

Abstract
Background
Intravenous broad–spectrum antibiotics are indicated for the treatment of severe infections. However, the emergence of infections caused by multiple‐drug resistant organisms in conjunction with a lack of novel antibiotics has prompted the investigation of alternative dosing strategies to improve clinical efficacy and tolerability. To optimise pharmacokinetic and pharmacodynamic antibiotic parameters, continuous antibiotic infusions have been compared with traditional intermittent antibiotic infusions.
Objectives
To compare the clinical efficacy and safety of continuous intravenous administration of concentration–dependent and time–dependent antibiotics with traditional intermittent intravenous administration in adults with severe acute bacterial infections.
Search methods
The following electronic databases were searched in September 2012: The Cochrane Injuries Group Specialised Register, the Cochrane Central Register of Controlled Trials (CENTRAL), The Cochrane Library, MEDLINE (OvidSP), EMBASE (OvidSP), CINAHL, ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED), and ISI Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S). The reference lists of all relevant materials, the Internet, and the trials registry www.clinicaltrials.gov for completed and ongoing trials were also searched.
Selection criteria
Randomised controlled trials in adults with a bacterial infection requiring intravenous antibiotic therapy comparing continuous versus intermittent infusions of antibiotics were included. Both time–dependent and concentration–dependent antibiotics were considered.
Data collection and analysis
Three independent authors performed data extraction for the included studies. All data were cross–checked and disagreements resolved by consensus. An intention‐to‐treat analysis was conducted using a random–effects model.
Main results
Twenty–nine studies met inclusion criteria with a combined total of more than 1600 participants. Most included studies were judged to be at unclear or high risk of bias with regard to randomisation sequence generation, allocation concealment, blinding, management of incomplete outcome data, selective outcome reporting, and other potential threats to validity. No studies were judged to be at low risk of bias for all methodological quality items assessed. No differences in all–cause mortality (n = 1241, risk ratio (RR) 0.89, 95% confidence interval (CI) 0.67 to 1.20, P = 0.45), infection recurrence (n = 398, RR 1.22, 95% CI 0.35 to 4.19, P = 0.76), clinical cure (n = 975, RR 1.00, 95% CI 0.93 to 1.08, P = 0.98), and super‐infection post–therapy (n = 813, RR 1.08, 95% CI 0.60 to 1.94, P = 0.79) were reported, nor were any differences observed in safety outcomes, including adverse events (n = 575, RR 1.02, 95% CI 0.94 to 1.12, P = 0.63), serious adverse events (n = 871, RR 1.36, 95% CI 0.80 to 2.30, P = 0.26), and withdrawals due to adverse events (n = 871, RR 2.03, 95% CI 0.52 to 7.95, P = 0.31). A difference was observed in subgroup analyses of clinical cure in septic versus non‐septic participants, in which intermittent antibiotic infusions were favoured for clinical cure in septic participants. However, this effect was not consistent between random‐effects and fixed‐effect analyses. No differences were noted in the sensitivity analyses conducted.
Authors' conclusions
No differences in mortality, infection recurrence, clinical cure, super‐infection post–therapy, and safety outcomes were reported when continuous infusions of intravenous antibiotics were compared with traditional intermittent infusions of antibiotics. However, the wide confidence intervals suggest that beneficial or harmful effects cannot be ruled out for all outcomes. Therefore, the current evidence is insufficient to recommend the widespread adoption of continuous infusion antibiotics in the place of intermittent infusions of antibiotics. Additinal large prospective randomised trials, with consistent and complete reporting of clinical outcome measures, conducted with concurrent pharmacokinetic and pharmacodynamic studies in special populations, are required to determine whether adoption of continuous antibiotic infusions is warranted in specific circumstances.
Keywords: Adult; Humans; Acute Disease; Anti‐Bacterial Agents; Anti‐Bacterial Agents/administration & dosage; Anti‐Bacterial Agents/pharmacokinetics; Bacterial Infections; Bacterial Infections/drug therapy; Bacterial Infections/metabolism; Bacterial Infections/mortality; Infusions, Intravenous; Infusions, Intravenous/methods; Randomized Controlled Trials as Topic; Recurrence
Plain language summary
Alternative dosing strategies for intravenous antibiotics to treat severe infections
Intravenous (through the vein) antibiotics are used to treat severe bacterial infections. Currently, the most common way to administer intravenous antibiotics is by intermittent infusion, whereby an antibiotic is infused into a patient over 30 minutes to 1 hour multiple times per day during the course of treatment. To optimise the efficacy and potentially the safety of these antibiotics, alternative dosing strategies have been studied. One proposed strategy is to administer intravenous antibiotics by continuous or extended infusions over 3 to 24 hours.
Twenty–nine randomised trials comprising more than 1600 participants were reviewed to study the effects of continuous infusion antibiotics versus intermittent infusion antibiotics. When mortality, infection recurrence, clinical cure, super‐infection after treatment, and safety concerns were considered, no differences between the two dosing strategies were noted.
The authors conclude that because continuous antibiotic infusions provide no benefit over standard intermittent infusions, they cannot recommend continuous antibiotic infusions for widespread use.
Summary of findings
Summary of findings for the main comparison. Continuous versus Intermittent Antibiotic Infusions for Treatment of Severe Bacterial Infections.
| Continuous versus Intermittent Antibiotic Infusions for Treatment of Severe Bacterial Infections | |||||
| Patients: Adults with Severe Bacterial Infections Setting: Hospital Intervention: Continuous Antibiotic Infusions Comparison: Intermittent Antibiotic Infusions | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Intermittent antibiotic Infusions | Continuous antibiotic Infusions | ||||
| All‐cause mortality | 131 per 1000 | 116 per 1000 (88 to 157) | RR 0.89 (0.67 to 1.20) | 1241 (19 studies) | ⊕⊕⊕⊝ moderate |
| Infection recurrence | 19 per 1000 | 24 per 1000 (7 to 81) | RR 1.22 (0.35 to 4.19) | 398 (8 studies) | ⊕⊕⊝⊝ low |
| Clinical cure | 584 per 1000 | 608 per 1000 (555 to 660) | RR 1.04 (0.95 to 1.13) | 975 (15 studies) | ⊕⊕⊝⊝ low |
| Super‐infection | 49 per 1000 | 53 per 1000 (29 to 95) | RR 1.08 (0.60 to 1.94) | 813 (12 studies) | ⊕⊕⊝⊝ low |
| Serious adverse events | 48 per 1000 | 65 per 1000 (38 to 110) | RR 1.36 (0.80 to 2.30) | 871 (10 studies) | ⊕⊕⊝⊝ low |
| Withdrawal due to adverse events | 7 per 1000 | 14 per 1000 (4 to 54) | RR 2.03 (0.52 to 7.95) | 871 (10 studies) | ⊕⊕⊝⊝ low |
| Adverse events | 441 per 1000 | 450 per 1000 (415 to 494) | RR 1.02 (0.94 to 1.12) | 575 (5 studies) | ⊕⊝⊝⊝ very low |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
Background
Description of the condition
Intravenous broad–spectrum antibiotics are indicated for the treatment of severe community–acquired or healthcare–associated infection. However, the emergence of multiple–drug resistant infections caused by organisms such as Staphylococcus aureus, Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, Enterococcus faecium, Enterococcus faecalis, and Enterobacteriaceae is growing worldwide (Rosenthal 2012; Ghafourian 2011; Meyer 2011; Neidell 2012). Such antimicrobial–resistant infections have been associated with poor outcomes, such as increases in length of hospital stay, healthcare costs, and mortality (Sunenshine 2007; Neidell 2012). Despite this growing problem, few novel antibiotics have been developed in recent years; therefore, alternative dosing strategies have been investigated to improve clinical efficacy while ensuring tolerability. To optimise the pharmacokinetic and pharmacodynamic properties of antibiotics, dosing strategies such as continuous or extended intravenous antibiotic infusions have been compared with traditional intermittent intravenous antibiotic dosing.
Description of the intervention
Antibiotics are divided into categories on the basis of the pharmacokinetic and pharmacodynamic parameters associated with antibacterial efficacy. Although these bacterial kill characteristics have been determined most often from in vitro studies, this information remains important in optimising antibiotic clinical efficacy. For example, aminoglycosides, fluoroquinolones, and metronidazole are classified as concentration–dependent antibiotics, for which efficacy is determined by peak plasma drug concentration over minimum inhibitory concentration (Cmax/MIC) (Roberts 2006; Craig 1998; Moore 1987). Conversely, beta–lactams, carbapenems, clindamycin, linezolid, and clarithromycin are time–dependent or concentration–independent antibiotics for which the time the drug serum concentration remains above the minimum inhibitory concentration (T > MIC) is the best predictor of efficacy (Roberts 2006; McKinnon 2008; Craig 1998; van Zanten 2009). Although it is commonly accepted that antibiotics are divided into two main classifications, some antimicrobials exhibit more complex bacterial kill characteristics. Fluoroquinolones, azithromycin, glycopeptides, and tetracyclines are concentration–dependent antibiotics with time dependence, for which efficacy is best predicted by the area under the serum concentration–time curve during 24 hours over the minimum inhibitory concentration (AUC24/MIC) (Roberts 2006; Craig 1998).
Studies evaluating the efficacy of continuous or extended antibiotic infusions generally involve time–dependent antibiotics (Buck 2005; Roberts 2008; Roberts 2009a). Several time–dependent antibiotics are known to have a short half‐life; therefore, concern has arisen that the drug serum concentration will drop below the minimum inhibitory concentration (MIC) before the next scheduled intermittent infusion (Lipman 2001). To optimise antibiotic kill characteristics, extended (> 3‐hour intermittent infusions) or continuous (24‐hour fixed–rate infusions) administration is thought to prolong T > MIC and to improve clinical efficacy (Tamma 2011; Lodise 2006). Although T > MIC targets vary between antibiotic classes (20% to 40% carbapenems, 50^ to 60% penicillins, 60% to 70% cephalosporins, 50% to 60% monobactams) (Craig 1998; Drusano 2004), improved clinical cure rates and bacteriologic eradication were observed in critically ill participants when T > MIC was maintained at 100% (McKinnon 2008). It has also been suggested in comparison with intermittent infusions of vancomycin and beta–lactams, continuous infusions may reduce time to achieve therapeutic drug serum concentrations (Roberts 2008). In contrast, for concentration–dependent antibiotics that exhibit post–antibiotic effects, it is not known whether extended or continuous infusions would be of additional benefit because large, infrequent infusions would maximize Cmax/MIC, resulting in peak efficacy. However, for select concentration‐dependent antibiotics in which efficacy is also characterized by AUC24/MIC (e.g. fluoroquinolones), extended or continuous infusions may result in favourable clinical outcomes.
Why it is important to do this review
Previous reviews have suggested that continuous or extended infusion of time–dependent antibiotics results in more favourable pharmacodynamic outcomes. A systematic review of 17 randomised clinical trials performed by Kasiakou et al compared the pharmacokinetic and pharmacodynamic parameters of intermittent and continuous infusions of time–dependent antibiotics in hospitalised adults and in healthy volunteers (Kasiakou 2005a). It was found that the mean Cmax of the intermittent infusion group was 5.5 times greater than the steady–state serum concentration (Css) of the continuous infusion group (range 1.9 to 11.2). Additionally, the Css of the continuous infusion group was 5.8 times higher than the trough serum concentration (Cmin) of the intermittent infusion group (range 1.2 to 15.6). Investigators also observed that the T > MIC was longer in the continuous infusion group in three of the six studies included.
Although it is of theoretical advantage to administer time–dependent antibiotics by continuous or extended infusion, several systematic reviews investigating this issue have not confirmed these proposed clinical benefits. A meta–analysis of nine randomised controlled trials (RCTs) performed by Kasiakou et al to compare continuous versus intermittent administration of beta–lactams, aminoglycosides, and vancomycin showed no difference in clinical failure (odds ratio (OR) 0.73, 95% CI 0.53 to 1.01) or mortality (OR 0.89, 95% CI 0.48 to 1.64) (Kasiakou 2005b). However, a subgroup analysis of those RCTs that used the same daily antibiotic dose in both intervention groups showed reduced clinical failure in the continuous infusion arm (OR 0.70, 95% CI 0.50 to 0.98) (Kasiakou 2005b). A systematic review of 14 RCTs conducted by Tamma et al in hospitalised patients showed that prolonged beta–lactam infusions rather than intermittent infusions did not affect mortality (n = 982, RR 0.92, 95% CI 0.61 to 1.37) or clinical cure (n = 1380, RR 1.00, 95% CI 0.94 to 1.06) (Tamma 2011). Another systematic review of 14 RCTs including 846 hospitalised patients from 9 countries also showed that continuous or extended infusion beta–lactam infusions versus bolus dosing led to no improvement in clinical cure (n = 745, OR 1.04, 95% CI 0.74 to 1.46, P = 0.83) or mortality (n = 541, OR 1.00, 95% CI 0.48 to 2.06, P = 1.00) (Roberts 2009a). These systematic reviews were generally well conducted; however, they were limited by small sample sizes, clinical heterogeneity in participants and infections studied, and study designs that were not blind.
Investigators reported some improvement in pharmacodynamic outcomes with weak evidence supporting clinical benefits of continuous or extended infusion antibiotics. Therefore, the purpose of this review will be to determine whether any advantage is derived from using this alternative dosing strategy rather than conventional dosing strategies in patients with severe infection. In addition to evaluating clinical and safety outcomes of continuous versus intermittent dosing of time–dependent antibiotics, this review will be extended to include concentration–dependent antibiotics.
Objectives
To compare the clinical efficacy and safety of continuous intravenous administration of concentration‐dependent and time‐dependent antibiotics with traditional intermittent intravenous administration in patients with severe acute bacterial infection. Continuous intravenous infusions included extended and continuous infusions. Severe acute infection was defined as any infection requiring intravenous antibiotics.
Methods
Criteria for considering studies for this review
Types of studies
Open‐label or blinded parallel‐group RCTs comparing continuous versus intermittent intravenous infusions of the same antibiotic were included. Cross‐over studies were excluded.
Types of participants
Study participants were male or non‐pregnant female adults (18 years or older) with a bacterial infection requiring intravenous antibiotic therapy. Investigators considered different infections in this review, and no restrictions were placed on the anatomical site of infection, participant baseline risk, or co‐morbid conditions.
Types of interventions
Included studies compared continuous versus intermittent infusions of the same intravenous antibiotic. Both time‐dependent and concentration‐dependent antibiotics were included.
Types of outcome measures
Primary outcomes
All‐cause mortality.
Infection recurrence within 14 days of resolution of primary infection.
Secondary outcomes
Clinical cure (any pre‐defined criteria specific to the infection being studied that address signs and symptoms of infection, such as fever, leukocyte counts, bacterial culture results, vital signs and visual signs or symptoms of infection, such as sputum production or inflammation, redness, or size of skin lesion).
Secondary/super‐infections post‐therapy (new infection with different organisms from those observed in the primary infection).
-
Safety:
Number of participants who experienced at least one serious adverse event (results in death, is life threatening, places the participant at immediate risk of death, requires or prolongs hospitalisation, causes permanent/significant disability or incapacity, or is another condition that investigators judge to represent significant hazards).
Number of participants who withdrew as the result of adverse events.
Number of participants with at least one adverse event.
Search methods for identification of studies
The searches were not restricted by date, language, or publication status.
Electronic searches
The following electronic databases were searched;
Cochrane Injuries Group Specialised Register (13 September 2012).
Cochrane Central Register of Controlled Trials (CENTRAL)(The Cochrane Library) 2012, Issue 8 of 12.
MEDLINE (OvidSP) 1946 to September Week 1 2012.
EMBASE (OvidSP) 1980 to 2012 Week 37.
CINAHL (EBSCO) (1982 to September 2012).
ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED) (1970 to 13 September 2012).
ISI Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S) (1990 to 13 September 2012).
Searches were based on the MEDLINE search strategy reported in the protocol, and amendments were made, when necessary, to adapt it for the other databases. All search strategies are reported in full in Appendix 1.
Searching other resources
A manual search of reference lists of all relevant material was performed to identify additional potentially eligible studies. The Internet was searched using the Google search engine (www.google.com), with selected terms from the MEDLINE strategy, to identify any further unpublished or grey literature. An online clinical trials register (www.clinicaltrials.gov) was searched for completed and ongoing trials, and authors were contacted for information about ongoing or recently completed trials.
Data collection and analysis
The Injuries Group Trials Search Co‐ordinator conducted the search using the methods described and collated the results before sending them to the authors. The review was conducted according to the previously published protocol (Yu 2010).
Selection of studies
Four independent authors (JS, EW, AT, MW) screened the titles and abstracts of the search results. Studies not meeting the pre‐defined inclusion criteria were excluded. Reasons for excluding studies that seemed to meet the inclusion criteria, but then were subsequently excluded, were documented. Studies that met the inclusion criteria were further examined. The full text of all studies that were potentially relevant was retrieved and, where necessary, was translated into English. Studies with more than one publication were examined closely to ensure that each study was counted only once, and that multiple references were included by study.
Data extraction and management
Three independent authors (JS, EW, AT) used a pre‐formed standardised data extraction sheet to record study characteristics and outcomes considered for this review. All data were cross‐checked, and differences were resolved with further examination until a consensus was reached. The data extracted from each study included the following:
Participant characteristics (e.g. gender, age, ethnicity, co‐morbid conditions).
Methods (e.g. random allocation procedures; allocation concealment; blinding of participants, healthcare providers, and outcome assessors).
Losses to follow‐up, how they were handled, and follow‐up duration.
Interventions (including antibiotic used, dose, and duration).
Outcome measures as listed previously.
All data were collected, regardless of compliance or completion of follow‐up, to allow for an intention‐to‐treat analysis.
Assessment of risk of bias in included studies
Three independent authors (JS, EW, AT) assessed the methodological quality of each study using the following parameters as an evaluation tool and transcribed information from each included study into the 'Risk of bias' tables. Criteria for assessing risk of bias included evaluating the sequence generation, allocation concealment, blinding, management of incomplete data, selective outcome reporting, and other potential threats to the validity of the studies (Higgins 2009).
Sequence generation
Was sequence generation adequate?
Low risk of bias: A random component of sequence generation is described (e.g. computer‐generated random sequence, random number table).
High risk of bias: A non‐random component of sequence generation is described (e.g. allocation by clinician's judgement, allocation by participant's preference, sequence generated by admission date).
Unclear: insufficient information to conclude 'low risk of bias' or 'high risk of bias.'
Allocation concealment
Was allocation concealment adequate?
Low risk of bias: Participants and investigators enrolling participants could not predict assignment (e.g. sequentially numbered opaque sealed envelopes, telephone randomisation).
High risk of bias: Participants and investigators enrolling participants could predict assignment (e.g. unsealed envelopes, alternation).
Unclear: Information is insufficient to allow conclusion of 'low risk of bias' or 'high risk of bias.'
Blinding of participants, personnel, and outcome assessors
Was blinding of individuals involved in the study (participants, personnel, and outcome assessors) to the treatment allocation adequate?
-
Low risk of bias—any of the following:
No blinding, but the review authors judge that the outcome and the outcome measurement are not likely to be influenced by lack of blinding.
Blinding of participants and key study personnel ensured; unlikely that the blinding could have been broken.
Either participants or some key study personnel were not blinded, but outcome assessment was blinded and the non‐blinding of others unlikely to introduce bias.
-
High risk of bias—any of the following:
No blinding or incomplete blinding, and the outcome or outcome measurement is likely to be influenced by lack of blinding.
Blinding of key study participants and personnel attempted, but likely that the blinding could have been broken.
Either participants or some key study personnel were not blinded, and the non‐blinding of others is likely to introduce bias.
Unclear: insufficient information allow conclusion of 'low risk of bias' or 'high risk of bias.'
Incomplete outcome data
Were incomplete outcome data adequately addressed?
-
Low risk of bias-any of the following:
No missing outcome data.
Reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias).
Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups.
For dichotomous outcomes data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate.
For continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes not enough to have a clinically relevant impact on observed effect size.
Missing data have been imputed using appropriate method.
-
High risk of bias-any of the following:
Reason for missing outcome data likely to be related to true outcome, with imbalance in numbers of or reasons for missing data across intervention groups.
For dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate.
For continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size.
'As‐treated' analysis done with substantial departure of the intervention received from that assigned at randomisation.
Potentially inappropriate application of simple imputation.
Unclear: insufficient reporting of attrition/exclusions to conclude 'low risk of bias' or 'high risk of bias'
Selective outcome reporting
Are reports of the study free of selective outcome reporting?
-
Low risk of bias-any of the following:
Study protocol is available and all pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way.
Study protocol is not available, but it is clear that published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon).
-
High risk of bias-any of the following:
Not all pre‐specified primary outcomes of the study have been reported.
One or more primary outcomes are reported using measurements, analysis methods, or subsets of the data (e.g. subscales) that were not pre‐specified.
One or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect).
One or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis.
Study report fails to include results for a key outcome that would be expected to have been reported for such a study.
Unclear: information is insufficient to allow investigators to conclude 'low risk of bias' or 'high risk of bias'
Other potential threats to validity
Was the study free from other problems that could put it at risk of bias?
Low risk of bias: Study appears to be free from other sources of bias.
High risk of bias: One or more important risks of bias are included (e.g. extreme baseline imbalance).
Unclear: Information is insufficient to allow investigators to assess whether there is an important risk of bias.
An overall assessment of the level of bias in the included trials was performed to determine the reliability and validity of the data.
Measures of treatment effect
All outcomes were dichotomous; therefore, the measure of treatment effect calculated was risk ratio (RR) with an associated 95% confidence interval (95% CI) using a random‐effects model. A random‐effects model was conducted for all analyses to account for the underlying heterogeneity of included studies, in which different participant populations, antibiotics, and infections were studied. Also, most of the studies were small; therefore, it was thought that a random‐effects model would be less likely to diminish the importance of an observed effect because the weights assigned to each study would be more balanced.
Unit of analysis issues
The participant was the unit of analysis. Data from all randomly assigned participants were used for analyses. Scenarios in which censoring or exclusion of data was possible and whether results were presented as the total number of events or the total number of participants with a first event were examined closely. Authors of the studies were contacted regarding any ambiguity.
Dealing with missing data
Authors of the studies were contacted via e‐mail to clarify and provide any missing data.
Assessment of heterogeneity
The I2 statistic and the Chi2 test were used to test for heterogeneity in the included studies. The threshold for the I2 statistic was > 50% for important heterogeneity to be considered. The threshold for the Chi2 statistic was P < 0.10 for important heterogeneity to be considered. When heterogeneity was detected (Chi2 test value of P < 0.10 or I2 > 50%), a random‐effects model was used to confirm whether a statistically significant difference between the effects of continuous infusion and those of intermittent infusion could be noted. Clinical and methodological sources of heterogeneity were investigated, including baseline risk factors for outcome measures, study duration, age, race, and sex distribution of participants across studies.
Initially, a fixed‐effect meta‐analysis was planned because the default for all analyses and a random‐effects model would be used only when heterogeneity was detected. However, a random‐effects model was used as the default because a large number of small studies with between‐study heterogeneity (e.g. different infections and antibiotics between studies) were used. Fixed‐effect analyses were conducted to determine if there were differences between models because it is possible that small positive studies may drive the meta‐analysis to look more positive as small studies may be given more weight when a random‐effects model is used.
Assessment of reporting biases
Funnel plots were produced to detect potential publication bias. Any asymmetry noted in the plot was investigated to identify possible reasons for the asymmetry (i.e. true heterogeneity of treatment effect, play of chance, poor methodology in included studies).
Data synthesis
Review Manager 5 (RevMan 2008) was used to perform all data syntheses and analyses. Risk ratios for dichotomous clinical outcomes were calculated and are presented with 95% confidence intervals. GRADEpro (GRADEpro 2008) was used to generate the 'summary of findings' table.
Subgroup analysis and investigation of heterogeneity
Any heterogeneity detected was investigated for possible reasons (Higgins 2003). Aspects of trials assessed included antibiotic choice, infections being treated, use of other open‐label antibiotics, and dose/duration of antibiotic therapy. A subgroup analysis of trials that included septic participants versus non‐septic participants was conducted to determine whether differences in effect were based on this variable.
Sensitivity analysis
Sensitivity analyses were performed for primary outcomes only. Sensitivity analyses were performed to determine the impact of the presence or absence of appropriate allocation concealment procedures on all‐cause mortality and infection recurrence effect estimates. Sensitivity analyses were also performed to determine the impact of trials studying extended interval infusions compared with intermittent infusions and the impact of the use of other open‐label antibiotics in studies.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies.
Results of the search
The searches conducted in January 2010 identified 2935 records and 6 additional records from other sources. After removal of 1133 duplicate records, 1808 records were screened. The titles and abstracts for these results were screened by four appraisers; initially, 1752 studies were excluded for not meeting the pre‐defined inclusion criteria. The full texts of the remaining 56 potentially eligible studies were retrieved for further assessment. Of 56 potentially eligible studies, 26 studies met the pre–specified inclusion criteria and were included in this review (Figure 1). Two trials were translated from French into English. However, only a partial translation of 1 trial from Japanese into English could be obtained. An updated search conducted in September 2012 identified 326 records and 3 additional records from other sources. After removal of 102 duplicate records, 227 records were screened and 4 full text articles were retrieved for further assessment. Of the 4 potentially eligible studies, 3 studies were included in this review (Figure 1). No trials from the updated search required translation into English. A total of 29 studies were included in this review from searches conducted in January 2010 and September 2012. A search of an online clinical trials register (www.clinicaltrials.gov) and the Internet identified 8 ongoing studies as of November 2012 that may meet inclusion criteria for this review.
1.
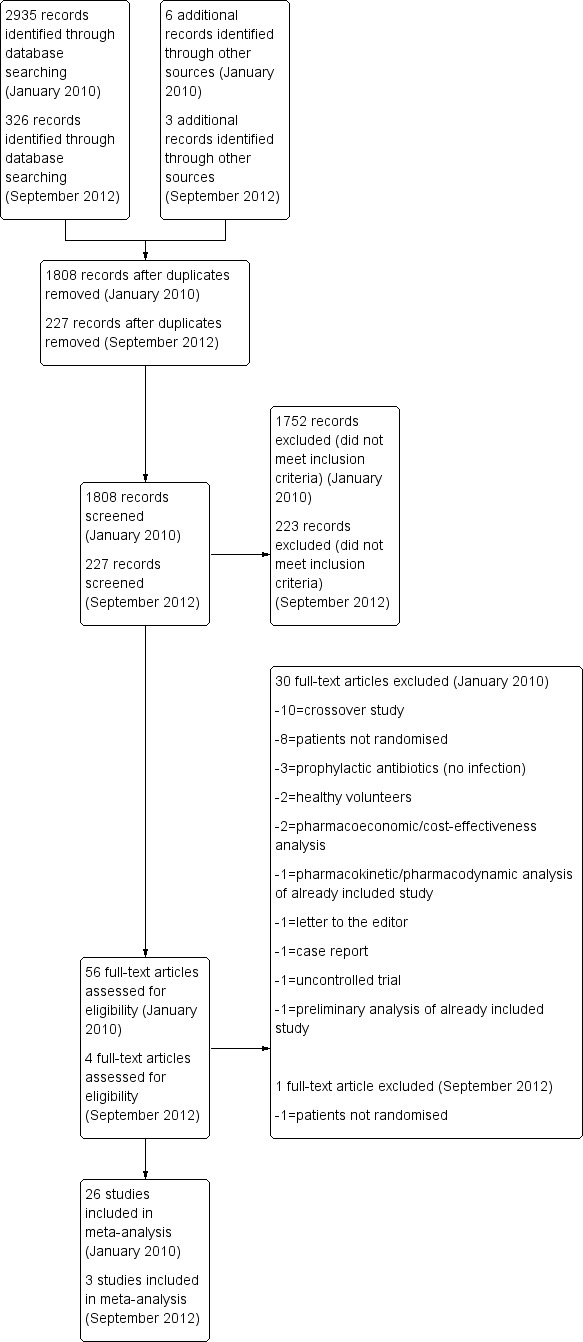
Continuous vs intermittent study flow diagram.
Included studies
Of the 29 included studies, 25 studies compared continuous antibiotic infusions with traditional intermittent infusions, and 4 studies compared extended antibiotic infusions with intermittent infusions. Hospitalised patients were studied in the 29 included trials, and 19 of these trials studied patients admitted to a critical care unit. The population size of each study ranged from 7 to 262 patients; 4 studies had a study sample size greater than 100 patients. The studies included adults between the ages of 18 and 80 years, with the exception of one trial (Feld 1977), which included patients aged 15 years and older. This trial was included in the analysis because the median participant age was 43 to 46 years; therefore, it was thought that very few patients younger than 18 years participated in the study. The percentage of males in the included studies ranged from 43% to 82%.
Types of infections studied in the included trials were pneumonia (n = 16), septicemia (n = 13), urinary tract infection (n = 3), skin and soft tissue infection (n = 2), peritonitis (n = 2), acute exacerbation of chronic obstructive pulmonary disease (n = 2), acute exacerbation of chronic bronchitis (n = 1), fever of unknown origin (n = 1), cholangitis (n = 1), sinusitis (n = 1), perirectal infection (n = 1), intra–abdominal or periappendiceal abscess (n = 1), complicated perforated diverticulitis (n = 1), shock lung (n = 1), endocarditis (n = 1), melioidosis (n = 1), catheter‐related infection (n = 1), mediastinitis (n = 1), post‐operative surgical infection (n = 1), and meningitis or central nervous system infection (n = 1). Organisms isolated included Enterococcus faecalis, Enterococcus faecium, coagulase‐negativeStaphylococcus (CoNS), Staphylococcus aureus, methicillin–resistantStaphylococcus aureus (MRSA), Burkholderia, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Enterobacteriaceae, Proteus mirabilis, Citrobacter, Serratia marcescens, Acinetobacter, Clostridium, Haemophilus influenzae, Aeromonas, Moraxella catarrhalis, Morganella morganii, Enterobacter, Salmonella, Streptococcus viridans, Streptococcus milleri, Streptococcus mitis, Streptococcus pneumoniae, Stenotrophomonas, Bacteroides, and Peptostreptococcus. Antibiotics studied included ceftazidime (n = 8), piperacillin‐tazobactam (n = 5), meropenem (n = 3), tobramycin (n = 2), piperacillin (n = 1), linezolid (n = 1), carbenicillin (n = 1), cefamandole (n = 1), temocillin (n = 1), sisomicin (n = 1), cefepime (n = 1), cefoperazone (n = 1), ceftriaxone (n = 1), imipenem (n = 1), cefotaxime (n = 1), gentamicin (n = 1), and vancomycin (n = 1). Open–label antibiotics were permitted in 17 studies, although the remaining included studies did not indicate whether any additional antibiotics were permitted. Antibiotic treatment duration ranged from 4 to 14 days. Follow‐up duration ranged from 24 hours to 28 days but was not reported in 18 studies.
Of studies meeting inclusion criteria, 3 studies (Lipman 1999; Nicolau 1999a; Nicolau 1999b) did not report any outcome data. One study (Bodey 1979) reported outcomes expressed as episodes and not by participant. This unit of analysis issue could not be reconciled. Therefore, 4 studies that met inclusion criteria did not contribute data to this meta‐analysis.
Excluded studies
Of 60 potentially eligible studies, 31 were excluded after closer examination by 4 independent study appraisers. Studies excluded were cross‐over studies (n = 10), were not randomised (n = 9), used prophylactic antibiotics (n = 3), and included healthy volunteers (n = 2), pharmacoeconomic or cost‐effectiveness analyses (n = 2), pharmacokinetic/pharmacodynamic analyses of already included studies (n = 1), letters to the editor (n = 1), case reports (n = 1), uncontrolled (n = 1), or preliminary analyses of an already included study (n = 1).
Risk of bias in included studies
Most included studies were judged to be at unclear or high risk of bias with regard to randomisation sequence generation, allocation concealment, blinding, management of incomplete outcome data, selective outcome reporting, and other potential threats to validity (86%, 76%, 97%, 69%, 76%, and 90% of studies, respectively). No studies were judged to be at low risk of bias for all methodological quality items assessed.
See 'Risk of bias' tables and 'Risk of bias' graphs (Figure 2, Figure 3) for additional details regarding evaluation of the included studies.
2.
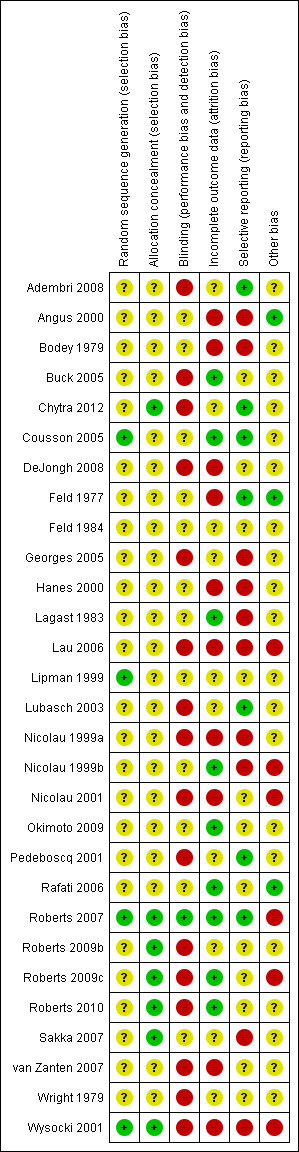
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
3.
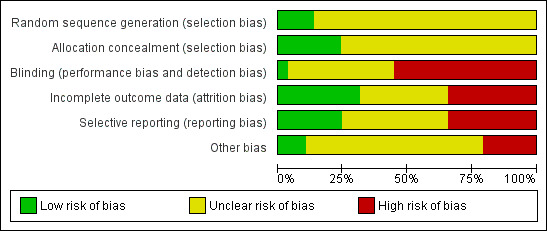
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
Allocation
Generation of randomisation sequence
Four trials (Cousson 2005; Lipman 1999; Roberts 2007; Wysocki 2001) adequately described generation of allocation sequence procedures and were judged to be at low risk of bias. Four trials (Nicolau 1999b; Nicolau 2001; Rafati 2006; Roberts 2009c) did not describe randomisation methods or generation of allocation sequence methods and were judged to have an unclear risk of bias. The remaining 21 studies were randomised, but randomisation methods were not reported or could not be translated into English, and these studies were judged to have an unclear risk of bias. For additional details, refer to information presented in 'Risk of bias' tables.
Allocation concealment
Six studies (Chytra 2012; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001) used sealed, opaque envelopes to conceal randomisation allocation and were judged to be at low risk of bias. Five trials (Adembri 2008; Bodey 1979; Buck 2005; Feld 1984; Sakka 2007) used sealed envelopes to conceal randomisation allocation, but opacity of envelopes was not described, and these studies were judged to be at unclear risk of bias. Allocation concealment was not clear in 1 study (Okimoto 2009) because only a partial translation from Japanese into English was available. The remaining 17 studies did not describe allocation concealment methods and were judged to be at unclear risk of bias. For additional details, refer to information presented in 'Risk of bias' tables.
Blinding
Fourteen studies (Adembri 2008; Buck 2005; Chytra 2012; DeJongh 2008; Georges 2005; Lau 2006; Lubasch 2003; Nicolau 2001; Pedeboscq 2001; Roberts 2009b; Roberts 2009c; Roberts 2010; van Zanten 2007; Wysocki 2001) were not blind and were judged to be at high risk of bias. One study (Nicolau 1999a) was single blind and was judged to be at high risk of bias. Another study (Roberts 2007) was double blind and was judged to be at low risk of bias. Blinding was not clear in 1 study (Okimoto 2009) because only a partial translation from Japanese into English was available. The remaining 12 studies did not comment on blinding and were judged to be at unclear risk of bias. For additional details, refer to information presented in 'Risk of bias' tables.
Incomplete outcome data
Participants were lost to follow‐up or were censored in 12 trials (Angus 2000; Chytra 2012; DeJongh 2008; Feld 1977; Feld 1984; Georges 2005; Hanes 2000; Lau 2006; Lubasch 2003; Nicolau 1999a; Nicolau 2001; Wysocki 2001) and were judged to be at unclear or high risk of bias. One trial (Adembri 2008) included participants who did not complete the study and was judged to be at unclear risk of bias. Two studies (Bodey 1979; van Zanten 2007) did not report all pre–specified outcomes for all participants and were judged to be at high risk of bias. Nine studies (Buck 2005; Cousson 2005; Lagast 1983; Nicolau 1999b; Okimoto 2009; Rafati 2006; Roberts 2007; Roberts 2009c; Roberts 2010) yielded complete outcome data and were judged to be at low risk of bias. Five studies (Lipman 1999; Pedeboscq 2001; Roberts 2009b;Sakka 2007; Wright 1979) did not report on attrition bias and were judged to be at unclear risk of bias. For additional details, refer to information presented in 'Risk of bias' tables.
The corresponding authors of 25 studies were contacted via e‐mail to clarify and provide missing outcome data. Nine authors replied with the requested missing data, which were incorporated into the analyses. Five authors stated that the requested missing data were no longer available, and the remaining authors did not respond.
No authors of the 8 ongoing studies were contacted, and no data from these unpublished or ongoing trials were included, because it was not clear from the current information provided whether these trials will meet inclusion criteria for this review. When the full texts of these studies become available, they will be considered for future updates.
It is important to note that a substantial amount of information was not available for the pre‐specified outcome measures of this review. Data from all 29 RCTS were not reported for any outcome measures. See table below.
| Outcome | Number of trials reporting outcomes (of a total of 29 possible trials) |
| All‐cause mortality | 19 |
| Infection recurrence | 8 |
| Clinical cure | 15 |
| Super‐infection | 12 |
| Adverse events | 5 |
| Serious adverse events | 10 |
| Withdrawal due to adverse events | 10 |
Selective reporting
Seven studies (Adembri 2008; Chytra 2012; Cousson 2005; Feld 1977; Lubasch 2003; Pedeboscq 2001; Roberts 2007) reported all pre–specified outcomes and were judged to be at low risk of bias. Seven studies (Angus 2000; DeJongh 2008; Georges 2005; Lipman 1999; Nicolau 1999a; Sakka 2007; Wright 1979) reported outcomes that were not pre–specified and were judged to be at unclear or high risk of bias. Five studies (Hanes 2000; Lagast 1983; Lau 2006; Nicolau 1999b; van Zanten 2007) did not report all pre–specified outcomes and were judged to be at unclear or high risk of bias. Three studies (Bodey 1979; Feld 1984; Wysocki 2001) did not report the total numbers of participants or participants in each group and were judged to be at high or unclear risk of bias. Five studies (Nicolau 2001; Rafati 2006; Roberts 2009b; Roberts 2009c; Roberts 2010) did not describe methods, including outcomes, and were judged to be at unclear risk of bias. One study (Buck 2005) did not assess clinical outcomes and was judged to be at unclear risk of bias. Selective reporting was not clear in 1 study (Okimoto 2009) because only a partial translation from Japanese into English was available. For additional details, refer to information presented in 'Risk of bias' tables.
Other potential sources of bias
No other potential sources of bias were identified in 3 studies (Angus 2000; Feld 1977; Rafati 2006), which were judged to be at low risk of bias. Other potential sources of bias were not clear in 1 study (Okimoto 2009) because only a partial translation from Japanese into English was available. The remaining 25 studies included participant baseline imbalances, unclear use of open–label antibiotics, or pharmaceutical company funding and were judged to be at unclear or high risk of bias. For additional details, refer to information presented in 'Risk of bias' tables.
Effects of interventions
See: Table 1
Intention‐to‐treat analyses were conducted for all outcomes using the total numbers of participants randomly assigned to each intervention for each included study. All analyses were conduced using a random‐effects model and a fixed‐effect model. One difference between the 2 models in the subgroup analysis of septic versus non‐septic participants reporting clinical cure data was noted. However, no differences between the 2 models were observed in all other analyses, suggesting no bias due to small study effects.
Worst‐case scenarios were conducted for primary and secondary outcomes for participants lost to follow‐up or censored. Analyses were conducted only when exact numbers of missing participants were known.
All–cause mortality (Analysis 1.1)
1.1. Analysis.
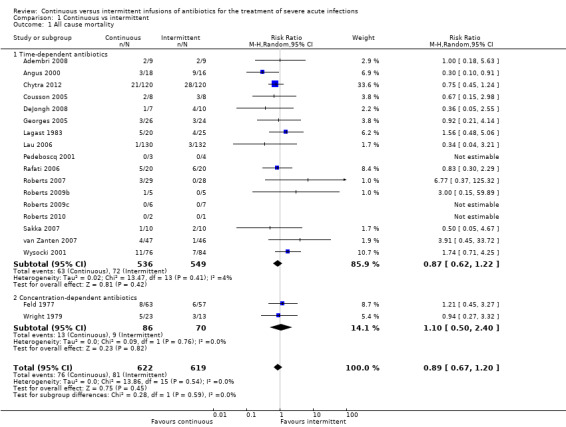
Comparison 1 Continuous vs intermittent, Outcome 1 All cause mortality.
Nineteen studies reported mortality data (Adembri 2008; Angus 2000; Chytra 2012; Cousson 2005; DeJongh 2008; Feld 1977; Georges 2005; Lagast 1983; Lau 2006; Pedeboscq 2001; Rafati 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Sakka 2007; van Zanten 2007; Wright 1979; Wysocki 2001). No statistically significant differences in all–cause mortality were noted when time–dependent antibiotics and concentration–dependent antibiotics were analysed together (n = 1241, pooled relative risk (RR) 0.89, 95% CI 0.67 to 1.20, P = 0.45). No evidence of statistical heterogeneity was found (Chi2 = 13.86, degrees of freedom (df) = 15, P = 0.54; I2 = 0%).
Seventeen studies compared time–dependent antibiotics (Adembri 2008; Angus 2000; Chytra 2012; Cousson 2005; DeJongh 2008; Georges 2005; Lagast 1983; Lau 2006; Pedeboscq 2001; Rafati 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Sakka 2007; van Zanten 2007; Wysocki 2001). No statistically significant differences in all–cause mortality were reported with time–dependent antibiotics (n = 1085, RR 0.87, 95% CI 0.62 to 1.22, P = 0.42). No evidence of statistical heterogeneity was obtained (Chi2 = 13.47, df = 13, P = 0.41; I2 = 4%).
Two studies compared concentration–dependent antibiotics (Feld 1977; Wright 1979). No statistically significant differences in all–cause mortality were found with concentration–dependent antibiotics (n = 156, RR 1.10, 95% CI 0.50 to 2.40, P = 0.82). No evidence of statistical heterogeneity was noted (Chi2 = 0.09, df = 1, P = 0.76; I2 = 0%).
When the worst–case scenario was calculated for time‐dependent and concentration‐dependent antibiotics, when all missing participants were assumed to have died, no statistically significant differences in all–cause mortality were observed (n = 1241, RR 0.94, 95% CI 0.80 to 1.11, P = 0.49).
Infection recurrence within 14 days of resolution of primary infection (Analysis 1.2)
1.2. Analysis.
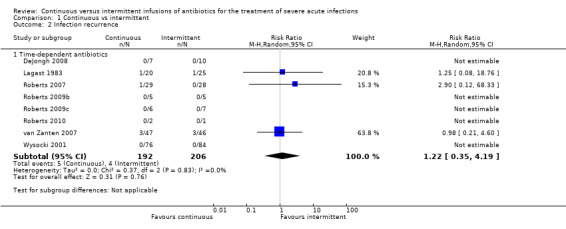
Comparison 1 Continuous vs intermittent, Outcome 2 Infection recurrence.
Eight studies reported infection recurrence data; all of these trials compared time–dependent antibiotics (DeJongh 2008; Lagast 1983; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; van Zanten 2007; Wysocki 2001). No statistically significant differences in infection recurrence (n = 398, RR 1.22, 95% CI 0.35 to 4.19, P = 0.76) were described, and no evidence of statistical heterogeneity was found (Chi2 = 0.37, df = 2, P = 0.83; I2 = 0%).
When the worst–case scenario was calculated for time‐dependent antibiotics, and when all missing participants were assumed to have infection recurrence, no statistically significant differences in infection recurrence were observed (n = 398, RR 0.88, 95% CI 0.46 to 1.66, P = 0.69).
Clinical cure (Analysis 1.3)
1.3. Analysis.
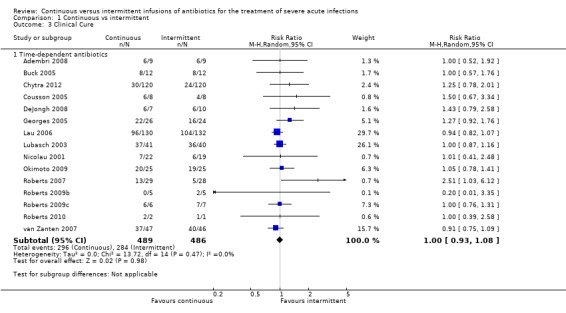
Comparison 1 Continuous vs intermittent, Outcome 3 Clinical Cure.
Fifteen studies reported clinical cure; all of these studies compared time–dependent antibiotics (Adembri 2008; Buck 2005; Chytra 2012; Cousson 2005; DeJongh 2008; Georges 2005; Lau 2006; Lubasch 2003; Nicolau 2001; Okimoto 2009; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; van Zanten 2007). No statistically significant differences in clinical cure were reported (n = 975, RR 1.00, 95% CI 0.93 to 1.08, P = 0.98). No evidence of statistical heterogeneity was found (Chi2 = 13.72, df = 14, P = 0.47; I2 = 0%).
A worst‐case scenario is not estimable because it is already assumed that all missing participants did not attain clinical cure.
Superinfections post–therapy (Analysis 1.4)
1.4. Analysis.
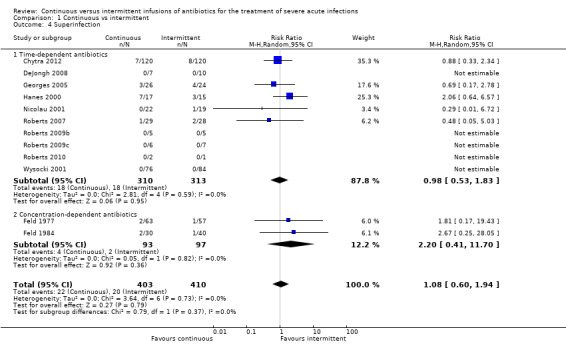
Comparison 1 Continuous vs intermittent, Outcome 4 Superinfection.
Twelve studies reported secondary super‐infections post–therapy (Chytra 2012; DeJongh 2008; Feld 1977; Feld 1984; Georges 2005; Hanes 2000; Nicolau 2001; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001). No statistically significant differences in superinfections post–therapy when time–dependent antibiotics and concentration–dependent antibiotics were analysed together (n = 813, RR 1.08, 95% CI 0.60 to 1.94, P = 0.79). No evidence of statistical heterogeneity was found (Chi2 = 3.64, df = 6, P = 0.73; I2 = 0%).
Ten studies reported secondary super‐infections in studies comparing time–dependent antibiotics (Chytra 2012; DeJongh 2008; Georges 2005; Hanes 2000; Nicolau 2001; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001). No statistically significant differences in super‐infections with time–dependent antibiotics were reported (n = 623, RR 0.98, 95% CI 0.53 to 1.83, P = 0.95). No evidence of statistical heterogeneity was found (Chi2 = 2.81, df = 4, P = 0.59; I2 = 0%).
Two studies reported secondary super‐infections when concentration–dependent antibiotics were compared (Feld 1977; Feld 1984). No statistically significant differences in secondary super‐infections were seen with concentration–dependent antibiotics (n = 190, RR 2.20, 95% CI 0.41 to 11.70, P = 0.36). No evidence of statistical heterogeneity was found (Chi2 = 0.05, df = 1, P = 0.82; I2 = 0%).
When the worst–case scenario was calculated for time‐dependent and concentration‐dependent antibiotics, and when all missing participants were assumed to have secondary super‐infections, no statistically significant differences in infection recurrence were reported (n = 813, RR 1.01, 95% CI 0.73 to 1.40, P = 0.96).
Serious adverse events (Analysis 1.5)
1.5. Analysis.
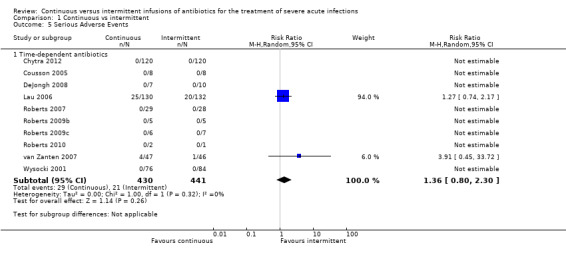
Comparison 1 Continuous vs intermittent, Outcome 5 Serious Adverse Events.
Ten studies reported serious adverse events; all of these studies were conducted to compare time–dependent antibiotics (Chytra 2012; Cousson 2005; DeJongh 2008; Lau 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; van Zanten 2007; Wysocki 2001). No statistically significant differences in participants experiencing at least one serious adverse event were observed (n = 871, RR 1.36, 95% CI 0.80 to 2.30, P = 0.26), and no evidence of statistical heterogeneity was found (Chi2 = 1.00, df = 1, P = 0.32; I2 = 0%).
When the worst–case scenario was calculated for time‐dependent antibiotics, and when all missing participants were assumed to have had a serious adverse event, no statistically significant differences in serious adverse events were noted (n = 871, RR 1.04, 95% CI 0.67 to 1.62, P = 0.85).
Withdrawals due to adverse events (Analysis 1.6)
1.6. Analysis.
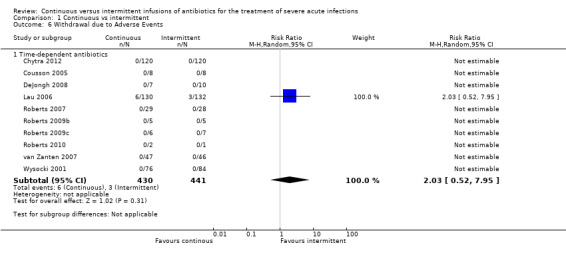
Comparison 1 Continuous vs intermittent, Outcome 6 Withdrawal due to Adverse Events.
Ten studies reported withdrawals due to adverse events; all of these studies compared time–dependent antibiotics (Chytra 2012; Cousson 2005; DeJongh 2008; Lau 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; van Zanten 2007; Wysocki 2001). No statistically significant differences in withdrawals due to adverse events were described (n = 871, RR 2.03, 95% CI 0.52 to 7.95, P = 0.31). Statistical heterogeneity could not be calculated because only one study reported participant withdrawals due to adverse events.
When the worst–case scenario was calculated, and when all missing participants were assumed to have withdrawn because of an adverse event, no statistically significant differences in withdrawals due to adverse events were seen (n = 871, RR 0.99, 95% CI 0.66 to 1.50, P = 0.98).
Adverse events (Analysis 1.7)
1.7. Analysis.
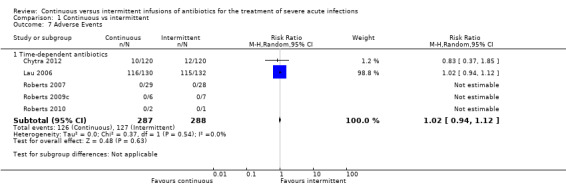
Comparison 1 Continuous vs intermittent, Outcome 7 Adverse Events.
Five studies reported adverse events; all of these studies were performed to compare time–dependent antibiotics (Chytra 2012; Lau 2006; Roberts 2007; Roberts 2009c; Roberts 2010). No statistically significant differences in adverse events were observed (n = 575, RR 1.02, 95% CI 0.94 to 1.12, P = 0.63), and no evidence of statistical heterogeneity was found (Chi2 = 0.37, df = 1, P = 0.54; I2 = 0%).
When the worst‐case scenario was calculated, in which all missing participants were assumed to have had an adverse event, no statistically significant differences in adverse events were noted (n = 575, RR 1.02, 95% CI 0.94 to 1.12, P = 0.60).
Subgroup analysis: septic versus non–septic participants
Twenty studies included participants with sepsis (met criteria for systemic inflammatory response syndrome with a documented or suspected infection and/or critically unwell admitted to an intensive care unit) (Adembri 2008; Angus 2000; Chytra 2012; Cousson 2005; DeJongh 2008; Georges 2005; Hanes 2000; Lipman 1999; Nicolau 1999a; Nicolau 1999b; Nicolau 2001; Pedeboscq 2001; Rafati 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Sakka 2007; Wright 1979; Wysocki 2001).
Fifteen studies reported mortality in septic participants (Adembri 2008; Angus 2000; Chytra 2012; Cousson 2005; DeJongh 2008; Georges 2005; Pedeboscq 2001; Rafati 2006; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Sakka 2007; Wright 1979; Wysocki 2001). No statistically significant differences in all–cause mortality were reported when time–dependent antibiotics and concentration–dependent antibiotics were analysed together (n = 721, RR 0.82, 95% CI 0.59 to 1.13, P = 0.23). Four studies reported mortality in non–septic participants (Feld 1977; Lagast 1983; Lau 2006; van Zanten 2007). No statistically significant differences in all–cause mortality were noted when time–dependent antibiotics and concentration–dependent antibiotics were analysed together (n = 520, RR 1.32, 95% CI 0.67 to 2.61, P = 0.43).
Six studies reported infection recurrence in septic participants; all of these trials were conducted to compare time–dependent antibiotics (DeJongh 2008; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001). No statistically significant differences in infection recurrence were observed (n = 260, RR 2.90, 95% CI 0.12 to 68.33, P = 0.51). Two studies reported infection recurrence in non–septic participants; all of these trials compared time–dependent antibiotics (Lagast 1983; van Zanten 2007). No statistically significant differences in infection recurrence were described (n = 138, RR 1.04, 95% CI 0.27 to 3.99, P = 0.95).
Ten studies reported clinical cure in septic participants; all of these studies compared time–dependent antibiotics (Adembri 2008; Chytra 2012; Cousson 2005; DeJongh 2008; Georges 2005; Nicolau 2001; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010). No statistically significant differences in clinical cure were reported (n = 465, RR 1.17, 95% CI 0.99 to 1.37, P = 0.06) when a random‐effects model was used. However, a statistically significant difference favouring intermittent antibiotic infusions was seen when a fixed‐effect model was used (n = 465, RR 1.26, 95% CI 1.02 to 1.55, P = 0.03). Five studies reported clinical cure in non–septic participants; all of these trials compared time–dependent antibiotics (Buck 2005; Lau 2006; Lubasch 2003; Okimoto 2009; van Zanten 2007). No statistically significant differences in clinical cure were observed (n = 510, RR 0.96, 95% CI 0.89 to 1.04, P = 0.36).
Ten studies reported super‐infection in septic participants; all of these studies compared time‐dependent antibiotics (Chytra 2012; DeJongh 2008; Georges 2005; Hanes 2000; Nicolau 2001; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001). No statistically significant differences in super‐infection were described (n = 623, RR 0.98, 95% CI 0.53 to 1.83, P = 0.95). Two studies reported super‐infection in non–septic participants; all of these studies compared concentration‐dependent antibiotics (Feld 1977; Feld 1984). No statistically significant differences in super‐infection were reported (n = 190, RR 2.20, 95% CI 0.41 to 11.70, P = 0.36).
Eight studies addressed serious adverse events and withdrawals due to adverse events in septic participants; all of these compared time‐dependent antibiotics (Chytra 2012; Cousson 2005; DeJongh 2008; Roberts 2007; Roberts 2009b; Roberts 2009c; Roberts 2010; Wysocki 2001). However, no serious adverse events or withdrawals due to adverse events were recorded in these studies; therefore, comparison with non–septic participants could not be performed.
Four studies reported adverse events in septic participants; all of these studies compared time‐dependent antibiotics (Chytra 2012; Roberts 2007; Roberts 2009c; Roberts 2010). No statistically significant differences in adverse events were observed (n = 313, RR 0.83, 95% CI 0.37 to 1.85, P = 0.66). One study, which compared time‐dependent antibiotics, reported adverse events in non‐septic participants (Lau 2006). No statistically significant differences in adverse events were noted (n = 262, RR 1.02, 95% CI 0.94 to 1.12, P = 0.60).
Sensitivity analysis
When studies that did not report allocation concealment procedures were removed from analysis, no statistically significant differences in all–cause mortality were reported (n = 521, RR 1.00, 95% CI 0.63 to 1.58, P = 1.00) nor were differences in infection recurrence described (n = 243, RR 2.90, 95% CI 0.12 to 68.33, P = 0.51).
When studies that compared extended interval infusions with intermittent infusions were removed from analysis, no statistically significant differences in all–cause mortality (n = 1187, RR 0.90, 95% CI 0.65 to 1.25, P = 0.53) or infection recurrence were found (n = 398, RR 1.22, 95% CI 0.35 to 4.19, P = 0.76).
When studies that stated that open–label antibiotics were permitted were removed from analysis, no statistically significant differences in all–cause mortality (n = 546, RR 1.37, 95% CI 0.71 to 2.67, P = 0.35) or infection recurrence were described (n = 164, RR 1.04, 95% CI 0.27 to 3.99, P = 0.95).
The Feld 1977 study included some participants younger than 18 years; therefore, it was removed from analysis. When removed, no statistically significant differences in all‐cause mortality were seen (n = 1121, RR 0.87, 95% CI 0.64 to 1.18, P = 0.37). This study did not contribute data to the infection recurrence outcome.
The Lau 2006 and Lubasch 2003 studies reported clinical success as a composite outcome of clinical cure and clinical improvement, and these studies were removed from the analysis. When they were removed, no statistically significant differences in clinical cure were reported (n = 632, RR 1.06, 95% CI 0.94 to 1.19, P = 0.36).
Discussion
Summary of main results
No differences in all–cause mortality, infection recurrence, clinical cure, or super‐infection post–therapy were found between continuous antibiotic infusions and intermittent antibiotic infusions. Nor were differences reported when safety outcomes (serious adverse events, withdrawals due to adverse events, and adverse events) were compared. When comparisons between time–dependent and concentration–dependent antibiotics were made, no differences in all–cause mortality or super‐infection post–therapy were noted.
Subgroup analyses revealed no differences in all–cause mortality, infection recurrence, or super‐infection post–therapy when septic or critical care participants were compared with non–septic participants. A difference was observed in the subgroup analyses of clinical cure in septic versus non‐septic participants. However, this result was not robust because it was not observed in both random‐effects and fixed‐effect models. Additionally, this result is difficult to interpret because clinical cure was a subjective outcome, and no standard definition was used in the included studies. Clinical cure was defined by clinician judgement as improvement in signs and symptoms of infection, which included assessment of some or all of the following factors: bacteriological eradication, leukocyte counts, vital signs, inflammation, and sputum production. In addition, clinical success was defined as a composite outcome of clinical cure and clinical improvement in two studies. Therefore, it cannot be concluded from these data that greater clinical cure was seen in the intermittent antibiotic infusion group. It is also unclear whether any clinically meaningful differences in clinical cure were noted between continuous antibiotic infusions and intermittent antibiotic infusions because of identified risks of bias due to outcome subjectivity and lack of a robust statistical finding.
No differences between continuous and intermittent antibiotic infusions were reported for any sensitivity analyses performed.
Overall completeness and applicability of evidence
A wide range of antibiotics, infections, and organisms were included in this review, and this may allow the results to be broadly generalized. Although continuous infusions are thought to optimise the pharmacokinetic and pharmacodynamic parameters of time–dependent antibiotics, both time–dependent and concentration–dependent antibiotics were included in this analysis, because there is some thought that a lower total daily antibiotic dose could be used during continuous infusions (Nicolau 1996). This would affect the peak concentration and the area under the curve (estimate of drug exposure) for a drug; therefore, concentration–dependent antibiotics were included to investigate whether any adverse clinical outcomes or effects related to continuous infusions of concentration–dependent antibiotics were noted. Additionally, because the continuous infusion of concentration–dependent antibiotics has not been widely studied, it is not clear whether this dosing strategy would be beneficial or harmful to patients. The results of this study are applicable only to hospitalised patients and may not be applied to outpatient parenteral therapy programs based on the types of patients and settings of trials included in this review.
Several other concerns surround the applicability of these results. One concern is the heterogeneous definition of clinical cure in each study. Measuring an outcome such as clinical cure is especially difficult when clinicians or outcome assessors are not blinded, which was the case in most of the studies included in this review. Also, the use of open–label antibiotics in many of the studies may bias the effect of the intervention to show no difference. However, it would be unethical to limit antibiotic use in patients with severe bacterial infection. Another factor not considered in this review was pathogen susceptibility. It is possible that a difference between interventions was not observed as more highly susceptible pathogens were studied. Outcomes would theoretically be more similar between groups if the pathogens were highly susceptible (i.e. organisms were very sensitive to the effects of study antibiotics) because any suitable antibiotic would be effective no matter the dosing strategy. It could be hypothesized that continuous antibiotic infusions would be of greater benefit in cases of less susceptible organisms. Other confounding factors are the numerous other therapeutic interventions in hospitalised and critically ill patients (such as fluid resuscitation, vasopressor and inotrope use, and blood transfusion) that could also have a substantial impact on mortality, infection recurrence, and clinical cure. This is an important issue despite the fact that only randomised studies were included, because the validity of randomisation is affected by the small size of the included studies.
Although all of the trials included were randomised, it is of note that most of the studies had small participant populations. Generally, fewer than 100 participants were included in each study, and in one case, as few as seven participants were included. This is a potential problem because it raises the issue of whether the pooling of small studies for this review has sufficient power to detect a difference between continuous and intermittent antibiotic infusions. It is not clear whether the sample size as a result of pooling would be able to detect differences between the two antibiotic dosing strategies. This becomes an important issue when the safety outcomes of this review are interpreted because only two studies analysed reported participants experiencing an adverse effect or a serious adverse event, and only one study reported participant withdrawal due to an adverse event. This potential under–reporting of safety outcomes makes it difficult to assess the safety of continuous antibiotic infusions and intermittent antibiotic infusions in severe bacterial infection.
Furthermore, for each outcome, the calculated effect estimates in this review are associated with wide confidence intervals and show no particular consistent trend in beneficial or harmful effects with either continuous or intermittent administration. If data were available for outcomes from the trials that did not report on these outcomes, more precise effect estimates could have been calculated and could possibly show advantages or disadvantages for either continuous or intermittent antibiotic administration. It is important to refrain from concluding that outcomes for continuous and intermittent administration are equal, given the quantity of missing information and the possibility that the meta‐analyses are underpowered.
In addition to the limitations already described, many logistical concerns have been raised regarding the administration of continuous infusion antibiotics. Some beta–lactams, such as carbapenems, are thought to be too unstable for continuous infusion (Viaene 2002). Continuous infusion pumps may increase nursing workload and may limit patient mobility on medical wards, and additional intravenous lines may be required if the antibiotic chosen is not compatible with other medications (Ariano 2010). Extending the infusion also occupies an intravenous line that may be essential for other therapies, especially in critically ill patients who have limited intravenous access. As well, it is not known whether continuous infusions would result in increased dosing and administration errors by physicians, pharmacists, and nursing staff. However, several studies have suggested that continuous infusion of antibiotics is more cost–effective compared with intermittent infusions (McNabb 2001; Florea 2003; Hitt 1997; Grant 2002). Although intermittent antibiotic infusions are the current standard of therapy, some disadvantages merit consideration. Intermittent antibiotic infusions may increase nursing workload for those antibiotics that require multiple daily doses compared with continuous infusion pumps. In the preparation of these multiple antibiotic doses, the chance of dispensing and mixing errors by pharmacy and nursing staff may be increased. Intermittent infusions typically also result in higher peak concentrations of antibiotics, which could lead to an increase in adverse effects related to drug toxicity. Finally, intermittent antibiotic infusions could put the patient at increased infection risk compared with continuous infusions because more frequent access to intravenous lines is required to give multiple daily doses of antibiotics.
Quality of the evidence
The quality of evidence included in this review was very low to moderate according to GRADE considerations (GRADEpro 2008; Balshem 2011; Guyatt 2011). Although all 29 studies were randomised, procedures for both generation of the randomisation sequence and allocation concealment were described in only two studies. Only two studies were blinded, and the remaining studies were not blinded, or blinding was not reported and the studies appeared to be unblinded as judged by the authors. Additionally, accounting of missing participants and missing outcomes was not well described in the studies reviewed. It is interesting to note that many included studies were published before the CONSORT guidelines for reporting of RCTs were available.
Potential biases in the review process
This review had a focused objective and used a systematic search strategy to identify studies for potential inclusion. Biases in the review process were minimized by using pre–defined inclusion and exclusion criteria for study selection and standardized data extraction forms to gather data and appraise studies. Although no evidence of statistical heterogeneity was observed for all outcomes studied, several potential sources of heterogeneity (such as variation in outcomes studied, open–label antibiotic use, infection type, and patient co–morbidities) should be considered. Also, the potential for publication bias exists, although the funnel plots for all–cause mortality, infection recurrence, clinical cure, and super‐infection do not seem to indicate this (Figure 4, Figure 5, Figure 6, Figure 7). Publication bias increases the potential for adverse events, serious adverse events, and withdrawals due to adverse events as a result of the fact that funnel plots could not be analysed because the number of trials reporting these outcomes was insufficient.
4.
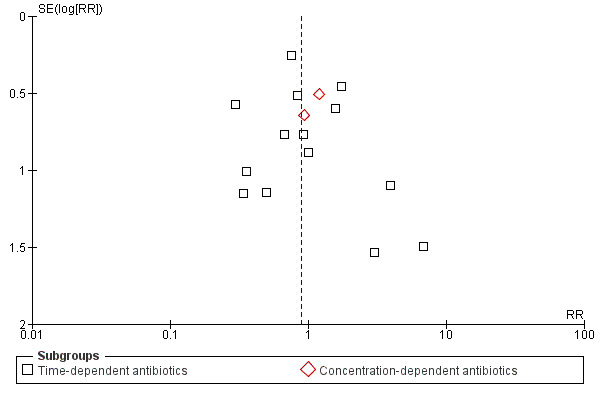
Funnel plot of comparison: continuous vs intermittent antibiotic infusions, outcome. All‐cause mortality.
5.
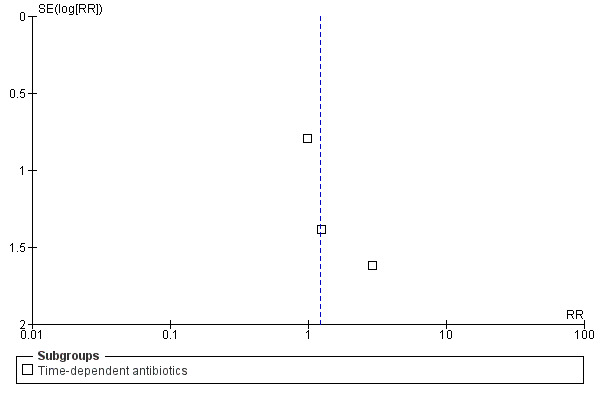
Funnel plot of comparison: continuous vs intermittent antibiotic infusions. Outcome: infection recurrence.
6.
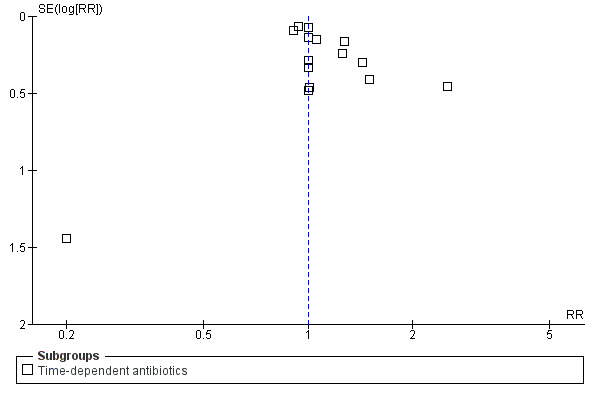
Funnel plot of comparison: continuous vs intermittent antibiotic infusions. Outcome: clinical cure.
7.
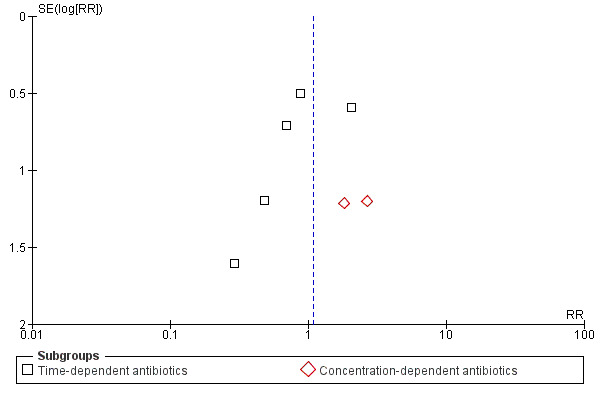
Funnel plot of comparison: continuous vs intermittent antibiotic infusions. Outcome: super‐infection.
Agreements and disagreements with other studies or reviews
This systematic review is consistent with previously published reviews. A meta–analysis of nine RCTs did not show any statistically significant differences in mortality (OR 0.89, 95% CI 0.48 to 1.64) (Kasiakou 2005b). A potential explanation for the similarity of results is that seven of the nine RCTs in the Kasiakou et al study also met inclusion criteria for this review. The remaining two studies were excluded from this review because one study was a cost‐effectiveness re‐analysis of an already included study, and the other study included nonーrandomly assigned participants. Moreover, four of the five RCTs that reported mortality data were included in this review. Similar antibiotics, such as beta–lactams, aminoglycosides, and vancomycin, were compared in both reviews. Similar participant populations, such as critical care participants, were compared in both reviews. Although no differences in mortality were reported by Kasiakou et al, these authors still concluded that continuous antibiotic infusions have a clinical advantage compared with intermittent infusions. This conclusion was based on a subgroup analysis in which only trials comparing the same total antibiotic dose showed that clinical failure rates were lower in the continuous infusion group (OR 0.70, 95% CI 0.50 to 0.98) (Kasiakou 2005b). Conversely, a difference favouring intermittent antibiotic infusions for clinical cure in septic participants was observed in this review. However, as has been discussed, the difference observed was not a robust statistical finding. A larger systematic review of 14 RCTs conducted by Roberts et al also did not find any differences in mortality (OR 1.00, 95% CI 0.48 to 2.06) or clinical cure (OR 1.04, 95% CI 0.74 to 1.46) (Roberts 2009a). Again, this similarity is likely related to the fact that 13 of the 14 RCTs in the review by Roberts et al are included in this review. The remaining study was not included in this review because it was not randomised. Therefore, both reviews had similar patient populations, study settings, and antibiotics studied. Tamma et al compared only beta–lactam continuous infusions versus intermittent infusions when conducting another systematic review of 14 RCTs. Similar to this review, Tamma et al did not find any differences in mortality (RR 0.92, 95% CI 0.61 to 1.37) or clinical cure (RR 1.00, 95% CI 0.94 to 1.06) (Tamma 2011). Once again, with 12 of the 14 RCTs included in both reviews, this consistency is likely explained. The remaining 2 studies were not included in this review because 1 study was not randomised, and the other study compared different antibiotics, which was an exclusion criterion for this review.
Generally, those studies that have shown a trend toward improved outcomes with continuous infusion antibiotics were investigating more resistant organisms in critically ill participants. A cohort study performed by Lodise et al compared 194 participants with a mean APACHE II score of 16, who received piperacillin‐tazobactam extended infusions versus intermittent infusions to treat Pseudomonas aeruginosa (Lodise 2007). Lodise et al found that participants with an APACHE II score > 17 receiving piperacillin‐tazobactam extended infusions had a statistically significant benefit in 14–day mortality (P = 0.04) (Lodise 2007). However, when the overall 14–day mortality was calculated, no statistically significant difference was noted (P = 0.17) (Lodise 2007). Therefore, it could be hypothesized that continuous antibiotic infusions provide clinically meaningful benefit only in the critically ill. However, the subgroup analysis comparing septic or critically ill participants with non–septic participants in this review did not find any differences except for clinical cure in septic participants, which was not robust. A potential explanation for this is that all studies included in this review were RCTs, and the Lodise et al study was a nonーrandomly assigned retrospective cohort.
Authors' conclusions
Implications for practice.
No differences in mortality, infection recurrence, clinical cure, and super‐infection post–therapy were found when continuous infusions of intravenous antibiotics were compared with traditional intermittent antibiotic infusions. However, the wide confidence intervals suggest that beneficial or harmful effects cannot be ruled out for all outcomes. Although no evidence of statistical heterogeneity was found, some clinically meaningful heterogeneity between studies is likely and should be considered. Also, no differences in safety outcomes between the two interventions were apparent. Because several trials did not report data for clinically important outcomes, and because confidence intervals for effect estimates were wide, it is possible that the analyses in this review are underpowered because of lack of data. Therefore, the current available evidence is insufficient to recommend the widespread adoption of continuous infusion antibiotics in the place of standard intermittent antibiotic infusions.
Implications for research.
Large, prospective randomised trials looking at additional outcomes, such as length of hospital stay, and reporting on all outcomes of interest as outlined in this review, would add to the findings of this review. It would also be helpful if these large trials were conducted with concurrent pharmacokinetic and pharmacodynamic studies. Trials investigating the effects of continuous infusion antibiotics in critically ill participants should be considered, because this population is theoretically more likely to benefit from this alternate dosing strategy based on subgroup data from previous retrospective studies. It is also not clear whether there would be additional therapeutic efficacy if continuous antibiotic infusions were used to treat more resistant organisms. Additional pharmacoeconomic studies are required to confirm whether there are other reasons to support the use of continuous infusion antibiotics.
What's new
| Date | Event | Description |
|---|---|---|
| 29 May 2013 | Amended | Copy edits made. |
Acknowledgements
We would like to thank the Cochrane Injuries Group for its support in the preparation of the protocol and review. We would also like to thank Ivane Stephanie Yu for contributions to search result screening and data extraction.
Appendices
Appendix 1. Search strategy
Cochrane Injuries Group Specialised Register (anti‐biotic* or antibiotic* or anti‐infect* or antiinfect* or antibacteria* or anti‐bacteria* or microbicide* or anti‐microbi* or antimicrobi*) AND (infusion* or intravenous* or drip or drips) AND (infection* or infectious or Sepsis or pneumonia* or mening*) AND ((drug* and schedule*) or continuous* or discontinu* or intermittent* or interval*)
Cochrane Central Register of Controlled Trials (The Cochrane Library) #1MeSH descriptor Anti‐Infective Agents explode all trees with qualifier: AD #2MeSH descriptor Anti‐Bacterial Agents explode all trees with qualifier: AD #3(anti‐biotic* or antibiotic* or anti‐infect* antiinfect* or antibacteria* or anti‐bacteria*) #4(microbicide* or anti‐microbi* or antimicrobi* or microbi*) #5(beta‐lactam* or betalactam* or B‐lactam* or aminoglycoside* or vancomycin) #6(#1 OR #2 OR #3 OR #4 OR #5) #7MeSH descriptor Infusions, Intravenous explode all trees #8(infusion* or intravenous* or drip or drips) #9 MeSH descriptor Infection explode all trees #10 MeSH descriptor Soft Tissue Infections explode all trees #11 MeSH descriptor Pneumonia, Viral explode all trees #12 MeSH descriptor Meningitis explode all trees #13 MeSH descriptor Urinary Tract Infections explode all trees #14 MeSH descriptor Sepsis explode all trees #15 (infected or infection* or infectious or infectious disease* or (infect* near disease*)) #16 (Sepsis or pneumonia* or mening*) #17 (urine or urinary tract) near3 (infect*) #18 ((skin or soft tissue) near3 infect*) #19 (#7 OR #8) #20 (#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18) #21 (#19 AND #20) #22 (#6 AND #21) #23 MeSH descriptor Drug Administration Schedule explode all trees #24 (continuous* or discontinu* or intermittent* or interval*) #25 (#23 OR #24) #26 (#22 AND #25)
MEDLINE (OvidSP) 1.exp anti‐infective agents/ad [Administration & Dosage] 2.exp anti‐bacterial agents/ad [Administration & Dosage] 3.(anti‐biotic* or antibiotic* or anti‐infect* antiinfect* or antibacteria* or anti‐bacteria*).ab,ti. 4.(microbicide* or anti‐microbi* or antimicrobi* or microbi*).ab,ti. 5.(beta‐lactam* or betalactam* or B‐lactam* or aminoglycoside* or vancomycin).ab,ti. 6.or/1‐5 7.exp Infusions, Intravenous/ 8.(infusion* or intravenous* or drip or drips).ab,ti. 9.7 or 8 10.6 and 9 11.exp Infection/ 12.exp Soft Tissue Infections/ 13.exp Pneumonia, Viral/ 14.exp Meningitis/ 15.exp Urinary Tract Infections/ 16.exp Sepsis/ 17.(infected or infection* or infectious or infectious?disease* or (infect* adj disease*)).ab,ti. 18.(Sepsis or pneumonia* or mening*).ab,ti. 19.((urine or urinary tract) adj3 infect*).ab,ti. 20.((skin or soft tissue) adj3 infect*).ab,ti. 21.or/11‐20 22.10 and 21 23.(continuous* or discontinu* or intermittent* or interval*).ab,ti. 24.exp Drug administration schedule/ 25.23 or 24 26.22 and 25 27.randomi?ed.ab,ti. 28.randomized controlled trial.pt. 29.controlled clinical trial.pt. 30.placebo.ab. 31.clinical trials as topic.sh. 32.randomly.ab. 33.trial.ti. 34.27 or 28 or 29 or 30 or 31 or 32 or 33 35.(animals not (humans and animals)).sh. 36.34 not 35 37.26 and 36
EMBASE (OvidSP) 1.exp antibiotic agent/ad, do [Drug Administration, Drug Dose] 2.exp antiinfective agent/ad, do [Drug Administration, Drug Dose] 3.(anti‐biotic* or antibiotic* or anti‐infect* antiinfect* or antibacteria* or anti‐bacteria*).ab,ti. 4.(microbicide* or anti‐microbi* or antimicrobi* or microbi*).ab,ti. 5.(beta‐lactam* or betalactam* or B‐lactam* or aminoglycoside* or vancomycin).ab,ti. 6.or/1‐5 7.exp intravenous drug administration/ 8.(infusion* or intravenous* or drip or drips).ab,ti. 9.7 or 8 10.6 and 9 11.exp Infection/ 12.exp infection control/ 13.exp Soft Tissue Infection/ 14.exp pneumonia/ 15.exp virus pneumonia/ 16.exp Meningitis/ 17.exp virus meningitis/ 18.exp Urinary Tract Infection/ 19.exp intrauterine infection/ 20.exp Sepsis/ 21.(infected or infection* or infectious or infectious?disease* or (infect* adj disease*)).ab,ti. 22.(Sepsis or pneumonia* or mening*).ab,ti. 23.((urine or urinary tract) adj3 infect*).ab,ti. 24.((skin or soft tissue) adj3 infect*).ab,ti. 25.or/11‐24 26.10 and 25 27.(continuous* or discontinu* or intermittent* or interval*).ab,ti. 28."dosage schedule comparison"/ 29.exp drug intermittent therapy/ 30.27 or 28 or 29 31.26 and 30 32.exp Randomized Controlled Trial/ 33.exp controlled clinical trial/ 34.randomi?ed.ab,ti. 35.placebo.ab. 36.*Clinical Trial/ 37.randomly.ab. 38.trial.ti. 39.32 or 33 or 34 or 35 or 36 or 37 or 38 40.exp animal/ not (exp human/ and exp animal/) 41.39 not 40 42.31 and 41
CINAHL (EBSCOhost) S25 S13 and S24 S24 S14 or S15 or S16 or S17 or S18 or S19 or S20 or S21 or S22 or S23 S23 skin infect* or soft tissue infect* S22 urine infec* or urinary tract infec* S21 Sepsis or pneumonia* or mening* S20 infected or infection* or infectious or infectious disease* S19 (MH "Sepsis+") S18 (MH "Urinary Tract Infections+") S17 (MH "Meningitis+") S16 (MH "Pneumonia, Viral") Interface ‐ S15 (MH "Soft Tissue Infections") S14 (MH "Infection+") S13 S9 and S12 S12 S10 or S11 S11 (continuous* or discontinu* or intermittent* or interval*) S10 (MH "Drug Administration Schedule") S9 S5 and S8 S8 S6 or S7 S7 (infusion* or intravenous* or drip or drips) S6 (MH "Administration, Intravenous+") or (MH "Infusions, Intravenous") S5 S1 or S2 or S3 or S4 S4 (beta‐lactam* or betalactam* or B‐lactam* or aminoglycoside* or vancomycin) S3 (microbicide* or anti‐microbi* or antimicrobi* or microbi*) Search S2 (anti‐biotic* or antibiotic* or anti‐infect* antiinfect* or antibacteria* or anti‐bacteria) S1 (MH "Antibiotics+")
ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED), ISI Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S) #1 TS=(anti‐biotic* or antibiotic* or anti‐infect* antiinfect* or antibacteria* or anti‐bacteria* or microbicide* or anti‐microbi* or antimicrobi*) AND TS=(infusion* or intravenous* or drip or drips) AND TS=(infection* or infectious or Sepsis or pneumonia* or mening*) AND TS=((drug* same schedule*) or continuous* or discontinu* or intermittent* or interval*) #2 TS=(clinical OR control* OR placebo OR random OR randomised OR randomized OR randomly OR random order OR random sequence OR random allocation OR randomly allocated OR at random) SAME TS=(trial* or group* or study or studies or placebo or controlled) #3 #1 and #2
Data and analyses
Comparison 1. Continuous vs intermittent.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All cause mortality | 19 | 1241 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.67, 1.20] |
| 1.1 Time‐dependent antibiotics | 17 | 1085 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.62, 1.22] |
| 1.2 Concentration‐dependent antibiotics | 2 | 156 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.50, 2.40] |
| 2 Infection recurrence | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Time‐dependent antibiotics | 8 | 398 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.35, 4.19] |
| 3 Clinical Cure | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Time‐dependent antibiotics | 15 | 975 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.08] |
| 4 Superinfection | 12 | 813 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.60, 1.94] |
| 4.1 Time‐dependent antibiotics | 10 | 623 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.53, 1.83] |
| 4.2 Concentration‐dependent antibiotics | 2 | 190 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [0.41, 11.70] |
| 5 Serious Adverse Events | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Time‐dependent antibiotics | 10 | 871 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.80, 2.30] |
| 6 Withdrawal due to Adverse Events | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Time‐dependent antibiotics | 10 | 871 | Risk Ratio (M‐H, Random, 95% CI) | 2.03 [0.52, 7.95] |
| 7 Adverse Events | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Time‐dependent antibiotics | 5 | 575 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.94, 1.12] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adembri 2008.
| Methods | Prospective, open‐label, randomised trial | |
| Participants | 18 septic ICU participants (mean age range 57 to 64 years; 69% male) with microbiologically documented infection caused by glycopeptide‐resistant or ‐sensitive gram‐positive strains without clinical improvement after 5 days of glycopeptide therapy. Exclusion: age < 18 years, pregnancy, previous known allergic reaction to linezolid, creatinine clearance < 40 mL/min, platelet count < 80 000, and simultaneous administration of other drugs (such as erythromycin) capable of interfering with the linezolid assay. |
|
| Interventions | Linezolid 600 mg i.v. q12h versus linezolid 300 mg i.v. loading dose followed by 900 mg continuous infusion on day 1, then 1200 mg i.v. continuous infusion daily; mean treatment duration 10 days | |
| Outcomes | Global response to therapy (clinical success or clinical failure). Microbiological result (eradication, failure, or not able to be evaluated). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, but randomisation method not stated (pg.123). |
| Allocation concealment (selection bias) | Unclear risk | Via closed envelope method, but opacity of envelope not stated (pg.123). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label, not blinded (pg.123). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 18 participants randomly assigned but only 16 participants completed the study: "One patient died before completing serum sample collection and one was excluded because he developed renal failure during sampling period" (pg.124). |
| Selective reporting (reporting bias) | Low risk | Pre‐specified outcomes reported as appropriate. |
| Other bias | Unclear risk | Simultaneous use of antibiotics against gram‐negative strains and/or fungi was not considered an exclusion criterion. No comment on how many in each group were taking additional antibiotics. |
Angus 2000.
| Methods | Prospective, randomised trial. | |
| Participants | 34 participants (age range 18 to 73 years; 47% male) with septicaemic melioidosis. Exclusion: pregnant women, participants who had already received effective antimicrobial therapy, and those with known hypersensitivity to beta‐lactam antibiotics. |
|
| Interventions | Ceftazidime 40 mg/kg i.v. q8h versus ceftazidime 12 mg/kg i.v. loading dose, followed by 4 mg/kg/h; treatment duration at least 10 days. | |
| Outcomes | No outcomes explicitly stated. | |
| Notes | Objective was to study the "pharmacokinetics and in vivo bacterial killing rates" of both regimens. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned, but randomisation method was not stated (pg. 446). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 34 participants were enrolled in the study and received study drug, but 13 were excluded (8 continuous, 5 intermittent) from the PK/PD analysis. Reason for exclusion from analysis was not clear ("data from 21 patients were suitable for PK analysis" pg. 447). Mortality status of these 13 participants is known, but not sure how many in continuous or intermittent group (11/13 died). |
| Selective reporting (reporting bias) | High risk | No outcomes stated. The objective stated in the discussion does not match that stated in the introduction. The authors state, "the original objective of the study to compare bacterial clearance rates between the two regimens could not be fulfilled because the overall mortality was so high in pour plate positive patients." |
| Other bias | Low risk | Female to male ratio for continuous infusion group is 1:9 and for bolus group is 7:4. Blood cultures were negative for 4 participants in continuous infusion group and for none in bolus group. More in bolus group were pour plate positive compared with the infusion group (N = 7 vs N = 4). Maintenance oral treatment with amoxicillin/clavulanic acid or the combination of cotrimoxazole, chloramphenicol, and doxycycline permitted. Funded by the Wellcome Trust for Great Britain. |
Bodey 1979.
| Methods | Prospective, randomised trial. | |
| Participants | 490 febrile episodes (number of participants not stated) in participants with malignant diseases (42% > 50 y; 56% male) with neutropenia and suspected or proven infection (some afebrile participants with proven infections also included). Exclusion: participants whose fever was related to the transfusion of blood products or to the administration of known pyrogens (e.g. immunotherapeutic agents), documented penicillin allergy. |
|
| Interventions | All participants received carbenicillin 5 g i.v. q4h (each dose given over 2 h) plus one of the following 3 regimens: Tobramycin given as a loading dose of 90 mg/m2 i.v. (over 30 minutes) followed by 360 mg/m2 i.v. daily as a continuous infusion. Cefamandole given as a loading dose of 3 g i.v. (given over 30 minutes) followed by 12 g i.v. daily as a continuous infusion. Cefamandole 3 g i.v. q6h (each dose given over 30 minutes). Treatment duration: minimum of seven days or four days after becoming afebrile, whichever was longer, unless untoward reactions, death, or definite clinical deterioration occurred. |
|
| Outcomes | Cure. Relapse. Super‐infection. |
|
| Notes | Outcomes all expressed as episodes and not on a per participant basis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned to receive one of three regimens by selecting cards placed in sealed envelopes, which were compiled from a table of random numbers" (pg. 609). |
| Allocation concealment (selection bias) | Unclear risk | Via sealed envelopes, but opacity of envelopes not described (pg. 609). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 490 febrile episodes were included in the study, but only 460 episodes were evaluable, of which 216 were febrile episodes with 235 episodes of documented infection; 19 participants had 2 infectious episodes (pg. 610). 68% of 234 febrile episodes, in which infection could not be demonstrated as the cause of fever, received antibiotics. Data on these participants were not included in the analysis (pg. 610). Participants who received less than 12 h of antibiotic therapy were not evaluated (pg. 610). |
| Selective reporting (reporting bias) | High risk | Total number of participants in the study not stated, but 490 febrile episodes were screened, of which 460 episodes could be evaluated. Reported on all pre‐specified outcomes of interest; however, reported outcomes as number with cure per episode of documented infection. Outcomes reported on 204 participants with 235 documented infections (approximately double this number received antibiotics). Other reported outcomes (not pre‐specified): response according to type of infection, organism, sensitivity to carbenicillin/cephalosporin/aminoglycoside, sensitivity to carbenicillin and initial neutrophil counts/trend, incidence of azotemia. |
| Other bias | Unclear risk | Funding source not stated. Cefamandole was supplied by Eli Lilly (pg. 609). |
Buck 2005.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 24 hospitalised participants (age range 41 to 76 years; 58% male) with community‐ or hospital‐acquired infections (late‐onset hospital‐acquired pneumonia, severe community‐acquired pneumonia, severe urinary tract infection, cholangitis in participants with risk factors, complicated peritonitis, participants at risk with fever of unknown origin). Exclusion: lack of informed consent, pregnancy or lactation in women, known hypersensitivity or intolerance to piperacillin‐tazobactam, and epilepsy. |
|
| Interventions | Piperacillin/tazobactam 2 g/0.5 g i.v. bolus loading dose, given over 1 h, followed by 8 g/1 g i.v. over 23 h on day 1, then over 24 h vs piperacillin 4 g + tazobactam 0.5 g i.v. q8h (doses were adjusted in those with renal dysfunction); mean treatment duration not stated. | |
| Outcomes | Pharmacokinetic analysis: serum concentration‐time profiles. Clinical assessment and response (clinical or bacteriological success). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned, but method of randomisation was not stated. |
| Allocation concealment (selection bias) | Unclear risk | Concealment via envelopes, but opacity of envelopes not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Participants and clinicians were not blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomly assigned participants were accounted for at study end. |
| Selective reporting (reporting bias) | Unclear risk | Clinical assessment was not the primary outcome (not assessed systematically in every participant). Reported all pre‐specified outcomes of interest. |
| Other bias | Unclear risk | Other antibiotics were permitted. Sponsored by Wyeth Lederle. |
Chytra 2012.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 240 intensive care unit patients (mean age 45 to 47 years; 67% male) with severe infection treated with meropenem (with a predicted treatment duration > 4 days). Exclusion: < 18 years, pregnancy, acute or chronic renal failure, immunodeficiency or immunosuppressant medication, neutropenia, meropenem hypersensitivity. |
|
| Interventions | Meropenem 2 g i.v. loading dose, then 4 g i.v. continuous infusion over 24 h versus meropenem 2 g i.v. infused over 30 minutes q8h; mean treatment duration 7 to 8 days. | |
| Outcomes | Clinical and microbiological outcomes. Safety. Meropenem‐related length of ICU and hospital stay. Meropenem‐related length of mechanical ventilation. Duration of meropenem treatment. Total dose of meropenem. ICU and in‐hospital mortality. |
|
| Notes | All participants included met sepsis criteria. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were "randomised using sealed opaque envelopes in one‐to‐one proportion without stratification" (pg. 3), but no sequence generation was mentioned. |
| Allocation concealment (selection bias) | Low risk | Participants were "randomised using sealed opaque envelopes in one‐to‐one proportion without stratification" (pg. 3). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open‐label. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Clinical cure rate, concomitant antibiotic therapy, microbiological findings, and bacteriological success rate were evaluated only in the per protocol population" (pg. 4). "14 patients from continuous group and 12 patients from intermittent group excluded from per protocol analysis because of death or other protocol violation" (pg. 4). |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | Unclear risk | Other antibiotics were permitted and were used in more than 50% of the clinically evaluable participants. More than half were trauma or post‐surgical patients. |
Cousson 2005.
| Methods | Single‐centre, prospective, randomised trial. | |
| Participants | 16 intensive care patients (median age 61 years; 75% male) with severe nosocomial gram‐negative pneumonia requiring mechanical ventilation. Exclusion: weight > 100 kg, pregnant or breastfeeding, beta‐lactam allergy, creatinine clearance < 60 mL/min, pulmonary fibrosis. |
|
| Interventions | Ceftazidime 20 mg/kg i.v. loading dose, then 60 mg/kg i.v. continuous infusion vs ceftazidime 20 mg/kg i.v. over 30 minutes q8h; treatment duration not stated. | |
| Outcomes | Pharmacodynamic profile of ceftazidime (duration plasma concentration > 20 mg/L). | |
| Notes | Pharmacokinetic study, no clinical outcomes. Article in French translated into English. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned using a random numbers table (pg. 547). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomly assigned participants were accounted for at end of study. |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | Unclear risk | Tobramycin use permitted in both groups (pg. 548). |
DeJongh 2008.
| Methods | Prospective, unblinded, randomised trial. | |
| Participants | 17 participants (mean age 57 years; 75% male) with a high probability of infection from nosocomial origin, and no suspicion of infection ofPseudomonas spp. or other temocillin‐resistant bacteria. Exclusion: age < 18 or > 75 years, weight < 50 or > 100 kg, renal insufficiency (estimated clearance < 45 mL/min), hemodialysis, estimated survival < 5 days, documentation of temocillin‐resistant organism, meningitis or other proven infection of the CNS, IgE‐mediated allergy to penicillins, severe granulocytopenia, pregnancy, participants having participated in another study < 30 days before, and marked deterioration of renal function during the study period. |
|
| Interventions | Temocillin 2 g i.v. loading dose given over 30 minutes, followed by 4 g i.v. infused at a rate of 2 mL/min vs temocillin 2 g i.v. given over 30 minutes q12h (all participants received flucloxacillin (six times 1 g/day)); mean treatment duration 8.5 to 8.8 days. | |
| Outcomes | PK/PD breakpoints. Stability and compatibility studies. MIC with E. coli. PK analyses. Population PK. Probability of target attainment rate. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned, but method of randomisation was not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 participants in the intermittent i.v. group were considered clinically not evaluable; no information on their outcome is provided, and it is unclear whether they were considered failure or cure. |
| Selective reporting (reporting bias) | Unclear risk | Reported on all pre‐specified outcomes of interest (except clinical outcomes were not specified a priori-were reported as favourable). |
| Other bias | Unclear risk | SC is supported by a First‐Entreprise grant awarded by the Direction Generale de la Recherche et des Technologies of the Region Wallonne; also supported by the Belgian Fonds de la Recherche Scientifique Medicale and by a grant‐in‐aid from Eumedica S.A., Brussels, Belgium. SC is working under contract with Eumedica s.a., Brussels, Belgium, and RDJ and PMT are unpaid advisors to Eumedica s.a., Brussels, Belgium. |
Feld 1977.
| Methods | Prospective, randomised trial. | |
| Participants | 120 cancer patients (age range 15 to 76; 48% male) who did not show signs of improvement within 48 to 72 hours after starting carbenicillin and cephalosporin for presumed or proven infection caused by gram‐negative bacilli. Exclusion: not stated. |
|
| Interventions | Sisomicin 30 mg/m2 i.v. loading dose given over 30 minutes, followed by 120 mg/m2 i.v. daily continuous infusion vs sisomicin 30 mg/m2 i.v. given over 30 minutes q6h; treatment duration: minimum of seven days or five days after the participant became afebrile (whichever was longer). | |
| Outcomes | Complete response. Super‐infection. |
|
| Notes | Some participants enrolled were < 18 years old. Difficult to assess clinical response because outcomes are listed as numbers of cases instead of numbers of participants. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned, but method of randomisation was not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Eighteen of 139 episodes were considered to be not evaluable for response, although all were considered evaluable for toxicity. Eleven of these were infections caused by organisms which would not be expected to respond to aminoglycoside antibiotics." "During the remaining seven inevaluable episodes the patients received other antibiotics in addition to sisomicin which were active against the infecting organisms." Do not know why these seven were inevaluable or why they required other antibiotics (pg. 181). |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | Low risk | |
Feld 1984.
| Methods | Prospective, randomised trial. | |
| Participants | 70 afebrile participants with neutropenia and malignant neoplasms (mean age 54 years; % male not stated) with presumed or documented infection due to gram‐negative bacilli. Exclusion: participants with poor veins, known to be allergic to cephalosporins or aminoglycoside antibiotics, pregnant or lactating, and those with abnormal renal function. |
|
| Interventions | Tobramycin 60 mg/m2 i.v. loading dose given over 30 minutes, followed by 300 mg/m2 i.v. daily as a continuous infusion (adjusted to maintain a serum concentration of approximately 4 to 5 mg/L) vs tobramycin 75 mg/m2 i.v. given over 30 minutes q6h (adjusted to a peak serum concentration of approximately 6 to 7 mg); minimum treatment duration of seven days or five days after the participant became afebrile. | |
| Outcomes | Clinical cure. Partial response. Clinical failure. Mortality. Superinfection. Nephrotoxicity. |
|
| Notes | Focused on nephrotoxicity and ototoxicity. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was determined by a series of random allocations (stratified by hospital), but do not know how these random allocations were generated. |
| Allocation concealment (selection bias) | Unclear risk | Via sealed envelopes, but did not state the opacity of the envelopes. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 6 patients could not be evaluated for efficacy, although all were included in the adverse event analysis. |
| Selective reporting (reporting bias) | Unclear risk | Super‐infection was mentioned but did not mention how many in each group. |
| Other bias | Unclear risk | Outcomes were reported as episodes instead of numbers of participants with at least 1 episode. |
Georges 2005.
| Methods | Prospective, open‐label, parallel‐group, randomised trial. | |
| Participants | 50 critically ill participants (mean age range 46 to 50 years; 82% male) with nosocomial pneumonopathy or nosocomial bactermia thought to be sensitive to cefepime. Exclusion: history of allergy to the beta‐lactams, isolation of a bacterium resistant to cefepime and/or amikacin, presence of renal insufficiency (creatinine clearance estimated according to Cockcroft‐Gault, < 30 mL/min), administration of antibiotics in the 3 preceding days except in cases of clinical failure or isolation of a resistant bacterium, septic shock. |
|
| Interventions | Cefepime 2 g i.v. continuous infusion over 12 hours twice daily vs cefepime 2 g i.v over 30 min twice daily (amikacin given simultaneously in both groups); mean treatment duration 12 days. | |
| Outcomes | Bacterial MIC. Pharmacokinetic and pharmacodynamic parameters (AUCss, AUCss > MIC, AUCss/MIC, AUICss, t > MIC, t > five‐fold MIC, t > French breakpoint). Clinical, laboratory and bacterial efficacy. Tolerance. Mortality. Clinical cure. |
|
| Notes | Counted over‐infection as super‐infection in outcomes (p.364). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg.123). |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label, not blinded (pg. 361)/ |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Three participants were excluded from analysis (2 in continuous infusion group, 1 in intermittent infusion group withdrew for allergy, shock, and death independent of the infection) (pg. 363). Imputation of data for 4 participants in continuous infusion group and 5 participants in intermittent infusion group (presumed bacteriological eradication if patient extubated during treatment) (pg. 363). |
| Selective reporting (reporting bias) | High risk | Outcomes not specified a priori reported (mean duration of treatment, mean duration ventilation, mean duration ICU hospitalisation, clinical failure, bacteriological cure, no eradication, over‐infection) (pg. 364). Mortality outcome does not include all deaths (participants withdrawn from the study for death independent of infection) (pg. 363). |
| Other bias | Unclear risk | Amikacin given simultaneously in both groups and "other authorized antimicrobial treatment included the glycopeptides (1 patient in each group), antifungal agents (1 patient in intermittent group) and imidazoles" (pg. 361, 363). Bristol‐Myers Squibb acknowledged for helpful discussions (pg. 367). |
Hanes 2000.
| Methods | Randomised trial. | |
| Participants | 32 critically ill trauma participants (mean age range 33 to 36 years; 81% male) with gram‐negative nosocomial pneumonia occurring more than 48 h after admission. Exclusion: known sensitivity to cephalosporins, estimated creatinine clearance of < 30 mL/min, or causative bacterial pathogen resistant to ceftazidime. |
|
| Interventions | Ceftazidime 2 g i.v. bolus followed by 60 mg/kg continuous infusion daily vs ceftazidime 2 g i.v. infused over 30 minutes q8h; mean treatment duration not stated. | |
| Outcomes | Pharmacokinetic parameters. Clinical response (cure, improvement, failure, intermediate). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 436). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 15 participants randomly assigned to the intermittent group (14 participants reported) and 17 patients randomly assigned to the continuous group (17 participants reported) (pg.437‐8). 2 participants excluded from analysis (1 participant from each group) (pg. 437). Super‐infection not reported in absolute numbers, only in percentages ("pneumonia superinfection occurred in 22% versus 44% in all patients for intermittent and continuous ceftazidime regimens, respectively") (pg. 438). |
| Selective reporting (reporting bias) | High risk | Did not report rates of clinical response (cure, improvement, failure, intermediate). Outcomes not specified a priori reported (proportion of patients with normalization of white blood cell count and temperature, duration of leukocytosis and pyrexia, duration of mechanical ventilation, duration of intensive care unit stay, duration of hospital stay) (pg.438). |
| Other bias | Unclear risk | Funded by GlaxoWellcome, Inc (pg.436). |
Lagast 1983.
| Methods | Randomised trial | |
| Participants | 45 patients (age not described; 44% male) with demonstrated aerobic gram‐negative bacillary septicemia. Exclusion: high likelihood of death from non‐infectious causes, history of allergy to penicillins or cephalosporins, those with impaired hepatic function or renal function |
|
| Interventions | Cefoperazone 2 g i.v. infused over 15 minutes twice daily vs cefoperazone 1 g i.v. loading dose infused over 15 minutes, followed by 3 g infused over the remainder of the first 24 h, then 4 g infused continuously over 24 h daily; mean treatment duration not stated. | |
| Outcomes | Cure. Failure (death, clinical deterioration requiring a change in antimicrobial therapy). Super‐infection. Bacterial colonisation. Bacteriological cure. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 555). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcomes reported for all 45 participants (ITT analysis) (pg. 556). |
| Selective reporting (reporting bias) | High risk | Cure not reported. Super‐infection, bacterial colonisation, and bacteriological cure were not reported by intervention group. |
| Other bias | Unclear risk | Baseline characteristics of patients not stated. Funded by Pfizer Laboratories (pg. 558). |
Lau 2006.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 262 hospitalised participants (mean age 50 years; 60% male) with complicated intra‐abdominal infection. Exclusion: Underlying immunodeficiency or receiving immunosuppressant medications (including > 5 mg prednisone or equivalent per day); other infections requiring systemic antibiotic or antifungal treatment; infections caused by organisms resistant to piperacillin‐tazobactam; active or treated leukemia or a systemic malignancy that required chemotherapy, immunotherapy, radiation therapy, or antineoplastic therapy within the past year; known hypersensitivity to beta‐lactams; infected pancreatic or peripancreatic necrosis in association with necrotising pancreatitis; severe renal dysfunction; neutropenia; thrombocytopenia; high levels of liver enzymes; INR > 2 x upper limit of normal; multiorgan system failure; irreversible shock; or anticipated discharge from the hospital in less than 4 days. |
|
| Interventions | Piperacillin‐tazobactam 2 g/0.250 g i.v. bolus infused over 30 minutes, followed by 12 g/1.5 g infused continuously over 24 h vs piperacillin‐tazobactam 3 g/0.375 g i.v. infused over 30 minutes q6h; mean treatment duration 4 to 14 days. | |
| Outcomes | Clinical success at the test of cure (cure, improvement). Bacteriological response at the test of cure (success, failure). Time to defervescence. Time to WBC normalisation. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 3557). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label, not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | After randomisation, 167 participants of 262 randomly assigned were included in the primary analysis (clinically evaluable population), 114 participants of 262 randomly assigned were analysed as the bacteriologically evaluable population (pg. 3557, Figure 1). Two patients in each group of the modified all‐treated population excluded from analysis (pg. 3559). |
| Selective reporting (reporting bias) | High risk | Did not report outcomes for all‐treated population (all patients who were randomly assigned). |
| Other bias | High risk | Did not meet pre‐specified sample size calculation of 180 participants in the clinically evaluable population (potentially a type 2 error) (pg. 3557). Baseline imbalance, sicker patients in continuous infusion group (7 vs 0 patients with APACHE II score > 20) (pg. 3558). Study funded by Wyeth Pharmaceuticals (pg. 3560), Authors affiliated with Wyeth and Merck Research Laboratories (pg. 3560). |
Lipman 1999.
| Methods | Randomised trial. | |
| Participants | 18 critical care patients (mean age range 53 to 64 years; proportion of males/females not stated) with normal renal function requiring ceftazidime according to usual clinical practice (pg. 309). Exclusion: not explicitly stated. |
|
| Interventions | Ceftazidime 12 mg/kg i.v. loading dose infused over 2 minutes, followed by 2 g infused over 478 minutes, then 2 g infusion q8h versus ceftazidime 12 mg/kg i.v. loading dose infused over 2 minutes, followed by 2 g infused over 28 minutes, then 2 g over 30 minutes q8h; treatment duration not stated. | |
| Outcomes | Total time plasma ceftazidime concentrations below 40 mg/L. | |
| Notes | Pharmacokinetic trial, no clinical outcomes reported. Not included in meta‐analysis because number of patients randomly assigned into each group not reported. Did not report any outcomes of interest except "no ceftazidime‐related adverse reactions were noted" (pg. 310). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomly assigned with “computer‐generated random numbers” (pg. 309). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes not specified a priori reported (range of plasma ceftazidime concentrations achieved, number of patients with plasma ceftazidime concentrations > 40 mg/L). |
| Other bias | Unclear risk | Baseline imbalances, members of infusion group were older (mean age 64 years vs 53 years) and had higher APACHE II scores (20.5 vs 15.5) (pg. 310). Funding source not stated. |
Lubasch 2003.
| Methods | Multicentre, randomised trial. | |
| Participants | 81 hospitalised patients (mean age 65 years; 69% male) with acute exacerbation of severe chronic bronchitis, Exclusion: pregnancy or lactation period, allergy to beta‐lactams and/or aminoglycosides, other infection requiring systemic antibiotics, last dose of antibiotic < 72 h, impaired renal function. |
|
| Interventions | Ceftazidime 2 g i.v. q8h vs ceftazidime 2 g i.v. loading dose, followed by 2 g i.v. infused over 7 h q12h; treatment duration 8 to 14 days. | |
| Outcomes | Clinical and bacteriological responses at day 8 or 9, and 72 h after the end of therapy. Pharmacokinetic and pharmacodynamic parameters of ceftazidime, |
|
| Notes | Extended interval. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 670). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Outcomes reported for clinically evaluable population, but number of patients in clinically evaluable population not stated (pg. 661). |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | Unclear risk | Not all patients were assessed for clinical outcomes, reason for exclusion of patients from clinically evaluable population not stated. Clinical success was defined as cure or improvement. |
Nicolau 1999a.
| Methods | Prospective, single‐blind, randomised trial. | |
| Participants | 34 critical care patients (mean age range 43 to 51 years; 65% male) with nosocomial pneumonia (pg. 134). Exclusion: not stated. |
|
| Interventions | Ceftazidime 2 g i.v. q8h infused over 30 minutes vs ceftazidime 1 g i.v. loading dose over 30 minutes, followed by 3 g continuous infusion over 24 h (both groups received concomitant tobramycin 7 mg/kg i.v. daily); treatment duration not stated. | |
| Outcomes | Pharmacokinetic parameters (Cmax, Cmin, K, t1/2, AUC, clearance). Pharmacodynamic profile. Follow‐up between days 2 and 5 of therapy. |
|
| Notes | Pharmacokinetic study, clinical outcomes not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 134). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Single blind, but unclear which group was blinded (pg. 134). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data were acquired from 41 participants, but only 34 participants were included in analysis; reasons for exclusion not stated (pg. 134). |
| Selective reporting (reporting bias) | High risk | Outcomes not specified a priori reported (Cmean, isolated pathogens, T > MIC) (pg. 136‐7). "All patients tolerated the continuous infusions with no infusion‐related adverse effects (e.g. phlebitis)" (pg. 136); however, adverse events for the intermittent group were not reported. |
| Other bias | Unclear risk | Baseline imbalance, intermittent participants older (mean age 51 years vs 43 years). Funded by Glaxo Pharmaceuticals (pg. 139). |
Nicolau 1999b.
| Methods | Prospective, randomised, controlled trial. | |
| Participants | 24 ICU patients (mean age range 37 to 45 years; 63% male) suspected of having bacterial pneumonia. Exclusion: not stated, |
|
| Interventions | Tobramycin 7 mg/kg i.v. once daily plus one of the following, depending on renal function: Creatinine clearance > 50 mL/min: ceftazidime 3 g i.v given over 24 h vs ceftazidime 2 g i.v q8h. Creatinine clearance 31 to 50 mL/min: ceftazidime 2.5 g i.v given over 24 h vs ceftazidime 2 g i.v. q12h. Creatinine clearance 20 to 30 mL/min: ceftazidime 2 g i.v. given over 24 h vs ceftazidime 2 g i.v. q24h. Mean treatment duration not stated. |
|
| Outcomes | Pharmacokinetic parameters. Follow‐up duration unclear; however, it was stated that pharmacokinetic analyses were conducted on the basis of samples drawn during second week of hospitalisation. |
|
| Notes | No clinical outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Listed only pharmacokinetic parameter results for patients with normal renal function (n = 10 for continuous infusion and n = 11 for intermittent infusion) (pg. 47). |
| Selective reporting (reporting bias) | High risk | Reported "no adverse events were attributed to the dosing regimen of ceftazidime"; however, total adverse events were not reported (pg. 47). |
| Other bias | High risk | Funded by Glaxo Pharmaceuticals. |
Nicolau 2001.
| Methods | Prospective, open‐label, randomised controlled trial. | |
| Participants | 41 ICU patients (mean age range 46 to 56 years; 56% male) with nosocomial acquired pneumonia with a clinical suspicion of bacterial etiology. Exclusion: documented active tuberculosis, cystic fibrosis, viral pneumonia, infection with a microorganism known to be resistant to study medication, or use of antimicrobial therapy with activity against suspected pathogen for longer than 48 h before enrolment without a persistently positive culture. |
|
| Interventions | Tobramycin 7 mg/kg i.v. once daily plus one of the following, depending on renal function: Creatinine clearance > 50 mL/min (normal renal function): ceftazidime 3 g i.v. over 24 h vs ceftazidime 2 g i.v given over 30 minutes q8h. Creatinine clearance 31 to 50 mL/min: ceftazidime 2.5 g i.v. over 24 h vs ceftazidime 2 g i.v. q12h. Creatinine clearance 20 to 30 mL/min: ceftazidime 2 g i.v. over 24 h versus ceftazidime 2 g i.v q24h. Mean treatment duration 16 to 18 h. |
|
| Outcomes | Clinical outcome at 14 to 21 days post‐therapy or at the time of institutional discharge (cure, improved, failure). Microbiological outcome at 14 to 21 days post‐therapy or at the time of institutional discharge (eradication, presumed eradication, persistence). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Participants who received 5 or more days of therapy were considered for inclusion in the final data analysis. 6 patients were subsequently declared clinically non‐evaluable because of their short duration of therapy (5 days). 5 patients were withdrawn from continuous group and 1 patient from the intermittent group; these patients were not included in the analyses. |
| Selective reporting (reporting bias) | Unclear risk | "Of the 41 patients enrolled, 21 (51%) experience at least one adverse event attributable to the study agent" (pg. 501); however, total adverse events were not reported. No protocol cited. |
| Other bias | High risk | All patients may have received open‐label metronidazole or vancomycin, but the number of patients in each group who needed open‐label therapy was not reported. Funded by Glaxo Wellcome Inc. |
Okimoto 2009.
| Methods | Prospective, randomised trial. | |
| Participants | 50 elderly patients (mean age 80 years; 60% male) with community‐acquired pneumonia. | |
| Interventions | Meropenem 500 mg i.v. q12h vs meropenem 1.0 g/day i.v. continuous 24 h infusion; mean treatment duration 12 to 13 days. | |
| Outcomes | Clinical efficacy. Bacteriological efficacy. |
|
| Notes | Able to obtain only partial translation of this trial from Japanese into English. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but unclear randomisation method. |
| Allocation concealment (selection bias) | Unclear risk | Unclear, complete translation not available. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Unclear, complete translation not available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients assessed for clinical cure, but not for bacteriological efficacy. |
| Selective reporting (reporting bias) | Unclear risk | Unclear; complete translation not available. |
| Other bias | Unclear risk | Unclear; complete translation not available. |
Pedeboscq 2001.
| Methods | Open‐label, randomised trial. | |
| Participants | 7 gastrointestinal intensive care patients (mean age 58 years; 43% male) with severe sepsis. Exclusion: severe liver dysfunction, severe renal impairment, shock, suspected infection not susceptible to piperacillin‐tazobactam, previous antibiotic use in past 15 days. |
|
| Interventions | Piperacillin‐tazobactam 4 g/0.5 g a day i.v. over 30 minutes q8h vs piperacillin‐tazobactam 12 g/1.5 g a day i.v. continuous 24 h infusion; mean treatment duration not stated. | |
| Outcomes | Time > MIC for Enterobacteria, Pseudomonas. | |
| Notes | Mortality reported in Roberts 2009a review. Article in French translated into English. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but randomisation method not stated (pg. 542). |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated whether any patients were lost to follow‐up or withdrew from the study. |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | Unclear risk | Concomitant fluoroquinolone use permitted (pg.541). |
Rafati 2006.
| Methods | Prospective, randomised controlled trial. | |
| Participants | 40 general ICU patients (mean age range 48 to 50 years; 68% male) with sepsis with systemic inflammatory response syndrome and known or documented infection. Exclusion: patients with renal dysfunction. |
|
| Interventions | Piperacillin 2 g i.v. loading dose given over 0.5 h, followed by 8 g i.v. over 24 h daily vs piperacillin 3 g i.v. given over 0.5 h q6h (all patients received amikacin 15 mg/kg daily); mean treatment duration 5 to 6 days. | |
| Outcomes | Pharmacokinetic parameters. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients were accounted for in terms of mortality, but no mention was made of those lost to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Mortality reported as an outcome, but it was not stated a priori. No protocol cited. |
| Other bias | Low risk | |
Roberts 2007.
| Methods | Prospective, single‐blind, randomised controlled trial. | |
| Participants | 57 critically ill patients (mean age range 43 to 52 years; 58% male) with sepsis for whom clinicians deemed ceftriaxone as appropriate empirical therapy (needed to be on at least 4 days of ceftriaxone before randomisation). Exclusion: history of organ transplant or recent treatment with cytotoxic drugs. |
|
| Interventions | Ceftriaxone 2 g i.v. continuously infused over 24 h vs ceftriaxone 2 g i.v daily (ceftriaxone 500 mg i.v. loading dose given in both groups); mean treatment duration 6 days. | |
| Outcomes | Clinical response. Clinical cure. Bacteriological response. Bacteriological cure. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised into two groups...using sequential opaque sealed envelopes (sequence generated from a table of random numbers) which were opened by the treating physician after consent was gained from the patient or legally authorized representative." |
| Allocation concealment (selection bias) | Low risk | "...using sequential opaque sealed envelopes..." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "Clinical and bacteriological outcomes were assessed at the cessation of ceftriaxone treatment by a critical care physician blinded to the groupings and with no role in the management of the subjects" (pg. 286). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Analysis of data was primarily performed on an intention‐to‐treat (ITT) basis. However, as this was a pilot study, a priori, we also elected to analyse patients that received at least 4 days of antibiotic therapy." "The clinical outcomes of the other patients classified as failures were not assessable and were included as failures to be conservative." |
| Selective reporting (reporting bias) | Low risk | Reported on all pre‐specified outcomes of interest. |
| Other bias | High risk | 43 patients used open‐label antibiotics, and authors state no statistical differences (P = 0.66) between the groups, but no details were given. |
Roberts 2009b.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 10 critically ill patients (mean age range 55 to 57 years; 70% male) with a clinical indication for meropenem, normal renal function, and known or suspected sepsis. Exclusion: not stated. |
|
| Interventions | Meropenem 500 mg i.v. infused over 3 min, followed by 3000 mg continuous infusion over 24 h (given as three 1000‐mg infusions over 8 h) vs meropenem 1500 mg i.v. infused over 5 min, followed by 1000 mg infused over 3 min q8h. | |
| Outcomes | Subcutaneous tissue concentration‐time profiles. Plasma concentration‐time profiles. Pharmacokinetic variability. Plasma pharmacokinetic‐pharmacodynamic profile. Expected probability of target attainment. |
|
| Notes | No clinical outcomes specified. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomised using random numbers concealed in opaque sealed envelopes" (pg. 143); however, no sequence generation mentioned. |
| Allocation concealment (selection bias) | Low risk | "Patients were randomised using random numbers concealed in opaque sealed envelopes" (pg. 143). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not stated whether any patients were lost to follow‐up or withdrew from the study. |
| Selective reporting (reporting bias) | Unclear risk | No protocol cited. |
| Other bias | Unclear risk | Open‐label use of other antibiotics not stated. |
Roberts 2009c.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 13 septic ICU patients (median age range 24 to 42 years; 77% male) with known or suspected sepsis, in whom the clinician deemed piperacillin‐tazobactam to be appropriate therapy, and who had normal renal function. Exclusion: not stated. |
|
| Interventions | Piperacillin‐tazobactam 4 g/0.5 g i.v. over 20 minutes, followed by 8 g/1 g continuous infusion over 24 h (piperacillin 333 mg/h) on day 1, then 12 g/1.5 g continuous infusion over 24 h (piperacillin 500 mg/h) vs piperacillin‐tazobactam 4 g/0.5 g i.v. q6h or q8h; mean treatment duration not stated. | |
| Outcomes | Clinical outcomes (resolution, improvement, failure). In vivo microdialysis of plasma and tissue to determine pharmacokinetics and pharmacodynamics. |
|
| Notes | Clinical outcomes determined by unblinded, treating physician. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated. |
| Allocation concealment (selection bias) | Low risk | "Patients were randomised using opaque sealed envelopes..." (pg. 927). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reported clinical "cure" for all 13 randomised patients. |
| Selective reporting (reporting bias) | Unclear risk | No clinical trial protocol cited, although stated assessments in the methods were conducted. |
| Other bias | High risk | Continuous group was younger. Patients had normal renal function and were "young" for sepsis patients. Results may not be generalisable to typical sepsis patients. Pre‐specified definitions of clinical outcomes were not used in the reporting of clinical results (e.g. reported "cure" with no definitions given). Clinical outcome definitions were subjective, and it would have been useful to state consistency in categorization of clinical outcomes (not reported). Open‐label use of other antibiotics not stated. |
Roberts 2010.
| Methods | Randomised trial. | |
| Participants | 16 critically ill patients (mean age range 30 to 41 years; 61% male) with known or suspected sepsis and normal renal function. Exclusion: not stated. |
|
| Interventions | Piperacillin‐tazobactam 4 g/0.5 g i.v. over 20 minutes, followed by 8 g/1 g continuous infusion over 24 h (piperacillin 333 mg/h) on day 1, then 12 g/1.5 g continuous infusion over 24 h (piperacillin 500 mg/h) vs piperacillin‐tazobactam 4 g/0.5 g i.v. q6h or q8h; mean treatment duration not stated. | |
| Outcomes | Plasma‐concentration time profiles for first dose and steady state. Probability of target attainment by MIC against bacterial pathogens commonly encountered in critical care units. |
|
| Notes | Same data set as Roberts 2009c, with 3 additional patients (all met sepsis criteria). Will include only outcomes of interest for the 3 additional patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned "using random numbers selected from an opaque sealed envelope" (pg. 157); however, no sequence generation mentioned. |
| Allocation concealment (selection bias) | Low risk | "Random numbers selected from an opaque sealed envelope" (pg. 157). |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reported mortality for all 16 randomly assigned participants (pg. 159, table 1). |
| Selective reporting (reporting bias) | Unclear risk | No clinical trial protocol cited. |
| Other bias | Unclear risk | Open‐label use of other antibiotics not stated. All patients except 1 in the intermittent group received q6h dosing. |
Sakka 2007.
| Methods | Prospective, randomised trial. | |
| Participants | 20 surgical intensive care unit patients (mean age range 59 to 62 years; 55% male) with ICU‐acquired pneumonia (duration of endotracheal intubation and mechanical ventilation > 3 days). Exclusion: not stated. |
|
| Interventions | Imipenem‐cilastatin 1 g/1 g i.v. given over 40 minutes, followed by 2 g/2 g continuously infused q24h for 3 days (thereafter, 1 g/1 g i.v q8h) vs imipenem‐cilastatin 1 g/1 g i.v. given over 40 minutes three times daily for 3 days; mean treatment duration 12 to 14 days. | |
| Outcomes | Pharmacokinetic analysis. Pharmacodynamic analysis. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, but method of randomisation is unclear. |
| Allocation concealment (selection bias) | Low risk | "The randomisation code was provided to the clinical investigator in sealed envelopes" (pg. 3306), but there is no mention of the opaqueness of envelopes. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not stated, likely not blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Participants lost to follow‐up or withdrawn from study not stated (although mortality reported over a denominator of 20 participants). |
| Selective reporting (reporting bias) | High risk | Reported on outcomes not specified a priori (imipenem‐related adverse reactions, mortality). |
| Other bias | Unclear risk | "Antibiotic pretreatment was given to eight patients in the short‐term infusion group (four patients pretreated with ceftriaxone, one with cefuroxime, two with piperacillin‐tazobactam, and one with moxifloxacin). For comparison, nine patients in the continuous group received antibiotic therapy before administration of imipenem‐cilastatin (four patients pretreated with ceftriaxone, two with cefuroxime, two with piperacillin‐tazobactam, and one with cefepime) (pg. 3306). Funded by MDS Sharp & Dohme (pg. 3309). Author conflict of interest not stated. |
van Zanten 2007.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 93 hospitalised patients (mean age range 65 to 69 years; 69% male) who required antibiotic treatment for moderate to severe acute exacerbations of COPD (GOLD class 2 to 4). Exclusion: suspected or proven resistance to cefotaxime, administration of antibiotics in the preceding 48 h, allergy to beta‐lactam antibiotics, bilirubin concentrations > 20 umol/L, serum creatinine > 120 umol/L, and whole blood count < 3.0 x 10^9/L. |
|
| Interventions | Cefotaxime 1 g i.v. given over 30 minutes, followed by 2 g i.v. continuous infusion q24h vs cefotaxime 1 g i.v. given over 30 minutes q8h; mean treatment duration 9 to 10 days. | |
| Outcomes | Clinical assessment (successful treatment, treatment failure, non‐evaluable). Pharmacokinetic variables (t1/2, AUC, CL, Vss). Pharmacodynamic variables (MIC, numbers of patients with serum drug concentrations below MIC, numbers of patients with serum drug concentrations below 5 x MIC, average time concentrations below 5 x MIC). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients randomly assigned, but method of randomisation not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not all deaths were counted. "Of the 93 patients initially enrolled, 10 were excluded for the following reasons (after randomisation): death due to cardiac failure (n = 5); antibiotic treatment in the 48‐h period before initiation of cefotaxime therapy (n = 2); final diagnosis of squamous cell carcinoma instead of infection (n = 1); and protocol violations (n = 2)" (pg. 103). Pharmacokinetic evaluation "was not measured in six patients, because three patients in group 1 died before blood samples could be drawn, and technical errors, such as lost blood samples, occurred in three patients in group 2." Pharmacodynamic evaluation had the same denominator as pharmacokinetic evaluation. |
| Selective reporting (reporting bias) | Unclear risk | Only mention adverse events that were drug related, not total adverse events. However, did not state a priori that they were going to report on any adverse events. |
| Other bias | Unclear risk | Hoechst Marion Roussel provided a restricted research grant for analysing serum cefotaxime concentrations and for assessing MIC values. |
Wright 1979.
| Methods | Prospective, randomised trial. | |
| Participants | 36 patients (mean age range 33 to 53 years; % male not stated) with severe respiratory infection (pneumonia, chronic obstructive airways disease, or shock lung), Exclusion: not stated. |
|
| Interventions | Gentamicin 60 mg/m2 i.v. continuously infused over 8 hours vs gentamicin 60 mg/m2 i.v. given over 30 minutes q8h (all patients received penicillin 5 000 000 units i.v. q6h). | |
| Outcomes | None stated. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients randomly allocated, but method of randomisation not stated. |
| Allocation concealment (selection bias) | Unclear risk | Not stated. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not stated, but assumed not blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Analysis via intention‐to‐treat, number who withdrew or were lost to follow‐up not reported. |
| Selective reporting (reporting bias) | Unclear risk | No outcomes specified a priori. |
| Other bias | Unclear risk | Number of patients in each group who used open‐label cloxacillin unclear: "Cloxacillin was added to the dose in 3 patients from whom Staphylococcus aureus was isolated during the course of their illness" (pg. 198), Funding source and author conflict of interest not stated, |
Wysocki 2001.
| Methods | Prospective, open‐label, randomised trial. | |
| Participants | 160 medico‐surgical ICU patients (mean age 63 years; 65% male) given vancomycin for suspected or well‐established methicillin‐resistant staphylococcal infection acquired 72 h after admission Exclusion: received vancomycin 72 h before current infection, beta‐lactam allergy, previously included in the same protocol, or currently in another protocol. |
|
| Interventions | Vancomycin 15 mg/kg i.v. infused over 60 minutes, followed by 30 mg/kg continuous infusion vs vancomycin 15 mg/kg i.v. infused over 60 minutes q12h; mean treatment duration range 13 to 14 days. | |
| Outcomes | Efficacy: clinical failures at treatment end, clinical failures at treatment day 10, deaths while in intensive care, microbiological failures at treatment day 5. Safety: side effects attributed to vancomycin or that resulted in treatment discontinuation. Pharmacokinetics, treatment adjustment, and monitoring (AUC24h, time required to reach targeted concentrations, number of samples needed to adjust the treatment). Cost. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation was stratified by centre using a random‐number table and a block randomisation method with a block size of 8" (pg. 2461). |
| Allocation concealment (selection bias) | Low risk | "The infusion mode was contained in sealed opaque envelopes labelled consecutively with the randomisation numbers" (pg. 2461). |
| Blinding (performance bias and detection bias) All outcomes | High risk | "Clinical failure was first evaluated by local investigators, and since the treatment was not administered in a blinded fashion, a committee blinded to the infusion mode reviewed the charts from patients with clinical failure, as well as those of all of the study patients who died in the ICU" (pg. 2462). Patients and clinicians were not blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 160 participants randomly assigned, but 119 participants analysed. Figure 1: patients were excluded after randomisation (differential number of patients were excluded from each group); no intention‐to‐treat analysis was conducted. |
| Selective reporting (reporting bias) | High risk | "After reviewing clinical, laboratory, radiological, and pathological findings, the committee decided by consensus if death could reasonably be attributed to the staphylococcal infection" (pg. 2462); unsure whether all‐cause mortality was reported (unclear if people who died from other types of infections or other causes included). Authors state: "Outcomes were evaluated in all included patients, and mortality was also evaluated in an intent‐to‐treat analysis" (pg. 2462), but this was not done as patients were selectively excluded after randomisation, and these people were not included in the analysis. |
| Other bias | High risk | Administration of non‐glycopeptide antibiotics in combination with vancomycin was permitted (and was only reported if > 5 d use). Authors state: "All side‐effects attributed to vancomycin or which resulted in treatment discontinuation were reported" (pg. 2463), which implies that the authors preferentially screened for side effects (unsure whether authors included all side effects or all adverse events), Author conflict of interest not stated. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adembri 2010 | Prophylactic antibiotics (no infection) |
| Ambrose 1998 | Patients not randomly assigned |
| Benko 1996 | Cross‐over study |
| Bosso 1999 | Cross‐over study |
| Buijk 2002 | 6 continuous infusion patients were not randomly assigned |
| Burgess 2002 | Healthy volunteers |
| DeRyke 2006 | Pharmacoeconomic analysis of Lau 2006 study |
| Georges 1999 | Preliminary analysis of Georges 2005 study |
| Grant 2002 | Patients not randomly assigned |
| Hutschala 2009 | Patients not randomly assigned |
| James 1996 | Cross‐over study |
| Jaruratanasirikul 2002 | Cross‐over study |
| Jaruratanasirikul 2005 | Cross‐over study |
| Jaruratanasirikul 2009 | Cross‐over study |
| Jaruratanasirikul 2010 | Cross‐over study |
| Kirkpatrick 2001 | Letter to the editor about Pass 2001 study |
| Klepser 1998 | Healthy volunteers |
| Kojika 2005 | Patients not randomly assigned |
| Langgartner 2007 | Cross‐over study |
| Li 2005 | Pharmacokinetic and pharmacodynamic analysis of Lau 2006 study |
| Lorente 2006 | Patients not randomly assigned |
| Martin 1998 | Prophylactic antibiotics (no infection) |
| McNabb 2001 | Cost‐effectiveness analysis of Nicolau 2001 study |
| Nicasio 2007 | Case report |
| Pass 2001 | Uncontrolled study |
| Schuster 2009 | Patients not randomly assigned |
| Seguin 2009 | Patients not randomly assigned |
| Thalhammer 1999 | Cross‐over study |
| Vinks 2003 | Cross‐over study |
| Vuagnat 2004 | Patients not randomly assigned |
| Waltrip 2002 | Prophylactic antibiotics (no infection) |
Characteristics of ongoing studies [ordered by study ID]
Cousson 2010.
| Trial name or title | Comparison of pharmacodynamic interest of ceftazidime continuous infusion vs intermittent bolus administration in epithelial lining fluid concentrations of patients with severe nosocomial pneumonia |
| Methods | Prospective, randomised trial |
| Participants | Enrolment = 32, critically ill patients |
| Interventions | Ceftazidime 20 mg/kg i.v. bolus, then 60 mg/kg/24 h continuous infusion vs ceftazidime 20 mg/kg i.v. over 30 minutes q8h (combined with tobramycin 5 mg/kg i.v in both groups) |
| Outcomes | Ceftazidime epithelial lining fluid concentrations MIC for causative organisms |
| Starting date | Not stated |
| Contact information | Joel Cousson (jcousson@chu‐reims.fr) |
| Notes | Data not published, information obtained from poster presentation |
NCT00891423.
| Trial name or title | A randomised controlled cross‐over pilot study of meropenem standard 30‐minute infusion vs prolonged 3‐hour infusion in intensive care patients |
| Methods | Randomised, open‐label trial |
| Participants | Enrolment = 10, adults in intensive care |
| Interventions | Meropenem 1 g i.v. infused over 30 minutes q8h (if creatinine clearance > 50 mL/min) OR q12h (if creatinine clearance < 50 mL/min) vs meropenem 500 mg i.v. infused over 3 h q8h (if creatinine clearance > 50 mL/min) OR q12h (if creatinine clearance < 50 mL/min) |
| Outcomes | Primary outcome: time above MIC |
| Starting date | April 2009 |
| Contact information | Katherine Langan (Katherine.Langan@monash.edu.au) |
| Notes | Study completed January 2010, data not published (no clinical outcomes measured) |
NCT01158937.
| Trial name or title | Pharmacokinetic study of extended infusion meropenem in adult cystic fibrosis patients with exacerbation of pulmonary infection |
| Methods | Randomised, open‐label, cross‐over, pharmacokinetics/pharmacodynamics trial |
| Participants | Estimated enrolment = 12, adults with cystic fibrosis experiencing new or exacerbation of active pulmonary infection with recent sputum culture positive for Pseudomonas aeruginosa and/or Burkholderia cepacia at a prior visit |
| Interventions | Meropenem 2 g i.v. infused over 30 minutes loading dose vs meropenem 2 g i.v. infused over 3 h; 2 x 8‐h pharmacokinetic monitoring periods |
| Outcomes | Primary outcome: pharmacokinetic profile of extended infusion meropenem in cystic fibrosis |
| Starting date | May 2010 |
| Contact information | Daniel Cortes (cortesd@smh.ca), Jonah Crespo (crespoj@smh.ca) |
| Notes | Estimated study completion date August 2012 |
NCT01198925.
| Trial name or title | Assessment of the optimal dosing of piperacillin‐tazobactam in intensive care unit patients: extended vs continuous infusion |
| Methods | Randomised, open‐label, pharmacokinetic/pharmacodynamic trial |
| Participants | Estimated enrolment = 30, adults admitted to the intensive care unit |
| Interventions | Piperacillin‐tazobactam 4 g i.v. loading dose infused over 30 minutes, then 4 x 4‐g i.v. infused over 3 h (extended infusion) vs piperacillin‐tazobactam 4 g i.v. loading dose infused over 30 minutes, then 16 g i.v. infused over 24 h (continuous infusion) |
| Outcomes | Primary outcome: pharmacokinetics of piperacillin continuous infusion compared with piperacillin extended infusion Secondary outcome: 95% probability of target attainment vs MIC of different organisms |
| Starting date | September 2010 |
| Contact information | Johan Decruyenaere (johan.decruyenaere@ugent.be) |
| Notes | Did not report estimated study completion date |
NCT01379157.
| Trial name or title | The pharmacodynamics of imipenem in critically ill patients with ventilator‐associated pneumonia following administration by 4 h or 0.5 h infusion |
| Methods | Randomised, open‐label pharmacokinetics/pharmacodynamics trial |
| Participants | Estimated enrolment = 12, critically ill patients > 20 years old with ventilator‐associated pneumonia with gram‐negative bacilli infections |
| Interventions | Imipenem 0.5 g i.v. infused over 0.5 h q6h vs imipenem 1 g i.v. infused over 4 h q8h; treatment duration 3 to 5 days |
| Outcomes | Primary outcome: pharmacokinetic/pharmacodynamic parameters |
| Starting date | November 2011 |
| Contact information | Sutep Jaruratanasirikul (jasutep@medicine.psu.ac.th) |
| Notes | Estimated study completion date December 2012 |
NCT01577368.
| Trial name or title | Efficacy and safety of piperacillin‐tazobactam continuous infusion vs intermittent infusion for complicated nosocomialPseudomonas aeruginosa infection or suspected infection |
| Methods | Randomised, double‐blind, multicentre trial |
| Participants | Estimated enrollment = 400, adults with complicated or nosocomial infection with or without isolation of Pseudomonas aeruginosa |
| Interventions | Piperacillin‐tazobactam 2 g i.v. loading dose, then continuous infusion 8 g i.v. q24h vs piperacillin‐tazobactam intermittent infusion 4 g i.v. q8h; treatment duration 14 days |
| Outcomes | Primary outcome: proportion of participants with satisfactory clinical response (cure or improvement) at the end of piperacillin‐tazobactam treatment Secondary outcomes: proportion of participants with clinical response at 3 days, proportion of participants with microbiological response, time to defervescence, time to clinical cure, mortality, proportion of participants with adverse effects |
| Starting date | May 2011 |
| Contact information | Maria V Gil‐Navarro (maria.gil.sspa@juntadeandalucia.es) Roberto Marin‐Gill (roberto.marin.sspa@juntadeandalucia.es) |
| Notes | Estimated study completion date October 2012 |
NCT01667094.
| Trial name or title | Continuous infusion anti‐pseudomonal beta‐lactams for the treatment of acute, infective pulmonary exacerbations in cystic fibrosis: a prospective randomised controlled trial |
| Methods | Randomised, open‐label trial |
| Participants | Estimated enrolment = 120, adults with cystic fibrosis with Pseudomonas aeruginosa isolated in sputum within the past 12 months with an acute infective exacerbation |
| Interventions | Short infusion over 30 minutes of either cefepime 1 g i.v. q8h OR ceftazidime 2 g i.v. q8h OR meropenem 1 g i.v. q8h OR piperacillin‐tazobactam 4.5 g i.v. q8h OR ticarcillin‐clavulanate 3.1 g i.v. q6h vs continuous infusion of either cefepime 500 mg i.v. loading dose, then 1.5 g i.v. infused over 12 h q12h OR ceftazidime 1 g i.v. loading dose, then 3 g i.v. infused over 12 h q12h OR meropenem 500 mg i.v. loading dose, then 1.5 g i.v. infused over 12 h q12h OR piperacillin‐tazobactam 1.55 g i.v. loading dose, then 13.5 g i.v. infused over 24 h |
| Outcomes | Primary outcome: cystic fibrosis questionnaire‐revised respiratory component respiratory symptom score Secondary outcomes: change in cystic fibrosis questionnaire‐revised respiratory symptom score, lung function testing/FEV1, C‐reactive peptide, quantitative bacterial load in sputum, time above MIC, antibiotic stability, Pseudomonas aeruginosa virulence gene determinants |
| Starting date | September 2012 |
| Contact information | Katherine Langan (Katherine.Langan@monash.edu.au) |
| Notes | Estimated study completion date January 2015 |
NCT01720940.
| Trial name or title | Reducing nephrotoxicity of vancomycin: a prospective, randomised study of continuous vs intermittent infusion of vancomycin |
| Methods | Randomised, open‐label trial |
| Participants | Estimated enrolment = 220, adults aged 21 to 80 years with documented infection requiring prolonged vancomycin therapy |
| Interventions | Vancomycin 24 h continuous i.v. infusion vs vancomycin intermittent infusion |
| Outcomes | Primary outcome: nephrotoxicity as defined by the acute kidney injury network criteria using only serum creatinine criteria Secondary outcome: biomarkers for detection of early nephrotoxicity (serum and urine NGAL and cystatin C) |
| Starting date | October 2012 |
| Contact information | Shire Yang Tan (shire_yang_tan@nuhs.edu.sg) |
| Notes | Estimated study completion date September 2015 |
Differences between protocol and review
Revised wording of review objective.
Added clarification in the review objective for the term continuous intravenous infusions.
Added clarification that cross‐over studies were excluded from the review.
Revised definition of adult to age 18 or older instead of older than 18 years.
Four independent authors (JS, EW, AT, MW) screened the titles and abstracts of the search results.
Data extraction and assessment of risk of bias of included studies were performed by JS, EW, and AT.
Default analysis was conducted with a random‐effects model instead of a fixed‐effect model (more conservative to assume underlying heterogeneity in included studies when different antibiotics, participant populations, and infection types were reviewed).
Added that GRADEpro was used to generate the 'Summary of findings' table.
Performed additional sensitivity analyses to determine the impact of studies using extended interval antibiotic infusions (instead of continuous infusions) and the impact of the use of open‐label antibiotics.
Contributions of authors
Jennifer Shiu selected trials for inclusion, extracted data, critically appraised included studies, analysed data, and wrote and revised the final report.
Erica Wang selected trials for inclusion, extracted data, critically appraised included studies, and revised the final report.
Aaron Tejani selected trials for inclusion, extracted data, critically appraised included studies, and revised the final report.
Michael Wasdell selected trials for inclusion and revised the final report.
Declarations of interest
All authors: none known.
Aaron Tejani: no direct or indirect association with the pharmaceutical industry in the past 8 years.
Edited (no change to conclusions)
References
References to studies included in this review
Adembri 2008 {published data only}
- Adembri C, Fallani S, Cassetta MI, Arrigucci S, Ottaviano A, Pecile P, Mazzei T, Gaudio R, Novelli A. Linezolid pharmacokinetic/pharmacodynamic profile in critically ill septic patients: intermittent versus continuous infusion. International Journal of Antimicrobial Agents 2008;31(2):122‐9. [DOI] [PubMed] [Google Scholar]
Angus 2000 {published data only}
- Angus BJ, Smith MD, Suputtamongkol Y, Mattie H, Walsh AL, Wuthiekanun V, Chaowagul W, White NJ. Pharmacokinetic‐pharmacodynamic evaluation of ceftazidime continuous infusion vs intermittent bolus injection in septicaemic melioidosis. British Journal of Clinical Pharmacology 2000;49(5):445‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bodey 1979 {published data only}
- Bodey GP, Ketchel SJ, Rodriguez V. A randomized study of carbenicillin plus cefamandole or tobramycin in the treatment of febrile episodes in cancer patients. American Journal of Medicine 1979;67(4):608‐16. [DOI] [PubMed] [Google Scholar]
Buck 2005 {published data only}
- Buck C, Bertram N, Ackermann T, Sauerbruch T, Derendorf H, Paar WD. Pharmacokinetics of piperacillin‐tazobactam: intermittent dosing versus continuous infusion. International Journal of Antimicrobial Agents 2005;25(1):62‐7. [DOI] [PubMed] [Google Scholar]
Chytra 2012 {published and unpublished data}
- Chytra I, Stepan M, Benes J, Pelnar P, Zidkova A, Bergerova T, Pradl R, Kasal E. Clinical and microbiological efficacy of continuous versus intermittent application of meropenem in critically ill patients: a randomized open‐label controlled trial. Critical Care 2012;16:R113 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
Cousson 2005 {published and unpublished data}
- Cousson J, Floch T, Vernet‐Garnier V, Appriou M, Petit JS, Jovenin N, Lamiable D, Hoizey G. Pharmacodynamic interest of ceftazidime continuous infusion vs intermittent bolus administration in patients with severe nosocomial pneumonia (French). Pathologie Biologie 2005;53:546‐50. [DOI] [PubMed] [Google Scholar]
DeJongh 2008 {published and unpublished data}
- Jongh R, Hens R, Basma V, Mouton JW, Tulkens PM, Carryn S. Continuous versus intermittent infusion of temocillin, a directed spectrum penicillin for intensive care patients with nosocomial pneumonia: stability, compatibility, population pharmacokinetic studies and breakpoint selection. Journal of Antimicrobial Chemotherapy 2008;61(2):382‐8. [DOI] [PubMed] [Google Scholar]
Feld 1977 {published data only}
- Feld R, Valdivieso M, Bodey GP, Rodriguez V. A comparative trial of sisomicin therapy by intermittent versus continuous infusion. The American Journal of the Medical Sciences 1977;274(2):179‐88. [DOI] [PubMed] [Google Scholar]
Feld 1984 {published data only}
- Feld R, Rachlis A, Tuffnell PG, Duncan I, Moran L, Pinfold P, DeBoer G. Empiric therapy for infections in patients with granulocytopenia. Continuous v interrupted infusion of tobramycin plus cefamandole. Archives of Internal Medicine 1984;144(5):1005‐10. [PubMed] [Google Scholar]
Georges 2005 {published data only}
- Georges B, Conil JM, Cougot P, Decun JF, Archambaud M, Seguin T, Chabanon G, Virenque C, Houin G, Saivin S. Cefepime in critically ill patients: continuous infusion vs. an intermittent dosing regimen. International Journal of Clinical Pharmacology and Therapeutics 2005;43(8):360‐9. [DOI] [PubMed] [Google Scholar]
Hanes 2000 {published data only}
- Hanes SD, Wood GC, Herring V, Croce MA, Fabian TC, Pritchard E, Boucher BA. Intermittent and continuous ceftazidime infusion for critically ill trauma patients. The American Journal of Surgery 2000;179(6):436‐40. [DOI] [PubMed] [Google Scholar]
Lagast 1983 {published data only}
- Lagast H, Meunier‐Carpentier F, Klastersky J. Treatment of gram‐negative bacillary septicemia with cefoperazone. European Journal of Clinical Microbiology 1983;2(6):554‐8. [DOI] [PubMed] [Google Scholar]
Lau 2006 {published data only}
- Lau WK, Mercer D, Itani KM, Nicolau DP, Kuti JL, Mansfield D, Dana A. Randomized, open‐label, comparative study of piperacillin‐tazobactam administered by continuous infusion versus intermittent infusion for treatment of hospitalized patients with complicated intra‐abdominal infection. Antimicrobial Agents and Chemotherapy 2006;50(11):3556‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lipman 1999 {published data only}
- Lipman J, Gomersall CD, Gin T, Joynt GM, Young RJ. Continuous infusion ceftazidime in intensive care: a randomized controlled trial. Journal of Antimicrobial Chemotherapy 1999;43(2):309‐11. [DOI] [PubMed] [Google Scholar]
Lubasch 2003 {published data only}
- Lubasch A, Luck S, Hartmut L, Mauch H, Lorenz J, Bolcskei P, Welte T. Optimizing ceftazidime pharmacodynamics in patients with acute exacerbation of severe chronic bronchitis. Journal of Antimicrobial Chemotherapy 2003;51:659‐64. [DOI] [PubMed] [Google Scholar]
Nicolau 1999a {published data only}
- Nicolau DP, McNabb J, Lacy MK, Li J, Quintiliani R, Nightingale CH. Pharmacokinetics and pharmacodynamics of continuous and intermittent ceftazidime during the treatment of nosocomial pneumonia. Clinical Drug Investigation 1999;18(2):133‐9. [Google Scholar]
Nicolau 1999b {published data only}
- Nicolau DP, Lacy MK, McNabb J, Quintiliani R, Nightingale CH. Pharmacokinetics of continuous and intermittent ceftazidime in intensive care unit patients with nosocomial pneumonia. Infectious Diseases in Clinical Practice 1999;8(1):45‐9. [Google Scholar]
Nicolau 2001 {published data only}
- Nicolau DP, McNabb J, Lacy MK, Quintiliani R, Nightingale CH. Continuous versus intermittent administration of ceftazidime in intensive care unit patients with nosocomial pneumonia. International Journal of Antimicrobial Agents 2001;17(6):497‐504. [DOI] [PubMed] [Google Scholar]
Okimoto 2009 {published data only}
- Okimoto N, Ishiga M, Nanba F, Kibayashi T, Kishimoto M, Kurihara T, Honda Y, Asaoka N, Tamada S. Clinical effects of continuous infusion and intermittent infusion of meropenem on bacterial pneumonia in the elderly (Japanese). Nihon Kokyuki Gakkai Zasshi 2009;47(7):553‐7. [PubMed] [Google Scholar]
Pedeboscq 2001 {published and unpublished data}
- Pedeboscq S, Dubau B, Frappier S, Hernandez V, Veyssieres D, Winnock S, Pometan JP. Comparison of 2 administration protocols (continuous or discontinuous) of a time‐dependent antibiotic, Tazocin (French). Pathologie Biologie (Paris) 2001;49(7):540‐7. [DOI] [PubMed] [Google Scholar]
Rafati 2006 {published data only}
- Rafati MR, Rouini MR, Mojtahedzadeh M, Najafi A, Tavakoli H, Gholami K, Fazeli MR. Clinical efficacy of continuous infusion of piperacillin compared with intermittent dosing in septic critically ill patients. International Journal of Antimicrobial Agents 2006;28(2):122‐7. [DOI] [PubMed] [Google Scholar]
Roberts 2007 {published and unpublished data}
- Roberts JA, Boots R, Rickard CM, Thomas P, Quinn J, Roberts DM, Richards B, Lipman J. Is continuous infusion ceftriaxone better than once‐a‐day dosing in intensive care? A randomized controlled pilot study. Journal of Antimicrobial Chemotherapy 2007;59(2):285‐91. [DOI] [PubMed] [Google Scholar]
Roberts 2009b {published and unpublished data}
- Roberts JA, Kirkpatrick CMJ, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Meropenem dosing in critically ill patients with sepsis and without renal dysfunction: intermittent bolus versus continuous administration? Monte Carlo dosing simulations and subcutaneous tissue distribution. Journal of Antimicrobial Chemotherapy 2009;64(1):142‐50. [DOI] [PubMed] [Google Scholar]
Roberts 2009c {published and unpublished data}
- Roberts JA, Roberts MS, Robertson TA, Dalley AJ, Lipman J. Piperacillin penetration into tissue of critically ill patients with sepsis‐bolus versus continuous administration?. Critical Care Medicine 2009;37(3):926‐33. [DOI] [PubMed] [Google Scholar]
Roberts 2010 {published and unpublished data}
- Roberts JA, Kirkpatrick CMJ, Roberts MS, Dalley AJ, Lipman J. First‐dose and steady‐state population pharmacokinetics and pharmacodynamics of piperacillin by continuous or intermittent dosing in critically ill patients with sepsis. International Journal of Antimicrobial Agents 2010;35:156‐63. [DOI] [PubMed] [Google Scholar]
Sakka 2007 {published data only}
- Sakka SG, Glauner AK, Bulitta JB, Kinzig‐Schippers M, Pfister W, Drusano GL, Sorgel F. Population pharmacokinetics and pharmacodynamics of continuous versus short‐term infusion of imipenem‐cilastatin in critically ill patients in a randomized, controlled trial. Antimicrobial Agents and Chemotherapy 2007;51(9):3304‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Zanten 2007 {published and unpublished data}
- Zanten ARH, Oudijk M, Nohlmans‐Paulssen MKE, Meer YG, Girbes ARJ, Polderman KH. Continuous vs. intermittent cefotaxime administration in patients with chronic obstructive pulmonary disease and respiratory tract infections: pharmacokinetics/pharmacodynamics, bacterial susceptibility and clinical efficacy. British Journal of Clinical Pharmacology 2007;63(1):100‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wright 1979 {published data only}
- Wright JP, Potgieter PD, Forder AA, Botha P, Brinkworth G, Elisha G, Ferguson AD. Gentamicin and penicillin in the treatment of severe respiratory infections. South African Medical Journal 1979;55(6):197‐200. [PubMed] [Google Scholar]
Wysocki 2001 {published and unpublished data}
- Wysocki M, Delatour F, Faurisson F, Rauss A, Pean Y, Misset B, Thomas F, Timsit JF, Similowski T, Mentec H, Mier L, Dreyfuss D, and the study group. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: prospective multicenter randomized study. Antimicrobial Agents and Chemotherapy 2001;45(9):2460‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Adembri 2010 {published data only}
- Adembri C, Ristori R, Chelazzi C, Arrigucci S, Cassetta MI, Gaudio AR, Novelli A. Cefazolin bolus and continuous administration for elective cardiac surgery: improved pharmacokinetic and pharmacodynamic parameters. Journal of Thoracic and Cardiovascular Surgery 2010;140:471‐5. [DOI] [PubMed] [Google Scholar]
Ambrose 1998 {published data only}
- Ambrose PG, Quindliani R, Nightingale CH, Nicolau DP. Continuous vs. intermittent infusion of cefuroxime for the treatment of community‐acquired pneumonia. Infectious Diseases in Clinical Practice 1998;7(9):463‐70. [Google Scholar]
Benko 1996 {published data only}
- Benko A, Cappelletty D, Kruse J, Rybak M. Continuous infusion versus intermittent administration of ceftazidime in critically ill patients with suspected gram‐negative infections. Antimicrobial Agents and Chemotherapy 1996;40(3):691‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bosso 1999 {published data only}
- Bosso J, Bonapace C, Flume P, White R. A pilot study of the efficacy of constant‐infusion ceftazidime in the treatment of endobronchial infections in adults with cystic fibrosis. Pharmacotherapy 1999;19(5):620‐26. [DOI] [PubMed] [Google Scholar]
Buijk 2002 {published data only}
- Buijk SLCE, Gyssens IC, Mouton JW, Vliet A, Verbrugh HA. Pharmacokinetics of ceftazidime in serum and peritoneal exudate during continuous versus intermittent administration to patients with severe intra‐abdominal infections. Journal of Antimicrobial Chemotherapy 2002;49(1):121‐8. [DOI] [PubMed] [Google Scholar]
Burgess 2002 {published data only}
- Burgess D, Waldrep T. Pharmacokinetics and pharmacodynamics of piperacillin/tazobactam when administered by continuous infusion and intermittent dosing. Clinical Therapeutics 2002;24(7):1090‐1104. [DOI] [PubMed] [Google Scholar]
DeRyke 2006 {published data only}
- DeRyke CA, Kuti JL, Mansfield D, Dana A, Nicolau DP. Pharmacoeconomics of continuous versus intermittent infusion of piperacillin‐tazobactam for the treatment of complicated intraabdominal infection. American Journal of Health System Pharmacy 2006;63(8):750‐5. [DOI] [PubMed] [Google Scholar]
Georges 1999 {published data only}
- Georges B, Archambaud M, Saivin S, Decun J, Cougot P, Mazerolles M, Andrieu P, Suc C, Houin G, Chabanon G, Virenque C. Continuous versus intermittent cefepime infusion in critical care. Preliminary results (French). Pathologie Biologie 1999;47(5):483‐85. [PubMed] [Google Scholar]
Grant 2002 {published data only}
- Grant E, Kuti J, Nicolau D, Nightingale C, Quintiliani R. Clinical efficacy and pharmacoeconomics of a continuous‐infusion piperacillin‐tazobactam program in a large community teaching hospital. Pharmacotherapy 2002;22(4):471‐83. [DOI] [PubMed] [Google Scholar]
Hutschala 2009 {published data only}
- Hutschala D, Kinstner C, Skhirdladze K, Thalhammer F, Muller M, Tschernko E. Influence of vancomycin on renal function in critically ill patients after cardiac surgery. Anesthesiology 2009;111:356‐65. [DOI] [PubMed] [Google Scholar]
James 1996 {published data only}
- James J, Palmer S, Levine D, Rybak M. Comparison of conventional dosing versus continuous‐infusion vancomycin therapy for patients with suspected or documented gram‐positive infections. Antimicrobial Agents and Chemotherapy 1996;40(3):696‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaruratanasirikul 2002 {published data only}
- Jaruratanasirikul S, Sriwiriyajan S, Ingviya N. Continuous infusion versus intermittent administration of cefepime in patients with gram‐negative bacilli bacteraemia. Journal of Pharmacy and Pharmacology 2002;54:1693‐96. [DOI] [PubMed] [Google Scholar]
Jaruratanasirikul 2005 {published data only}
- Jaruratanasirikul S, Sriwiriyajan S, Punyo J. Comparison of the pharmacodynamics of meropenem in patients with ventilator‐associated pneumonia following administration by 3‐hour infusion or bolus injection. Antimicrobial Agents and Chemotherapy 2005;49(4):1337‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaruratanasirikul 2009 {published data only}
- Jaruratanasirikul S, Sudsai T. Comparison of the pharmacodynamics of imipenem in patients with ventilator‐associated pneumonia following administration by 2 or 0.5h infusion. Journal of Anitimicrobial Chemotherapy 2009;63:560‐63. [DOI] [PubMed] [Google Scholar]
Jaruratanasirikul 2010 {published data only}
- Jaruratanasirikul S, Julamanee J, Sudsai T, Saengsuwan P, Jullangkoon M, Ingviya N, Jarumanokul R. Comparison of continuous infusion versus intermittent infusion of vancomycin in patients with methicillin‐resistant Staphylococcus aureus. Journal of the Medical Association of Thailand 2010;93(2):172‐6. [PubMed] [Google Scholar]
Kirkpatrick 2001 {published data only}
- Kickpatrick C, Howard G, Vella‐Brincat J. Comment: serum concentrations of cefuroxime after continuous infusion in coronary bypass graft patients. The Annals of Pharmacotherapy 2001;53:1295‐96. [DOI] [PubMed] [Google Scholar]
Klepser 1998 {published data only}
- Klepser M, Patel K, Nicolau D, Quintiliani R, Nightingale C. Comparison of bactericidal activities of intermittent and continuous infusion dosing of vancomycin against methicillin‐resistant staphylococcus aureus and enterococcus faecalis. Pharmacotherapy 1998;18(5):1069‐74. [PubMed] [Google Scholar]
Kojika 2005 {published data only}
- Kojika M, Sato N, Hakozaki M, Suzuki Y, Takahasi G, Endo S, Suzuki K, Wakabayasi G. A preliminary study of the administration of carbapenem antibiotics in sepsis patients on the basis of administration time. The Japanese Journal of Antibiotics 2005;58(5):452‐7. [PubMed] [Google Scholar]
Langgartner 2007 {published data only}
- Langgartner J, Lehn N, Gluck T, Herzig H, Kees F. Comparison of the pharmacokinetics of piperacillin and sulbactam during intermittent and continuous intravenous infusion. Chemotherapy 2007;53:370‐77. [DOI] [PubMed] [Google Scholar]
Li 2005 {published data only}
- Li C, Kuti JL, Nightingale CH, Mansfield DL, Dana A, Nicolau DP. Population pharmacokinetics and pharmacodynamics of piperacillin/tazobactam in patients with complicated intra‐abdominal infection. Journal of Antimicrobial Chemotherapy 2005;56(2):388‐95. [DOI] [PubMed] [Google Scholar]
Lorente 2006 {published data only}
- Lorente L, Lorenzo L, Martin MM, Jimenez A, Mora ML. Meropenem by continuous versus intermittent infusion in ventilator‐associated pneumonia due to gram‐negative bacilli. Annals of Pharmacotherapy 2006;40:219‐23. [DOI] [PubMed] [Google Scholar]
Martin 1998 {published data only}
- Martin C, Cotin A, Giraud A, Beccani‐Argeme M, Alliot P, Mallet MN, Argeme M. Comparison of concentrations of sulbactam‐ampicillin administered by bolus injections or bolus plus continuous infusion in tissues of patients undergoing colorectal surgery. Antimicrobial Agents and Chemotherapy 1998;42(5):1093‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
McNabb 2001 {published data only}
- McNabb JJ, Nightingale CH, Quintiliani R, Nicolau DP. Cost‐effectiveness of ceftazidime by continuous infusion versus intermittent infusion for nosocomial pneumonia. Pharmacotherapy 2001;21(5):549‐55. [DOI] [PubMed] [Google Scholar]
Nicasio 2007 {published data only}
- Nicasio A, Quintiliani R, DeRyke C, Kuti J, Nicolau D. Treatment of serratia marcescens meningitis with prolonged infusion of meropenem. The Annals of Pharmacotherapy 2007;41:1077‐81. [DOI] [PubMed] [Google Scholar]
Pass 2001 {published data only}
- Pass SE, Miyagawa CI, Healy DP, Ivey TD. Serum concentrations of cefuroxime after continuous infusion in coronary bypass graft patients. Annals of Pharmacotherapy 2001;35:409‐13. [DOI] [PubMed] [Google Scholar]
Schuster 2009 {published data only}
- Schuster KM, Wilson D, Schulman CI, Pizano LR, Ward CG, Namias N. Continuous infusion oxacillin for the treatment of burn wound cellulitis. Surgical Infections 2009;10(1):41‐5. [DOI] [PubMed] [Google Scholar]
Seguin 2009 {published data only}
- Seguin P, Verdier M, Chanavaz C, Engrand C, Laviolle B, Donnio P, Malledant Y. Plasma and peritoneal concentration following continuous infusion of cefotaxime in patients with secondary peritonitis. Journal of Antimicrobial Chemotherapy 2009;63:564‐67. [DOI] [PubMed] [Google Scholar]
Thalhammer 1999 {published data only}
- Thalhammer F, Traunmuller F, Menyawi I, Frass M, Hollenstein U, Locker G, Stoiser B, Staudinger T, Thalhammer‐Scherrer R, Burgmann H. Continuous infusion versus intermittent administration of meropenem in critically ill patients. Journal of Antimicrobial Chemotherapy 1999;43:523‐27. [DOI] [PubMed] [Google Scholar]
Vinks 2003 {published data only}
- Vinks A, Hollander J, Overbeek S, Jelliffe R, Mouton J. Population pharmacokinetic analysis of nonlinear behavior of piperacillin during intermittent of continuous infusion in patients with cystic fibrosis. Antimicrobial Agents and Chemotherapy 2003;47(2):541‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vuagnat 2004 {published data only}
- Vuagnat A, Stern R, Lotthe A, Schuhmacher H, Duong M, Hoffmeyer P, Bernard L. High dose vancomycin for osteomyelitis: continuous vs. intermittent infusion. Journal of Clinical Pharmacy and Therapeutics 2004;29:351‐57. [DOI] [PubMed] [Google Scholar]
Waltrip 2002 {published data only}
- Waltrip T, Lewis R, Young V, Farmer M, Clayton S, Myers S, Gray L, Galandiuk S. A pilot study to determine the feasibility of continuous cefazolin infusion. Surgical Infections 2002;3(1):5‐9. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Cousson 2010 {unpublished data only}
- Cousson J, Floch T, Hoizey G, Nicolai F, Guillard T, Vernet‐Garnier V. Comparison of pharmacodynamic interest of ceftazidime continuous infusion vs intermittent bolus administration in epithelial lining fluid concentrations of patients with severe nosocomial pneumonia. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (poster) 2010.
NCT00891423 {unpublished data only}
- Johnson P. A Randomised controlled crossover pilot study of meropenem standard 30 minute infusion versus prolonged 3 hour infusion in intensive care patients. Available at http://www.clinicaltrials.gov, last updated July 2012. Accessed November 14, 2012.
NCT01158937 {unpublished data only}
- Cortes D. Pharmacokinetic study of extended infusion meropenem in adult cystic fibrosis patients with exacerbation of pulmonary infection. Available at http://www.clinicaltrials.gov, last updated June 2012. Accessed November 14, 2012.
NCT01198925 {unpublished data only}
- Decruyenaere J. Assessment of the optimal dosing of piperacillin‐tazobactam in intensive care unit patients: extended versus continuous infusion. Available at http://www.clinicaltrials.gov, last updated July 2012. Accessed November 14, 2012.
NCT01379157 {unpublished data only}
- Jaruratanasirikul S. The pharmacodynamics of imipenem in critically ill patients with ventilator‐associated pneumonia following administration by 4h or 0.5h infusion. Available at http://www.clinicaltrials.gov, last updated December 2011. Accessed November 14, 2012.
NCT01577368 {unpublished data only}
- Gil‐Navarro MV. Efficacy and safety of piperacillin‐tazobactam continuous infusion vs intermittent infusion for complicated or nosocomial Pseudomonas aeruginosa infection or suspected infection. Available at http://www.clinicaltrials.gov, last updated April 2012. Accessed November 14, 2012.
NCT01667094 {unpublished data only}
- Peleg A. Continuous infusion anti‐pseudomonal beta‐lactams for the treatment of acute, infective pulmonary exacerbations in cystic fibrosis: a prospective randomised controlled trial. Available at http://www.clinicaltrials.gov, last updated August 2012. Accessed November 14, 2012.
NCT01720940 {unpublished data only}
- Fisher D. Reducing nephrotoxicity of vancomycin: a prospective, randomised study of continuous versus intermittent infusion of vancomycin. Available at http://www.clinicaltrials.gov, last updated November 2012. Accessed November 14, 2012.
Additional references
Ariano 2010
- Ariano R, Zelenitsky S, McCormack J. Should IV antibiotics be administered by prolonged infusion?. Canadian Journal of Hospital Pharmacy 2010;63(3):246‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Balshem 2011
- Balshem H, Helfand M, Schunemann HJ, Oxman AD, Kunz R, Brozek J, Vist GE, Flack‐Ytter Y, Meerpohl J, Norris S, Guyall GH. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64:401‐06. [DOI] [PubMed] [Google Scholar]
Craig 1998
- Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clinical Infectious Diseases 1998;26(1):1‐10. [DOI] [PubMed] [Google Scholar]
Drusano 2004
- Drusano GL. Antimicrobial pharmacodynamics: critical interactions of "bug and drug". Nature Reviews Microbiology 2004;2(4):289‐300. [DOI] [PubMed] [Google Scholar]
Florea 2003
- Florea NR, Kotapati S, Kuti JL, Geissler EC, Nightingale CH, Nicolau DP. Cost analysis of continuous versus intermittent infusion of piperacillin–tazobactam: a time–motion study. American Journal of Health System Pharmacy 2003;60(22):2321‐7. [DOI] [PubMed] [Google Scholar]
Ghafourian 2011
- Ghafourian S, Sekawi Z, Sadeghifard N, Mohebi R, Neela VK, Maleki A, Hematian A, Rahbar M, Raftari M, Ranjbar R. The prevalence of ESBLs producing Klebsiella pneumoniae isolates in some major hospitals, Iran. The Open Microbiology Journal 2011;5:91‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- Version 3.2 for Windows. Brozek J, Oxman A, Schunemann H. GRADEpro (GRADEprofiler).. The Cochrane Collaboration, 2008.
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, Norris S, Falck‐Ytter Y, Glasziou P, deBeer H, Jaeschke R, Rind D, Meerpohl J, Dahm P, Schunemann HJ. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64:383‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. British Medical Journal 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2009. Available from www.cochrane‐handbook.org.
Hitt 1997
- Hitt CM, Nighingale CH, Quintilliani R, Nicolau DP. Cost comparison of single daily IV doses of ceftriaxone versus continuous infusion of cefotaxime. American Journal of Health System Pharmacy 1997;54(14):1614‐8. [DOI] [PubMed] [Google Scholar]
Kasiakou 2005a
- Kasiakou SK, Lawrence KR, Choulis N, Falagas ME. Continuous versus intermittent intravenous administration of antibacterials with time‐ dependent action: a systematic review of pharmacokinetic and pharmacodynamic parameters. Drugs 2005;65(17):2499‐511. [DOI] [PubMed] [Google Scholar]
Kasiakou 2005b
- Kasiakou SK, Sermaides GJ, Michalopoulos A, Soteriades ES, Falagas ME. Continuous versus intermittent intravenous administration of antibiotics: a meta‐analysis of randomised controlled trials. Lancet Infectious Diseases 2005;5(9):581‐9. [DOI] [PubMed] [Google Scholar]
Lipman 2001
- Lipman J, Wallis SC, Rickard CM, Fraenkel D. Low cefpirome levels during twice daily dosing in critically ill septic patients: pharmacokinetic modelling calls for more frequent dosing. Intensive Care Medicine 2001;27(2):363‐70. [DOI] [PubMed] [Google Scholar]
Lodise 2006
- Lodise TP, Lomaestro BM, Drusano GL, Society of Infectious Diseases Pharmacists. Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on B‐lactam antibiotics. Pharmacotherapy 2006;26(9):1320‐32. [DOI] [PubMed] [Google Scholar]
Lodise 2007
- Lodise TP Jr, Lomaestro B, Drusano GL. Piperacillin‐tazobactam for pseudomonas aeruginosa Infection: clinical implications of an extended‐infusion dosing strategy. Clinical Infectious Diseases 2007;44(3):357‐63. [DOI] [PubMed] [Google Scholar]
McKinnon 2008
- McKinnon PS, Paladino JA, Schentag JJ. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. International Journal of Antimicrobial Agents 2008;31(4):345‐51. [DOI] [PubMed] [Google Scholar]
Meyer 2011
- Meyer E, Ziegler R, Mattner F, Schwab F, Gastmeier P, Martin M. Increase of patients co‐colonised or co‐infected with methicillin‐resistant Staphylococcus aureus, vancomycin‐resistant Enterococcus faecium or extended‐spectrum B‐lactamase‐producing Enterobacteriaceae. Infection 2011;39(6):501‐6. [DOI] [PubMed] [Google Scholar]
Moore 1987
- Moore RD, Lietman PS, Smith CR. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. Journal of Infectious Diseases 1987;155(1):93‐9. [DOI] [PubMed] [Google Scholar]
Neidell 2012
- Neidell MJ, Cohen B, Furuya Y, Hill J, Jeon CY, Glied S, Larson EL. Costs of healthcare‐ and community‐associated infections with antimicrobial‐resistance versus susceptible organisms. Clinical Infectious Diseases 2012;Jun 14:(epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
Nicolau 1996
- Nicolau DP, Nightingale CH, Banevicius MA, Fu Q, Quintiliani R. Serum bactericidal activity of ceftazidime: continuous infusion versus intermittent infections. Antimicrobial Agents and Chemotherapy 1996;40(1):61‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Roberts 2006
- Roberts JA, Lipman J. Antibacterial dosing in intensive care: pharmacokinetics, degree of disease and pharmacodynamics of sepsis. Clinical Pharmacokinetics 2006;45(8):755‐73. [DOI] [PubMed] [Google Scholar]
Roberts 2008
- Roberts JA, Lipman J, Blot S, Rello J. Better outcomes through continuous infusion of time‐dependent antibiotics to critically ill patients?. Current Opinion in Critical Care 2008;14(4):390‐6. [DOI] [PubMed] [Google Scholar]
Roberts 2009a
- Roberts JA, Webb S, Paterson D, Ho KM, Lipman J. A systematic review on clinical benefits of continuous administration of B‐lactam antibiotics. Critical Care Medicine 2009;37(6):2071‐8. [DOI] [PubMed] [Google Scholar]
Rosenthal 2012
- Rosenthal VD, Bijie H, Maki DG, Mehta Y, Apisarnthanarak A, Medeiros EA, Leblebicioglu H, Fisher D, Alvarez‐Moreno C, Khader IA, Martinez M, Cuellar LE, Navoa‐Ng JA, Abouqal R, Garcell HG, Mitrev Z, Garcia MCP, Hamdi A, Duenas L, Cancel E, Gurskis V, Rasslan O, Ahmed A, Kanj SS, Ugalde OC, Mapp T, Raka L, Meng CY, Thu LA, Ghazal S, Gikas A, Narvaez LP, Mejia N, Hadjieva N, Elanbya MOG, Siritt MEG, Jayatilleke K, INICC members. International nosocomial infection control consortium (INICC) report, data summary of 36 countries, for 2004‐2009. American Journal of Infection Control 2012;40(5):396‐407. [DOI] [PubMed] [Google Scholar]
Sunenshine 2007
- Sunenshine RH, Wright MO, Maragakis LL, Harris AD, Song X, Hebden J, Cosgrove SE, Anderson A, Carnell J, Jernigan DB, Kleinbaum DG, Peri TM, Standiford HC, Srinivasan A. Multidrug‐resistant Acinetobacter infection mortality rate and length of hospitalization. Emerging Infectious Diseases 2007;13(1):97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tamma 2011
- Tamma PD, Putcha N, Suh YD, Arendonk KJ, Rinke ML. Does prolonged B‐lactam infusions improve clinical outcomes compared to intermittent infusions? A meta‐analysis and systematic review of randomized, controlled trials. BMC Infectious Diseases 2011;11:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Zanten 2009
- Zanten AR. The jury is still out on continuous infusion of B‐lactam antibiotics in intensive care patients. Critical Care Medicine 2009;37(6):2137‐8. [DOI] [PubMed] [Google Scholar]
Viaene 2002
- Viaene E, Chanteux H, Servais H, Mingeot‐Leclercq M, Tulkens P. Comparative stability studies of antipseudomonal B‐Lactams for potential administration through portable elastomeric pumps (home therapy for cystic fibrosis patients) and motor‐operated syringes (intensive care units). Antimicrobial Agents and Chemotherapy 2002;46(8):2327‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Yu 2010
- Yu IS, Tejani AM, Wasdell M. Continuous versus intermittent infusions of antibiotics for the treatment of severe acute infections (Protocol). Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD008481] [DOI] [PMC free article] [PubMed] [Google Scholar]