

Significance
The cerebral accumulation of amyloid β (Aβ) is a hallmark of Alzheimer’s disease (AD). While type 2 diabetes mellitus is known to be a risk factor for AD, the underlying mechanisms remain unclear. In the present study, we demonstrate that plasma Aβ is produced from glucose- and insulin-susceptible peripheral tissues, such as the pancreas, adipose tissues, skeletal muscles, and liver, to inhibit insulin secretion from islet β-cells. Our findings suggest a physiological role of peripheral Aβ in glucose and insulin metabolism and a possible mechanism linking diabetes to AD. In addition, although plasma Aβ levels are currently used as a diagnostic biomarker of AD, our data suggest they should be used with caution.
Keywords: diabetes, Alzheimer’s disease, plasma Aβ, insulin, glucose
Abstract
Type 2 diabetes mellitus is known to be a risk factor for Alzheimer’s disease (AD), but the underlying mechanisms remain unclear. In AD, the cerebral accumulation of amyloid β (Aβ) triggers a pathological cascade leading to neurodegeneration. Plasma Aβ levels are thought to reflect the brain amyloid pathology and currently used as a diagnostic biomarker of AD. However, amyloid precursor protein and Aβ-generating enzymes, β- and γ-secretases, are widely expressed in various peripheral tissues. Previous reports have shown that glucose and insulin loading cause a transient increase of plasma Aβ in mice and humans. These findings led us to speculate that plasma Aβ is produced from glucose- and insulin-susceptible peripheral tissues to play a role in glucose and insulin metabolism. To test this hypothesis, we investigated the effects of glucose and insulin on Aβ secretion and the effect of Aβ on insulin secretion in vivo, ex vivo, and in vitro. Aβ was found to be secreted from β-cells of the pancreas along with insulin upon glucose stimulation. Upon insulin stimulation, Aβ was secreted from cells of insulin-targeted organs, such as adipose tissues, skeletal muscles, and the liver, along with their organokines. Furthermore, Aβ inhibited the glucose-triggered insulin secretion from β-cells, slowing down glucose clearance from the blood. These results suggest that peripheral Aβ acts as a negative modulator of insulin secretion. Our findings provide a possible mechanism linking diabetes to AD and call attention to how plasma Aβ levels are used in AD diagnosis.
Type 2 diabetes mellitus is known to be a risk factor for Alzheimer’s disease (AD) (1, 2), but the underlying mechanisms remain unclear. Amyloid β (Aβ), a main component of amyloid plaques and cerebrovascular amyloids in AD, plays a pivotal role in the pathogenesis of AD (3). There are several isoforms of Aβ: the minor isoform Aβ42 is more aggregable than the major isoform Aβ40, and amyloid plaques are primarily composed of Aβ42. While Aβ is secreted from neurons and accumulates in the brain, it is detected not only there but also in the blood. In parallel with the formation of amyloid plaques, the levels of Aβ, particularly Aβ42, in the blood as well as in the cerebrospinal fluid (CSF) tends to decrease during the disease progression. Since blood Aβ is assumed to be mostly derived from the brain and its level likely reflects amyloid pathology in the brain, the Aβ42/Aβ40 ratio in plasma is currently used as a diagnostic biomarker of AD (3). However, Aβ is produced not only in the brain but also in peripheral tissues. Aβ is generated from amyloid precursor protein (APP) by the function of two enzymes, β- and γ-secretases (4). APP is a ubiquitous protein that is expressed widely in the body (4). Beta-site APP cleaving enzyme 1 (BACE1), the enzyme of β-secretase, and presenilin 1 and 2 (PS1 and PS2), which are catalytic subunits of γ-secretase, are also expressed in various tissues (5, 6). For example, adipose tissues express APP, BACE1, and PS1/2 and secrete Aβ in response to glucose and insulin loading in vitro (7). Skeletal muscles also express these proteins and accumulate Aβ within abnormal muscle fibers in inclusion-body myositis (8, 9). Finally, the pancreas (10) and liver (11, 12) express these proteins, with the liver suggested to be a major source of plasma Aβ and also a source of brain Aβ (13).
However, the physiological role of peripherally generated Aβ is unknown. We previously reported that intraperitoneal glucose injection into wild-type and APP transgenic mice caused a transient increase in the levels of blood glucose, plasma insulin, and plasma Aβ (14). Blood glucose and plasma insulin reached their peaks at 15 min after the glucose injection, but the peak of plasma Aβ was slightly later: Aβ40 peaked at 30–60 min in wild-type mice and 60–120 min in APP transgenic mice. We later reported that oral glucose loading induced a significant increase in plasma Aβ40 and Aβ42 levels in AD patients but not in non-AD dementia patients (15). On the other hand, a different group found that the intravenous infusion of insulin raised plasma Aβ42 levels in AD patients but not in normal adults (16). Another group reported that in healthy young adults, the intravenous infusion of insulin increased plasma Aβ42 levels but not Aβ40 levels (17). Although glucose and insulin loading affected plasma Aβ40 and Aβ42 levels in AD and non-AD individuals differently, these findings led us to speculate that plasma Aβ is produced from peripheral tissues that are susceptible to changes in blood glucose or insulin levels and that peripheral Aβ may contribute to glucose and insulin metabolism. If true, the high levels of blood glucose and insulin in diabetes would persistently increase peripheral Aβ production, which may affect brain Aβ levels. To test this hypothesis, here we investigated the effects of glucose and insulin on Aβ secretion and the effect of Aβ on insulin secretion in vivo, ex vivo, and in vitro. Our findings suggest a physiological role of peripheral Aβ, a mechanism that links diabetes to AD, and, furthermore, raise caution in using plasma Aβ levels in AD diagnosis.
Results
Glucose and Insulin Loading Increases Plasma Aβ Levels.
Initially, we studied the effects of glucose and insulin on plasma Aβ levels in mice. All mice were fasted for 16 h prior to glucose and insulin loading. Saline containing 0.06 g glucose was injected intraperitoneally (approximate dose of 2 g/kg) into 10-mo-old wild-type mice, and the blood was collected from the tail vein at 0, 15, 30, 45, 60, 120, and 180 min after the injection. Glucose injection caused a transient increase in blood glucose, plasma insulin, and plasma Aβ40 and Aβ42 levels, which all peaked at 30 min, but glucose and Aβ decreased gradually and insulin fell rapidly thereafter (Fig. 1A). Next, saline containing 0.015 U insulin was injected intraperitoneally (approximate dose of 0.5 U/kg) into 11-mo-old wild-type mice, and the blood was collected at 0, 15, 30, 45, and 60 min after the injection. The insulin injection induced a transient increase in plasma insulin levels with a peak at 15 min and a transient decrease in blood glucose levels with the lowest level at 30 min (Fig. 1B). Plasma Aβ40 and Aβ42 levels transiently increased after the insulin injection, with a peak at 30 min and a gradual decrease thereafter. These results are consistent with previous reports that found glucose and insulin loading affect plasma Aβ levels in mice and humans (14–17), suggesting a possible role of plasma Aβ in glucose and insulin metabolism.
Fig. 1.

In vivo effects of glucose and insulin loading on plasma Aβ and of Aβ loading on plasma insulin in mice. (A) Glucose was injected intraperitoneally into fasted wild-type mice, which caused a transient increase in blood glucose, plasma insulin, and plasma Aβ40 and Aβ42 levels. (B) Insulin was injected intraperitoneally into fasted wild-type mice, which induced a transient increase in the plasma insulin and plasma Aβ40 and Aβ42 levels and a transient decrease in the blood glucose level. (C) Glucose was injected intraperitoneally into fasted APP23 (green) and APP knockout mice (red). The data of wild-type mice (blue) are the same as in (A). In APP23 mice, glucose injection elicited a marked increase in the plasma Aβ40, Aβ42 and blood glucose levels and a rapid decrease in the plasma insulin level. On the contrary, in APP knockout mice, which showed no detectable plasma Aβ level, glucose injection caused a marked increase in the plasma insulin level and a rapid decrease in the blood glucose level. AUC (area under the curve) values were calculated and are shown below the corresponding graphs. (D) Aβ40 peptide (orange open square) or saline only (red closed circle) was injected intraperitoneally into fasted APP knockout mice, which was followed by subsequent intraperitoneal glucose injection. The Aβ40 injection suppressed the glucose-induced increase in the plasma insulin level. (E) Aβ42 (orange open square), scramble Aβ42 (violet open square) or saline only (red closed circle) was injected intraperitoneally into APP knockout mice. The Aβ42 injection but not scramble Aβ42 injection suppressed the glucose-induced increase in the plasma insulin level. (F) A Western blot shows the injected Aβ40 and Aβ42 peptides were predominantly monomers. M, MagicMark XP Western Protein Standard (Invitrogen, Carlsbad, CA).
Aβ Affects Blood Glucose and Plasma Insulin Levels.
To further investigate the in vivo relationship between Aβ, insulin, and glucose in the blood, we repeated the glucose injection experiments in 10-mo-old APP23 mice, an APP transgenic line which overproduces Aβ (18), and 12- to 15-mo-old APP knockout mice, which lack Aβ production (19). The fasting levels of blood glucose, plasma insulin, and plasma Aβ40 and Aβ42 in each mouse group are shown in Table 1. In APP23 mice, the glucose injection elicited a transient increase in blood glucose, plasma insulin, and plasma Aβ40 and Aβ42 levels similar to wild-type mice (Fig. 1C). However, while the levels of glucose and Aβ40 and Aβ42 peaked at 30 min for both mice, they were higher in APP23 mice at all time points. Additionally, the area under the curve (AUC) for glucose and Aβ40 and Aβ42 in APP23 mice was significantly greater than in wild-type mice. In contrast, the levels of insulin increased quickly with a peak at 15 min and thereafter decreased rapidly to levels lower than in wild-type mice. Although the difference in AUC was not significant between the two groups, insulin secretion in APP23 mice appeared to be suppressed immediately after its initiation. On the other hand, in APP knockout mice, glucose injection induced a transient increase in blood glucose and plasma insulin levels, whereas plasma Aβ40 and Aβ42 levels remained undetectable for 180 min (Fig. 1C). The levels of glucose increased quickly with a peak at 15 min and thereafter decreased rapidly to levels lower than in wild-type mice, and the AUC was significantly lower in APP knockout mice. In contrast, the level of insulin peaked at 30 min and was higher than in wild-type mice at all time points. Further, the AUC was significantly greater than in wild-type mice. These results collectively suggest that plasma Aβ inhibits glucose-triggered insulin secretion to slow down glucose clearance from the blood.
Table 1.
Fasting and peak levels of blood glucose, plasma insulin, and plasma Aβ40 and Aβ42 after glucose injection into mice
| Wild-type mice | APP23 mice | APP knockout mice | |
|---|---|---|---|
| Glucose (mg/dL) | |||
| Fasting | 61.5 ± 2.2 | 67.8 ± 8.5 | 75.0 ± 9.0 |
| Peak | 326.8 ± 24.6 | 436.8 ± 19.2 | 353.5 ± 34.7 |
| Insulin (ng/mL) | |||
| Fasting | 0.467 ± 0.060 | 0.383 ± 0.083 | 0.605 ± 0.085 |
| Peak | 1.434 ± 0.238 | 1.668 ± 0.386 | 1.854 ± 0.266 |
| Aβ40 (pM) | |||
| Fasting | 67.0 ± 13.7 | 242.4 ± 32.8 | Not detected |
| Peak | 131.5 ± 16.5 | 395.0 ± 43.0 | Not detected |
| Aβ42 (pM) | |||
| Fasting | 12.1 ± 5.0 | 59.2 ± 3.6 | Not detected |
| Peak | 43.1 ± 3.5 | 94.9 ± 14.9 | Not detected |
Values represent the mean ± SEM, n = 6 in each group.
Next, we examined the effect of Aβ on blood glucose and plasma insulin levels. Saline containing 6 pmol synthetic Aβ40 peptide or saline alone was injected intraperitoneally into 12- to 15-mo-old APP knockout mice. Five minutes after the injection, 0.06 g glucose was injected intraperitoneally, and the blood was collected 0, 15, and 30 min later. In the saline-injected group, blood glucose transiently increased by glucose loading with a peak at 15 min and thereafter began to decrease, while plasma insulin increased over time (Fig. 1D). In the Aβ40-injected group, plasma Aβ40 immediately increased to a peak of 70 pM at 15 min but was rapidly cleared at 30 min (Fig. 1D, Right). Plasma insulin was suppressed by Aβ injection at 15 min and maintained a low levels at 30 min (Fig. 1D, Middle). Blood glucose showed a similar pattern as the saline-injected group, but Aβ injection slightly slowed down the glucose clearance at 30 min (Fig. 1D, Left). We repeated the experiments with synthetic Aβ42 and scramble Aβ42 peptides. Similar to Aβ40, plasma Aβ42 immediately increased at 15 min but was rapidly cleared at 30 min in the Aβ42-injected group (Fig. 1E, Right). Plasma insulin and blood glucose also showed similar patterns to those in Aβ40-injected mice (Fig. 1E). In contrast, scramble Aβ42 did not affect glucose or insulin levels. Western blot analysis revealed that the injected Aβ40 was primarily monomers, whereas the injected Aβ42 consisted predominantly of monomers with some dimers (Fig. 1F). The changing profiles of blood glucose and plasma insulin in the Aβ- and saline-injected groups resemble those in APP23 mice and APP knockout mice after glucose injection. Thus, the present results support our hypothesis that plasma Aβ inhibits glucose-triggered insulin secretion.
Aβ Is Secreted from the Pancreas and Insulin-Sensitive Organs to Inhibit Insulin Secretion.
We next searched for the origin of plasma Aβ. Given the fact that plasma Aβ levels changed immediately upon glucose and insulin loading, we hypothesized that plasma Aβ comes from glucose- and insulin-sensitive peripheral organs. Accordingly, we carried out ex vivo experiments using live peripheral tissues isolated from wild-type mice after 16-h fasting. The pancreas, abdominal white adipose tissues, anterior tibial muscles, liver, and kidneys were collected after removing the blood by cold phosphate-buffered saline (PBS) infusion and chopped into small pieces on ice. The minced tissues were dispensed into tubes and stimulated with glucose or insulin at 37 °C for 60 min with rotation. After a brief centrifugation, the levels of Aβ secreted from the tissues into the media were measured by enzyme-linked immunosorbent assay (ELISA). Stimulation with high-glucose (4,500 mg/L) medium caused a significant increase in Aβ40 and Aβ42 secretion from the pancreatic tissues (Fig. 2A) but not the adipose (Fig. 2B), muscle (Fig. 2C), or liver tissues (Fig. 2D). In contrast, the addition of 200 nM insulin into the media elicited a significant increase in Aβ40 and Aβ42 secretion from the adipose, muscle, and liver tissues, but not the pancreatic tissues. In either condition, Aβ secretion was not detected from the kidney tissues (Fig. 2E), which are not involved in glucose or insulin metabolism.
Fig. 2.

Ex vivo effects of glucose and insulin loading on Aβ secretion and of Aβ loading on insulin secretion in live peripheral tissues isolated from mice. The pancreas (A), abdominal white adipose tissues (B), anterior tibial muscles (C), liver (D), and kidneys (E) were collected from fasted wild-type mice. The minced tissues were stimulated with high-glucose or insulin for 60 min. High-glucose stimulation significantly increased Aβ40 and Aβ42 secretion from the pancreatic tissues, but not the adipose, muscle, or liver tissues. In contrast, insulin stimulation significantly enhanced Aβ40 and Aβ42 secretion from the adipose, muscle, and liver tissues but not the pancreatic tissues. In either condition, Aβ secretion was not detected from the kidney tissues. (F) The pancreatic tissues were stimulated with high-glucose with or without Aβ40, Aβ42, or scramble Aβ42 peptide for 60 min. Glucose-induced insulin secretion was significantly attenuated in the presence of Aβ40 and Aβ42 but not of scramble Aβ42 peptide.
To evaluate the effects of Aβ on insulin secretion, the pancreatic tissues were stimulated with high-glucose medium with or without 100 pM Aβ40, Aβ42, or scramble Aβ42 peptide for 60 min. Glucose-induced insulin secretion was significantly attenuated in the presence of Aβ40 and Aβ42 but not scramble Aβ42 peptide (Fig. 2F). These results suggest that plasma Aβ is secreted from the pancreas upon glucose stimulation and from insulin-sensitive organs, such as adipose tissues, skeletal muscles, and liver, upon insulin stimulation to inhibit insulin secretion from the pancreas.
Aβ Is Localized in Pancreatic β-Cells and Endocrine-Containing Cells of Insulin-Targeted Organs and Released with Tissue Organokines.
To examine the localization of Aβ in the peripheral tissues, we performed immunohistochemistry with antibodies for Aβ and specific markers of cells in the pancreas and insulin-targeted organs. First, we tested the pancreas. Ten-month-old freely fed wild-type mice were fixed by 4% paraformaldehyde perfusion under anesthesia. Pancreas tissue sections were prepared and stained for Aβ, insulin (β-cells), glucagon (α-cells), and somatostatin (δ-cells). As shown in Fig. 3A, Aβ was detected only in β-cells in the islet of Langerhans in the pancreas and at least in part, if not entirely, colocalized with insulin. We confirmed that Aβ immunoreactivity was absent in sections from APP knockout mice (SI Appendix, Fig. S1), indicating the Aβ-specificity of the antibody we used. Our data appear to support another report that found APP, BACE1, and PS1/2 colocalize in β-cells but not in other islet cells in the pancreas (10). Then we investigated the effect of glucose loading on insulin and Aβ levels in β-cells. Mice were fasted for 16 h, and some of them received intraperitoneal glucose injections. Fifteen minutes after the injection, the mice were fixed, and pancreas tissue sections were stained. The glucose injection markedly reduced the immunoreactivities of both insulin and Aβ in β-cells (Fig. 3B). These results suggest that in the fasting state, Aβ and insulin are stored in β-cells and that upon glucose stimulation they are simultaneously released into the circulation.
Fig. 3.

Immunohistochemical analysis of Aβ localization in the pancreas, adipose tissues, skeletal muscles, and liver and the effects of glucose and insulin loading. (A) Pancreas tissue sections from freely fed wild-type mice were stained for Aβ (green), insulin (red), glucagon (red), and somatostatin (red). Aβ was detected only in β-cells in the islet of Langerhans and partially colocalized with insulin. The far left panel shows combined images of insulin, glucagon, and somatostatin. (B) Pancreas tissue sections from glucose-injected fasted wild-type mice were stained for Aβ (green) and insulin (red). Glucose injection markedly reduced the immunoreactivities of both insulin and Aβ in β-cells. Tissue sections from insulin-injected fasted wild-type mice were prepared from abdominal white adipose tissues (C), anterior tibial muscles (D), and the liver (E). The sections were stained for Aβ (green) and the adipokine Acrp30 (red) (C), the myokine IL-6 (red) (D), and the hepatokine IGF1 (red) (E). Aβ colocalized largely with IL-6 and partially with Acrp30 and IGF1. Insulin injection caused a marked decrease in the immunoreactivities of both Aβ and these organokines.
For insulin-targeted organs, wild-type mice were fasted for 16 h and some received intraperitoneal insulin injections. Thirty minutes after the injection, the mice were fixed, and tissue sections were prepared from abdominal white adipose (Fig. 3C), anterior tibial muscles (Fig. 3D), and the liver (Fig. 3E). Tissue sections were stained for Aβ, and their own endocrine factors called organokines: the adipokine adiponectin/Acrp30 for adipocytes (20), the myokine interleukin (IL)-6 for myocytes (21), and the hepatokine insulin-like growth factor-1 (IGF1) for hepatocytes (22). Aβ was detected in all tissues tested and showed large colocalization with IL-6 and partial colocalization with Acrp30 and IGF1. Insulin injection resulted in a marked decrease in the immunoreactivities of both Aβ and organokines. These results suggest that in the fasting state, Aβ and organokines are stored in each cell and that upon insulin stimulation they are simultaneously released into the circulation.
Aβ Is Secreted from Cultured Islet β-Cells, Adipocytes, Myocytes, and Hepatocytes upon Glucose and Insulin Stimulation to Inhibit Insulin Secretion.
To validate the findings from the ex vivo experiments and immunohistochemistry, we next examined the effects of glucose and insulin on Aβ secretion in cultured cells, which include β-TC-6 cells as islet β-cells, 3T3-L1 cells as adipocytes, C2C12 cells as myocytes, and FL83B cells as hepatocytes. Prior to use, the 3T3-L1 and C2C12 cells were allowed to differentiate into mature adipocytes and myocytes, which was confirmed by the formation of intracellular lipid drops and myotubes, respectively. All the cells were initially incubated in serum-free no-glucose medium for 120 min.
β-TC-6 cells were stimulated with high glucose medium for 60 min in the presence or absence of 10 nM glucagon-like peptide-1 (GLP-1). GLP-1, a member of the metabolic hormones called incretins, is released from enteroendocrine L cells in response to food intake and acts on islet β-cells to promote glucose-dependent insulin secretion (23). Culture media were collected, and the concentrations of Aβ40, Aβ42, and insulin were measured by ELISA. High-glucose stimulation caused a significant increase in the Aβ40, Aβ42, and insulin levels in the media, an effect further enhanced by GLP-1 (Fig. 4A). Then we examined the effect of Aβ on the glucose-induced insulin secretion. β-TC-6 cells were stimulated with high-glucose medium for 60 min in the presence or absence of 50 pM Aβ40. Aβ40 significantly inhibited glucose-induced insulin secretion (Fig. 4B). This observation suggests that Aβ secreted from β-cells acts on the same cells to inhibit insulin secretion in an autocrine manner. Thus, we treated the β-TC-6 cells with 1 μM of a γ-secretase inhibitor, L685,458, for 120 min to inhibit the endogenous Aβ production. The cells were then stimulated with high-glucose medium containing 1 μM L685,458 for 60 min. The addition of L685,458 significantly enhanced both the steady state and glucose-induced insulin secretion from β-TC-6 cells (Fig. 4C), supporting the autocrine function of Aβ.
Fig. 4.

In vitro effects of glucose and insulin loading on Aβ secretion and of Aβ loading on insulin and GLP-1 secretion and glucose uptake in cell culture. All cells were cultured in no-glucose medium for 120 min prior to the glucose and insulin stimulation. (A) β-TC-6 cells (islet β-cells) were stimulated with high-glucose for 60 min in the presence or absence of GLP-1. High-glucose stimulation caused a significant increase in Aβ40, Aβ42, and insulin levels in the media, which was further enhanced by GLP-1. (B) β-TC-6 cells were stimulated with high glucose for 60 min in the presence or absence of Aβ40. Aβ40 significantly inhibited the glucose-induced insulin secretion. (C) β-TC-6 cells were stimulated with high glucose in the presence or absence of a γ-secretase inhibitor, L685,458. L685,458 significantly enhanced both the steady state and glucose-induced insulin secretion. Differentiated 3T3-L1 cells (adipocytes) (D), differentiated C2C12 cells (myocytes) (E), and FL83B cells (hepatocytes) (F) were stimulated with high glucose for 60 min with or without insulin. High-glucose stimulation did not affect Aβ40 or Aβ42 secretion, but insulin stimulation significantly enhanced it in these cells. (G) NCI-H716 cells were stimulated with high glucose for 120 min in the presence or absence of Aβ40 or Aβ42. High-glucose stimulation caused a significant increase in the GLP-1 level in the media, while the addition of Aβ40 and Aβ42 had no effect. (H) Differentiated 3T3-L1 cells were stimulated with insulin in the presence or absence of Aβ40 or Aβ42 for 20 min. The cells were further incubated for 10 min in the presence of 3H-labeled glucose. Insulin stimulation significantly increased the glucose uptake, but the addition of Aβ40 or Aβ42 had no effect.
Next, we stimulated 3T3-L1, C2C12, and FL83B cells with high-glucose medium or 200 nM insulin for 60 min. High-glucose stimulation did not affect Aβ40 or Aβ42 secretion from the cells, but insulin stimulation significantly enhanced it (Fig. 4 D–F).
Aβ Affects Neither GLP-1 Secretion Nor Glucose Uptake.
Our data suggest that plasma Aβ directly acts on islet β-cells to inhibit insulin secretion. However, there remains the possibility that Aβ affects insulin secretion by suppressing incretin secretion from the intestine. Thus, we examined the effect of Aβ on GLP-1 secretion from NCI-H716 cells, which were originated from human enteroendocrine L cells. The cells were stimulated with high-glucose medium for 120 min in the presence or absence of 100 pM Aβ40 or Aβ42, and GLP-1 concentrations in the culture media were measured by ELISA. High-glucose stimulation caused a significant increase in the GLP-1 level in the media, an effect unchanged by the addition of Aβ40 or Aβ42 (Fig. 4G). These results support our notion that Aβ directly inhibits insulin secretion from β-cells.
We assumed that plasma Aβ slows down blood glucose clearance by inhibiting insulin secretion. However, Aβ could alternatively act on insulin-targeted cells directly to prevent the glucose uptake, since Aβ is known to interact with insulin receptors and impair insulin signaling (24, 25). Such an impairment may disturb the insulin-dependent translocation of glucose transporter 4 (GLUT4) from cytoplasmic storage vesicles to the plasma membrane (26). Thus, we examined the effect of Aβ on glucose uptake by 3T3-L1 cells. The cells were stimulated with 1 or 10 nM insulin-containing buffer in the presence or absence of 100 pM Aβ40 or Aβ42 for 20 min. Then, 3H-labeled glucose was added to the medium. Ten minutes later, the amounts of internalized radiolabeled glucose were measured using a liquid scintillation counter. Insulin stimulation significantly increased the glucose uptake by 3T3-L1 cells, an effect again unchanged by the addition of Aβ40 or Aβ42 (Fig. 4H).
Taken together, these results suggest that Aβ and insulin are secreted from islet β-cells upon glucose stimulation, and Aβ and organokines are secreted from cells of insulin-targeted organs upon insulin stimulation. The secreted Aβ acts on islet β-cells to negatively modulate insulin secretion without affecting GLP-1 secretion from the intestine, thereby slowing down blood glucose clearance without directly inhibiting insulin-dependent glucose uptake by peripheral tissues.
Discussion
To elucidate the relationship between Aβ, insulin, and glucose, we performed in vivo (mice), ex vivo (live tissues), and in vitro (cultured cells) experiments. Our results suggest a role of peripheral Aβ in insulin and glucose metabolism. Peripheral tissues, including the pancreas, adipose tissues, skeletal muscles, and liver, serve as sources of plasma Aβ. Increased levels of blood glucose after food intake trigger insulin secretion from islet β-cells. According to our data, glucose-triggered insulin secretion is accompanied by Aβ secretion from the same β-cells. Thus, we named this glucose-dependent Aβ secretion the “primary secretion” of Aβ (Fig. 5). Secreted insulin acts on insulin-targeted organs to promote glucose uptake by adipose tissues and muscles, glycogen synthesis in muscles and the liver, and lipogenesis in adipose tissues and the liver. These tissues release their own endocrine factors called organokines, such as adipokines, myokines, and hepatokines, upon insulin stimulation to maintain glucose and lipid homeostasis and insulin sensitivity in an autocrine, paracrine, and endocrine manner (27). Our data suggest that the insulin-dependent secretion of organokines coincides with Aβ secretion from the same cells. Thus, we named this insulin-mediated Aβ secretion the “secondary secretion” of Aβ (Fig. 5). The secreted Aβ from β-cells, adipocytes, myocytes, and hepatocytes acts on β-cells to negatively modulate insulin secretion. Insulin secretion is primarily regulated by blood glucose. Therefore, we hypothesize that insulin secretion is finely adjusted by plasma Aβ. Further, we speculate that Aβ secreted from islet β-cells promptly adjusts insulin secretion in an autocrine manner and that Aβ secreted from insulin-targeted organs suppresses insulin secretion in an endocrine manner. This two-step adjustment of insulin secretion contributes to keeping proper levels of blood glucose. Our results therefore suggest that peripherally secreted Aβ functions as an organokine, which describes molecules involved in crosstalk between peripheral tissues and play a role in glucose and insulin homeostasis. Our hypothesis falls in line with Tu et al. (28), who, using an integrative analysis of a cross-loci regulation network, identified APP as a top candidate gene that regulates insulin secretion from pancreatic islets. Furthermore, they showed that the loss of APP in mice leads to increased insulin secretion from islets in response to glucose by an unexplained mechanism. The present study addresses this question by showing that APP knockout mice have up-regulated insulin secretion because insulin secretion is negatively regulated by APP-derived Aβ. The molecular mechanism by which Aβ inhibits insulin secretion remains to be studied.
Fig. 5.
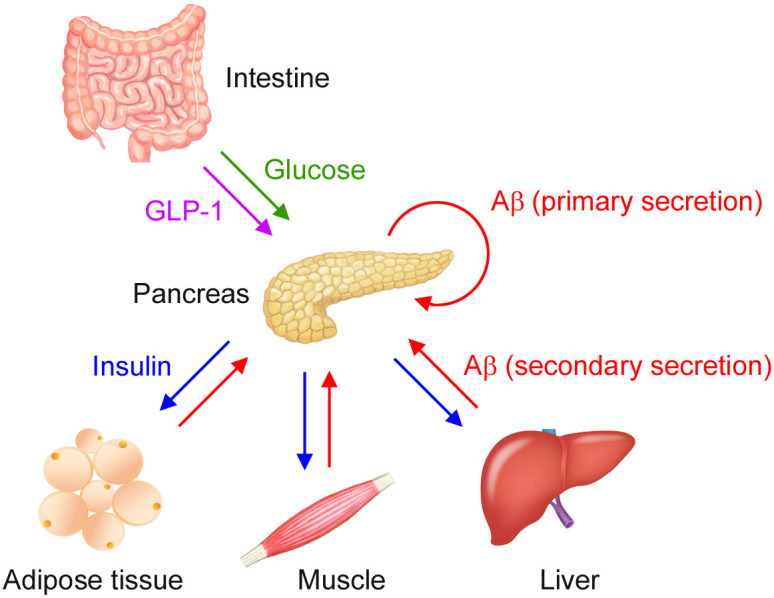
Aβ-mediated crosstalk of peripheral tissues in glucose and insulin homeostasis. Increased levels of blood glucose (green) after food intake trigger insulin (blue) secretion from islet β-cells. This glucose-triggered insulin secretion is accompanied with Aβ (red) secretion from the same β-cells, a phenomenon we call ‘the primary secretion’ of Aβ. Secreted insulin acts on insulin-targeted organs to promote glucose uptake by adipose tissues and muscles and glycogen synthesis in muscles and the liver. These tissues release their own endocrine factors (i.e., organokines, including adipokines, myokines, and hepatokines) upon insulin stimulation to maintain glucose and lipid homeostasis and insulin sensitivity in an autocrine, paracrine, and endocrine manner. This insulin-dependent secretion of organokines coincides with Aβ secretion from the same cells, a phenomenon we called ‘the secondary secretion’ of Aβ. The secreted Aβ acts on β-cells in an autocrine and endocrine manner to negatively modulate insulin secretion.
Our findings also suggest a mechanism linking diabetes to AD. It has been suggested that the level of brain Aβ is regulated by receptor-mediated transport at the blood brain barrier (BBB) (29). Aβ efflux from the brain into the circulation is mediated by low density lipoprotein receptor-related protein 1 (LRP1), whereas Aβ influx from the circulation into the brain is mediated by the receptor for advanced glycation end products (RAGE). Hyperglycemia and hyperinsulinemia in diabetes cause a prolonged production of peripheral Aβ. The resulting high levels of plasma Aβ may affect the equilibrium between brain Aβ and peripheral Aβ to suppress Aβ efflux from the brain. Plasma insulin can enter the brain across the BBB by receptor-mediated transcytosis (30), and hyperinsulinemia may also promote insulin influx from the circulation into the brain. The resulting high levels of insulin in the brain possibly compete with Aβ for insulin-degrading enzyme (31), causing insufficient Aβ digestion. Consequently, Aβ becomes abundant in the brain by reduced excretion into the circulation and/or insufficient enzymatic degradation in diabetes. The enriched Aβ likely forms soluble oligomers, which elicit synaptic and cognitive dysfunction and initiate the pathological cascade of AD (32, 33). This may explain why diabetes could be a risk factor for AD. In fact, it has been shown that diabetes accelerates brain Aβ pathology including Aβ oligomer accumulation in rabbits (34), nonhuman primates (35), APP transgenic mice (36–38), and humans (39). It is unclear whether glucose- and insulin-dependent Aβ production also occurs in the brain. Additionally, we hypothesize that high levels of plasma Aβ may exacerbate diabetes by affecting the balance of blood glucose and insulin levels toward insufficient insulin versus excess glucose (i.e., insulin resistance). The long-lasting insulin production and sustained contradictory signals from glucose and Aβ (promotive versus suppressive for insulin secretion) may cause the perturbation and exhaustion of islet β-cells, leading to pancreatic dysfunction in late-stage diabetes.
On the other hand, several cohort studies have shown results opposed to our hypothesis that brain Aβ increases in diabetes. In humans, diabetes was associated with cerebrovascular lesions, neurodegeneration, and cognitive dysfunction, but not brain amyloid burden, which was assessed by amyloid PET imaging with Pittsburgh Compound B (40–42). We previously reported that when APP23 mice are crossbred with diabetic ob/ob mice, cerebrovascular amyloid deposition is accelerated and cognitive dysfunction is exacerbated without an increase in brain Aβ levels (43). These findings suggest an alternative mechanism linking diabetes to AD, in which diabetes accelerates dementia onset via cerebrovascular damage. Interestingly, APP23 x ob/ob mice showed an up-regulation of RAGE in blood vessels before cerebrovascular amyloid deposition (43). This observation implies that diabetes promotes Aβ transport from the circulation into the brain. The flux of Aβ at the BBB may result in amyloid deposition at the vascular wall. A different pathway of brain Aβ flux has recently been proposed in which brain Aβ is discharged into the circulation by the glymphatic system (44). This pathway consists of the influx of CSF from the para-arterial space into the interstitial space of the brain, the flow of CSF and interstitial fluid (ISF) through the brain parenchyma toward the veins, and the efflux of ISF into the paravenous space reaching the cervical lymph nodes. The driving force of this system is arterial pulsation, and the flow is facilitated by perivascular aquaporin 4 water channels on astrocytic endfeet. Diabetes-induced amyloid deposition at the arterial wall may affect the arterial pulsation and thereby impair this system. Moreover, the transport across the BBB and the glymphatic system may be the major systems for brain waste clearance (45). Thus, the stagnation of Aβ efflux at the BBB or dysfunction of the glymphatic system would lead to Aβ accumulation in the brain. Although several reports have shown that brain amyloid burden does not increase in diabetes (40–43), soluble Aβ oligomers, an important molecule in AD pathogenesis, cannot be detected by amyloid PET or immunohistochemistry and ELISA with conventional Aβ antibodies. In addition, diabetes may influence brain Aβ levels differently depending on the disease status by enhancing Aβ deposition at the vasculature early and at the brain parenchyma later. Further studies are necessary to ascertain whether brain Aβ increases or not in diabetes.
Last, our findings call attention to using plasma Aβ levels as a diagnostic biomarker of AD. Our data suggest that plasma Aβ levels rapidly change upon food intake. Our previous report showed that the fasting levels of plasma Aβ40 and Aβ42 in AD patients were 110 pM and 6.9 pM, respectively, while the levels at 30 min after oral glucose loading were approximately 113 pM and 8.8 pM, respectively (15). The Aβ42/Aβ40 ratio was changed from 0.062 to 0.078 by glucose loading. On the other hand, in non-AD dementia patients, the Aβ42/Aβ40 ratio in fasting and glucose-loaded states were reported as 0.074 and 0.074, respectively (15). Thus, the Aβ42/Aβ40 ratio in AD patients becomes indistinguishable from that in non-AD dementia patients by glucose intake. In the present study, we used APP23 mice at 10 mo of age, at which time they scarcely display brain amyloid plaques, preventing us from comparing their plasma Aβ42/Aβ40 ratios with AD patients. A different study demonstrated that insulin infusion raised plasma Aβ42 levels in AD patients by 15% but lowered it in normal adults by 15% (16). Although the study did not mention plasma Aβ40 levels, the results suggest that the plasma Aβ42/Aβ40 ratio is inconclusive in hyperinsulinemia. In addition, another study has reported that insulin infusion significantly increased plasma Aβ42 levels but not Aβ40 levels in healthy adults (17), demonstrating that hyperinsulinemia affects the plasma Aβ42/Aβ40 ratio. Some metabolic syndromes, such as obesity, are also suggested to be associated with insulin resistance and glucose intolerance (46), in which the plasma Aβ42/Aβ40 ratio may be affected. Thus, when using plasma Aβ levels for the diagnosis of AD, blood should be taken in the fasting state. Furthermore, it has been shown that in sporadic inclusion-body myositis, the levels of BACE1, PS1, and sAPPβ in plasma are increased (47). Although that study did not describe plasma Aβ levels, it is likely that Aβ is increased in those patients. We previously showed that hypercholesterolemia induces cerebral Aβ production and subsequent Aβ efflux from the brain to the periphery along with excess brain cholesterol (48). These findings suggest the possibility that other systemic degenerative and metabolic disorders also affect plasma Aβ levels.
Materials and Methods
Mice.
C57BL/6 wild-type mice were purchased from Japan SLC, Inc. (Hamamatsu, Japan), APP23 mice were kindly provided by Novartis Pharma, Inc., and APP knockout mice were purchased from the Jackson Laboratory (Bar Harbor, ME). APP23 and APP knockout mice were mated with C57BL/6 mice and maintained as heterozygotes in our animal facility. Hetero-knockout mice were crossbred to produce homo-knockout mice when used. Thus, our APP23 and APP knockout mice have the same genetic background as C57BL/6 mice. All animal experiments were approved by the ethics committee of Osaka City University and performed in accordance with the Guide for Animal Experimentation, Osaka City University.
Glucose, Insulin, and Aβ Injection to Mice.
All mice were fasted for 16 h prior to glucose, insulin, or Aβ injection. Glucose and insulin doses were determined according to Vinué and González-Navarro (49), who recommended an intraperitoneal injection dose for mice of 2.0 g glucose/kg and 0.5 U insulin/kg in the glucose and insulin tolerance tests. Assuming a mouse body weight of 30 g, these injection doses correspond to 0.06 g glucose/head and 0.015 U insulin/head, respectively. Saline containing 0.06 g glucose was injected intraperitoneally into 10-mo-old wild-type mice (n = 6: 3 male and 3 female), 10-mo-old APP23 mice (n = 6: 3 male and 3 female), and 12- to 15-mo-old APP knockout mice (n = 6: 3 male and 3 female), and the blood was collected from the tail vein at 0, 15, 30, 45, 60, 120, and 180 min after the injection. Blood glucose levels were determined using a blood glucose meter, Glutest Ai (Sanwa Kagaku Kenkyusho, Nagoya, Japan), while plasma insulin, Aβ40, and Aβ42 levels were measured by ELISA using the Ultra Sensitive Mouse Insulin ELISA Kit (Morinaga Institute of Biological Science, Yokohama, Japan), Human/Rat β Amyloid ELISA Kit Wako II (measuring range: 1.0–100 pmol/L), and Human/Rat β Amyloid ELISA Kit Wako, High Sensitive (measuring range: 0.1–20 pmol/L) (Fujifilm-Wako, Osaka, Japan), respectively. No cross reactivities with amylin peptide (Peptide Institute, Ibaraki, Japan) were observed. In ELISA, sodium dodecyl sulfate (SDS) was added to the plasma samples at a concentration of 0.05% to dissociate possible complexes of Aβ and insulin with other molecules that may disturb the immune reaction. Then, the samples were properly diluted with buffers included in the ELISA kits. Saline containing 0.015 U (∼0.58 μg) mouse insulin I (Peptide Institute) was injected intraperitoneally into 11-mo-old wild-type mice (n = 8: 1 male and 7 female), and blood glucose and plasma Aβ40 and Aβ42 levels were measured at 0, 15, 30, 45, and 60 min after the injection.
The Aβ injection dose was determined based on our data. Fig. 1 indicates that the maximum plasma concentration of Aβ40 in APP23 mice was ∼400 pM. Assuming a mouse total blood volume of 2.4 mL (50), the blood contains at most 1 pmol Aβ40. We considered the possibility that Aβ injected into the abdominal cavity is adhered to, adsorbed, and degraded by the surrounding tissues, and only a portion enters the blood circulation. Thus, we used an Aβ40 injection dose of 6 pmol/head. Saline containing 6 pmol synthetic Aβ40 peptide (Peptide Institute) or saline alone was injected intraperitoneally into 12- to 15-mo-old APP knockout mice (n = 6: 3 male and 3 female). Five minutes after the injection, 0.06 g glucose was injected intraperitoneally into the mice. Blood glucose and plasma insulin and Aβ40 levels were measured at 0, 15, and 30 min after the glucose injection. The Aβ injection experiments were repeated with synthetic Aβ42 and scramble Aβ42 peptides. Saline containing 6 pmol synthetic Aβ42 peptide, 6 pmol scramble Aβ42 peptide (Peptide Institute), or saline alone was injected intraperitoneally into 11- to 12-mo-old APP knockout mice (n = 4: 2 male and 2 female for saline; n = 3: 1 male and 2 female for Aβ). Five minutes after the injection, 0.06 g glucose was injected intraperitoneally. Blood glucose and plasma insulin and Aβ42 levels were measured. The Aβ inoculums were examined for their aggregation state by Western blot analysis with an anti-Aβ antibody, 6E10 (Covance Research Products, Dedham, MA).
Aβ Secretion from Isolated Mouse Pancreas, Adipose, Skeletal Muscle, Liver, and Kidney.
After fasted for 16 h, 10-mo-old wild-type mice (n = 3: all male for pancreas, adipose, liver; n = 4: all female for muscle; and n = 3: 1 male and 2 female for kidneys) were anesthetized, and the blood was removed by cold PBS infusion. The pancreas, abdominal white adipose tissues, anterior tibial muscles, liver, and kidneys were collected and cut into small pieces with surgery scissors and further chopped into fine pieces with a razor edge on ice. The minced tissues were dispensed into tubes and washed once with cold Hanks’ balanced salt solution. After centrifugation at 100 × g, 4 °C for 1 min, the weight of the tissue pellets in each tube was measured. Then, 1 mL of high-glucose (4,500 mg/L) Dulbecco's modified Eagle's medium (DMEM) (Fujifilm-Wako), 200 nM insulin-containing no-glucose DMEM (Fujifilm-Wako), or no-glucose DMEM alone was added to the tubes, which were incubated at 37 °C for 60 min with rotation. After centrifugation at 100 × g, 4 °C for 1 min again, the supernatants were collected, and the levels of Aβ in the media were measured by ELISA using the Wako Aβ kits described above. We did not use collagenase or mesh filters for the cell preparation to avoid possible cell damage. The experiments were performed twice.
Aβ Effect on Insulin Secretion from Isolated Mouse Pancreas.
The pancreas was collected from 16-h fasted 10-mo-old wild-type mice (n = 3: 1 male and 2 female). The minced tissues were dispensed into tubes and stimulated with high-glucose medium with or without 100 pM Aβ40, Aβ42, or scramble Aβ42 peptide for 60 min. This Aβ dose was selected based on our data in Fig. 1. After centrifugation, the levels of insulin in the media were measured by ELISA using the LBIS Mouse Insulin ELISA Kit (Fujifilm-Wako-Shibayagi, Gunma, Japan).
Immunohistochemistry for Aβ in the Pancreas, Adipose, Skeletal Muscle, and Liver of Mice.
The antibodies used are listed in Table 2. The Aβ40-specific antibody, Ter40, was generated in our laboratory against the C terminus of Aβ40 (51), while other antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). First, 10-mo-old freely fed wild-type mice (n = 3: 2 male and 1 female) were anesthetized and fixed by 4% paraformaldehyde perfusion. The pancreas was removed, and 5-μm tissue sections were prepared. To expose the antigens, sections were boiled in 10 mM citrate buffer, pH6 for 30 min. After blocking with 20% calf serum for 60 min, the sections were stained with antibodies for Aβ, insulin, glucagon, and somatostatin at 4 °C overnight and then with FITC- or rhodamine-labeled second antibodies (Jackson Laboratory) at room temperature for 60 min. The stained sections were mounted with Vectashield with DAPI (Vector Laboratories, Burlingame, CA) and viewed under a BZ-X800 fluorescence microscope (Keyence, Osaka, Japan).
Table 2.
Antibodies used in the immunohistochemistry
| Target | Antibody | Species | Supplier | Working dilution |
|---|---|---|---|---|
| Aβ40 | Ter40 | Rabbit | Our laboratory | 500× |
| Insulin | 2D11-H5 | Mouse | Santa Cruz | 0.4 μg/mL |
| Somatostatin | H-11 | Mouse | Santa Cruz | 0.4 μg/mL |
| Glucagon | C-11 | Mouse | Santa Cruz | 0.4 μg/mL |
| Acrp30 | 31 | Mouse | Santa Cruz | 0.4 μg/mL |
| IL-6 | 10E5 | Mouse | Santa Cruz | 0.2 μg/mL |
| IGF1 | H-9 | Mouse | Santa Cruz | 0.4 μg/mL |
Next, 10-mo-old wild-type mice (n = 3: 1 male and 2 female) were fasted for 16 h and divided into three groups: one for intraperitoneal glucose injection, one for intraperitoneal insulin injection, and the third for nontreatment. Fifteen minutes after the glucose injection, the mice were fixed, and the pancreas tissue sections were prepared. After pretreatment in pH6 buffer, the sections were stained for Aβ and insulin. Thirty minutes after the insulin injection, the mice were fixed, and the tissue sections were prepared from abdominal white adipose tissues, anterior tibial muscles, and the liver. After pretreatment in pH6 buffer, the sections were stained for Aβ and each organokine: adipokine Acrp30 for adipocytes, myokine IL-6 for myocytes, and hepatokine IGF1 for hepatocytes. Images were compared between nontreated and glucose- or insulin-injected groups. The experiments were performed 5 times, and staining was done with one section per animal.
Cells.
The mouse pancreatic β-cell line β-TC-6, mouse embryonic fibroblast line 3T3-L1, mouse myoblast line C2C12, and mouse hepatocyte line FL83B were obtained from the ATCC/American Type Culture Collection. β-TC-6, 3T3-L1, and C2C12 cells were cultured in low-glucose DMEM (Fujifilm-Wako) supplemented with 15% fetal bovine serum (FBS), 10% bovine serum, and 10% FBS, respectively, while FL83B cells were maintained in Kaighn’s Modification of Ham’s F-12 medium containing 10% FBS. For the experiments, 3T3-L1 cells were allowed to differentiate into mature adipocytes by stimulating them with insulin, dexamethasone, and 3-isobutyl-1-methylxanthine using AdipoInducer reagent (Takara Bio, Kusatsu, Japan) essentially as described previously (52). C2C12 cells were differentiated into mature myocytes by reducing FBS from 10 to 1% essentially as described previously (53). The maturation of these cells was confirmed by the formation of intracellular lipid drops for adipocytes and of myotubes for myocytes.
Aβ Secretion from Cultured β-Cells, Adipocytes, Myocytes, and Hepatocytes.
Prior to the glucose and insulin stimulation, β-TC-6 cells, differentiated 3T3-L1 and C2C12 cells, and FL83B cells were cultured in serum-free no-glucose DMEM for 120 min. β-TC-6 cells were then stimulated with serum-free high-glucose DMEM in the presence or absence of 10 nM GLP-1 (Peptide Institute) for 60 min. Culture media were collected, and the concentrations of Aβ40, Aβ42, and insulin were measured by ELISA using the Wako Aβ kits and LBIS Insulin kit, respectively. After washing with PBS, the cells were harvested, and the protein concentrations in cell lysates were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Meanwhile, the 3T3-L1, C2C12, and FL83B cells were stimulated with 200 nM insulin-containing serum-free no-glucose or high-glucose DMEM for 60 min. The concentrations of Aβ40 and Aβ42 in the culture media and the protein concentrations in the cell lysates were measured.
Aβ Effect on Insulin Secretion from Cultured β-Cells.
β-TC-6 cells were incubated in serum-free no-glucose DMEM for 120 min. The cells were then stimulated with serum-free high-glucose DMEM in the presence or absence of 50 pM Aβ40. After a 60-min incubation, insulin concentrations in the culture media and protein concentrations in the cell lysates were measured by ELISA and the BCA protein assay, respectively.
To evaluate the influence of endogenous Aβ, β-TC-6 cells were incubated in serum-free no-glucose DMEM with or without 1 μM of the γ-secretase inhibitor L685,458 (Bachem, Bubendorf, Switzerland) for 120 min. The cells were then stimulated with serum-free high-glucose DMEM in the presence or absence of 1 μM L685,458. After a 60-min incubation, insulin concentrations in the culture media and protein concentrations in the cell lysates were measured.
Aβ Effect on GLP-1 Secretion from Cultured Enteroendocrine L Cells.
Human colon cancer NCI-H716 cells were obtained from ATCC and cultured in RPMI-1640 medium supplemented with 10% FBS. To study GLP-1 secretion, the cells were cultured on a BD Matrigel Basement Membrane Matrix (BD Biosciences, Billerica, MA), which promotes cell attachment and differentiation (54). After washing with no-glucose 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Hepes) buffer (20 mM Hepes, 146 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 1 mM MgSO4, pH 7.4), the cells were stimulated with high-glucose Hepes buffer in the presence or absence of 100 pM Aβ40 or Aβ42. After a 120-min incubation, the culture media were collected, and the GLP-1 concentrations were measured using the LBIS GLP-1 (Active) ELISA kit (Fujifilm-Wako-Shibayagi). After washing with PBS, the cells were harvested and counted.
Aβ Effect on Glucose Uptake by Cultured Adipocytes.
Glucose uptake experiments were performed essentially as described previously (52). To study the glucose uptake, differentiated 3T3-L1 cells were starved in serum-free DMEM for 5 h. After washing with Krebs-Ringer Hepes buffer (20 mM Hepes, 5 mM KH2PO4, 136 mM NaCl, 4.7 mM KCl, 1 mM CaCl2, 1 mM MgSO4, pH 7.4), the cells were stimulated with 1 or 10 nM insulin-containing buffer in the presence or absence of 100 pM Aβ40 or Aβ42 for 20 min. Then, unlabeled 2-deoxy-D-glucose (2-DG) (Sigma-Aldrich) and 3H-labeled 2-DG (PerkinElmer, Waltham, MA) were added to the medium at a final concentration of 1 mM and 1 μCi/mL, respectively. Ten minutes after the incubation, the cells were washed with cold PBS and lysed in 50 mM NaOH. The amount of internalized 3H-labeled 2-DG in the cell lysates was measured using a liquid scintillation counter (PerkinElmer). Protein concentrations were also measured by a BCA protein assay.
Statistical Analysis.
All experiments and data analyses were performed under unblinded conditions. Comparisons of means among more than two groups were performed using ANOVA (for single time point measurements and AUC) or two-factor repeated measures ANOVA (for multiple time point measurements), followed by Fisher’s PLSD test. Differences with a P value of <0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Tsuneo Nakajima, Yuki Ito, and Hikari Takeshita for technical advice and Peter Karagiannis for reading the manuscript. This study was supported by a grant from the Research Foundation for Dementia of Osaka, Osaka, Japan.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission. S.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2117723119/-/DCSupplemental.
Data Availability
All study data are included in the article and/or supporting information.
References
- 1.Rojas M., et al. , Alzheimer’s disease and type 2 diabetes mellitus: Pathophysiologic and pharmacotherapeutics links. World J. Diabetes 12, 745–766 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei Z., Koya J., Reznik S. E., Insulin resistance exacerbates Alzheimer disease via multiple mechanisms. Front. Neurosci. 15, 687157 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hampel H., et al. , The amyloid-beta pathway in Alzheimer's disease. Mol. Psychiatry. 26, 5481–5503 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Y., Wang Q., Chen S., Xu C., Functions of amyloid precursor protein in metabolic diseases. Metabolism 115, 154454 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Decourt B., Sabbagh M. N., BACE1 as a potential biomarker for Alzheimer's disease. J. Alzheimers Dis. 24 (suppl. 2), 53–59 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vetrivel K. S., Zhang Y. W., Xu H., Thinakaran G., Pathological and physiological functions of presenilins. Mol. Neurodegener. 1, 4 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tharp W. G., et al. , Effects of glucose and insulin on secretion of amyloid-β by human adipose tissue cells. Obesity (Silver Spring) 24, 1471–1479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vattemi G., et al. , BACE1 and BACE2 in pathologic and normal human muscle. Exp. Neurol. 179, 150–158 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Sakuma K., Nakao R., Yamasa Y., Yasuhara M., Normal distribution of presenilin-1 and nicastrin in skeletal muscle and the differential responses of these proteins after denervation. Biochim. Biophys. Acta 1760, 980–987 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Figueroa D. J., Shi X. P., Gardell S. J., Austin C. P., Abetapp secretases are co-expressed with Abetapp in the pancreatic islets. J. Alzheimers Dis. 3, 393–396 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Meakin P. J., et al. , The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat. Commun. 9, 1306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Y., et al. , Modulation of the gamma-secretase activity as a therapy against human hepatocellular carcinoma. J. Cancer Res. Ther. 14 (suppl.), S473–S479 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Sagare A. P., Winkler E. A., Bell R. D., Deane R., Zlokovic B. V., From the liver to the blood-brain barrier: An interconnected system regulating brain amyloid-β levels. J. Neurosci. Res. 89, 967–968 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Takeda S., et al. , Elevation of plasma beta-amyloid level by glucose loading in Alzheimer mouse models. Biochem. Biophys. Res. Commun. 385, 193–197 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Takeda S., et al. , Oral glucose loading modulates plasma β-amyloid level in Alzheimer’s disease patients: Potential diagnostic method for Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 34, 25–30 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Kulstad J. J., et al. , Differential modulation of plasma beta-amyloid by insulin in patients with Alzheimer disease. Neurology 66, 1506–1510 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Karczewska-Kupczewska M., et al. , The influence of insulin infusion on the metabolism of amyloid β peptides in plasma. Alzheimers Dement. 9, 400–405 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Sturchler-Pierrat C., et al. , Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng H., et al. , Beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Parrettini S., Cavallo M., Gaggia F., Calafiore R., Luca G., Adipokines: A rainbow of proteins with metabolic and endocrine functions. Protein Pept. Lett. 27, 1204–1230 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Chen W., Wang L., You W., Shan T., Myokines mediate the cross talk between skeletal muscle and other organs. J. Cell. Physiol. 236, 2393–2412 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Jensen-Cody S. O., Potthoff M. J., Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 44, 101138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nauck M. A., Meier J. J., Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 20 (suppl. 1), 5–21 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Townsend M., Mehta T., Selkoe D. J., Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 282, 33305–33312 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Zhao W. Q., et al. , Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 22, 246–260 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Klip A., McGraw T. E., James D. E., Thirty sweet years of GLUT4. J. Biol. Chem. 294, 11369–11381 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Oliveira Dos Santos A. R., et al. , Adipokines, myokines, and hepatokines: Crosstalk and metabolic repercussions. Int. J. Mol. Sci. 22, 2639 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu Z., et al. , Integrative analysis of a cross-loci regulation network identifies App as a gene regulating insulin secretion from pancreatic islets. PLoS Genet. 8, e1003107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deane R., Zlokovic B. V., Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 4, 191–197 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Blázquez E., Velázquez E., Hurtado-Carneiro V., Ruiz-Albusac J. M., Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer's disease. Front. Endocrinol. (Lausanne) 5, 161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sousa L., Guarda M., Meneses M. J., Macedo M. P., Vicente Miranda H., Insulin-degrading enzyme: An ally against metabolic and neurodegenerative diseases.J. Pathol. 255, 346–361 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Tomiyama T., et al. , A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 30, 4845–4856 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S., Selkoe D. J., A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 154, 583–597 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bitel C. L., Kasinathan C., Kaswala R. H., Klein W. L., Frederikse P. H., Amyloid-β and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J. Alzheimers Dis. 32, 291–305 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Okabayashi S., Shimozawa N., Yasutomi Y., Yanagisawa K., Kimura N., Diabetes mellitus accelerates Aβ pathology in brain accompanied by enhanced GAβ generation in nonhuman primates. PLoS One 10, e0117362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S., et al. , Chronic diabetic states worsen Alzheimer neuropathology and cognitive deficits accompanying disruption of calcium signaling in leptin-deficient APP/PS1 mice. Oncotarget 8, 43617–43634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh S. H., et al. , A high-sucrose diet aggravates Alzheimer’s disease pathology, attenuates hypothalamic leptin signaling, and impairs food-anticipatory activity in APPswe/PS1dE9 mice. Neurobiol. Aging 90, 60–74 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Imamura T., et al. , Insulin deficiency promotes formation of toxic amyloid-β42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model. Neurobiol. Dis. 137, 104739 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Matsuzaki T., et al. , Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 75, 764–770 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Thambisetty M., et al. , Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 70, 1167–1172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts R. O., et al. , Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J. Nucl. Med. 55, 759–764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frison E., et al. ; MEMENTO Cohort Study Group, Diabetes mellitus and cognition: Pathway analysis in the MEMENTO cohort. Neurology 97, e836–e848 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeda S., et al. , Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. U.S.A. 107, 7036–7041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rasmussen M. K., Mestre H., Nedergaard M., The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarasoff-Conway J. M., et al. , Clearance systems in the brain—Implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neeland I. J., Poirier P., Després J. P., Cardiovascular and metabolic heterogeneity of obesity: Clinical challenges and implications for management. Circulation 137, 1391–1406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Catalán-García M., et al. , BACE-1, PS-1 and sAPPβ levels are increased in plasma from sporadic inclusion body myositis patients: Surrogate biomarkers among inflammatory myopathies. Mol. Med. 21, 817–823 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Umeda T., Mori H., Zheng H., Tomiyama T., Regulation of cholesterol efflux by amyloid beta secretion. J. Neurosci. Res. 88, 1985–1994 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Vinué Á., González-Navarro H., Glucose and insulin tolerance tests in the mouse. Methods Mol. Biol. 1339, 247–254 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Harkness J. E., Turner P. V., VandeWoude S., Wheler C. L., Harkness and Wagner's Biology and Medicine of Rabbits and Rodents (Wiley-Blackwell, ed. 5, 2010). [Google Scholar]
- 51.Nishitsuji K., et al. , Cerebral vascular accumulation of Dutch-type Abeta42, but not wild-type Abeta42, in hereditary cerebral hemorrhage with amyloidosis, Dutch type. J. Neurosci. Res. 85, 2917–2923 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ishibashi K., Nehashi K., Oshima T., Ohkura N., Atsumi G., Differentiation with elaidate tends to impair insulin-dependent glucose uptake and GLUT4 translocation in 3T3-L1 adipocytes. Int. J. Food Sci. Nutr. 67, 99–110 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Lesmana R., et al. , Short communication: Optimazing culture and differentiation L6 cell, C2C12 cell and primary myoblast cells culture. Cell Biol. Dev. 2, 51–54 (2018). [Google Scholar]
- 54.de Bruïne A. P., et al. , Extracellular matrix components induce endocrine differentiation in vitro in NCI-H716 cells. Am. J. Pathol. 142, 773–782 (1993). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or supporting information.