

Abstract
Recent advances in sample preparation enable label-free mass spectrometry (MS)-based proteome profiling of small numbers of mammalian cells. However, specific devices are often required to downscale sample processing volume from the standard 50–200 μL to sub-μL for effective nanoproteomics, which greatly impedes the implementation of current nanoproteomics methods by the proteomics research community. Herein, we report a facile one-pot nanoproteomics method termed SOPs-MS (surfactant-assisted one-pot sample processing at the standard volume coupled with MS) for convenient robust proteome profiling of 50–1000 mammalian cells. Building upon our recent development of SOPs-MS for label-free single-cell proteomics at a low μL volume, we have systematically evaluated its processing volume at 10–200 μL using 100 human cells. The processing volume of 50 μL that is in the range of volume for standard proteomics sample preparation has been selected for easy sample handling with a benchtop micropipette. SOPs-MS allows for reliable label-free quantification of ∼1200–2700 protein groups from 50 to 1000 MCF10A cells. When applied to small subpopulations of mouse colon crypt cells, SOPs-MS has revealed protein signatures between distinct subpopulation cells with identification of ∼1500–2500 protein groups for each subpopulation. SOPs-MS may pave the way for routine deep proteome profiling of small numbers of cells and low-input samples.
Keywords: SOPs-MS, nanoproteomics, small numbers of cells, surfactant DDM, colon crypt cells
Graphical Abstract:
INTRODUCTION
Cells are the building blocks and functional units of life. Cell heterogeneity is a characteristic feature of all living organisms and is inherent in cellular processes.1,2 Due to current limitations of proteomics technologies, a large number of mixed populations of cells (i.e., bulk cells) are needed to generate comprehensive quantitative proteome data.3–5 However, such bulk measurements average out stochastic variations of individual cells and thus obscure important cell-to-cell variability (i.e., cell heterogeneity).6,7 Furthermore, precious clinical samples are often available in low amounts (e.g., hundreds of tumor cells from fine-needle aspiration or microdissected cells), which cannot be readily analyzed by current proteomics platforms. Therefore, there is an urgent need for nanoproteomics platforms capable of quantitative proteome profiling of small numbers of cells and low-input samples (10 ng of protein mass equivalent to 100 human cells).
Mass spectrometry (MS)-based proteomics has emerged as a powerful technique for providing genome-scale quantitative proteome profiling, but it suffers from the typical requirement of large amounts of starting materials (e.g., ∼100–1000 μg or ∼1–10 million human cells) for comprehensive proteomic analysis.3–5,8 Thus, it cannot be used for many potentially important proteomic studies where only low-input samples (e.g., ng levels of starting materials) are available. With this recognition, there are great efforts focusing on the development of effective preparation of low-input samples for MS-based nanoproteomics. For example, the SP3 protocol allows reproducible quantification of 500 proteins from 100 HeLa cells.9 With improvements in reducing surface adsorption losses, a conventional in-solution sample handling method can identify ∼829 proteins from 1000 U2OS cells.10 A miniaturized filter-aided sample preparation (micro-FASP) allows reliable identification of ∼500 proteins from 100 MCF7 cells with the MS/MS spectra only.11 However, all these easily implementable nanoproteomics methods do not provide reliable deep proteome profiling, such as identification of >1000 proteins from 100 cells based on the MS/MS spectra only that is the cornerstone of MS-based proteomics.12 Furthermore, these methods involve several sample transfer steps that can lead to surface adsorption losses. In parallel, MS-based nanoproteomics for analysis of small numbers of cells can be achieved either by reducing the sample processing volume (e.g., using nanoPOTS,13 OAD,14 iPAD-1,15 and ISPEC16 devices downscaling the processing volume from the standard ∼50–100 μL to ∼2–2000 nL) or by using excessive amounts of exogenous bovine serum albumin (BSA) as the carrier protein (e.g., carrier-assisted liquid chromatography (LC)–SRM termed cLC–SRM for targeted proteomics).17–19 However, all these approaches have drawbacks, which greatly impede the progress of current nanoproteomics. NanoPOTS, OAD, iPAD-1, and ISPEC are not easily adoptable for broad benchtop applications.13–16 Exogenous BSA protein carrier approaches are more suitable for targeted proteomics since BSA peptides are frequently sequenced by MS/MS, which greatly reduces the chance for sequencing low-abundant endogenous peptides for global proteomics.17–19
To alleviate the shortcomings of existing nanoproteomics approaches, we and others have made significant progress in the development of broadly adoptable, convenient nanoproteomics methods for label-free proteome profiling of small numbers of cells including single cells.20,21 For example, very recently, we have developed an easily implementable nonionic surfactant n-dodecyl β-d-maltoside (DDM)-assisted one-pot sample preparation (SOP) at the low μL processing volume for label-free single-cell proteomics.20 To further improve its conveniency and robustness, we have systematically optimized the SOP processing volume. We have demonstrated that 100 human cells can be effectively processed in the range of volume for standard proteomics sample preparation with identification of >1200 protein groups with the MS/MS spectra only. When we applied it to analyze small subpopulations of mouse colon crypt cells, this facile one-pot nanoproteomics method revealed distinct protein signatures between any two subpopulations (i.e., cellular heterogeneity) with identification of ∼1500– 2500 protein groups from ∼140 to 1700 subpopulation cells. These results demonstrate the potential of this method for broad applications in the biomedical research.
EXPERIMENTAL SECTION
Cell Culture
The MCF10A breast cell line was obtained from the American Type Culture Collection (Manassas, VA) and was grown in culture media, as previously described.22 Briefly, MCF10A cells were cultured and maintained in 15 cm dishes in ATCC-formulated Eagle’s minimum essential medium (Thermo Fisher Scientific) supplemented with 0.01 mg/mL human recombinant insulin and a final concentration of 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) with 1% penicillin/streptomycin (Thermo Fisher Scientific). Cells were grown at 37 °C in 95% O2 and 5% CO2. Cells were seeded and grown until near confluence.
Mouse Colons
All mouse work was performed in accordance with NIH guidelines and was approved by the Institutional Animal Care and Use Committee of the University of California, Irvine (approval numbers AUP-17–053). Male C57BL/6N(NJ) mice aged 5–7 weeks were obtained from the Knockout Mouse Project repository.23 Colonic crypt cells were isolated as previously described.24 In brief, mouse colons (cecum to rectum) were removed, flushed, and linearized. Tissue was dissociated at a slow rotation at 4 °C for 1 h in a solution of 2 mM ethylenediamine tetraacetic acid and 10 μM Rock inhibitor. Aggressive shaking of the tissue solution, filtering (using 100 μm followed by 40 μm filters), and centrifugation (500–1000g for 5–10 min at 4 °C depending on the step) were performed for tissue dissociation into single cells.
Small Numbers of Cells Sorted by Fluorescence-Assisted Cell Sorting
Prior to cell collection, polymerase chain reaction (PCR) tubes or 96-well PCR plates were pretreated with 0.1% DDM for surface coating followed by the removal of DDM solution. The pretreated PCR tubes or 96-well PCR plates were air-dried in the fume hood. MCF10A cells were detached from the culture dish and dispersed into a single-cell suspension to avoid cell clumping by passing through a 25-gauge needle three times. The cells were then suspended in phosphate-buffered saline (PBS) and collected by centrifugation for 5 min at 500g. This process was repeated five times to remove the remaining PBS and trypsin. The cells were then resuspended in PBS and passed through a 35 μm mesh cap (BD Biosciences, Canaan, CT) to remove large aggregates. A BD Influx flow cytometer (BD Biosciences, San Jose, CA) was used to sort 100 MCF10A cells into the precoated PCR tubes and small numbers of MCF10A cells into the precoated 96-well PCR plates with the 1-drop single sort mode. After isolation of the desired number of cells, the PCR tubes and 96-well plates were immediately centrifuged at 1000g for 10 min at 4 °C to keep the cells at the bottom of the well to avoid potential cell loss. For the PCR tubes with 100 MCF10A cells, the sample volume was not adjusted with ∼2 μL per tube from the droplets of fluorescence-assisted cell sorting (FACS) isolation. For small numbers of cells collected in the 96-well PCR plates, various volumes of 25 mM ammonium bicarbonate (ABC) were added to bring up the final volume to ∼50 μL for each well (e.g., the addition of ∼30 μL of 25 mM ABC to the well with 1000 MCF10A cells because of ∼20 μL from the droplets of FACS isolation). The 96-well plates were stored in a −80 °C freezer until further analysis.
Mouse colon crypt cells were sorted on a BD FACS Aria Fusion using a 100 μm nozzle (20 PSI) at a flow rate of 2.0 with a maximum threshold of 5000 events/sec. The sample chamber and collection tubes were kept at 4 °C. Following exclusion of debris and singlet/doublet discrimination, small populations were sorted into PCR tubes containing 50 μL of 100 mM ABC. At least 100 cells were sorted for each subpopulation, and tubes were promptly spun down and frozen at −80 °C until further proteomic processing.
Cell Lysis and Trypsin Digestion
For proteome profiling of small numbers of cells in a more convenient robust manner, 100 FACS-sorted MCF10A cells in the PCR tubes were used for optimization of sample processing volume in the range of 10–200 μL. 2 μL of 0.2% DDM in 25 mM ABC was added to each PCR tube followed by the addition of 25 mM ABC to bring up the final processing volume to 10, 20, 50, 100, and 200 μL (e.g., the addition of 46 μL of 25 mM ABC to the tube with the processing volume of 50 μL because of ∼2 μL from the droplets for FACS isolation of 100 cells). Three replicates per processing volume were analyzed with the total of 15 samples. The samples were gently shaken for mixing for 5 min at ∼500g and then centrifugated for 3 min at 3000g. After incubation with DDM for overnight at 4 °C, intact cells were sonicated at 1 min intervals for five times over ice for cell lysis and centrifuged for 3 min at 3000g. Samples were incubated at 75 °C using the thermocycler for 1 h for protein denaturation and then centrifugated for 3 min at 3000g. 1 μL of 10 ng/μL trypsin (Promega) in 25 mM ABC was added to each tube with a total amount of 10 ng. Samples were digested overnight (∼16 h) at 37 °C with gentle shaking at ∼500g. After digestion, 2 μL of 5% formic acid (FA) was added to the tube to stop enzyme reaction. The final sample volume was adjusted to ∼20 μL either by the addition of 25 mM ABC or by SpeedVac to reduce the sample volume for direct LC injection. Samples were then centrifuged for 1 h at 6000g. The sample PCR tube was inserted into the LC vial. They were either analyzed directly or stored at −20 °C for later LC–MS analysis.
50–1000 cells were collected into DDM-treated PCR wells of the 96-well plate with the total sample volume of ∼50 μL for each well. 2 μL of 0.2% DDM in 25 mM ABC was added to the PCR tube or each well of the 96-well plate. The same procedure as described above for cell lysis, and protein denaturation was used for analysis of a wide range of small numbers of cells. For the cell number <500, 1 μL of 10 ng/μL trypsin (Promega) in 25 mM ABC was added with a total amount of 10 ng. For the cell number ≥ 500, 2 μL of 10 ng/μL trypsin (Promega) in 25 mM ABC was added with a total amount of 20 ng. Samples were digested overnight (∼16 h) at 37 °C with gentle shaking at ∼500g. After digestion, 2 μL of 5% FA was added to the well to stop the enzyme reaction. The sample volume was reduced to ∼20 μL by SpeedVac for direct LC injection. Samples were then centrifuged for 1 h at 6000g. The 96-well PCR plate was sealed with a matt. They were either analyzed directly or stored at −20 °C for later LC-MS analysis.
LC–MS/MS Analysis
Peptide digests from small numbers of cells were analyzed using a commonly available Q Exactive Plus Orbitrap MS (Thermo Scientific, San Jose, CA). The standard LC system consisted of a PAL autosampler (CTC ANALYTICS AG, Zwingen, Switzerland), two Cheminert six-port injection valves (Valco Instruments, Houston, USA), a binary nanoUPLC pump (Dionex UltiMate NCP-3200RS, Thermo Scientific), and an HPLC sample loading pump (1200 Series, Agilent, Santa Clara, USA). Both SPE precolumn (150 μm i.d., 4 cm length) and LC column (50 μm i.d., 70 cm Self-Pack PicoFrit column, New Objective, Woburn, USA) were slurry-packed with 3 μm C18 packing material (300 Å pore size) (Phenomenex, Terrence, USA). The sample was fully injected into a 20 μL loop and loaded onto the SPE column using buffer A (0.1% formic acid in water) at a flow rate of 5 μL/min for 20 min. Parameters for LC gradient and MS data acquisition have been previously described.20
Data Analysis
The freely available open-source MaxQuant software25 was used for protein identification and quantification. The MS raw files were processed with MaxQuant (Version 1.6.2.0), and MS/MS spectra were searched by Andromeda search engine against the human or mouse UniProt database (fasta file dated November 5, 2019) [with the following parameters: tryptic peptides with up to two missed cleavage sites; 10 ppm parent ion tolerance; 0.6 Da fragment ion mass tolerance; variable modifications (methionine oxidation)]. Search results were processed with MaxQuant and filtered with a false discovery rate ≤ 1% at both protein and peptide levels. For label-free quantification (LFQ), the match between runs (MBR) function was activated with a matching window of 0.4 min and the alignment window of 20 min. Protein quantification was performed by using the LFQ function.
The search result files “peptides.txt” and “proteinGroups.txt” were used for further data processing. For each sample, the number of unique peptides/proteins identified by MS/MS only was obtained by counting these with “By MS/MS” in the identification type column. To count the identification number when the MBR is enable, peptides/proteins identified “By MS/MS” and “By matching” were both accepted. Statistical significance in peptide/protein identification among different groups was determined by one-way ANOVA. The coefficient of variation (CV) was calculated using the raw LFQ intensities among biological replicates. Quantification correlation was determined by Pearson’s correlation using the log 2 transformed intensities. Proteins quantified in all samples were used for unsupervised hierarchical clustering with the Ward’s method in R (Version 4.0.2). Comparative proteome analysis was performed by first conducting Student’s t-test between two groups and then plotting the resulting difference (as log 2 fold change on the x axis) and statistical significance (as −log 10 p value) in a volcano plot. Significant proteins were defined as those showing >50% in fold change and p < 0.01.26
RESULTS AND DISCUSSION
Facile One-Pot Nanoproteomics at the Standard Processing Volume
The major sample loss in nanoproteomics can be attributed to contact surface adsorption of proteins and/or peptides for low-input samples during sample processing. Standard proteomics preparation work flow for analysis of bulk samples is problematic for nanoproteomics because it involves multiple sample transfer steps, and surface adsorption losses are negligible for large starting materials (e.g., less than 0.5% assuming 0.5 μg of sample loss equivalent to ∼5000 human cells for ∼100 μg). We previously used BSA as a protein carrier to reduce surface adsorption losses for targeted proteomics analysis of small numbers of cells including single cells.17–19 Unfortunately, the addition of BSA is not suitable for global proteomics analysis.
Surfactants are an alternative to BSA for effective prevention of protein surface adsorption. Very recently, we have developed surfactant-based SOP-MS at the low μL processing volume for label-free single-cell proteomics.20 SOP-MS combines all steps into one pot (e.g., a single PCR tube or a single well from a multi-well PCR plate routinely used for low-input genomics and transcriptomics) including collection of single cells, multistep sample processing, and elimination of all sample transfer steps with direct sample loading for LC–MS analysis. This “all-in-one” SOP-MS method maximizes sample recovery by greatly reducing possible surface absorption losses.20
With its demonstration for single-cell proteomics analysis at the low μL volume, it is feasible to operate SOP-MS at the standard sample processing volume (e.g., ∼50–100 μL) with a benchtop micropipette for more convenient robust nanoproteomics. To test this assumption, we have systemically evaluated the SOP processing volume in the range of ∼10–200 μL for the development of facile SOPs-MS (surfactant-assisted one-pot sample processing at the standard volume coupled with MS) (Figure 1). A commonly accessible Q Exactive Plus MS platform was used for optimization and evaluation of SOPs-MS and its application demonstration.
Figure 1.

Schematic diagram of the SOPs-MS work flow at the standard sample processing volume (∼50 μL). Small subpopulations of cells are isolated by FACS into either PCR tubes or 96-well PCR plates (top panel). Surfactant DDM-assisted one-pot sample processing is conducted in either a single PCR tube or a PCR well without sample transfer including cell lysis, protein denaturation, reduction and alkylation (these two steps are optional), and trypsin digestion (middle panel). Prior to LC–MS analysis, the sample volume is reduced to ∼20 μL for full injection. The cap of the PCR tube is removed, and the tube is inserted into the LC vial to avoid transfer loss, and the 96-well cap matt is used to cover the 96-well plate for automatic injection without sample transfer (bottom panel). Samples are analyzed by standard LC–MS platforms for quantitative label-free proteomic analysis.
Optimization of Sample Processing Volume Using FACS-Isolated 100 MCF10A Cells
100 MCF10A cells were sorted directly into single low-bind PCR tubes by FACS (100 cells per tube). Under the same digestion conditions (e.g., the same amounts of DDM and trypsin), a wide range of sample processing volumes (10–200 μL) were selected to optimize SOPs-MS for label-free proteome profiling. Surprisingly, higher processing volume does not lead to a significant drop in proteome coverage (Figure 2A,B), which can be attributed to the DDM additive for effectively reducing contact surface adsorption losses.20 When compared to an average of 5106 unique peptides (1362 protein groups) identified with the MS/MS spectra only for 10 μL processing volume, only 3% (3%) and 16% (11%) reduction in the peptide (proteome) coverage was observed with an average of 4966 unique peptides (1326 protein groups) and 4295 unique peptides (1208 protein groups) for 50 and 200 μL processing volumes, respectively. One-way ANOVA analysis indicated that there is no significant difference for the peptide identification number among different volumes (p = 0.43 for both the MS/MS spectra only and the combined MS/MS spectra and MBR function). Similar results were observed for the protein identification number (p = 0.66 and 0.73 for the MS/MS spectra only and the combined MS/MS spectra and MBR function, respectively). However, higher processing volume resulted in improved quantitation reproducibility, as reflected by lower median CVs (7.1% for 50 μL, 8.4% for 100 μL, and 7.8% for 200 μL) and higher median CVs (12.8% for 10 μL and 12.4% for 20 μL) (Figure 2D). This strongly suggests that the standard proteomics sample preparation volume of 50–200 μL can be used for effective processing of 100 cells with high reproducibility and minimal sample loss compared to the low μL volume.
Figure 2.
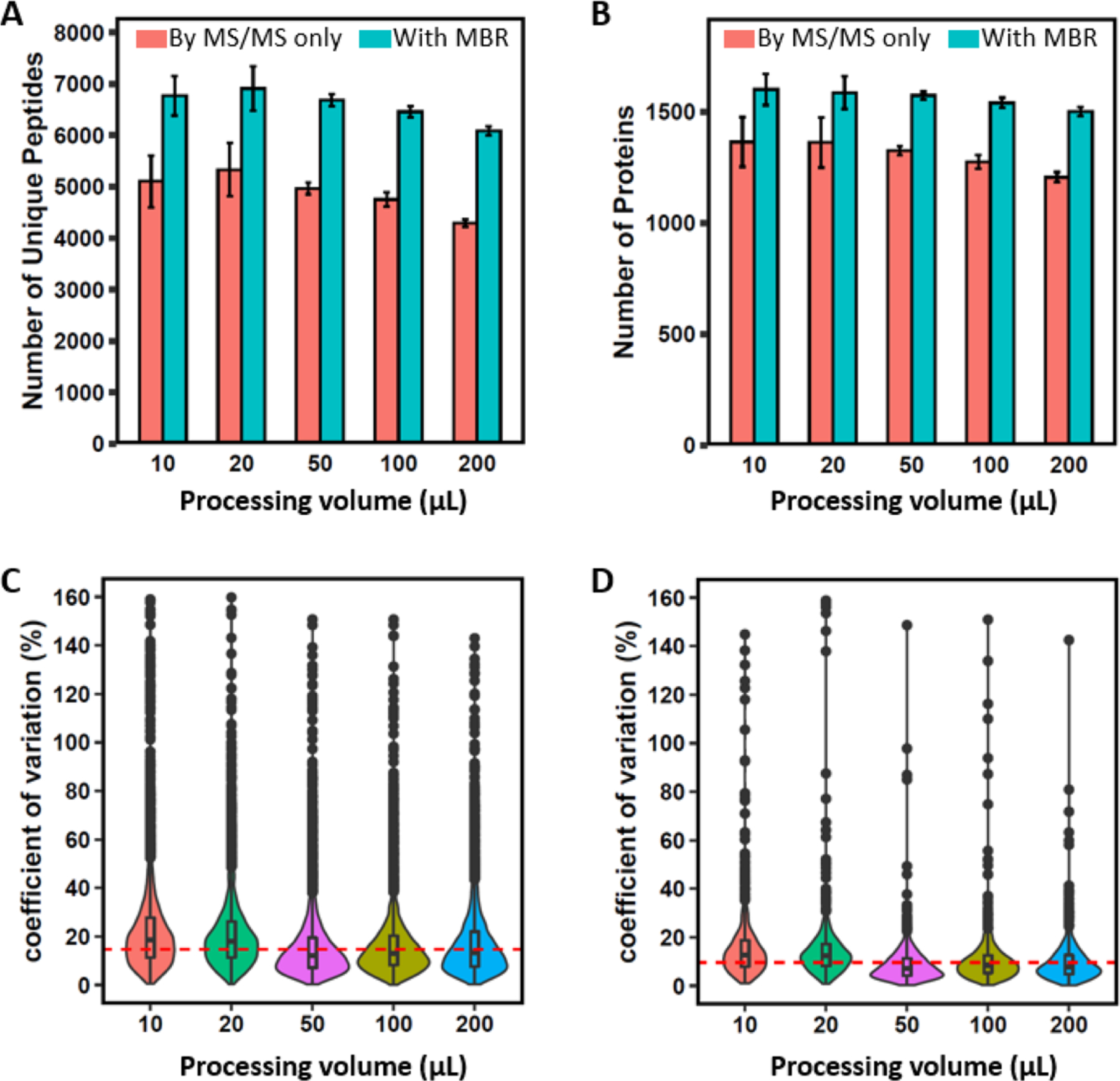
Evaluation of SOP performance at different processing volumes. (A) Number of identified unique peptides with MaxQuant. (B) Number of identified protein groups with MaxQuant. (C) Violin plots showing the distribution of the CVs of LFQ intensities at the peptide level. (D) Violin plots showing the distribution of CVs of LFQ intensities at the protein level. Red bar: MS/MS spectra only; blue bar: the combined MS/MS and MBR. Data are shown as the mean value ± SD. In (C,D), red horizontal lines indicate the median CVs of 14.7% for the peptides and 9.9% for the protein groups across different processing volumes.
To further confirm this observation as well as to increase the proteome coverage, other proteomic algorithms were used to reanalyze the 100-cell data. With the use of the MBR function in MaxQuant, the average numbers of identified protein groups were increased to 1590 for 10 μL, 1564 for 50 μL, and 1492 for 200 μL (Figure 2B). When compared to the number of protein groups for 10 μL, 2% and 6% reduction in the proteome coverage were observed for 50 and 200 μL, respectively, consistent with the above observation using the MS/MS spectra only for identification. By using the recently developed MSFragger algorithm,27 >500 more protein groups relative to the MS/MS spectra only using MaxQuant were identified with 1881 for 10 μL, 1908 for 50 μL, and 1749 for 200 μL (Figure S1B). By comparison with the number of identified protein groups at the 10 μL processing volume, a similar number and only 7% reduction in the proteome coverage was observed for 50 and 200 μL, respectively (Figure S1B). These results have further confirmed that SOPs-MS enables robust reproducible processing of 100 cells at the standard proteomics sample preparation volume. The processing volume of ∼50 μL was selected for SOPs-MS analysis because of high reproducibility and high numbers of identified peptides and protein groups. Unless otherwise mentioned, the most commonly used MaxQuant software tool was employed for quantitative analysis of the following nanoproteomics data.
Performance Evaluation Using FACS-Isolated 50–1000 MCF10A Cells
To evaluate its performance, SOPs-MS was used for label-free proteome profiling of 50–1000 FACS-isolated MCF10A cells. Protein groups were identified by both MS/MS spectra only and the combined MS/MS spectra and MBR function. As the cell number increased from 50 to 1000 cells, a steady increase in the number of unique peptides and protein groups was observed (Figure 3A,B). With the MBR function, the number of identified protein groups can be increased by ∼50% for 50 cells and only ∼10% for 1000 cells (Figure S2), suggesting that the lack of MS/MS spectra is more prevalent for low numbers of cells, and the increased identification number is governed by the size of the spectra library. Interestingly, a higher correlation between the number of identified peptides/protein groups and the cell number was observed for the MS/MS spectra only with r = 0.91 for peptides and r = 0.87 for protein groups when compared to the combined MS/MS spectra and MBR method (r = 0.85 for peptides and r = 0.83 for protein groups).
Figure 3.

Evaluation of SOPs-MS using FACS-sorted MCF10A cells. (A) Number of identified unique peptides for 50–1000 cells. (B) Number of identified protein groups for 50–1000 cells. (C) Venn diagram showing the number of protein groups identified from each of 3 biological replicates for 50 cells. (D) Pairwise correlation of protein LFQ intensities between any two replicates with the Pearson correlation coefficient. Red bar: MS/MS spectra only; blue bar: combined MS/MS and MBR. Data are shown as the mean value ± SD.
With the use of the LFQ algorithm, a total of 1026 protein groups can be quantified in 50 cells with three biological replicates and an average of ∼54% of protein groups overlapped between any two biological replicates (Figure 3C and Table S1). As expected, for large numbers of cells (e.g., 500 and 1000 cells) the overlapping percentage was greatly increased, which can be attributed to the reduced sample variation between any replicates and the increased detection reproducibility. For example, an average of ∼69% overlap was observed for 500 cells with the total 2348 quantifiable protein groups (Figure S3). We next evaluated the reproducibility of SOPs-MS for quantitative analysis of small numbers of cells. High reproducibility was observed with Pearson correlation between any two replicates (an average of ∼0.95 for 50 cells and ∼0.99 for 100–1000 cells) (Figures 3D and S4). Taken together, these results demonstrate that the SOPs-MS approach can operate at the standard processing volume and be used for deep label-free proteome profiling of low numbers of cells.
Application of SOPs-MS to Small Subpopulations of Colon Crypt Cells
We next applied SOPs-MS for label-free proteome profiling of small subpopulations of epithelial crypt cells from dissociated colon tissues of wild-type C57BL/6-N mice. A recently developed new flow sorting protocol was used to purify small numbers of colon stem cells (Stem), their immediate daughters (SecPDG, AbsPro), and their differentiated cell types, including tuft cells (Tuft) and enterocytes (Ent) (Figure S5).23 The cell number collected for each subpopulation ranged from 141 to 1691, and subpopulations of cells were collected into PCR tubes with the total volume of ∼50 μL. The number of identified protein groups correlates well with the cell number (Figure 4A). With the combined MS/MS spectra and the MBR function, ∼1500, ∼1900, and ∼2500 protein groups were identified from 141 Tuft cells, 480 SecPDG cells, and 1691 AbsPro cells, respectively. For the same number of cells, the number of identified protein groups from dissociated mouse tissues is slightly lower than that from MCF10A cells (e.g., ∼1900 for 480 SecPDG cells and ∼2500 for 500 MCF10A cells) (Figures 3B and 4A). This may be attributed to different cell origins and cell isolation procedures between dissociated mouse tissues and the cultured MCF10A cell line.
Figure 4.
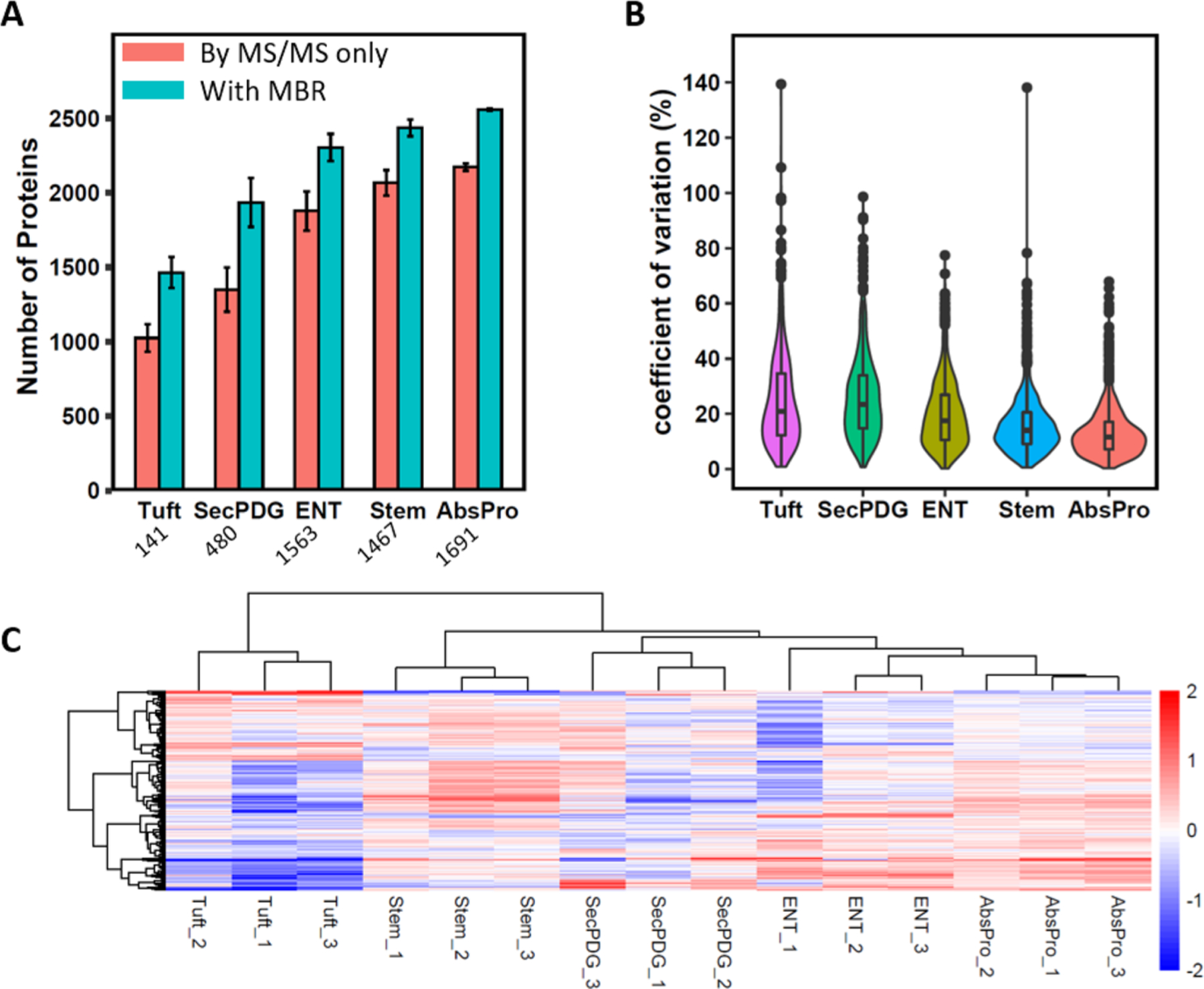
Application of SOPs-MS for analysis of small subpopulations of colon crypt cells. (A) Number of identified protein groups for the five small subpopulation cells. Red bar: MS/MS spectra only; blue bar: combined MS/MS and MBR. Data are shown as the mean value ± SD. Below is the average cell number for each subpopulation. (B) Violin plots of the distribution of CVs for proteomic analysis of each of the subpopulation cells. (C) Heatmap with unsupervised clustering to show relative abundance for each protein group identified by the MaxQuant MBR for all the five small subpopulations with three biological replicates per subpopulation.
We then evaluated the quantitation reproducibility for analysis using three replicates for each subpopulation. The median CVs of ∼20 and ∼10% were observed for the lowest number of Tuft cells and the highest number of AbsPro cells, respectively (Figure 4B and Table S2). This result is consistent with the Pearson correlation analysis of small numbers of MCF10A cells (50–1000) (Figures 3D and S4). The high quantitation reproducibility allows reliable evaluation of proteome expression difference among the five subpopulations of colon crypt cells. Based on protein expression alone from LFQ, unsupervised clustering analysis has shown that the three replicates from the same type of subpopulation cells are clustered together with distinct protein signatures compared to other cell types. This reflects cellular heterogeneity for these five subpopulations of colon crypt cells, which is consistent with the transcriptomic analysis.23,28 We also performed comparative analysis of protein abundance from each of the four subpopulations (immediate daughters: SecPDG and AbsPro; differential cell types: Tuft and ENT) against stem cells (Figure S6). As expected, using the highly stringent criteria (p ≤ 1% and >50% in fold change), the immediate daughters were found to be more similar to stem cells with less differentially expressed proteins (23 for SecPDG and 16 for AbsPro) as compared to mature, differentiated cell types (74 for Tuft and 44 for ENT) (Figure S6). This further demonstrates the quantitation reliability of SOPs-MS for proteomic analysis of small subpopulations of cells.
Comparison with Existing MS-Based Nanoproteomics Methods
SOPs-MS at the standard processing volume is a robust, easily implementable method for label-free nanoproteomics. Small numbers of cells or low-input samples are processed in either low-bind single tubes or multiwell plates that are routinely used for genomics and transcriptomics. The performance of SOPs-MS has been demonstrated by label-free MS analysis of small numbers of mammalian cells (MCF10A and subpopulations of colon crypt cells). Based on the actual MS/MS spectra for reliable protein identification (without using the MBR function), SOPs-MS can identify ∼1100–1360 protein groups from 100 human cells, at least 2-fold higher than that from other easily accessible methods (e.g., autoSP3,9 micro-FASP,11 and SLIPS10) (Table S3). When compared to specific device-based nanoproteomics methods, SOPs-MS has comparable performance in terms of the number of identified protein groups (e.g., ∼1100 for ULF LC–FAIMS–MS,29 ∼1360 for OAD,14 and ∼1350 for ISPEC16) (Table S3). One exception is nanoPOTS-MS13 and its benchtop version μPOTS,30 which can identify ∼1.5-fold more protein groups than SOPs-MS. This may be attributed to somewhat more effective sample processing and the use of a more advanced LC–MS platform (Table S3).13 It should be noted that comparison of nanoproteomics methods by using the number of identified protein groups at 100 mammalian cells may not be ideal because different LC–MS platforms and searching tools as well as different cell types were used for data generation and analysis. For example, analysis of 100-cell data using MSFragger identified >500 more protein groups than MaxQuant with the MS/MS spectra only (Figure S1B). Nonetheless, we have used the commonly accessible LC–MS platform and MaxQuant data analysis tool for SOPs-MS. Furthermore, SOPs-MS can be readily operated with ∼50 μL sample processing volume using a benchtop micropipette, and automation of SOPs can be achieved by using most commercially available liquid handlers. Therefore, SOPs-MS can be easily implemented and widely adopted by the proteomics research community for broad applications.
Future developments will focus on improvements in detection sensitivity and sample throughput for rapid deep proteome profiling of small numbers of cells.31 Enhancing detection sensitivity could be achieved by effective integration of ultralow-flow LC or capillary electrophoresis (CE) and a high-efficiency ion source/ion transmission interface32 with the most advanced MS platform. Sample throughput could be increased using ultrafast high-resolution ion mobility-based gas-phase separation (e.g., SLIM33) to replace current slow liquid-phase (LC or CE) separation and effective integration of liquid- and gas-phase separations (e.g., SLIM33 or FAIMS34) for greatly reducing separation time but without trading off separation resolution. Alternatively, sample multiplexing with isobaric barcoding can also be considered to increase sample throughput.35–38
CONCLUSIONS
The SOPs-MS method at the standard processing volume was demonstrated to enable effective processing of small numbers of cells for quantitative nanoproteomics. With the use of a nonionic surfactant DDM additive, the “all-in-one” SOPs sample preparation greatly reduces surface adsorption losses that allows for identification of ∼900–2400 protein groups from 50 to 1000 human cells with the MS/MS spectra only and the correlation coefficients of 0.95–0.99 depending on the cell number. SOPs-MS has better performance in the number of identified protein groups than other reported readily accessible methods and comparable performance as other specific device-based nanoproteomics methods. The application of SOPs-MS for analysis of small subpopulations of colon crypt cells demonstrates its power for proteome characterization of cellular heterogeneity and discovery of distinct protein signatures for small subpopulations. With improvements in detection sensitivity and sample throughput, we believe that SOPs-MS will have broad applicability in the biological and biomedical research for deep quantitative proteome profiling of small subpopulations of cells as well as other mass-limited samples that cannot be readily accessible by current proteomic platforms.
Supplementary Material
ACKNOWLEDGMENTS
Portions of the research were supported by NIH UG3CA256967 (to T.S.), NIH R21CA223715 (to T.S.), NIH P41GM103493 (to R.D.S.), NIH U01CA227544 (to H.S.W.), NCI EDRN Interagency Agreement ACN20007–001 (to T.L.), and DOD BCRP W81XWH-16–1-0021 (to W.-J.Q.). Work at the University of California, Irvine was supported by NSF GRFP grant DGE-1321846 (to A.N.H.), an NCI training grant to the Cancer Research Institute at the University of California, Irvine (T32CA009054) (to A.N.H.), NIH P30CA062203 (to M.L.W.), and NIH U54CA217378 (to M.L.W.). The cellular work with murine colon crypts was made possible via access to the UC Irvine Flow Cytometry Core in the Optical Biological Center (OBC), the Experimental Tissue Resource (ETR), and the Genomics High Throughput Facility (GHTF), all of which are Shared Resources supported by the Cancer Center Support Grant (P30CA062203) at the University of California, Irvine. The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory, Pacific North-west National Laboratory, a national scientific user facility sponsored by the United States of America Department of Energy under Contract DE-AC05–76RL0 1830. We thank N. Johnson for generous assistance with graphics.
Footnotes
Complete contact information is available at:
The authors declare no competing financial interest.
The RAW global MS data and the identified protein groups from MaxQuant have been deposited in Japan ProteOme STandard Repository (jPOST: https://repository.jpostdb.org/).39 The accession codes are JPST001175 for jPOST and PXD026009 for ProteomeXchange.
Contributor Information
Kendall Martin, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States; Bioproducts, Sciences & Engineering Laboratory, Department of Biological Systems Engineering, Washington State University, Richland, Washington 99354, United States.
Tong Zhang, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Tai-Tu Lin, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Amber N. Habowski, Department of Microbiology and Molecular Genetics, University of California Irvine, Irvine, California 92697, United States
Rui Zhao, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Chia-Feng Tsai, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
William B. Chrisler, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Ryan L. Sontag, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Daniel J. Orton, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States
Yong-Jie Lu, Centre for Cancer Biomarker and Biotherapeutics, Barts Cancer Institute, Queen Mary University of London, London EC1M 6BQ, United Kingdom.
Karin D. Rodland, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Bin Yang, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States; Bioproducts, Sciences & Engineering Laboratory, Department of Biological Systems Engineering, Washington State University, Richland, Washington 99354, United States.
Tao Liu, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Richard D. Smith, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Wei-Jun Qian, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Marian L. Waterman, Department of Microbiology and Molecular Genetics, University of California Irvine, Irvine, California 92697, United States
H. Steven Wiley, Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
Tujin Shi, Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99354, United States.
REFERENCES
- (1).Altschuler SJ; Wu LF Cellular Heterogeneity: Do Differences Make a Difference? Cell 2010, 141, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hughes AJ; Spelke DP; Xu Z; Kang C-C; Schaffer DV; Herr AE Single-cell western blotting. Nat. Methods 2014, 11, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; Wang X; Qiao JW; Cao S; Petralia F; Kawaler E; Mundt F; Krug K; Tu Z; Lei JT; Gatza ML; Wilkerson M; Perou CM; Yellapantula V; Huang K.-l.; Lin C; McLellan MD; Yan P; Davies SR; Townsend RR; Skates SJ; Wang J; Zhang B; Kinsinger CR; Mesri M; Rodriguez H; Ding L; Paulovich AG; Fenyö D; Ellis MJ; Carr SA Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhang H; Liu T; Zhang Z; Payne SH; Zhang B; McDermott JE; Zhou J-Y; Petyuk VA; Chen L; Ray D; Sun S; Yang F; Chen L; Wang J; Shah P; Cha SW; Aiyetan P; Woo S; Tian Y; Gritsenko MA; Clauss TR; Choi C; Monroe ME; Thomas S; Nie S; Wu C; Moore RJ; Yu K-H; Tabb DL; Fenyö D; Bafna V; Wang Y; Rodriguez H; Boja ES; Hiltke T; Rivers RC; Sokoll L; Zhu H; Shih I-M; Cope L; Pandey A; Zhang B; Snyder MP; Levine DA; Smith RD; Chan DW; Rodland KD; Carr SA; Gillette MA; Klauser KR; Kuhn E; Mani DR; Mertins P; Ketchum KA; Thangudu R; Cai S; Oberti M; Paulovich AG; Whiteaker JR; Edwards NJ; McGarvey PB; Madhavan S; Wang P; Chan DW; Pandey A; Shih I-M; Zhang H; Zhang Z; Zhu H; Cope L; Whiteley GA; Skates SJ; White FM; Levine DA; Boja ES; Kinsinger CR; Hiltke T; Mesri M; Rivers RC; Rodriguez H; Shaw KM; Stein SE; Fenyo D; Liu T; McDermott JE; Payne SH; Rodland KD; Smith RD; Rudnick P; Snyder M; Zhao Y; Chen X; Ransohoff DF; Hoofnagle AN; Liebler DC; Sanders ME; Shi Z; Slebos RJC; Tabb DL; Zhang B; Zimmerman LJ; Wang Y; Davies SR; Ding L; Ellis MJC; Townsend RR Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang B; Wang J; Wang X; Zhu J; Liu Q; Shi Z; Chambers MC; Zimmerman LJ; Shaddox KF; Kim S; Davies SR; Wang S; Wang P; Kinsinger CR; Rivers RC; Rodriguez H; Townsend RR; Ellis MJC; Carr SA; Tabb DL; Coffey RJ; Slebos RJC; Liebler DC Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Macaulay IC; Ponting CP; Voet T Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet 2017, 33, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gaudet S; Miller-Jensen K Redefining Signaling Pathways with an Expanding Single-Cell Toolbox. Trends Biotechnol 2016, 34, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wisniewski JR; Zougman A; Nagaraj N; Mann M Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- (9).Müller T; Kalxdorf M; Longuespée R; Kazdal DN; Stenzinger A; Krijgsveld J Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol 2020, 16, No. e9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wu R; Xing S; Badv M; Didar TF; Lu Y Step-Wise Assessment and Optimization of Sample Handling Recovery Yield for Nanoproteomic Analysis of 1000 Mammalian Cells. Anal. Chem 2019, 91, 10395–10400. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Z; Dubiak KM; Huber PW; Dovichi NJ Miniaturized Filter-Aided Sample Preparation (MICRO-FASP) Method for High Throughput, Ultrasensitive Proteomics Sample Preparation Reveals Proteome Asymmetry in Xenopus laevis Embryos. Anal. Chem 2020, 92, 5554–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aebersold R; Mann M Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [DOI] [PubMed] [Google Scholar]
- (13).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun 2018, 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li Z-Y; Huang M; Wang X-K; Zhu Y; Li J-S; Wong CCL; Fang Q Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem 2018, 90, 5430–5438. [DOI] [PubMed] [Google Scholar]
- (15).Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal. Chem 2018, 90, 14003–14010. [DOI] [PubMed] [Google Scholar]
- (16).Hata K; Izumi Y; Hara T; Matsumoto M; Bamba T In-Line Sample Processing System with an Immobilized Trypsin-Packed Fused-Silica Capillary Tube for the Proteomic Analysis of a Small Number of Mammalian Cells. Anal. Chem 2020, 92, 2997–3005. [DOI] [PubMed] [Google Scholar]
- (17).Zhang P; Gaffrey MJ; Zhu Y; Chrisler WB; Fillmore TL; Yi L; Nicora CD; Zhang T; Wu H; Jacobs J; Tang K; Kagan J; Srivastava S; Rodland KD; Qian W-J; Smith RD; Liu T; Wiley HS; Shi T Carrier-Assisted Single-Tube Processing Approach for Targeted Proteomics Analysis of Low Numbers of Mammalian Cells. Anal. Chem 2019, 91, 1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Martin K; Zhang T; Zhang P; Chrisler WB; Thomas FL; Liu F; Liu T; Qian W-J; Smith RD; Shi T Carrier-assisted One-pot Sample Preparation for Targeted Proteomics Analysis of Small Numbers of Human Cells. J. Visualized Exp 2020, No. e61797. [DOI] [PMC free article] [PubMed]
- (19).Shi T; Gaffrey MJ; Fillmore TL; Nicora CD; Yi L; Zhang P; Shukla AK; Wiley HS; Rodland KD; Liu T; Smith RD; Qian W-J Facile carrier-assisted targeted mass spectrometric approach for proteomic analysis of low numbers of mammalian cells. Commun. Biol 2018, 1, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tsai C-F; Zhang P; Scholten D; Martin K; Wang Y-T; Zhao R; Chrisler WB; Patel DB; Dou M; Jia Y; Reduzzi C; Liu X; Moore RJ; Burnum-Johnson KE; Lin M-H; Hsu C-C; Jacobs JM; Kagan J; Srivastava S; Rodland KD; Wiley HS; Qian W-J; Smith RD; Zhu Y; Cristofanilli M; Liu T; Liu H; Shi T Surfactant-assisted one-pot sample preparation for label-free single-cell proteomics. Commun. Biol 2021, 4, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liang Y; Acor H; McCown MA; Nwosu AJ; Boekweg H; Axtell NB; Truong T; Cong Y; Payne SH; Kelly RT Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Anal. Chem 2021, 93, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shi T; Niepel M; McDermott JE; Gao Y; Nicora CD; Chrisler WB; Markillie LM; Petyuk VA; Smith RD; Rodland KD; Sorger PK; Qian W-J; Wiley HS Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci. Signaling 2016, 9, rs6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Habowski AN; Flesher JL; Bates JM; Tsai C-F; Martin K; Zhao R; Ganesan AK; Edwards RA; Shi T; Wiley HS; Shi Y; Hertel KJ; Waterman ML Transcriptomic and proteomic signatures of stemness and differentiation in the colon crypt. Commun. Biol 2020, 3, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Habowski AN; Bates JM; Flesher JL; Edwards RA; Waterman ML Isolation of Murine Large Intestinal Crypt Cell Populations with Flow Sorting. Protocol Exchange; Research Square, 2020. [Google Scholar]
- (25).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (26).Zhang T; Schneider JD; Lin C; Geng S; Ma T; Lawrence SR; Dufresne CP; Harmon AC; Chen S MPK4 Phosphorylation Dynamics and Interacting Proteins in Plant Immunity. J. Proteome Res 2019, 18, 826–840. [DOI] [PubMed] [Google Scholar]
- (27).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Baulies A; Angelis N; Li VSW Hallmarks of intestinal stem cells. Development 2020, 147, dev182675. [DOI] [PubMed] [Google Scholar]
- (29).Greguš M; Kostas JC; Ray S; Abbatiello SE; Ivanov AR Improved Sensitivity of Ultralow Flow LC-MS-Based Proteomic Profiling of Limited Samples Using Monolithic Capillary Columns and FAIMS Technology. Anal. Chem 2020, 92, 14702–14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Xu K; Liang Y; Piehowski PD; Dou M; Schwarz KC; Zhao R; Sontag RL; Moore RJ; Zhu Y; Kelly RT Benchtop-compatible sample processing workflow for proteome profiling of < 100 mammalian cells. Anal. Bioanal. Chem 2019, 411, 4587–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kagan J; Moritz RL; Mazurchuk R; Lee JH; Kharchenko PV; Rozenblatt-Rosen O; Ruppin E; Edfors F; Ginty F; Goltsev Y; Wells JA; LaCava J; Riesterer JL; Germain RN; Shi T; Chee MS; Budnik BA; Yates JR; Chait BT; Moffitt JR; Smith RD; Srivastava S National Cancer Institute Think-Tank Meeting Report on Proteomic Cartography and Biomarkers at the Single-Cell Level: Interrogation of Premalignant Lesions. J. Proteome Res 2020, 19, 1900–1912. [DOI] [PubMed] [Google Scholar]
- (32).Marginean I; Page JS; Tolmachev AV; Tang K; Smith RD Achieving 50% ionization efficiency in subambient pressure ionization with nanoelectrospray. Anal. Chem 2010, 82, 9344–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ibrahim YM; Hamid AM; Deng L; Garimella SVB; Webb IK; Baker ES; Smith RD New frontiers for mass spectrometry based upon structures for lossless ion manipulations. Analyst 2017, 142, 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach J-J; Brademan DR; Westphall MS; Coon JJ Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem 2018, 90, 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dou M; Clair G; Tsai C-F; Xu K; Chrisler WB; Sontag RL; Zhao R; Moore RJ; Liu T; Pasa-Tolic L; Smith RD; Shi T; Adkins JN; Qian W-J; Kelly RT; Ansong C; Zhu Y High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem 2019, 91, 13119–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tsai C-F; Zhao R; Williams SM; Moore RJ; Schultz K; Chrisler WB; Pasa-Tolic L; Rodland KD; Smith RD; Shi T; Zhu Y; Liu T An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) Strategy for Precise Quantitative Single-cell Proteomics. Mol. Cell. Proteomics 2020, 19, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yi L; Tsai C-F; Dirice E; Swensen AC; Chen J; Shi T; Gritsenko MA; Chu RK; Piehowski PD; Smith RD; Rodland KD; Atkinson MA; Mathews CE; Kulkarni RN; Liu T; Qian W-J Boosting to Amplify Signal with Isobaric Labeling (BASIL) Strategy for Comprehensive Quantitative Phosphoproteomic Characterization of Small Populations of Cells. Anal. Chem 2019, 91, 5794–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Okuda S; Watanabe Y; Moriya Y; Kawano S; Yamamoto T; Matsumoto M; Takami T; Kobayashi D; Araki N; Yoshizawa AC; Tabata T; Sugiyama N; Goto S; Ishihama Y jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res 2017, 45, D1107–D1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.