

Abstract
In postmitotic neurons, several tumor suppressor genes (TSGs), including p53, Rb, and PTEN, modulate the axon regeneration success after injury. Particularly, PTEN inhibition is a key driver of successful CNS axon regeneration after optic nerve or spinal cord injury. In contrast, in peripheral neurons, TSG influence in neuronal morphology, physiology, and pathology has not been investigated to the same depth. In this study, we conditionally deleted PTEN from mouse facial motoneurons (Chat-Cre/PtenloxP/loxP) and analyzed neuronal responses in vivo with or without peripheral facial nerve injury in male and female mice. In uninjured motoneurons, PTEN loss induced somatic, axonal, and nerve hypertrophy, synaptic terminal enlargement and reduction in physiological whisker movement. Despite these morphologic and physiological changes, PTEN deletion positively regulated facial nerve regeneration and recovery of whisker movement after nerve injury. Regenerating PTEN-deficient motoneurons upregulated P-CREB and a signaling pathway involving P-Akt, P-PRAS40, P-mTOR, and P-4EBP1. In aged mice (12 months), PTEN deletion induced hair loss and facial hyperplasia of the epidermis. This suggests a time window in younger mice with PTEN loss stimulating axon growth after injury, however, at the risk of hyperplasia formation at later time points in the old animal. Overall, our data highlight a dual TSG function with PTEN loss impairing physiological neuron function but furthermore underscoring the positive effects of PTEN ablation in axon regeneration also for the PNS.
SIGNIFICANCE STATEMENT Tumor suppressor genes (TSGs) restrict cell proliferation and growth. TSG inhibition, including p53 and PTEN, stimulates axon regeneration after CNS injury. In contrast, in PNS axon regeneration, TSGs have not been analyzed in great depth. Herein we show enhanced peripheral axon regeneration after PTEN deletion from facial motoneurons. This invokes a signaling cascade with novel PTEN partners, including CREB and PRAS40. In adult mice, PTEN loss induces hyperplasia of the skin epidermis, suggesting detrimental consequences when reaching adulthood in contrast to a beneficial TSG role for regeneration in young adult mice. Thus, our data highlight the double-edged sword nature of interfering with TSG function.
Keywords: Akt, CREB, facial nerve, motoneuron, nerve regeneration, PTEN
Introduction
In recent years, several tumor suppressor genes (TSGs) were demonstrated to have functions beyond regulation of cell cycle and proliferation (Park et al., 2008; Duraikannu et al., 2019). In the nervous system, modulation of axon growth after injury is one such common TSG function, including p53 (Di Giovanni et al., 2006), Rb (Christie et al., 2014), and PTEN (Park et al., 2008; for review, see Duraikannu et al., 2019).
Particularly PTEN deletion in mouse CNS regeneration models, such as optic nerve (Park et al., 2008; Kurimoto et al., 2010; Leibinger et al., 2019) and spinal cord (K. Liu et al., 2010), injury resulted in robust stimulation of axon growth. This suggests a suppressive growth function of PTEN in WT mice (Gutilla and Steward, 2016). Through its phosphatase activity, PTEN counteracts growth-promoting signaling by the PI3K/Akt pathway. Upon PTEN deletion, Akt phosphorylation, and thereby activity, is increased. Subsequently, Akt modulates downstream effectors, including inhibition of the growth inhibitor GSK3β (Gutilla and Steward, 2016). In addition, Akt stimulates axon growth through mTOR with its effectors S6 kinase and 4EBP1 (Wiza et al., 2012). Despite positive functions of PTEN ablation in axon regeneration, PTEN-deficient mice bear the risk of tumor formation (e.g., in the skin) (Suzuki et al., 2003; Zagni et al., 2017).
In addition to its crucial function in axon growth after injury, PTEN regulates CNS development, morphology, and physiological functions in both neurons and glia (Skelton et al., 2020). PTEN-deficient neurons develop hypertrophy in soma, axonal, and dendritic compartments pointing at a WT PTEN function in restricting cell growth (Fraser et al., 2008; Williams et al., 2015; Gutilla et al., 2016; Gallent and Steward, 2018). Such exuberant growth of cellular compartments was further corroborated in PTEN-deficient oligodendrocytes and Schwann cells (Figlia et al., 2017; Goebbels et al., 2017).
Currently, most PTEN functions were reported in central neurons, whereas our understanding of PTEN functions in peripheral neurons is more limited. Reports showed enhanced sciatic nerve regeneration by genetic or pharmacological PTEN inhibition in mice (Christie et al., 2010; Gallaher and Steward, 2018; Holland et al., 2019; Zhou et al., 2020). Functional nerve regeneration has not been analyzed in all cases (Christie et al., 2010; Zhou et al., 2020) or revealed no differences when assessing sensory or sensorimotor recovery (Gallaher and Steward, 2018; Holland et al., 2019). In a mouse model of diabetic axon regeneration, Singh et al. (2014) demonstrated morphologic, electrophysiological, and behavioral recovery after Pten knockdown. Furthermore, PTEN depletion rescued an axonal growth defect in a spinal muscular atrophy model (Ning et al., 2010). In addition to those injury-associated PTEN functions, there are only few data available addressing consequences of PTEN deletion on peripheral neurons under physiological conditions.
In this study, we used motoneuron (MN)-restricted conditional PTEN ablation in mice through Cre recombinase expression driven by the choline acetyltransferase (Rossi et al., 2011) promoter in MNs (Chat-Cre/PtenloxP/loxP). We focused on facial MNs residing in brainstem facial motor nuclei (FMNs) that are connected via the facial nerve (FN) with several facial muscles (Moran and Graeber, 2004). Thus, this model system consists of centrally located MNs and axons projecting into the periphery, thereby covering aspects of CNS and PNS regeneration. This model system allows for monitoring somatic and axonal changes in MNs and FN as well as functional assessment of whisker movement both in the absence and presence of an FN injury (Gey et al., 2016; Wanner et al., 2017).
Our data revealed peripheral PTEN functions in MN physiology of control (Ctr.) mice, including restriction of MN soma size, axonal and nerve diameter, neuromuscular junction (NMJ) area, and control of whisker movement. After FN injury, MN regeneration was enhanced after PTEN ablation. We identified novel (CREB, PRAS40) and established PTEN signaling partners (Akt, mTOR, 4EBP1) potentially mediating the positive effects on axon regeneration after PTEN deletion. In adult PTEN-deficient mice, facial hyperplasia of the epidermis and hair loss was evident. This suggests a critical “window of opportunity” in young adult mice where PTEN ablation has positive effects on de novo axon growth after injury.
Materials and Methods
Mouse model
All experiments were performed using B6;129S6-Chat-Cre/PtenloxP/loxP mice of both sexes (Groszer et al., 2001; Rossi et al., 2011). As Ctr. mice, both homozygous WT (Chat-Cre/PtenWT/WT) and heterozygous (Chat-Cre/PtenWT/loxP) mice were used since we did not observe differences between the two genotypes. Pten KO animals had the following genotype: (Chat-Cre/PtenloxP/loxP). All mice (Ctr. and KO) were positive for the Chat-Cre allele. Mice were maintained in groups with free access to food and water in the animal facility (12 h light-dark cycle) of Ulm University. All experiments were in accordance with institutional guidelines and German animal protection laws and were approved by the regional government authority (1389; Regierungspräsidium, Tübingen, Germany). All procedures with human material were approved by the ethical committee of Ulm University (208/16) and in compliance with the guidelines of the Federal Government of Germany.
FN transection
FN transection was performed as described previously (Stern et al., 2013; Anastasiadou and Knoll, 2016). Mice of either sex (10-12 weeks old) were anesthetized by inhalation of isoflurane, a skin incision was made behind the left ear, and the FN was exposed. The nerve was transected with microscissors 2 mm posterior to the foramen stylomastoideum. Absence of eyelid closure and whisker movement proved successful nerve transection on the ipsilateral nerve. The contralateral nerve was left intact, and responses in this FN and FMN served as intra-animal control. Regeneration of the FN was quantified by retrograde axonal tracing with fluorogold (FG, Fluorochrome). For this, 4 × 1 μl of FG (4% in H2O) was injected with a Hamilton syringe at multiple positions in each whisker pad 21 d after injury. After another 36 h, brains were dissected. Before and after FN transection, animals were housed in groups in standard cages.
Histology
Freshly isolated brains and nerves were fixed in 4% formaldehyde followed by preparation of 5 μm paraffin microtome slices. Primary antibodies used included the following: anti-PTEN (rabbit, 1:200, Cell Signaling, 9559), anti-Akt (rabbit, 1:200, Cell Signaling, 9272), anti-P-Akt (rabbit, 1:200, Cell Signaling, 4060), anti-FG (rabbit, 1:5000, Millipore, AB153-I), anti-IBA (rabbit, 1:600, Wako, 019-19741), anti-GFAP (mouse, 1:1000, Santa Cruz Biotechnology, sc-21867), anti CD45 (rat, 1:100, BD Pharmingen, 550539), anti-βIIITUB (rabbit, 1:5000, BioLegend, 801201), anti-MBP (mouse, 1:2000, BioLegend, 836504), anti-P-CREB (rabbit, 1:800, Cell Signaling, 9198), anti-P-PRAS40 (rabbit, 1:400, Cell Signaling, 2997), anti-mTOR (rabbit, 1:200, Cell Signaling, 2972), anti-P-mTOR (rabbit, 1:200, Cell Signaling, 2974), anti-4EBP1 (rabbit, 1:200, Cell Signaling, 9644), anti-P-4EBP1 (rabbit, 1:200, Cell Signaling, 2855), anti-P-S6 (rabbit, 1:1000, Cell Signaling, 5364), anti-Ki67 (rabbit, 1:200, Thermo Fisher Scientific, MA5-14520), anti-K14 (chicken, 1:1000, BioLegend, 906004), anti-F4/80 (rat, 1:500, Bio-Rad, MCA497GA), anti-cJUN (rabbit, 1:500, Cell Signaling, #9165), and anti-EGR2 (rabbit, 1:500, Novusbio, NB-92327). Immunohistochemistry was performed using Biotin-conjugated secondary antibodies (1:500; BA-1000, Vectorlabs) and a peroxidase-based detection system using the ABC kit (PK-6100, Vectorlabs) and DAB as substrate. Alternatively, anti-mouse, -rabbit, or -chicken Alexa-488 and Alexa-546-conjugated secondary antibodies (1:1500, Thermo Fisher Scientific, A-11003, A-11008/A-11001/A-11003/A-11071/A-11039) were used.
Nissl staining was performed on paraffin sections using the following incubation protocol: 100% xylene 2 × 2 min, 100% isopropanol 2 × 2 min, 96% isopropanol 1 × 2 min, 75% isopropanol 1 × 2 min, dip in H2O, 0.1% cresyl violet solution 3-5 min, dip in 96% isopropanol 3×, 100% isopropanol 2 × 2 min, 100% xylene 2 × 2 min.
For NMJ staining, lower lips were collected, fixed in 4% PFA and cryoprotected in 30% sucrose followed by preparation of 10 µm cryotome sections. For staining, α-bungarotoxin (BTX, 1:500, Molecular Probes, B35451) was applied together with the primary antibody anti-synaptophysin (guinea pig, 1:500, Synaptic System, 101004) overnight. As a secondary antibody anti-guinea pig CF 633 (1:500, Biotium, 20171) was used. In the case of NMJs, confocal images were acquired using an LSM-700 (Carl Zeiss AG) inverted microscope.
Imaging quantification
Quantification of histologic fluorescent and bright field images was performed using the ImageJ software. For each staining, a threshold was used to set a constant brightness or intensity threshold to differentiate between specific staining and background. Depending on the staining, either the number or the area of stained objects (as indicated in each graph) was quantified using the automated “analyze particles” function of ImageJ. For axon number quantification (see Fig. 5), the ImageJ function “find maxima” was used in each time point. For NMJ quantification, 20 junctions per animal and uninjured/injured side were quantified. Synapse reinnervation was assumed when at least 5% of BTX-stained area colocalized with the SYP staining.
Figure 5.
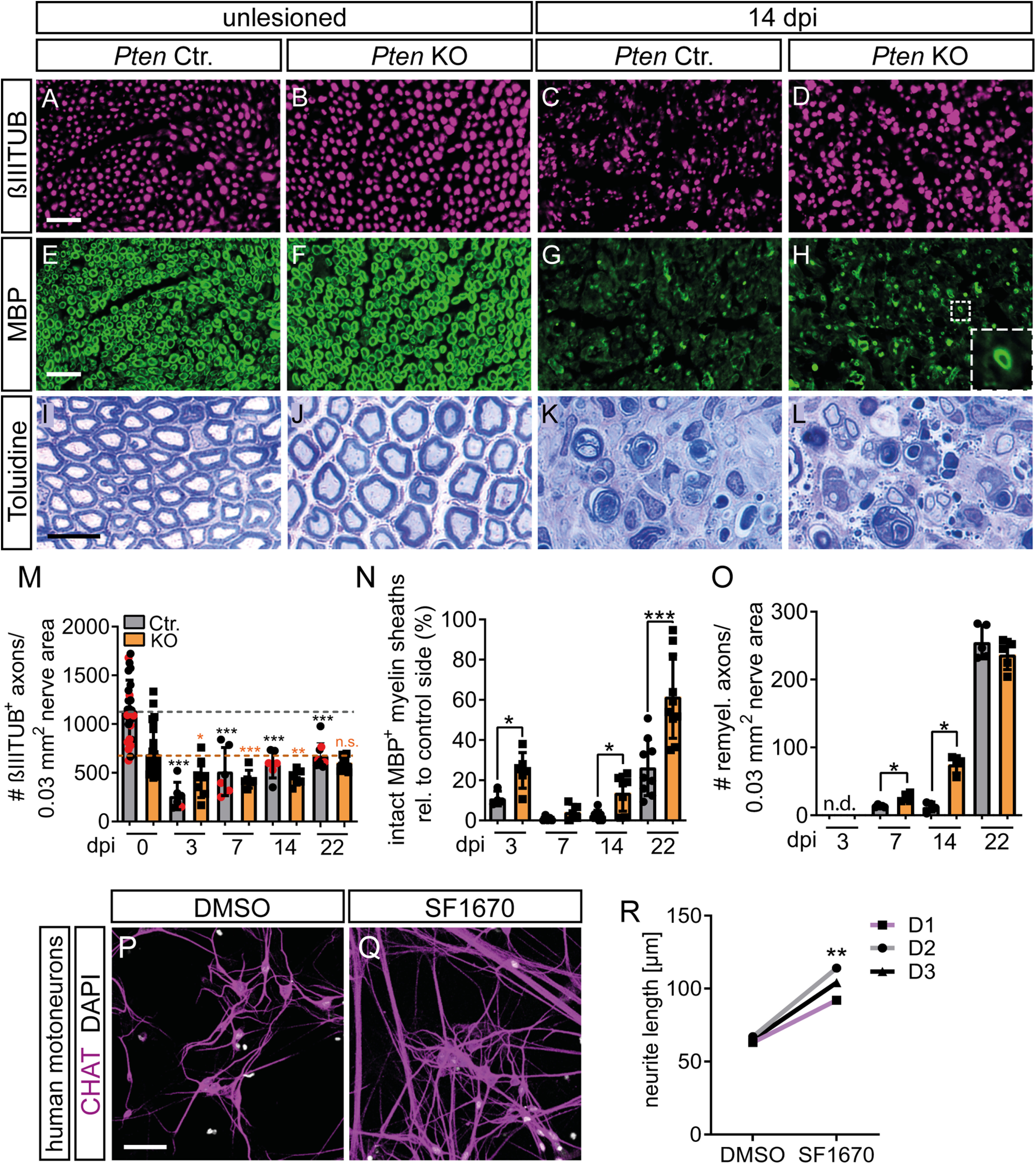
PTEN deletion induces axon and myelin regeneration after FN injury. A-D, M, In the absence of injury, βIII tubulin-positive numbers were identical between Ctr. and KO nerves. At 14 dpi, axon numbers in the FN decreased in Ctr. (C) but not as much in Pten KO animals (D). M, Axon numbers per 0.03 mm2 nerve area. Because of excess connective tissue and larger axon diameter, Pten KO animals have lower axon density in uninjured nerves. After injury, axon numbers decreased in both genotypes but recovered more quickly in PTEN-deficient animals. Dashed lines indicate axonal numbers in uninjured nerves in Ctr. (gray) or Pten KO (orange) animals (n = 6, 7, 6, 6, 6, 6, 9, 10 for each bar, respectively). R, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. Black asterisks indicate significant differences of each bar to the uninjured (0 dpi) Ctr. bar, orange asterisks to the uninjured (0 dpi) Pten KO bar. E-H, N, Myelin was detected by MBP. In Pten KO nerves, numbers of properly myelinated axons (see inset in H) were increased compared with Ctr. nerves (G; quantified in N; n = 4, 6, 5, 5, 8, 7, 9, 10 for each bar, respectively). I-L, O, Myelination was stained with toluidine and more myelinated axons were observed after injury in KO (L) compared with Ctr. (K) animals (quantified in O; n = 0, 0, 5, 4, 4, 4, 5, 5 for each bar, respectively). Note the increased axon diameters in KO animals in the unlesioned condition (J). P-R, MNs differentiated from hiPSCs showed increased neurite outgrowth when treated the PTEN inhibitor SF1670 (Q) compared with control treatment with DMSO (P). R, Quantification of neurite outgrowth of MNs from tree different donors (D1-D3). M-O, Each dot indicates 1 animal. Data are mean ± SD. M-O, *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test. R, *p < 0.05; **p < 0.01; ***p < 0.001; two-sided t test. Scale bars: A-H, 25 µm; I-L, 10 µm; P, Q, 50 µm.
Human induced pluripotent stem cells (hiPSCs) and MN differentiation
hiPSC lines used in this study are listed in Table 1. The lines generated at Ulm University have been previously published and characterized (Catanese et al., 2021). The third line has been commercially purchased from the iPSC Core facility of Cedars Sinai (Los Angeles). hiPSCs were cultured at 37°C (5% CO2, 5% O2) on Matrigel-coated (Corning, 354277) 6-well plates using mTeSR1 medium (Stem Cell Technologies, 83850). When colonies reached 80% of confluence, they were detached using Dispase (Stem Cell Technologies, 07923) and passaged in a 1:6 split ratio.
Table 1.
hiPSC donors used for MN differentiation
| hiPSC line | Gender | Age (yr) | Source |
|---|---|---|---|
| Donor 1 | Female | 45 | Ulm University |
| Donor 2 | Male | 64 | Ulm University |
| Donor 2 | Male | 49 | Cedars-Sinai (CS0YX7iCTR) |
Human MNs were generated as previously described (Catanese et al., 2021). Briefly, hiPSC colonies were detached and cultured in suspension using ultra-low attachment flasks T75 for 3 d. This allowed the formation of embryoid bodies in hESC medium (DMEM/F12 + 20% KO serum replacement + 1% NEAA + 1% β-mercaptoethanol + 1% antibiotic-antimycotic + SB-431542 10 μm + dorsomorphin 1 μm + CHIR 99021 3 μm + pumorphamine 1 μm + ascorbic acid 200 ng/µl + cAMP 500 μm + 1% B27 + 0.5% N2). On the fourth day, culturing medium was switched to MN medium (DMEM/F12 + 24 nm sodium selenite + 16 nm progesterone + 0.08 mg/ml apotransferrin + 0.02 mg/ml insulin + 7.72 μg/ml putrescin + 1% NEAA, 1% antibiotic-antimycotic + 50 mg/ml heparin + 10 μg/ml of the neurotrophic factors BDNF, GDNF, and IGF-1, SB-431542 10 μm, dorsomorphin 1 μm, CHIR 99021 3 μm, pumorphamine 1 μm, ascorbic acid 200 ng/µl, retinoic acid 1 μm, cAMP 500 μm, 1% B27, 0.5% N2). After 5 further days, embryoid bodies were dissociated using Accutase (Sigma-Aldrich) and plated onto μPlates (Ibidi) precoated with Growth factor Reduced Matrigel (Corning). The treatment with the PTEN inhibitor SF1670 (stock of 10 mm diluted in DMSO; final concentration used for treatment 10 nm) was started at the fourth day after plating and conducted until DIV 14 by replacing half of the medium every second day.
Immunocytochemistry and neurite outgrowth quantification
hiPSC-derived MNs were fixed with 4% PFA (containing 10% sucrose) blocked and incubated with primary antibody against CHAT (rabbit, 1:100, Abcam, ab181023) for 24 h at 4°C. Secondary antibody (Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, AlexaFluor-568; Invitrogen A-11036) was used for 2 h at room temperature. Cells were mounted with ProLong Gold Antifade mountant with DAPI (Thermo Fisher Scientific) and Mounting Medium (Ibidi).
Fluorescence microscopy was performed with a Thunder imaging system (Leica Microsystems) equipped with a DFC9000 sCMOS camera and an HC PL Apo 40× (NA 1.10) water immersion objective using the LasX software (Leica Microsystems). The neurite length of human MN was calculated using the semiautomated framework SNT contained in the Neuroanatomy plug-in for ImageJ (Catanese et al., 2021). Data were collected from analyzing a minimum of 21 neurons from three independent differentiations for each donor. Plotted data represent the average value obtained from the independent treatments from each donor.
Electron microscopy (EM)
Nerves were fixed overnight in 4% PFA, postfixed in 2.5% glutaraldehyde for at least 24 h. Then semithin sections (300 nm) were prepared, stained with toluidine by applying a 0.05% toluidine solution for 10-20 s, and the areas of interest were used for the preparation of ultrathin sections (80 nm). For quantification of axon diameter and G-ratio, axon and myelin perimeters were measured and used to calculate the diameter. G-ratio was calculated using the formula g-ratio = diameteraxon/diameterfiber. For each animal, 30-40 random axons and myelin sheaths in 3 frames were quantified (as indicated in the figure legends). The mean of each animal was then built and compared with the mean of the other animals. For the parameter “connective tissue spread,” we used three independent frames per animal and the ImageJ function “nearest distance” to calculate the nearest distance of each myelinated fiber to neighboring fibers in one frame. The mean of each frame was used to calculate a mean value per animal. Those values where then used to perform statistics.
Whisker movement analysis
Whisker movement analysis was performed as reported previously (Wanner et al., 2017). Mice were subjected to daily handling, and training starting 5 d before injury. One day before first whisker movement recording, mice were anesthetized and all whiskers except the C row were clipped. Hand restraint mice were videotaped for 51 s by a high-speed camera (Basler acA1300-60gc) at 100 Hz. Video sequences were reviewed, and 1 s fragments were further processed in Templo Software (Contemplas). The selected video sequences were analyzed by Vicon Motus 2D software (Contemplas).
The parameters “acceleration” and “velocity” of the whiskers were calculated by the Vicon Motus 2D software. The parameter “angular sum” was calculated by a self-written MATLAB program (developed by Hans-Georg Glöckler, Institute of Physiological Chemistry, Ulm University). Hereby, deflections of the whisker ≥10°, 20°, 30°, 40°, and 50° were incorporated into the calculations. For each parameter, the ratio of the value of the whisker on the injured side/uninjured side was calculated to eliminate inter- and intra-individual variance.
Experimental design and statistical analyses
For all experiments, at least three biological replicates were analyzed and (n) numbers are indicated in figure bars or figure legends. For statistical analysis of data and graph generation, GraphPad Prism software (GraphPad Software) was used. Sample groups were tested for normality using the D'Agostino-Pearson omnibus normality test. Since some groups were not normally distributed, or groups were too small to be tested for normality (n < 10), the nonparametric unpaired Mann–Whitney test (two-sided) was chosen to calculate significance if not mentioned otherwise in the figure legend. For multiple comparisons, one-way or two-way ANOVA was performed as mentioned in the figure legends. SD is provided if not mentioned otherwise in the figure legends.
Results
PTEN deletion induces hypertrophy of facial MN cell body, axon, and nerve
In a first step, we analyzed whether PTEN ablation from facial MNs interfered with neuronal morphology and function under physiological uninjured conditions in young adult mice aged 2-3 months (Fig. 1). To prove downregulated PTEN expression in the conditional Chat-Cre/PtenloxP/loxP mice (from now on referred to as “KO” mice), brainstem sections harboring the FMN with facial MNs of Ctr. or KO mice were stained for PTEN (Fig. 1A,B,K). FMN of Ctr. mice were positive for PTEN expression (Fig. 1A), whereas this was strongly reduced in Pten KO mice (Fig. 1B,K). Congruent with this finding, phosphorylated Akt (P-Akt) was upregulated in MNs of Pten KO mice (Fig. 1D), and lower P-Akt levels were observed in Ctr. mice (Fig. 1C,L).
Figure 1.
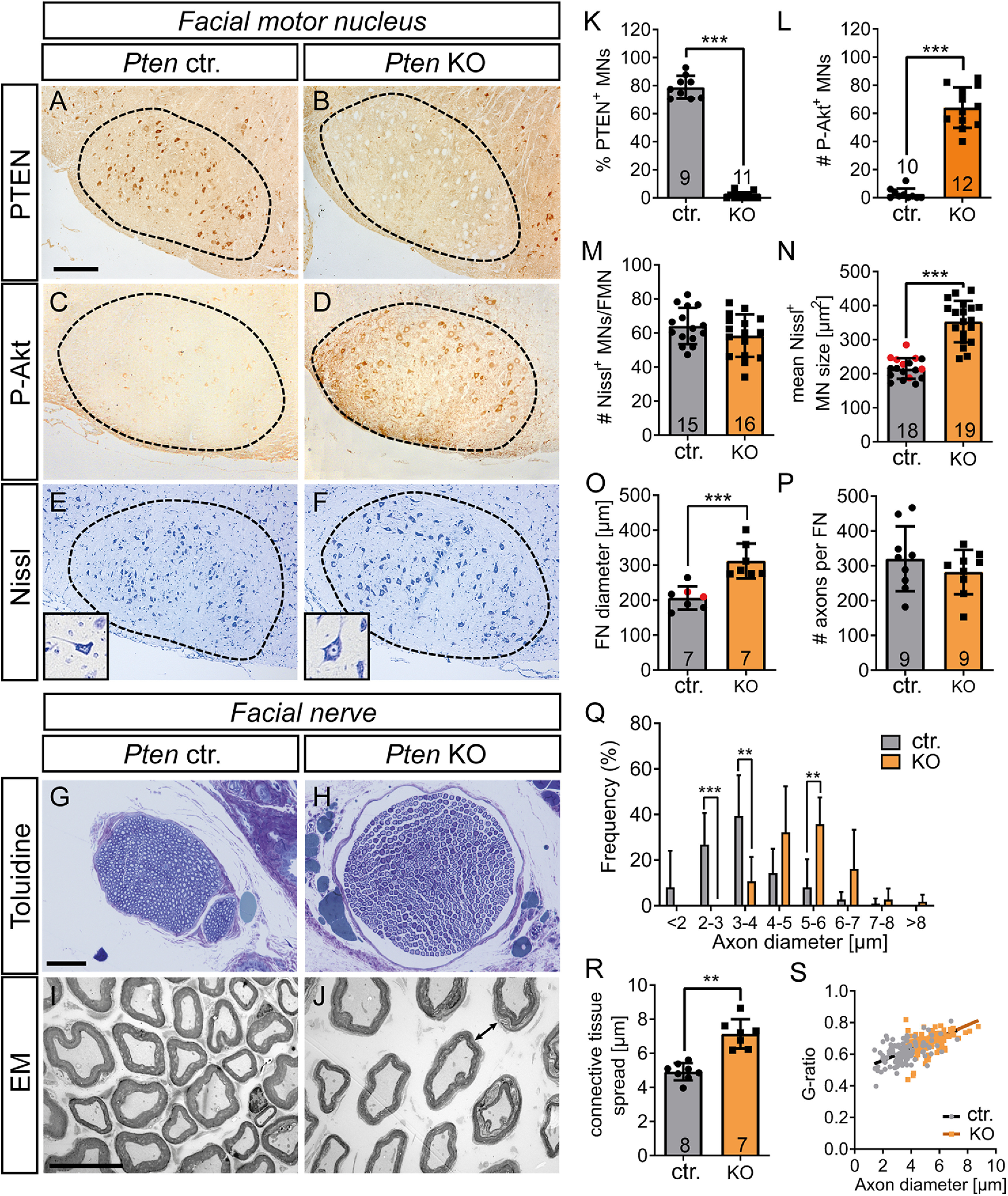
PTEN deficiency in intact MNs results in soma, axon, and nerve hypertrophy. A-F, The FMN (A-F) and FN (G-J) were stained in uninjured animals. A, B, K, Ctr. MNs expressed PTEN (A), whereas PTEN expression was absent in Pten KO neurons (B; quantified in K). C, D, L, In Ctr. mice, MNs were only weakly P-Akt positive (C). In contrast, PTEN deletion resulted in strong P-Akt induction (D; quantified in L). E, F, M, N, The size of MNs was higher in Pten KO neurons (F; see inset) compared with Ctr. neurons (E). The total MN number was not changed by PTEN ablation (M), whereas area/MN was elevated (N). N, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. G, H, O, P, The average FN diameter stained with toluidine was higher in Pten KO (H) compared with Ctr. animals (G; quantified in O). O, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. Axon numbers per FN remained unaltered (P). I, J, Q-S, EM pictures of Ctr. nerves (I) revealed smaller axon diameter compared with PTEN deleted animals (J). The axon diameter was plotted against the frequency for Ctr. and KO animals and showed more large-diameter axons after PTEN deletion (Q; 30 arbitrary axons for each of 7 Ctr. and 7 KO animals were analyzed). Ctr. animals also had less connective tissue between individual axons (arrow in J indicates larger distance between axons; quantified in R). The G-ratio was not altered between genotypes (S; 30 arbitrary axons for each of 7 Ctr. and 7 KO animals were analyzed). Each dot in (K-P, R) indicates 1 animal. Numbers in the bars represent numbers of animals analyzed. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test. Scale bars: A-F, 200 µm; G, H, 50 µm; I, J, 20 µm.
Since PTEN regulates cell size in central neurons (Skelton et al., 2020), we analyzed a potential hypertrophy associated with PTEN deletion in the PNS by Nissl staining (Fig. 1E,F,M,N), toluidine staining (Fig. 1G,H,O,P), and EM (Fig. 1I,J,Q–S). Indeed, Nissl staining revealed an ∼60% increase of MN soma size on PTEN ablation (Fig. 1E,F,N) without total MN numbers in the FMN being changed (Fig. 1M). MN axons establish the FN with several branches running along the facial muscles (see Fig. 4A). When inspecting the FN diameter, a 50% increase in PTEN-deficient mice compared with Ctr. mice was observed (Fig. 1G,H,O). This increase was most likely caused by a concomitant increase in axonal diameter (Fig. 1I,J,Q). In addition, elevated connective tissue abundance resulting in more separation between individual axons in PTEN-deficient mice (arrow Fig. 1J) compared with Ctrl. mice (Fig. 1I,R) was noticed. Changes in total MN numbers (Fig. 1M) or axon number/nerve (Fig. 1P) do most likely not account for this nerve diameter enlargement on PTEN ablation. Also, myelin thickness determined by the G-ratio was not altered between genotypes (Fig. 1S). Since we used both Pten homozygous and heterozygous animals as WTs (see also Materials and Methods), we analyzed whether heterozygous mice differed from their homozygous WT littermates to rule out any gene-dose dependent effects of PTEN. Comparing several parameters, including MN size and nerve diameter (Fig. 1N,O), numbers of FG-positive MNs (see Fig. 4R), and P-Akt and P-mTOR levels (see Fig. 9O and Fig. 10C, respectively), did not reveal differences between both genotypes. Thus, Pten heterozygous and homozygous WT animals behaved similarly and were combined in one “control” group throughout the manuscript.
Figure 4.
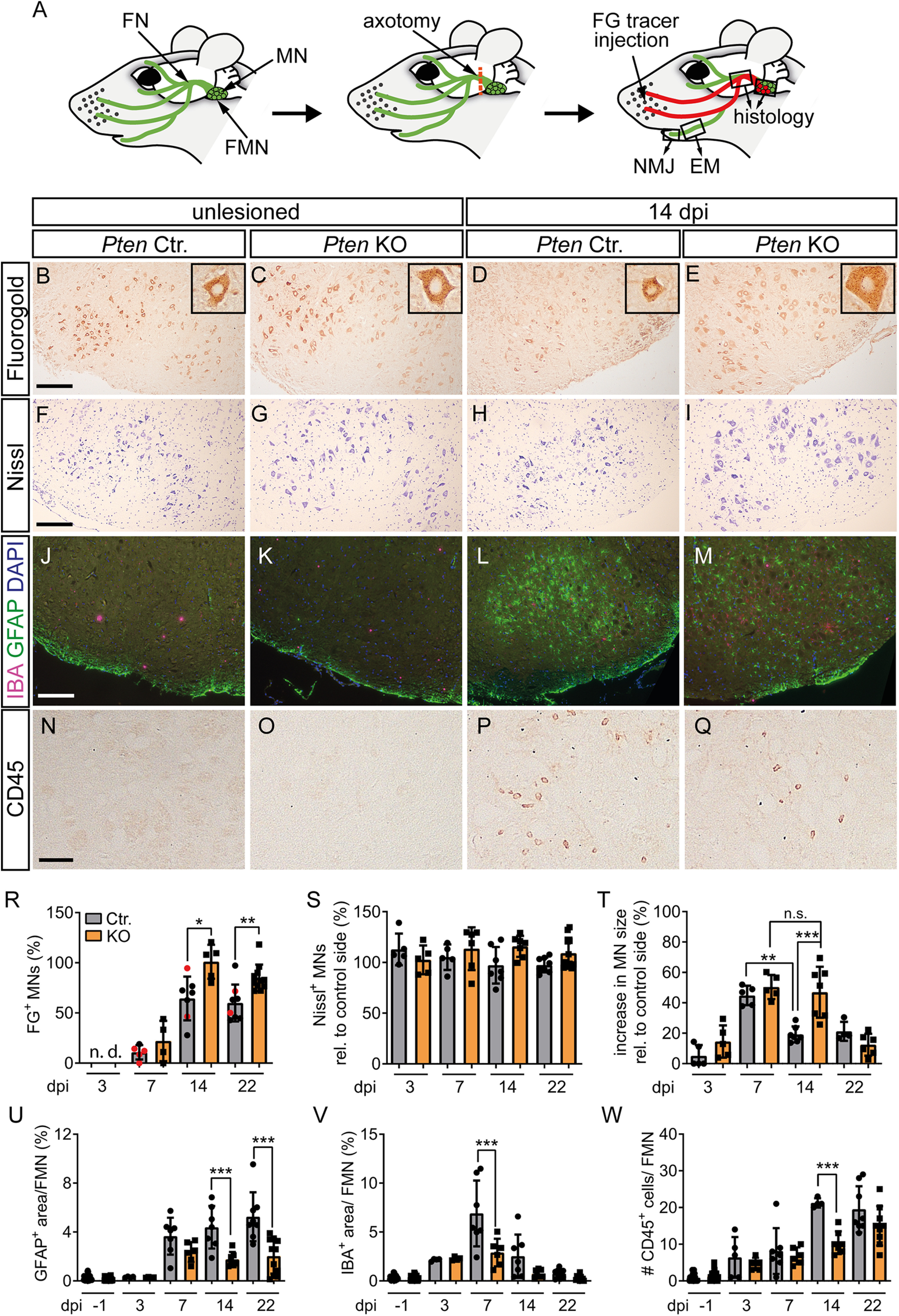
PTEN deletion stimulates FN regeneration and decreases injury-associated neuroinflammation. A, Overview of the FN injury model. MNs connected to several FN branches (green; FN) are localized to the FMN in the hindbrain. Injury of the FN (red dashes line) was performed at the central branch. Upon successful regeneration, FG tracer was retrogradely transported from the whisker pad along the axons to the MNs (labeled in red). Right, Squares represent the parts of the nerve that were analyzed in histology or EM. B-E, R, The number of FG-positive neurons on the unlesioned side was identical between Ctr. (B) and KO (C) neurons. At 14 dpi, FG numbers in the Ctr. FMN (D) were lower compared with KO animals (E; quantified in R; n = 0, 0, 5, 4, 7, 5, 8, 11 for each bar, respectively). R, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. Insets, Higher magnification of individual FG-positive neurons. F-I, S, T, The number of Nissl-positive MNs was not affected by injury or genotype (F–I; quantified in S; n = 5, 5, 5, 6, 7, 7, 8, 11 for each bar, respectively). At 14 dpi, the size of PTEN-deficient MNs was larger compared with Ctr. MNs (T; n = 5, 5, 5, 5, 7, 7, 4, 6 for each bar, respectively). J-M, U, V, In unlesioned FMN, no changes between Ctr. (J) and KO (K) were observed for astrocytes (GFAP, green) and microglia (IBA, red). In Ctr. FMN, astrocytes and microglia were increased after injury (L). In Pten KO FMN, such neuroinflammation was decreased (M; quantified in U, V; n = 27, 24, 4, 4, 7, 6, 7, 7, 8, 11 for each bar, respectively). N-Q, W, Peripheral leukocytes were labeled with anti CD45 antibody. After injury, more CD45-positive cells were visible in Ctr. (P) compared with KO (Q) animals (quantified in W; n = 21, 24, 4, 4, 7, 6, 7, 7, 8, 11 for each bar, respectively). R-W, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA test. Scale bars: B-M, 200 µm; N-Q, 50 µm.
Figure 9.
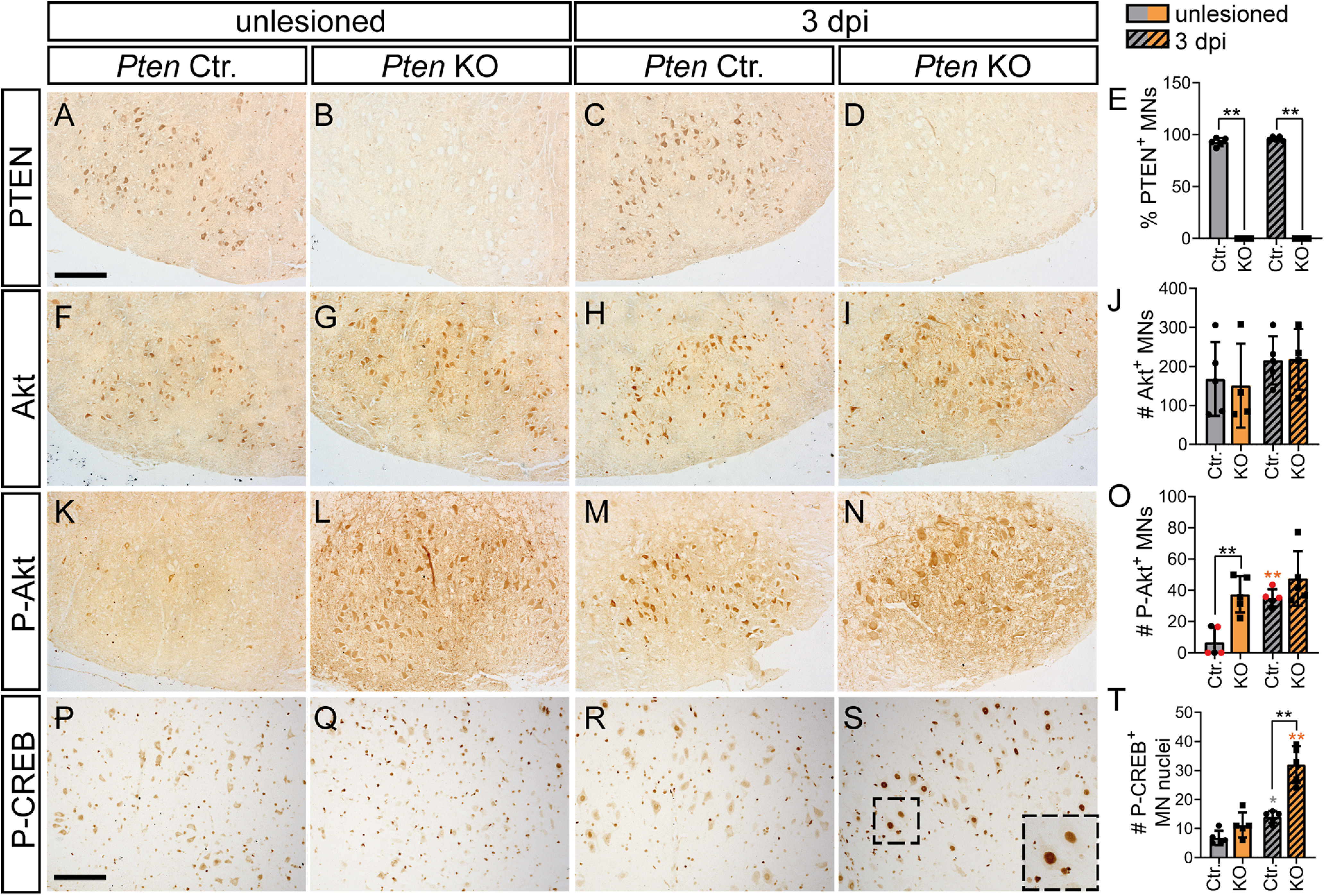
PTEN deletion enhances P-Akt and P-CREB levels in injured MNs. Unlesioned and injured FMN of Ctr. and Pten KO animals were stained for proteins indicated at 3 dpi. A-E, PTEN abundance in Ctr. MNs did not change after injury (A,C). In PTEN-deficient animals, almost no remaining PTEN levels were present in MNs (B,D; quantified in E; n = 5 for each bar). F-J, Total Akt levels were unchanged by injury and PTEN deletion (n = 5 for each bar in J). K-N, O, In Ctr. animals, levels of P-Akt were upregulated after FN injury (K,M). In PTEN-deficient animals, P-Akt levels were increased already before injury (L) and slightly higher after injury (N; quantified in O; n = 5, 4, 5, 4 for each bar, respectively). O, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. P-T, Nuclear P-CREB abundance was enhanced in injured PTEN-deficient MNs (S; insets indicate higher magnifications of individual MNs) compared with other conditions (P-R; quantified in T; n = 5 for each bar). E, J, O, T, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test. Scale bars: A-N, 200 µm; P-S, 150 µm.
Figure 10.
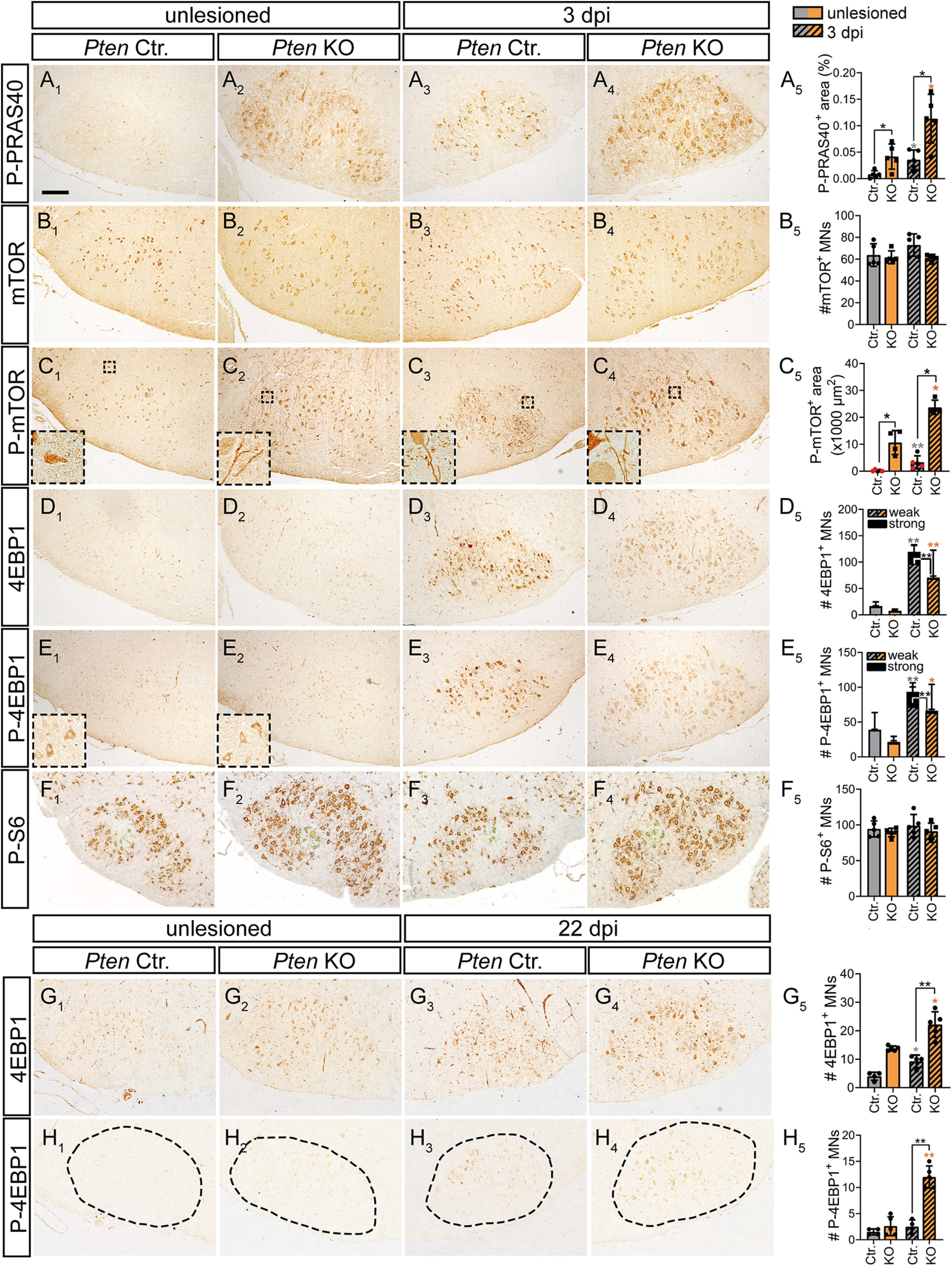
Induction of a PRAS40-mTOR pathway by PTEN deletion in injured MNs. A1-A5, In unlesioned Ctr. MNs, no P-PRAS40 was visible (A1) in contrast to 3 dpi (A3). In KO neurons, P-PRAS40 abundance was higher in FMNs before (A2) and after injury (A4; quantified in A5; n = 5 for each bar). B1-B5, Total mTOR abundance was not affected by genotype or injury status at 3 dpi (quantified in B5; n = 5 for each bar). C1-C5, P-mTOR levels were slightly upregulated in injured Ctr. neurons (C3) at 3 dpi. After PTEN ablation, P-mTOR expression was elevated before (C2) and after injury (C4; quantified in C5; n = 5, 4, 5, 4 for each bar, respectively). Insets, P-mTOR localization on neuronal protrusions. C5, Black dots in gray bars indicate Pten homozygous WT mice. Red dots indicate heterozygous mice. D1-D5, At 3 dpi, total 4EBP1 abundance was higher in Ctr. (D3) compared with injured KO (D4) MNs (quantified in D5; n = 4 for each bar). E1-E5, P-4EBP1 levels were upregulated in injured Ctr. (E3) and KO (E4) neurons compared with the uninjured side (E1,E2) at 3 dpi. The number of strongly positive P-4EBP1 MNs was higher in Ctr. compared with injured KO MNs (quantified in E5; n = 4 for each bar). F1-F5, P-S6 levels were similar in unlesioned and lesioned Ctr. and KO neurons (quantified in F5; n = 5 for each bar). G1-G5, At 22 dpi, total 4EBP1 levels were elevated after FN injury in Ctr. MNs (G1,G3). In PTEN-deficient animals, 4EBP1 levels were higher both on the unlesioned (G2) and injured (G4) side (quantified in G5; n = 4, 5, 4, 5 for each bar, respectively). H1-H5, At 22 dpi, highest P-4EBP1 levels were quantified in KO animals after injury (H4). In contrast, in all other conditions (H1-H3), only weak P-4EBP1 levels were measured (H5; n = 4, 5, 4, 5 for each bar, respectively). A5, B5, C5, F5, G5, H5, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test. Scale bars: A-H, 200 µm.
FN axons ultimately terminate in facial muscles, including the lip muscles where they form neuromuscular synapses (Moran and Graeber, 2004). Thus, in a next step, we analyzed PTEN-associated morphologic synapse changes by labeling postsynaptic NMJ structures with BTX. Indeed, these terminal axon structures also showed signs of hypertrophy, that is, increased total synapse area from 171.7 µm2 (±22.3 µm2) in Ctr. mice to 352.6 µm2 (±46.9 µm2) in Pten KO animals. However, we also observed an elevated fragmentation within individual NMJs of PTEN-deficient mice, which might be indicative of altered NMJ functionality. In agreement with this, when PTEN KO mice reached an age of ∼14 weeks (14 d after injury in lesioned mice), whisker movement was compromised even on the uninjured face side of KO mice compared with Pten Ctr. mice. This resulted in a reduction of the sum of angles exceeding 20° during 51 s of whisking from 1395.3° (±194.8°) in Ctr. mice to 1025.5° (±289.7°) in KO mice. This suggests a function of PTEN in Ctr. mice ensuring NMJ functionality associated with triggering physiological whisking.
In addition to analyzing young adult mice (Fig. 1), we also investigated whether PTEN's role in controlling size of neuronal compartments was maintained in aged mice (12 months; Fig. 2A–M). Indeed, PTEN-dependent hypertrophy of peripheral neuron cell soma, axon, and nerve observed in young adult mice (Fig. 1) was preserved in these old mice (Fig. 2A-K). In contrast, the separation of individual axons by more connective tissue observed in younger mice (Fig. 1I,J) was not observed in aged Pten KO mice (Fig. 2E–H,L). However, EM data suggested higher mitochondria numbers in axons of PTEN ablated compared with Ctr. MNs (Fig. 2G,H,M).
Figure 2.
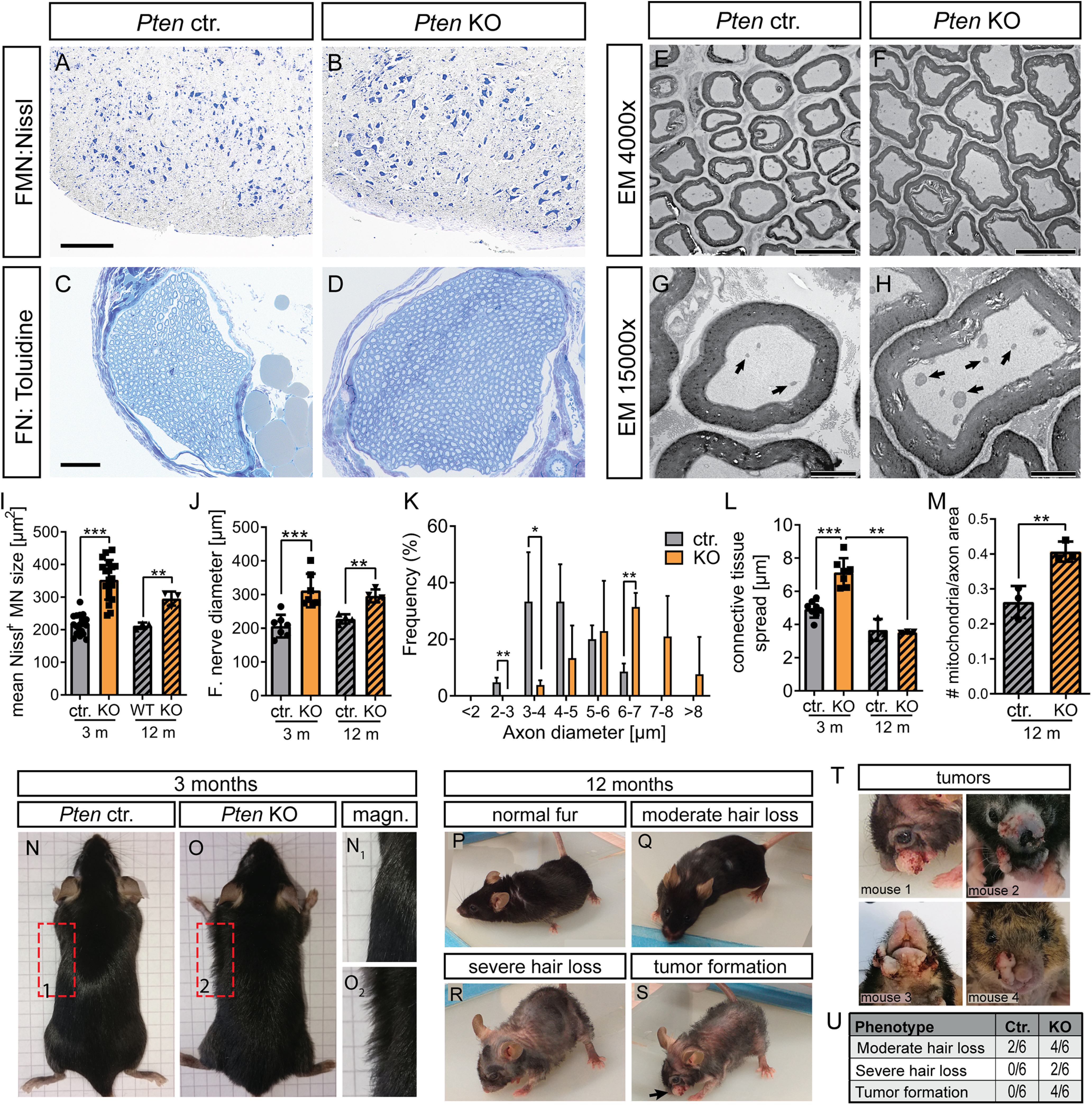
Twelve-month-old PTEN-deficient mice show MN hypertrophy and skin abnormalities. A, B, I, Nissl staining was performed in the facial nucleus of Ctr. (A) and KO (B) animals aged 10-12 months. Similar to results obtained in mice aged 3 months, the MN size was enhanced in PTEN deleted compared with Ctr. animals (I; n = 18, 19, 3, 3 for each bar, respectively). C, D, J, The nerve diameter analyzed with toluidine staining was increased in Pten KO (D) compared with Ctr (C) mice (quantified in J; n = 7, 7, 4, 4 for each bar, respectively). E-H, K-M, EM pictures at lower (E,F) and higher (G,H) magnification in the FN of Ctr. (E,G) and KO (F,H) animals at the age of 10-12 months. In PTEN-deficient animals, the diameter of individual axons was increased compared with Ctr. animals (K; n = 4 Ctr. and 4 KO animals). The connective tissue present between individual axons was elevated by PTEN ablation at 3 months of age, however not at 12 months (L; n = 8, 7, 3, 3 for each bar, respectively). The number of mitochondria present in axons was higher in KO (arrows in H) compared with axons derived from Ctr. mice (arrows in G) animals (quantified in M; n = 3 for each bar). N, O, Ctr. (N) and KO (O) mice aged 3 months are depicted. In Pten KO animals (O), a “frizzy” hair phenotype was visible, whereas in Ctr. animals, hair was smoothly aligned to the body (N). P-U, PTEN-deficient mice at the age of 10-12 months showed more severe skin phenotype (moderate to severe hair loss; see R and S) in addition to facial hyperplasia/tumor formation of the epidermis (arrow in S). T, Four different Pten KO animals are depicted showing different types and sizes of neoplasms in the facial region. Frequency of observed skin phenotype in Ctr. and Pten KO mice is shown in U. I-M, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; t test. Scale bars: A, B, 200 µm; C, D, 50 µm; E, F, 10 µm; G, H, 2 µm.
Interestingly, Pten KO mice developed a phenotype of frizzy fur by the age of 3 months (Fig. 2N,O). By the age of 10-12 months, we observed moderate to severe hair loss and facial tumor formation in 4/6 Pten KO mice animals (Fig. 2P–U). Such hyperplasia was never observed in the younger animals used for FN injury experiments. Interestingly, histologic analysis of the skin of young Pten KO mice revealed reduced PTEN expression in follicular keratinocytes (Fig. 3A,B,E). This PTEN downregulation was accompanied by P-Akt upregulation (Fig. 3C,D,F). In keratinocytes of the outer epidermal layer of Ctr. mice, PTEN expression was in general lower and showed no further reduction in Pten KO mice (Fig. 3A,B). At first glance, epidermal PTEN depletion was somewhat surprising given that ChAT promotor-dependent Cre recombinase expression was considered to be largely MN-specific. However, previous literature already described ChAT expression in non-neuronal tissue, including skin keratinocytes (Wessler and Kirkpatrick, 2008). In addition, previous reports show that keratinocyte-specific PTEN deletion induced skin tumor formation (Suzuki et al., 2003; Zagni et al., 2017), which is an obvious risk of abrogating TSG activity in animals as done with PTEN in this study. Microscopic analysis of the neoplasms of old Pten KO animals showed a thickened stratum corneum (H&E staining in Fig. 3G–J), increased inflammation, hyperproliferation of keratinocytes, and a drastically thickened keratinocyte layer (CD45+, Ki67+, and K14+ cells, respectively, in Fig. 3K,L) in the epidermis of Pten KO compared with Ctr. mice. Nevertheless, there was no disrupted cytoplasm/nucleus ratio or atypical mitosis in the basal layer of the epidermis, and the basement membrane did not seem to be disrupted (Fig. 3I,J). This argues for a benign character of the epidermal hyperplasia. In addition, no macroscopically visible metastases were observed in other organs, so that an aggressive squamous cell carcinoma can most likely be ruled out.
Figure 3.
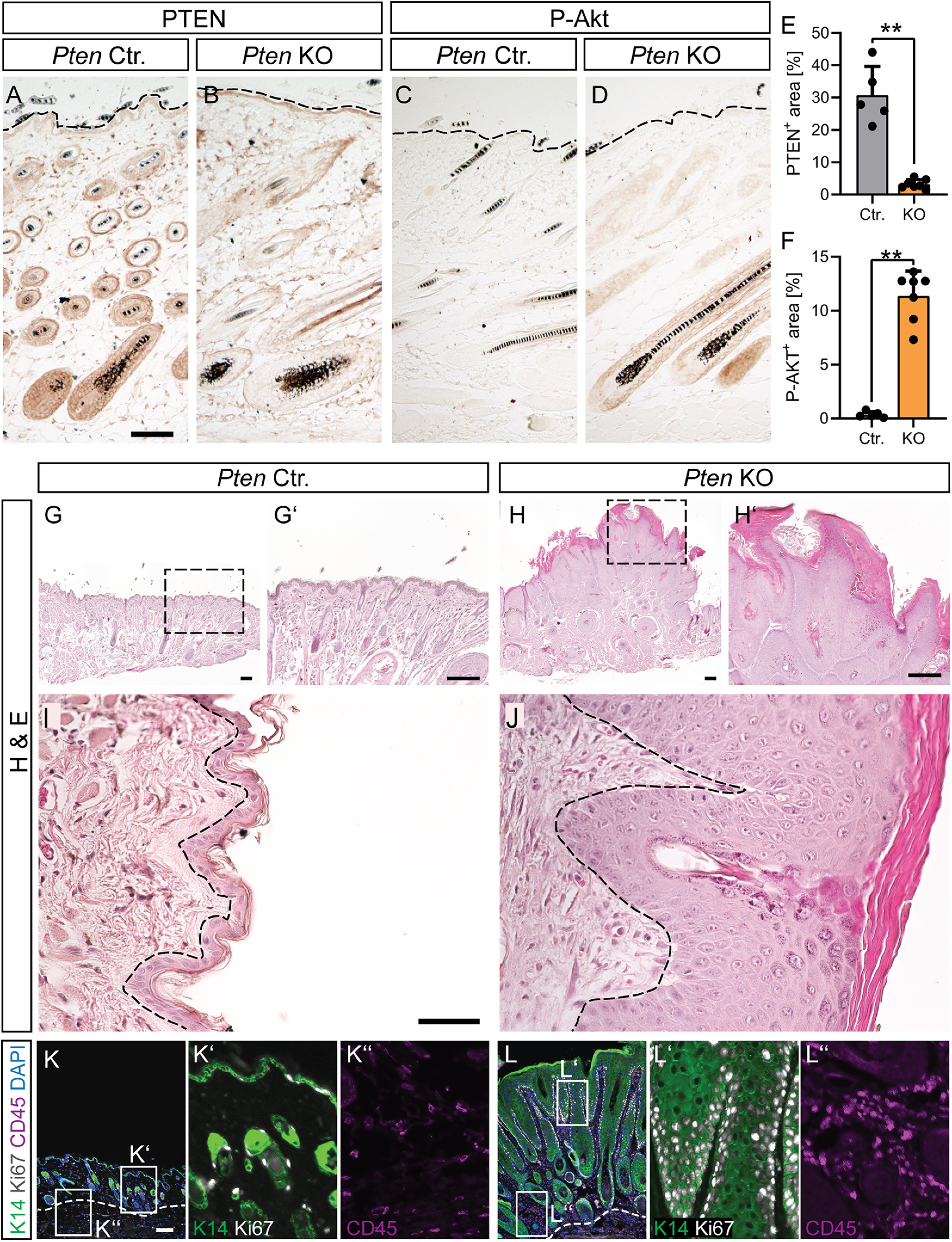
Epidermal depletion of PTEN results in epidermal hyperplasia. A-F, Skin sections of the epidermal layer of young adult Ctr. (A,C) and Pten KO (B,D) mice stained with PTEN (A,B; quantified in E; n = 5 Ctr. and 7 KO animals) and P-Akt (C,D; quantified in F; n = 5 Ctr. and 7 KO animals) directed antibodies. Dashed line indicates the outermost edge of the epidermis. G-J, H&E staining of skin sections in 12-month-old Ctr. (G,G′,I) and Pten KO (H,H′,J) mice. Pten KO mice show epidermal hyperplasia and clearly thickened stratum corneum but no disruption of the basement membrane. Dashed lines indicate the edge of the basal epidermal layer. ChAT-mediated Cre recombinase activity also deleted PTEN from follicular keratinocytes resulting in PTEN loss and concomitant P-Akt upregulation. K, L, K14 (green), Ki67 (white), and CD45 (magenta) staining of Ctr. (K; magnifications in K′ and K′′) and KO skin (L; magnifications in L′ and L′′) reveals hyperproliferation of keratinocytes (K14+), hyperkeratosis, and increased inflammation (CD45+) in Pten KO skin sections. E, F, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney. Scale bars: A-D, 100 µm; G, H, K, L, 200 µm; I, J, 50 µm.
In summary, similar to central neurons, PTEN regulates neuronal soma and axon size as well physiological neuron function (here: whisking) in “young” and “old” peripheral neurons, but its deletion in keratinocytes results in skin abnormalities.
PTEN inhibition facilitates facial MN regeneration
After having addressed PTEN deletion in intact MNs (Figs. 1 and 2), we analyzed PTEN function in nerve regeneration after FN injury (Figs. 4–10). For this, one of the two FNs of Ctr. and Pten KO mice was completely dissected. The contralateral FN on the other side of the face was left intact and served as control within the same animal (Fig. 4A). Subsequently, animals were analyzed at 3, 7, 14, and 22 d post injury (dpi) with several read-outs, including histology/EM (Figs. 4–7) and functional whisker movement (see Fig. 8).
Figure 7.

PTEN deficiency does not affect immune cell infiltration and Schwann cell dedifferentiation in the nerve after injury. A-D, Q, FN cross-sections of Ctr. (A,C) and KO (B,D) animals were stained for CD45 expression. After injury, CD45-positive cells go up without difference between Ctr. (C) and KO mice (D; quantified in Q; n = 26, 26, 6, 7, 5, 4, 6, 6, 9, 10 for each bar, respectively). E-H, R, F4/80-positive cells were present in the injured FN without noticeable difference between Ctr. (G) and KO mice (H; quantified in R; n = 26, 26, 3, 5, 5, 6, 5, 6, 9, 10 for each bar, respectively). I-L, S, cJUN was upregulated by FN injury, however, in a similar manner between Ctr. (K) and PTEN-deficient (L) animals (quantified in S; n = 26, 26, 5, 5, 7, 6, 4, 7, 9, 10 for each bar, respectively). M-P, T, After injury, more EGR2-positive Schwann cells were present in the nerve. No differences between Ctr. (O) and KO mice (P) were discernible (quantified in T; n = 21, 21, 0, 0, 7, 6, 5, 5, 9, 10 for each bar, respectively). Q-T, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test. Scale bars: A-P, 50 µm.
Figure 8.
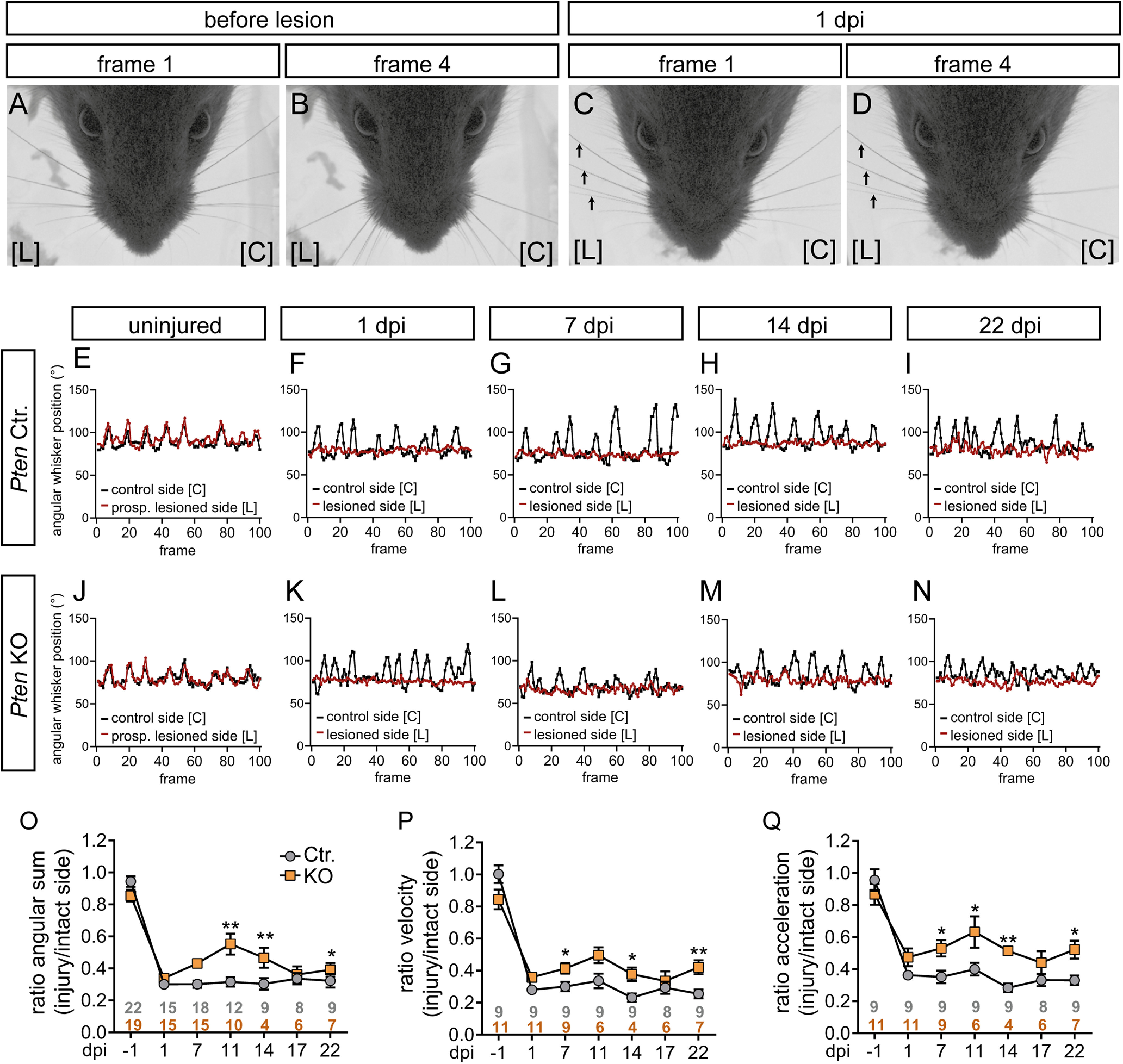
Recovery of whisker movement after injury is improved by PTEN deletion. A-D, Before lesion, whiskers move synchronously backward and forward (A,B). At 1 dpi, whiskers on the lesioned side [L] did not move anymore (arrows in C,D), while on the control side [C] movement was still possible. Frames 1 and 4 show a time span of 4 ms derived from a 100 ms (100 frame; see E-N) video sequence. E-N, Typical sequences of whisker movement in Ctr. (E-I) and KO animals (J-N) before (E,J) and after 1, 7, 14, 22 dpi (F-I,K-N). Before injury (E,J) both whiskers (red; prospective lesioned side, and black, control side) moved in a synchronous pattern. At 1 dpi, only the uninjured whisker moved (black in F,K) in contrast to the whisker on the lesioned side (red in F,K). Starting from 7 dpi, some movement of the injured whisker (red in G-I, L-N) was observed. O-Q, Three whisking parameters, angular sum (O), velocity (P), and acceleration (Q), are depicted for Ctr. and Pten KO animals along several postinjury time points. Data are ratio between the injured and intact side. This ratio is close to 1 at −1 dpi, since both sides move equally. O-Q, Gray and orange numbers indicate animal numbers at respective time points. For all three parameters, whisker movement in PTEN-deficient animals was improved at several dpi compared with Ctr. animals. Data are mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; two-sided Mann–Whitney test.
For quantification of FN regeneration at the histologic level, FG, a fluorescent tracer, was injected into the whisker pad muscles on both sides (Fig. 4A). Axons successfully reconnecting to the whisker muscles can transport FG retrogradely to the MN soma where numbers in the FMN can be quantified (Fig. 4A). On the unlesioned side, FG-positive numbers were identical between Ctr. and Pten KO animals (Fig. 4B,C). In contrast, after injury starting already at 7 dpi and statistically significant at 14 and 22 dpi, more FG-positive MNs were observed in PTEN-deficient animals (Fig. 4D,E,R). This suggests a stimulatory role of PTEN ablation in facial MNs for axon regeneration. This finding was not indirectly affected by different total MN numbers between genotypes as quantified by Nissl (Fig. 4F–I,S). Further, injured MNs in both Pten Ctr. and KO mice reacted to injury by developing a hypertrophic phenotype (Fig. 4H,I,T). Nevertheless, this hypertrophy lasted up to 2 weeks after injury in Pten KO mice, while it was already reverted in Ctr. animals at this time point (Fig. 4T).
Next, potential cellular processes involved in PTEN's role in FN regeneration were assessed starting with neuroinflammation (Fig. 4J–Q,U–W). For this, FMNs were stained for astrocyte (GFAP), microglia (IBA1), and peripheral monocytes (CD45) abundance at various stages post injury. All three neuroinflammatory cell types were almost absent in uninjured FMNs regardless of genotype (Fig. 4J,K,N,O). In contrast, astrocytes, microglia, and monocytes were strongly present in the injured FMN (Fig. 4L,M,P,Q). Of note, the extent of neuroinflammation for all three markers was consistently decreased in the FMN of PTEN-depleted compared with Ctr. animals (quantified in Fig. 4U–W).
Overall, our findings indicate a function of PTEN in Ctr. mice to induce injury-associated neuroinflammation, which might impinge on PNS regeneration.
PTEN deletion evokes changes in the FN after injury
Above we analyzed PTEN-associated changes connected to MN cell bodies after injury (Fig. 4). Since MN axons join to form the FN, a subsequent step was to investigate whether PTEN inhibition induced changes directly on FN axon regeneration (Figs. 5–7).
To analyze whether axonal regeneration is stimulated by PTEN deletion, FNs were analyzed at several time points after injury for axon number (βIII tubulin, Fig. 5A–D,M; EM analysis, Fig. 6A–H,M,N), myelination (MBP and toluidine, Fig. 5E–L,N,O; EM analysis, Fig. 6A–H,O), and NMJ formation (BTX and synaptophysin, SYP; Fig. 6I–L,P).
Figure 6.
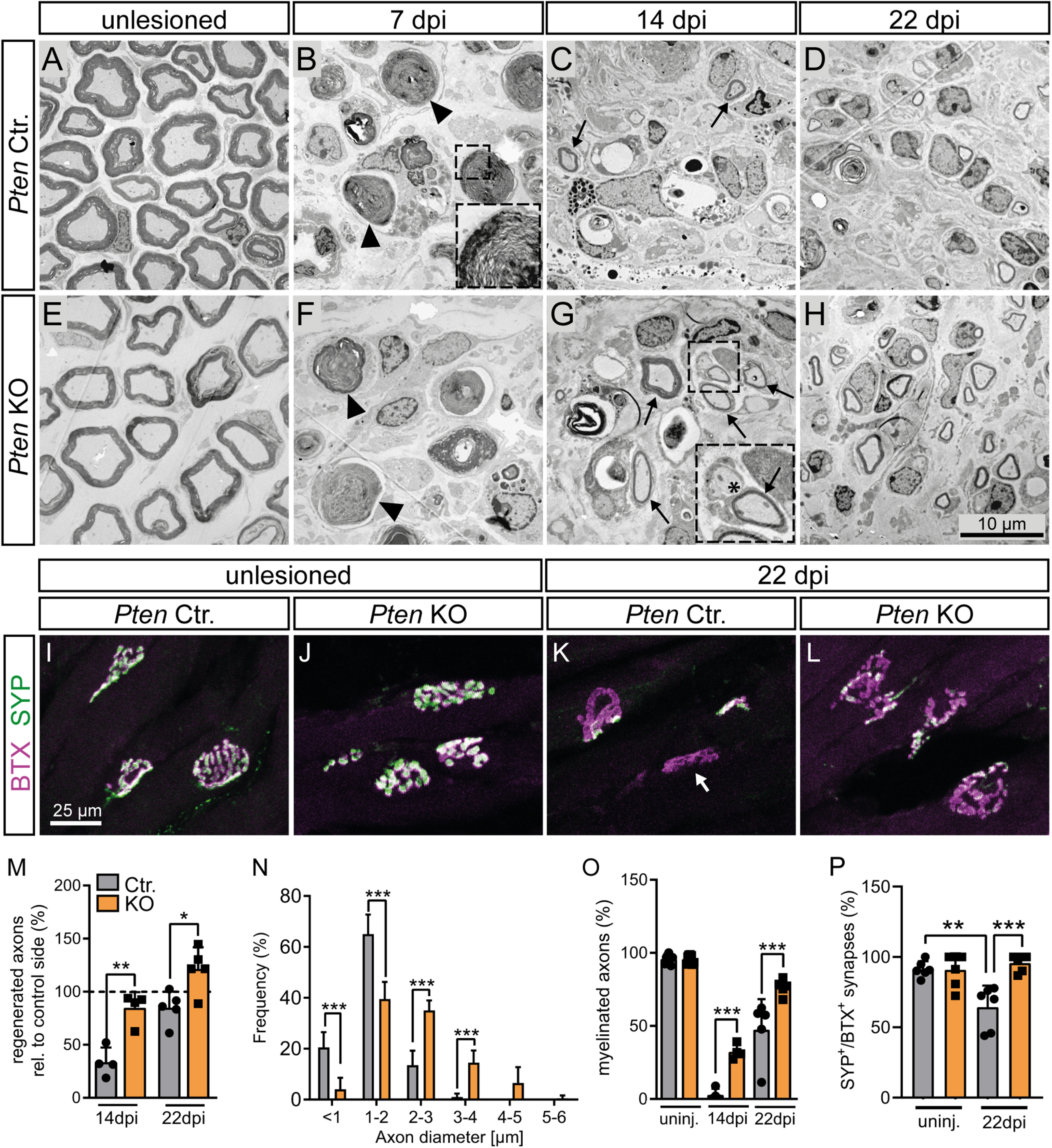
PTEN deletion enhances regeneration of myelinated fibers and neuromuscular synapses after FN injury. A-H, M-O, EM pictures of FN of Ctr. (A-D) and KO (E-H) mice at different time points after injury show improved axon regeneration and remyelination in Pten KO animals. B, F, Arrows indicate degenerating myelin sheaths. B, Inset, Such a degenerating myelin sheath in higher magnification. C, G, Arrows indicate remyelinated axons. G, Inset, Asterisk indicates an unmyelinated axon. M, Quantification of axon numbers relative to the corresponding control side. Axon numbers in the unlesioned nerve were set to 100% (dashed line; n = 4, 4, 5, 5 for each bar, respectively). After injury, axon numbers regenerated more efficiently in PTEN-deficient animals. N, Distribution of axon diameter in regenerating FNs at 22 dpi. Pten KO animals show enlarged axons compared with Ctr. animals (40 arbitrary axons for each of 5 Ctr. and 5 KO animals were analyzed). O, Quantification of the percentage of myelinated axons at each time point. Pten KO animals show a substantial amount of remyelinated axons already at 14 dpi and even more at 22 dpi. Remyelination in Ctr. animals seems to be significantly slower (n = 4, 4, 5, 5 for each bar, respectively). I-L, P, Neuromuscular junctions were stained with BTX (magenta) and synaptophysin (SYP; green) at 22 dpi. In Ctr. mice, FN injury decreased presynaptic innervation as revealed by reduced SYP levels (K) compared with PTEN-deficient animals (L; quantified in P; n = 6, 6, 6, 6 for each bar, respectively). K, Arrow indicates a non-reinnervated synapse. M-P, Each dot indicates 1 animal. Data are mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 (M, O, P, one-way ANOVA test; N, two-way ANOVA test). Scale bars: A-H, 10 µm; I-L, 25 µm.
In the absence of injury, total axon number/area was identical between genotypes, although larger axon calibers were also obvious in PTEN-deficient animals (Fig. 5A,B,I,J). After injury, Wallerian degeneration, a mechanism of programmed axon degeneration, is induced (Coleman and Hoke, 2020). Indeed, also in our injury model, the number of axons expectedly decreased compared with an uninjured nerve (quantified in Fig. 5M). However, with increasing regeneration time, axon numbers gradually increase; and here we observed a faster recovery in axon numbers at all time points for Pten KO compared with Ctr. animals, with KO animals almost reaching pre-injury axon numbers at 22 d of regeneration (Fig. 5M). Of note, as already shown in Figure 1, we observed a lower compaction grade of axons in Pten KO compared with Ctr. mice because of excess connective tissue and larger axon diameter. This resulted in lower axon numbers/area in Pten KO mice (Fig. 5M). Nevertheless, quantification of axons in the whole nerve had revealed similar total axon numbers in Ctr. and Pten KO animals (Fig. 1P). Related to axonal changes, the number of intact MBP-positive myelin sheaths recovered more quickly after injury in PTEN-deficient animals (Fig. 5G,H,N). This was corroborated by toluidine staining of SC myelin sheaths in semithin sections (Fig. 5I–L,O). In order to further confirm the role of PTEN in axonal outgrowth, we used hiPSCs, differentiated them into MNs, and treated them with the PTEN inhibitor SF1670. As expected, inhibition of PTEN resulted in increased neurite outgrowth (Fig. 5P–R), which is also in line with previous reports (Zhou et al., 2020).
EM analysis confirmed the observations of improved axonal regrowth (Fig. 6A–H,M) and remyelination in Pten KO animals (Fig. 6A–H,O). Of note, although regenerated axons were still thinner than the original uninjured axons (compare Fig. 1Q vs Fig. 6N), Pten KO mice again showed thicker axons than Ctr. mice (Fig. 6N). Finally, NMJ analysis after injury revealed that, at 22 dpi, the percentage of NMJs with high levels of both presynaptic synaptophysin and postsynaptic acetylcholine receptors (stained with BTX) was reduced in Ctr. (Fig. 6I,K,P) compared with Pten KO animals (Fig. 6J,L,P).
In addition to axonal regrowth and myelination, we also inspected immune cell abundance in the FN (Fig. 7A–H,Q,R). Similar to the FMN (Fig. 4), immune cells were upregulated by injury in the FN. However, in contrast to the FMN, immune cell abundance in the injured FN was not altered between Ctr. and Pten KO animals (Fig. 7A–H,Q,R). Thus, PTEN deletion evokes reduced neuroinflammation around the MN cell bodies but not in the distal axons.
After injury, SCs dedifferentiate from a myelinating to a repair phenotype, which is accompanied by changes in transcription factors, such as cJUN and EGR2, and is crucial for axonal regeneration (Jessen et al., 2015). However, neuronal PTEN deletion did not obviously affect this SC transition as shown by identical cJUN and EGR2 numbers in the FN after injury (Fig. 7I–P,S,T). This might be expected given that PTEN was deleted from MNs and not SCs.
In summary, axonal regeneration is improved after PTEN deletion, while the response of SCs was comparable.
PTEN deletion improves whisker function after FN injury
Following demonstration that PTEN inhibition enhances PNS regeneration at the histologic level, we assessed regeneration at the functional level (Fig. 8). Here, we took advantage of a well-established functional parameter affected by FN injury (i.e., whisker movement) (Moran and Graeber, 2004; Wanner et al., 2017).
For monitoring whisker movement, single whiskers of the intact and injured side of each animal were investigated before and at several time points after injury with a high-speed camera recording 100-ms-long video sequences at 100 Hz (i.e., 100 picture frames; see Fig. 8). Before lesion, whisker movement showed a typical synchronized and rhythmic pattern between control and prospective lesion side (Fig. 8A,B,E,J). In contrast, after lesion whiskers on the uninjured side still moved, whereas this was impaired on the lesion side (Fig. 8C,D, red line; Fig. 8F,K). At ∼7 d after injury, first whisker movements on the injury side were noticeable, which became more evident at 22 d after injury (Fig. 8G–I,L–N, red line).
We compared whisker movement between Ctr. and Pten KO animals at six time points after injury with several parameters, including angular sum (Fig. 8O), velocity (Fig. 8P), and acceleration (Fig. 8Q). Data revealed a consistent increase in all parameters for PTEN-deficient animals, particularly at 7, 11, 14, and 22 dpi (Fig. 8O–Q).
This suggests that PTEN deficiency stimulates functional nerve regeneration. Of note, although molecular regeneration (evidenced by FG tracer incorporation) was already quite successful and also whisker movement partially reappeared within 3 weeks after injury (Figs. 4–6, 8), functional regeneration is still not complete, since whisking does not reach initial levels neither in Ctr. nor in Pten KO animals (Fig. 8E–Q).
Akt and CREB phosphorylation are induced by PTEN deletion after nerve injury
In a next step, we analyzed possible changes in signaling pathways which might be modulated downstream of PTEN and contribute to the positive growth effects after PTEN deletion (Figs. 9 and 10).
For this, we first analyzed PTEN and P-Akt abundance before injury and at 3 dpi (Fig. 9A–O). In Ctr. mice, PTEN total abundance was not altered at 3 dpi (Fig. 9A,C,E). PTEN was largely confined to the MN cytoplasm, and only weak levels were observed in neurites (Fig. 9A). This is in line with previous reports also showing no changes in total PTEN levels after sciatic nerve injury but decreased P-PTEN (Christie et al., 2010). Expectedly, in Pten KO animals MNs in the FMN were not expressing PTEN anymore (Fig. 9B,D,E). So far, changes in P-Akt abundance after PNS injury in vivo have not been investigated in great detail. Total Akt levels were unchanged between conditions and genotypes (Fig. 9F–I,J). P-Akt levels in MNs were upregulated by FN injury in Ctr. mice at 3 dpi (Fig. 9K,M,O). After PTEN deletion, P-Akt levels were higher compared with Ctr. mice at 3 dpi, both on the uninjured and somewhat more on the injured FMN (Fig. 9L,N,O).
Next, we analyzed P-CREB abundance, a key neuronal activity regulated transcription factor known to enhance axon regeneration (Gao et al., 2004). So far, an interaction between PTEN/Akt signaling and CREB has not been addressed after axon injury; however, outside the nervous system, PTEN can de-phosphorylate CREB (Gu et al., 2011) or CREB can be phosphorylated by Akt at serine 133 for activation (Walton and Dragunow, 2000). At 3 dpi, P-CREB signals in the nucleus of MNs were slightly but not significantly elevated by injury in Ctr. mice (Fig. 9P,R,T). In Pten KO animals, nuclear P-CREB levels in injured FMN were almost doubled compared with Ctr. animals (Fig. 9S,T). Thus, PTEN deficiency upregulates P-CREB and most likely thereby activates CREB in injured MNs.
A PRAS40-mTOR-4EBP1 pathway is induced in PTEN-deficient mice
In previous experiments (Fig. 9), CREB was investigated as a first PTEN/Akt downstream target. PTEN/Akt can stratify signaling to many targets; however, an established pathway, primarily analyzed in non-neuronal cells, involves the PRAS40-mTOR1 complex (Wiza et al., 2012). Here, PRAS40 phosphorylation by Akt and by mTOR1 itself triggers PRAS40 dissociation from mTOR1 and can activate mTORC1 activity.
In Ctr. mice at 3 dpi, P-PRAS40 was upregulated by FN injury in MN cell bodies (Fig. 10A1,A3,A5). Since PRAS40 is phosphorylated by Akt, elevated P-PRAS40 levels might be expected on Pten KO mice. Indeed, P-PRAS40 levels were strongly augmented compared with Ctr. mice on both the uninjured and injured FMN (Fig. 10A2,A4,A5).
Next, total mTOR and P-mTOR levels were inspected at 3 dpi (Fig. 10B,C). Total mTOR levels were almost not altered by injury or PTEN deficiency (Fig. 10B). In contrast, P-mTOR levels were induced by injury at 3 dpi (Fig. 10C1,C3,C5) in Ctr. mice. In Pten KO mice, P-mTOR levels exceeded Ctr. mice levels with stronger effects observed at the lesion site (Fig. 10C2,C4,C5). P-mTOR was present in MN cell bodies and on neuronal protrusions in Ctr. and PTEN-deficient MNs (Fig. 10C, insets).
Downstream of mTOR, we inspected activation of two prototypical mTOR targets (i.e., P-4EBP1 and P-S6). Starting with 4EBP1, we observed only weak signals for total 4EBP1 (Fig. 10D1,D2) and P-4EBP1 (Fig. 10E1,E2) for both Ctr. and KO in the uninjured FMN. After injury at 3 dpi, total 4EBP1 (Fig. 10D3,D4) and P-4EBP1 (Fig. 10E3,E4) were elevated in both Ctr. and KO animals. Interestingly, quantification revealed as stronger total 4EBP1 (Fig. 10D5) and P-4EBP1 (Fig. 10E5) induction in Ctr. animals at 3 dpi compared with PTEN-deficient animals. In contrast, at 22 dpi, total 4EBP1 and P-4EBP1 abundance was higher in injured KO compared with Ctr. FMNs (Fig. 10G,H). The latter result is in agreement with elevated P-Akt in KO animals, resulting in more P-4EBP1. In contrast to 4EBP1 phosphorylation, levels of P-S6 were not altered by FN injury or PTEN deletion (Fig. 10F).
In summary, PTEN deletion evoked upregulation of P-CREB as well as P-PRAS40, P-mTOR, and, in a time-dependent manner, also P-4EBP1.
Discussion
PTEN's dual function in neuronal physiology and pathology
In the uninjured brain, PTEN deletion generates a hypertrophic state of neurons during development (Skelton et al., 2020). This includes enlarged cell soma, ectopic cell migration, overgrowth of axons and dendrites, increased synaptic spine density accompanied by behavioral alterations, macrocephaly, and epileptic seizures. Thus, in WT mice, PTEN ensures physiological CNS development and function.
So far, data on physiological PTEN function in PNS neurons, particularly MNs, are lacking behind CNS reports. We expanded on this knowledge by using facial MNs residing in the CNS brainstem and extending axons in the periphery. In this study, somatic and axonal hypertrophy were uncovered in facial MNs (Fig. 1). Hypertrophy also extended to whole nerves that were larger after PTEN deletion (Fig. 1). This might be caused by enhanced axon diameter (rather than MN number). Furthermore, we made the novel observation of more “spacing” between individual axons that might contribute to enlarged nerves in PTEN-deficient mice (Fig. 1J). This enhanced space, resulting in more segregation between individual axons, might be explained by more connective tissue deposition separating single axons (Fig. 1). The latter could reflect an exacerbated fibrosis in nerves since PTEN deletion is known to activate fibroblasts and extracellular matrix deposition (White et al., 2006).
PTEN deletion switches neurons in a growth-promoting state already in the intact nervous system before injury onset. Once a mechanical injury occurs, this growth-promoting state evoked by PTEN inhibition is further augmented and axon regeneration is stimulated in neurons. During development, such exuberant axon and dendritic growth might be detrimental, resulting in overactive synaptic networks and, for example, seizures in PTEN-deficient animals (Williams et al., 2015; Skelton et al., 2020). In contrast, after axonal injury, such strong de novo axon growth induced by PTEN deletion is beneficial and stimulates axonal re-connectivity, thereby allowing for functional regeneration.
Our data support a mechanism by which PTEN in WT mice is a growth brake also on PNS axon regeneration. So far, data available on PTEN in PNS regeneration are confined to the sciatic nerve and DRG neurons (Christie et al., 2010; Gallaher and Steward, 2018; Holland et al., 2019; Zhou et al., 2020). Herein, we expanded on this by investigating PTEN in the facial MNs. Similar to others (Christie et al., 2010; Gallaher and Steward, 2018; Holland et al., 2019; Zhou et al., 2020), we showed improved nerve regeneration on the histologic level (Figs. 4–6). So far, functional PNS regeneration in Pten KO mice was not observed in all tests (Christie et al., 2010; Gallaher and Steward, 2018; Holland et al., 2019; Zhou et al., 2020). Herein, improved whisker movement after FN injury in PTEN-deficient mice was observed (Fig. 8), showing that PTEN inhibition can improve functionality of injured MNs. We identified reduced neuroinflammation as a process by which PTEN deletion might enhance PNS axon regeneration. Neuroinflammatory responses of astrocytes, microglia, and peripheral macrophages were reduced on PTEN deletion (Fig. 4). So far, a role of PTEN in neuroinflammation during axon regeneration has not been described in great depth. In traumatic brain injury (R. Liu et al., 2019) and Tau-associated neurodegeneration (Benetatos et al., 2020), PTEN inhibition decreased microglia activation, a result in accordance with our data. Microglia support or inhibit axon regeneration depending on, for example, context and timing (Zigmond and Echevarria, 2019). In this study, enhanced regeneration after PTEN deficiency correlated with decreased neuroinflammatory responses. This indicates a regeneration-inhibiting role of neuroinflammation in our experimental settings.
Finally, previous reports analyzed PTEN-deficient animals mainly for short regeneration periods (typically 1-3 months after injury). Herein, animals were followed over 1 year. This time span covered neurodevelopment and regeneration in young adult animals but also epidermal hyperplasia development associated with PTEN deficiency in the aged animal (Figs. 2 and 3). Together with previously described PTEN functions, our data suggest that PTEN deficiency has a dual function during an animal's lifetime.
In young adult mice, PTEN inhibition sets neurons in a pro-growth state, which might have detrimental implications on neuronal morphology, circuits, and behavior during development and neuronal physiology (Skelton et al., 2020). However, after an injury in such young aged animals, the neuronal pro-growth state evoked by PTEN inhibition is clearly beneficial for axons to regenerate as shown by others (Duraikannu et al., 2019) and herein.
In the aged animals (12 months), PTEN is tumor-suppressive and tumor development is a major risk when deleting TSGs, such as PTEN.
Those diverse PTEN functions along the animal's age have to be considered when attempting pharmacological manipulation of PTEN activity for therapeutic purposes.
Growth promoting signaling pathways switched on by PTEN inhibition
We identified several signaling molecules whose activation, as analyzed by phosphorylation status, was altered on PTEN deletion (Figs. 9 and 10). A first target regulated by PTEN deficiency was CREB, a neuronal activity regulated transcription factor involved in regulating neuronal excitability and survival (Walton and Dragunow, 2000). So far, the interaction of PTEN with CREB has only sparsely been investigated in neurons. Herein, we demonstrated that P-CREB levels were elevated by PTEN inhibition, suggesting enhanced CREB activity in injured facial MNs (Fig. 9). Interestingly, this novel finding in neurons was corroborated in PTEN-deficient mouse embryonic fibroblasts (Wan et al., 2021). Furthermore, complementary to PTEN deletion, PTEN overexpression was associated with decreased CREB phosphorylation in the lung tissue of asthmatic mice (Wu et al., 2020). In our study, such elevated P-CREB levels might be accomplished in PTEN-deficient neurons through the absence of a direct PTEN-mediated dephosphorylation (Gu et al., 2011) or indirectly through elevated levels of activated Akt (P-Akt).
Activated CREB is known to enhance regeneration-associated gene expression (e.g., Atf3; see Fig. 11) (Ma et al., 2014), circuit formation (Bradley et al., 2019), and regeneration after CNS injury (Gao et al., 2004). In contrast, in the PNS, no evidence for P-CREB in axon regeneration is reported. Current literature shows conflicting results as to whether P-CREB is induced in the severed sciatic nerve (Herdegen et al., 1994; Li et al., 2009). Herein, a mild P-CREB upregulation in injured Ctr. and strongly enhanced induction in Pten KO facial MN neurons was observed (Fig. 9). Thus, our data support a functional implication for a PTEN-CREB signaling unit in PNS regeneration (Fig. 11).
Figure 11.

Summary of findings observed in Pten KO animals. Nerve injury in PTEN-deficient MNs results in P-Akt upregulation (green arrow), which was accompanied by elevated P-CREB levels. P-CREB reflects activated CREB, which might enhance axonal regeneration through RAG expression. In addition, P-Akt may phosphorylate and thereby inhibit PRAS40, which in turn prevents PRAS40 from inhibiting mTOR. Thus, overall activation of growth-promoting mTOR achieved by PTEN deletion might also contribute to enhanced peripheral nerve regeneration. For this, P-mTOR might activate its effector 4EBP1, whereas no effects were seen on the S6 pathway. Created with BioRender.com.
In the CNS, an established PTEN downstream pathway connected to axon regeneration is enhanced Akt-mTOR signaling with its targets 4EBP1 and S6 (Yang et al., 2014; Miao et al., 2016; Huang et al., 2019). Functional roles of P-Akt have not been addressed in the PNS in contrast to CNS injury (Miao et al., 2016; Huang et al., 2019). However, we (Fig. 9) and others (Shi et al., 2009) show P-Akt upregulation in injured PNS neurons supporting an Akt function also during PNS regeneration. Downstream of Akt, growth-promoting signaling can be diverged to several signaling partners, including pro-growth molecules, such as CREB (see above), or by suppression of anti-growth signals, such as PRAS40 (Zhou et al., 2021) (see below) or GSK3β (Leibinger et al., 2019) (see Fig. 11). Our data suggest that, during FN injury, growth-promoting signals, such as P-CREB (see above) and P-mTOR, operate to enhance regeneration (Fig. 10).
In the PNS, some studies have shown that axon regeneration is inhibited by mTOR deletion (Miao et al., 2016), whereas others did not confirm such an mTOR function (Christie et al., 2010). In agreement with an mTOR role during PNS regeneration, we observed P-mTOR induction in both injured Ctr. and more pronounced in PTEN ablated facial MNs (Figs. 10 and 11). Downstream of mTOR, we observed no obvious regulation of P-S6 levels by FN injury (Figs. 10 and 11) in contrast to reports in sciatic nerve injury (Chen et al., 2016). In opposite to S6, total and P-4EBP1 were upregulated in FN injury in accordance with a previous report on sciatic nerve injury (Kalinski et al., 2015). Notably, for total and P-4EBP1, we observed shift depending on postinjury time with lower and higher 4EBP1/P-4EBP1 levels in injured KO compared with Ctr. MNs at 3 and 22 dpi, respectively (Fig. 10). The results at 3 dpi, but not 22 dpi, were contrary to expectation since we expected elevated P-Akt levels in KO animals (Fig. 9K–N,O), resulting in more P-4EBP1 in KO FMNs. A previous report in fibroblasts also showed such decreased P-4EBP1 levels in PTEN-deficient cells (Goo et al., 2012). Nevertheless, we currently do not have an explanation for this.
In this study, we analyzed PRAS40, a signaling molecule connecting Akt with mTOR activation (Wiza et al., 2012), which on phosphorylation by either Akt or mTOR releases its inhibition on mTOR signaling (Fig. 11). PRAS40 is a novel signaling intermediate in axon regeneration. So far, only one report documented enhanced P-PRAS40 levels after spinal cord injury in WT rats (Yu et al., 2008). In this study, we corroborate this finding in Ctr. animals for PNS injury (Fig. 10). Furthermore, in Pten KO mice, a strong P-PRAS40 abundance was observed after injury (Figs. 10 and 11). Since phosphorylated PRAS40 fails to interact with mTOR, this might facilitate activation of the growth-enhancing mTORC1 complex, one mechanism by which PTEN inhibition might enhance axon growth (Fig. 11). This points at PRAS40 as a novel signaling partner to be shut off by PTEN inhibition during axon regeneration.
In conclusion, data provided in this study on a mouse PNS regeneration model point at an important function of PTEN as a regenerative brake in the PNS.
Footnotes
This work by B.K. and S.M.z.R. was supported by Deutsche Forschungsgemeinschaft Project ID 251293561-SFB 1149. S.M.z.R. was supported by Deutsche Forschungsgemeinschaft Grant ME 5415/2-1. We thank Prof. Dr. Karin Scharffetter-Kochanek and Dr. Pallab Maity (Clinic of Dermatology and Allergology, Ulm University) for support in skin tumor characterization; and Valerie Huhle for excellent work during her bachelor thesis in the laboratory.
The authors declare no competing financial interests.
References
- Anastasiadou S, Knoll B (2016) The multiple sclerosis drug fingolimod (FTY720) stimulates neuronal gene expression, axonal growth and regeneration. Exp Neurol 279:243–260. 10.1016/j.expneurol.2016.03.012 [DOI] [PubMed] [Google Scholar]
- Benetatos J, Bennett RE, Evans HT, Ellis SA, Hyman BT, Bodea LG, Gotz J (2020) PTEN activation contributes to neuronal and synaptic engulfment by microglia in tauopathy. Acta Neuropathol 140:7–24. 10.1007/s00401-020-02151-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley PM, Denecke CK, Aljovic A, Schmalz A, Kerschensteiner M, Bareyre FM (2019) Corticospinal circuit remodeling after central nervous system injury is dependent on neuronal activity. J Exp Med 216:2503–2514. 10.1084/jem.20181406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese A, Rajkumar S, Sommer D, Freisem D, Wirth A, Aly A, Massa-López D, Olivieri A, Torelli F, Ioannidis V, Lipecka J, Guerrera IC, Zytnicki D, Ludolph A, Kabashi E, Mulaw MA, Roselli F, Böckers TM (2021) Synaptic disruption and CREB-regulated transcription are restored by K(+) channel blockers in ALS. EMBO Mol Med 13:e13131. 10.15252/emmm.202013131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Lu N, Ding Y, Wang Y, Chan LT, Wang X, Gao X, Jiang S, Liu K (2016) Rapamycin-resistant mTOR activity is required for sensory axon regeneration induced by a conditioning lesion. eNeuro 3:ENEURO.0358-16.2016. 10.1523/ENEURO.0358-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie KJ, Krishnan A, Martinez JA, Purdy K, Singh B, Eaton S, Zochodne D (2014) Enhancing adult nerve regeneration through the knockdown of retinoblastoma protein. Nat Commun 5:3670. 10.1038/ncomms4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW (2010) PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci 30:9306–9315. 10.1523/JNEUROSCI.6271-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Hoke A (2020) Programmed axon degeneration: from mouse to mechanism to medicine. Nat Rev Neurosci 21:183–196. 10.1038/s41583-020-0269-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, Knights CD, Rao M, Yakovlev A, Beers J, Catania J, Avantaggiati ML, Faden AI (2006) The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J 25:4084–4096. 10.1038/sj.emboj.7601292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraikannu A, Krishnan A, Chandrasekhar A, Zochodne DW (2019) Beyond trophic factors: exploiting the intrinsic regenerative properties of adult neurons. Front Cell Neurosci 13:128. 10.3389/fncel.2019.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlia G, Norrmen C, Pereira JA, Gerber D, Suter U (2017) Dual function of the PI3K-Akt-mTORC1 axis in myelination of the peripheral nervous system. Elife 6:e29241. 10.7554/eLife.29241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ (2008) Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151:476–488. 10.1016/j.neuroscience.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallaher ZR, Steward O (2018) Modest enhancement of sensory axon regeneration in the sciatic nerve with conditional co-deletion of PTEN and SOCS3 in the dorsal root ganglia of adult mice. Exp Neurol 303:120–133. 10.1016/j.expneurol.2018.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallent EA, Steward O (2018) Neuronal PTEN deletion in adult cortical neurons triggers progressive growth of cell bodies, dendrites, and axons. Exp Neurol 303:12–28. 10.1016/j.expneurol.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44:609–621. 10.1016/j.neuron.2004.10.030 [DOI] [PubMed] [Google Scholar]
- Gey M, Wanner R, Schilling C, Pedro MT, Sinske D, Knoll B (2016) Atf3 mutant mice show reduced axon regeneration and impaired regeneration-associated gene induction after peripheral nerve injury. Open Biol 6:160091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, Karram K, Renier N, Tessier-Lavigne M, Rossner MJ, Káradóttir RT, Nave KA (2017) A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat Neurosci 20:10–15. 10.1038/nn.4425 [DOI] [PubMed] [Google Scholar]
- Goo CK, Lim HY, Ho QS, Too HP, Clement MV, Wong KP (2012) PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS One 7:e45806. 10.1371/journal.pone.0045806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294:2186–2189. 10.1126/science.1065518 [DOI] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y (2011) CREB is a novel nuclear target of PTEN phosphatase. Cancer Res 71:2821–2825. 10.1158/0008-5472.CAN-10-3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutilla EA, Steward O (2016) Selective neuronal PTEN deletion: can we take the brakes off of growth without losing control? Neural Regen Res 11:1201–1203. 10.4103/1673-5374.189160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutilla EA, Buyukozturk MM, Steward O (2016) Long-term consequences of conditional genetic deletion of PTEN in the sensorimotor cortex of neonatal mice. Exp Neurol 279:27–39. 10.1016/j.expneurol.2016.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdegen T, Gass P, Brecht S, Neiss WF, Schmid W (1994) The transcription factor CREB is not phosphorylated at serine 133 in axotomized neurons: implications for the expression of AP-1 proteins. Brain Res Mol Brain Res 26:259–270. 10.1016/0169-328X(94)90098-1 [DOI] [PubMed] [Google Scholar]
- Holland SD, Ramer LM, McMahon SB, Denk F, Ramer MS (2019) An ATF3-CreERT2 knock-in mouse for axotomy-induced genetic editing: proof of principle. eNeuro 6:ENEURO.0025-19.2019. 10.1523/ENEURO.0025-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Miao L, Yang L, Liang F, Wang Q, Zhuang P, Sun Y, Hu Y (2019) AKT-dependent and -independent pathways mediate PTEN deletion-induced CNS axon regeneration. Cell Death Dis 10:203. 10.1038/s41419-018-1289-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R, Lloyd AC (2015) Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol 7:a020487. 10.1101/cshperspect.a020487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinski AL, Sachdeva R, Gomes C, Lee SJ, Shah Z, Houle JD, Twiss JL (2015) mRNAs and protein synthetic machinery localize into regenerating spinal cord axons when they are provided a substrate that supports growth. J Neurosci 35:10357–10370. 10.1523/JNEUROSCI.1249-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kugler S, Benowitz LI (2010) Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci 30:15654–15663. 10.1523/JNEUROSCI.4340-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibinger M, Hilla AM, Andreadaki A, Fischer D (2019) GSK3-CRMP2 signaling mediates axonal regeneration induced by Pten knockout. Commun Biol 2:318. 10.1038/s42003-019-0524-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MY, Lai FJ, Hsu LJ, Lo CP, Cheng CL, Lin SR, Lee MH, Chang JY, Subhan D, Tsai MS, Sze CI, Pugazhenthi S, Chang NS, Chen ST (2009) Dramatic co-activation of WWOX/WOX1 with CREB and NF-kappaB in delayed loss of small dorsal root ganglion neurons upon sciatic nerve transection in rats. PLoS One 4:e7820. 10.1371/journal.pone.0007820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z (2010) PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13:1075–1081. 10.1038/nn.2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Liao XY, Tang JC, Pan MX, Chen SF, Lu PX, Lu LJ, Zhang ZF, Zou YY, Bu LH, Qin XP, Wan Q (2019) BpV(pic) confers neuroprotection by inhibiting M1 microglial polarization and MCP-1 expression in rat traumatic brain injury. Mol Immunol 112:30–39. 10.1016/j.molimm.2019.04.010 [DOI] [PubMed] [Google Scholar]
- Ma TC, Barco A, Ratan RR, Willis DE (2014) cAMP-responsive element-binding protein (CREB) and cAMP co-regulate activator protein 1 (AP1)-dependent regeneration-associated gene expression and neurite growth. J Biol Chem 289:32914–32925. 10.1074/jbc.M114.582460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao L, Yang L, Huang H, Liang F, Ling C, Hu Y (2016) mTORC1 is necessary but mTORC2 and GSK3beta are inhibitory for AKT3-induced axon regeneration in the central nervous system. Elife 5:e14908. 10.7554/eLife.14908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LB, Graeber MB (2004) The facial nerve axotomy model. Brain Res Brain Res Rev 44:154–178. 10.1016/j.brainresrev.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Ning K, Drepper C, Valori CF, Ahsan M, Wyles M, Higginbottom A, Herrmann T, Shaw P, Azzouz M, Sendtner M (2010) PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum Mol Genet 19:3159–3168. 10.1093/hmg/ddq226 [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322:963–966. 10.1126/science.1161566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK (2011) Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13:195–204. 10.1016/j.cmet.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi TJ, Huang P, Mulder J, Ceccatelli S, Hokfelt T (2009) Expression of p-Akt in sensory neurons and spinal cord after peripheral nerve injury. Neurosignals 17:203–212. 10.1159/000210400 [DOI] [PubMed] [Google Scholar]
- Singh B, Singh V, Krishnan A, Koshy K, Martinez JA, Cheng C, Almquist C, Zochodne DW (2014) Regeneration of diabetic axons is enhanced by selective knockdown of the PTEN gene. Brain 137:1051–1067. 10.1093/brain/awu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelton PD, Stan RV, Luikart BW (2020) The role of PTEN in neurodevelopment. Mol Neuropsychiatry 5:60–71. 10.1159/000504782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Haverkamp S, Sinske D, Tedeschi A, Naumann U, Di Giovanni S, Kochanek S, Nordheim A, Knöll B (2013) The transcription factor serum response factor stimulates axon regeneration through cytoplasmic localization and cofilin interaction. J Neurosci 33:18836–18848. 10.1523/JNEUROSCI.3029-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Itami S, Ohishi M, Hamada K, Inoue T, Komazawa N, Senoo H, Sasaki T, Takeda J, Manabe M, Mak TW, Nakano T (2003) Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res 63:674–681. [PubMed] [Google Scholar]
- Walton MR, Dragunow I (2000) Is CREB a key to neuronal survival? Trends Neurosci 23:48–53. 10.1016/s0166-2236(99)01500-3 [DOI] [PubMed] [Google Scholar]
- Wan X, Zhou M, Huang F, Zhao N, Chen X, Wu Y, Zhu W, Ni Z, Jin F, Wang Y, Hu Z, Chen X, Ren M, Zhang H, Zha X (2021) AKT1-CREB stimulation of PDGFRalpha expression is pivotal for PTEN-deficient tumor development. Cell Death Dis 12:172. 10.1038/s41419-021-03433-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner R, Gey M, Abaei A, Warnecke D, de Roy L, Dürselen L, Rasche V, Knöll B (2017) Functional and molecular characterization of a novel traumatic peripheral nerve-muscle injury model. Neuromolecular Med 19:357–374. 10.1007/s12017-017-8450-1 [DOI] [PubMed] [Google Scholar]
- Wessler I, Kirkpatrick CJ (2008) Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154:1558–1571. 10.1038/bjp.2008.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD, Toews GB (2006) Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med 173:112–121. 10.1164/rccm.200507-1058OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, DeSpenza T Jr, Li M, Gulledge AT, Luikart BW (2015) Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci 35:943–959. 10.1523/JNEUROSCI.3144-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiza C, Nascimento EB, Ouwens DM (2012) Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab 302:E1453–E1460. 10.1152/ajpendo.00660.2011 [DOI] [PubMed] [Google Scholar]
- Wu Y, Lu Y, Zou F, Fan X, Li X, Zhang H, Chen H, Sun X, Liu Y (2020) PTEN participates in airway remodeling of asthma by regulating CD38/Ca(2+)/CREB signaling. Aging (Albany NY) 12:16326–16340. 10.18632/aging.103664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Miao L, Liang F, Huang H, Teng X, Li S, Nuriddinov J, Selzer ME, Hu Y (2014) The mTORC1 effectors S6K1 and 4E-BP play different roles in CNS axon regeneration. Nat Commun 5:5416. 10.1038/ncomms6416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Narasimhan P, Saito A, Liu J, Chan PH (2008) Increased expression of a proline-rich Akt substrate (PRAS40) in human copper/zinc-superoxide dismutase transgenic rats protects motor neurons from death after spinal cord injury. J Cereb Blood Flow Metab 28:44–52. 10.1038/sj.jcbfm.9600501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagni C, Almeida LO, Balan T, Martins MT, Rosselli-Murai LK, Papagerakis P, Castilho RM, Squarize CH (2017) PTEN mediates activation of core clock protein BMAL1 and accumulation of epidermal stem cells. Stem Cell Reports 9:304–314. 10.1016/j.stemcr.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LY, Han F, Qi SB, Ma JJ, Ma YX, Xie JL, Zhang HC, Fu XY, Chen JQ, Li B, Yang HL, Zhou FSaijilafu (2020) Inhibition of PTEN activity promotes IB4-positive sensory neuronal axon growth. J Cell Mol Med 24:11012–11017. 10.1111/jcmm.15648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Tang S, Zhang X, Chen L (2021) Targeting PRAS40: a novel therapeutic strategy for human diseases. J Drug Target 29:703–715. [DOI] [PubMed] [Google Scholar]
- Zigmond RE, Echevarria FD (2019) Macrophage biology in the peripheral nervous system after injury. Prog Neurobiol 173:102–121. 10.1016/j.pneurobio.2018.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]