

SUMMARY
Activated macrophages must carefully calibrate their inflammatory responses to balance efficient pathogen control with inflammation-mediated tissue damage, but the molecular underpinnings of this “balancing act” remain unclear. Using genetically engineered mouse models and primary macrophage cultures, we show that Toll-like receptor (TLR) signaling induces the expression of the transcription factor Spic selectively in patrolling monocytes and tissue macrophages by a nuclear factor κB (NF-κB)-dependent mechanism. Functionally, Spic downregulates pro-inflammatory cytokines and promotes iron efflux by regulating ferroportin expression in activated macrophages. Notably, interferon-gamma blocks Spic expression in a STAT1-dependent manner. High levels of interferon-gamma are indicative of ongoing infection, and in its absence, activated macrophages appear to engage a “default” Spic-dependent anti-inflammatory pathway. We also provide evidence for the engagement of this pathway in sterile inflammation. Taken together, our findings uncover a pathway wherein counter-regulation of Spic by NF-κB and STATs attune inflammatory responses and iron metabolism in macrophages.
In Brief
Activated macrophages must fine-tune their inflammatory responses to promote host defense while limiting tissue damage. Alam et al. find that the transcription factor Spic restrains inflammatory responses and promotes iron efflux from activated macrophages, thereby calibrating macrophage responses during the resolution of inflammation.
Graphical Abstract
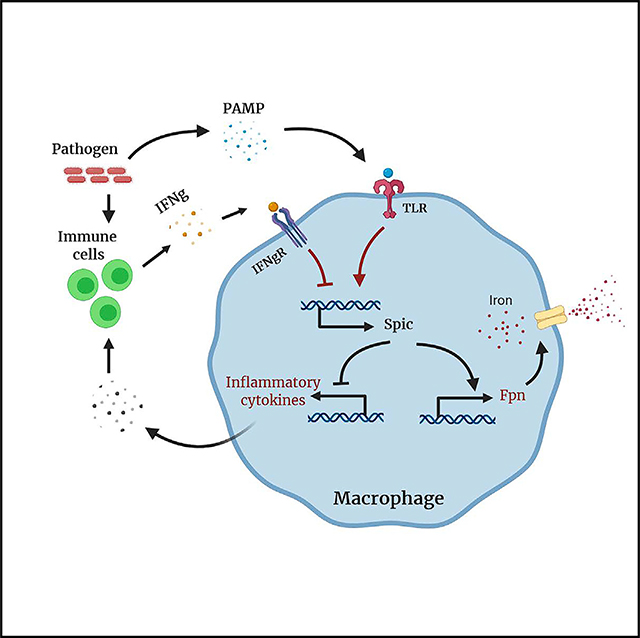
INTRODUCTION
Macrophages are widely distributed with impressive functional diversity (Gordon et al., 2014; Haldar and Murphy, 2014). At steady state, tissue macrophages help maintain local tissue homeostasis (Gordon et al., 2014; Haldar and Murphy, 2014). Injury or infection leads to the recruitment of circulating monocytes that can locally differentiate into macrophages (monocytes-derived macrophages [Mo-MACs]) that produce cytokines and other factors that shape the ensuing immune response. Resolution of inflammation is facilitated by reduced pro-inflammatory and increased anti-inflammatory cytokine production by Mo-MACs (Murray, 2017; Oishi and Manabe, 2018; Wynn and Vannella, 2016). Excessive or prolonged inflammatory responses can impair tissue repair, whereas suboptimal responses lead to poor pathogen control (Murray and Wynn, 2011). Therefore, macrophage inflammatory responses are dynamically regulated, but the molecular underpinnings are unclear.
Macrophages have receptors that detect pathogen-associated or endogenous danger-associated molecular patterns (PAMPs and DAMPs, respectively) (Mukhopadhyay et al., 2009; Zhang and Mosser, 2008). Lipopolysaccharide (LPS), the prototypical PAMP, activates Toll-like receptor 4 (TLR4), which induces large-scale transcriptional changes. LPS-induced genes can be classified as primary and secondary response genes (Medzhitov and Horng, 2009). The induction of primary response genes does not require new protein synthesis and occurs curs within minutes by activation of pre-existing transcription factors, such as nuclear factor κB (NF-κB) and AP-1. Primary response genes mainly promote inflammation; however, a subset set of these genes encode transcription factors that mediate the expression of secondary response genes with more diverse function (Medzhitov and Horng, 2009). Temporally and mechanistically, the regulation of secondary response genes provides a convenient fulcrum for calibrating macrophage inflammatory responses. As an example, the transcription factor C/EBPβ is a secondary response gene that counteracts the pro-inflammatory actions of NF-κB(Kaneda et al., 2016; Ruffell et al., 2009). Nonetheless, how secondary response genes regulate macrophage inflammatory responses is not fully understood.
PAMP recognition induces cytokine production by macrophages that alert the rest of the immune system to the presence of pathogens. The subsequent influx and activation of other immune cells at the site of inflammation change the cytokine milieu. Sensing this evolving cytokine milieu is one way through which macrophages can assess the status of local inflammation to regulate their own function accordingly (Oishi and Manabe, 2018; Wynn and Vannella, 2016). T cells and natural killer (NK) cells produce high levels of interferon-gamma (IFNγ) at sites of infection, which dissipates upon resolution of infection (Thäle and Kiderlen, 2005). Hence, IFNγ can serve as a “second signal” for PAMP-activated macrophages, corroborating the presence of pathogens. Consistent with this notion, IFNγ augments inflammatory and microbicidal functions of macrophages (Hu and Ivashkiv, 2009). However, how activated macrophages respond to falling IFNγ levels in the resolution phase of pathogen-induced inflammation and how IFNγ affects macrophage function during sterile inflammation remain unclear.
Iron enters macrophages through diverse pathways and is either stored inside the cell or released back into the surrounding environment by the iron exporter ferroportin (Fpn, Slc40a1) (Alam et al., 2017; Soares and Hamza, 2016). Because pathogens require iron to thrive in the host, macrophages sequester it during infection (Ganz and Nemeth, 2015). A key mechanism controlling iron availability is the regulation of macrophage Fpn during inflammation (Drakesmith et al., 2015). Toll-like receptor (TLR) activation rapidly downregulates Fpn transcription (Guida et al., 2015). Hepcidin, a peptide hormone produced by hepatocytes during inflammation, also causes internalization and degradation of FPN in macrophages (Drakesmith and Prentice, 2012). Systemically, prolonged or excessive iron sequestration can lead to iron deficiency, whereas locally this can impair wound repair (Ganz and Nemeth, 2015; Recalcati et al., 2019). Therefore, macrophages must release trapped iron during the resolution phase of inflammation, but pathways linking the resolution of inflammation to iron efflux in macrophages are unclear.
The transcription factor Spic was previously shown to be required for the development of iron-recycling macrophages (Haldar et al., 2014; Kohyama et al., 2009). Here, we show that Spic is also induced in PAMP- or DAMP-activated macrophages where it reduces the inflammatory response and promotes iron efflux. In this setting, the mechanism of Spic induction is distinct from the previously reported Bach1 and heme-dependent pathway. Importantly, IFNγ blocks Spic expression, providing insight into how Spic-dependent functions are differentially engaged in the presence or absence of an infectious threat.
RESULTS
TLR Ligands Selectively Induce Spic Expression in Patrolling Monocytes and Tissue Macrophages
Spic regulates the development of iron-recycling macrophages in the spleen and bone marrow (Haldar et al., 2014; Kohyama et al., 2009). Because monocytes and macrophages alter iron metabolism during inflammation (Ganz and Nemeth, 2015), we examined whether inflammation regulates Spic by treating SpicGFP/GFP reporter mice (Haldar et al., 2014) with intraperitoneal LPS. In the blood, LPS induced Spic selectively in monocytes (Figures 1A–1C). Ly6C expression marks two major subseets of murine monocytes (Geissmann et al., 2003). Notably, Spic was induced in Ly6CloTremL4hi (patrolling) but not Ly6ChiTremL4lo (classical) monocytes (Figures 1B and 1C). To test whether blood monocyte subsets inherently differ in their capacity to express Spic, we analyzed Spic expression in Bach1-deficient SpicGFP/GFP reporter (Bach1−/−: SpicGFP/GFP) mice. We previously showed that the transcription factor Bach1 constitutively represses Spic expression in monocytes (Haldar et al., 2014). Bach1 deficiency, similar to LPS treatment, promoted Spic expression in Ly6CloTremL4hi patrolling monocytes (Figure 1D). Therefore, the two major subsets of circulating monocytes differ in their ability to express Spic.
Figure 1. TLR Activation Induces Spic in Monocytes and Macrophages.

(A–C) LPS (50 μg) or PBS (control) was injected intraperitoneally (i.p.) into mice of indicated genotypes (headers). Peripheral blood was collected 3 days later. Shown are the flow cytometry plots (FCSs) with indicated markers. (A) Cells pre-gated for CD45+ singlets expresses SPIC (GFP+) selectively in CD11B+ myeloid cells. (B) Expression of indicated markers on cells gated on SPIC expression (A, arrow). (C) FCS showing SPIC expression in monocyte subsets defined by Ly6C (pre-gated for CD45+ singlets).
(D) FCS of circulating monocytes (CD45+LY6G-CD115+) from mice of indicated genotypes (header) showing Spic expression in circulating leukocytes.
(E) Zbtb46GFP/GFP bone marrow cells were cultured in GM-CSF for 7 days. F4/80hiZbtb46GFP− macrophages (MAC) and F4/80loZbtb46GFP+ DCs were purified by fluorescence-assisted cell sorting (FACS), cultured in media without cytokines, and treated with LPS. Cells were harvested 24 h later, and qRT-PCR was performed for indicated genes (y axis, normalized to 18S rRNA). MACs but not DCs induced Spic while both cell types increased Tnfα expression with LPS.
FCS, numbers represent percentage of cells within indicated gate. (A–D) Represents ≥3 experiments with ≥3 mice per group. qRT-PCR, data representative of ≥3 independent experiments; and graphs show a single experiment with n ≥2 per group. Results are expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figure S1.
Monocytes can differentiate into macrophages or dendritic cells (DCs), and we previously showed that TREML4 expression marks the loss of DC differentiation potential in circulating monocytes (Briseño et al., 2016). The selective expression of Spic in TremL4+ monocytes suggests that the capacity to express Spic is linked to macrophage identity. To further test this, we obtained Zbtb46GFP reporter mice in which GFP expression is restricted to DCs (Satpathy et al., 2012). We generated a mixed population of macrophages and DCs in vitro by culturing Zbtb46GFP bone marrow cells with granulocyte-macrophage colony-stimulating factor (GM-CSF) (Helft et al., 2015). LPS treatment in this setting strongly induced Spic in ZBTB46GFP−F4/80hi macrophages but not ZBTB46-GFP+F4/80− DCs, confirming the specificity of Spic to the macrophage lineage (Figure 1E).
We next examined whether tissue macrophages induce Spic during inflammation. High levels of heme can induce Spic in iron-recycling macrophages of the spleen, bone marrow, and liver (Haldar et al., 2014). Hence, we excluded these organs from our initial analyses to disentangle the impact of heme from TLR activation on Spic. Intraperitoneal LPS induced Spic in monocytes and macrophages of various tissues but not in DCs (Figures 2A and S1A). Macrophages in the intestinal tract are exposed to TLR ligands derivedp from gut microbiota (Bain et al., 2013). Correspondingly, we detected Spic in macrophages of the large intestine at steady state (Figure. 2B). Gut macrophages are further divided into three major subsets: (1) CD4+Tim4+ subset that is maintained by local proliferation, (2) CD4+Tim4- subset with slow turnover from monocytes, and (3) CD4-Tim4- subset with rapid turnover from circulating monocytes (Shaw et al., 2018). We detected Spic in all three subsets (Figure 2B). Our findings are consistent with a recent report that also cited Spic expression in gut macrophages (Kayama et al., 2018). Notably, the level of Spic was higher in the colon than in the small intestine, which likely reflects the higher density of microbiota in the colon (Figure 2C). Correspondingly, Spic expression was lower in the colon of germ-free than in conventionally housed mice (Figure 2D).
Figure 2. TLR Activation Induces Spic in the Tissue Macrophages.

(A) SpicGFP/GFP mice were treated with i.p. LPS (75 μg in PBS) or control (PBS), and lungs were harvested 48 h after treatment. Top: FCS with indicated markers on singlets, highlighting SPIC expression. Bottom: distribution of indicated markers on SPIC+ and SPIC- cells (gating shown in top panels, arrow).
(B) FCS with indicated markers in CD45+ live (7AAD-) singlets from large intestine of SpicGFP/GFP and wild-type (WT) mice, showing Tim4 and CD4 expression largely restricted to SPIC+ cells.
(C) Published gene expression profiles (microarray based) of murine ileum and colon were downloaded from a public database (GEO: GSE32513). Shown are expression values (linear scale) of Spic.
(D) qRT-PCR-based expression of Spic (normalized to Hprt) in colon obtained from conventionally raised (Cont) and germ-free (GF) mice.
(E) Lung macrophages (CD45+CD64+CD11C+) were isolated by FACS, cultured with M-CSF for 12 h, and treated with LPS (1 μg/ml). RNA was extracted 10 h after LPS treatment, and the expression (normalized to Hprt)ofSpic was measured by qRT-PCR.
(F) Peritoneal cells (PECs) were cultured with M-CSF for 12 h, followed by LPS (1 μg/ml) treatment. RNA was extracted 20 h after treatment, and the expression (relative to Hprt)ofSpic was measured by qRT-PCR.
(G) CD45+F4/80+GFP+ and CD45+F4/80+GFP- liver macrophages were purified by FACS, cultured with M-CSF for 12 h, and treated with LPS (1 μg/ml). RNA was extracted 14 h after LPS treatment, and the expression (normalized to 18S rRNA)ofSpic was measured by qRT-PCR.
(H) RPMs from SpicGFP/GFP spleen were purified by FACS, cultured with M-CSF for 12 h, and treated with LPS (1 μg/ml). RNA was extracted 16 h after LPS treatment, and the expression (normalized to Hprt)ofSpic was measured by qRT-PCR.
FCS, numbers represent percentage of cells within indicated gate. (A and B) Represent ≥3 experiments with ≥3 mice per group. qRT-PCR, data representative of ≥ 3 independent experiments; and graphs show a single experiment with n ≥ 2 per group. Results expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figures S1, S6, and S7
TLR activation promotes monocyte differentiation into macrophages (Krutzik et al., 2005). Hence, TLR-induced Spic in tissues may represent macrophages newly differentiated from infiltrating monocytes, or they may represent pre-existing tissue-resident macrophages. To test whether tissue-resident macrophages can induce Spic, we purified lung and peritoneal resident macrophages and exposed them to LPS in vitro, finding robust Spic induction (Figures 2E and 2F). Next, we asked whether macrophages that already express high levels of Spic at the steady state (splenic red pulp macrophages [RPMs] and liver Kupffer cells) can further induce it upon TLR activation. We isolated Spic-high and Spic-negative macrophages from spleen and liver of SpicGFP/GFP mice and exposed them to LPS ex vivo. Although LPS further increased Spic expression in Spic-high macrophages, the level of induction was significantly less (2× versus >10×) than that in macrophages that did not express Spic prior to LPS exposure (Figures 2G, 2H, and S1B). Hence, TLR-induced Spic is a conserved feature of macrophages.
Spic Downregulates Inflammatory Responses in Activated Macrophages
The spleen contains Spic-high RPMs and their Spic-low precursors (PreRPM) (Haldar et al., 2014). Spic−/− mice lack RPM but not PreRPMs (Figure 3A). A microarray-based gene expression comparison of wild-type (WT) and Spic−/− PreRPM revealed a prominent inflammatory signature in the latter, suggesting an anti-inflammatory function of Spic (Figure 3B). Indeed, a recent study suggested that heme-induced Spic downregulates inflammation in a murine model of dextran sodium sulfate(DSS)-induced colitis (Kayama et al., 2018). Therefore, we further examined whether TLR-induced Spic serves an anti-inflammatory function. We treated SpicGFP/GFP bone-marrow-derived macrophages (BMDMs) with LPS in vitro and isolated SPIC+ and SPIC- macrophages. In this setting, SPIC+ macrophages expressed lower pro- and higher anti-inflammatory cytokines (Figures 3C). These differences were maintained when SPIC+ and SPIC- macrophages were re-exposed to LPS (Figure S2A). Hence, high levels of Spic expression marks macrophages with lower inflammatory responses. Next, we compared LPS responses of WT and Spic−/− BMDMs. Consistent with the above observations, Spic deficiency engendered higher pro-inflammatory cytokine expression (Figures 3D and 3E). Correspondingly, Spic−/− mice showed higher body temperature, higher levels of circulating tumor necrosis factor α (TNF-α), and increased lung Nos2 than WT mice upon intraperitoneal LPS exposure (Figures 3F and S2B).
Figure 3. Spic Controls Inflammatory Response in Macrophages.

(A) FCS (pre-gated on singlets) on splenocytes from WT and Spic−/− mice show drastically reduced RPMs but relatively normal PreRPMs in Spic-deficient spleen.
(B) PreRPM from WT and Spic−/− spleen (two mice per genotype) were purified by FACS and subjected to microarray-based gene expression profiling (Affymetrix, mouse gene_2.0ST). Shown are the gene set enrichment analyses (GSEAs) for the signature associated with inflammatory response (left) and the corresponding heatmap based on differentially expressed genes (nominal p = 0.504) between the two genotypes (right).
(C) BMDMs from SpicGFP/GFP mice were treated with LPS (1 μg/ml). After 48 h, GFP+(SPIC-expressing) and GFP-(SPIC-) cells were purified by FACS and RNA extracted, and the expression (normalized to Hprt) of indicated genes (y axis, relative to GFP-cells) was measured by qRT-PCR.
(D) Mo-MACs from WT and Spic−/− mice were treated with LPS (1 μg/ml), RNA was extracted 24 h later, and the expression (normalized to Hprt) of indicated genes (y axis, relative to WT no treatment) was measured by qRT-PCR.
(E) BMDMs from WT and Spic−/− mice were treated with LPS (1 μg/ml). After 16 h, the amount (y axis) of indicated cytokines released into the media was measured by ELISA.
(F) WT, Spic−/−, and Spic+/− mice were treated (i.p.) with LPS (7.5 μg/gm). Rectal temperature (right graph) and plasma TNF-α (ELISA, left graph) were measured 24 h after treatment.
(G) BMDMs from WT, Bach1−/−, and Bach1−/−: Spic−/− double knockout (DKO) were treated with LPS (1 μg/ml). RNA was extracted 24 h later, and the expression (normalized to Hprt) of indicated genes was measured (y axis, relative to WT non-treated group) by qRT-PCR.
(H) Mo-MACs from WT mice were treated with LPS (1 μg/ml) with or without PI3Kγ inhibitor, IPI549 (100 nM). RNA was extracted 20 h later, and the expression (normalized to Hprt) of indicated genes was measured (y axis, relative to no treatment group) by qRT-PCR.
FCS, numbers represent percentage of cells within indicated gate. (A) Represents ≥3 experiments with ≥3 mice per group. qRT-PCR, data representative of ≥3 independent experiments; and graphs show single experiment with n ≥2 per group. Results expressed as mean ± SEM. p% 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figure S2
Bach1−/− macrophages have been shown to display an anti-inflammatory phenotype (Harusato et al., 2013). Because Bach1−/macrophages also express high levels of Spic, we asked whether Spic might drive anti-inflammatory properties of Bach1−/− macrophages. We generated Bach1−/−: Spic−/− (double knockout [DKO]) mice and compared inflammatory responses of DKO and Bach1−/− BMDMs. In the setting of LPS exposure, we found that the loss of Spic reversed the anti-inflammatory phenotype of Bach1−/− BMDMs (Figure 3G). Finally, a recent study showed that phosphatidylinositol 3 kinase (PI3K)-γ signaling promotes a “switch” from a pro- to anti-inflammatory phenotype in actimacrophages (Kaneda et al., 2016). Inhibiting PI3K-γ in TLR-activated vated macrophages reduced Spic and increased pro-inflammatory cytokine expression (Figure 3H). Taken together, these findings suggest that Spic downregulates the transcription of pro-inflammatory cytokines in activated macrophages.
Spic Promotes Iron Export in Activated Macrophages
To identify the genetic targets of Spic in inflammatory settings, we compared the gene expression profile (microarray based) of patrolling monocytes from Spic+/− and Spic−/− mice treated with intraperitoneal LPS. Gene set enrichment analysis (GSEA) of differentially expressed genes revealed a hallmark for heme metabolism in Spic−/− monocytes, which is typically assocated with cells containing high levels of heme and iron (Figure 4A). Fpn is the only known mammalian exporter of iron and, remarkably, was one of the most downregulated genes in LPS-exposed Spic−/− monocytes (Figure 4B). This was independently validated by measuring Fpn expression in classical and patrolling monocytes from mice treated with LPS (Figure S2C). We found a similar trend in vitro, where LPS-treated Spic−/− macrophages expressed higher levels of genes involved in heme metabolism (Figure 4C) and lower levels of Fpn (Figure 4D). Correspondingly, lung macrophages and cells from the peritoneal cavity of Spic−/− mice showed lower FPN protein expression after LPS exposure in vivo (Figures 4E and S2D). Finally, splenic PreRPM expressed lower Fpn than their WT counterpart (Figure 4F). These findings suggest that Spic promotes Fpn expression and are consistent with previous observations of higher splenic iron in Spic−/− mice (Kohyama et al., 2009).
Figure 4. Spic Regulates Ferroportin-Mediated Iron Export in Macrophages.

(A) Spic+/− and Spic−/− mice (three mice per group) were treated with LPS (30 μg/mouse). Patrolling monocytes (CD45+CD11B+CD115+LY6C-) were purified 72 h after treatment and subjected to microarray-based (affymetrix, mouse gene 2.0_ST) gene expression profiling. Shown are the GSEA plots for the hallmark of heme metabolism (left) and corresponding heatmap based on differentially expressed genes (nominal p < 0.01) between the two genotypes (right).
(B) Expression (y axis, linear scale) of Fpn from the microarray data.
(C) WT and Spic−/− BMDMs (two mice per group) were treated with LPS (100 ng/ml), RNA was extracted 24 h later, and microarray-based (affymetrix, mouse gene 2.0_ST) gene expression profiling was performed. Shown are the GSEA plots for the hallmark of heme metabolism (left) and the corresponding heatmap based on differentially expressed genes (nominal p = 0.457) between the two genotypes (right).
(D) Mo-MACs from WT and Spic−/− mice were treated with LPS (1 μg/ml), RNA was extracted 16 h ater, and the expression (normalized to 18S rRNA)ofFpn was measured (y axis relative to a non-treated group) by qRT-PCR.
(E) WT or Spic−/− mice were treated (i.p.) with LPS (150 μg/mouse) twice 48 h apart. Lungs were harvested 48 h after the final LPS treatment, and levels of FPN protein measured by flow cytometry (left). The bar graph (right) shows quantification of mean fluorescent intensity (MFI) of FPN staining.
(F) PreRPM (CD11BhiF4/80lo) from WT and Spic−/− spleen were purified by FACS, RNA was extracted, and the levels (normalized to 18S rRNA)ofSpic and Fpn were measured (y axis, relative a WT PreRPM) by qRT-PCR.
(G) BMDMs from WT mice were treated with LPS (100 ng/ml), RNA was extracted at indicated time points (x axis), and the levels (normalized to 18S rRNA) of Fpn were measured (y axis, relative to non-treated group) by qRT-PCR.
(H) BMDMs of indicated genotypes were cultured for 7 days, after which the medium was removed, and fresh medium containing 100 mM of FeSO4 was added, followed by treatment with LPS (1 mg/mL) or PBS (control). Intracellular iron was measured 24 h later. Graph is representative of five independent experiments.
The replicates for each individual experiment are technical replicates for the assay.
qRT-PCR, data representative of ≥3 independent experiments. Plots show a single experiment with n ≥2 per group. Results are expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figures S2 and S3.
LPS strongly downregulates Fpn transcription in macrophages to sequester iron during inflammation (Abreu et al., 2018). Its expression gradually recovers during the resolution of inflammation, presumably to facilitate an efflux of the sequestered iron (Figure 4G). Our findings suggest that the “recovery” of Fpn expression in activated macrophages is regulated by Spic. In contrast, the transcription of ferritin heavy and light chains, which are key players in intracellular iron storage, did not show significant alterations with Spic deficiency (Figure S2E). Hence, Spic may selectively impact iron efflux by FPN without directly affecting other elements of cellular iron homeostasis.
Most studies of macrophage iron sequestration during inflammation have focused on the paracrine circuit involving Hepcidin (Hamp), an inflammation-induced hormone produced by hepatocytes that mediates the degradation of surface FPN on macrophages (Ganz and Nemeth, 2015). Our findings show a cell-intrinsic transcriptional circuitry that may regulate macrophage iron efflux during the resolution of inflammation. This is reminiscent of RPM, suggesting that TLR activation induces an RPM-like phenotype in macrophages. Indeed, a recent study showed that chronic TLR7/9 signaling induces an RPM-like macrophage differentiation from circulating monocytes (Akilesh et al., 2019). We found that Spic deficiency is associated with a trend toward higher liver Hamp upon TLR exposure (Figure S2F). Therefore, the impact of Spic on FPN-mediated iron regulation is 2-fold: (1) regulation of Fpn transcription within macrophages and (2) regulation of FPN protein stability by Hamp. However, Hamp expression within macrophages itself did not change significantly with Spic deficiency, suggesting that higher Hamp in the liver likely reflects a higher production by hepatocytes in response to higher inflammation in Spic−/− mice (Figure S2G).
Spic−/− macrophages upregulate Fpn in response to heme, much like their WT counterparts (Haldar et al., 2014). Hence, Spic is not required for Fpn expression, instead promoting Fpn transcription in specific contexts, such as PAMP-activated macrophages This raises the question of what impact, if any, does Spic-regulated Fpn have on macrophage iron storage during inflammation. To address this, we devised an in vitro assay where WT, Bach1−/−, and Spic−/− BMDMs were loaded with iron (ferrous sulfate) prior to LPS exposure, followed by measurement of total intracellular iron. As expected, LPS led to increased intracellular iron in WT and Spic−/− macrophages (Figure 4H). Bach1 is a negative regulator of Fpn, and Bach1−/macrophages express high levels of Fpn. Correspondingly, Bach1−/− macrophages did not show significant increases in intracellular iron with LPS (Figure 4H). Importantly, LPS-exposed Spic−/− macrophages displayed higher intracellular iron than their WT counterparts (Figure 4H). Taken together, these findings show that transcriptional fine-tuning of Fpn expression by Spic regulates macrophage iron storage during inflammation.
The dramatic downregulation of Fpn with LPS appears to be an important driver of iron accumulation in activated macrophages. Nonetheless, the underlying molecular mechanism is unclear. Bach1 is a negative regulator of Fpn (Igarashi and Watanabe-Matsui, 2014). However, LPS downregulated Fpn in Bach1−/− macrophages to the same extent as in the WT (Figure S3A). The transcription factor Nrf2 is known to promote Fpn and heme oxygenase 1 (Ho1) expression ( Ma,2013). LPS did not significantly alter Nrf2 expression, and Ho1 levels increased with LPS, ruling out Nrf2 transcriptional downregulation as a mediator of LPS-induced suppression of Fpn (Figures S3B and S3C). Blocking new protein synthesis with cycloheximide did not affect Fpn downregulation by LPS, indicating the role of a preformed factor (Figure S3D). Activation of preformed components of the NF-κB pathway play a critical role in LPS signaling, but both pharmacological inhibition and genetic disruption of NF-κB signaling failed to block Fpn downregulation (Figures S3E and S3F). Hence, LPS-induced activation of a preformed factor likely mediates Fpn downregulation, and Spic may facilitate Fpn recovery by suppressing this “unidentified factor.” This idea is also consistent with previous observations that Spic generally acts as a transcriptional repressor.
TLR Activation Induces Spic by a Heme-Independent and NF-κB-Dependent Mechanism
Heme can induce Spic by proteasome-dependent degradation of BACH1 (Haldar et al., 2014; Kayama et al., 2018). Therefore, hemophagocytosis or heme accumulation by activated macrophages may explain TLR-induced Spic. However, in-vitro-cultured BMDMs treated with TLR agonists strongly induced Spic, suggesting a heme-independent mechanism (Figure 5A). LPS treatment of Bach1−/− BMDMs (which constitutively express press Spic) further increased Spic, supporting a BACH1-independent pathway for Spic induction (Figure 5B). LPS induced Spic at a later time point than heme (Figure 5C), and LPS treatment did not reduce Bach1 transcript levels in macrophages (Figure 5D). These results suggest that TLR activation induces Spic by a mechanism distinct from the previously described heme and BACH1-dependent pathway.
Figure 5. NF-κB Is Required for Spic Expression.

(A) BMDMs from WT mice were treated with LPS (100 ng/ml) or TLR9 ligand CpG (30 μg/ml), RNA was extracted 8 h later, and the expression (normalized to 18S rRNA) of Spic was measured (y axis, relative to no treatment) by qRT-PCR.
(B) WT and Bach1−/− Mo-MACs were treated with LPS (100 ng/ml), RNA was harvested 24 h later, and the expression (normalized to Hprt)of Spic was measured (y axis, relative to WT no treatment) by qRT-PCR.
(C) WT BMDMs were treated with LPS (100 ng/ml) or hemin (80 μM), RNA was extracted at indicated (x axis, hours) time points, and the expression (normalized to 18S rRNA)of Spic was measured (y axis, relative to no treatment) by qRT-PCR.
(D) WT Mo-MACs were treated with LPS (1 μg/ml), RNA was extracted at indicated time points (x axis), and expression of Bach1 (normalized to 18S rRNA) was measured (y axis, relative to no treatment) by qRT-PCR.
(E) WT Mo-MACs were treated with LPS (1 μg/ml) with or without an Iκκ-2 inhibitor, Bot64 (10 μM). RNA was extracted 18 h later, and the expression (normalized to 18S rRNA)of Spic was measured (y axis, relative to no treatment) by qRT-PCR.
(F) Mice of indicated genotypes (header) were treated with LPS with or without Bot64 (i.p.). Bot64 treatment (60 mg/kg/day for 3 days) started 24 h before LPS (single dose, 100 μg/mouse) treatment. Peripheral blood and lungs were collected 24 h after LPS treatment. FCS plots (left) show the distribution of indicated markers in peripheral blood (cells pre-gated for CD45+ Ly6G- singlets). The expression (relative to Hprt)of Spic in the lungs (measured by qRT-PCR) is shown in the right plot.
(G) WT or c-rel−/− Mo-MACs were treated with LPS (1 μg/ml), RNA was extracted 16 h later, and the expression (normalized to Hprt)of Spic was measured (y axis, relative to WT no treatment) by qRT-PCR.
(H) WT and Bach1−/− Mo-MACs were treated with an Iκκ-2 inhibitor, Bot64 (10 μM), RNA was extracted 14 h later, and the expression of Spic (normalized to 18S rRNA) was measured (y axis, relative to WT no treatment) by qRT-PCR.
(I) WT Mo-MACs were treated with LPS (1 μg/ml) alone or with cycloheximide (10 μg/ml, added 1 h before LPS) to inhibit new protein synthesis. RNA was harvested 8 h later, and the expression (normalized to Hprt)of Spic was measured (y axis, relative to no treatment) by qRT-PCR.
FCS, numbers represent percentage of cells within indicated gate. (F) Represents 2 experiments with ≥3 mice per group. qRT-PCR, representative of ≥3 independent experiments and graphs show single experiment with n ≥2 per group. Results expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figure S4.
TLR4 activates several latent transcription factors, including NF-κB and AP-1. Although AP-1 inhibition did not affect Spic expression, blocking NF-κB by bot 64, a small molecule inhibitor of the inhibitor of NF-κB kinase beta (Iκκ-2), abrogated Spic induction by LPS in vitro and reduced it in vivo (Figures 5E, 5F, and S4A). Rel knockout BMDMs also showed reduced Spic induction with LPS, further confirming a role of NF-κB(Figure 5G). Notably, NF-κB blockade also reduced Spic in Bach1-deficient macrophages, suggesting a central role for NF-κB in Spic regulation (Figure 5H). Correspondingly, heme-mediated Spic expression also showed dependence on NF-κB activity (Figure S4B). NF-κB may directly promote Spic transcription (primary response gene) or indirectly by transcribing another factor (secondary response gene). To address this, we blocked new protein synthesis by treatment with cycloheximide, which completely blocked Spic induction by LPS (Figure 5I). Taken together, these results show that Spic is a TLR-induced and NF-κB-dependent secondary response gene in activated macrophages.
IFNγ Signaling Suppresses Spic Expression
The aforementioned findings show that Spic downregulates inflammatory responses and promotes iron-efflux in macrophages. Although this is beneficial during the resolution of inflammation, it is detrimental to host defense against pathogens. Infection that activates macrophages is unlikely to be fully resolved within the period of Spic induction (6–8 h after TLR activation;Figure 5C). Hence, induction of Spic in the setting of a true infection appears counter-intuitive, and we wondered whether there are additional constraints on Spic expression. Local IFN levels are elevated during infection. Therefore, we examined whether the presence of IFNs affect Spic expression. Remarkably, IFNγ strongly inhibited LPS-mediated Spic expression in BMDMs, which was dependent on STAT1 activity (Figure 6A). Type-1 IFNs also showed a similar trend, albeit to a much lesser extent (Figure S5A). Correspondingly, pretreatment of mice with IFNγ prior to LPS exposure suppressed Spic induction in vivo (Figures 6B and S5B).
Figure 6. IFNγ Suppresses Spic Expression.

(A) WT BMDMs were treated with LPS (1 μg/ml), IFNγ (50 ng/ml), or STAT1 blocker fludarabine (50 μM). RNA was extracted 20 h later, and the expression (normalized to Hprt)of Spic was measured (y axis, compared to no treatment group) by qRT-PCR.
(B) Mice were treated (i.p.) with LPS (once, 100 μg/mouse) with or without recombinant IFNγ (20 μg/mouse per dose for a total of five doses). IFNγ treatment was started 1 day before LPS injection. Lungs were harvested within 24 h of LPS treatment, RNA extracted, and the expression of Spic was measured (relative to Hprt) by qRT-PCR. Five mice per treatment group.
(C) Anti-IFNγ antibody (200 μg/mouse) was injected i.p. every 24 h for 3 days. At 24 h after the last treatment, mice were euthanized, and indicated organs were harvested. RNA was extracted, and the levels (normalized to Hprt)of Spic (y axis, relative to a non-treated mouse) were measured by qRT-PCR.
(D) WT mice were treated (i.p.) with LPS (150 μg/mouse, three treatments 48 h apart) with or without anti-IFNγ antibody (200 mg/mouse, daily, starting with LPS treatment). Lungs were harvested 24 h after the last LPS treatment, RNA was extracted, and the expression (normalized to Hprt) of Spic (y axis, relative to non-treated mice) was measured by qRT-PCR.
(E) WT and Spic−/− mice were treated (i.p.) with LPS (150 μg/mouse, 5 doses, 48 h apart) with or without anti-IFNγ antibody (200 μg/mouse, daily, starting with LPS treatment). Lungs were harvested 24 h after the final LPS treatment, RNA was extracted, and the levels (normalized to Hprt) of the indicated genes (y axis, relative to non-treated mice) were measured using qRT-PCR (first two graphs). TNF-α levels were also measured using ELISA (final graph) in plasma from blood collected 24 h before sacrificing the mice.
(F) SpicGFP/GFP mice were treated with recombinant IFNγ (20 μg/mouse per dose, every 12 h, for 3 days), followed by purification of splenic RPMs by FACS (using GFP expression). RPMs from untreated SpicGFP/GFP mice served as controls. RNA was extracted, and the expression (relative to Hprt) of indicated genes was measured by qRT-PCR.
(G) RPMs purified from SpicGFP/GFP splenocytes by FACS were cultured with macrophage colony stimulating factor (M-CSF). After 6 h, cells were treated with vehicle, LPS (1 μg/ml), IFNγ (50ng/ml), or IFNγ + LPS. 16 h after treatment, RNA was extracted, and the expression (relative to Hprt)of Spic was measured by qRT-PCR.
(H) WT BMDMs were treated with heme (80 μM) with or without IFNγ (50 ng/ml). 4 h after treatment, RNA was extracted, and expression (relative to Hprt)of Spic was measured by qRT-PCR.
qRT-PCR, data representative of ≥3 independent experiments. Plots show a single experiment with n ≥2 per group. Results expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figure S5.
We showed above that gut macrophages express Spic in response to local microbiota. Consistent with the suppressive effects of IFNγ, treatment with an anti-IFNγ antibody led to increased Spic in colonic macrophages but not lungs, an organ we used as a control due to lower exposure to TLR ligands at steady state than that in the gut (Figure 6C). Furthermore, IFNγ blockade augmented LPS-induced Spic in lungs (Figure 6D). Based on these findings, we wondered whether some of the known anti-inflammatory effects of blocking IFNγ during inflammation might be mediated by higher Spic. Therefore, we compared inflammatory markers in WT and Spic−/− mice treated with LPS and anti- IFNγ and found higher levels of inflammatory markers in the absence of Spic (Figure 6E).
We next asked whether IFNγ could also suppress Spic in macrophages that already express high levels of this transcription factor at the steady state (non-TLR induced). Mice treated with recombinant IFNγ showed a reduced expression of Spic and Fpn in RPMs (Figure 6F). To confirm that a lower Spic expression in this setting reflects the direct action of IFNγ within macrophages, we isolated RPM and exposed them to IFNγ ex vivo, which reduced Spic (Figure 6G). IFNγ also suppressed heme-induced Spic in BMDMs (Figure 6H). Indeed, IFNγ alone further suppressed the very low levels of basal Spic and Fpn in BMDMs (Figure S5C). Hence, the suppressive effects of IFNγ on Spic is independent of the stimuli inducing Spic expression.
Spic Induction in Sterile Inflammation
NF-κB is also activated in macrophages in PAMP-independent sterile inflammation. Because sterile inflammation is usually not associated with high IFNs, we wondered whether this might also be a relevant setting of Spic induction and function. We first examined whether Spic is induced in lung macrophages upon bleomycin exposure, a commonly used model of sterile lung inflammation and fibrosis (Liu et al., 2017). We found significantly elevated Spic in lungs of bleomycin-treated mice compared to controls (Figure 7A). Spic expression was higher at later time points, suggestive of its role during the resolution stage of the injury (Figure 7A). We also examined Spic induction in a different type of sterile inflammation within a different organ, namely, ischemia-reperfusion injury in kidney (Aufhauser et al., 2016). Consistent with our observation in lungs, Spic was significantly elevated in the kidneys 30 days post-injury (Figure 7B). These findings show that sterile inflammation can also induce Spic.
Figure 7. Spic Expression Is Induced in Sterile Inflammation.

(A) WT (C57BL/6J) mice were treated (intratracheal [i.t.]) with bleomycin sulfate (3 U/kg) or water (control). Mice were euthanized 1 or 2 weeks after treatment, RNA was extracted from lungs, and the expression (relative to Hprt)of Spic was measured using qRT-PCR.
(B) Kidneys were harvested from control or 30 days after inducing renal ischemia in WT (C56BL/6J) mice, RNA was extracted, and the expression (relative to Hprt)of Spic was measured by qRT-PCR. Experiment representative of two experiments with n ≥3 mice per group per experiment.
(C) Mice of indicated genotypes were treated (i.t.) with bleomycin sulfate (3 U/kg) and were euthanized 3 weeks later, lungs were harvested and the expression (normalized to Hprt) of indicated genes was measured by qRT-PCR. Data are representative of four independent experiments with ≥ 2 mice per genotype/condition.
(D) Representative model for the regulation and function of Spic. PAMP, pathogen-associated molecular pattern; DAMP, damage-associated molecular pattern.
qRT-PCR, data representative of ≥2 independent experiments. Plots show a single experiment with n ≥2 per group. Results are expressed as mean ± SEM. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), and p ≤ 0.0001 (****). See also Figures S6 and S7.
Tissue macrophages are heterogeneous in origin (monocyte derived versus embryonic) and phenotype. As an example, lungs contain SiglecFhiCD11chiCD11B− alveolar macrophages (embryonic origin), SiglecF−CD11c−CD11B+ MHCIIhi interstitial macrophages (monocyte derived), and SiglecF−CD11c−CD11B+ MHCIIlow interstitial macrophages (monocyte derived) (Chakarov et al., 2019). We next examined whether pathogen-associated and sterile inflammation induce Spic in specific macrophage subsets within the same tissue by using lung as the model. Intraperitoneal LPS induced Spic predominantly in interstitial macrophages, whereas intra-tracheal bleomycin induced it in both alveolar and interstitial macrophages in the lungs (Figure S6). A confounding factor in this analysis is that exposure to PAMPs and DAMPs may alter the expression of key surface markers on macrophages, which can make it difficult to identify the different macrophage subsets. To circumvent this limitation, we used a genetic model where Spic expression does not rely on PAMP or DAMP activation. As described above, Bach1 is a negative regulator of Spic, and Bach1-deficient mice constitutively express high levels of Spic in macrophages. Examination of lung macrophages in Bach1−/− SpicGFP/GFP mice clearly showed Spic in both alveolar and interstitial macrophages (Figures S7A and S7B). Hence, all major macrophage subsets in the lungs appear capable of inducing Spic with appropriate stimuli.
Next, we examined the pathophysiological implications of Spic induction in sterile inflammation. Bleomycin-induced lung injury appeared to engender a stronger fibrotic response in Spic−/− mice than in WT mice based on the expression of collagen 1a1 and Tenascin C, two markers of lung fibrosis (Figure 7C). Finally, we asked whether sterile-inflammation-associated human pathological conditions may be associated with macrophage Spic expression by analyzing a public dataset of single-cell RNA sequencing of renal immune cells from normal and lupus nephritis patients (Arazi et al., 2019). Consistent with our observations in mice, a subset of monocyte and Mo-MAC in nephritic, but not normal, kidney expressed high levels of Spic (Figure S7C).
In summary, we provide evidence of a transcriptional circuitry by which macrophages sense and respond to their inflammatory milieu (Figure 7D). At the core of this mechanism lies counter-regulation of Spic by NF-κB and STAT (IFN signaling). NF-κBis activated in myriad settings in various cell types; yet, the induction of Spic is highly restricted to patrolling monocytes and macrophages, highlighting a lineage-restricted role of this pathway.
DISCUSSION
Activated macrophages release effector molecules that not only control infection but also cause tissue damage. Therefore, macrophage inflammatory responses are downregulated after elimination of the infectious threat. Indeed, macrophages undergo a switch from a pro- to anti-inflammatory phenotype during the resolution of inflammation. Our findings support a role of the transcription factor Spic in facilitating this switch. Although the role of macrophages in systemic iron homeostasis is well known, there is also a growing appreciation of their importance in regulating local iron availability (Winn et al., 2020). Iron is an essential element in many key biological processes, and hence, local iron availability can affect tissue homeostasis. As an example, iron efflux from macrophages was shown to influence tissue repair in the skin (Recalcati et al., 2019). How tissue repair is regulated by local macrophage iron efflux likely depends on the tissue type and/or the nature of the injury, and our study shows that the transcription factor Spic may have a key role in this process.
Two recent studies reported the induction of Spic in inflammation-induced hemophagocytes, which are monocyte-derived cells that phagocytose red cells and other leukocytes (Akilesh et al., 2019; Wang et al., 2019). These hemophagocytes are thought to drive inflammation and cytopenia. Our findings are congruent with these recent reports and extend the field by uncovering the function and regulation of Spic in these inflammatory settings. Furthermore, Spic induction in sterile inflammation and its negative regulation by IFNγ indicate a general role of this transcriptional circuit within activated macrophages.
An intriguing observation in our study is the highly restricted nature of Spic expression. The capacity to induce this transcription factor was restricted to macrophages but not DCs, whereas its expression in monocytes was restricted to the patrolling subset. Although the molecular basis of this specific expression pattern awaits further studies, it underscores the functional distinction between monocyte subsets.
The requirement of NF-κB for macrophage Spic expression is consistent with a previous study that describes a role of NF-κB in Spic expression during B cell development (Bednarski et al., 2016). Macrophages activate NF-κB in response to many other stimuli besides TLR activation. Therefore, it was somewhat surprising that the role of Spic in activated macrophages has not been widely reported. One explanation is the existence of counter-regulatory mechanisms, one of which (IFN dependent) we describe here. It is likely that such inhibitory pathways allow Spic expression only in situations where the threat of infection is very low. This type of counter-regulatory mechanism for Spic expression allows fine-tuning of macrophage inflammatory responses.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for additional information about the manuscript or for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Malay Haldar (mhaldar@pennmedicine.upenn.edu).
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
The accession number for the micarroarray data described in this manuscript isGEO: GSE150520.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Spic−/− and SpicGFP/GFP mice were described before (Haldar et al., 2014; Kohyama et al., 2009). Bach1−/− were generated by the European Conditional Mouse Mutagenesis Program (EUCOMM). c-rel−/− mice were kindly provided by Dr. Youhai H. Chen from the University of Pennsylvania. Spic−/− mice are in 129/SvEv and SpicGFP/GFP mice in C57/6J background. Both male and female mice between 2–12 months of age were used in the experiments.
Mice were genotyped using published primer sets and PCR protocol. Germ-free C57BL/6J mice were obtained from the PennCHOP Microbiome Program Gnotobiotic Core facility. The university of Pennsylvania Institutional Animal Care and Use Committee approved all mouse experiments.
Inflammation models
For pathogen-associated inflammation, Escherichia coli-derived lipopolysaccharides were injected intraperitoneally (100–150 μg of LPS in sterile1X PBS, 200 μL total volume). For IFNγ blockade, 200 μg of anti-mouse IFNγ antibody in 200 μL of total volume was injected (intraperitoneal).
For sterile pulmonary inflammation, bleomycin (at 3U/kg; Fresenius Kabi) or water was instilled intratracheally in wild-type C57BL/6 mice. The mice were euthanized at various time points after injury and the lungs harvested and processed for downstream experiments.
For inducing kidney ischemia reperfusion injury (IRI), mice were anesthetized with pentobarbital sodium (65 mg/kg IP) and placed in a temperature-controlled operative apparatus. Core body temperature was continuously measured and maintained at 36.0 ± 0.5° C. Under an operating microscope, the left renal pedicle exposed and clamped for 28 min with a microvascular clip (Roboz Surgical Instrument, Gaithersburg, MD). After the clamp was released, the right kidney was exposed and removed. After closure, animals were subcutaneously injected with 100 mL/kg of warm saline after the operation to ensure hydration. Animals were kept in an incubator (37° C) until awake. Mice were given access to water ad-lib post-procedure. All animal protocols adhered to the NIH Guide for the Care and Use of Laboratory Animals and were performed in an AAALAC accredited facility.
Cell culture
Bone marrow derived macrophage (BMDM): Total bone marrow cells were flushed out of femur, red cells removed using RBC lysis buffer, and remaining cells cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 10% FCS and supplemented with 20 ng/ml M-CSF. Macrophages were generated after 7–9 days in this culture.
Monocyte-derived macrophages (Mo-MACs): Monocytes were isolated from bone marrow cells using the ‘monocyte-isolation kit (BM)’ from Miltenyi Biotech and following manufacturer’s protocol. Monocytes were then cultured in IMDM + 10%FCS supplemented with M-CSF to generate Mo-MACs after 3–5 days in culture.
In vitro treatments: Cell culture media from BMDMs or Mo-MACs were removed and replaced with fresh media (without M-CSF) containing TLR ligands and/or drugs at indicated doses. TLR ligands: LPS (100 to 1000 ng/ml) and CpG (30 μg/ml), IKK-2 inhibitor (10 μM), cycloheximide (10 μg/ml), AP1 inhibitor (10 μM), STAT1 blocker (50 μM), and PI3K-gamma inhibitor (100 nM).
METHOD DETAILS
Tissue harvest and flow cytometry
Organs were harvested from euthanized mice, washed with sterile PBS, and cut into small pieces (1–3 mm). Tissue pieces were then digested with an enzyme cocktail (5 ml) comprised of DMEM (with 10% FBS) containing collagenase at 0.25 mg/ml (Roche) and DNase I at 30 U/ml (Sigma-Aldrich). Tissue digestion occurred at 37° C for 45 min with constant stirring. After the digestion, the materials were filtered through 70-μm nylon filter (Celltreat Scientific Product), RBC lysed, and single-cell suspensions.
For flow cytometry, cells were counted and incubated with fluorescently tagged antibodies in MACS buffer (1X PBS, 0.5 mM EDTA, and 0.5% BSA).
Gene expression profiling by microarray
Microarray was performed at the UPenn Molecular Profiling Facility, including quality control tests of the total RNA samples by agilent bioanalyzer and nanodrop spectrophotometry. All protocols were conducted as described in the Affymetrix WT Pico Reagent Kit Manual and the Affymetrix GeneChip Expression Analysis Technical Manual. Gene expression data were normalized and values modeled using ArrayStar4 (DNASTAR). Microarray reported here is deposited in Gene Expression Omnibus (GEO: GSE150520).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from tissues and cells using the GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich) or RNeasy Mini Kit (QIAGEN). Reverse transcription of mRNA was performed using the High-Capacity RNA-to-cDNA Kit (Life Technologies). qRT-PCR was performed using a ViiA7 Real-Time PCR system. All probes were obtained from TaqMan.
Iron Quantitation
BMDMs were generated in 75 mm flask in I-10F media (10 mL), containing 20 ng/mL of M-CSF. After 7 days of culture, the media was replaced with 10 mL of fresh I-10F media containing 100 mM of FeSO4 (Sigma Aldrich F8633). Cells were then treated with LPS (1000 ng/mL) or control (PBS). 24 h later the media was removed and the adherent BMDMs were washed 3X with sterile ice-cold PBS. BMDMs were then detached with trypsin (GIBCO, 0.25%) and pelleted by centrifugation at 450xg for 5 min. Supernatant was removed and the cells re-suspended in 1 mL of IL-10F media and cell numbers counted to ensure similar numbers of cells in each assay condition. The cell suspension were spun down again (450xg for 5 min) and re-suspended in 400 uL of Iron assay buffer. Cells were next sonicated (1 min/sample) and spun down at 1300xg for 5 min. The supernatant was collected and stored in −80 freezer. Iron assay was performed on the stored supernatant using the Iron Assay Kit (Abcam, catalog no: 83366) and following the manufacturer’s protocol.
Measurement of cytokines by ELISA
Cell culture supernatant were collected and stored in −80°C until cytokines concentrations were quantified by ELISA. By following the protocol as provided by the manufacturer, the concentrations of TNFα and IL1β were measured using the mouse TNF alpha and IL1 beta ELISA Kit.
QUANTIFICATION AND STATISTICAL ANALYSIS
To calculate the significance for two individual groups, unpaired t test were performed. To compare the mean of three or more groups, one-way ANOVA with Tukey’s multiple comparison tests were used. p values of < 0.05 (*), < 0.01 (**), < 0.001 (***) and < 0.0001 (****) were considered statistically significant. Statistically non-significant is indicated as ns. Data were analyzed using the GraphPad Prism Software (Prism 5).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse-PECY7-CD11b antibody | Invitrogen | Cat# 25–0112–82 |
| Anti-mouse-APC/CY7-CD115 antibody | Biolegend | Cat# 135531 |
| Anti-mouse-AF647-CD64 antibody | BD Bioscience | Cat# 558539 |
| Anti-mouse-PE-CD103 antibody | Biolegend | Cat# 121406 |
| Anti-mouse-BV510-Ly6G antibody | Biolegend | Cat# 127633 |
| Anti-mouse-APC-CD115 antibody | eBioscience | Cat# 17–1152–82 |
| Anti-mouse-APC/CY7-CD11C antibody | Biolegend | Cat# 117324 |
| Anti-mouse-BV510-CD45 antibody | Biolegend | Cat# 103138 |
| Anti-mouse-APC/CY7-Ly6G antibody | Biolegend | Cat# 127624 |
| Anti-mouse-BV510-MHC II antibody | Biolegend | Cat# 107635 |
| Anti-mouse-APC-CD11b antibody | Biolegend | Cat# 101212 |
| Anti-mouse-AF488-F4/80 antibody | Bio Rad | Cat# MCA497A488 |
| Anti-mouse-Percpcy5.5-Ly6G antibody | Biolegend | Cat# 127615 |
| Anti-mouse-APC-F4/80 antibody | Invitrogen | Cat# MF48005 |
| Anti-mouse-PECY7-MerTK antibody | eBioscience | Cat# 25–575–82 |
| Anti-mouse-BV605-CD45 antibody | Biolegend | Cat# 103139 |
| Anti-mouse-PECY7-CD301 antibody | Biolegend | Cat# 145706 |
| Anti-mouse-AF488-CD45 antibody | Biolegend | Cat# 103122 |
| Purified from rabbit-PE- FPN- antibody | Novus | Cat# F-1–062918-PE |
| Anti-mouse-PE-TremL4-antibody | Biolegend | Cat# 143304 |
| Anti-mouse-BV421-CD206 antibody | Biolegend | Cat# 141717 |
| Anti-mouse-BV605-Ly6C antibody | Biolegend | Cat# 128036 |
| Anti-mouse-Percpcy5.5-CD45 antibody | Biolegend | Cat# 103132 |
| Anti-mouse-BV605-CD11b antibody | Biolegend | Cat# 101257 |
| Anti-mouse-APC/CY7-Ly6C antibody | Biolegend | Cat# 128026 |
| Anti-mouse-BV421-MHC II antibody | Biolegend | Cat# 107631 |
| Anti-mouse-BV421 -SiglecF antibody | BD Biosciences | Cat# 562681 |
| Anti-mouse-APC-SiglecF antibody | Biolegend | Cat# 155508 |
| Anti-mouse-PECY7-Tim-4 antibody | Biolegend | Cat# 130009 |
| Anti-mouse-BV421-CD4 antibody | Biolegend | Cat# 100437 |
| Ultra-LEAF Purified anti-mouse IFN-γ Antibody | BioLegend | Cat# 505847 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipopolysaccharide | Sigma-Aldrich | Cat# L4391–10X1 MG |
| Bleomycin | Fresenius Kabi | DIN: 02265982 |
| BOT-64, IKK-2 inhibitor | Fisher Scientific | Cat# AAJ64555LB0 |
| T-5224 (AP-1 Inhibitor) | Cayman Chemical | Cat# 22904 |
| Phosphate Buffered Saline (PBS) | Life Technologies | Cat# 13151014 |
| UltraPure 0.5M EDTA | Invitrogen | Cat# 15575–038 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher | Cat# 15140122 |
| L-Glutamine | Thermo Fisher | Cat# 25030081 |
| GIBCO-Non Essential Amino Acid | Fisher Scientific | Cat# 11140050 |
| Sodium Pyruvate (100 mM) | Thermo Fisher | Cat# 11360070 |
| 2-mercaptoethanol | Fisher Scientific | Cat# 21985023 |
| Fludarabine 10mM | Selleck chemicals | Cat# S1491 |
| 7AAD Viability staining | Biolegend | Cat# 420404 |
| Murine GM-CSF | PeproTech | Cat# 315–03–20ug |
| Murine M-CSF | PeproTech | Cat# 315–02–50ug |
| Murine INF-gamma | PeproTech | Cat# 315–05–1 OOug |
| TaqMan Universal Master Mix | Thermo Fisher | Cat# 4304437 |
| Critical Commercial Assays | ||
| TNF alpha ELISA Kit, Mouse | Thermo Fisher | Cat# BMS607–3 |
| Monocyte Isolation Kit (BM), mouse | Miltenyi Biotec | Cat# 130–100–629 |
| High Capacity RNA to cDNA kit | Thermo Fisher | Cat# 4387406 |
| Genelute Mammalian Total RNA isolation kit | Sigma-Aldrich | Cat# RTN70–1KT |
| Iron assay kit | Abeam | Cat# ab83366 |
| Deposited Data | ||
| Microarray data | This study | GSE150520 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Spic-Knockout | Kohyama et al., 2009 | PMID: 19037245 |
| Mouse: Spic-GFP | Haidar et al., 2014 | PMID: 24630724 |
| Mouse: Bach 1-Knockout | EUCOMM | MGL5009633 |
| Mouse: cREL-Knockout | Dr. Hsiou-Chi Liou | PMID: 12235116 |
| Oligonucleotides | ||
| SpiC (TaqMan primers and probe) | Thermo Fisher | Mm00488428_m1 |
| Arg1 (TaqMan primers and probe) | Thermo Fisher | Mm00475988_m1 |
| IL10 (TaqMan primers and probe) | Thermo Fisher | Mm01288386_m1 |
| TNFalpha (TaqMan primers and probe) | Thermo Fisher | Mm00443258_m1 |
| NOS2 (TaqMan primers and probe) | Thermo Fisher | Mm00440502_m1 |
| IL-1beta (TaqMan primers and probe) | Thermo Fisher | Mm00434228_m1 |
| IL-6 (TaqMan primers and probe) | Thermo Fisher | Mm00446190_m1 |
| Bachl (TaqMan primers and probe) | Thermo Fisher | Mm01344527_m1 |
| Ferroportin (Slc4Qa1) (TaqMan primers and probe) | Thermo Fisher | Mm01254822_m1 |
| Fth1 (TaqMan primers and probe) | Thermo Fisher | Mm00850707_g1 |
| Ftl1 (TaqMan primers and probe) | Thermo Fisher | Mm03030144_g1 |
| Nrf2 (TaqMan primers and probe) | Thermo Fisher | Mm04231240_s1 |
| Hamp (TaqMan primers and probe) | Thermo Fisher | Mm00477784_m1 |
| Hmoxl (TaqMan primers and probe) | Thermo Fisher | Mm00516005_m1 |
| Col1a1 (TaqMan primers and probe) | Thermo Fisher | Mm00801666_g1 |
| Tnc (TaqMan primers and probe) | Thermo Fisher | Mm00495662_m1 |
| HPRT (TaqMan primers and probe) | Thermo Fisher | Mm03024075_m1 |
| Euk 18S rRNA (TaqMan primers and probe) | Life Technologies | Cat# 4333760F |
| Software and Algorithms | ||
| GraphPad Prism 8 | Graph Pad Software | N/A |
| FlqwJo LLOVI0.1 | FlowJo LLC | N/A |
| Gene Set Enrichment Analysis (GSEA) | Broad institute | N/A |
| Arraystar 4 | DNASTAR | N/A |
| Other | ||
| Iscove’s Modified Dulbecco’s Medium (IMDM) | Thermo Fisher | Cat# 12440053 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher | Cat# 10567014 |
| Bovine Serum Albumin (BSA) | VWR | Cat# 97061–420 |
| Fetal Bovine Serum (FBS) | GeminiBio | Cat#100–500 |
| Collagenase B | Roche | Cat# 11088831001 |
| DNase I | Sigma-Aldrich | Cat# D4527–40KU |
Highlights.
The transcription factor Spic restrains inflammatory responses in macrophages
Spic promotes the expression of the iron exporter ferroportin in activated macrophages
NF-κB activity is required for the expression of Spic in activated macrophages
Interferon-gamma suppresses Spic expression in activated macrophages
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health grant NIAID-KO8AI106953 (M.H.), a Burroughs Wellcome Fund (BWF) Career Awards for Medical Scientists (CAMS) (M.H.), and an American Society of Hematology (ASH) scholar award (M.H.). We thank Dr. Hsiou-chi Liou for providing the c-Rel knockout mice.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107825.
REFERENCES
- Abreu R, Quinn F, and Giri PK (2018). Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. 2, 1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, Gessay G, Whalen E, Mason M, Presnell SR, et al. (2019). Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 363, eaao5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam MZ, Devalaraja S, and Haldar M (2017). The Heme Connection: Linking Erythrocytes and Macrophage Biology. Front. Immunol. 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoov Chicoine A, Eisenhaure TM, Jonsson AH, Li S, et al. ; Acce Medicines Partnership in SLE network (2019). The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 20, 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufhauser DD Jr., Wang Z, Murken DR, Bhatti TR, Wang Y, Ge G, Redfield RR III, Abt PL, Wang L, Svoronos N, et al. (2016). Improved renal ischemia tolerance in females influences kidney transplantation out-comes. J. Clin. Invest. 126, 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, and Mowat AM (2013). Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarski JJ, Pandey R, Schulte E, White LS, Chen B-R, Sandoval GJ, Kohyama M, Haldar M, Nickless A, Trott A, et al. (2016). RAG-mediated DNA double-strand breaks activate a cell type-specific checkpoint to inhibit pre-B cell receptor signals. J. Exp. Med. 213, 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, Durai V, Grajales-Reyes GE, Iwata A, Bagadia P, et al. (2016). Distinct Transcription Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep. 15, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang X-M, Foo S, Nakamizo S, Duan K, et al. (2019). Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964. [DOI] [PubMed] [Google Scholar]
- Drakesmith H, and Prentice AM (2012). Hepcidin and the iron-infection axis. Science 338, 768–772. [DOI] [PubMed] [Google Scholar]
- Drakesmith H, Nemeth E, and Ganz T (2015). Ironing out Ferroportin. Cell Metab. 22, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T, and Nemeth E (2015). Iron homeostasis in host defence and inflammation. Nat. Rev. Immunol. 15, 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, and Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Gordon S, Pluddemann A, and Martinez Estrada F (2014). Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol. Rev. 262, 36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guida C,Altamura S,Klein FA,Galy B,Boutros M,Ulmer AJ,Hentze MW, and Muckenthaler MU (2015). A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 125, 2265–2275.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, and Murphy KM (2014). Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 262, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar M, Kohyama M, So AY-L, Kc W, Wu X, Briseno CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, et al. (2014). Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, Higashimura Y, Katada K, Handa O, Ishikawa T, et al. (2013). BTB and CNC homolog 1 (Bach1) deficiency ameliorates TNBS colitis in mice: role of M2 macrophages and heme oxygenase-1. Inflamm. Bowel Dis. 19, 740–753. [DOI] [PubMed] [Google Scholar]
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, and Reis e Sousa C (2015). GM-CSF Mouse Bone Marrow Cultures Comprise a Heterogeneous Population of CD11c(+)MHCII(+) Macrophages and Dendritic Cells. Immunity 42, 1197–1211. [DOI] [PubMed] [Google Scholar]
- Hu X, and Ivashkiv LB (2009). Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K, and Watanabe-Matsui M (2014). Wearing red for signaling: the heme-bach axis in heme metabolism, oxidative stress response and iron immunology. Tohoku J. Exp. Med. 232, 229–253. [DOI] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. (2016). PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayama H, Kohyama M, Okuzaki D, Motooka D, Barman S, Okumura R, Muneta M, Hoshino K, Sasaki I, Ise W, et al. (2018). Heme ameliorates dextran sodium sulfate-induced colitis through providing intestinal macrophages with noninflammatory profiles. Proc. Natl. Acad. Sci. USA 115, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, Frazier WA, Murphy TL, and Murphy KM (2009). Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 457, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu Y-J, Rea TH, et al. (2005). TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat. Med. 11, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, De Los Santos FG, and Phan SH (2017). The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 1627, 27–42. [DOI] [PubMed] [Google Scholar]
- Ma Q (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, and Horng T (2009). Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Plüddemann A, and Gordon S (2009). Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. Adv. Exp. Med. Biol. 653, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ (2017). Macrophage Polarization. Annu. Rev. Physiol. 79, 541–566. [DOI] [PubMed] [Google Scholar]
- Murray PJ, and Wynn TA (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, and Manabe I (2018). Macrophages in inflammation, repair and regeneration. Int. Immunol. 30, 511–528. [DOI] [PubMed] [Google Scholar]
- Recalcati S, Gammella E, Buratti P, Doni A, Anselmo A, Locati M, and Cairo G (2019). Macrophage ferroportin is essential for stromal cell proliferation in wound healing. Haematologica 104, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, and Nerlov C (2009). A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 106, 17475–17480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 209, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T, Strangward P, Ridley AJL, Wang P, Tamoutounour S, et al. (2018). Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 215, 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MP, and Hamza I (2016). Macrophages and Iron Metabolism. Immunity 44, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thäle C, and Kiderlen AF (2005). Sources of interferon-gamma (IFN-gamma) in early immune response to Listeria monocytogenes. Immunobiology 210, 673–683. [DOI] [PubMed] [Google Scholar]
- Wang A, Pope SD, Weinstein JS, Yu S, Zhang C, Booth CJ, and Medzhitov R (2019). Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice. Proc. Natl. Acad. Sci. USA 116, 2200–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn NC, Volk KM, and Hasty AH (2020). Regulation of tissue iron homeostasis: the macrophage “ferrostat”. JCI Insight 5, e132964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, and Vannella KM (2016). Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Mosser DM (2008). Macrophage activation by endogenous danger signals. J. Pathol. 214, 161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the micarroarray data described in this manuscript isGEO: GSE150520.