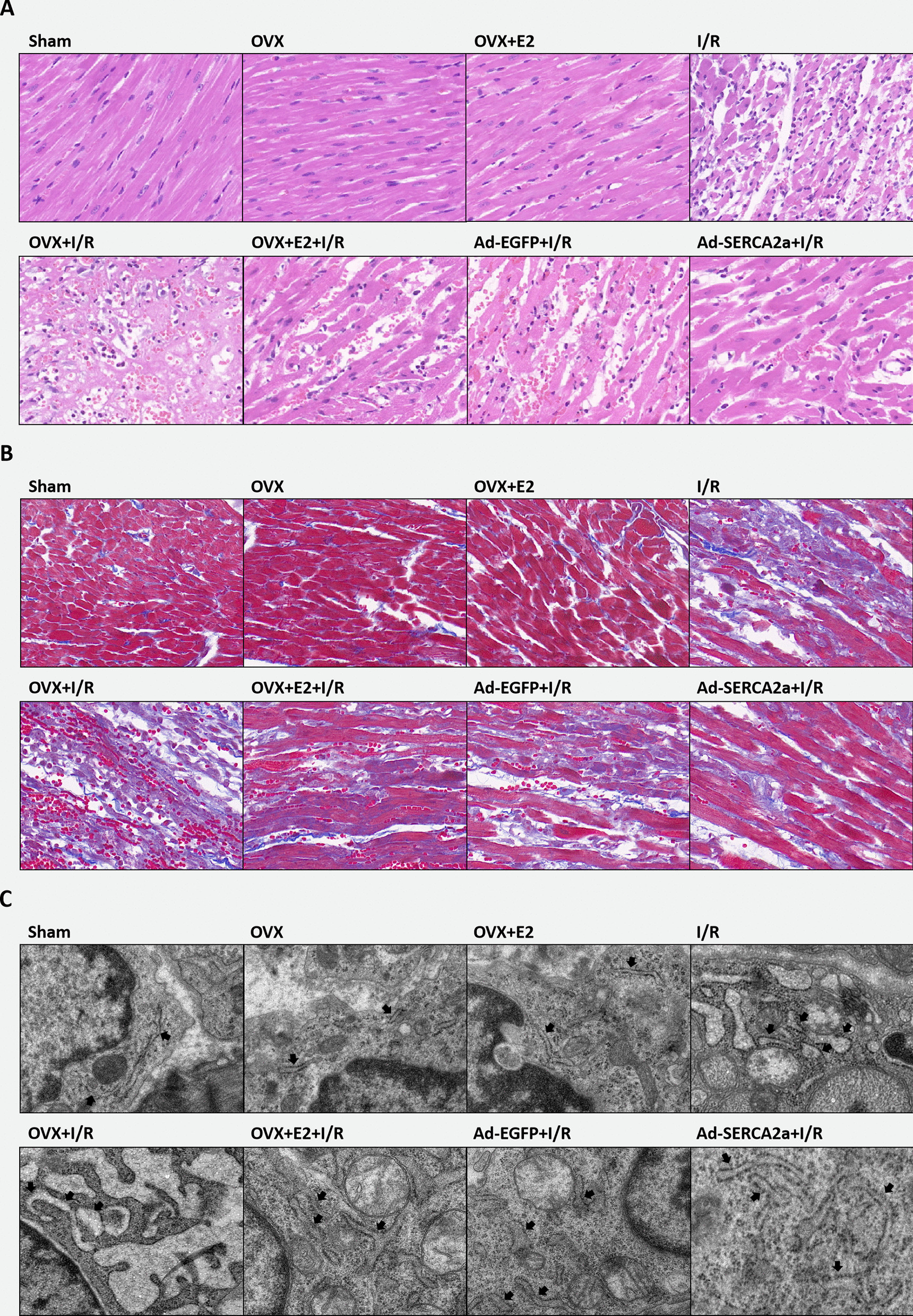
E2 relieved pathological myocardial damage, fibrosis, and ER swelling via increasing SERCA2a expression. a Cardiomyocytes HE staining of each group (n = 5). b Masson staining of every group (n = 5). Collagenous fibers showed as blue. c Representative TEM images of cardiomyocytes (× 11,000; n = 3). Black arrows indicated the ER
Masson staining was used to observe myocardial fibrosis. Collagenous fibers in myocardial tissue were blue (Fig. 6b). Fibrosis was severe in each I/R group, especially in the OVX + I/R group. The OVX + E2 + I/R and Ad-SERCA2a groups had fewer collagenous fibers than the OVX + I/R group.
As shown by TEM analysis, the ER was extended and swollen in the OVX + I/R group. The OVX + E2 + I/R and Ad-SERCA2a + I/R groups exhibited reduced ER swelling compared with the OVX + I/R group (Fig. 6c).
Discussion
This study established the MI/R model in vivo and H/R model in vitro to verify that estrogen could increase SERCA2a expression, inhibit ER stress, and alleviate MI/R injury. We confirmed the causal relationship between increased SERCA2a expression and decreased ER stress through adenoviral infection and siRNA transfection of SERCA2a. These results indicated that estrogen has a protective effect on the myocardium by promoting SERCA2a expression.
Our experimental results showed that SERCA2a expression was significantly reduced in the myocardium of rats after bilateral OVX. However, after supplementing E2, SERCA2a protein level almost returned to normal. This result indicates that estrogen has a significant influence on SERCA2a expression in cardiomyocytes. Previous experiments in our laboratory showed that MI/R cause decreased SERCA2a expression in cardiomyocytes [31, 41, 42], which was further confirmed in this study. We also found that SERCA2a expression was more significantly decreased in the OVX + I/R group than in the I/R group. This result may be the superimposition of the effects of OVX and I/R on SERCA2a, but it does not rule out the possibility that after bilateral OVX, the myocardium, which has now lost the protective effects of estrogen, is more sensitive to I/R injury, causing a more severe decrease in SERCA2a expression. After transfection with Ad-SERCA2a, SERCA2a expression was significantly increased, especially at the transcript level. Although I/R caused decreased SERCA2a expression, SERCA2a levels were higher in the Ad-SERCA2a + I/R group than in the sham group. Transfecting si-SERCA2a resulted in a significant decrease of SERCA2a expression in NRCMs. Even with E2 intervention, SERCA2a levels did not increase. This indicated that E2 might affect the expression of SERCA2a at the transcription level.
ER stress is one of the crucial mechanisms of myocardial I/R injury [37–39, 43]. We determined ER stress levels in each experimental group by detecting the levels of ER stress-related proteins, including GRP78, CRT, and CHOP, all of which are increased under ER stress [44]. We found no significant difference in the non-I/R group, indicating OVX does not cause severe ER stress in cardiomyocytes. Under I/R, the ER stress-related proteins increased sensitively and significantly. Among these groups, ER stress in the OVX + I/R group was most severe; however, it was relieved by E2 treatment. This result indicated that E2 could inhibit ER stress during myocardial I/R. The effect of overexpressing SERCA2a on ER stress was consistent with E2 intervention. ER stress was more severe in the si-SERCA2a + H/R group than in the H/R group due to the lack of SERCA2a. However, ER stress was not relieved in the si-SERCA2a + E2 + H/R group. This indicated that the ability of estrogen to relieve ER stress was related to its ability to increase SERCA2a expression.
ER stress can induce apoptosis [21, 45]. Unlike the death receptor signaling pathway or mitochondrial pathway, apoptosis caused by ER stress specifically requires caspase 12 activation [46]. Therefore, we examined the expression of caspase 12 to observe ER stress-related apoptosis. The results showed that caspase 12 expression was consistent with that of ER stress-related proteins. We also demonstrated that the OVX + I/R group in vivo and the si-SERCA2a + H/R group in vitro showed more apoptotic cells along with experiencing severe ER stress. E2 administration and Ad-SERCA2a transfection could reduce apoptosis levels. TUNEL assays and flow cytometric analyses also showed similar results.
The protective effect of estrogen on the myocardium was also reflected in the area of myocardial infarction, serum cTn I levels, and EF%. Meanwhile, conditions that caused more severe ER stress were associated with more pronounced morphological changes to the ER, which showed varying degrees of swelling and expansion.
Currently, there have been few studies focused on the effects of estrogen on SERCA2a expression in cardiomyocytes. However, the results of this study consistently showed that estrogen promoted SERCA2a expression [32, 47–52]. SERCA2a is an essential protein that maintains intracellular Ca2+ homeostasis and is a crucial protein for cell excitation–contraction coupling [28]. Decreased SERCA2a expression in cardiomyocytes is one of the leading causes of calcium overload in MI/R injury [53]. Disturbing the Ca2+ environment is also crucial for generating ER stress [20]. Therefore, we hypothesized that increasing SERCA2a expression would decrease the ER stress of cardiomyocytes during I/R. This hypothesis was confirmed in this study. Estrogen also affects other calcium handling proteins in cardiomyocytes, such as L-type Ca2+ channel, ryanodine receptor, and sodium-calcium exchanger [54]. The roles of estrogen are pervasive, and its protective effects on the cardiovascular system have been previously confirmed [55–57]. The inhibition of ER stress by estrogen has been studied in many diseases related to the nervous system and reproductive system [58, 59], but not in cardiomyocytes. ER stress is an important mechanism of CHD [60]. This study confirmed the inhibitory effect of estrogen on ER stress during MI/R injury. We found that overexpressing SERCA2a can effectively inhibit ER stress. After transfecting si-SERCA2a, even E2 intervention could not relieve the ER stress and cell damage of H/R. Therefore, we believe that part of estrogen's inhibitory effect on ER stress is through increasing SERCA2a expression.
We found that levels of ER stress-related proteins in the OVX + E2 + I/R group were higher than in the I/R group. We suspect that this might because the E2 supplementation did not fully reach the normal levels in rats due to the absorption efficiency of subcutaneous injection. Hormones secreted by the ovaries include estrogen, progesterone, androgens, and other polypeptide hormones and growth factors. Estrogens include estrone, estradiol, and estriol, of which estradiol has the highest activity [61]. We only supplemented with estradiol, but after removing the ovaries, other ovarian hormones were also decreased in the rats. This may be why simply supplementing E2 could not completely restore the MI/R injury of rats to normal.
Previous studies have shown that estrogen can exert myocardial protection through various mechanisms. Estrogen could inhibit nuclear factor-κB (NF-κB), reducing the infarcted area and neutrophil accumulation [62, 63]. It also acts on PI3K/Akt pathway to reduce oxidative stress, ATP depletion, mPTP opening and increase the eNOS activity [64–66]. Additionally, other reports indicated that estrogen protect against I/R injury by activation of the mitochondrial and sarcolemma ATP-sensitive K+ channels [13], suppressing Ca2+/calmodulin-dependent protein kinase II [54], and modulating manganese superoxide dismutase phosphorylation by mitochondrial p38β at Threonine 79 and Serine 106 [9]. Furthermore, protein kinase C was demonstrated involved in 17β-estradiol-induced protection against MI/R injury and cardiomyocyte apoptosis [11]. Our experimental results further explained the mechanism of myocardial protection of estrogen.
Some downstream pathways of ER stress are also affected by E2. First of all, one of the essential effects of ER stress is to induce specific apoptosis, which has a characteristic increased expression of CHOP and caspase 12. Our experimental results confirmed it. Guo et al. proved that E2 enhances the induction of GRP78 and inhibits the activation of caspase 12 and caspase 3 [24]. It was reported that the anti-apoptotic effect of E2 is dependent on estrogen receptor-mediated upregulation of the expression of anti-apoptotic proteins Bcl-2 and Bcl-xL [67]. Estrogen also rescued pancreatic β-cells from high glucose-induced apoptosis via decreased ER stress followed by downregulation of CHOP [23]. Studies have found that ER stress and inflammatory signaling pathways are coupled through a variety of mechanisms, including NF-κB, activator protein 1 (AP1), reactive oxygen species (ROS), and the release of Ca2+ in the ER [68, 69]. Each of the three pathways of UPR can activate NF-κB through different mechanisms [70]. It has been proven that estrogen can inhibit NF-κB [71], such as in I/R injury [72], atherosclerotic tissues [73], lung cancer [74], and other diseases (Additional file 1).
Moreover, autophagy is one of the effective mechanisms proposed for UPR in recent years. Autophagy is the process of cellular self-degradation, which occurs at a low basal level and helps to remove damaged organelles, cytoplasmic proteins, and pathogens. However, various stress conditions, such as hypoxia, may amplify the incidence of autophagy and destroy cell homeostasis, leading to cell death and apoptosis through excessive self-digestion [75]. ER stress signals can lead to an increase in general autophagy [76]. Some studies have shown that estrogen can induce autophagy through specific pathways to protect cells or tissues from damage [77, 78]. However, other studies demonstrated that estrogen could suppress autophagy activation [79–82]. Whether E2 exerts a protective effect through autophagy remains unknown in our experiments. We confirmed that E2 could promote the expression of SERCA2a, which is upstream of ER stress. It is also significant to investigate the effects of E2 on the downstream pathways of ER stress.
There are still many problems that need to be solved. For example, the mechanism through which estrogen acts on SERCA2a is unclear. It is known that estrogen acts on specific receptors through genomic and non-genomic signaling mechanisms. Genomic actions of estrogen are mediated by classic nuclear estrogen receptors alpha and beta (ERα and ERβ). It is now recognized that many of the rapid, non-genomic effects of estrogen are mediated by G-protein coupled estrogen receptors (GPER). In direct genomic regulation, the binding of E2 to the estrogen receptors promotes homo/hetero dimers that translocate to the nucleus to bind directly to gene Estrogen Response Elements, regulating gene transcription [83]. Estrogen receptors at the plasma membrane or in the cytosol can interact with non-genomic targets like kinases and scaffolding molecules modulating multiple signal pathways [65]. All the estrogen receptor subtypes are widely expressed in cardiovascular tissues in various species, ranging from mice to humans [8]. The ways that E2 acts on the expression of SERCA2a are currently unknown. We suspect that both genomic and non-genomic pathways are involved, which requires further research.
In addition, the decline of SERCA2a will cause Ca2+ disorders and trigger UPR and ER stress, but how changes in the Ca2+ environment trigger UPR is currently unknown.
Conclusions
Our study demonstrated that estrogen alleviates MI/R injury in rats and inhibits ER stress by upregulating SERCA2a. We also confirmed that inhibiting ER stress is one of the mechanisms through which estrogen protects the cardiovascular system. These results suggest an urgent need to study the downstream targets of SERCA2a. Finally, we provided specific references and the basis for further clinical research on the protective effects of estrogen on cardiovascular disease.
Acknowledgements
We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Abbreviations
- CHD
Coronary heart disease
- MI/R
Myocardial ischemia/reperfusion
- E2
17β-Estradiol
- SERCA2a
Sarcoplasmic reticulum Ca2+ ATPase pump
- ER
Endoplasmic reticulum
- UPR
Unfolded protein response
- IRE1α
Inositol-requiring enzyme 1α
- PERK
Protein kinase RNA-like ER kinase
- ATF
Activating transcription factor
- CHOP
CCAAT/enhancer-binding protein homologous protein
- SR
Sarcoplasmic reticulum
- OVX
Bilateral ovariectomy
- NRCMs
Neonatal rat cardiomyocytes
- SD
Sprague–Dawley
- H/R
Hypoxia/reoxygenation
- LAD
Left anterior descending artery
- TTC
2,3,5-Triphenyltetrazolium chloride
- AAR
Area at risk
- EF%
Ejection fraction
- CRT
Calreticulin
- NF-κB
Nuclear factor-κB
- AP1
Activator protein 1
- ROS
Reactive oxygen species
- GPER
G-protein coupled estrogen receptors
Authors' contributions
JWC, DFP, TDX, HZ, and DYL generated the hypothesis and experimental design. JWC, YL and DYL prepared the manuscript. JWC, YL, YYL, WLW, and PW conducted experiments and helped with data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81570326) and Natural Science Foundation of Jiangsu Province (Grants No. BK 20171177).
Availability of data and materials
The datasets used/analyzed to support the conclusion of article are available from the corresponding author upon reasonable request.
Declarations
Ethical Approval and Consent to participate
All the animal experiments were carried out in accordance with the approval of the Animal Ethics Committee of Xuzhou Medical University.
Consent for publication
Not applicable.
Competing interests
The authors report no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jingwen Chen, Email: cjwxyfy@163.com.
Yang Liu, Email: yangliu@xzhmu.edu.cn.
Defeng Pan, Email: xzdefengpan@yahoo.com.
Tongda Xu, Email: xutongda3004@163.com.
Yuanyuan Luo, Email: luoyuanyuanxyfy@126.com.
Wanling Wu, Email: wanlingrose@126.com.
Pei Wu, Email: wupei1995@126.com.
Hong Zhu, Email: hongzhumao@sohu.com.
Dongye Li, Email: dongyeli@xzhmu.edu.cn.
References
- 1.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khamis RY, Ammari T, Mikhail GW. Gender differences in coronary heart disease. Heart. 2016;102(14):1142–1149. doi: 10.1136/heartjnl-2014-306463. [DOI] [PubMed] [Google Scholar]
- 3.Barrett-Connor E. Gender differences and disparities in all-cause and coronary heart disease mortality: epidemiological aspects. Best Pract Res Clin Endocrinol Metab. 2013;27(4):481–500. doi: 10.1016/j.beem.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011;109(6):687–696. doi: 10.1161/CIRCRESAHA.110.236687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson MS, Moore RL, Brown DA. Sex differences in myocardial infarct size are abolished by sarcolemmal KATP channel blockade in rat. Am J Physiol Heart Circ Physiol. 2006;290(6):H2644–H2647. doi: 10.1152/ajpheart.01291.2005. [DOI] [PubMed] [Google Scholar]
- 6.Ross JL, Howlett SE. Age and ovariectomy abolish beneficial effects of female sex on rat ventricular myocytes exposed to simulated ischemia and reperfusion. PLoS ONE. 2012;7(6):e38425. doi: 10.1371/journal.pone.0038425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lujan HL, Dicarlo SE. Sex differences to myocardial ischemia and beta-adrenergic receptor blockade in conscious rats. Am J Physiol Heart Circ Physiol. 2008;294(4):H1523–H1529. doi: 10.1152/ajpheart.01241.2007. [DOI] [PubMed] [Google Scholar]
- 8.Bell JR, Porrello ER, Huggins CE, et al. The intrinsic resistance of female hearts to an ischemic insult is abrogated in primary cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294(4):H1514–H1522. doi: 10.1152/ajpheart.01283.2007. [DOI] [PubMed] [Google Scholar]
- 9.Luo T, Liu H, Kim JK. Estrogen protects the female heart from schemia/reperfusion injury through manganese superoxide dismutase phosphorylation by mitochondrial p38β at threonine 79 and serine 106. PLoS ONE. 2016;11(12):e0167761. doi: 10.1371/journal.pone.0167761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Q, Zhao Z, Sun H, et al. Oestrogen changed cardiomyocyte contraction and beta-adrenoceptor expression in rat hearts subjected to ischaemia–reperfusion. Exp Physiol. 2008;93(9):1034–1043. doi: 10.1113/expphysiol.2007.041939. [DOI] [PubMed] [Google Scholar]
- 11.Morkuniene R, Arandarcikaite O, Ivanoviene L, Borutaite V. Estradiol-induced protection against ischemia-induced heart mitochondrial damage and caspase activation is mediated by protein kinase G. Biochim Biophys Acta. 2010;1797(6–7):1012–1017. doi: 10.1016/j.bbabio.2010.03.027. [DOI] [PubMed] [Google Scholar]
- 12.Kolodgie FD, Farb A, Litovsky SH, et al. Myocardial protection of contractile function after global ischemia by physiologic estrogen replacement in the ovariectomized rat. J Mol Cell Cardiol. 1997;29(9):2403–2414. doi: 10.1006/jmcc.1997.0476. [DOI] [PubMed] [Google Scholar]
- 13.Lauro FV, Francisco DC, Elodia GC, et al. Evaluation of activity of an estrogen-derivative as cardioprotector drug using an ischemia–reperfusion injury model. Int J Clin Exp Med. 2015;8(8):12041–12055. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang ZL, Qin P, Liu Y, et al. Alleviation of ischaemia-reperfusion injury by endogenous estrogen involves maintaining Bcl-2 expression via the ERα signalling pathway. Brain Res. 2017;1661:15–23. doi: 10.1016/j.brainres.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Hutchens MP, Fujiyoshi T, Komers R, et al. Estrogen protects renal endothelial barrier function from ischemia–reperfusion in vitro and in vivo. Am J Physiol Renal Physiol. 2012;303(3):F377–F385. doi: 10.1152/ajprenal.00354.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricardo-da-Silva FY, Fantozzi ET, Rodrigues-Garbin S, et al. Estradiol prevented intestinal ischemia and reperfusion-induced changes in intestinal permeability and motility in male rats. Clinics (Sao Paulo) 2021;76:e2683. doi: 10.6061/clinics/2021/e2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Shi Y, Li Z, et al. BPA disrupts 17-estradiol-mediated hepatic protection against ischemia/reperfusion injury in rat liver by upregulating the Ang II/AT1R signaling pathway. Mol Med Rep. 2020;22(1):416–422. doi: 10.3892/mmr.2020.11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. 2016;7(1):113–170. doi: 10.1002/cphy.c160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73(1):79–94. doi: 10.1007/s00018-015-2052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107(9):1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 21.Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 22.Choi JY, Jo MW, Lee EY, et al. Ovarian steroid dependence of endoplasmic reticulum stress involvement in endometrial cell apoptosis during the human endometrial cycle. Reproduction. 2018;155(6):493–503. doi: 10.1530/REP-17-0713. [DOI] [PubMed] [Google Scholar]
- 23.Kooptiwut S, Mahawong P, Hanchang W, et al. Estrogen reduces endoplasmic reticulum stress to protect against glucotoxicity induced-pancreatic β-cell death. J Steroid Biochem Mol Biol. 2014;139:25–32. doi: 10.1016/j.jsbmb.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Guo YS, Sun Z, Ma J, et al. 17β-Estradiol inhibits ER stress-induced apoptosis through promotion of TFII-I-dependent Grp78 induction in osteoblasts. Lab Investig. 2014;94(8):906–916. doi: 10.1038/labinvest.2014.63. [DOI] [PubMed] [Google Scholar]
- 25.Shapiro DJ, Livezey M, Yu L, et al. Anticipatory UPR activation: a protective pathway and target in cancer. Trends Endocrinol Metab. 2016;27(10):731–741. doi: 10.1016/j.tem.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajapaksa G, Thomas C, Gustafsson JÅ. Estrogen signaling and unfolded protein response in breast cancer. J Steroid Biochem Mol Biol. 2016;163:45–50. doi: 10.1016/j.jsbmb.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 27.Avila MF, Cabezas R, Torrente D, et al. Novel interactions of GRP78: UPR and estrogen responses in the brain. Cell Biol Int. 2013;37(6):521–532. doi: 10.1002/cbin.10058. [DOI] [PubMed] [Google Scholar]
- 28.Zhai Y, Luo Y, Wu P, Li D. New insights into SERCA2a gene therapy in heart failure: pay attention to the negative effects of B-type natriuretic peptides. J Med Genet. 2018;55(5):287–296. doi: 10.1136/jmedgenet-2017-105120. [DOI] [PubMed] [Google Scholar]
- 29.Liu XH, Zhang ZY, Andersson KB, et al. Cardiomyocyte-specific disruption of Serca2 in adult mice causes sarco(endo)plasmic reticulum stress and apoptosis. Cell Calcium. 2011;49(4):201–207. doi: 10.1016/j.ceca.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Xin W, Lu X, Li X, et al. Attenuation of endoplasmic reticulum stress-related myocardial apoptosis by SERCA2a gene delivery in ischemic heart disease. Mol Med. 2011;17(3–4):201–210. doi: 10.2119/molmed.2010.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du Y, Liu P, Xu T, et al. Luteolin modulates SERCA2a leading to attenuation of myocardial ischemia/reperfusion injury via sumoylation at lysine 585 in mice. Cell Physiol Biochem. 2018;45(3):883–898. doi: 10.1159/000487283. [DOI] [PubMed] [Google Scholar]
- 32.Bupha-Intr T, Wattanapermpool J. Regulatory role of ovarian sex hormones in calcium uptake activity of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2006;291(3):H1101–H1108. doi: 10.1152/ajpheart.00660.2005. [DOI] [PubMed] [Google Scholar]
- 33.Liu CG, Xu KQ, Xu X, et al. 17Beta-oestradiol regulates the expression of Na+/K+-ATPase beta1-subunit, sarcoplasmic reticulum Ca2+-ATPase and carbonic anhydrase iv in H9C2 cells. Clin Exp Pharmacol Physiol. 2007;34(10):998–1004. doi: 10.1111/j.1440-1681.2007.04675.x. [DOI] [PubMed] [Google Scholar]
- 34.Zheng D, Wang G, Li S, et al. Calpain-1 induces endoplasmic reticulum stress in promoting cardiomyocyte apoptosis following hypoxia/reoxygenation. Biochim Biophys Acta. 2015;1852(5):882–892. doi: 10.1016/j.bbadis.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wahlquist C, Jeong D, Rojas-Muñoz A, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508(7497):531–535. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Souza VR, Mendes E, Casaro M, et al. Description of ovariectomy protocol in mice. Methods Mol Biol. 2019;1916:303–309. doi: 10.1007/978-1-4939-8994-2_29. [DOI] [PubMed] [Google Scholar]
- 37.Ren L, Wang Q, Chen Y, et al. Involvement of microRNA-133a in the protective effect of hydrogen sulfide against ischemia/reperfusion-induced endoplasmic reticulum stress and cardiomyocyte apoptosis. Pharmacology. 2019;103(1–2):1–9. doi: 10.1159/000492969. [DOI] [PubMed] [Google Scholar]
- 38.Zhong CJ, Chen MM, Lu M, et al. Astrocyte-specific deletion of Kir6.1/K-ATP channel aggravates cerebral ischemia/reperfusion injury through endoplasmic reticulum stress in mice. Exp Neurol. 2019;311:225–233. doi: 10.1016/j.expneurol.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, Wang Z, Ge N, et al. Sodium pump alpha-2 subunit (ATP1A2) alleviates cardiomyocyte anoxia-reoxygenation injury via inhibition of endoplasmic reticulum stress-related apoptosis. Can J Physiol Pharmacol. 2018;96(5):515–520. doi: 10.1139/cjpp-2017-0349. [DOI] [PubMed] [Google Scholar]
- 40.Andersson KB, Birkeland JA, Finsen AV, et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47(2):180–187. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 41.Hu Y, Zhang C, Zhu H, et al. Luteolin modulates SERCA2a via Sp1 upregulation to attenuate myocardial ischemia/reperfusion injury in mice. Sci Rep. 2020;10(1):15407. doi: 10.1038/s41598-020-72325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu S, Xu T, Luo Y, et al. Luteolin enhances sarcoplasmic reticulum Ca2+-ATPase activity through p38 MAPK signaling thus improving rat cardiac function after ischemia/reperfusion. Cell Physiol Biochem. 2017;41(3):999–1010. doi: 10.1159/000460837. [DOI] [PubMed] [Google Scholar]
- 43.Zhu P, Hu S, Jin Q, et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toth A, Nickson P, Mandl A, et al. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets. 2007;7(3):205–218. doi: 10.2174/187152907781745260. [DOI] [PubMed] [Google Scholar]
- 45.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rana SVS. Endoplasmic reticulum stress induced by toxic elements-a review of recent developments. Biol Trace Elem Res. 2020;196(1):10–19. doi: 10.1007/s12011-019-01903-3. [DOI] [PubMed] [Google Scholar]
- 47.Alencar AK, da Silva JS, Lin M, et al. Effect of age, estrogen status, and late-life GPER activation on cardiac structure and function in the Fischer344×Brown Norway female rat. J Gerontol A Biol Sci Med Sci. 2017;72(2):152–162. doi: 10.1093/gerona/glw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunay GA, Paragi P, Sára L, et al. Depressed calcium cycling contributes to lower ischemia tolerance in hearts of estrogen-deficient rats. Menopause. 2015;22(7):773–782. doi: 10.1097/GME.0000000000000377. [DOI] [PubMed] [Google Scholar]
- 49.Ogunbayo OA, Michelangeli F. Related flavonoids cause cooperative inhibition of the sarcoplasmic reticulum Ca2+ ATPase by multimode mechanisms. FEBS J. 2014;281(3):766–777. doi: 10.1111/febs.12621. [DOI] [PubMed] [Google Scholar]
- 50.Kravtsov GM, Kam KW, Liu J, et al. Altered Ca(2+) handling by ryanodine receptor and Na(+)–Ca(2+) exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol. 2007;292(5):C1625–C1635. doi: 10.1152/ajpcell.00368.2006. [DOI] [PubMed] [Google Scholar]
- 51.Liu YH, Qi J, Hou YX, Wang F. Effects of sex hormones on genioglossal muscle contractility and SR Ca2+-ATPase activity in aged rat. Arch Oral Biol. 2008;53(4):353–360. doi: 10.1016/j.archoralbio.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Liu YH, Li W, Song WH. Effects of oestrogen on sarcoplasmic reticulum Ca2+-ATPase activity and gene expression in genioglossus in chronic intermittent hypoxia rat. Arch Oral Biol. 2009;54(4):322–328. doi: 10.1016/j.archoralbio.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Luo Y, Wang S, et al. Roles and mechanisms of SUMOylation on key proteins in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2019;134:154–164. doi: 10.1016/j.yjmcc.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Jiao L, Machuki JO, Wu Q, et al. Estrogen and calcium handling proteins: new discoveries and mechanisms in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2020;318(4):H820–H829. doi: 10.1152/ajpheart.00734.2019. [DOI] [PubMed] [Google Scholar]
- 55.Ostadal B, Ostadal P. Sex-based differences in cardiac ischaemic injury and protection: therapeutic implications. Br J Pharmacol. 2014;171(3):541–554. doi: 10.1111/bph.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Wang IW, Turrentine M, Wang M. Postischemic application of estrogen ameliorates myocardial damage in an in vivo mouse model. J Surg Res. 2018;231:366–372. doi: 10.1016/j.jss.2018.05.076. [DOI] [PubMed] [Google Scholar]
- 57.Menazza S, Sun J, Appachi S, et al. Non-nuclear estrogen receptor alpha activation in endothelium reduces cardiac ischemia–reperfusion injury in mice. J Mol Cell Cardiol. 2017;107:41–51. doi: 10.1016/j.yjmcc.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu F, Ma R, Zhang G, et al. Estrogen and propofol combination therapy inhibits endoplasmic reticulum stress and remarkably attenuates cerebral ischemia–reperfusion injury and OGD injury in hippocampus. Biomed Pharmacother. 2018;108:1596–1606. doi: 10.1016/j.biopha.2018.09.167. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Lin P, Li Y, et al. Brucella suis vaccine strain 2 induces endoplasmic reticulum stress that affects intracellular replication in goat trophoblast cells in vitro. Front Cell Infect Microbiol. 2016;6:19. doi: 10.3389/fcimb.2016.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Binder P, Fang Q, et al. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br J Pharmacol. 2018;175(8):1293–1304. doi: 10.1111/bph.13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leeners B, Geary N, Tobler PN, Asarian L. Ovarian hormones and obesity. Hum Reprod Update. 2017;23(3):300–321. doi: 10.1093/humupd/dmw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pelzer T, Schumann M, Neumann M, et al. 17beta-estradiol prevents programmed cell death in cardiac myocytes. Biochem Biophys Res Commun. 2000;268(1):192–200. doi: 10.1006/bbrc.2000.2073. [DOI] [PubMed] [Google Scholar]
- 63.Pelzer T, Neumann M, de Jager T, et al. Estrogen effects in the myocardium: inhibition of NF-kappaB DNA binding by estrogen receptor-alpha and -beta. Biochem Biophys Res Commun. 2001;286(5):1153–1157. doi: 10.1006/bbrc.2001.5519. [DOI] [PubMed] [Google Scholar]
- 64.Pedram A, Razandi M, Aitkenhead M, Levin ER. Estrogen inhibits cardiomyocyte hypertrophy in vitro. Antagonism of calcineurin-related hypertrophy through induction of MCIP1. J Biol Chem. 2005;280(28):26339–26348. doi: 10.1074/jbc.M414409200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patten RD, Pourati I, Aronovitz MJ, et al. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res. 2004;95(7):692–699. doi: 10.1161/01.RES.0000144126.57786.89. [DOI] [PubMed] [Google Scholar]
- 66.Patten RD, Karas RH. Estrogen replacement and cardiomyocyte protection. Trends Cardiovasc Med. 2006;16(3):69–75. doi: 10.1016/j.tcm.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 68.McGuckin MA, Eri RD, Das I, et al. Intestinal secretory cell ER stress and inflammation. Biochem Soc Trans. 2011;39(4):1081–1085. doi: 10.1042/BST0391081. [DOI] [PubMed] [Google Scholar]
- 69.Garg AD, Kaczmarek A, Krysko O, et al. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol Med. 2012;18(10):589–598. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 70.Hu P, Han Z, Couvillon AD, et al. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26(8):3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalaitzidis D, Gilmore TD. Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab. 2005;16(2):46–52. doi: 10.1016/j.tem.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 72.Guo H, Yang J, Liu M, et al. Selective activation of estrogen receptor β alleviates cerebral ischemia neuroinflammatory injury. Brain Res. 2020;1726:146536. doi: 10.1016/j.brainres.2019.146536. [DOI] [PubMed] [Google Scholar]
- 73.Chen Y, Zhao H, Ren X. Estrogen and progestogen inhibit NF-κB in atherosclerotic tissues of ovariectomized ApoE (-/-) mice. Climacteric. 2016;19(4):357–363. doi: 10.3109/13697137.2016.1167867. [DOI] [PubMed] [Google Scholar]
- 74.Deng S, Ramos-Castaneda M, Velasco WV, et al. Interplay between estrogen and Stat3/NF-κB-driven immunomodulation in lung cancer. Carcinogenesis. 2020;41(11):1529–1542. doi: 10.1093/carcin/bgaa064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhesi P, Tehrani F, Fuess J, Schwarz ER. How does the heart (not) die? The role of autophagy in cardiomyocyte homeostasis and cell death. Heart Fail Rev. 2010;15(1):15–21. doi: 10.1007/s10741-009-9137-y. [DOI] [PubMed] [Google Scholar]
- 76.Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61(6):625–635. doi: 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gavali S, Gupta MK, Daswani B, et al. Estrogen enhances human osteoblast survival and function via promotion of autophagy. Biochim Biophys Acta Mol Cell Res. 2019;1866(9):1498–1507. doi: 10.1016/j.bbamcr.2019.06.014. [DOI] [PubMed] [Google Scholar]
- 78.Mohapatra S, Chakraborty T, Shimizu S, et al. Estrogen and estrogen receptors chauffeur the sex-biased autophagic action in liver. Cell Death Differ. 2020;27(11):3117–3130. doi: 10.1038/s41418-020-0567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hsieh DJ, Kuo WW, Lai YP, et al. 17β-estradiol and/or estrogen receptor β attenuate the autophagic and apoptotic effects induced by prolonged hypoxia through HIF-1α-mediated BNIP3 and IGFBP-3 signaling blockage. Cell Physiol Biochem. 2015;36(1):274–284. doi: 10.1159/000374070. [DOI] [PubMed] [Google Scholar]
- 80.Choi S, Shin H, Song H, Lim HJ. Suppression of autophagic activation in the mouse uterus by estrogen and progesterone. J Endocrinol. 2014;221(1):39–50. doi: 10.1530/JOE-13-0449. [DOI] [PubMed] [Google Scholar]
- 81.Wang F, Xiao J, Shen Y, et al. Estrogen protects cardiomyocytes against lipopolysaccharide by inhibiting autophagy. Mol Med Rep. 2014;10(3):1509–1512. doi: 10.3892/mmr.2014.2365. [DOI] [PubMed] [Google Scholar]
- 82.Chen BC, Weng YJ, Shibu MA, Han CK, Chen YS, et al. Estrogen and/or estrogen receptor α inhibits BNIP3-induced apoptosis and autophagy in H9c2 cardiomyoblast cells. Int J Mol Sci. 2018;19(5):1298. doi: 10.3390/ijms19051298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–170. doi: 10.1016/bs.apcsb.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used/analyzed to support the conclusion of article are available from the corresponding author upon reasonable request.