

Abstract
The glutathione (GSH) redox control is critical to maintain redox balance in the body’s internal environment, and its perturbation leads to a dramatic increase in reactive oxygen species (ROS) levels and oxidative stress which have negative impacts on human health. Although ionizing radiation increases mitochondrial ROS generation, the mechanisms underlying radiation-induced late ROS accumulation are not fully understood. Here we investigated the radiation effect on GSH redox reactions in normal human diploid lung fibroblasts TIG-3 and MRC-5. Superoxide anion probe MitoSOX-red staining and measurement of GSH peroxidase (GPx) activity revealed that high dose single-radiation (SR) exposure (10 Gy) increased mitochondrial ROS generation and overall oxidative stress in parallel with decrease in GSH peroxidase (GPx) activity, while GSH redox control was effective after exposure to moderate doses under standard serum conditions. We used different serum conditions to elucidate the role of serum on GSH redox reaction. Serum starvation, serum deprivation and DNA damage response (DDR) inhibitors-treatment reduced the GPx activity and increased mitochondrial ROS generation regardless of radiation exposure. Fractionated-radiation was used to evaluate the radiation effect on GSH reactions. Repeated fractionated-radiation induced prolonged oxidative stress by down-regulation of GPx activity. In conclusion, radiation affects GSH usage according to radiation dose, irradiation methods and serum concentration. Radiation affected the GPx activity to disrupt fibroblast redox homeostasis.
Keywords: Glutathione (GSH), glutathione peroxidase, radiation, ROS, redox homeostasis
INTRODUCTION
The redox balance is maintained through an intricate network in the body’s internal environment among all living organisms, but its perturbation leads to adverse effects in human health [1]. The resulting high levels of reactive oxygen species (ROS) induce oxidative stress that has been linked to the main cause of age-related diseases, including cancer, cardiovascular disease and diabetes [2]. The source of intracellular ROS has been proposed to be mitochondria where adenosine triphosphate (ATP) is synthesized by oxidative phosphorylation (OXPHOS). Superoxide anions (O2−) are mainly produced from complexes I and III at the mitochondrial electron transport chain by electron leakage and are mostly released to the mitochondrial matrix [1, 3, 4]. Cells harbor mitochondrial antioxidant defense systems in which glutathione (GSH) has a critical role for the regulation of intracellular redox homeostasis. Manganese superoxide dismutase (MnSOD) and GSH peroxidase (GPx) are the major ROS detoxifying enzymes. These enzymes are localized in the mitochondrial matrix where MnSOD converts O2− to H2O2. H2O2 is less reactive than O2− and can diffuse from the mitochondria to mediate oxidative signaling [5]. Lastly, H2O2 oxidizes reduced GSH by the enzyme activity of GPx to from GSH disulfide (GSSG) and H2O [6–8]. GSSG is then reduced back to GSH by NADPH-dependent GSH reductase. GSH exists in the cytoplasm, nucleus and mitochondria. Mitochondrial GSH stability and turn-over are regulated by cytoplasmic GSH synthesis and transport to mitochondria [9]. Mitochondrial GSH is a key molecule in the control of oxidative stress within mitochondria.
Radiation causes direct damage to DNA, such as single- and double-strand breaks (DSBs), clustered DNA damage, and reacts with water to generate short-lived ROS. Later effects of exposure to radiation include activation of OXPHOS as a response to radiation-induced DNA damage, and this also generates mitochondrial ROS as by-product [4]. ROS and subsequent activation of cellular antioxidant enzymes are involved in radiation-induced adaptive responses [10, 11]. Transiently increased levels of GSH following whole-body irradiation to mouse is associated with the elevation of natural killer (NK) activity in mouse splenocytes [12]. Radiation induces mitochondrial change, including activation of OXPHOS, generation of ROS, mitochondrial Parkin staining and induction of mitochondrial biogenesis in terms of oxidative stress responses in human fibroblasts [4, 13–15]. Mitochondria are vulnerable to radiation-induced effects because they are the sites of ROS generation. Deleterious ROS levels cause indirect DNA damage and inflict oxidative stresses in irradiated cells [16]. However, the role of GSH redox reaction on radiation response are not fully understood.
ROS are key mediators for the development of cancer by many mechanisms, including ROS-mediated genomic instability, tumor cell proliferation and chronic inflammation [16, 17]. Reduce in serum concentration enhanced radiation-induced late ROS accumulation which was detected by using 2′, 7′-dichlorofluorescin diacetate (DCFDA) in human fiboroblasts. ROS appeared by exposure to irradiation with any doses even at low doses in the growth-restricted conditions in which cells were cultured in 0.5% or 0% fetal calf serum (FCS), while this occurred only by exposure to ≥5 Gy of single-radiation (SR) under standard serum condition (10% FCS) [13]. Thus, serum starvation or serum deprivation elevates baseline oxidative stress compared to that in standard serum condition (10% FCS) [13]. Interaction between tumor-associated stromal cells and tumor cells makes tumor microenvironment formation which plays an important role in the initiation and progression of cancer [18–20]. The purpose of this study is to clarify the mechanism of radiation carcinogenesis by analyzing the radiation-induced oxidative stress response of stromal cells. Therefore, we used un-transformed normal human fibroblasts TIG-3 and MRC-5 cells.
Here, we investigated radiation effect on GSH reaction to identify the radiation target regarding perturbation of cellular redox homeostasis. We used different serum conditions to elucidate the role of serum on GSH redox reaction. We further examined the correlation between radiation sensitivity and intracellular GSH levels in human fibroblasts.
MATERIALS AND METHODS
Cell culture conditions and drugs
Normal human diploid lung fibroblasts TIG-3 and MRC-5 were purchased from the Health Science Research Resources Bank and the Riken Cell Bank, respectively. Cells were maintained in T25 flasks with alpha minimum essential medium (Nacalai Tesque) containing 10% heat-inactivated FCS. Medium was changed from 10% to 0 or 0.5% at once time 24 h before irradiation. Cells were further cultured in medium with 0% or 0.5% FCS for 24 h until we collect. ATM inhibitor KU55933 and DNA-PK inhibitor NU7026 were purchased from Sigma. AKT inhibitor API-2 was purchased from Millipore. KU55933 (5 μM), NU7026 (10 μM) and API-2 (20 μM) were added to the media 2 h before irradiation.
Irradiation experiments
Cells were not allowed to reach to confluent state. Cells were seeded in a new flask one day before irradiation, irradiated at the indicated doses at a dose rate of 0.5 Gy/min using a 150-kVp X-ray generator (Model MBR-1505R2, Hitachi) with 0.5-mm Cu and 0.1-mm Al filters and were further incubated for 24 h. For fractionated radiation (FR), X-ray fractions (0.01 or 0.05 Gy) were administered twice a day for 5 days/week. Cells were not irradiated on Saturdays or Sundays. The total doses delivered for 31 days were 0.46 (= 0.01 Gy/fraction × 2 times × 23 days) and 2.3 Gy (= 0.05 Gy/fraction × 2 times × 23 days) for cells exposed to FR of 0.01 and 0.05 Gy, respectively. Cells were sub-cultured in a new flask twice in a week during FR for 31 days to maintain non-confluent conditions.
Mitochondrial ROS measurements
Cells were stained with 2.5 μM MitoSOX-red (Thermo Fisher Scientific) for 10 min in alpha minimum essential medium without serum. MitoSOX-red fluoresennce were measured with FACScan (Becton Dickinson). The data were shown by normalizing fluorescence intensity values of MitoSOX-red staining to non-irradiated controls.
GSH staining
GSH quantitative-imaging probe QuicGSH was purchased from Goryo Chemical [21]. Cells (1 × 105) were seeded onto 35-mm glass-bottom dishes. Cells were stained with this probe following the manufacturer’s protocol at 24 h after irradiation. QuicGSH has a fluorescence maximum wavelength at 625 nm (red color) and is shifted to 582 nm (green color) by binding with GSH (QuicGSH3.0-GSH). The fluorescent intensity ratio (R = F582/F625) was changes as the GSH concentration changes. Image acquisition and evaluation were conducted in at least three independent samples with a Keyence BZ-X700 fluorescence microscope and Hybrid Cell Count software (BZ-II Analyzer; Keyence Corporation).
GPx assay
Cell extracts were collected at 24 h after irradiation. GPx activity was calculated according to the manufacturer’s instructions using a GPx activity assay kit (Biovision).
Western blotting
Western blotting was performed as previously described [22]. Cell extracts were collected 24 h after irradiation. Primary antibodies against GPx1 (#3206, Cell signaling), glutamate-cysteine ligase catalytic (GCLC) (ab41463, Abcam), and β-tubulin (10068-1-AP, Proteintech) and secondary anti-rabbit antibodies conjugated to horseradish peroxidase (GE Healthcare) were used. Protein bands were visualized using Chemi-Lumi One L Western blotting substrate (Nacalai Tesque), and band intensities were measured using Image Lab software (Bio-Rad). Protein expression levels were normalized to β-tubulin and are expressed relative to the control value of non-irradiated cells.
Statistical analysis
Error bars represent standard deviations. All experiments were repeated at least three times using independent samples. Following one-way analysis of variance (ANOVA), the Dunnett test was used to detect significant differences among the means of three or more independent groups. Double and single asterisks indicate significant differences with p-values of <0.01 and < 0.05, respectively.
RESULTS
Radiation-induced mitochondrial ROS in different culture conditions
The fluorescence of DCFDA indicates oxidation by hydrogen peroxide, peroxynitrite or hydroxyl radical. The effect of serum concentration on the radiation-induced oxidative stress was repored by using DCFDA staining in human fiboroblasts [13]. However, DCFDA staining cannot distinguish specific types of ROS [23, 24]. We here examined the effect of radiation on generation of intracellular mitochondrial ROS in human normal fibroblasts (TIG-3 and MRC-5) by measuring the amounts of O2− using commercial probe MitoSOX-red. Mitochondrial O2− levels increased by exposure to high dose of SR (10 Gy) in TIG-3 and MRC-5 cells in standard serum condition (10% FCS) or serum deprivation conditions (0.5% FCS) compared to non-irradiated control cells (0 Gy) in 10% FCS (Fig. 1). In addition to 10% FCS and 0.5% FCS, cells were cultured for 24 h in medium with 0% FCS before irradiation to achieve a dormant state as that in vivo (Fig. 1). These cells showed high levels of mitochondrial ROS compared to non-irradiated control cells in 10% FCS regardless of radiation exposure. The results of MitoSOX-red staining after exposure of FR in TIG-3 and MRC-5 cells were reported previously. Strong intensity of MitoSOX-red staining was evident in 31FR TIG-3 cells treated with 0.01Gy/fraction, but not in non-irradiated control cells [15].
Fig. 1.
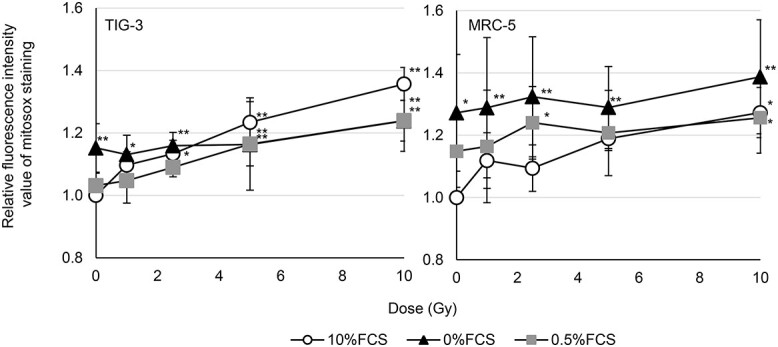
Mitochondrial ROS levels following radiation. The relative fluorescence intensity values of MitoSOX-red staining were normalized to nonirradiated control cells in 10% FCS. Cells were examined at 24 h after irradiation. Asterisk indicates a significant increase in fluorescence intensity value of MitoSOX-red staining compared with nonirradiated control cells in 10% FCS. *P < 0.05, **P < 0.01.
Radiation effect on intracellular GSH levels
To elucidate the mechanism of radiation action on GSH redox reaction, intracellular reduced GSH was visualized in living cells using the GSH quantitative-imaging probe QuicGSH (Fig. 2A) [21]. The GSH amount in cells growing with 10% FCS was increased by 10 Gy of acute SR at 24 h after irradiation compared with that in non-irradiated control cells (Fig. 2A and B). In contrast, cells in medium with 0% or 0.5% FCS maintained constant GSH levels regardless of radiation exposure (Fig. 2A and B). Different radiation exposure methods were used to evaluate the effect of FR on the GSH levels in cells growing with 10% FCS. Fractions with 2 Gy were delivered in clinical standard radiation therapy for cancer treatment [25]. However, a previous report demonstrated that exposure to ≥0.5 Gy per fraction prevented cell growth of human fibroblasts. These cells can grow only at 0.01 or 0.05 Gy per fraction [26]. Therefore, we used low-doses of FR for further analysis. Low doses of FR for 31 days increased GSH levels both in TIG-3 and MRC-5 cells (Fig. 2C).
Fig. 2.
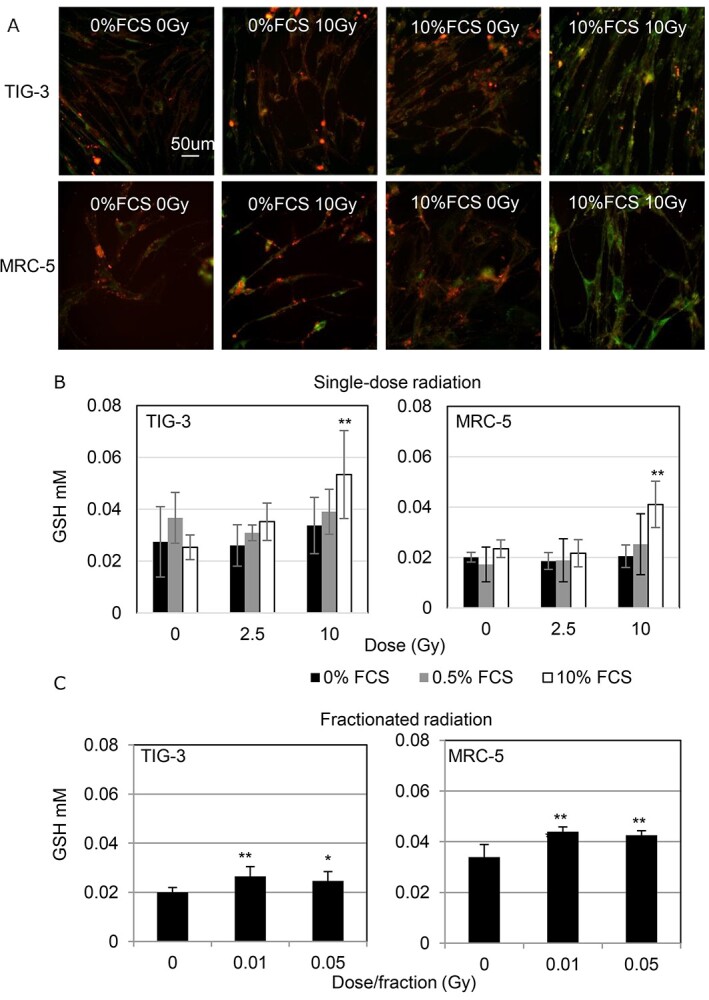
Changes in intracellular GSH levels following radiation. (A) Images of reduced GSH staining obtained using GSH quantitative-imaging probe QuicGSH in the indicated cells. Scale bar = 50 μm. Cells were examined at 24 h after irradiation. (B) GSH levels in SR cells. Asterisk indicates a significant increase in GSH levels in indicated samples compared with those in non-irradiated control cells. (C) GSH levels in FR cells. Asterisk indicates a significant increase in GSH levels compared with non-FR cells at the indicated day. *P < 0.05, **P < 0.01.
Association between intracellular GSH levels and DNA damage responses
Nuclear DNA damage responses (DDR) are linked to radiation response of mitochondria where ROS-meditated oxidative stress are generated [4]. DDR are likely to be involved in GSH-redox reaction in mitochondria. We examined the role of GSH on radiation sensitivity in human fibroblasts. ATM inhibitor KU55933 [27] and DNA-PK inhibitor NU7026 [28] were used to abrogate the DDR. The AKT inhibitor API-2 has antitumor activity [29] and is unable to activate cell survival signal after irradiation [15, 30]. Pre-radiation treatment with all inhibitors enhanced radiation sensitivity in TIG-3 and MRC-5 cells [13] and cancelled increases in the amounts of intracellular reduced GSH after 10 Gy in both TIG-3 and MRC-5 cells (Fig. 3).
Fig. 3.
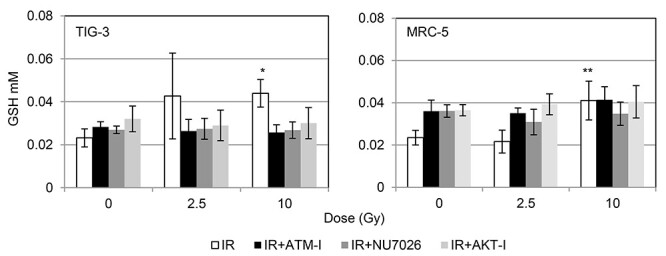
Effect of pre-radiation treatment with DDR inhibitors on GSH levels. (A) The results of QuicGSH staining in TIG-3 and MRC-5 cells with IR, IR + ATM-I, IR + NU7026 and IR + AKT-I. Cells were stained with this probe at 24 hours after irradiation. Asterisk indicates a significant increase in GSH levels in irradiated cells compared with those in non-irradiated control cells. *P < 0.05, **P < 0.01.
Effect of radiation on the GPx activity
Effects of radiation on the GPx enzyme were examined in different culture conditions and different irradiation condition. Intracellular GPx activity was measured in indicated samples using a GPx activity assay kit. Removal of serum from medium resulting in a lowering of the GPx activity in 0% or 0.5% FCS and the low GPx activity was maintained even after irradiation compared to non-irradiated cells in 10% FCS (Fig. 4A). Both SR and FR suppressed the GPx activity compared with that in non-irradiated cells under standard conditions (10% FCS) (Fig. 4A and B). Treatment with all DDR inhibitors significantly attenuated the GPx activity in TIG-3 and MRC-5 cells regardless of radiation exposure (Fig. 4C).
Fig. 4.

Effect of radiation on GPx activity. Cells were collected for the GPx activity measurement at 24 h after irradiation or last FR. (A) GPx activity following SR in cells cultured with different concentrations of FCS. Asterisk indicates a significant decrease in the GPx activity in irradiated cells compared with that in non-irradiated control cells grown with 10% FCS. (B) GPx activity in FR cells. Asterisk indicates a significant decrease in the GPx activity in FR cells compared with that in non-FR cells. (C) GPx activity in IR cells, ATM-I-treated IR cells, NU7026-treated IR cells and AKT-I-treated IR cells. Asterisk indicates a significant decrease in the GPx activity in irradiated cells compared with that in non-irradiated control cells. *P < 0.05, **P < 0.01.
Protein expression associated with GSH redox reaction after irradiation
To identify the molecular target affected by radiation, we used western blotting to quantify the expression of proteins associated with GSH redox reaction including GPx1 and GCLC. Serum starvation or serum deprivation by culturing the cells with 0% or 0.5% FCS lowered GPx1 expression and elevated GCLC expression compared with the expressions of these enzymes in control cells in medium with 10% FCS. GPx1 expression was decreased by 10 Gy of SR in 10% FCS compared with that in non-irradiated cells (Fig. 5). We further examined protein expression in irradiated cells treated with either DDR inhibitor. GPx1 expression tends to decrease by DDR inhibitor-treatment compared to non-treated control cells (Fig. 6).
Fig. 5.

Protein expression of GPx1, GCLC and tubulin following SR. Cells extracts were collected for the western blotting at 24 h after irradiation. (A) Western blotting for GPx1, GCLC and tubulin in MRC-5 cells among the indicated treatment groups. (B) Protein expression level was expressed relative to the untreated control value. Asterisk indicates a significant change in expression in the indicated sample compared with that in non-irradiated control cells grown with 10% FCS. *P < 0.05, **P < 0.01.
Fig. 6.
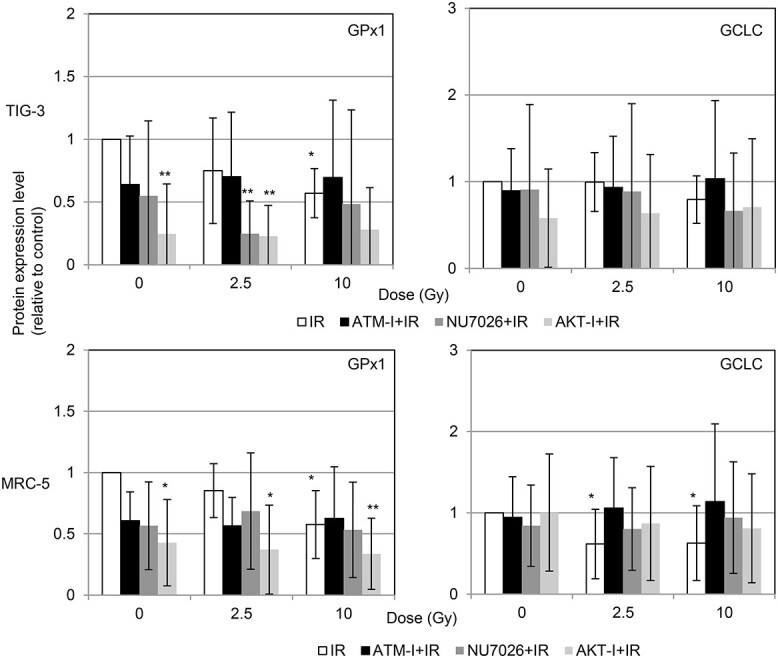
Protein expression level in IR cells, ATM-I-treated irradiated (IR) cells, NU7026-treated IR cells and AKT-I-treated IR cells. Cells extracts were collected for the western blotting at 24 h after irradiation. Protein expression level was expressed relative to the non-irradiated control value. Asterisk indicates a significant change in expression in the indicated sample compared with that in non-irradiated control cells. *P < 0.05, **P < 0.01.
DISCUSSION
Ionizing radiation-induced delayed production of mitochondrial ROS is implicated in the extended effects of radiation known as persistent oxidative stress [4]. Table 1 shows the radiation effect on GSH redox control among different culture conditions and different irradiation condition. Although reduced GSH remained, GPx activity remarkably decreased with its low protein expression following a 10-Gy dose of SR under standard serum conditions (10%). Therefore, intrinsic antioxidant regulation contributes to maintain the redox homeostasis after exposure to moderate doses of ionizing radiation, but this system is useless after exposure to high-dose radiation in standard serum condition. Serum starvation or serum deprivation conditions change a threshold for radiation-induced oxidative stress [13]. Serum starvation enhanced mitochondrial ROS generation without radiation exposure. The GPx activity was constant low under poor growth conditions in 0% FCS and 0.5% FCS. Serum produces various metabolites to promote cell growth, this may have an impact that can affect the effect of IR. Selenium, which is absent under serum starved conditions [31], is associated with stimulation of GPx1 protein expression and activity [32, 33]. Selenium levels also decline in senescent human fibroblasts [34]. Aging is related to decreased expression and activity of GPx1 in human cells [35]. Radiation is known to induce cellular senescence in human fibroblasts [15]. Thus, reduced selenium levels are likely to be associated with decreased GPx activity by serum starvation or radiation (Fig. 7). FR was used to evaluate the long-term effect of low-dose radiation on GSH reactions. Repeated FR induces persistent oxidative stress at least 24 h after FR in FR-treated cells [15]. GSH radiation responses were differ between SR and FR. Relatively inactive GPx toward ROS causes chronic oxidative stress in FR-treated cells. The aforementioned results support the decrease in GPx activity is associated with defects in ROS elimination. Growth inhibition was achieved in human fibroblasts by serum starvation, serum deprivation or high doses of radiation (10 Gy) in 10% FCS-culture [13]. Low-dose FR induced cellular senescence in irradiated cells [15]. These results suggested that growth arrest is associated with attenuation of GPx activity among these treatment groups. Some studies suggests that whole-body irradiation of mouse or in vitro irradiation of blood cells causes inactivation of GPx1 enzyme activity in bone marrow cells [36, 37], while another study suggests that GPx1 expression increased after irradiation in lung mouse tissues [38]. Furthere investigation was needed to clarify the GPx1 radiation responses in tissue. Radiation induces alteration in ROS levels [13] and GSH redox reaction according to radiation dose, irradiation condition and cell proliferating ability in human fibroblasts.
Table 1.
Radiation effect on GSH redox control among different culture conditions.
| Single-radiation | Fractionated radiation | |||
|---|---|---|---|---|
| Culture condition | serum starvation (0% FCS) |
serum deprivation (0.5% FCS) |
standard serum (10% FCS) |
standard serum (10% FCS) |
| Cell Growth | growth arrest | growth retardation | exponential growth | exponential growth |
| Overall oxidative stress 1 | ↑ at ≧2.5Gy | ↑ at 1, 2.5 Gy | ↑ at ≧5 Gy | ↑ |
| Mitochondrial O 2 − generation | constant high | ↑ at 10 Gy | ↑ at 10 Gy | No data |
| Reduced GSH levels | → | → | ↑ at 10 Gy | ↑ |
| GPx activity | constant low | constant low | ↓ | ↓ |
| GPx1 expression | constant low | constant low | ↓ at 10Gy | No data |
1 Cell Cycle 2020;19 [23]:3375-3385.
Fig. 7.
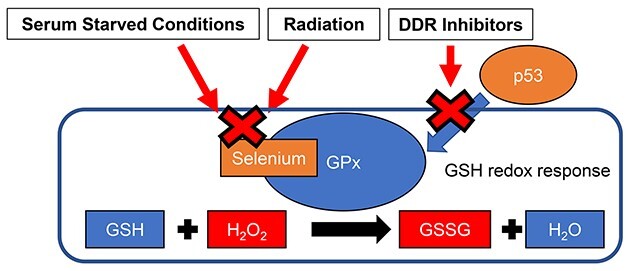
Schematic representation of decreased GPx activity by serum starvation, radiation and DDR inhibitors.
Pre-radiation treatment with DDR inhibitors such as the ATM inhibitor KU55933, the DNA-PK inhibitor NU7026 and the AKT inhibitor API-2 enhanced radiation sensitivity in human fibroblasts [13]. Treatment with KU55933 prevented an increase in ROS levels following radiation exposure [13]. Loss of ATM function leads to defects in ROS-mediated antioxidant defense systems including mitochondrial ROS generation and oxidative stress response such as GSH radiation response. ATM is essential for DNA damage signaling from the damaged nucleus to mitochondria [4]. In contrast, preradiation treatment with both NU7026 and API-2 increases radiation-induced ROS levels compared to that in cells exposed to radiation alone [13]. NU7026 or API-2 treatments prevented recovery from radiation damage and sustained DNA damage signaling to mitochondria. Here, we demonstrated that DDR inhibitors attenuated the GPx activity regardless of radiation exposure in human fibroblasts. GPx1 expression is shown to be under the control of p53 [39]. Inhibition of ATM, DNA-PK and AKT function by using those inhibitors may reduce GPx1 expression by affecting p53 stability (Fig. 7). However, further investigation is necessary to clarify the mechanism underlying the function of DDR inhibitor on the alteration of GPx activity. DDR is associated with GSH redox reaction in human fibroblasts.
In conclusions, IR-induced accumulation of ROS is attiributed to a decrease in GPx activity. The GPx is key mediator for perturbation of redox homeostasis in irradiated human fibroblasts.
ACKNOWLEDGEMENTS
The author thanks the staff of the National Institute of Public Health for assistance with the research.
Contributor Information
Tsutomu Shimura, Department of Environmental Health; National Institute of Public Health 2-3-6 Minami; Wako, Saitama, 351-0197, Japan.
Chinami Nakashiro, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo, 204-8588, Japan.
Kazusi Fujiwara, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo, 204-8588, Japan.
Rina Shiga, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo, 204-8588, Japan.
Megumi Sasatani, Department of Experimental Oncology; Research Center for Radiation Genome Medicine; Research Institute for Radiation Biology and Medicine (RIRBM); Hiroshima University, Hiroshima, 734-8551, Japan.
Kenji Kamiya, Department of Experimental Oncology; Research Center for Radiation Genome Medicine; Research Institute for Radiation Biology and Medicine (RIRBM); Hiroshima University, Hiroshima, 734-8551, Japan.
Akira Ushiyama, Department of Environmental Health; National Institute of Public Health 2-3-6 Minami; Wako, Saitama, 351-0197, Japan.
CONFLICT OF INTEREST
The author declare they have no conflicts of interest.
FUNDING
This work was supported by a grant from the JSPS KAKENHI Grant Number 18H03377, Research on Health effects of radiation organized by the Japanese Ministry of the Environment, Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labour, and Welfare and in part by National Institute for Fusion Science Collaborative Research Program (NIFS13KOBA028). This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University and the Program of the network-type joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University.
References
- 1. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol 2014;24:R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005;120:483–95. [DOI] [PubMed] [Google Scholar]
- 3. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol 2020;37:101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimura T, Sasatani M, Kawai H et al. ATM-mediated mitochondrial damage response triggered by nuclear DNA damage in normal human lung fibroblasts. Cell Cycle 2017;16:2345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H(2)O(2) signaling. Antioxid Redox Signal 2011;14:459–68. [DOI] [PubMed] [Google Scholar]
- 6. Holley AK, Bakthavatchalu V, Velez-Roman JM et al. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci 2011;12:7114–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marí M, de Gregorio E, de Dios C et al. Mitochondrial glutathione: recent insights and role in disease. Antioxidants (Basel) 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Panfili E, Sandri G, Ernster L. Distribution of glutathione peroxidases and glutathione reductase in rat brain mitochondria. FEBS Lett 1991;290:35–7. [DOI] [PubMed] [Google Scholar]
- 9. Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci U S A 1985;82:4668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bravard A, Luccioni C, Moustacchi E et al. Contribution of antioxidant enzymes to the adaptive response to ionizing radiation of human lymphoblasts. Int J Radiat Biol 1999;75:639–45. [DOI] [PubMed] [Google Scholar]
- 11. de Toledo SM, Asaad N, Venkatachalam P et al. Adaptive responses to low-dose/low-dose-rate gamma rays in normal human fibroblasts: the role of growth architecture and oxidative metabolism. Radiat Res 2006;166:849–57. [DOI] [PubMed] [Google Scholar]
- 12. Kojima S, Ishida H, Takahashi M et al. Elevation of glutathione induced by low-dose gamma rays and its involvement in increased natural killer activity. Radiat Res 2002;157:275–80. [DOI] [PubMed] [Google Scholar]
- 13. Shimura T, Ando T, Narao M et al. Mechanism of turnover or persistence of radiation-induced myofibroblast in vitro. Cell Cycle 2020;19:3375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimura T, Kobayashi J, Komatsu K et al. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016;15:1099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimura T, Sasatani M, Kamiya K et al. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016;7:3559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szumiel I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol 2015;91:1–12. [DOI] [PubMed] [Google Scholar]
- 17. Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–2629 discussion 30-2. [PubMed] [Google Scholar]
- 18. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16:582–98. [DOI] [PubMed] [Google Scholar]
- 19. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 20. Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol 2006;1:119–50. [DOI] [PubMed] [Google Scholar]
- 21. Umezawa K, Yoshida M, Kamiya M et al. Rational design of reversible fluorescent probes for live-cell imaging and quantification of fast glutathione dynamics. Nat Chem 2017;9:279–86. [DOI] [PubMed] [Google Scholar]
- 22. Shimura T, Kakuda S, Ochiai Y et al. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3 beta-mediated cyclin D1 overexpression. Oncogene 2010;29:4826–37. [DOI] [PubMed] [Google Scholar]
- 23. Chen X, Zhong Z, Xu Z et al. 2′,7'-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radic Res 2010;44:587–604. [DOI] [PubMed] [Google Scholar]
- 24. Kalyanaraman B, Darley-Usmar V, Davies KJ et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med 2012;52:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer 2005;5:516–25. [DOI] [PubMed] [Google Scholar]
- 26. Shimura T, Hamada N, Sasatani M et al. Nuclear accumulation of cyclin D1 following long-term fractionated exposures to low-dose ionizing radiation in normal human diploid cells. Cell Cycle 2014;13:1248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hickson I, Zhao Y, Richardson CJ et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004;64:9152–9. [DOI] [PubMed] [Google Scholar]
- 28. Nutley BP, Smith NF, Hayes A et al. Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. Br J Cancer 2005;93:1011–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L, Dan HC, Sun M et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 2004;64:4394–9. [DOI] [PubMed] [Google Scholar]
- 30. Shimura T, Toyoshima M, Adiga SK et al. Suppression of replication fork progression in low-dose-specic p53-dependent S-phase DNA damage checkpoint. Oncogene 2006;25:5921–32. [DOI] [PubMed] [Google Scholar]
- 31. Mehdi Y, Dufrasne I. Selenium in cattle: a review. Molecules 2016;21:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schnabel R, Lubos E, Messow CM et al. Selenium supplementation improves antioxidant capacity in vitro and in vivo in patients with coronary artery disease the SElenium therapy in coronary artery disease patients (SETCAP) study. Am Heart J 2008;156:1201 e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goldson AJ, Fairweather-Tait SJ, Armah CN et al. Effects of selenium supplementation on selenoprotein gene expression and response to influenza vaccine challenge: a randomised controlled trial. PLoS One 2011;6:e14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Legrain Y, Touat-Hamici Z, Chavatte L. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J Biol Chem 2014;289:6299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He T, Joyner MJ, Katusic ZS. Aging decreases expression and activity of glutathione peroxidase-1 in human endothelial progenitor cells. Microvasc Res 2009;78:447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu G, Wu H, Zhang J et al. Metformin ameliorates ionizing irradiation-induced long-term hematopoietic stem cell injury in mice. Free Radic Biol Med 2015;87:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H, Yang YL, Zhang H et al. Administration of the resveratrol analogues isorhapontigenin and heyneanol-a protects mice hematopoietic cells against irradiation injuries. Biomed Res Int 2014;2014:282657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian X, Wang F, Luo Y et al. Protective role of nuclear factor-erythroid 2-related factor 2 against radiation-induced lung injury and inflammation. Front Oncol 2018;8:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tan M, Li S, Swaroop M et al. Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem 1999;274:12061–6. [DOI] [PubMed] [Google Scholar]