

Abstract
For over 100 years, the fruit fly Drosophila melanogaster has been one of the most studied model organisms. Here we present a single cell atlas of the adult fly, Tabula Drosophilae, that includes 580k nuclei from 15 individually dissected sexed tissues as well as the entire head and body, annotated to >250 distinct cell types. We provide an in-depth analysis of cell type-related gene signatures and transcription factor markers, as well as sexual dimorphism, across the whole animal. Analysis of common cell types between tissues, such as blood and muscle cells, reveals rare cell types and tissue-specific subtypes. This atlas provides a valuable resource for the entire Drosophila community and serves as a reference to study genetic perturbations and disease models at single-cell resolution.
One Sentence Summary:
A single-nucleus transcriptomic map of the entire adult Drosophila melanogaster
Drosophila melanogaster has a fruitful history in biological research, dating back to experiments of Thomas Hunt Morgan a century ago (1) and has been at the basis of many key biological discoveries. The highly collaborative nature of the Drosophila community contributed to many of these successes, and led to the development of essential research resources, including a high-quality genome (2), a large collection of genetic and molecular tools, and important databases such as Flybase (3), FlyMine (4), FlyLight (5), VirtualFlyBrain (6) and ModERN (7). The fly genome contains about 17,000 genes, including 13,968 protein-coding genes of which ~63% have human orthologues. Studies such as ModENCODE (8) and FlyAtlas (9) explored expression patterns in different tissues, but lacked cell type resolution. Recent advances in single-cell technologies have enabled the transcriptomic profiling of thousands of cells at once, facilitating the creation of tissue-wide atlases. Several studies have already applied single-cell RNA sequencing (scRNA-seq) to multiple Drosophila tissues and developmental stages (10). However, these data were generated by different laboratories on different genetic backgrounds, with different dissociation protocols and sequencing platforms, hindering systematic comparison of gene expression across cells and tissues.
Here, we present a single cell transcriptomic atlas of the entire adult Drosophila, separately analyzing male vs female samples, using a uniform genotype and a unified single-nucleus RNA-seq (snRNA-seq) platform (11) with two sequencing strategies: droplet-based 10x Genomics (12) and plate-based Smart-seq2 (13). The resulting Tabula Drosophilae, the first dataset within the Fly Cell Atlas consortium (FCA), contains over 580k cells, resulting in >250 distinct cell types annotated by >100 experts from 40 laboratories. This atlas reports cellular signatures for each tissue, providing the entire Drosophila community a reference for studies that probe the effects of genetic perturbations and disease models at single-cell resolution. All data and annotations can be accessed through multiple visualization and analysis portals from https://flycellatlas.org (fig. S1–S3).
Sampling single cells across the entire adult fly
We used a unified snRNA-seq platform for all samples, because it is difficult to isolate intact cells from many adult Drosophila tissues, especially cuticular ones (e.g., antenna, wing) and adipocyte-enriched ones (e.g., fat body). In addition, snRNA-seq can be applied to large multinucleated cells (e.g., muscle) and facilitates (frozen) tissue collection from different laboratories. Finally, 70–90% of transcriptomic information is preserved from snRNA-seq compared to scRNA-seq of the same fly cell types (11).
To achieve a comprehensive sampling, we used two complementary strategies. First, we dissected 12 individual tissues from both males and females, plus 3 sex-specific tissues (Fig. 1A). For tissues that are localized across the body (fat body, oenocytes, and trachea) and cannot be directly dissected, we used specific GAL4 lines driving nuclear-GFP to label and collect nuclei using FACS. In addition, two rare cell types were sequenced only with Smart-seq2: insulin-producing cells (IPCs) and corpora cardiaca cells (CCs). Second, we sorted and profiled nuclei from the entire head and body, aiming to detect cell types not covered by the selected tissues. In total, we obtained 580k high-quality nuclei: 570k from 10x Genomics and 10k from Smart-seq2 (Fig. 1A).
Figure 1. Overview of the Fly Cell Atlas.
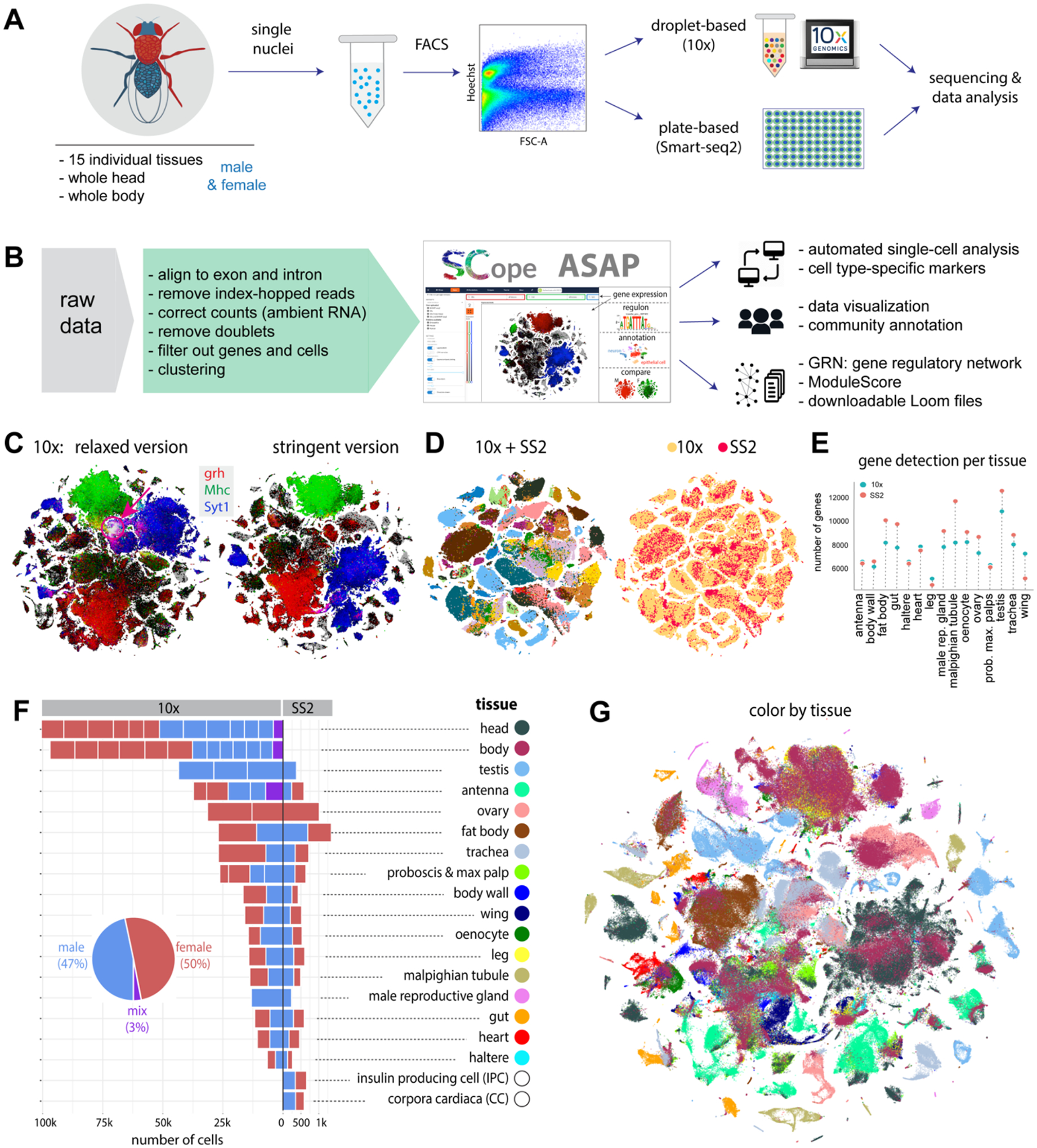
(A) Experimental platform of snRNA-seq using 10x Genomics and Smart-seq2 (SS2).
(B) Data analysis pipeline and data visualization using SCope (17) and ASAP (18).
(C) Two versions of 10x datasets: Relaxed and Stringent. tSNE colors based on gene expression: grh (epithelia, red), Mhc (muscle, green) and Syt1 (neuron, blue). Red arrow denotes an artefactual cluster with co-expression of all three markers in the Relaxed dataset.
(D) tSNE visualization of cells from the Stringent 10x dataset and Smart-seq2 (SS2) cells. 10x cells are from individual tissues. Integrated data is colored by tissue (left) and platform (right).
(E) Tissue-level comparison of the number of detected genes between 10x and Smart-seq2 platforms.
(F) Number of cells for each tissue by 10x and Smart-seq2. Male and female cells are indicated. Mixed cells are from pilot experiments where flies were not sexed. Different batches are separated by vertical white lines.
(G) All 10x cells from the Stringent dataset clustered together; cells are colored by tissue type. Tissue names and colors are indexed in F.
To analyze the 10x Genomics data in a reproducible manner, we used the automated VSN pipeline (14) (Methods, Table S1), which takes the raw sequencing data as input and performs preprocessing (e.g., normalization, doublet removal, batch effect correction) to produce LoomX formatted files with expression data, embeddings and clusterings (Fig. 1B and fig. S4). A presumed artifactual cluster showed expression of nearly all genes, so we added an additional preprocessing step that models and subtracts ambient RNA signals (15) to remove this cluster, resulting in a Stringent dataset of 510k cells (see Methods and Fig. 1C). However, since adjusting the gene expression values per cell can introduce other biases (e.g., overcorrection, removal of non-doublet cells), we also retained the original Relaxed dataset of 570k cells. In the analyses below, unless mentioned otherwise (e.g., Fig. 2C), the Stringent dataset was used.
Figure 2: Cell type annotation for dissected tissues.
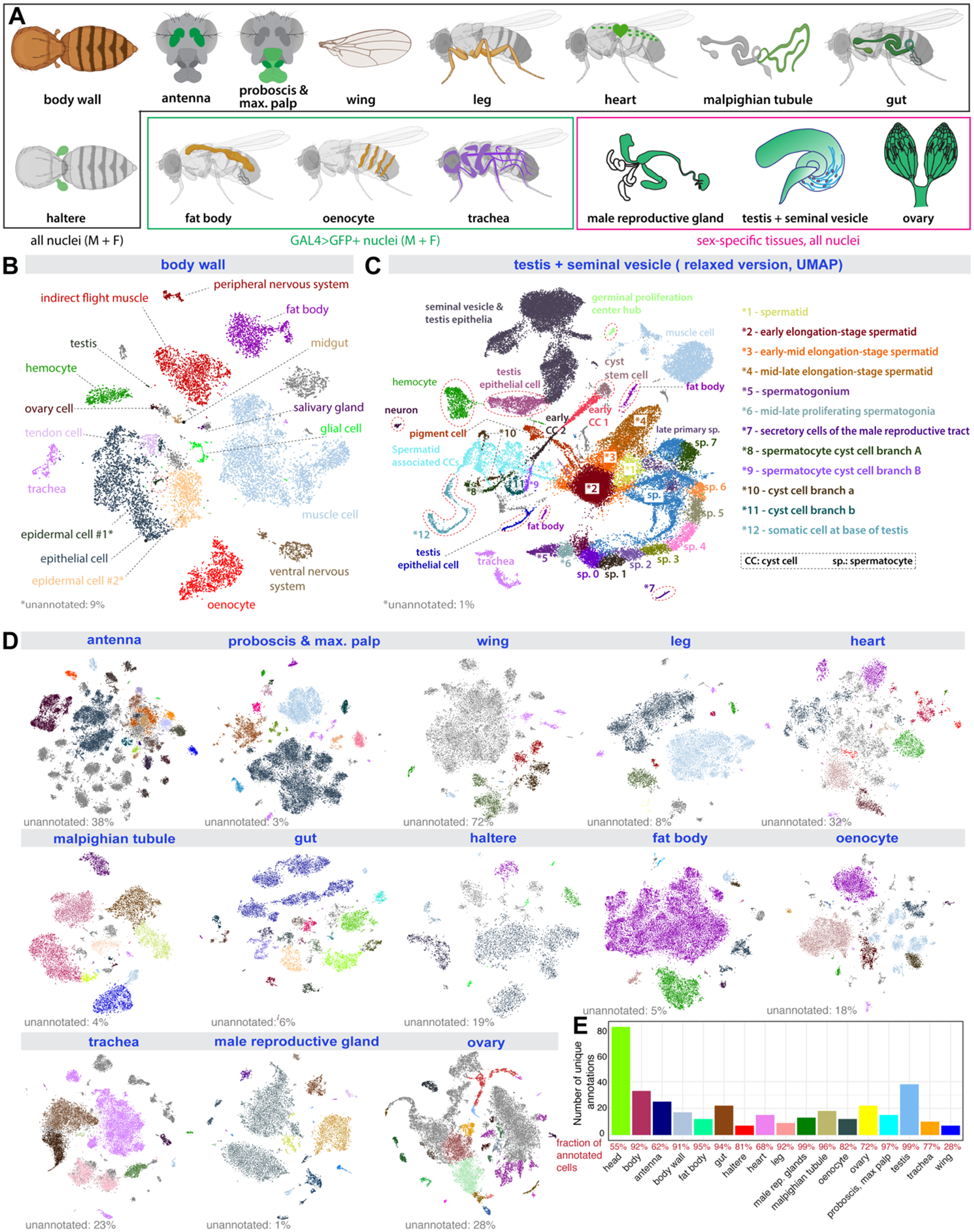
(A) Illustration of 15 individual tissues. 12 sequenced separately from males and females, 3 sex-specific. Fat body, oenocyte, and tracheal nuclei were labeled using a tissue-specific GAL4 driving UAS-nuclearGFP.
(B) tSNE plot with annotations for body wall from the Stringent 10x dataset. *1, epidermal cells of the abdominal posterior compartment. *2, epidermal cells specialized in antimicrobial response.
(C) UMAP plot with annotations for the testis from the Relaxed 10x dataset.
(D) tSNE plots of the other 13 tissues from the Stringent 10x dataset. Detailed annotations are in fig. S6–S18.
(E) Number of unique annotations for each tissue. Fractions of annotated cells over all analyzed cells from the Relaxed dataset are indicated in red.
Cells from 10x Genomics and Smart-seq2 were well integrated after batch correction using Harmony (16) (Fig. 1D). Smart-seq2 yielded a higher number of detected genes for most tissues (Fig. 1E) as cells were sequenced to a higher depth. We analyzed each tissue separately, combining the male and female runs, which yielded between 6.5k (haltere) and 100k (head) cells and a median of 16.5k cells per tissue for 10x and between 263 (male reproductive gland) and 1,349 (fat body) cells and a median of 534 cells per tissue for Smart-seq2 (Fig. 1F). We obtained similar numbers of male and female cells for non-sex-specific tissues with on average 1895 unique molecular identifiers (UMIs) and 828 genes per cell (fig. S5). Next, all cells were combined in a meta-analysis, showing tissue-specific clusters like the germline cells of the testis and ovary, and shared clusters of common cell types (Fig. 1G; see fig. S24, 25).
Crowd-based cell type annotation by tissue experts
Experts from 40 laboratories collaborated on cell type annotation for 15 individual tissues, including 12 tissues for both sexes: antenna, body wall, fat body, haltere, heart, gut, leg, Malpighian tubule, oenocyte, proboscis with maxillary palp, trachea, and wing; and 3 sex-specific tissues: male reproductive gland, testis, and ovary (Fig. 2A). We developed a consensus-voting strategy within the SCope web application (https://flycellatlas.org/scope) (17), where curators annotated clusters at multiple resolutions (ranging from 0.8 to 8, fig. S6A), with additional analysis performed in ASAP (https://flycellatlas.org/asap) (18). To ensure that cell type annotations are consistent with previous literature and databases and to allow a posteriori computational analyses at different anatomical resolutions, we used Flybase anatomy ontology terms (19).
Since some cell types are annotated at low, and others at high resolutions, we collapsed all annotations across resolutions and retained the annotation with the highest number of up-votes. All initial annotations were performed on the Relaxed dataset, and were then exported to the Stringent dataset, where field experts verified the accuracy of the annotation transfer (Fig. 2A–E and fig. S6–S18). Overall, we annotated 251 cell types in the Stringent dataset (262 cell types if combining Relaxed and Stringent datasets, Table S2), with a median of 15 cell types per tissue.
Our dataset provides a single-cell transcriptomic profiling for several adult tissues not profiled previously, including the haltere, heart, leg, Malpighian tubule, proboscis, maxillary palp, trachea, and wing (fig. S6–S18). In these tissues, all major expected cell types were identified. In the proboscis and maxillary palp (fig. S7A, B), we could annotate gustatory and olfactory receptor neurons, mechanosensory neurons, and several glial clusters. All 7 olfactory receptors expressed in the maxillary palp were detected. In the wing (fig. S8), we could identify four different neuronal types – gustatory receptor neurons, pheromone-sensing neurons, nociceptive neurons, mechanosensory neurons, as well as three glial clusters. In the leg (fig. S9), we could distinguish gustatory receptor neurons from two clusters of mechanosensory neurons. In the heart (fig. S10), we found a large proportion of resident hemocytes and muscle cells, with the cardiac cells marked by the genes Hand and tinman constituting a small proportion. In the Malpighian tubule (fig. S11), 15 cell types were identified, including the different principal cells of the stellate and main segments. In the haltere (fig. S13), we identified two clusters of neurons, three clusters of glial cells, and a large population of epithelial cells. In some tissues, cell types formed a big cluster instead of being split into distinct populations. In these cases, we identified genes or pathways that showed a gradient or compartmentalized expression. For example, in the fat body (fig. S14 and S19), the main fat body cells formed one big cluster, but our metabolic pathway enrichment analysis performed through ASAP (18) revealed that fatty acid biosynthesis and degradation are in fact compartmentalized, highlighting possible fat body cell heterogeneity in metabolic capacities.
Our crowd annotations with tissue experts also revealed cell types that had not been profiled previously, such as multinucleated muscle cells (Fig. 2B) and two distinct types of nuclei among the main cells in the male accessory gland (fig. S17), a cell type that was previously thought to be uniform. The high number of nuclei analyzed allowed identification of rare cell types. For example, in the testis (Fig. 2C), we identified 25 unique cell types, covering all expected cell types, including very rare cells, such as germinal proliferation center hub cells (79 nuclei in the Relaxed version, out of 44,621 total testis nuclei).
Next, we compared the distribution of cells between 10x and Smart-seq2, finding a good match based on a co-clustering analysis (fig. S20 and S21). Since Smart-seq2 cells only account for a small fraction, our previous annotations focused on 10x cells. The cell-matched co-clustering analysis allowed us to transfer annotations from 10x to Smart-seq2 datasets (fig. S20E), using cluster-specific markers as validation (fig. S20F). We also identified genes that were specifically detected using Smart-seq2 thanks to its higher gene detection rate (fig. S20G and Fig. 1E). In summary, the high-throughput 10x datasets form the basis for identifying cell types while the Smart-seq2 datasets facilitate the detection of lowly expressed genes and enable future exploration of cell-specific isoform information.
Correspondence between dissected tissues and whole head and body
To generate a complete atlas of the fly, we next performed snRNA-seq experiments on whole-head and whole-body samples. Whole-body single-cell experiments were previously performed on less complex animals (20, 21). Full head and body sequencing provides a practical means to assess the impact of mutations or to track disease mechanisms, without having to focus on specific tissues. In addition, it could yield cell types that are not covered by any of the targeted tissue dissections.
In the head, we annotated 81 mostly neuronal cell types (Fig. 3A and S22). In the body, we annotated the top 33 most abundant cell classes, including epithelia, muscle, and ventral nerve cord and peripheral neurons, followed by fat cells, oenocytes, germ line cells, glia, and tracheal cells (Fig. 3B and S23). Many of these cell classes can be further divided into cell types for further annotation (see Fig. 2 and fig. S6–S18).
Figure 3: Whole-head and whole-body sequencing leads to full coverage of the entire fly.
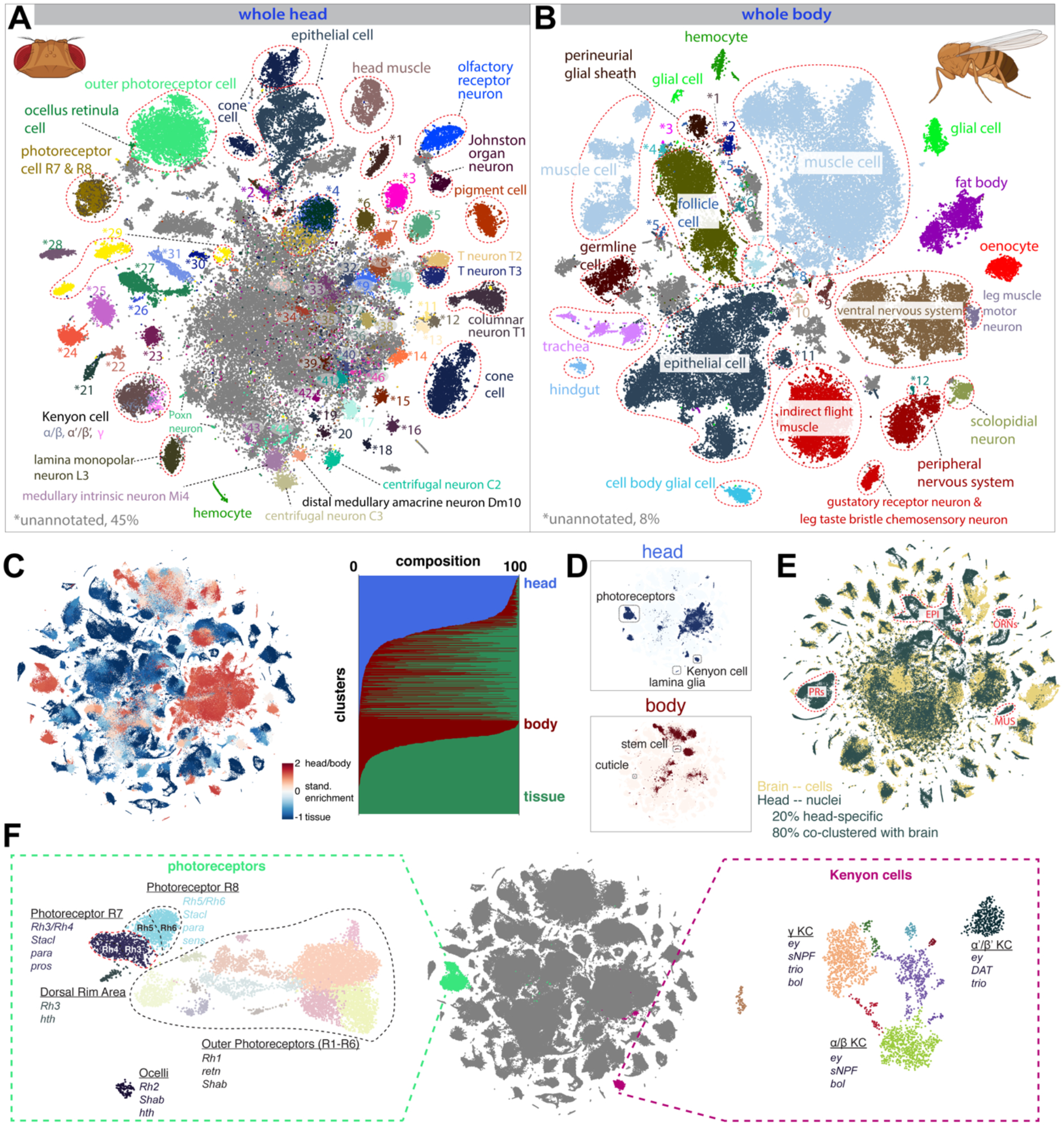
(A) tSNE of the whole-head sample with 81 annotated clusters. See fig. S22 for full cell types. Many cells in the middle (gray) are unannotated, most of which are central brain neurons.
(B) tSNE of the whole-body sample with 33 annotated clusters, many of which can be further divided into sub-clusters. Cells in gray are unannotated. See fig. S23 for full cell types.
(C) (left) tSNE of the entire dataset colored by standardized tissue enrichment, leading to the identification of head- and body-specific clusters. (right) Bar plots showing tissue composition (head, body, or dissected tissues) for different clusters at Leiden resolution 50.
(D) Examples of head- and body-specific clusters.
(E) Integration of a brain scRNA-seq dataset with the head snRNA-seq for label transfer. Outlined are example clusters revealed by the head snRNA-seq dataset but not by the brain scRNA-seq datasets, including epithelial cells (EPI), photoreceptors (PRs), olfactory receptor neurons (ORNs), and muscle cells (MUS).
(F) Subclustering analysis reveals types of photoreceptors, including inner and outer photoreceptors, with the inner photoreceptors further splitting into R7 and R8 types, and mushroom body Kenyon cells comprising three distinct types: α/β, α’/β’ and γ.
Next, we examined how well the head and body samples covered the cell types from the dissected tissues. We analyzed head, body, and tissue samples together, with most of the selected tissues clustering together with the body. We also detected head and body enriched clusters (Fig. 3C). One body-specific cluster contained cuticle cells, likely from connective tissue (Fig. 3D). Others were relatively rare cell types in their respective tissues, such as adult stem cells. Conversely, most tissue clusters contained body cells, with only a small number being completely specific to dissected tissues. As tissue-specific clusters were mostly observed in tissues with high cell coverage, such as the testis and Malpighian tubule, we anticipate that these clusters would also be identified in the body upon sampling a larger number of cells.
For the head, antenna and proboscis with maxillary palp were dissected for tissue sequencing. Cell types from those two tissues largely overlapped with head cells. Many other cell types, such as central brain cells, including Kenyon cells (ey, prt) and lamina glia (repo, Optix), were only detected in the head sample.
To compare our data with existing datasets, we integrated our head snRNA-seq dataset (“head” hereafter) with published brain single-cell RNA-seq data (“brain” hereafter) (17, 22–24) (Fig. 3E). Head unique clusters made up 20% of the cells, including the antennae, photoreceptors, muscle, cone cells and cuticular cell types, whereas the other 80% were present in clusters containing both head- and brain-derived cells covering the neuronal and glial cell types of the brain. This co-clustering across genotypes and protocols underscores the quality and utility of our snRNA-seq data compared to scRNA-seq data. Next, we used machine learning models to predict annotations per cluster, followed by manual curation (22). Given the high number of neuron types, additional subclustering was performed on each cluster, identifying subtypes of peptidergic neurons (dimm, Pdf) and olfactory projection neurons based on oaz, c15, and kn. Finally, we identified many cell types in the optic lobe, including lamina (e.g. L1–L5), medulla (e.g. Mi1, Mi15), lobula (e.g. LC), and lobula plate (e.g. LPLC). Using acj6 and SoxN, we identified the T4/T5 neurons of the optic lobe that split in T4/T5a-b and T4/T5c-d subtypes by subclustering. A big clump of neurons remained unannotated (Fig. 3A), indicating that our dataset cannot resolve the complexity of the central brain, which may contain hundreds to thousands of neuron types.
Subclustering in the combined dataset separated inner and outer photoreceptors from dorsal rim area and ocellar photoreceptors, with the inner photoreceptors further splitting into R7 and R8 types, each with pale and yellow types based on rhodopsin expression (Fig. 3F). Additionally, Kenyon cells were split into three types: α/β, α’/β’ and γ (17). These cases highlight the resolution in our dataset and the potential of using subclustering to discover rare cell types.
Cross-tissue analyses allow comparison of cell types by location
Using the whole body and head sequencing data, we assigned cells to major cell classes (e.g., epithelial cells, neurons, muscle cells, hemocytes), allowing us to compare common classes across tissues (Fig. 4A–C and fig. S24, S25). First, we compared blood cells across tissues by selecting all Hml-positive cells, a known marker for hemocytes (Fig. 4D). Combining hemocytes across tissues revealed a major group of plasmatocytes, the most common hemocyte type (~56%), crystal cells (1.5%, PPO1, PPO2), and several unknown types (fig. S26A, B). Looking deeper into the plasmatocytes, we uncovered gradients based on the expression of Pxn, LysX, Tep4, trol and Nplp2 that can be linked to maturation and plasticity with Pxn positive cells showing the highest Hml expression, while Tep4, trol and Nplp2 are prohemocyte markers (25). Furthermore, different antimicrobial peptide (AMP) families such as the Attacins and Cecropins were expressed in different subgroups indicating specialization. Finally, expression of acetylcholine receptors was specific for a subset of hemocytes, relating to the cholinergic anti-inflammatory pathway as described in humans and mice (26). Lamellocytes were not observed in adults as previously suggested (27). On the contrary, an unknown hemocyte type expressed Antp and kn (43 cells, 0.5%) reminiscent of the posterior signaling center in the lymph gland, an organization center previously thought to be absent in the adult (28, 29) (fig. S26B). These findings highlight the value of performing a whole organism-level single cell analysis and constitute a foundation to investigate the fly immune system in greater detail.
Figure 4: Cross-tissue analyses of common cell classes.
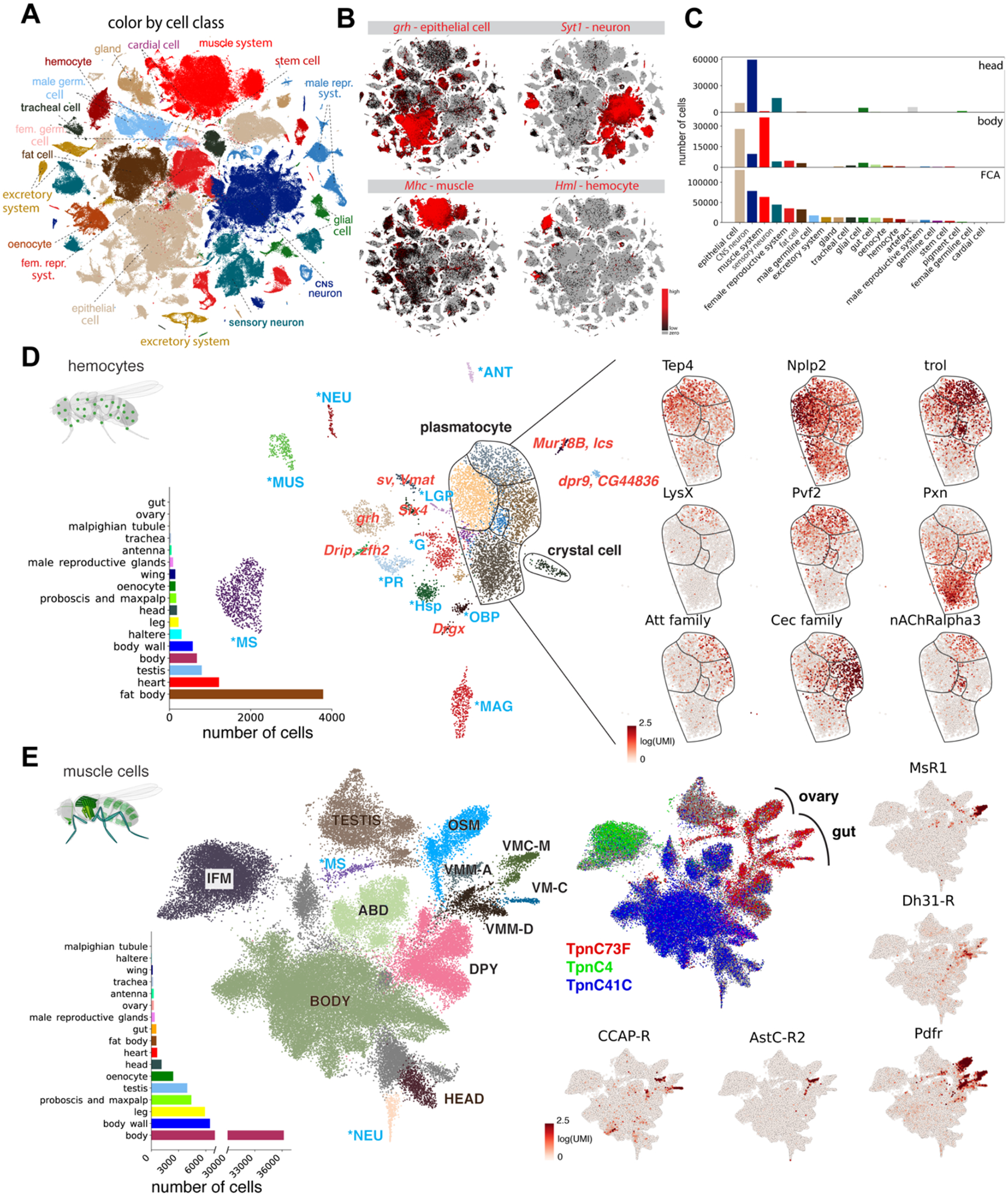
(A) Overview of main cell classes identified throughout the fly cell atlas. Som. pre., somatic precursor cells; male repr. and fem. repr., male and female reproductive system; male germ. and fem. germ., male and female germline cells.
(B) tSNE plots showing expression of four markers in four common cell classes.
(C) Composition of whole head and body samples, showing a shift from neurons to epithelial and muscle cells. Composition of the entire fly cell atlas shows enrichment for rarer cell classes compared to the whole-body sample.
(D) Cross-tissue analysis of hemocytes reveals different cell states of plasmatocytes. Annotations marked as blue are hemocytes containing markers of different cell types, including lymph gland posterior signaling center (LGP), muscle (MUS), antenna (ANT), neurons (NEU), photoreceptor (PR), male accessory glands (MAG), glia (G), male testis and spermatocyte (MS), olfactory-binding proteins (OBP), and heat-shock proteins (Hsp). Other abbreviations show top marker gene(s) in red. Plasmatocytes and crystal cells are indicated. On the right are genes showing compartmentalized expression patterns within the plasmatocyte cluster.
(E) Cross-tissue analysis of muscle cells reveals subdivision of the visceral muscle cells based on neuropeptide receptors. Annotations marked as blue are muscle cells containing markers of different cell types, including neuron (NEU) and male testis and spermatocyte (MS). Muscle cells from three body parts are indicated: head muscle (HEAD), body muscle (BODY), and testis muscle (TESTIS). Other annotated muscle types include indirect flight muscle (IFM), ovarian sheath muscle (OSM), abdominal visceral muscle (ABD), dpy expressing muscle (DPY), visceral muscle of the midgut AstC-R2 (VMM-A), visceral muscle of the crop MsR1 (VMC-M), visceral muscle of the midgut Dh31-R (VMM-D), and visceral muscle CCAP-R (VM-C). Pdfr is expressed in all visceral muscle cells, including the ovarian sheath muscle; other four receptor genes (AstC-R2, MsR1, Dh31-R, CCAP-R) are expressed in different gut visceral muscle types.
Second, we compared the muscle cells of the different tissues (Fig. 4E and fig. S26C, D). Muscle cells are syncytia—individual cells containing many nuclei, and to our knowledge have not been profiled by single-cell sequencing prior to our study. With snRNA-seq, we recovered all known muscle cell types, with specific enrichment in the body, body wall, and leg. This comprehensive view of the fly muscular system highlights a separation of visceral, skeletal, and indirect flight muscle based on the expression of different troponins. Specifically, we discovered gradients of dysf and fln in the indirect flight muscle, which may indicate regional differences in these very large cells (>1000 nuclei) (fig. S26E). We identified four types of visceral muscle in the gut based on expression of the AstC, Ms, Dh31 and CCAP neuropeptide receptors, indicating potential modulators for muscle contraction (30). Ms and Dh31 have been described to function in spatially restricted domains (30, 31, 32), suggesting similar domains for AstC and CCAP. All visceral muscle cells are enriched for the receptor of Pdf, a neuropeptide involved in circadian rhythms, pointing towards a function in muscle contraction as well (33).
Transcription factors and cell type specificity
Our data allow the comparison of gene expression across the entire fly. Clustering cell types showed the germline cells as the most distinct group, followed by neurons (fig. S27–S32). We calculated marker genes for every cell type using the whole FCA data as background, with 14,240 genes found as a marker for at least one cell type and a median of 638 markers per cell type [min: visceral muscle (94), max: spermatocyte (7736)]. Notably, markers specific for cell types in a tissue were not always specific in the whole body (fig. S33).
Next, we calculated the tau score of tissue specificity (34) for all predicted transcription factors (TFs) (3), identifying 500 TFs with a score > 0.85, indicating a high specificity for one or very few cell types (Fig. 5A, Table S3). 127 of these TFs were “CGs” (computed genes), indicating that their functions are poorly studied. We found that the male germline stands out in showing expression of a great number of cell type-specific TFs. This may be related to the broad activation of many genes in late spermatocytes, as discussed below.
Figure 5: Transcription factor (TF) pleiotropy versus cell-type specificity.
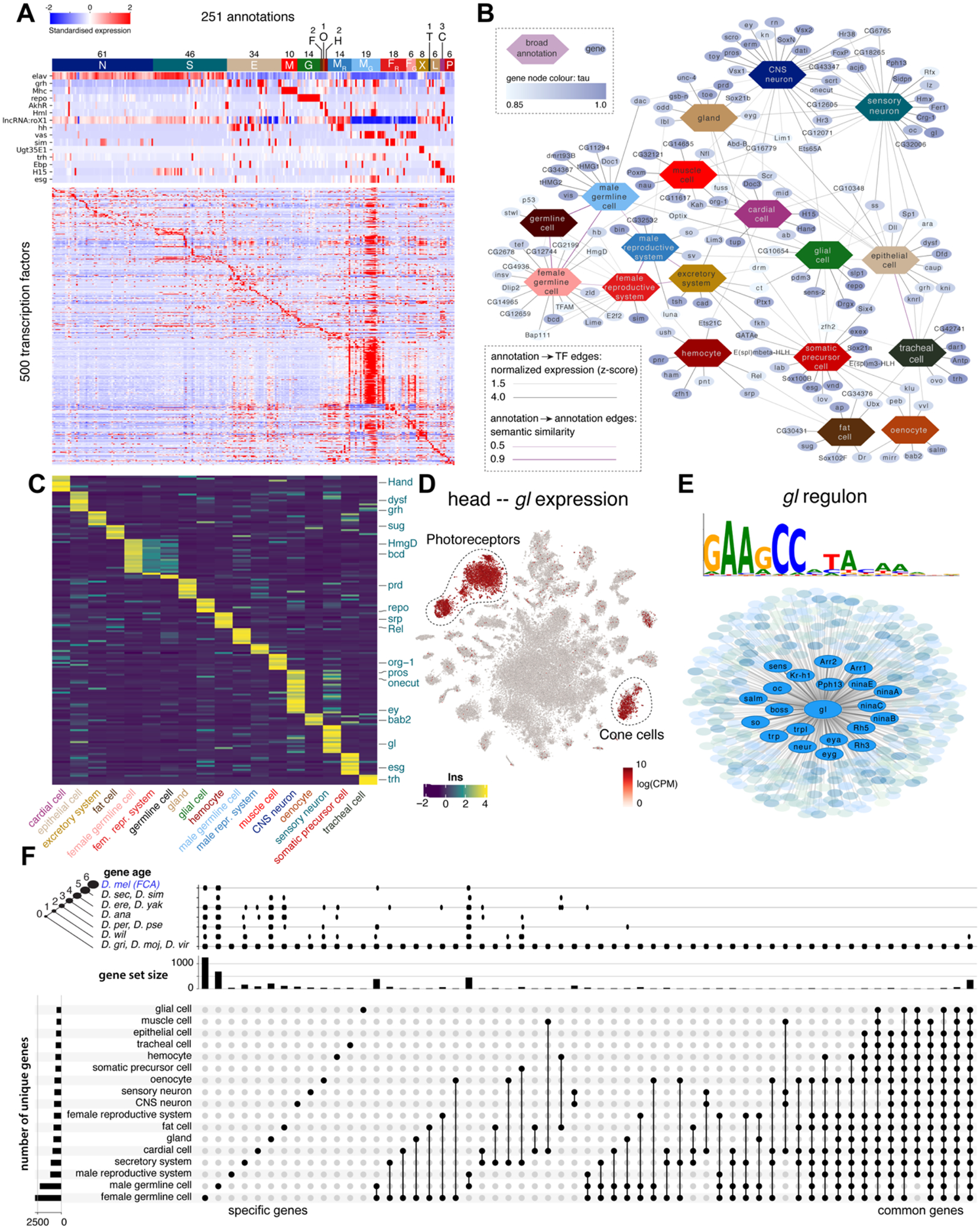
(A) Heatmap showing the expression of key marker genes and unique TF profiles for each of the annotated cell types. TFs were selected based on tau score. Cell types were grouped based on hierarchical terms: CNS neurons (N), sensory organ cells (S), epithelial cells (E), muscle cells (M), glia (G), fat cells (F), oenocytes (O), hemocytes (H), (fe)male reproductive system and germline (MR, MG, FR, FG), excretory system (X), tracheal cell (T), gland (L), cardiac cell (C), somatic precursor cell (P).
(B) A network analysis of TFs and cell classes based on similarity of ontology terms, reveals unique and shared TFs across the individual tissues.
(C) Heatmap showing the expression of unique TFs per cell class. Factors from the literature are highlighted.
(D) Glass is uniquely expressed in photoreceptors and cone cells in the head.
(E) Overview of the Glass regulon of 444 target genes, highlighting known photoreceptor marker genes.
(F) Gene expression comparison across broad cell types. Only sets with more than 10 genes are shown. The left bar graph shows the number of uniquely expressed genes for each tissue. The top bar graph shows the gene age in branches, ranging from the common ancestor to Drosophila melanogaster-specific genes (http://gentree.ioz.ac.cn). See fig. S34 for tissue-based comparison.
Similar analysis across broad cell types (Fig. 5B, C) identified 156 TFs with high tau scores, for example the known regulators grh for epithelial cells and repo for glia, as well as 24 uncharacterized genes. Network visualization shows the grouping of CNS neurons and sensory organ cells, including many sensory neurons, with shared pan-neuronal factors such as onecut and scrt but each cluster having a unique set of TFs, such as ey, scro and dati for CNS neurons and lz and gl for sensory neurons.
In addition to the specificity of TF expression, we predicted gene regulatory networks based on co-expression and motif enrichment using SCENIC (31). Because of the stochasticity of this network inference method, we ran SCENIC 100 times, ranking predicted target genes by their recurrence. This approach selected 6112 “regulons” for 583 unique TFs across all tissues, whereby each regulon consists of the TF, its enriched motif, and the set of target genes that are predicted in at least 5/100 runs. In fat cells, our analysis predicted a regulon for sugarbabe (sug), a sugar-sensitive TF necessary for the induction of lipogenesis (32). In photoreceptors, the analysis identified a glass (gl) regulon, with key photoreceptor markers such as Arr1, eya and multiple rhodopsins as predicted target genes (Fig. 5D, E)(33). The SCENIC predictions for all cell types are available via SCope (https://flycellatlas.org/scope).
Comparative analysis of genes across broad cell types or tissues (Fig. 5F, fig. S34) identified common genes and specifically expressed genes, such as a shared set of 555 housekeeping genes that are expressed in all tissues. The testis has the highest number of uniquely expressed genes consistent with previous reports (34), followed by the Malpighian tubule and male reproductive glands (fig. S34). These tissue-specific genes seemed to be evolutionarily “younger” based on GenTree age compared to the set of commonly expressed genes that are all present in the common ancestor. This suggests that natural selection works on the tissue specialization level, with the strongest selection on testis, male reproductive tract, and Malpighian tubules (35). In addition, this analysis allowed an estimation of transcriptomic similarity or difference measured by the number of shared unique genes. For example, the two flight appendages, the haltere and wing, share a set of 16 uniquely expressed genes, reflecting the evolutionary origin of halteres as a modified wing (36) (fig. S34).
Analysis of sex-biased expression and sex-specialized tissues
To study sex-related differences, we compared male- versus female-derived nuclei for all common tissues (fig. S35), finding roX1/2 and Yp1/2/3 as the top male- and female-specific genes, respectively. Notably, a large fraction of genes with male-enriched expression were uncharacterized (37). The primary sex determination pathway in somatic cells leads to sex-specific splicing of doublesex (dsx) to encode female- or male-specific TFs (38) (Fig. 6A). Consistent with this, we found dsx expression in a largely non-sex-specific pattern, while many other genes showed sex-biased expression (Fig. 6B).
Figure 6. Sex-biased expression and trajectory analysis of testis cell lineages.
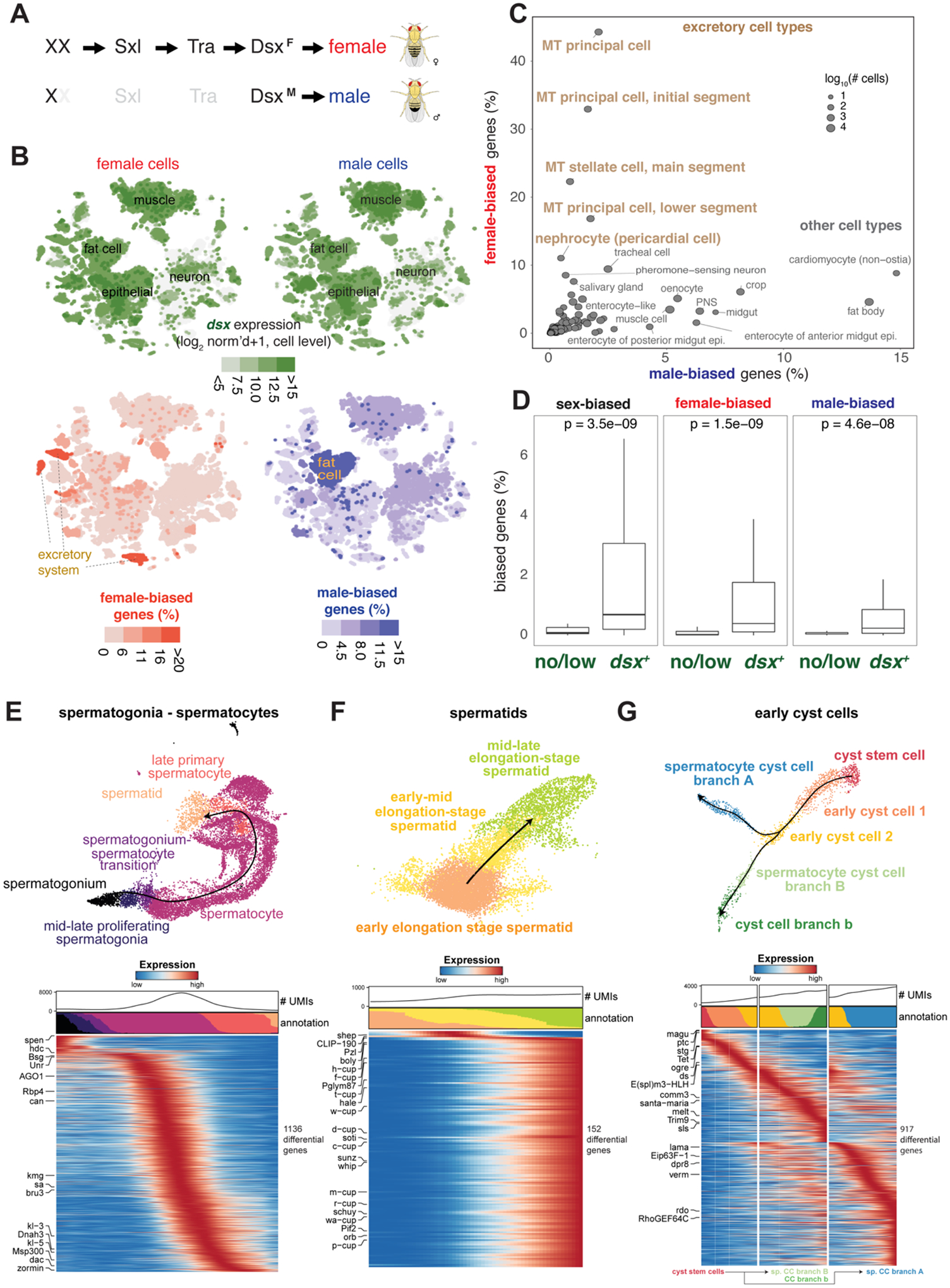
(A) Simplified sex determination pathway. Sex chromosome karyotype (XX) activates Sex-lethal (Sxl) which regulates transformer (Tra), resulting in a female Dsx isoform (DsxF). In XY (or X0) flies, Sxl and Tra are inactive (light gray) and the male-specific DsxM is produced.
(B) Top, Dsx expression and female- and male-biased expression projected onto tSNE plots of all female (left column) and male (right column) cells except reproductive tissue cells (Table S4 and S5). female- and male-biased expression measured as the percentage of genes in the cluster showing biased expression in favor of the respective sex (Table S6). These percentage values were computed for each annotated cluster and those cluster-level values were projected onto the individual cells in the corresponding clusters. For all four tSNE plots, values outside the scale in the heatmap key are represented by the closest extreme color (> and < signs in the scale).
(C) Scatter plot of female- and male-bias values across non-reproductive cell clusters defined as % sex-biased genes (at least 2-fold change with FDR < 0.05 on Wilcoxon test and BH correction) in the cluster (Table S6). Data point size indicatess cell numbers per cluster (key). Selected clusters are labeled, with those from excretory cells highlighted (brown). MT, Malpighian tubule.
(D) Box plots showing the relationship between dsx gene expression and sex-biased expression (Table S5). Clusters (B) were partitioned into the set of clusters with Dsx expression (dsx+) or not (no/low) using dsx expression in germ cells as an expression cut-off. Each box shows hinges at first and third quartiles and median in the middle. The upper whisker extends from the upper hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Outliers are not shown. p-values are based on Wilcoxon test.
(E–G) Trajectory of testis subsets. We used slingshot to infer a possibly branching trajectory for spermatogonia-spermatocytes (E), spermatids (F), and early cyst cells (G). Shown are the trajectories on a UMAP (top) and the expression patterns of the strongest differentially expressed genes, together with the smoothed proportions of annotated cells and average number of unique molecular identifiers (UMIs) along the trajectory (bottom).
Next, we performed differential expression between sexes for all cell types. Notably, cell types tended to show either high female- or male-bias, not both (Fig. 6B–C). We found strong female-bias in the excretory system, including the principal and stellate cells of the Malpighian tubule (MT) and in the pericardial nephrocytes (Fig. 6C). Female-biased genes (i.e., Ics and whe) were differentially expressed under high salt conditions, suggesting sex-bias in nephric ion transport. Across cell types, sex-biased expression strongly correlated with dsx expression (Fig. 6D) (39), consistent with the role of Dsx as a key regulator.
Among all tissues in the adult fly, those best characterized that have ongoing cellular differentiation are the gut, ovaries, and testis. Trajectory analysis has been performed on the gut and ovary stem cell lineages in previous studies (40–42), and our FCA data on gut and ovary accurately co-clustered with these published datasets (fig. S36, S37). Therefore, we focused on the testis plus seminal vesicle as a case study. The testis has two populations of stem cells, the somatic cyst stem cells (CySCs) that produce cell types with supporting roles essential to spermatogenesis, and the germline stem cells (GSCs) that produce haploid sperm (Fig. 2C). The main testis analysis (Fig. 2C) revealed transitions from GSCs and proliferating spermatogonia, spermatocytes, to maturing spermatids, and finally late elongation stage spermatids.
We further performed trajectory inference on spermatocytes and spermatids separately (Fig. 6E–F). As expected, the spermatocyte stage featured a continuous increase in the number of genes being transcribed (Fig. 6E), with many of the strongly upregulated genes (kmg, Rbp4, fzo, can, sa, and, for later spermatocytes, Y-linked fertility factors kl-3 and kl-5) not substantially expressed in any other cell type. Late spermatocytes, however, showed expression of marker genes from many other cell types like somatic cells (Upd1, eya)), epithelial cells (grh), muscle (Mhc) or hemocytes (Hml) (Fig. 5A), although their expression level was lower than in their marked cell type. Early spermatids are in transcriptional quiescence, as can be seen by a very low number of nuclear transcripts (Fig. 6F, low UMI), followed by a burst of new transcription in elongating spermatids including many cup genes (48).
In the somatic cyst cell lineage, we found CySCs expressing the cell cycle marker string, transitioning into post-mitotic (no string expression) early cyst cells, and branching into two related clusters of cyst cells likely associated with spermatocytes (Fig. 6G).
Discussion
Recent technological development has enabled single-cell transcriptomic atlases of C. elegans (21) and selected tissues in mice and humans (43–46). Here, we provide a single-cell transcriptomic map of the entire adult Drosophila melanogaster, a premier model organism for studies of fundamental and evolutionarily conserved biological mechanisms. The FCA provides a resource for the Drosophila community as a reference for studies of gene function at single-cell resolution.
A key challenge in large-scale cell atlas projects is the definition of cell types. We addressed this using a consensus-based voting system across multiple resolutions. An FCA cell type is thus defined as a transcriptomic cluster detected at any clustering resolution that could be separated by the expression of known marker genes from other clusters. Further, all annotations were manually curated by tissue experts, leading to a high-confidence dataset with over 250 annotated cell types. We note differences in annotation depth for different cell groups, with some cell types only linked to broad classes (e.g. epithelial cell), in contrast to other, more detailed cell types (e.g., different ORNs). We also note that while many marker genes are useful in identifying cell types, some marker gene expression was not congruent with cluster expression. This can be caused by discrepancies between mRNA and expression or by mistakes that were made in the literature. These examples highlight the need and the opportunities presented by Tabula Drosophilae to serve as the basis for future validation.
We have generated lists of marker genes per cell type with different levels of specificity, ranging from tissue-wide to animal-wide. This unique level of precision presents a blueprint for future integration with other data modalities such as single-cell ATAC-seq (47) and spatial omics, and for generating cell-type reporter lines to study new cellular functions. Furthermore, the large number of uncharacterized genes that show cell-type specific, sex-biased or trajectory-dependent expression provides the foundation for many follow-up studies. Our analysis also presents several technical novelties, including the use of reproducible Nextflow pipelines (VSN, https://github.com/vib-singlecell-nf), the availability of raw and processed datasets for users to explore, and the development of a crowd-annotation platform with voting, comments and references via SCope (https://flycellatlas.org/scope), linked to an online analysis platform in ASAP (https://asap.epfl.ch/fca). These elements may inspire future atlas projects. Given the work in other model organisms, we also envision a use for the FCA data in cross-species studies. Furthermore, Tabula Drosophilae is fully linked to existing Drosophila databases by a common vocabulary, benefitting its use and integration in future projects. Finally, all FCA data are freely available for further analysis via multiple portals and can be downloaded for custom analysis using other single cell tools (fig. S1; links available on https://www.flycellatlas.org).
Supplementary Material
ACKNOWLEDGMENTS:
We thank the entire fly community for the enthusiastic support for this project, Bill Burkholder, Cathryn Murphy, and Kathleen Vogelaers for coordinating FCA and all Jamboree meetings.
Funding:
The sequencing was supported by the Chan Zuckerberg Biohub (S. Quake), Genentech Inc (H. Jasper), National Institutes of Health (B. Oliver), and Howard Hughes Medical Institute and a National Institutes of Health grant (L. Luo). Computational work was supported by the KU Leuven and the Flemish Supercomputer Center (VSC) (S. Aerts) and EPFL (B. Deplancke). FCA Consortium Funding in the Supplemental Materials.
Footnotes
Competing interests: H. Jasper, N.S. Katheder and X.T. Cai are employees of Genentech, Inc. Other authors declare no competing interests.
Supplementary Materials
FCA Consortium Author Affiliations
FCA Consortium Author Affiliations
Figures S1 to S37
Data and materials availability:
All data are available for user-friendly querying via https://flycellatlas.org/scope and for custom analyses at https://flycellatlas.org/asap. For each tissue, a CellxGene portal is also available (www.flycellatlas.org). Raw data and count matrices can be downloaded from ArrayExpress (accession number E-MTAB-10519 for 10x, and E-MTAB-10628 for Smart-seq2; the same accession numbers are available at EBI Single Cell Expression Atlas https://www.ebi.ac.uk/gxa/sc). Files with expression data, clustering, embeddings, and annotation can be downloaded for each tissue, or all data combined, in h5ad and loomX formats from www.flycellatlas.org. Three Supplemental Figures describe how to access and explore FCA data: fig. S1 for summary of Data Availability, fig. S2 and S3 for how to use SCope and ASAP. We also include a video tutorial for using Scope (https://www.youtube.com/watch?v=yNETQVaSJYM&t=349s). Analysis codes are at Github (https://github.com/flycellatlas). Dataset access: GSE107451 (scRNA-seq adult fly brain), GSE120537 (scRNA-seq adult fly gut), GSE136162, GSE146040 and GSE131971 (scRNA-seq adult ovary). The neural network from (22) (Appendix 1).
References and Notes
- 1.Morgan TH, SEX LIMITED INHERITANCE IN DROSOPHILA. Science 32, 120–122 (1910). [DOI] [PubMed] [Google Scholar]
- 2.Adams MD et al. , The genome sequence of Drosophila melanogaster. Science 287, 2185–2195 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Larkin A et al. , FlyBase Consortium, FlyBase: updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res 49, D899–D907 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lyne R et al. , FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol 8, R129 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenett A et al. , A GAL4-driver line resource for Drosophila neurobiology. Cell Rep 2, 991–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milyaev N et al. , The Virtual Fly Brain browser and query interface. Bioinformatics 28, 411–415 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Kudron MM et al. , The ModERN Resource: Genome-Wide Binding Profiles for Hundreds of Drosophila and Caenorhabditis elegans Transcription Factors. Genetics 208, 937–949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.modENCODE Consortium et al. , Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chintapalli VR, Wang J, Dow JAT, Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet 39, 715–720 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Li H, Single-cell RNA sequencing in Drosophila: Technologies and applications. Wiley Interdiscip. Rev. Dev. Biol 10, e396 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLaughlin CN et al. , Single-cell transcriptomes of developing and adult olfactory receptor neurons in Drosophila. eLife 10 (2021), doi: 10.7554/eLife.63856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng GXY et al. , Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Picelli S et al. , Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 14.De Waegeneer M, Flerin CC, Davie K, Hulselmans G, vib-singlecell-nf/vsn-pipelines: v0.26.0 (v0.26.0). Zenodo. 10.5281/zenodo.5055627. Zenodo (2021). [DOI] [Google Scholar]
- 15.Yang S et al. , Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol 21, 57 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korsunsky I et al. , Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davie K et al. , A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell 174, 982–998.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David FPA, Litovchenko M, Deplancke B, Gardeux V, ASAP 2020 update: an open, scalable and interactive web-based portal for (single-cell) omics analyses. Nucleic Acids Res 48, W403–W414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costa M, Reeve S, Grumbling G, Osumi-Sutherland D, The Drosophila anatomy ontology. J. Biomed. Semantics 4, 32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy S et al. , A stony coral cell atlas illuminates the molecular and cellular basis of coral symbiosis, calcification, and immunity. Cell 184, 2973–2987.e18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J et al. , Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Özel MN et al. , Neuronal diversity and convergence in a visual system developmental atlas. Nature 589, 88–95 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H et al. , Classifying Drosophila Olfactory Projection Neuron Subtypes by Single-Cell RNA Sequencing. Cell 171, 1206–1220.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurmangaliyev YZ et al. , Transcriptional programs of circuit assembly in the drosophila visual system. Neuron 108, 1045–1057.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Cho B et al. , Single-cell transcriptome maps of myeloid blood cell lineages in Drosophila. Nat. Commun 11, 4483 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlov VA, Tracey KJ, The cholinergic anti-inflammatory pathway. Brain Behav. Immun 19, 493–499 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Sanchez Bosch P et al. , Adult drosophila lack hematopoiesis but rely on a blood cell reservoir at the respiratory epithelia to relay infection signals to surrounding tissues. Dev. Cell 51, 787–803.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krzemień J, L et al. , Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature 446, 325–328 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Mandal L et al. , A Hedgehog- and Antennapedia-dependent niche maintains Drosophila haematopoietic precursors. Nature 446, 320–324 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siviter RJ et al. , Expression and functional characterization of a Drosophila neuropeptide precursor with homology to mammalian preprotachykinin A. J. Biol. Chem 275, 23273–23280 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Aibar S et al. , SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattila J, Hietakangas V, Regulation of Carbohydrate Energy Metabolism in Drosophila melanogaster. Genetics 207, 1231–1253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moses K, Ellis MC, Rubin GM, The glass gene encodes a zinc-finger protein required by Drosophila photoreceptor cells. Nature 340, 531–536 (1989). [DOI] [PubMed] [Google Scholar]
- 34.Kaessmann H, Origins, evolution, and phenotypic impact of new genes. Genome Res 20, 1313–1326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao Y et al. , GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res 29, 682–696 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis EB, A gene complex controlling segmentation in Drosophila. Nature 276, 565–570 (1978). [DOI] [PubMed] [Google Scholar]
- 37.Andrews J et al. , Gene Discovery Using Computational and Microarray Analysis of Transcription in the Drosophila melanogaster Testis. Genome Res 10, 2030–2043 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salz HK, Erickson JW, Sex determination in Drosophila: The view from the top. Fly (Austin) 4, 60–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clough E et al. , Sex- and tissue-specific functions of Drosophila doublesex transcription factor target genes. Dev. Cell 31, 761–773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hung R-J et al. , A cell atlas of the adult Drosophila midgut. Proc Natl Acad Sci USA 117, 1514–1523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rust K et al. , A single-cell atlas and lineage analysis of the adult Drosophila ovary. Nat. Commun 11, 5628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jevitt A et al. , A single-cell atlas of adult Drosophila ovary identifies transcriptional programs and somatic cell lineage regulating oogenesis. PLoS Biol 18, e3000538 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabula Muris Consortium et al. , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han X et al. , Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 173, 1307 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Cao J et al. , A human cell atlas of fetal gene expression. Science 370 (2020), doi: 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han X et al. , Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Janssens J et al. , Decoding gene regulation in the fly brain. Nature (2022), doi: 10.1038/s41586-021-04262-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available for user-friendly querying via https://flycellatlas.org/scope and for custom analyses at https://flycellatlas.org/asap. For each tissue, a CellxGene portal is also available (www.flycellatlas.org). Raw data and count matrices can be downloaded from ArrayExpress (accession number E-MTAB-10519 for 10x, and E-MTAB-10628 for Smart-seq2; the same accession numbers are available at EBI Single Cell Expression Atlas https://www.ebi.ac.uk/gxa/sc). Files with expression data, clustering, embeddings, and annotation can be downloaded for each tissue, or all data combined, in h5ad and loomX formats from www.flycellatlas.org. Three Supplemental Figures describe how to access and explore FCA data: fig. S1 for summary of Data Availability, fig. S2 and S3 for how to use SCope and ASAP. We also include a video tutorial for using Scope (https://www.youtube.com/watch?v=yNETQVaSJYM&t=349s). Analysis codes are at Github (https://github.com/flycellatlas). Dataset access: GSE107451 (scRNA-seq adult fly brain), GSE120537 (scRNA-seq adult fly gut), GSE136162, GSE146040 and GSE131971 (scRNA-seq adult ovary). The neural network from (22) (Appendix 1).