

Abstract
We previously reported that a-disintegrin and metalloproteinase (ADAM)17 is a key protease regulating myelin formation. We now describe a role for ADAM17 during the Wallerian degeneration (WD) process. Unexpectedly, we observed that glial ADAM17, by regulating p75NTR processing, cell autonomously promotes remyelination, while neuronal ADAM17 is dispensable. Accordingly, p75NTR abnormally accumulates specifically when ADAM17 is maximally expressed leading to a downregulation of tissue plasminogen activator (tPA) expression, excessive fibrin accumulation over time, and delayed remyelination. Mutant mice also present impaired macrophage recruitment and defective nerve conduction velocity (NCV). Thus, ADAM17 expressed in Schwann cells, controls the whole WD process, and its absence hampers effective nerve repair. Collectively, we describe a previously uncharacterized role for glial ADAM17 during nerve regeneration. Based on the results of our study, we posit that, unlike development, glial ADAM17 promotes remyelination through the regulation of p75NTR-mediated fibrinolysis.
SIGNIFICANCE STATEMENT The α-secretase a-disintegrin and metalloproteinase (ADAM)17, although relevant for developmental PNS myelination, has never been investigated in Wallerian degeneration (WD). We now unravel a new mechanism of action for this protease and show that ADAM17 cleaves p75NTR, regulates fibrin clearance, and eventually fine-tunes remyelination. The results presented in this study provide important insights into the complex regulation of remyelination following nerve injury, identifying in ADAM17 and p75NTR a new signaling axis implicated in these events. Modulation of this pathway could have important implications in promoting nerve remyelination, an often-inefficient process, with the aim of restoring a functional axo-glial unit.
Keywords: ADAM17, fibrinolysis, p75NTR, remyelination, Schwann cell, Wallerian degeneration
Introduction
Tumor necrosis factor-α converting enzyme a-disintegrin and metalloproteinase (ADAM)17, also called TACE, is a transmembrane α-secretase belonging to the ADAM family (Gooz, 2010). ADAM17 cleaves several membrane–anchored substrates, among which growth factors and their receptors [e.g., TGF-α, Neuregulin-1 (NRG1), p75NTR, Notch1, Jagged], as well as inflammatory (e.g., TNF-α, TNF-RII, IL-1RII, IL-R) and adhesion molecules (e.g., L1-CAM, NCAM, ICAM-1; Scheller et al., 2011; Zunke and Rose-John, 2017). The biology of ADAM17 is complex as this protease regulates many physiological and pathologic processes, ranging from cardiac development to neuronal differentiation and survival, myelination, neovascularization, inflammation, and tumors' invasiveness (Yang et al., 2006; Gooz, 2010; Scheller et al., 2011; Monk et al., 2015; Zunke and Rose-John, 2017). We previously reported that axonal ADAM17 cleaves NRG1 Type III and inhibits its pro-myelinating activity by suppressing PNS myelination (La Marca et al., 2011). Further, we showed that ADAM17 expressed in Schwann cell is dispensable for myelin formation (La Marca et al., 2011).
After traumatic injury, peripheral nerves undergo a process called Wallerian degeneration (WD), a cascade of events following the disruption of the Schwann cell-axon unit, which lead to axon degeneration and myelin breakdown (Waller and Owen, 1850). Differently from the Central Nervous System, where the regenerative process is mostly inefficient, regeneration in the PNS allows axons regrowth and the re–establishment of a functional myelinated axo-glial unit (Arthur-Farraj et al., 2012; Jessen et al., 2015; Gerdts et al., 2016; Stassart et al., 2018; Pellegatta and Taveggia, 2019). Remyelination is a critical step to achieve a correct and functional nerve repair. One of the key mechanisms that regulates regenerative and remyelinating capacity of nerves is fibrin deposition and its clearance after injury. After a trauma, fibrin accesses the nerves via the vasculature (Akassoglou et al., 2002). Its accumulation contributes to maintain Schwann cells in a nonmyelinating state, with concomitant induction of p75 Neurotrophin receptor (p75NTR) expression, which activates ERK1/2 phosphorylation and inhibits myelin protein zero (P0) mRNA expression (Akassoglou et al., 2002). Fibrin is mainly cleared by the activity of the serine protease plasmin (Weisel and Litvinov, 2017), whose regulation is controlled by the tissue plasminogen activator (tPA) and the plasminogen activator inhibitor-1 (PAI-1; Lijnen, 2001). Plasmin activation is essential for fibrin clearance and peripheral nerves regeneration, as inhibition of fibrinolysis in mice deficient for plasminogen or tPA, exacerbates axonal damage (Akassoglou et al., 2002).
p75NTR, a reported substrate of ADAM17 (Zampieri et al., 2005), orchestrates a variety of cellular function in the nervous system, which are finely regulated by its interaction with specific ligands and co–receptors (Kraemer et al., 2014; Meeker and Williams, 2015). p75NTR is strongly upregulated in nerves after injury (Taniuchi et al., 1986) and its role in the repair program is complex. Along with maintenance of the nonmyelinating Schwann cells state (Mirsky et al., 2008), p75NTR supports neuronal survival and nerve regeneration and remyelination after injury (Song et al., 2006; Tomita et al., 2007; Chen et al., 2016). In addition, p75NTR blocks fibrin degradation after nerve injury by down–regulating tPA and activating PAI-1, ultimately leading to an overall decrease of the extracellular proteolytic activity (Sachs et al., 2007).
We now report that in WD neuronal ADAM17 is dispensable, while glial ADAM17 promotes remyelination by processing p75NTR thus regulating nerve fibrinolysis. Indeed, only transgenic mice lacking glial ADAM17 have impaired remyelination in the late WD. We also demonstrate that abnormal accumulation of p75NTR in injured mutant nerves resolves in downregulation of tPA expression and in fibrin accumulation over time, determining defective nerve repair. Collectively, we propose that during nerve regeneration glial ADAM17 promotes remyelination through the regulation of p75NTR-mediated fibrinolysis.
Materials and Methods
Mice and genotyping
Generation and genotyping of ADAM17 floxed (Adam17flx/flx, IMSR catalog #JAX:009597, RRID:IMSR_JAX:009597) mice have been previously reported (Horiuchi et al., 2007; La Marca et al., 2011). Homozygous ADAM17flx/flx mice were bred with Chat-CRE mice (Chat-IRES-CRE mice, The Jackson Laboratory stock #006410) to delete Adam17 in motor neurons or with P0-CRE mice to delete Adam17 in Schwann cells. Generation and characterization of Chat-CRE transgenic mice and of P0-CRE transgenic mice have been previously described (Feltri et al., 1999; Rossi et al., 2011). P0-CRE//ADAM17flx/flx mice were genotyped as in (La Marca et al., 2011). Chat-CRE mice were genotyped by PCR analysis on genomic DNA, using the following primers: 5′-GCAAAGAACCTCATCTGTGGA-3′ (wild-type allele forward), 5′-CAAAAGCGCTCTGAAGTTCCT-3′ (Chat-CRE allele forward), and 5′-CAGGGTTAGTAGGGGCTGAC-3′ (common reverse). Cycling conditions were: 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s (25 cycles), followed by a 10-min extension at 72°C. All amplified fragments were analyzed on a 2% agarose gel. To characterize Chat-CRE activation pattern, Chat-CRE mice were bred with ROSA-YFP transgenic mice (IMSR catalog #JAX:006148, RRID:IMSR_JAX:006148; Srinivas et al., 2001). ROSA-YFP mice were genotyped using the following primers: 5′-AAAGTCGCTCTGAGTTGTTAT-3′ (wild-type allele forward), 5′-AAGACCGCGAAGAGTTTGTC-3′ (ROSA-YFP allele forward) and 5′-GGAGCGGGAGAAATGGATATG-3′ (common reverse). Cycling conditions were: 94°C for 30 s, 58°C for 60 s and 72°C for 60 s (35 cycles), followed by a 10-min extension at 72°C. All amplified fragments were analyzed on a 2% agarose gel. For ADAM17flx/flx//Chat-CRE mice, the presence of the null allele was determined by PCR on genomic DNA extracted from spinal motor columns and from dorsal portions of the spinal cord using the following primers: 5′-TTACTCTTCTTACTAACAGTCCCCTG-3′ (forward) and 5′-GGGAGAGCCACACCTTGACC-3′ (reverse). Cycling conditions were: 94°C for 30 s, 60°C for 30 s and 72°C for 30 s (30 cycles), followed by a 5-min extension at 72°C. The expected 580-bp amplification product (null allele) has been detected on a 2% agarose gel. On the same samples Adam17 “floxed” allele (700 bp) and wild-type allele (553 bp) have been amplified using the following primers: 5′-TTACTCTTCTTACTAACAGTCCCCTG-3′ (forward) and 5′-AACTATCTCAAACAATAAGCTGAAGTG-3′ (reverse). Cycling conditions were: 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s (30 cycles), followed by a 5-min extension at 72°C. Amplified fragments were analyzed on a 2% agarose gel. All experiments involving animals were performed in strict accordance with protocols approved by the Animal Care and Use Committee of San Raffaele Scientific Institute and the Italian Minister of Health.
Sciatic nerve injury
Two-month-old mice were anesthetized with 2,2,2-tribromoethanol (Sigma-Aldrich; 10 mg/27 g of body weight) as described previously (Forese et al., 2020). Briefly, we exposed the sciatic nerve and caused a mechanical damage at the level of the sciatic notch using precooled forceps. The nerve was then repositioned under the muscle and the incision sutured. Crushed and uncrushed sciatic nerves were then harvested and used for morphologic and biochemical analyses.
Morphologic and morphometric analysis
Semithin and ultrathin sections were obtained as previously described (Quattrini et al., 1996); g ratio measurements were performed on nonoverlapping digitalized electron micrographs images as reported previously (La Marca et al., 2011). The number of remyelinated fibers and the number of degenerated fibers/myelin ovoids per area (mm2) have been determined on images of entire sections of crushed sciatic nerves from at least three transgenic mice and littermate controls. To this purpose we used the NIH ImageJ v1.45s software (NIMH Image Library, RRID:SCR_005588) on images acquired with the same settings (for reference, see Forese et al., 2020). Data were randomly collected and blindly assessed.
RNA extraction and reverse transcription
Total RNA was isolated from crushed and uncrushed mouse sciatic nerves using TriPure Isolation reagent (Roche) as per manufacturer's instructions. 300 ng of total mRNA was then reverse–transcribed to cDNA using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) according to the manufacturer's instruction.
Quantitative real-time PCR
cDNA aliquots from wild-type sciatic nerves uncrushed and harvested 3, 7, 14, 21, and 60 d after injury were tested in parallel by quantitative PCR to determine Adam17 expression using the following primers [5′-GGCAGATAACATCGTTGGGTCTG-3′ (forward) and 5′-CCATGCTGCTCAGCATCTCAATG-3′ (reverse)]. All experiments were performed in triplicate, using RNA preparations from three different mice/time point. The reaction was performed on the StepOne Plus Real-Time PCR System (Thermo Fisher Scientific) using PowerUp SYBR Green Master mix (Thermo Fisher Scientific). Cycling conditions were: 50°C for 2 min, 95°C for 2 min, followed by 95°C for 15 s, 57°C for 30 s, and 72°C for 1 min (39 cycles). Results were analyzed with the StepOne Software v2.3 (Thermo Fisher Scientific) by mean of the comparative Ct method (ΔΔCT). We used Hprt as a housekeeping gene [5′-CAGACTGAAGAGCTACTGTAATG-3′ (forward) and 5′-GGGCTGTACTGCTTAACCAGG-3′ (reverse)]. Statistical analyses were performed using the GraphPad Prism Software package (GraphPad version 8.0f GraphPad Prism, RRID:SCR_002798).
To analyze tPA, PAI-1, and P0 expression we purified RNA from 5 different preparations/genotype using pools of two sciatic nerves/preparation. qPCR experiments were performed using the following primers: for tPA 5′-GCCATGTGATGAAGGACCGA-3′ (forward) and 5′-GATCAGCACCCCTCCACAAA-3′ (reverse); for PAI-1 5′-TCAGTGGCCAATGGAAGACT-3′ (forward) and 5′-CGATGAACATGCTGAGGGTG-3′ (reverse); for P0 5′-CACAACCTAGACTACAGTGACAACG-3′ (forward) and 5′-TTCGAGGAGTCCTTCGAAGATTTG-3′ (reverse). Experiments were performed on the C1000 Thermal Cycler with CFX96 Real-Time System (Bio-Rad), using Sso FastTM EvaGreen Supermix (Bio-Rad) according to manufacturer's instruction. Cycling conditions were: 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min (40 cycles). Results were analyzed with Bio-Rad CFX Manager 3.1 software by mean of the comparative Ct method (ΔΔCT). As before, we used Hprt as a housekeeping gene. Statistical analyses were performed using the GraphPad Prism Software package.
Standard curves were performed to determine the right amount of cDNA template and the optimal cycling conditions to use for each gene analyzed. Genes of interest and the housekeeping gene Hprt were always amplified using the same cycling conditions.
In situ hybridization
Adam17 ribobrope was prepared from pBluescript-KS- plasmid containing cDNA encoding Adam17 mouse exon 2–3 and the beginning of exon 4. The cDNA was amplified using the following primers: 5′-GCGACTTGGGTACCACAACCTTGATTCTTT-3′ (forward) and 5′-TATGTGGCTAGCTCGAGAACCCTAGAGTCA-3′ (reverse). PCR cycling conditions were: 98°C for 30 s, 62°C for 30 s, 72°C 60 s (34 cycles). The amplified product was cloned in Kpn I and Xho I (New England Biolabs) cloning sites using standard molecular biology procedures. In situ hybridization was performed as previously described (Forese et al., 2020).
Preparation of protein lysates and immunoblotting
Crushed and uncrushed sciatic nerves were harvested from at least three transgenic mice and littermate controls and immediately frozen in liquid nitrogen. Total protein lysates from each sample, Western blotting procedures and quantitative analyses were conducted exactly as reported in (La Marca et al., 2011).
Antibodies
All antibodies were previously validated for the applications used. Primary antibodies used for Western blot analyses included: anti-ADAM17 (1:1000, Abcam, catalog #ab39162, RRID:AB_722565), anti-p–ErbB2 (1:50, Santa Cruz Biotechnology, catalog #sc-12352-R, RRID:AB_653115), anti-total ErbB2 (1:500, Cell Signaling Technology, catalog #2165, RRID:AB_10692490), anti-p-ERK1/2 (1:1000, Cell Signaling Technology, catalog #9101, RRID:AB_331646), anti-total ERK1/2 (1:1000, Cell Signaling Technology, catalog #9102, RRID:AB_330744), anti-p-AKT Ser473 (1:1000, Cell Signaling Technology, catalog #4060, RRID:AB_2315049), anti-total AKT (1:1000, Cell Signaling Technology, catalog #4691, RRID:AB_915783), anti-p75NTR (1:3000, a kind gift of Nicolò Zampieri), anti-Fibrinogen (1:3000, Chemicon, catalog #AB3236), anti-Vinculin (1:1000, Millipore, catalog #05-386; RRID:AB_11212640), anti-Calnexin (1:2000, Sigma-Aldrich, catalog #C4731, RRID:AB_476845). Primary antibodies for immunohistochemical analyses included anti-GFP (1:1000; Abcam catalog #ab13970 RRID:AB_300798), anti-Neurofilament H SMI 31 and SMI 32 (1:1000, BioLegend catalog #801601, RRID:AB_10122491; BioLegend catalog #801701, RRID:AB_2315331), anti-Neurofilament M (1:1000, BioLegend catalog #822701, RRID:AB_2564860), anti-Neu N (1:200, Millipore catalog #MAB377, RRID:AB_2298772), anti-F4/80 (1:200, Bio-Rad catalog #MCA497GA, RRID:AB_323806), anti-Fibrinogen (1:1000, DAKO, catalog #A0080, RRID:AB_2894406).
Secondary antibodies for Western blot analyses were all used 1:10,000 in PBS 1× and included: anti-mouse IRDye 680LT (LI–COR, catalog #926-68070, RRID:AB_10956588), anti-rabbit IRDye 800CW (LI–COR, catalog #926-32211, RRID:AB_621843), anti-rabbit IRDye 680LT (LI–COR, catalog #926-68021; RRID:AB_10706309). Secondary antibodies for immunohistochemistry were all used 1:500 and included: DyLight 549 (Jackson ImmunoResearch catalog #115-505-146, RRID:AB_2341133), and DyLight 488 (Jackson ImmunoResearch catalog #111-485-045, RRID:AB_2341130).
Immunohistochemistry
Postnatal mice were fixed by intracardiac perfusion with cooled 4% paraformaldehyde in 0.1 m phosphate buffer pH 7.2. Spinal cords and sciatic nerves were dissected and postfixed in 4% paraformaldehyde overnight at 4°C. P5 and embryonic tissues were collected without perfusion and directly fixed in 4% paraformaldehyde for 5 d. Tissues were cryoprotected in 30% sucrose in 0.1 m phosphate buffer pH 7.2 at 4°C overnight, embedded in OCT (Killik, Bio-Optica) and stored at –80°C until used. Sections (12 μm thick) were cryostat cut (Leica) at −20°C and collected on Superfrost glass slides (Thermo Scientific). Sections were permeabilized in cold acetone (Sigma-Aldrich) for 15 min at −20°C, blocked in 5% BSA (Sigma-Aldrich), 1% donkey serum (Jackson ImmunoResearch), 0.2% Triton X-100 (Sigma-Aldrich) in 1× PBS for 1 h at room temperature and incubated overnight at 4°C with primary antibodies. Sections were rinsed in 1× PBS, incubated for 1 h with secondary antibodies, stained with 1 µg/ml Hoechst (Sigma-Aldrich) and mounted with Vectashield. Images were acquired by confocal microscopy on a Leica TCS SP5. For cell counting, sciatic nerve sections from at least three animals/genotype were processed for immunohistochemistry and representative images were acquired with an epi-fluorescence microscope (Leica DM5500 B). Longitudinal sections of the whole nerve were acquired at low magnification and then matched to reconstruct the entire nerve. The number of F4/80 cells per area was counted using the NIH ImageJ v1.45s software on images acquired with the same settings and normalized by the area of the reconstructed nerve (mm2).
Behavioral analysis
Motor ability was assessed using the accelerating Rotarod (47650 Rota-Rod NG, Ugo Basile). Groups of two-month-old uncrushed (three mice/genotype) and 60 d postcrush (five to eight mice/genotype) transgenic and control animals were tested in two sessions of three trials each per day (3-h rest between the two daily sessions) for three consecutive days. During the test, the rod accelerated from 5 to 25 rotations per minute, and the time that the animal remained on the rod (maximum, 900 s) was measured, graphed and statistically analyzed using the Prism Software package.
Neurophysiology
Neurophysiology was performed as specified in (Triolo et al., 2006). Briefly, we analyzed groups of two-month-old uncrushed (three mice/genotype) and 60 d postcrush (6–10 mice/genotype) transgenic and control animals. Mice were anesthetized with 2,2,2-tribromoethanol and placed under a warming lamp to prevent hypothermia. Sciatic nerve conduction velocities (NCVs) were measured by stimulating the nerve with monopolar needle electrodes. A couple of stimulating electrodes was placed subcutaneously at the ankle. A second pair of electrodes was placed at the sciatic notch, to get two distinct sites of stimulation at the distal and proximal portion of the nerve, respectively. The muscular response to the electrical nerve stimulation (compound muscle action potentials; CMAPs) was measured with a pair of needle electrodes; the active electrode was placed in the middle of the paw, while the reference was placed between the first and the second digit. All measurements were graphed and statistically analyzed using the Prism Software package.
Statistical analyses
No statistical assays were used to predetermine the sample size. Data were collected randomly and assessed blindly. The data distribution was assumed to be normal, although we did not formally test it. Statistical analyses are reported in each figure legend (two tailed unpaired t test, Fisher's exact test, χ2 test) and were performed on at least three different experiments using the Prism Software package. A value of p ≤ 0.05 was considered statistically significant. Graphical data are represented as mean ± SEM.
Data availability
All relevant data of the present manuscript are available from the corresponding authors on reasonable request.
Results
ADAM17 is upregulated in sciatic nerves at T21
We have previously demonstrated that ADAM17 plays a key role in regulating myelin formation during PNS development (La Marca et al., 2011). However, it is not known whether this α-secretase is involved in peripheral nerve regeneration and remyelination after injury. To analyze the role of ADAM17 in WD, we first evaluated its expression levels in mouse sciatic nerves upon mechanical injury. We thus performed crush injury in two-month-old wild-type sciatic nerves and assessed Adam17 mRNA and protein expression at different time points by quantitative RT-PCR (qRT-PCR) and Western blot analyses. We determined ADAM17 expression soon after injury, when the nerve is undergoing extensive degeneration (T3–T7), during regeneration and early remyelination phases (T14–T21) and during late remyelination events (T60). Our results indicate that Adam17 mRNA levels decrease in sciatic nerves soon after injury (T3) but are progressively re–expressed paralleling nerve regeneration (T7–T14), suggesting that ADAM17 might have a role in remyelination. Interestingly, at T21 Adam17 mRNA levels were upregulated as compared with uncrushed nerves, further underscoring a possible role for this secretase in PNS remyelination (Fig. 1A). At protein level, we found that ADAM17 pro-protein (110 kDa) increases soon after injury (T3–T21), whereas the active form (∼80 kDa) peaks at T21 (Fig. 1B–D), during early remyelination phases, paralleling qRT-PCR data. During late phases of WD, ADAM17 levels decreased, although its expression persisted until T60. Collectively, these results suggest a precise fine–tuning in the expression of this secretase during specific stages of peripheral nerve regeneration and remyelination.
Figure 1.

ADAM17 is upregulated in injured sciatic nerves. A, Graph showing one out of three independent quantitative PCR analyses on mRNA prepared from wild-type uncrushed (Unc) and crushed sciatic nerves at T3, T7, T14, T21, and T60. Adam17 mRNA level is increased at T21, whereas it is significantly downregulated at all time points relative to uncrushed controls, arbitrarily set as 1.0. Error bars represent mean ± SEM (T3: unpaired t test **p = 0.0012 t = 8.249, df = 4; T7: unpaired t test **p = 0.0056 t = 5.415, df = 4; T14: unpaired t test *p = 0.0205 t = 3.719, df = 4; T21: unpaired t test ***p = 0.001 t = 8.645, df = 4; T60: unpaired t test ***p = 0.0005 t = 10.21, df = 4). N = 3 different RNA preparations and analyses. B, One out of three representative Western blot analyses showing ADAM17 protein levels (arrowhead pro-protein, arrow active form) and Vinculin, used as a loading control, in T3, T7, T14, T21, and T60 wild-type sciatic nerves relative to uncrushed controls (Unc). Active ADAM17 accumulates in injured nerves accumulates at T21. N = 3 different protein preparation/time point. C, Graph, average of three different experiments, showing levels of inactive ADAM17 (110 kDa) at T3, T7, T14, T21, and T60 wild-type sciatic nerves relative to uncrushed controls (Unc), arbitrarily set as 1.0, as determined using the LI–COR Odyssey® quantitative system. Levels of ADAM17 protein were normalized over Vinculin, used as loading control. Error bars represent mean ± SEM (T3: unpaired t test p = 0.1211, t = 1.963, df = 4; T7: unpaired t test *p = 0.0178, t = 3.882, df = 4; T14: unpaired t test *p = 0.0299, t = 3.302, df = 4; T21: unpaired t test p = 0.0679, t = 2.484, df = 4; T60: unpaired t test p = 0.3914, t = 0.9600, df = 4). N = 3 different protein preparation/time point. D, Graph, average of three different experiments, showing levels of active ADAM 17 (80 kDa) at T3, T7, T14, T21, and T60 wild-type sciatic nerves relative to uncrushed controls (Unc), arbitrarily set as 1.0, as determined using the LI–COR Odyssey® quantitative system. Levels of ADAM17 protein were normalized over Vinculin, used as loading control. Active ADAM17 accumulates at T21. Error bars represent mean ± SEM (T3: unpaired t test **p = 0.0076, t = 4.972, df = 4; T7: unpaired t test p = 0.0655, t = 2.518, df = 4; T14: unpaired t test p = 0.6799, t = 0.4441, df = 4; T21: unpaired t test *p = 0.0345, t = 3.150, df = 4; T60: unpaired t test p = 0.3465, t = 1.066, df = 4). N = 3 different protein preparation/time point.
Axonal ADAM17 is dispensable for peripheral nerve regeneration and remyelination
Our previous results indicate that ADAM17 is timely modulated after nerve injury, and suggest a role for this protein during regenerative events. Since both Schwann cells and neurons express ADAM17 (La Marca et al., 2011; Bolino et al., 2016), we sought to unravel its cell autonomous role in peripheral nerves after injury.
To evaluate whether axonal ADAM17 is required for nerve regeneration, as it occurs in development (La Marca et al., 2011), we generated mice specifically lacking Adam17 in motor neurons. We crossed Adam17flx/flx mutant mice (Horiuchi et al., 2007) with Chat-CRE transgenic mice, which have been previously used to drive cholinergic neurons specific recombination (Rossi et al., 2011).
Thus, we asked whether neuronal Adam17 is required for peripheral nerve regeneration and remyelination. We surgically exposed sciatic nerves of two-month-old controls and Chat-CRE//Adam17flx/flx mutants and performed crush injury by mechanical pressure. We then analyzed sciatic nerves morphology at T7, T14, T21, and T60 (Fig. 2). At T7 control nerves underwent extensive degeneration, showing collapsing axons and myelin debris. Between T14 and T21 the majority of axons in control nerves regenerated and Schwann cells started to remyelinate, a process almost complete at T60. A careful analysis revealed that Adam17 conditional null nerves morphologically resembled injured control nerves at all analyzed time points (Table 1), indicating that regeneration and remyelination are not impaired. These results suggest that neuronal Adam17 is dispensable for remyelination, differently from what we previously reported in development (La Marca et al., 2011). To further corroborate these data, we counted the number of remyelinated fibers in injured Chat-CRE//Adam17flx/flx versus control nerves and found no differences among these genotypes at all time points analyzed (Table 1). We also quantified myelination in regenerated fibers by determining g ratio values at T14, T21, and T60 (Table 1). In agreement with a dispensable role of ADAM17 in remyelination, the thickness of the myelin sheath was similar in mutant and control nerves.
Figure 2.
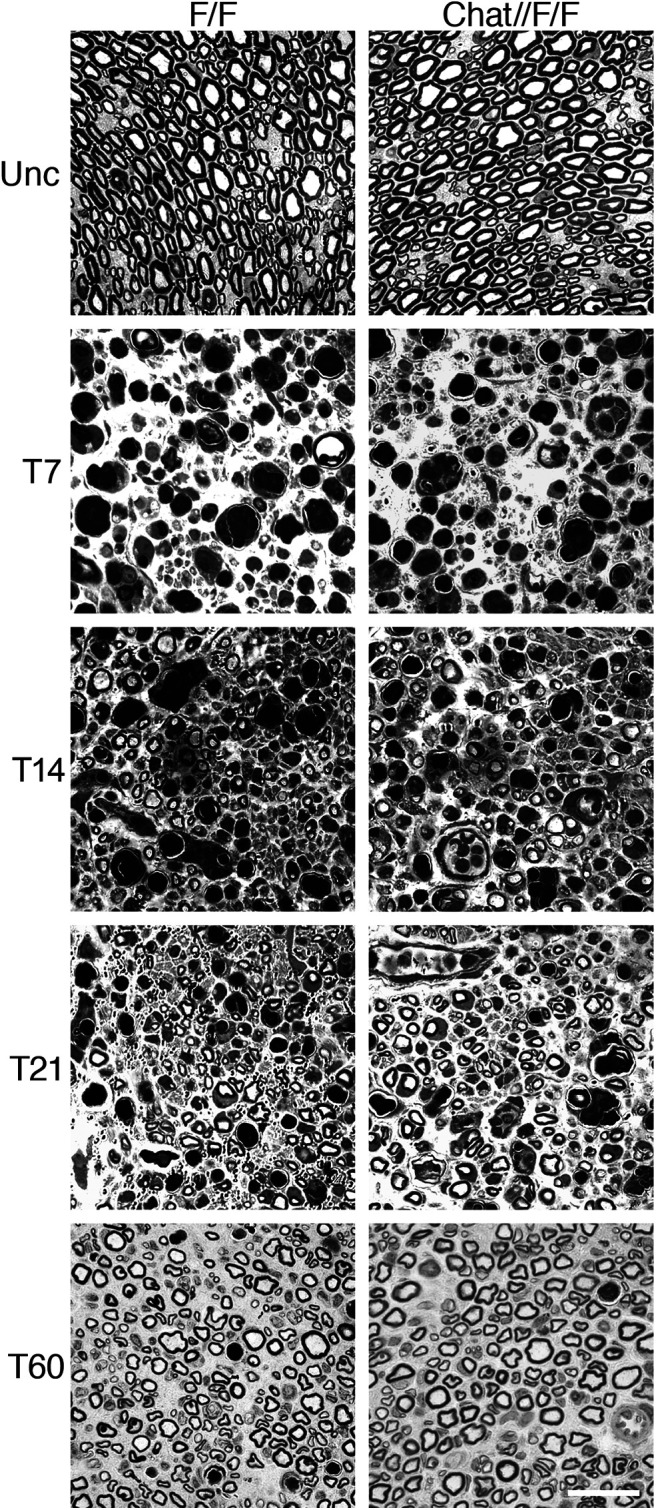
Axonal ADAM17 is dispensable for nerve regeneration and remyelination. Morphologic analyses of Chat-CRE//Adam17flx/flx and littermate controls injured sciatic nerves are comparable. Semithin sections of two-month-old uncrushed (Unc) controls and Chat-CRE//Adam17flx/flx sciatic nerves (top panels). Semithin sections of T7, T14, T21, and T60 controls and Chat-CRE//Adam17flx/flx crushed sciatic nerves. Scale bar: 20 µm.
Table 1.
Morphometric analyses of control (F/F) and Chat-CRE//Adam17flx/flx (Chat//F/F) nerves at different time points after crush injury
| P7 | ||||
| F/F | Chat//F/F | N | P | |
| g ratio | 0.690 ± 0.002 | 0.654 ± 0.004 | n = 3 | p = 0.0014 |
| P30 | ||||
| F/F | Chat//F/F | N | P | |
| g ratio | 0.669 ± 0.004 | 0.638 ± 0.002 | n = 3 | p = 0.0031 |
| T7 | ||||
| F/F | Chat//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 0.91 ± 0.16 | 0.86 ± 0.19 | n ≥ 4 | p = 0.856 |
| T14 | ||||
| F/F | Chat//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 5.04 ± 1.29 | 4.95 ± 1.51 | n ≥ 5 | p = 0.962 |
| g ratio | 0.792 ± 0.010 | 0.786 ± 0.014 | n = 7 | p = 0.778 |
| T21 | ||||
| F/F | Chat//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 11.27 ± 1.85 | 9.887 ± 2.77 | n = 5 | p = 0.688 |
| g ratio | 0.700 ± 0.009 | 0.697 ± 0.009 | n = 5 | p = 0.766 |
| T60 | ||||
| F/F | Chat//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 24.26 ± 1.21 | 22.72 ± 2.38 | n ≥ 4 | p = 0.557 |
| g ratio | 0.672 ± 0.007 | 0.664 ± 0.009 | n = 5 | p = 0.465 |
Summary of the morphometric analyses performed in control (F/F) and Chat-CRE//Adam17flx/flx (Chat//F/f) mice after sciatic nerve crush injury. Values represent mean ± SEM (t test analyses). Results with p-values < 0.05 have been considered significant. For g ratio analyses, more than 300 fibers/genotype/time point have been analyzed.
To validate this result and to exclude that inefficient recombination might disguise the analyses, we assessed recombination at the Adam17 locus on genomic DNA prepared from embryonic day (E)12.5 spinal motor columns and postnatal day (P)5 ventral spinal cord portions of Chat-CRE//Adam17flx/flx mice and littermate controls (Adam17flx/flx) by PCR analyses (Fig. 3A). As expected, we observed the 580-bp amplified band representing the null allele, only in Chat-CRE//Adam17flx/flx mice. Next, we determined Adam17 mRNA expression by in situ hybridization on P1 spinal cord sections harvested from Chat-CRE//Adam17flx/flx mice and littermate controls. Adam17 mRNA is highly expressed in neuronal bodies in control P1 spinal cord sections, and in particular in large motor neurons cell bodies in the ventral spinal cord (Fig. 3B). In turn, this specific signal is absent in spinal cords of mutant animals, corroborating ablation of Adam17 transcript specifically in Chat-CRE-expressing motor neurons. Further, we confirmed cellular and temporal specific recombination of the Chat-CRE using the ROSA-YFP transgene. Thus, we crossed Chat-CRE mutants with the ROSA-YFP reporter strain and monitored YFP expression by immunohistochemistry from E12.5 until P30 in motor neurons, ventral spinal roots and peripheral nerves (Fig. 3; data not shown). We observed specific and substantial YFP expression in both E18.5 ventral spinal cord neuronal cell bodies and axons (Fig. 3C) and in peripheral nerves (Fig. 3D), as well as in P30 sciatic nerves (Fig. 3E), indicating that inefficient recombination is unlikely the cause of the observed phenotype.
Figure 3.
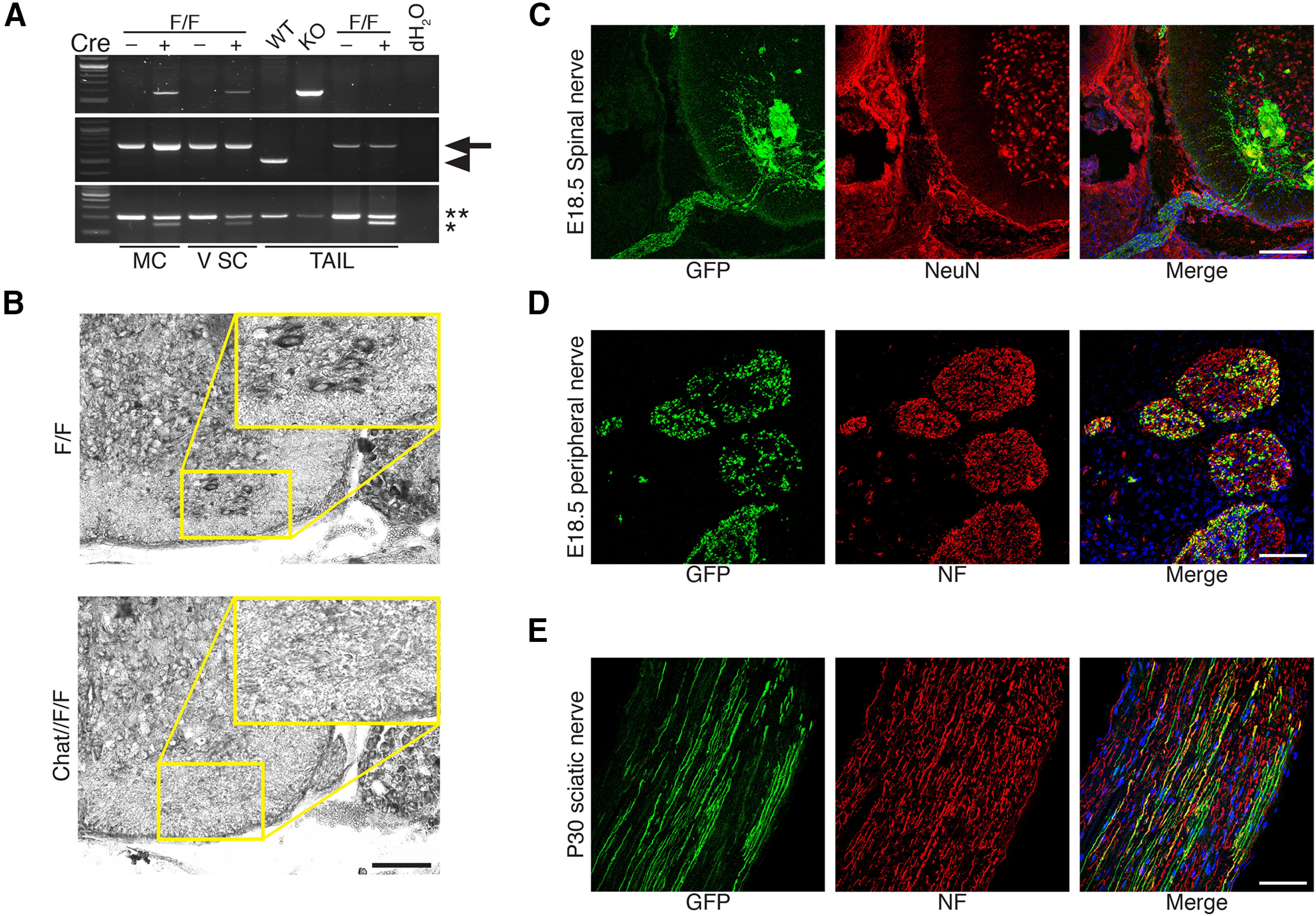
Recombination in Chat-CRE//Adam17flx/flx mice. A, Genotyping PCR for Adam17 and for Chat-CRE alleles on genomic DNA prepared from E12.5 spinal motor columns (MC) and P5 ventral spinal cord (V SC) regions of Chat-CRE//Adam17flx/flx mutants and littermate controls. The 580-bp Adam17 null allele (top panel) is present only in Adam17flox/flox mice expressing the CRE recombinase. We also detected the Adam17flx/flx allele (middle panel, 700 bp, arrow) and the corresponding wild-type allele (553 bp, arrowhead). The Chat-CRE allele (148 bp, asterisk) and the corresponding wild-type allele (200 bp, double asterisks) are also shown (bottom panel). To validate the results, we also included control samples of genomic DNA prepared from tails of wild-type (WT), complete Adam17 null (KO), Adam17flx/flx (F/F) with or without the CRE recombinase and no DNA (dH2O). B, In situ hybridization for Adam17 on frozen sections of P1 ventral spinal cords of Chat-CRE//Adam17flx/flx animals and littermate controls (Adam17flx/flx). Adam17 is highly expressed only in motor neurons of control ventral spinal cords (enlargement). Scale bar: 100 µm. C, Representative immunofluorescence of E18.5 Chat-CRE//ROSA–YFP ventral spinal cord stained for GFP (fluorescein), NeuN (rhodamine), and Hoechst (blue) to label nuclei. N = 3 mice per genotype. Scale bar: 100 µm. D, Representative immunofluorescence of E18.5 Chat-CRE//ROSA–YFP peripheral nerves stained for GFP (fluorescein), Neurofilament (rhodamine), and Hoechst (blue) to label nuclei. N = 3 mice per genotype. Scale bar: 50 µm. E, Representative immunofluorescence of P30 Chat-CRE//ROSA–YFP sciatic nerves stained for GFP (fluorescein), Neurofilament (rhodamine), and Hoechst (blue) to label nuclei. N = 3 mice per genotype. Scale bar: 50 µm.
Finally, we performed morphologic and ultrastructural analyses and g ratio measurements of P7 (Fig. 4A,B; Table 1) and P30 Chat-CRE//Adam17flx/flx (Fig. 4A,C; Table 1) sciatic nerves, confirming that similarly to our previous studies (La Marca et al., 2011), Adam17 ablation in neurons results in hypermyelination.
Figure 4.

Chat-CRE//Adam17flx/flx are hypermyelinated. A, Morphologic analyses of P7 (top panels) and P30 (bottom panels) Chat-CRE//Adam17flx/flx sciatic nerves and littermate controls showing hypermyelination in mutant nerves. Scale bar: 20 µm. B, C, g ratio as a function of axon diameter is decreased in Chat-CRE//Adam17flx/flx nerves (red line) as compared with littermate controls (black line) at P7 (B) and P30 (C). P7: unpaired t test analyses; **p = 0.014, t = 7.896, df = 4; P30: unpaired t test analyses; **p = 0.031 t = 6.378, df = 4. More than 300 myelinated fibers per genotype have been measured. N = 3 mice per genotype.
Collectively, these analyses indicate that unlike development, neuronal ADAM17 is dispensable for WD process.
Glial ADAM17 promotes late nerve remyelination
The above-described results suggest that neuronal ADAM17 does not contribute to PNS regeneration and remyelination. Since ADAM17 is highly expressed also in Schwann cells (La Marca et al., 2011), we next investigated whether glial ADAM17 might have a role in WD. Thus, we crossed Adam17flx/flx mice with P0-CRE transgenic mice, in which the CRE-recombinase is specifically induced in myelinating Schwann cells (Feltri et al., 1999).
We then performed sciatic nerve crush injuries in two-month-old conditional null mice (P0-CRE//Adam17flx/flx) and littermate controls and analyzed degeneration, regeneration and remyelination following the same experimental settings used in Chat-CRE//Adam17flx/flx mice. During degeneration and early regeneration phases, P0-CRE//Adam17flx/flx nerves were morphologically similar to control nerves (Fig. 5). Indeed, we did not observe any difference in the extent of fiber degeneration at T7. Further, the regenerative capacity of axons was well preserved in P0-CRE//Adam17flx/flx mice both at T14 and at T21, as confirmed also by the number of regenerated/remyelinated fibers that was comparable between mutants and controls at these time points (Table 2). Moreover, g ratio values did not change between P0-CRE//Adam17flx/flx nerves and controls at T14 and T21 (Table 2), further underscoring unaltered remyelinating capability of mutant Schwann cells during early phases of nerve regeneration.
Figure 5.
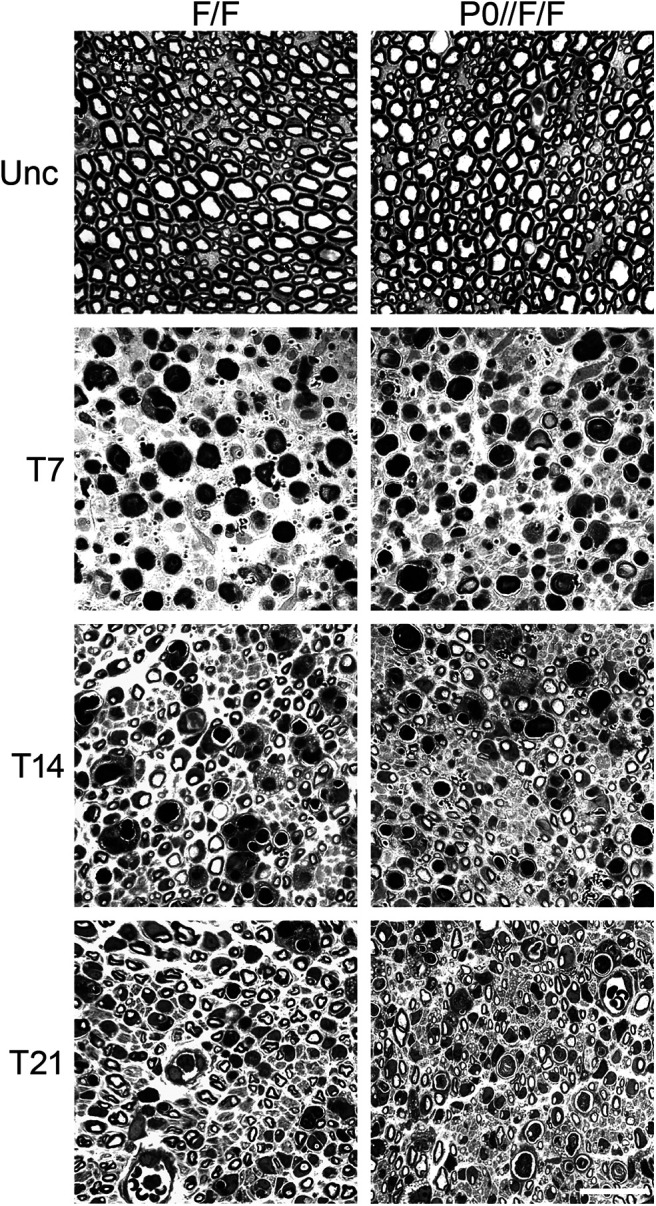
Glial ADAM17 is not required for early stages of nerve regeneration. Morphologic analyses of P0-CRE//Adam17flx/flx and littermate controls injured sciatic nerves are similar during early phases of WD. Semithin sections of two-month-old uncrushed (Unc) controls and P0-CRE//Adam17flx/flx sciatic nerves (top panels). Semithin sections of T7, T14, and T21 controls and P0-CRE//Adam17flx/flx crushed sciatic nerves. Scale bar: 20 µm.
Table 2.
Morphometric analyses of control (F/F) and P0-CRE//Adam17flx/flx (P0//F/F) nerves at different time points after crush injury
| T3 | ||||
| F/F | P0//F/F | N | P | |
| n. ovoids × 103/mm2 | 10.57 ± 1.16 | 10.47 ± 1.14 | n ≥ 3 | p = 0.956 |
| T7 | ||||
| F/F | P0//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 0.52 ± 0.13 | 0.63 ± 0.10 | n ≥ 8 | p = 0.514 |
| n. ovoids × 103/mm2 | 6.40 ± 0.18 | 7.08 ± 0.65 | n = 4 | p = 0.351 |
| n. F4/80+ cells/mm2 | 160.3 ± 26.5 | 182.3 ± 20.9 | n ≥ 3 | p = 0.536 |
| T14 | ||||
| F/F | P0//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 2.48 ± 0.80 | 2.88 ± 0.92 | n ≥ 7 | p = 0.766 |
| g ratio | 0.792 ± 0.008 | 0.783 ± 0.006 | n ≥ 10 | p = 0.38 |
| n. ovoids × 103/mm2 | 3.31 ± 0.22 | 3.38 ± 0.16 | n = 5 | p = 0.813 |
| n. F4/80+ cells/mm2 | 352.5 ± 55.6 | 170.6 ± 33.2 | n = 3 | p = 0.048 |
| T21 | ||||
| F/F | P0//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 11.11± 0.79 | 10.70 ± 1.42 | n = 8 | p = 0.804 |
| g ratio | 0.678 ± 0.007 | 0.690 ± 0.005 | n ≥ 5 | p = 0.235 |
| n. ovoids × 103/mm2 | 2.63 ± 0.21 | 3.29 ± 0.33 | n = 5 | p = 0.134 |
| n. F4/80+ cells/mm2 | 709.4 ± 105.4 | 219.4 ± 57.7 | n = 3 | p = 0.017 |
| T45 | ||||
| F/F | P0//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 27.01± 1.92 | 27.40 ± 1.39 | n ≥ 5 | p = 0.868 |
| g ratio | 0.667 ± 0.007 | 0.687 ± 0.008 | n ≥ 5 | p = 0.088 |
| n. ovoids × 103/mm2 | 1.11 ± 0.11 | 1.55 ± 0.12 | n ≥ 5 | p = 0.023 |
| n. F4/80+ cells/mm2 | 52.9 ± 2.9 | 43.8 ± 12.3 | n = 3 | p = 0.511 |
| T60 | ||||
| F/F | P0//F/F | N | P | |
| n. remyelinated fibers × 103/mm2 | 21.75 ± 0.57 | 21.82 ± 0.96 | n ≥ 9 | p = 0.949 |
| g ratio | 0.647 ± 0.007 | 0.681 ± 0.007 | n ≥ 9 | p = 0.002 |
| n. ovoids × 103/mm2 | 0.54 ± 0.04 | 0.62 ± 0.05 | n = 5 | p = 0.305 |
| n. F4/80+ cells/mm2 | 73 ± 2.6 | 74.5 ± 0.7 | n ≥ 3 | p = 0.646 |
Summary of the morphometric analyses and cell counting performed in control (F/F) and P0-CRE//Adam17flx/flx (P0//F/F) mice after sciatic nerve crush injury. Values represent mean ± SEM (t test analyses). Results with p-values < 0.05 have been considered significant. For g ratio analyses, more than 300 fibers/genotype/time points have been analyzed.
Interestingly, morphologic analyses performed at T60, when remyelination is complete, revealed that mutant nerves present a thinner myelin sheath as compared with controls (Fig. 6A), suggesting that deletion of Adam17 in Schwann cells could impair late remyelination. Although we did not observe any difference in the number of remyelinated fiber and in their distribution (Fig. 6C; Table 2), g ratio values were significantly higher in P0-CRE//Adam17flx/flx nerves at T60 (Fig. 6B).
Figure 6.

Glial ADAM17 promotes peripheral nerve remyelination after damage. A, Morphologic analyses of P0-CRE//Adam17flx/flx and littermate controls injured sciatic nerves at T60. Electron microscopy analyses sections show remyelination defects in mutant nerves. Scale bar: 2 µm. B, Graph showing g ratios as a function of axon diameter in P0-CRE//Adam17flx/flx nerves (red line) as compared with littermate controls (black line). Unpaired t test analyses; **p = 0.0024, t = 3.533, df = 18. More than 800 fibers per genotype have been measured. N = 9 mutants; N = 11 controls. C, Distribution of myelinated fibers is similar in mutants and controls. Fisher's exact test; p = 0.0955 (total vs 1–1.5 μm), χ2 test p = 0.9745 (total vs 1.5–2 μm), χ2 test p = 0.2791 (total vs 2–2.5 μm), χ2 test p = 0.7378 (total vs 2.5–3 μm), Fisher's exact test p = 0.0846 (total vs 3–3.5 μm), Fisher's exact test p = 1 (total vs 3.5–4 μm), Fisher's exact test p = 0.4397 (total vs > 4 μm). More than 800 fibers per genotype have been measured. N = 9 mutants; N = 11 controls.
To better characterize the hypomyelinating phenotype observed in T60 P0-CRE//Adam17flx/flx nerves and the possible causes, we assessed the number of infiltrating F4/80 positive macrophages and of degenerated fibers/myelin ovoids in injured mutant nerves and littermate controls at T7, T14, T21, T45, and T60. While in control animals, macrophages significantly increased at T14 and at T21, in P0-CRE//Adam17flx/flx nerves their overall number remain comparable to that observed at T7, suggesting a delay in myelin clearance and subsequent remyelination (Fig. 7A,B; Table 2). The increased number of degenerated fibers/myelin ovoids at T21 and at T45 (Fig. 7C,D; Table 2) in mutant nerves, further corroborates impaired myelin clearance. Moreover, electrophysiological and behavioral analyses strongly indicate that remyelination in the late phases of WD is impaired in P0-CRE//Adam17flx/flx mutants. Although rotarod tests did not reach significance, they are suggestive of a trend impairment in motor performances in T60 mutant mice (Fig. 7G). Of note, injured mutant nerves have reduced NCV and increased CMAP latency (Fig. 7E,F), but no changes in the amplitude (data not shown), in agreement with the comparable number of axons in P0-CRE//Adam17flx/flx mutants (Fig. 6C; Table 2).
Figure 7.
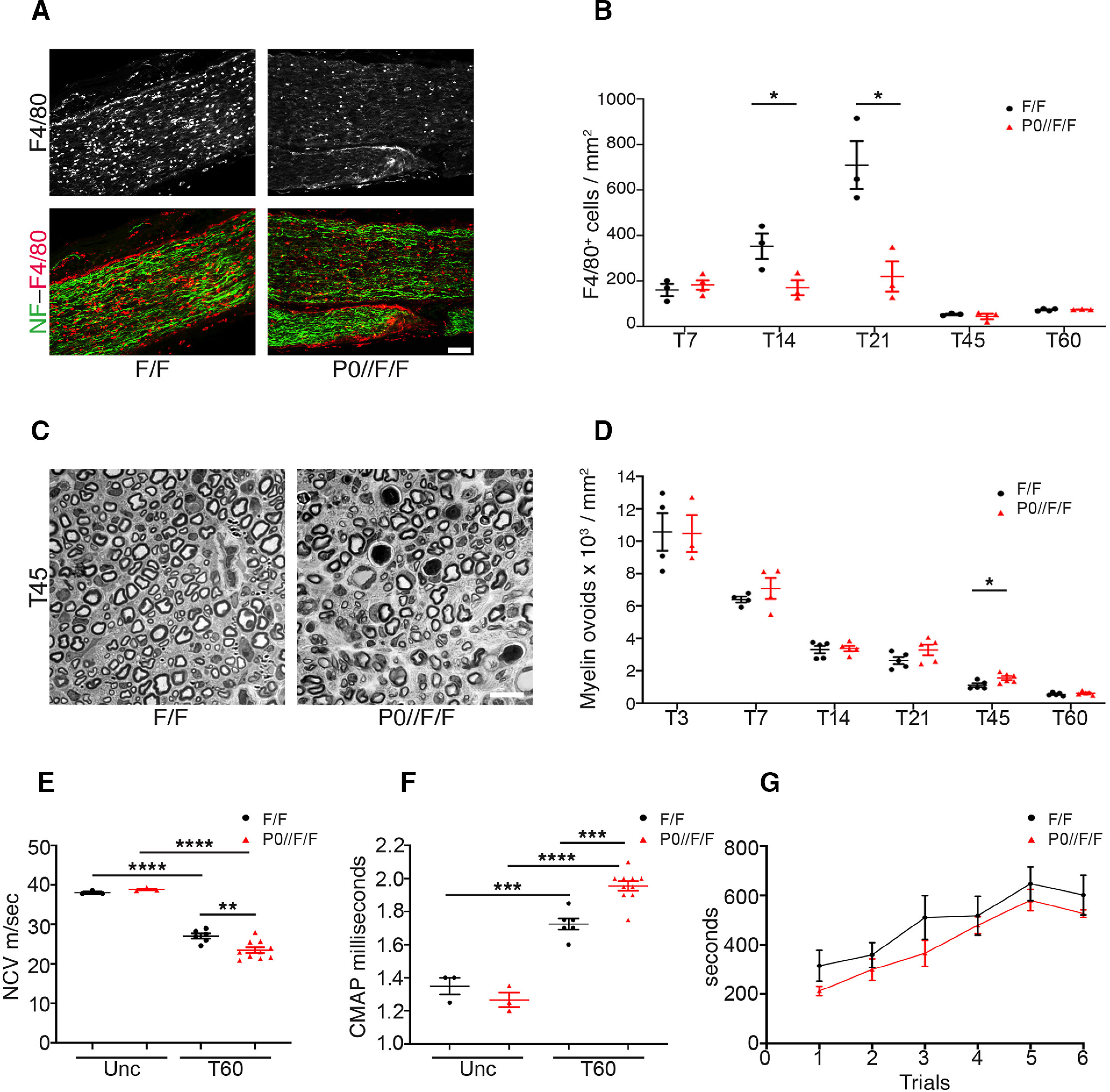
Glial ADAM17 primes the remyelinating process. A, Representative immunofluorescence images of macrophages in T21 P0-CRE//Adam17flx/flx and littermate controls sciatic nerves. Longitudinal sections have been stained for F4/80 (rhodamine), Neurofilament–M (NF; fluorescein). Scale bar: 75 µm. N = at least 3 mice per genotype. B, Graphs showing the density of F4/80+ cells in T7, T14, T21, T45, and T60 P0-CRE//Adam17flx/flx and littermate controls sciatic nerves. Macrophages accumulate only in injured control sciatic nerves. Error bars represent mean ± SEM (T7: unpaired t test p = 0.5363, t = 0.6636, df = 5; T14: unpaired t test *p = 0.0485, t = 2.806, df = 4; T21: unpaired t test *p = 0.0171, t = 3.931, df = 4; T45: unpaired t test p = 0.5109, t = 0.7208, df = 4; T60: unpaired t test p = 0.6457, t = 0.4888, df = 5). N = at least 3 mice per genotype. C, Representative semithin sections of T45 P0-CRE//Adam17flx/flx and littermate controls sciatic nerves showing not phagocytized degenerated fibers/myelin ovoids. Scale bar: 10 µm. D, Graphs showing the density of degenerated fibers/myelin ovoids in T3, T7, T14, T21, T45, and T60 P0-CRE//Adam17flx/flx and littermate controls sciatic nerves. Error bars represent mean ± SEM (T3: unpaired t test p = 0.9564, t = 0.05745, df = 5; T7: unpaired t test p = 0.3508, t = 1.011, df = 6; T14: unpaired t test p = 0.8131, t = 0.2443, df = 8; T21: unpaired t test p = 0.1336, t = 1.670, df = 8; T45: unpaired t test *p = 0.0229, t = 2.738, df = 9; T60: unpaired t test p = 0.3053, t = 1.095, df = 8). N = at least 3 mice per genotype. E, Graph showing NCV measurements in uncrushed (Unc) and T60 control and P0-CRE//Adam17flx/flx sciatic nerves. NCV is significantly reduced at T60 in mutant mice as compared with controls. Error bars represent mean ± SEM (unpaired t test Unc control versus Unc mutant p = 0.0919, t = 2.207, df = 4; unpaired t test Unc control versus T60 control ****p < 0.0001, t = 11.31, df = 7; unpaired t test Unc mutant vs T60 mutant ****p < 0.0001, t = 11.25, df = 11; unpaired t test T60 control vs T60 mutant **p = 0.0048, t = 3.346, df = 14). N = 3 uncrushed mice per genotype; N = 6 T60 Ctrl mice; N = 10 T60 mutant mice. F, Graph showing CMAPs measurements in uncrushed (Unc) and T60 control and P0-CRE//Adam17flx/flx sciatic nerves. CMAP is significantly increased at T60 in P0-CRE//Adam17flx/flx mice as compared with controls. Error bars represent mean ± SEM (unpaired t test Unc control vs Unc mutant p = 0.2794, t = 1.250, df = 4; unpaired t test Unc control vs T60 control ***p = 0.0004, t = 6.355, df = 7; unpaired t test Unc mutant vs T60 mutant ****p < 0.0001, t = 11.63, df = 11; unpaired t test T60 control vs T60 mutant ***p = 0.0002, t = 5.002, df = 14). N = 3 uncrushed mice per genotype; N = 6 T60 Ctrl mice; N = 10 T60 mutant mice. G, Graph showing Rotarod analyses of motor function (6 repeated motor trials) in T60 control and P0-CRE//Adam17flx/flx injured mice. Error bars represent mean ± SEM (trial 1: unpaired t test p = 0.0820, t = 1.914, df = 11; trial 2: unpaired t test p = 0.4027, t = 0.8704, df = 11; trial 3: unpaired t test p = 0.1587, t = 1.512, df = 11; trial 4: unpaired t test p = 0.6168, t = 0.5149, df = 11; trial 5: unpaired t test p = 0.4011, t = 0.8733, df = 11; trial 6: unpaired t test p = 0.2690, t = 1.164, df = 11). N = 8 mutants; N = 5 controls.
Collectively, these results indicate that although glial ADAM17 is not necessary for early regeneration, it is required in the entire WD process and promotes late phases of peripheral nerve remyelination after injury.
ADAM17 does not regulate remyelination through NRG1-ErbB signaling
We next sought to characterize the molecular mechanisms through which ADAM17 promotes remyelination. Among the various substrates of ADAM17 activity, we focused on those previously reported being involved in WD. We first verified NRG1, as glial NRG1 Type I promotes peripheral nerve regeneration (Stassart et al., 2013) and NRG1 is a direct substrate of ADAM17 (La Marca et al., 2011). Thus, we analyzed phosphorylation of ErbB2 (Fig. 8A,C), the cognate NRG1 receptor (Yarden and Sliwkowski, 2001), and of AKT and ERK1/2 (Fig. 8B,D,E), the main signaling transducers (Stassart et al., 2013), in lysates prepared from mutant and control nerves uncrushed and at T7, T14, T21, and T60. As shown in Figure 8, activation of ErbB2 signaling and of AKT and ERK1/2 pathways was comparable in P0-CRE//Adam17flx/flx and control nerves at all analyzed time points.
Figure 8.
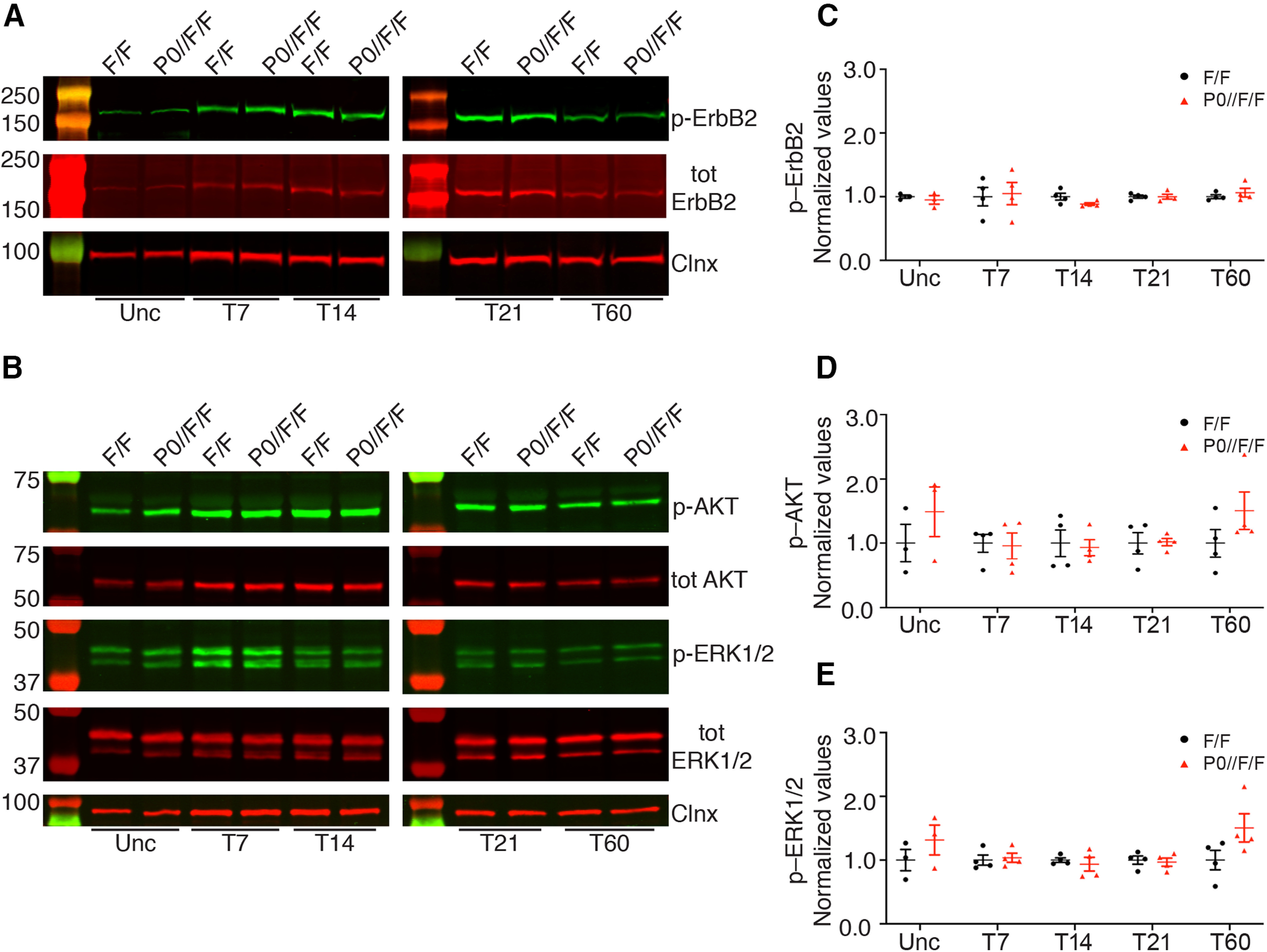
NRG1-ErbB signaling is not altered in P0-CRE//Adam17flx/flx injured nerves. A, Representative Western blot analyses of uncrushed (Unc), T7, T14, T21, and T60 P0-CRE//Adam17flx/flx and littermate controls injured sciatic nerves. Lysates were tested for phosphorylated ErbB2 (p-ErbB2), total ErbB2, and Calnexin, as loading control. N = at least 3 mice per genotype. B, Representative Western blot analyses of uncrushed (Unc), T7, T14, T21, and T60 P0-CRE//Adam17flx/flx and littermate controls injured sciatic nerves. Lysates were tested for phosphorylated AKT (p-AKT), total AKT, phosphorylated ERK1/2 (p-ERK1/2), total ERK1/2, and Calnexin as loading control. N = at least 3 mice per genotype. C, Graph showing levels of p-ErbB2 in controls versus P0-CRE//Adam17flx/flx nerves. All controls data were arbitrarily set as 1.0. We determined p-ErbB2 expression levels in uncrushed, T7, T14, T21, and T60 using the LI–COR Odyssey® quantitative system. Levels of total ErbB2 protein were first normalized over Calnexin, used as loading control. Levels of p–ErbB2 were averaged over the ratio of total ErbB2/Calnexin. Error bars represent mean ± SEM (Unc: unpaired t test p = 0.5168, t = 0.7102, df = 4; T7: unpaired t test p = 0.8386, t = 0.2127, df = 6; T14: unpaired t test p = 0.0881, t = 2.035, df = 6; T21: unpaired t test p = 0.9928, t = 0.009364, df = 6; T60: unpaired t test p = 0.4353, t = 0.8358, df = 6). N = at least 3 mice per genotype. D, Graph showing levels of p-AKT in controls versus P0-CRE//Adam17flx/flx nerves. All controls data were arbitrarily set as 1.0. We determined p-AKT expression levels in uncrushed, T7, T14, T21, and T60 using the LI–COR Odyssey® quantitative system. Levels of total AKT protein were first normalized over Calnexin, used as loading control. Levels of p-AKT were averaged over the ratio of total AKT/Calnexin. Error bars represent mean ± SEM (Unc: unpaired t test p = 0.3665, t = 1.017, df = 4; T7: unpaired t test p = 0.8716, t = 0.1687, df = 6; T14: unpaired t test p = 0.7909, t = 0.2773, df = 6; T21: unpaired t test p = 0.9191, t = 0.1059, df = 6; T60: unpaired t test p = 0.2111, t = 1.400, df = 6). N = at least 3 mice per genotype. E, Graph showing levels of p–ERK1/2 in controls versus P0-CRE//Adam17flx/flx nerves. All controls data were arbitrarily set as 1.0. We determined p–ERK1/2 expression levels in uncrushed, T7, T14, T21, and T60 using the LI–COR Odyssey® quantitative system. Levels of total ERK1/2 protein were first normalized over Calnexin, used as loading control. Levels of p–ERK1/2 were averaged over the ratio of total ERK1/2/Calnexin. Error bars represent mean ± SEM (Unc: unpaired t test p = 0.3340, t = 1.098, df = 4; T7: unpaired t test p = 0.7439, t = 0.3422, df = 6; T14: unpaired t test p = 0.5882, t = 0.5718, df = 6; T21: unpaired t test p = 0.7370, t = 0.3518, df = 6; T60: unpaired t test p = 0.1104, t = 1.872, df = 6). N = at least 3 mice per genotype.
Thus, glial ADAM17 is likely not targeting NRG1 during remyelination, as the activation of the NRG1/ErbB signaling pathway and of the main downstream effectors, were not affected in the absence of glial Adam17.
p75NTR accumulates in glial Adam17-deficient nerves at T21
Since NRG1 is unlikely a substrate for glial ADAM17 activity during remyelination, we next determined whether other molecules, previously implicated in nerve regeneration and target of ADAM17 activity, might be affected. We thus examined p75NTR, a known ADAM17 substrate (Zampieri et al., 2005) that promotes peripheral nerve remyelination (Song et al., 2006; Tomita et al., 2007). We first determined p75NTR relative abundance in lysates prepared from injured P0-CRE//Adam17flx/flx nerves collected at T21, when ADAM17 is specifically upregulated (Fig. 1). As previously reported (Taniuchi et al., 1986), full-length p75NTR was increased in injured sciatic nerves, and to a higher degree in mutant nerves (Fig. 9A,B), indicating that glial ADAM17 is likely implicated in p75NTR processing. Of note, p75NTR accumulates precisely at T21, when ADAM17 expression is mostly upregulated in injured nerves (Fig. 1).
Figure 9.
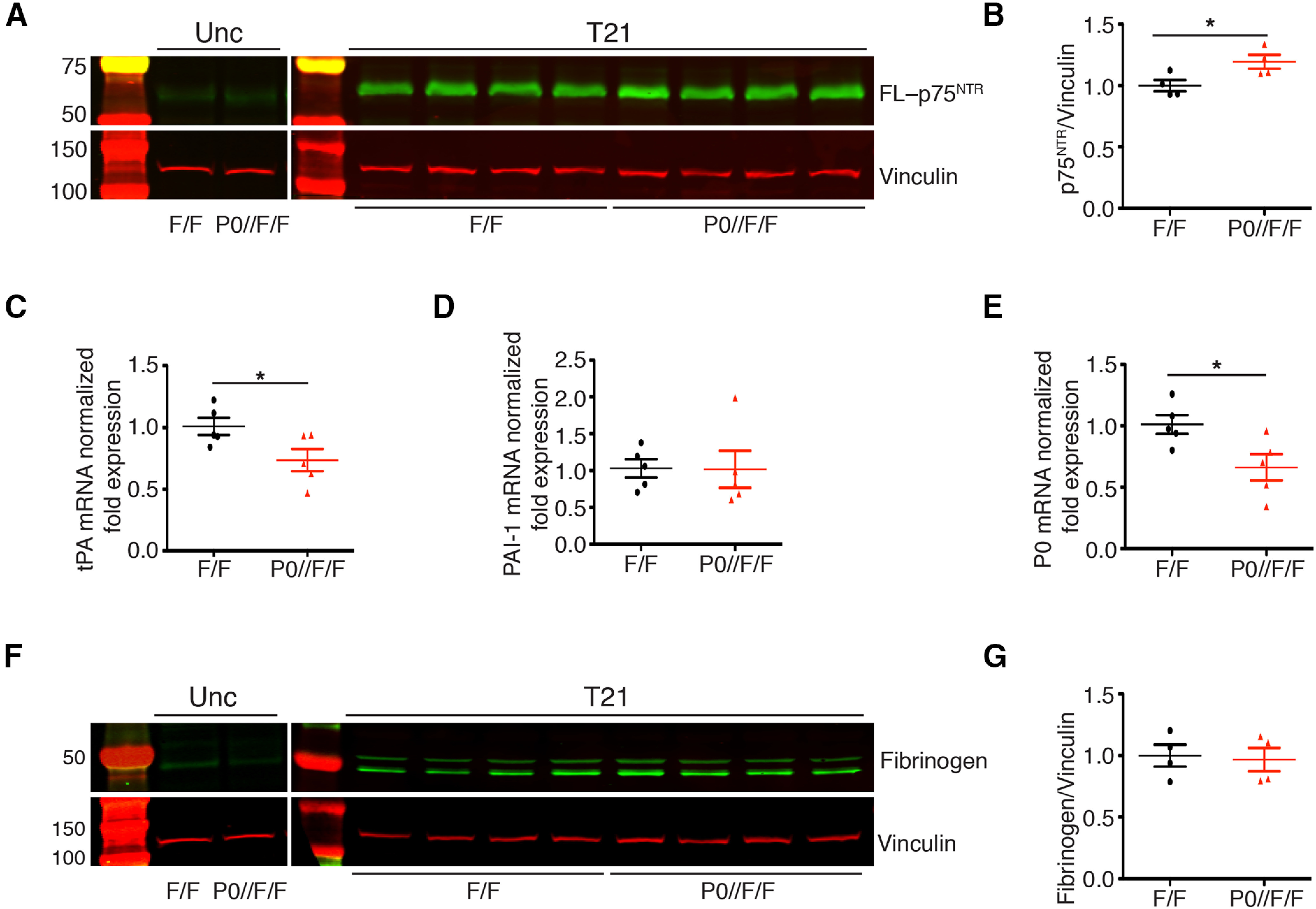
p75NTR accumulates in P0-CRE//Adam17flx/flx nerves at T21. A, Representative Western blot analyses of uncrushed and T21 P0-CRE//Adam17flx/flx sciatic nerves relative to controls. Lysates were tested for p75NTR and Vinculin as a loading control. N = 4 different mice per genotype. B, Graph showing full-length p75NTR accumulation in T21 mutant nerves as compared with control nerves, arbitrarily set as 1.0 as determined using the LI–COR Odyssey® quantitative system. Levels of full-length p75NTR were normalized over Vinculin, used as loading control. Error bars represent mean ± SEM (unpaired t test, * p = 0.037, t = 2.664, df = 6). N = 4 different mice per genotype. C–E, Quantitative tPA (C), PAI-1 (D), and P0 (E) PCR analyses on mRNA prepared from P0-CRE//Adam17flx/flx and control nerves harvested at T21. C, tPA mRNA levels are downregulated in mutant nerves as compared with control nerves, arbitrarily set as 1.0 (unpaired t test *p = 0.043, t = 2.398, df = 8) N = 5 different mice per genotype. D, PAI-1 mRNA levels are similar in P0-CRE//Adam17flx/flx and control nerves (unpaired t test p = 0.9637 t = 0.04693, df = 8). N = 5 different mice per genotype. E, P0 mRNA levels are downregulated in mutant nerves as compared with controls, arbitrarily set as 1.0 (unpaired t test *p = 0.0288, t = 2.661, df = 8). N = 5 different mice per genotype. F, Representative Western blot analyses of uncrushed and T21 P0-CRE//Adam17flx/flx and control sciatic nerves. Lysates were tested for fibrinogen expression and Vinculin as a loading control. N = 4 different mice per genotype. G, Graph showing that fibrinogen levels do not change in T21 mutant nerves as compared with control nerves, arbitrarily set as 1.0, as determined using the LI–COR Odyssey® quantitative system. Levels of fibrinogen were normalized over Vinculin, used as loading control. Error bars represent mean ± SEM (unpaired t test, p = 0.8165, t = 0.2424, df = 6). N = 4 different mice per genotype.
Since p75NTR inhibits nerve regeneration by blocking fibrin clearance (Akassoglou et al., 2002; Sachs et al., 2007), we next determined whether these events are linked also in P0-CRE//Adam17flx/flx nerves. Thus, we analyzed mRNA expression levels of tPA and PAI-1, key regulators of plasmin activity and fibrin clearance (Sachs et al., 2007), in T21 P0-CRE//Adam17flx/flx mutants and controls injured sciatic nerves. While tPA expression was specifically downregulated in mutant nerves (Fig. 9C), PAI-1 expression did not change (Fig. 9D). Interestingly, P0 expression was also significantly reduced in mutant nerves at T21 (Fig. 9E), further underscoring p75NTR accumulation (Akassoglou et al., 2002). Finally, to assess whether inhibition of tPA expression results in fibrin accumulation in mutant nerves, we also determined fibrinogen accumulation, as fibrinogen is the precursor form of fibrin (Petersen et al., 2018). Interestingly, fibrinogen did not accumulate in injured mutant nerves (Fig. 9F,G), suggesting that the observed downregulation of tPA is likely not sufficient to determine a significant accumulation of fibrin.
Fibrin accumulates in glial Adam17-deficient nerves at T45, impairing remyelination
Our results thus far indicate that in the absence of glial Adam17, p75NTR processing is impaired in injured nerves at T21, leading to a downregulation of tPA expression without fibrin accumulation. To determine whether fibrin might accumulate over time, we tested fibrinogen protein levels in mutant and control nerves at T45 by Western Blotting and immunofluorescence analyses. Intriguingly, we found significant fibrinogen accumulation in P0-CRE//Adam17flx/flx mutant nerves (Fig. 10A–C), possibly resulting from p75NTR accumulation and tPA downregulation at T21. Since fibrin accumulation inhibits remyelination (Akassoglou et al., 2002), we next analyzed sciatic nerve morphology of P0-CRE//Adam17flx/flx mice and littermate controls at T45. Although we found no differences in the number of remyelinated fibers (Table 2), we observed a slight not significant increase in g ratio in mutant nerves (Fig. 10D,E; Table 2). Accordingly, when we tested p75NTR protein levels at T45 days, we found no alterations as compared with control nerves (Fig. 11A,B). Similarly, also tPA and P0 expression levels did not change (Fig. 11C,D), suggesting that, during nerve regeneration, glial ADAM17 likely processes p75NTR at the peak of its expression levels.
Figure 10.
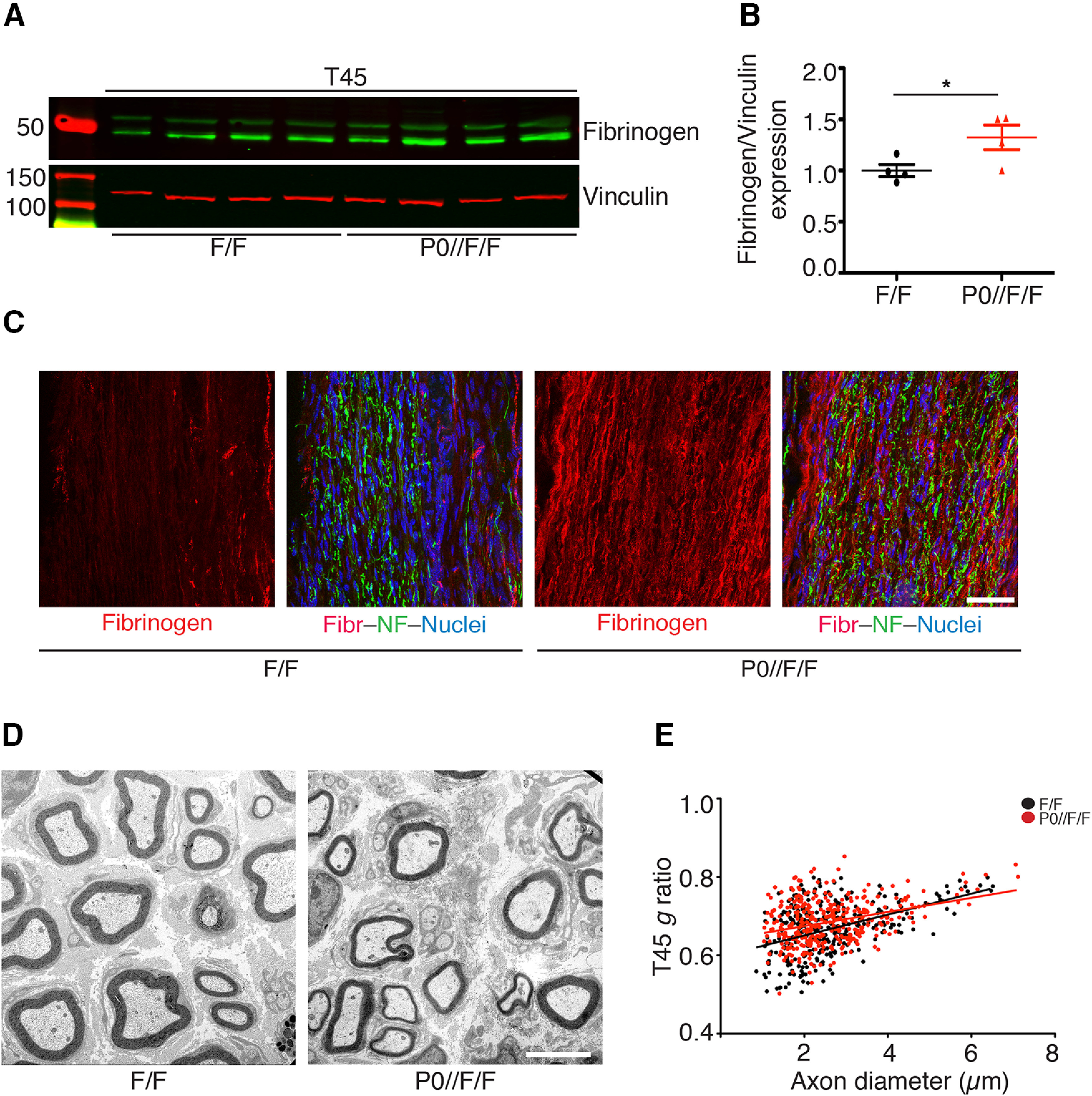
Fibrinogen accumulates in P0-CRE//Adam17flx/flx nerves at T45. A, Representative Western blot analyses of T45 P0-CRE//Adam17flx/flx and control sciatic nerves. Lysates were tested for fibrinogen and Vinculin as a loading control. N = 4 different mice per genotype. B, Graph showing fibrinogen accumulation in mutant nerves as compared with control nerves, arbitrarily set as 1.0, as determined using the LI–COR Odyssey® quantitative system. Levels of fibrinogen were normalized over Vinculin, used as loading control. Error bars represent mean ± SEM (unpaired t test, * p = 0.05, t = 2.427, df = 6). N = 4 different mice per genotype. C, Representative immunofluorescence images showing fibrinogen accumulation in T45 P0-CRE//Adam17flx/flx and littermate controls sciatic nerves. Longitudinal sections have been stained for fibrinogen (rhodamine), Neurofilament–M (NF; fluorescein), and Hoechst (blue) to identify cell nuclei. Scale bar: 50 µm. N = 3 mice per genotype. D, Morphologic analyses of P0-CRE//Adam17flx/flx and littermate controls injured sciatic nerves at T45. Electron microscopy analyses sections show slight initial remyelination impairment in mutant nerves. Scale bar: 5 µm. E, Graph showing g ratio as a function of axon diameter in T45 P0-CRE//Adam17flx/flx nerves (red line) as compared with littermate controls (black line; unpaired t test analyses; p = 0.0883, t = 1.911, df = 9). More than 400 fibers per genotype have been measured. N = 5 controls; N = 6 mutants.
Figure 11.

p75NTR processing is normal in T45 P0-CRE//Adam17flx/flx nerves. A, Representative Western blot analyses of T45 P0-CRE//Adam17flx/flx and control sciatic nerves. Lysates were tested for p75NTR and Vinculin as a loading control. N = 4 different mice per genotype. B, Graph showing that full-length p75NTR levels are similar at T45 in mutant and control nerves, arbitrarily set as 1.0, as determined using the LI–COR Odyssey® quantitative system. Levels of full-length p75NTR were normalized over Vinculin, used as loading control. Error bars represent mean ± SEM (unpaired t test, p = 0.9441, t = 0.0731, df = 6). N = 4 different mice per genotype. C, D, Quantitative tPA (C) and P0 (D) PCR analyses on mRNA prepared from T45 P0-CRE//Adam17flx/flx and control nerves. tPA (C) and P0 (D) mRNA levels are comparable in mutant and control nerves, arbitrarily set as 1.0 (tPA: unpaired t test p = 0.5111, t = 0.6813, df = 10; P0: unpaired t test p = 0.444, t = 0.797, df = 10). N = 6 different mice per genotype.
These observations suggest that at T45 fibrin accumulation might initiate a process, which culminates in the impaired remyelination observed at T60. Interestingly, at T60, p75NTR processing and fibrin accumulation were normal (data not shown), further underscoring that the observed remyelination impairment is likely the result of the molecular alterations occurring at previous time points.
Overall, our results suggest that, differently from development, ADAM17 expressed in Schwann cells promotes remyelination. After damage, in the absence of glial ADAM17, p75NTR processing is altered, resulting in the downregulation of tPA, an important activator of fibrin clearance, causing fibrin accumulation with time. These events eventually culminate in the impairment in later remyelination phases and demonstrate a novel unreported role for glial ADAM17 as a positive regulator of remyelination.
Discussion
In the current study, we describe a novel role for the α-secretase ADAM17 in nerve repair. Our results indicate that, in WD, ADAM17 regulates p75NTR processing and fibrin degradation and describe a previously uncharacterized mechanism of action for this protease in peripheral nerves. We thus propose that glial ADAM17 is involved in the regenerative process, differently from what happens in nerve development, in which neuronal ADAM17 regulates myelin formation. Our in vivo analyses in conditional mutants, in fact, strongly suggest that glial ADAM17 cell autonomously participates in PNS remyelination. Indeed, after damage, motor neuron mutants undergo physiological nerve repair, while Schwann cell mutants are impaired, mainly in the late phases of the regenerative process, indicating that the axonal form is dispensable for nerve repair.
The results observed in these studies are opposite to what we previously reported in development, in which axonal ADAM17 inactivates NRG1 Type III and consequently inhibits myelination, while conditional null mice in Schwann cells undergo physiological nerve maturation (La Marca et al., 2011). The fact that axonal ADAM17 is dispensable during regeneration agrees with previous studies, and further highlights that regeneration does not simply recapitulate nerve development (Fancy et al., 2011; Stassart et al., 2018; Pellegatta and Taveggia, 2019). During regeneration, in fact, axonal NRG1 does not control the myelinating fate of axons, as it occurs in development (Stassart et al., 2013), and it is dispensable for long–term remyelination (Fricker et al., 2011, 2013). Moreover, early after damage, when Schwann cells lose contacts with degenerating axons, the expression of axonal NRG1 Type III decreases, and it is re-expressed when axons re-innervate their targets (Bermingham-McDonogh et al., 1997; Stassart et al., 2013).
Previous studies have indicated NRG1 Type I, the NRG1 isoform expressed in Schwann cells, as the key molecule regulating the early events of WD (Stassart et al., 2013). Although we cannot rule out that ADAM17 might process NRG1 Type I, this is unlikely for several reasons. We did not observe any impairment in early events after injury, as we detected a remyelination defect only at the end of the regenerative process. Further, in injured P0-CRE//Adam17flx/flx nerves, the NRG1-ErbB pathway is not perturbed, as phosphorylation of ErbB2 and of downstream signaling molecules, previously implicated in modulating NRG1 Type I signaling after injury (Stassart et al., 2013), are unaltered. Thus, we propose that to coordinate nerve repair, ADAM17 relies on a different molecular machinery than canonical NRG1-ErbB signaling. Collectively, we posit that, after injury, signals coming from NRG1 might overlap or even be replaced by molecules that favor nerve regeneration through distinct mechanisms.
ADAM17 specificity for its substrates appears to be partly dependent on the structure of the extracellular region and on the amino acid sequence of the juxta–membrane domain of the target molecule, however, no conserved consensus sites have been identified (Horiuchi et al., 2007). Moreover, ADAM17 activity is controlled at different levels by a wide range of stimuli from the surrounding milieu (Horiuchi, 2013). Further, ADAM17 substrate recognition and activity can vary depending on the biological environment. Thus, the concomitant upregulation of ADAM17 and p75NTR in injured nerves, likely cooperates with the subset of signals involved in ADAM17 activation after damage. This results in the beneficial contribution of ADAM17 to nerve repair through the preferential processing of p75NTR, which has been previously implicated in remyelination and is highly induced in nerves after injury (Weskamp et al., 2004; Zampieri et al., 2005; Song et al., 2006; Sachs et al., 2007; Tomita et al., 2007).
During WD, p75NTR regulates fibrin clearance (Sachs et al., 2007). To physiologically facilitate nerve degeneration, p75NTR is upregulated in Schwann cells soon after injury, alongside to a massive influx of fibrin from the vasculature, ultimately leading to extra cellular matrix remodeling (Sachs et al., 2007). During regeneration, p75NTR expression is instead downregulated, to promote fibrin degradation and nerve repair (Akassoglou et al., 2002). We now report that in P0-CRE//Adam17flx/flx nerves, unprocessed full-length p75NTR accumulates at T21, precisely when ADAM17 is mostly upregulated in injured nerves. Further, we observed a concomitant downregulation of tPA and, with time, a consequent impairment in fibrin degradation and nerve remyelination.
Previous studies have reported that physiological fibrin degradation correlates with remyelination. More importantly, genetic or pharmacological depletion of fibrin accelerates remyelination of newly generated axons in the initial stages of the WD process (Akassoglou et al., 2002). However, whether fibrin influences the formation of new myelin at late time points after damage has never been analyzed. Our data demonstrate that accumulation of unprocessed p75NTR and the consequent tPA downregulation in P0-CRE//Adam17flx/flx nerves 21 d after damage, leads to progressive fibrin accumulation up to T45, resulting in a remyelination defect at T60. We therefore describe a novel correlation between the process of fibrin clearance and the timely regulated fine-tuning of Schwann cells remyelinating capability, which is controlled by ADAM17-mediated p75NTR processing. Overall, our results indicate that the extent of fibrin deposition, despite occurring during the initial phases of WD, affects also late remyelination events. Indeed, altered p75NTR processing and fibrin accumulation impact on already remyelinated axons, with a process starting three weeks after damage, and progressing with time as a gradual and cumulative impairment in remyelination.
Fibrin is abundantly deposited in a wide range of neurologic diseases (Petersen et al., 2018). Notably, in some human peripheral neuropathies there is a direct correlation between the extent of fibrinolytic activity and the degree of nerve regeneration attempts (Previtali et al., 2008; Rivellini et al., 2012). Importantly, ADAM17 expression is increased in preclinical models of some forms of Charcot Marie Tooth neuropathies (Bolino et al., 2016). In theory, it is thus possible to envisage therapeutic strategies involving modulators of ADAM17 activity in the treatment of peripheral neuropathies characterized by reduced fibrinolytic activity and impaired regeneration. Notably, Niaspan, a potent ADAM17 activator already employed in clinical practice (Lukasova et al., 2011), has been successfully used in preclinical trials for the treatment of hypermyelinating Charcot Marie Tooth neuropathies (Bolino et al., 2016). Whether and how this or other compounds enhancing ADAM17 activity could help in the resolution of fibrin clots boosting nerve repair, deserve additional studies.
In conclusion, understanding the molecular mechanisms underlying nerve regeneration and remyelination is of particular interest to identify new and effective strategies allowing complete recovery after damage. Although axons in the PNS regenerate and Schwann cells eventually re-establish a new functional axo-glial unit, the regenerative process is always inefficient as newly formed internodes are shorter and myelin is thinner (Fancy et al., 2011; Stassart et al., 2018). Moreover, conceiving treatments that could efficiently improve nerve repair could be beneficial also for human hereditary pathologies like Charcot Marie Tooth neuropathies characterized by axonal loss and unsuccessful repeated attempts of nerve regeneration.
Footnotes
We thank S. C. Previtali and C. Rivellini (San Raffaele Research Hospital, Milan, Italy) for helping with sciatic nerve crush injury and D. Bonanomi (San Raffaele Research Hospital, Milan, Italy) for motor column isolation. We also thank N. Zampieri (Max-Delbrück-Centrum for Molecular Medicine, Germany) for providing the p75NTR antibody and A. Trimarco (San Raffaele Research hospital, Milan, Italy), S. C. Previtali (San Raffaele Research Hospital, Milan, Italy), and F. Valtorta (UniSR: Vita -Salute San Raffaele University, Milan Italy) for critical reading of the manuscript and suggestions. This work was supported by the Italian Telethon Award Number GPP 15012 (to C.T.).
The authors declare no competing financial interests.
References
- Akassoglou K, Yu WM, Akpinar P, Strickland S (2002) Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron 33:861–875. 10.1016/s0896-6273(02)00617-7 [DOI] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R, Jessen KR (2012) c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75:633–647. 10.1016/j.neuron.2012.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham-McDonogh O, Xu YT, Marchionni MA, Scherer SS (1997) Neuregulin expression in PNS neurons: isoforms and regulation by target interactions. Mol Cell Neurosci 10:184–195. 10.1006/mcne.1997.0654 [DOI] [PubMed] [Google Scholar]
- Bolino A, Piguet F, Alberizzi V, Pellegatta M, Rivellini C, Guerrero-Valero M, Noseda R, Brombin C, Nonis A, D'Adamo P, Taveggia C, Previtali SC (2016) Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol Med 8:1438–1454. 10.15252/emmm.201606349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J, Wang X, Bi J (2016) Functional recovery from sciatic nerve crush injury is delayed because of increased distal atrophy in mice lacking the p75 receptor. Neuroreport 27:940–947. 10.1097/WNR.0000000000000635 [DOI] [PubMed] [Google Scholar]
- Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH (2011) Myelin regeneration: a recapitulation of development? Annu Rev Neurosci 34:21–43. 10.1146/annurev-neuro-061010-113629 [DOI] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L (1999) P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann NY Acad Sci 883:116–123. 10.1111/j.1749-6632.1999.tb08574.x [DOI] [PubMed] [Google Scholar]
- Forese MG, Pellegatta M, Canevazzi P, Gullotta GS, Podini P, Rivellini C, Previtali SC, Bacigaluppi M, Quattrini A, Taveggia C (2020) Prostaglandin D2 synthase modulates macrophage activity and accumulation in injured peripheral nerves. Glia 68:95–110. 10.1002/glia.23705 [DOI] [PubMed] [Google Scholar]
- Fricker FR, Lago N, Balarajah S, Tsantoulas C, Tanna S, Zhu N, Fageiry SK, Jenkins M, Garratt AN, Birchmeier C, Bennett DL (2011) Axonally derived neuregulin-1 is required for remyelination and regeneration after nerve injury in adulthood. J Neurosci 31:3225–3233. 10.1523/JNEUROSCI.2568-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker FR, Antunes-Martins A, Galino J, Paramsothy R, La Russa F, Perkins J, Goldberg R, Brelstaff J, Zhu N, McMahon SB, Orengo C, Garratt AN, Birchmeier C, Bennett DL (2013) Axonal neuregulin 1 is a rate limiting but not essential factor for nerve remyelination. Brain 136:2279–2297. 10.1093/brain/awt148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, DiAntonio A (2016) Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89:449–460. 10.1016/j.neuron.2015.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooz M (2010) ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol 45:146–169. 10.3109/10409231003628015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K (2013) A brief history of tumor necrosis factor α–converting enzyme: an overview of ectodomain shedding. Keio J Med 62:29–36. 10.2302/kjm.2012-0003-re [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP (2007) Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 179:2686–2689. 10.4049/jimmunol.179.5.2686 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R, Lloyd AC (2015) Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol 7:a020487. 10.1101/cshperspect.a020487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BR, Yoon SO, Carter BD (2014) The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb Exp Pharmacol 220:121–164. 10.1007/978-3-642-45106-5_6 [DOI] [PubMed] [Google Scholar]
- La Marca R, Cerri F, Horiuchi K, Bachi A, Feltri ML, Wrabetz L, Blobel CP, Quattrini A, Salzer JL, Taveggia C (2011) TACE (ADAM17) inhibits Schwann cell myelination. Nat Neurosci 14:857–865. 10.1038/nn.2849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijnen HR (2001) Elements of the fibrinolytic system. Ann NY Acad Sci 936:226–236. 10.1111/j.1749-6632.2001.tb03511.x [DOI] [PubMed] [Google Scholar]
- Lukasova M, Hanson J, Tunaru S, Offermanns S (2011) Nicotinic acid (niacin): new lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol Sci 32:700–707. 10.1016/j.tips.2011.08.002 [DOI] [PubMed] [Google Scholar]
- Meeker RB, Williams KS (2015) The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regen Res 10:721–725. 10.4103/1673-5374.156967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsky R, Woodhoo A, Parkinson DB, Arthur-Farraj P, Bhaskaran A, Jessen KR (2008) Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J Peripher Nerv Syst 13:122–135. 10.1111/j.1529-8027.2008.00168.x [DOI] [PubMed] [Google Scholar]
- Monk KR, Feltri ML, Taveggia C (2015) New insights on Schwann cell development. Glia 63:1376–1393. 10.1002/glia.22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegatta M, Taveggia C (2019) The complex work of proteases and secretases in Wallerian degeneration: beyond neuregulin-1. Front Cell Neurosci 13:93. 10.3389/fncel.2019.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MA, Ryu JK, Akassoglou K (2018) Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci 19:283–301. 10.1038/nrn.2018.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previtali SC, Malaguti MC, Riva N, Scarlato M, Dacci P, Dina G, Triolo D, Porrello E, Lorenzetti I, Fazio R, Comi G, Bolino A, Quattrini A (2008) The extracellular matrix affects axonal regeneration in peripheral neuropathies. Neurology 71:322–331. 10.1212/01.wnl.0000319736.43628.04 [DOI] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L (1996) Beta 4 integrin and other Schwann cell markers in axonal neuropathy. Glia 17:294–306. [DOI] [PubMed] [Google Scholar]
- Rivellini C, Dina G, Porrello E, Cerri F, Scarlato M, Domi T, Ungaro D, Del Carro U, Bolino A, Quattrini A, Comi G, Previtali SC (2012) Urokinase plasminogen receptor and the fibrinolytic complex play a role in nerve repair after nerve crush in mice, and in human neuropathies. PLoS One 7:e32059. 10.1371/journal.pone.0032059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13:195–204. 10.1016/j.cmet.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs BD, Baillie GS, McCall JR, Passino MA, Schachtrup C, Wallace DA, Dunlop AJ, MacKenzie KF, Klussmann E, Lynch MJ, Sikorski SL, Nuriel T, Tsigelny I, Zhang J, Houslay MD, Chao MV, Akassoglou K (2007) p75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J Cell Biol 177:1119–1132. 10.1083/jcb.200701040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J, Chalaris A, Garbers C, Rose-John S (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32:380–387. 10.1016/j.it.2011.05.005 [DOI] [PubMed] [Google Scholar]
- Song XY, Zhou FH, Zhong JH, Wu LL, Zhou XF (2006) Knockout of p75(NTR) impairs re-myelination of injured sciatic nerve in mice. J Neurochem 96:833–842. 10.1111/j.1471-4159.2005.03564.x [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. 10.1186/1471-213x-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassart RM, Fledrich R, Velanac V, Brinkmann BG, Schwab MH, Meijer D, Sereda MW, Nave KA (2013) A role for Schwann cell-derived neuregulin-1 in remyelination. Nat Neurosci 16:48–54. 10.1038/nn.3281 [DOI] [PubMed] [Google Scholar]
- Stassart RM, Möbius W, Nave KA, Edgar JM (2018) The axon-myelin unit in development and degenerative disease. Front Neurosci 12:467. 10.3389/fnins.2018.00467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi M, Clark HB, Johnson EM Jr (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA 83:4094–4098. 10.1073/pnas.83.11.4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Kubo T, Matsuda K, Fujiwara T, Yano K, Winograd JM, Tohyama M, Hosokawa K (2007) The neurotrophin receptor p75NTR in Schwann cells is implicated in remyelination and motor recovery after peripheral nerve injury. Glia 55:1199–1208. 10.1002/glia.20533 [DOI] [PubMed] [Google Scholar]
- Triolo D, Dina G, Lorenzetti I, Malaguti M, Morana P, Del Carro U, Comi G, Messing A, Quattrini A, Previtali SC (2006) Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J Cell Sci 119:3981–3993. 10.1242/jcs.03168 [DOI] [PubMed] [Google Scholar]
- Waller VA, Owen R (1850) Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond 140:423–429. [Google Scholar]
- Weisel JW, Litvinov RI (2017) Fibrin formation, structure and properties. Subcell Biochem 82:405–456. 10.1007/978-3-319-49674-0_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weskamp G, Schlöndorff J, Lum L, Becherer JD, Kim TW, Saftig P, Hartmann D, Murphy G, Blobel CP (2004) Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J Biol Chem 279:4241–4249. 10.1074/jbc.M307974200 [DOI] [PubMed] [Google Scholar]
- Yang P, Baker KA, Hagg T (2006) The ADAMs family: coordinators of nervous system development, plasticity and repair. Prog Neurobiol 79:73–94. 10.1016/j.pneurobio.2006.05.001 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137. 10.1038/35052073 [DOI] [PubMed] [Google Scholar]
- Zampieri N, Xu CF, Neubert TA, Chao MV (2005) Cleavage of p75 neurotrophin receptor by alpha-secretase and gamma-secretase requires specific receptor domains. J Biol Chem 280:14563–14571. 10.1074/jbc.M412957200 [DOI] [PubMed] [Google Scholar]
- Zunke F, Rose-John S (2017) The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res 1864:2059–2070. 10.1016/j.bbamcr.2017.07.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data of the present manuscript are available from the corresponding authors on reasonable request.