

Key Points
Sequential CAR T-cell therapy may result in a durable response and is safe in pediatric patients with R/R Burkitt lymphoma.
Sequential CAR T-cell therapy may benefit pediatric patients with R/R Burkitt lymphoma with CNS involvement.
Visual Abstract

Abstract
Single antigen–targeted chimeric antigen receptor (CAR) T-cell therapy may be insufficient to induce a durable response in pediatric aggressive B-cell lymphomas. This clinical trial examined the feasibility of sequential different B-cell antigen–targeted CAR T-cell therapy for pediatric relapsed/refractory (R/R) Burkitt lymphoma. Twenty-three patients received the first CD19 CAR T-cell infusion. The patients who did not achieve an ongoing complete response (CR) underwent 1 or more sequential infusions of CAR T-cell therapy that targeted CD22 followed by CD20 according to their disease status and CAR T-cell persistence after each infusion. The median time from the last infusion to the cutoff date was 17 months (range, 15-23 months). The estimated 18-month CR rate was 78% (95% confidence interval [CI], 54%-91%). The estimated 18-month progression-free survival rate was 78% (95% CI, 55%-90%), with 78% (95% CI, 37%-94%) in patients with bulky disease and 60% (95% CI, 25%-83%) in patients with central nervous system (CNS) involvement. During the first CD19 CAR T-cell infusion, grade ≥3 cytokine release syndrome (CRS) occurred in 34.8% and neurotoxicity occurred in 21.7% of all patients. During subsequent infusions, there were only a few incidences of grade >2 CRS and neurotoxicity. All adverse events were reversible. The severity of neurotoxicity was not significantly different between patients with CNS involvement and those who did not have CNS involvement. Sequential CAR T-cell therapy may result in a durable response and is safe in pediatric R/R Burkitt lymphoma. Patients with CNS involvement may benefit from sequential CAR T-cell therapy. This trial was registered at www.chictr.org.cn/index.aspx as #ChiCTR1800014457.
Introduction
Most pediatric patients with mature B-cell lymphoma can be cured with conventional chemotherapy.1-9 However, those with aggressive relapsed or refractory (R/R) B-cell lymphoma have a dismal prognosis and eventually die of the malignancy, even with continued conventional chemotherapy.10-14 Chimeric antigen receptor (CAR) T-cell therapy is among the most promising novel therapies that might be used to treat such patients.15,16 CD19-targeted CAR T-cell therapy has been shown to achieve a 6-month complete response (CR) rate of 29% to 41% in adult patients with R/R B-cell lymphoma.17-22 There are several actively recruiting trials evaluating CD19 CAR T-cell therapy for pediatric and young adult patients with R/R B-cell lymphomas.16
Despite this impressive efficacy, progressive disease (PD) and relapsed disease (RD) occur in a large proportion of patients who receive a CD19 CAR T-cell infusion. Tumor immune escape has emerged as a non-negligible obstacle to improving long-term survival. Several studies have addressed the potential mechanisms of resistance to CD19 CAR T-cell therapy with a primary focus on issues related to CD19– relapse.23,24 However, the CD19+ relapse rate seemed higher than the CD19– relapse rate in adult diffuse large B-cell lymphoma.17-21 The mechanisms underlying CD19+ relapse are multifactorial and are still poorly elucidated. Nevertheless, it has been postulated that CD19+ relapse may develop because of limited or a lack of CAR T-cell persistence.25
The feasibility and effectiveness of dual-antigen–targeted CAR T-cell therapy for mitigating CD19– relapse is being evaluated in several preclinical and clinical studies.26,27 The combination of CD19 CAR T-cell therapy with immune checkpoint inhibitors to improve CAR T-cell persistence in vivo is being tested in an ongoing clinical trial.28 Our team designed combinational and sequential administration strategies for different B-cell antigen–targeted CAR T-cell therapy to prevent tumor antigen escape and maintain CAR T-cell persistence. We have shown that sequential CD19-CD22 CAR T-cell infusions could induce sustained remission in pediatric patients with R/R B-cell acute lymphoblastic leukemia.29 We have also reported early responses to sequential murine CD19 (mCD19)-humanized CD22 (hCD22)-hCD20-hCD19 CAR T-cell infusion in pediatric patients with R/R Burkitt lymphoma.30 Here, we further investigated durable responses induced by this strategy in pediatric R/R Burkitt lymphoma.
Methods
Patient eligibility
The single-arm phase 1 clinical trial was approved by the institutional review board at Beijing Boren Hospital. Parents or legal guardians provided written informed consent for including their children in the study in accordance with the Declaration of Helsinki. Eligible patients were children and adolescents up to age 18 years with Burkitt lymphoma who had refractory (never obtaining a CR) or relapsed disease after first-line chemotherapy and had a partial response (PR) or no response (NR) as the best response to at least 2 cycles of salvage chemotherapy, including rituximab. The diagnosis of Burkitt lymphoma was verified by independent pathology review according to the 2016 World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissue.31 The only patients who qualified for this trial were whose lymphoma cells definitively expressed CD19 or more B-cell antigens (CD20 and CD22) as determined by immunohistochemical stain analysis of tumor biopsies obtained before enrollment. Additional details of inclusion and exclusion criteria are shown in supplemental Methods.
Constructing CARs
A lentiviral vector was constructed that contained a second-generation CAR construct with a 4-1BB costimulatory domain and a CD3-zeta domain. The antigen recognition domains of CARs targeting CD20 (YK-CD20BB-002) and CD22 (YK-CD22BB-002) were composed of a single-chain variable fragment obtained from a human antibody phage display library. The antigen recognition domains of CD19-targeted CARs were from 2 sources: one was derived from FMC63 monoclonal antibody (YK-CD19BB-001) and the other was obtained from a human antibody phage display library (YK-hCD19BB-002). The cytotoxicity of these CAR T cells was previously validated.32,33
Treatment procedures
Leukapheresis and lymphodepletion.
As shown in Figure 1, eligible patients underwent leukapheresis to obtain peripheral blood mononuclear cells. A week before leukapheresis, tumors were evaluated on the basis of the International Pediatric Non-Hodgkin Lymphoma Staging System and French-American-British poor-risk criteria.13,34 An additional portion of the leukapheresis products was routinely cryopreserved for potential sequential CAR T-cell generation. Manufacture of the first infused mCD19 CAR T cells started on the day of leukapheresis (day −6). Alternative CAR T cells were generated for sequential infusion when cryopreserved leukapheresis product was thawed for use. Peripheral blood mononuclear cells collected from patients were stimulated with magnetic beads coated with anti-CD3 and anti-CD28 antibodies, transduced with a lentiviral vector, and cultured for 5 to 8 days.32,33 Lymphodepleting (LD) chemotherapy consisting of fludarabine (30 mg/m2 per day) and cyclophosphamide (250 mg/m2 per day) was given on days −5, −4, and −3 before each CAR T-cell infusion on day 0 at a target dose of 2 × 106 cells per kg (range, 0.1 × 106 to 5 × 106 cells per kg), except in 1 patient with white blood cell counts <103/μL 1 week before infusion.
Figure 1.

Diagram of the sequential treatment procedure. PBMC, peripheral blood mononuclear cell; pts, patients.
Response assessment and CAR T-cell detection.
Response was assessed according to International Pediatric Non-Hodgkin Lymphoma Response Criteria35 on days 7, 15, 30, 45, and 60 and monthly from day 60 to month 6, every 3 months from month 6 to month 24, and as clinically indicated after each cycle of CAR T-cell infusion (Figure 1). CAR T-cell persistence was monitored at the same time points as response assessment by combining 3 different methods (supplemental Methods). Flow cytometry (FCM) was used to detect CAR+ T cells, but if CAR+ T cells were undetectable by FCM, real-time quantitative polymerase chain reaction (qPCR) was used to detect the copies of CAR transgenes in T cells. CAR T-cell levels below the FCM detection threshold (0.1%) could still be detected by qPCR. When CAR T-cell levels were below the qPCR detection threshold (0.01%), B-cell aplasia detected by FCM served as a surrogate marker for functional persistence of CAR T cells because polyclonal B-cell recovery occurs only when functional CAR T cells are no longer present in peripheral blood (PB).36-38 The combination of disease status and CAR T-cell persistence was used to determine the timing of subsequent infusions.
Sequential infusion of CAR T cells.
The sequential procedure is illustrated in Figure 1. All enrolled patients received the first cycle of CAR T-cell infusion using mCD19 cells. The patients who achieved an ongoing CR (defined as remaining in CR to the cutoff date) did not receive additional therapy.
For patients who maintained a PR or NR until mCD19 CAR T cells could no longer be detected in PB by FCM, the second cycle of CAR T-cell infusion using hCD22 cells was initiated at such a time that it did not prematurely interfere with the antitumor effect of mCD19 CAR T cells but could prevent PD in the setting of continuously decreasing levels of mCD19 CAR T cells. For patients who had PD or RD after responding to mCD19 CAR T-cell therapy, the second cycle of CAR T-cell infusion using hCD22 cells was initiated immediately, regardless of whether mCD19 CAR T cells were detected in PB by FCM, noting that the tumor had already effected immune escape from mCD19 CAR T cells.
After the second cycle of CAR T-cell infusion (with hCD22 cells), the patients who maintained a PR until hCD22 CAR T cells could no longer be detected in PB by FCM received the third cycle of CAR T-cell infusion using hCD20 cells. For patients who had RD after achieving the first CR (CR1) after the first infusion and then attained the second CR (CR2) by the second infusion, the third cycle of CAR T-cell infusion with hCD20 cells was given as consolidation therapy after the loss of hCD22 CAR T cells detectable in PB by FCM to prevent recurring immune escape by the tumor. Patients who had PD after having obtained a PR immediately received the third cycle of CAR T-cell infusion with hCD20 cells, even if hCD22 T cells were still detectable in PB by FCM.
After the third cycle of CAR T-cell infusion with hCD20 cells, the patients who maintained a PR until hCD20 CAR T cells could no longer be detected in PB by FCM were given the fourth cycle of CAR T-cell infusion using hCD19 cells. No patients were bridged to hematopoietic stem cell transplantation after completing sequential CAR T-cell treatment.
Assessment and management of toxicity
Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) were graded according to American Society for Transplantation and Cellular Therapy grading criteria.39 Based on the findings of Strati et al,40 caution is necessary when using glucocorticoids early in the management of toxicity. Dexamethasone or methylprednisolone or both were alternately administered to patients if either hypoxia requiring supplemental oxygen and/or hemodynamic instability requiring vasopressor support (despite intravenous fluids) occurred after the clinical signs of CAR T-cell expansion were evident. Dexamethasone was used in most cases, especially for patients with neurologic symptoms, whereas methylprednisolone was preferred for patients with pulmonary dysfunction. Dexamethasone was usually started at 6 mg/m2 per day; methylprednisolone was usually started at 2 mg/kg per day. If clinical deterioration occurred, dexamethasone could be escalated to as much as 10 mg/m2 per day or methylprednisolone could be escalated to as much as 6 mg/kg per day. Intrathecal dexamethasone was used in patients with severe (grade ≥3) ICANS in addition to systemic steroids. Tocilizumab was simultaneously administered to patients whose serum interleukin-6 (IL-6) levels were >1000 pg/mL. Upon clinical improvement, steroids were rapidly tapered.
End points
Follow-up time was defined as the time from the last CAR T-cell infusion to the cutoff date. The primary end points included safety and CR rate (defined as the proportion of participants who remained in CR to the cutoff date). Key secondary end points included duration of CR (defined as the time from the date of first CR after the last infusion to the date of first PD after the last infusion or death as a result of Burkitt lymphoma); progression-free survival (PFS) (defined as the time from the date of the last CAR T-cell infusion to the date of disease progression or death as a result of any reason); overall survival (OS) (defined as the time from the date of the last CAR T-cell infusion to the date of death as a result of any reason among all treated patients); duration of CAR T-cell persistence (defined as the time from the date of the first CAR T-cell infusion to the date of the first of 2 consecutive measurements to determine whether CAR T cells were undetectable in PB by qPCR); and duration of B-cell aplasia (defined as the time from the date of the first CAR T-cell infusion to the date of the first of 2 consecutive measurements to verify B-cell level recovery >3% by FCM).36
Statistical analysis
The sample size was approximated on the basis of a response rate of 60%, with 95% confidence in a true response rate of more than 39%. Quantitative and normally distributed variables were compared using Student t test. Otherwise, the Mann-Whitney U test/Wilcoxon rank-sum test was used to compare nonparametric quantitative variables. The χ2 test and Fisher’s exact test were used to assess independence between qualitative variables. Kaplan-Meier survival analysis was used to evaluate the duration of CR, duration of CAR T-cell persistence, duration of B-cell aplasia, OS, and PFS. All statistical analyses were two-sided, assumed a level of statistical significance of 0.05, and were performed using R software (version 3.6.2) and GraphPad Prism 8.0.2.
Results
Clinical characteristics of the enrolled patients
We enrolled 23 patients between January 17, 2018, and November 8, 2019. The baseline characteristics of all patients are listed in Table 1. The distribution among patients included staging classification at diagnosis for Burkitt lymphoma: stage III, 10 patients (43.5%), stage IV, 13 patients (56.5%); at study entry: stage II, 3 patients (13%), stage III, 10 patients (43.5%), and stage IV, 10 patients (43.5%). Also included was poor-risk classification: 16 patients (69.6%) who had R/R disease within 6 months from diagnosis, 20 patients (87%) with an elevated lactate dehydrogenase ≥2 times the upper limit of normal at diagnosis, and 4 patients (17.4%) who experienced a failure in the bone marrow. Overall, 21 patients (91%) had risk factors for treatment failure. Among 23 patients, 9 (39.1%) had bulky disease, and 10 (43.5%) showed evidence of central nervous system (CNS) involvement. All patients had positive expression of CD19, CD22, and CD20 on their lymphoma cells confirmed by immunohistochemical stain analysis on tumor biopsies before enrollment on the trial, except patients P5 and P21 who did not have CD20 expression (Table 2).
Table 1.
Baseline characteristics of the enrolled patients
| Patient | At diagnosis | At study entry | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (y) | Stage* | BM involved | CNS involved | LDH | First-line therapy | Time to failure (m) | Type of failure | Stage | Failure in BM | Failure in CNS | Bulky disease | Poor risk† | |
| P1 | 8 | III | No | No | ≥2 × ULN | FAB/LMB96-type | 5 | No CR | III | No | No | Yes | Yes |
| P2 | 8 | IV | Yes | No | ≥2 × ULN | FAB/LMB96-type | 5 | No CR | II | No | No | No | Yes |
| P3 | 10 | IV | Yes | No | ≥2 × ULN | FAB/LMB96-type | 5 | No CR | III | No | No | Yes | Yes |
| P4 | 11 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 5 | Failure after CR | III | No | No | No | Yes |
| P5 | 11 | IV | Yes | No | ≥2 × ULN | NHL/BFM95-type | 5 | Failure after CR | IV | No | Yes | No | Yes |
| P6 | 9 | III | No | No | <2 × ULN | NHL/BFM95-type | 7 | No CR | III | No | No | No | No |
| P7 | 9 | IV | No | Yes | ≥2 × ULN | FAB/LMB96-type | 8 | Failure after CR | IV | No | Yes | No | Yes |
| P8 | 10 | IV | Yes | No | ≥2 × ULN | FAB/LMB96-type | 8 | Failure after CR | III | No | No | No | Yes |
| P9 | 8 | IV | Yes | Yes | ≥2 × ULN | FAB/LMB96-type | 7 | Failure after CR | II | No | No | No | Yes |
| P10 | 7 | III | No | No | ≥2 × ULN | FAB/LMB96-type | 3 | No CR | III | No | No | Yes | Yes |
| P11 | 2.5 | IV | Yes | No | ≥2 × ULN | NHL/BFM95-type | 5 | Failure after CR | IV | Yes | Yes | Yes | Yes |
| P12 | 11 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 4 | No CR | III | No | No | Yes | Yes |
| P13 | 3 | IV | No | Yes | ≥2 × ULN | FAB/LMB96-type | 2 | No CR | IV | Yes | Yes | Yes | Yes |
| P14 | 6 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 3 | Failure after CR | III | No | No | No | Yes |
| P15 | 5 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 5 | No CR | IV | No | Yes | No | Yes |
| P16 | 7 | IV | Yes | Yes | ≥2 × ULN | NHL/BFM95-type | 9 | Failure after CR | IV | Yes | Yes | No | Yes |
| P17 | 7 | IV | No | Yes | <2 × ULN | NHL/BFM95-type | 3 | No CR | IV | No | Yes | No | Yes |
| P18 | 2 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 3 | No CR | III | No | No | Yes | Yes |
| P19 | 5 | IV | Yes | No | <2 × ULN | NHL/BFM95-type | 9 | Failure after CR | IV | No | Yes | No | No |
| P20 | 10 | IV | Yes | Yes | ≥2 × ULN | FAB/LMB96-type | 5 | Failure after CR | IV | No | Yes | No | Yes |
| P21 | 9 | IV | Yes | Yes | ≥2 × ULN | NHL/BFM95-type | 4 | No CR | IV | Yes | Yes | Yes | Yes |
| P22 | 8 | III | No | No | ≥2 × ULN | NHL/BFM95-type | 8 | Failure after CR | III | No | No | Yes | Yes |
| P23 | 12 | III | No | No | ≥2 × ULN | FAB/LMB96-type | 5 | No CR | II | No | No | No | Yes |
BFM, Berlin-Frankfurt-Muenster; BM, bone marrow; FAB, French‐American‐British; LDH, lactate dehydrogenase; LMB, Lymphome Malins B; NHL, non-Hodgkin lymphoma; ULN, upper limit of normal.
Based on the International Pediatric Non-Hodgkin Lymphoma Staging System.
Based on FAB poor-risk criteria.
Table 2.
B-cell antigen expression on lymphoma cells determined by IHC staining analysis of tumor biopsies from enrolled patients
| Patient | Before CAR T-cell infusion with:* | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| mCD19 | hCD22 | hCD20 | |||||||
| CD19 | CD22 | CD20 | CD19 | CD22 | CD20 | CD19 | CD22 | CD20 | |
| P1 | + | + | +++ | +++ | +++ | +++ | NA | NA | NA |
| P2 | ++ | ++ | +++ | ND | ND | ND | +++ | +++ | +++ |
| P3 | + | + | +++ | NA | NA | NA | NA | NA | NA |
| P4 | + | +++ | +++ | NA | NA | NA | NA | NA | NA |
| P5 | ++ | +++ | — | NA | NA | NA | NA | NA | NA |
| P6 | ++ | ++ | ++ | NA | NA | NA | NA | NA | NA |
| P7 | ++ | ++ | ++ | ++ | ++ | ++ | ND | ND | ND |
| P8 | + | + | + | +++ | +++ | +++ | ND | ND | ND |
| P9 | ++ | ++ | ++ | NA | NA | NA | NA | NA | NA |
| P10 | ++ | ++ | ++ | +++ | +++ | +++ | NA | NA | NA |
| P11 | ++ | ++ | ++ | — | ++ | ++ | ND | ND | ND |
| P12 | +++ | +++ | +++ | +++ | +++ | +++ | ND | ND | ND |
| P13 | +++ | +++ | +++ | +++ | +++ | +++ | NA | NA | NA |
| P14 | +++ | +++ | +++ | +++ | +++ | +++ | NA | NA | NA |
| P15 | +++ | +++ | +++ | NA | NA | NA | NA | NA | NA |
| P16 | +++ | +++ | +++ | NA | NA | NA | NA | NA | NA |
| P17 | ++ | ++ | ++ | ++ | ++ | ++ | NA | NA | NA |
| P18 | +++ | +++ | +++ | ND | ND | ND | ND | ND | ND |
| P19 | ++ | ++ | ++ | NA | NA | NA | NA | NA | NA |
| P20 | ++ | ++ | ++ | ND | ND | ND | NA | NA | NA |
| P21 | +++ | + | — | ND | ND | ND | NA | NA | NA |
| P22 | +++ | +++ | ++ | ND | ND | ND | NA | NA | NA |
| P23 | +++ | +++ | +++ | NA | NA | NA | NA | NA | NA |
IHC, immunohistochemical; NA, not applicable; ND, not done.
+, <30%; ++, 30% to ∼85%; +++, >85%.
Sequential infused CAR T-cell expansion and objective response
After leukapheresis, 23 patients received the first cycle of infusion with mCD19 CAR T cells at a median dose of 2.00 × 106 cells per kg (range, 0.11 × 106 to 3.00 × 106 cells per kg). The peak of mCD19 CAR T-cell expansion in PB occurred on day 7 after infusion with a median of 7.45% (range, 0.00%–59.40%) (supplemental Figure 1A). Through mCD19 CAR T-cell expansion, 15 patients achieved CR, 6 PR, 1 NR, and 1 PD.
Among the 15 patients who achieved CR, 9 patients (P3, P4, P5, P6, P9, P15, P16, P19, and P23) maintained an ongoing CR, whereas 6 patients (P7, P8, P11, P17, P18, and P21) developed RD at months 6, 6, 2, 3, 2, and 1.5, respectively, after infusion. Corresponding CAR T-cell levels at the time of relapse were 0.002%, 0.003%, 12.6%, 0.019%, 0.85%, and 39.6%, respectively. The tumors of 4 patients (P7, P8, P11, and P17) were biopsied again at the time of relapse (Table 2). In 1 patient (P11), the lymphoma cells lost CD19 expression, whereas the lymphoma cells of the remaining 3 patients (P7, P8, and P17) maintained CD19 expression.
Four patients (P7, P11, P17, and P21) with CNS involvement at enrollment had CNS disease relapse, and 1 patient (P17) died of rapid intracerebral mass (ICM) progression, which resulted in elevated intracranial pressure (ICP), a contraindication to further CAR T-cell therapy.
The remaining 5 patients (P7, P8, P11, P18, and P21) with RD, 6 patients (P1, P10, P13, P14, P20, and P22) with PR, 1 patient (P12) with NR, and 1 patient (P2) with PD received the second cycle of infusion with hCD22 CAR T cells at a median dose of 2.00 × 106 cells per kg (range, 0.17 × 106 to 4.13 × 106 cells per kg). The median interval between mCD19 and hCD22 CAR T-cell infusion was 80 days (range, 32-195 days). The peak of hCD22 CAR T-cell expansion in PB occurred on day 11 after infusion with a median expansion of 41.00% (range, 0.22%–73.60%) (supplemental Figure 1B).
Among 13 patients who received 2 cycles of CAR T-cell infusions, 5 patients (P1, P10, P13, P14, and P22) achieved an ongoing CR1; 2 patients (P20 and P21) died as a result of progression in the ICM who did not proceed with the third cycle of hCD20 CAR T-cell infusion as a result of elevated ICP and loss of CD20 in tumor cells, respectively; 3 patients (P7, P11, and P18) achieved a CR2; 2 patients (P8 and P12) attained a PR; and 1 patient (P2) developed PD after a transient PR, all of whom received the third cycle of infusion with hCD20 CAR T cells at a median dose of 1.29 × 106 cells per kg (range, 0.55 × 106 to 2.17 × 106 cells per kg). The median interval between hCD22 and hCD20 CAR T-cell infusions was 45 days (range, 26-70 days).
Although some tumor burdens were too low to be detected by magnetic resonance imaging scans after 2 cycles of CAR T-cell infusion, hCD20 CAR T cells still expanded in PB, and the expansion peak occurred on day 30 after infusion with a median of 2.97% (range, 0.60%–21.10%) (supplemental Figure 1C).
After the third cycle of CAR T-cell infusions, P2 obtained an ongoing CR1; P11 and P18 achieved an ongoing CR2; P8 who was in a PR received the fourth cycle of hCD19 CAR T-cell infusion and ultimately attained an ongoing CR2; P12 remained in CR for 5 months and then relapsed (the patient received alternative anticancer therapy demanded by the parents and sustained a PR). P7 had a CNS disease relapse again at month 3 after achieving a CR2 and finally died of rapid disease progression.
Persistence of sequentially infused CAR T cells and durable response
The median time from the last infusion to the cutoff date of March 8, 2021, was 17 months (range, 15-23 months). Twenty-one patients achieved a CR within 3 months after the last infusion. Among them, 9 patients had either NR or PR at 1 month that improved to CR in 0.5 to 1.5 months; 19 patients attained CR at 3 months; 18 patients remained in CR to the cutoff date. The probability of attaining a CR at 18 months after the last infusion was 78% (95% CI, 54%-91%) among all patients. The probability of attaining a durable CR at 18 months was 95% (95% CI, 62%-99%) among patients in CR at 3 months. Eighty-six percent (95% CI, 62%-95%) of patients were estimated to remain relapse free at 18 months after the first CR after the last infusion (Figure 2A). Durable CRs were observed for up to 34 months after the last infusion (Table 3). At 18 months after the last infusion, all patients had an estimated PFS rate of 78% (95% CI, 55%-90%) (Figure 2B) and OS rate of 83% (95% CI, 60%-93%) (Figure 2C). Fifty percent (95% CI, 28%-69%) of patients were estimated to have CAR T cells detectable in PB by qPCR at 6 months after the first infusion (Figure 3A). Consistently detectable levels of CAR transgenes were observed for up to 35 months after the first infusion (Table 3). B-cell aplasias were estimated to continue for 12 months among 51% (95% CI, 31%-79%) of patients (Figure 3B). Sustained B-cell aplasias were observed for up to 35 months after the first infusion (Table 3).
Figure 2.

Kaplan-Meier estimates of duration of CR, PFS, and OS. (A) Eighty-six percent (95% CI, 62%-95%) of patients are estimated to remain relapse-free at 18 months after the first CR after the last infusion. (B) All patients had an estimated PFS rate of 78% (95% CI, 55%-90%) at 18 months after the last infusion. (C) All patients had an estimated OS rate of 83% (95% CI, 60%-93%) at 18 months after the last infusion. Dotted lines indicate 95% CIs.
Table 3.
Durations of response, CAR T-cell persistence, and B-cell aplasia for each enrolled patient

|
Figure 3.
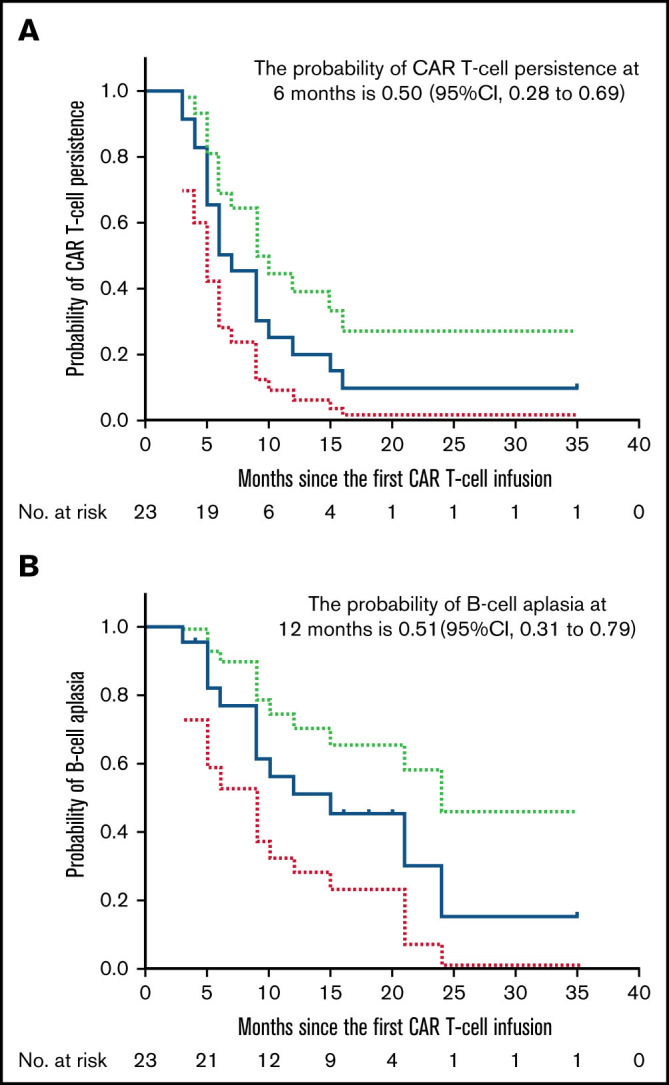
Kaplan-Meier estimates of duration of CAR T-cell persistence and B-cell aplasia. (A) Fifty percent (95% CI, 28%-69%) of patients are estimated to have CAR T cells detectable in PB by qPCR at 6 months after the first infusion. (B) B-cell aplasia is estimated to continue for 12 months among 51% (95% CI, 31%-79%) of patients. Dotted lines indicate 95% CIs.
Toxicity of sequentially infused CAR T cells
The toxicity of mCD19 CAR T-cell therapy is summarized in Table 4. CRS was reported in 16 (69.6%) of 23 patients. The median time to onset of CRS, as indicated by fever, hypotension, and/or hypoxia, was 1 day (range, 0-5 days). Peak toxicity developed between days 6 and 11. Eight patients (34.8%) experienced grade ≤2 CRS, which resolved completely by a median of 8 days (range, 2-10 days) with symptomatic support. Eight patients (34.8%) who developed grade 3 CRS received steroids, and their CRS entirely resolved by a median of 10.4 days (range, 8-17 days). Neurotoxicity occurred in 8 (34.8%) of 23 patients. The median time to development of neurotoxicity was 6.4 days (range, 5-10 days) in contemporary CRS settings. Grade 1 ICANS, as manifested by symptoms such as a delay in response, impaired handwriting, and mild drowsiness occurred in 3 (13.0%) of 23 patients. Five (21.7%) of 23 patients experienced grade 3 ICANS, as manifested by seizures, elevated ICP, and/or local edema shown by neuroimaging. All 5 patients received intravenous and intrathecal dexamethasone and attained remission with no clinical sequelae. The median duration of neurotoxicity was 5.5 days (range, 3-10 days). During the subsequent infusions, grade 3 CRS was observed in 2 (15.4%) of 13 patients who received CD22 CAR T-cell infusions. No treatment-related deaths occurred during the follow-up.
Table 4.
The incidence and kinetics of CRS and ICANS during mCD19 CAR T-cell therapy
| Patient | CRS grade | Onset of CRS (d) | Resolution of CRS (d) | CRS-specific symptoms | ICANS grade | Onset of ICANS (d) | Resolution of ICANS (d) | ICANS-specific symptom | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fever* | Hypotension† | Hypoxia‡ | Depressed level of consciousness | Seizure | Elevated ICP/cerebral edema | |||||||
| P1 | III | 2 | 8 | Yes | IV fluids | High-flow | NA | NA | NA | NA | NA | NA |
| P2 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P3 | III | 1 | 9 | Yes | 1 vasopressor | Low-flow | I | 7 | 4 | Awaken spontaneously | No | No |
| P4 | I | 3 | 9 | Yes | No | No | NA | NA | NA | NA | NA | NA |
| P5 | III | 1 | 9 | Yes | IV fluids | High-flow | NA | NA | NA | NA | NA | NA |
| P6 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P7 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P8 | I | 3 | 2 | Yes | No | No | NA | NA | NA | NA | NA | NA |
| P9 | I | 1 | 10 | Yes | No | No | NA | NA | NA | NA | NA | NA |
| P10 | III | 2 | 10 | Yes | IV fluids | High-flow | NA | NA | NA | NA | NA | NA |
| P11 | II | 1 | 10 | Yes | IV fluids | No | III | 7 | 10 | Awaken to voice | No | Local edema |
| P12 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P13 | III | 0 | 8 | Yes | 1 vasopressor | Low-flow | NA | NA | NA | NA | NA | NA |
| P14 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P15 | I | 1 | 7 | Yes | No | No | I | 5 | 3 | Awaken spontaneously | No | No |
| P16 | I | 0 | 9 | Yes | No | No | I | 5 | 5 | Awaken spontaneously | No | No |
| P17 | II | 5 | 2 | Yes | IV fluids | No | III | 5 | 10 | Awaken spontaneously | Yes | Elevated ICP |
| P18 | III | 0 | 17 | Yes | 1 vasopressor | Low-flow | III | 10 | 6 | Awaken spontaneously | Yes | Elevated ICP |
| P19 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P20 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P21 | III | 0 | 12 | Yes | 1 vasopressor | Low-flow | III | 6 | 6 | Awaken to voice | Yes | Elevated ICP |
| P22 | III | 0 | 10 | Yes | 1 vasopressor | Low-flow | III | 6 | 5 | Awaken spontaneously | Yes | Elevated ICP |
| P23 | I | 3 | 5 | Yes | No | No | NA | NA | NA | NA | NA | NA |
NA, not applicable; IV, intravenous.
Fever defined as temperature ≥38°C not attributable to any other cause.
Hypotension responding to IV fluids or managed with 1 or multiple vasopressors.
Hypoxia requiring supplemental oxygen (low flow ≤6 L/min nasal cannula or blow-by, high flow oxygen, or positive pressure).
Efficacy and safety of sequential CAR T-cell infusions in patients with bulky disease and CNS involvement
Of 9 patients with bulky disease, 4 patients received 2 cycles of CAR T-cell infusions, and 3 received 3 cycles. Seven of the 9 patients attained an ongoing CR. The median PFS rate was not reached, but an estimated 18-month PFS rate was 78% (95% CI, 37%-94%) in the patients with bulky disease. No significant difference was observed in the estimated 18-month PFS rate between the bulky disease group and the non-bulky disease group (P = .97) (Figure 4A). Patient 18 had rapidly progressing bulky disease before CAR T-cell treatment. On day 30 after mCD19 CAR T-cell infusion, an magnetic resonance imaging scan showed a CR (Figure 4B). However, on day 60, fluorodeoxyglucose-positron emission tomography/computed tomography revealed new lesions at multiple sites. Through sequential hCD22 and hCD20 CAR T-cell infusions, a metabolic CR was reached (Figure 4C). The patients with bulky disease experienced more severe CRS (7 of 9) than those without (1 of 14) (P = .001) (supplemental Figure 2A).
Figure 4.

Efficacy of sequential CAR T-cell infusion in 9 patients with bulky disease. (A) The median PFS rate was not reached with an estimated 18-month PFS rate of 78% (95% CI, 37%-94%) in the patients with bulky disease, and no significant difference was observed in the estimated 18-month PFS rate between the group of patients with bulky disease and the group of patients with non-bulky disease (P = .97). (B) Magnetic resonance imaging (MRI) scans for P18 demonstrated CR of the mass on day 30 after edema of the mass on day 15 following mCD19 CAR T-cell infusion. (C) Positron emission tomography/computed tomography scans of P18 revealed the emergence of new lesions at multiple sites on day 60 after mCD19 CAR T-cell infusion and then CR2 through sequential hCD22 and hCD20 CAR T-cell infusions. ADC, apparent diffusion coefficient; DWI, diffusion weighted imaging; T2WI, T2-weighted imaging.
To understand the potential mechanism, we measured the serum of these patients to determine levels of IL-6. We found significantly higher levels of serum IL-6 in patients with bulky disease (median peak, 147.60 pg/mL on day 7) than in those without bulky disease (median peak, 14.37 pg/mL on day 7) (P = .0069) (supplemental Figure 2B). The mCD19 CAR T cells expanded in the cerebrospinal fluids of 10 patients with CNS involvement (Figure 5A). The tumor beneath the leptomeningeum of P16 disappeared completely after mCD19 CAR T-cell infusion (Figure 5B). Six of 10 patients with CNS involvement achieved ongoing CR. The estimated 18-month PFS rate of 60% (95% CI, 25%-83%) in the patients with CNS involvement was not significantly lower than the rate of 92% (95% CI, 56%-99%) in the patients without CNS involvement (P = .28) (Figure 5C). We did not observe any statistically significant differences in the severity of CRS and neurotoxicity between patients with (P = 1.0) and without (P = 0.62) CNS involvement (supplemental Figure 3).
Figure 5.

Efficacy of sequential CAR T-cell infusions in 10 patients with CNS involvement. (A) CD19 CAR T-cell expansion in the cerebrospinal fluid (CSF) of 10 patients with CNS involvement. (B) The MRI scans demonstrate that the mass beneath the leptomeningeum of P16 disappeared completely after mCD19 CAR T-cell infusion. (C) PFS after sequential CAR T-cell infusion. The median PFS rate was not reached; the estimated 18-month PFS rate was 60% (95% CI, 25%-83%) in patients with CNS involvement, which was not significantly lower than that in patients with no CNS involvement (P = .28).
Discussion
To achieve their antitumor efficacy, CAR T cells must be able to reach tumor cells, interact with them through their intended antigen, proliferate, kill tumor cells, escape a hostile tumor microenvironment, and persist to provide durable tumor control.41 First, CAR T-cell expansion is a critical predictor of clinical response, as demonstrated in our study. The peak CAR T-cell expansion of mCD19, hCD22, and hCD20 occurred on days 7, 11, and 30 after infusion, respectively. These effective expansions resulted in significant responses. Fifteen (65.22%) of 23 patients with R/R Burkitt lymphoma achieved a CR after mCD19 CAR T-cell infusion; 8 (61.54%) of 13 patients with a PR, NR, PD, or RD achieved a CR after hCD22 CAR T-cell infusion; 2 (66.67%) of 3 patients with a PR or PD achieved a CR after hCD20 CAR T-cell infusion.
Second, CAR T-cell persistence is critical for preventing PD or relapse. Lymphoma cells may escape from immune surveillance by 2 mechanisms: they may hide themselves from immune recognition by losing or downregulating their target antigens, or they may defend themselves against immune eradication by expressing inhibitory ligands and inducing inhibitory cells to exhaust T cells.42,43 In our study, 6 patients who had attained a CR during mCD19 CAR T-cell expansion developed relapses during mCD19 CAR T-cell contraction. Biopsies are not always obtained from patients with lymphoma at the time of relapse. Four patients who had RD after mCD19 CAR T-cell infusion underwent biopsy. One of these patients had loss of CD19 in tumor cells, which suggests the first mechanism, whereas the remaining 3 patients had continued expression of CD19 in tumor cells, which suggests the second mechanism. In addition, we observed a loss of CAR T cells detectable in PB by FCM in the 3 patients at the time of relapse, which may support the second mechanism. The study of tisagenlecleucel in adult R/R diffuse large B-cell lymphoma demonstrated that 54% of patients with a PR or stable disease at 1 month after tisagenlecleucel infusion had improvement to a CR in a median of 2 months, and responses at 3 months were sustained through 6 months.18,19 In our study, 40% of the patients who achieved a CR after mCD19 CAR T-cell infusion developed relapses at a median of 2.5 months (range, 1.5-6 months). This finding indicates that mCD19 CAR T cells may not be able to be sustained long enough to provide persistent immune surveillance in these patients. We designed strategies for administering combinational and sequential CAR T cells to eradicate disease, maintain remission, and prevent relapse.
To minimize the risk of PD or relapse by maintaining CAR T-cell persistence in each patient, we attempted to identify the optimal timing for subsequent CAR T-cell infusions. If subsequent infusion is initiated when the tumor is still responding to the previous CAR T cells, LD chemotherapy before subsequent infusion may eradicate the previous CAR T cells that are still working, which may interrupt ongoing response before the tumor reaches the best response. If we do not proceed to the next infusion until we can observe loss of the previous CAR T cells detectable in PB by qPCR, the tumor may have progressed or recurred before the next infusion happens. We established a sequential strategy to treat patients who did not achieve an ongoing CR according to their disease status and CAR T-cell persistence. If a patient had a PR, NR, or CR2 with no CAR T cells being detectable in PB by FCM, or if a patient had PD or RD with or without CAR T cells being detectable in PB by FCM, an alternative CAR T-cell infusion was initiated. Akin to chemotherapy for Burkitt lymphoma, in this strategy, the first mCD19 CAR T-cell infusion provides induction therapy; this is followed by the second hCD22 CAR T-cell infusion, which provides intensification and consolidation therapy for the patients with PD, RD, NR, or PR; subsequently, the third hCD20 CAR T-cell infusion provides continued consolidation therapy for patients who still have PD, NR, or PR, and it provides maintenance therapy for patients with a CR2. Overall, this sequential strategy could play a preemptive and prophylactic role in eradicating disease, maintaining remission, and preventing relapse. We treated 13 patients with 2 cycles of infusion while they had a PR, NR, PD, or RD after the first infusion; we treated 6 patients with 3 cycles of infusion while they had PD, PR, or CR2 after the second infusion. Ninety-five percent of patients who attained a CR at 3 months after the last infusion were estimated to remain in CR at 18 months. The results suggest that CRs at 3 months after the last infusion may be durable in our patients. Durable responses may be partly attributable to retained functional CAR T-cell persistence, as indicated by 51% of patients estimated to have B-cell aplasia at 12 months after the first infusion.
We used sequential CAR T-cell infusion as a salvage therapy for patients in this trial who had bulky disease and/or CNS involvement. We observed that CAR T cells could be rapidly depleted from circulation and inactivated when they were exposed to a large number of target cancer cells. A high tumor burden before LD chemotherapy may impose a potential barrier to CAR T-cell persistence.25 However, in our study, no significant difference was observed in the estimated 18-month PFS rate between groups of patients with bulky disease and those with non-bulky disease, suggesting that sequential CAR T-cell infusions can overcome this potential CAR T-cell therapeutic barrier in these patients. A significant improvement has been made in the clinical outcomes of patients with CNS involvement who have an estimated 18-month PFS rate of 60%.
Regarding the safety of sequential CAR T-cell infusions, the toxicities of mCD19 CAR T cells seemed more common and severe than those of sequentially infused alternative CAR T cells, which may be attributable to a higher pretreatment tumor burden. Nevertheless, these toxicities did not have an adverse impact on subsequent CAR T-cell initiation because they were entirely resolved by 3 weeks after mCD19 CAR T-cell infusion. The severity of neurotoxicity was not significantly different between patients with CNS involvement and those without CNS involvement. These results suggest that sequential infusion of 2 to 4 CAR products is safe for pediatric patients.
Although we observed some promising results, this study had several limitations. First, this was a single-arm clinical study that lacked a control group for comparing clinical efficacy. This limitation was primarily attributed to distinct clinical characteristics of the patients. Second, we could not compare the outcome difference between sequential CAR T-cell infusions and dual-antigen CAR T-cell therapy. Third, the clinical trial excluded patients with CNS bulky disease and elevated ICP because of concerns regarding the increased risk of neurotoxicity. Finally, the cost-effectiveness of this treatment course imposed some practical constraints. The cost of sequential treatment with CAR T cells is higher than that of chemotherapy but not necessarily higher than that of stem cell transplantation. Moreover, compared with salvage chemotherapy and stem cell transplantation, sequential CAR T-cell therapy has a better safety profile and survival rate. In conclusion, our study demonstrates that sequential CAR T-cell therapy may result in a durable response and is safe in pediatric patients with R/R Burkitt lymphoma, and pediatric patients with R/R Burkitt lymphoma who also have CNS involvement may benefit from sequential CAR T-cell therapy.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all of the patients who participated in the study and their families, friends, and caregivers for their support; the study staff and health care providers at all of the clinical trial sites for their dedicated efforts in this trial; and Chen Dong, MD (Division of Hematopathology, Mayo Clinic, Rochester, MN) and Constance P. Chen (University of Notre Dame, Notre Dame, IN) for their critical review and editing the English for this manuscript.
This work was partially supported by grants from the National Key Basic Research Program of China (2016YFC1303403) and the National Natural Science Foundation of China (81272325).
Authorship
Contribution: Y.Z. and C.T. designed the study; A.H.C. and B.D. produced the CAR T cells; Y.Z., Ying Liu, B.H., W.Z., Q. Zhu, Yang Liu, S.W., Y.Y., Junhan Yang, P.Z., Jing Yang, and L.J. enrolled and treated patients and gathered data; Z.G., C.Z., Q. Zheng, and X.Y. performed the morphology, immunology, cytogenetics, and molecular biology integrated diagnoses of tumor tissues; B.D., Q. Zheng, and X.Y. monitored CAR T-cell expansion level, serum cytokines concentration, and B-cell level; W.H. and Ying Liu analyzed and interpreted data; Ying Liu wrote the manuscript; and all authors carefully read and approved the final manuscript.
Conflict-of-interest disclosure: A.H.C. is a founding member of Shanghai YaKe Biotechnology Ltd, a biotechnology company focused on research and development of tumor cellular immunotherapy. The remaining authors declare no competing financial interests.
Correspondence: Yonghong Zhang, Department of Pediatric Lymphoma, Beijing Boren Hospital, No. 6, South Zhengwangfen, Fengtai District, Beijing 100070, China; e-mail: zhangyongh@gobroadhealthcare.com; and Alex H. Chang, Clinical Translational Research Center, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai 200438, China; e-mail: alexhchang@yahoo.com.
References
- 1.Patte C, Auperin A, Michon J, et al. ; Société Française d’Oncologie Pédiatrique . The Société Française d’Oncologie Pédiatrique LMB89 protocol: highly effective multiagent chemotherapy tailored to the tumor burden and initial response in 561 unselected children with B-cell lymphomas and L3 leukemia. Blood. 2001;97(11):3370-3379. [DOI] [PubMed] [Google Scholar]
- 2.Woessmann W, Seidemann K, Mann G, et al. ; BFM Group . The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. Blood. 2005;105(3):948-958. [DOI] [PubMed] [Google Scholar]
- 3.Cairo MS, Gerrard M, Sposto R, et al. ; FAB LMB96 International Study Committee . Results of a randomized international study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109(7):2736-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patte C, Auperin A, Gerrard M, et al. ; FAB/LMB96 International Study Committee . Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007;109(7):2773-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerrard M, Cairo MS, Weston C, et al. ; FAB LMB96 International Study Committee . Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Br J Haematol. 2008;141(6):840-847. [DOI] [PubMed] [Google Scholar]
- 6.Cairo MS, Sposto R, Gerrard M, et al. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (≥ 15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB LMB 96 study. J Clin Oncol. 2012;30(4):387-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldman S, Smith L, Anderson JR, et al. Rituximab and FAB/LMB 96 chemotherapy in children with stage III/IV B-cell non-Hodgkin lymphoma: a Children’s Oncology Group report. Leukemia. 2013;27(5):1174-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frazer JK, Li KJ, Galardy PJ, et al. Excellent outcomes in children and adolescents with CNS+ Burkitt lymphoma or other mature B-NHL using only intrathecal and systemic chemoimmunotherapy: results from FAB/LMB96 and COG ANHL01P1. Br J Haematol. 2019;185(2):374-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minard-Colin V, Aupérin A, Pillon M, et al. ; Children’s Oncology Group . Rituximab for high-risk, mature B-cell non-Hodgkin’s lymphoma in children. N Engl J Med. 2020;382(23):2207-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita N, Mori T, Mitsui T, Inada H, Horibe K, Tsurusawa M; Lymphoma Committee of the Japanese Pediatric Leukemia/Lymphoma Study Group . The role of hematopoietic stem cell transplantation with relapsed or primary refractory childhood B-cell non-Hodgkin lymphoma and mature B-cell leukemia: a retrospective analysis of enrolled cases in Japan. Pediatr Blood Cancer. 2008;51(2):188-192. [DOI] [PubMed] [Google Scholar]
- 11.Anoop P, Sankpal S, Stiller C, et al. Outcome of childhood relapsed or refractory mature B-cell non-Hodgkin lymphoma and acute lymphoblastic leukemia. Leuk Lymphoma. 2012;53(10):1882-1888. [DOI] [PubMed] [Google Scholar]
- 12.Jourdain A, Auperin A, Minard-Colin V, et al. Outcome of and prognostic factors for relapse in children and adolescents with mature B-cell lymphoma and leukemia treated in three consecutive prospective “Lymphomes Malins B” protocols. A Société Française des Cancers de l’Enfant study. Haematologica. 2015;100(6):810-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cairo M, Auperin A, Perkins SL, et al. Overall survival of children and adolescents with mature B cell non-Hodgkin lymphoma who had refractory or relapsed disease during or after treatment with FAB/LMB 96: a report from the FAB/LMB 96 study group. Br J Haematol. 2018;182(6):859-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woessmann W, Zimmermann M, Meinhardt A, et al. Progressive or relapsed Burkitt lymphoma or leukemia in children and adolescents after BFM-type first-line therapy. Blood. 2020;135(14):1124-1132. [DOI] [PubMed] [Google Scholar]
- 15.Harker-Murray PD, Pommert L, Barth MJ. Novel therapies potentially available for pediatric B-cell non-Hodgkin lymphoma. J Natl Compr Canc Netw. 2020;18(8):1125-1134. [DOI] [PubMed] [Google Scholar]
- 16.Hsieh EM, Rouce RH. Chimeric antigen receptor T cells for mature B-cell lymphoma and Burkitt lymphoma. Hematology Am Soc Hematol Educ Program. 2020;2020(1):487-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuster SJ, Bishop MR, Tam CS, et al. ; JULIET Investigators . Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45-56. [DOI] [PubMed] [Google Scholar]
- 20.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20(1):31-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839-852. [DOI] [PubMed] [Google Scholar]
- 22.Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol. 2019;10:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie Y, Lu W, Chen D, et al. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res. 2020;8(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin H, Ramakrishna S, Nguyen S, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics. 2018;11:127-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong C, Zhang Y, Liu Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136(14):1632-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacobson CA, Locke FL, Miklos DB, et al. End of phase 1 results from Zuma-6: axicabtagene ciloleucel (axi-cel) in combination with atezolizumab for the treatment of patients with refractory diffuse large B cell lymphoma [abstract]. Blood. 2018;132(suppl 1). Abstract 4192. [Google Scholar]
- 29.Pan J, Zuo S, Deng B, et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood. 2020;135(5):387-391. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Yang J, Zhou C, et al. Early response observed in pediatric patients with relapsed/refractory Burkitt lymphoma treated with chimeric antigen receptor T cells. Blood. 2020;135(26):2425-2427. [DOI] [PubMed] [Google Scholar]
- 31.Cazzola M. Introduction to a review series: the 2016 revision of the WHO classification of tumors of hematopoietic and lymphoid tissues. Blood. 2016;127(20):2361-2364. [DOI] [PubMed] [Google Scholar]
- 32.Pan J, Yang JF, Deng BP, et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia. 2017;31(12):2587-2593. [DOI] [PubMed] [Google Scholar]
- 33.Pan J, Niu Q, Deng B, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33(12):2854-2866. [DOI] [PubMed] [Google Scholar]
- 34.Rosolen A, Perkins SL, Pinkerton CR, et al. Revised international pediatric non-Hodgkin lymphoma staging system. J Clin Oncol. 2015;33(18):2112-2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandlund JT, Guillerman RP, Perkins SL, et al. International pediatric non‐Hodgkin lymphoma response criteria. J Clin Oncol. 2015;33(18):2106-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yakoub-Agha I, Chabannon C, Bader P, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 2020;105(2):297-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lulla PD, Hill LC, Ramos CA, Heslop HE. The use of chimeric antigen receptor T cells in patients with non-Hodgkin lymphoma. Clin Adv Hematol Oncol. 2018;16(5):375-386. [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strati P, Ahmed S, Furqan F, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood. 2021;137(23):3272-3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev. 2018;8:210-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fowler NH, Cheah CY, Gascoyne RD, et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica. 2016;101(5):531-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Charette M, Houot R. Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica. 2018;103(8):1256-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.