

Abstract
Amelogenesis imperfecta (AI) is a collection of rare genetic disorders affecting the quantity and/or quality of the tooth enamel. AI can be classified into three major types according to the clinical phenotype: hypoplastic, hypocalcified, and hypomatured. Among them, the hypocalcified type shows the weakest physical properties, leaving rough and discolored enamel surfaces after tooth eruption. To date, mutations in the FAM83H gene are responsible for the autosomal-dominant hypocalcified AI. In this study, we recruited a four-generation Colombian family with hypocalcified AI and identified a recurrent nonsense mutation in the FAM83H gene (NM_198488.5:c.1289C>A, p.(Ser430 *)) by candidate gene sequencing. Cephalometric analyses revealed the anterior open bite that occurred in the proband is not correlated with the AI in this family.
Keywords: hereditary enamel defects, FAM83H, hypocalcified, anterior open bite, tooth impaction, crown resorption
1. Introduction
Amelogenesis imperfecta (AI) is a rare genetic disorder affecting the tooth enamel [1]. AI is heterogeneous in terms of etiology and clinical phenotype. To date, more than 20 genes are known to be involved in the molecular pathogenesis of AI [2,3]. Clinically, AI can be classified as one or mixed types of hypoplastic, hypomature, and hypocalcified enamel [4]. Various surface characteristics make it even more heterogeneous.
FAM83H mutations have been found to cause autosomal-dominant hypocalcified AI (ADHCAI) [5,6]. Other candidate genes for ADHCAI have not been discovered. FAM83H mutations causing ADHCAI are nonsense and frameshift mutations in the last exon, which generate truncated proteins that escape from nonsense-mediated mRNA decay [7]. Interestingly, the normal FAM83H is shown to be dispensable for proper enamel formation [8]. The truncated protein translocates into the nucleus, while its wild-type counterpart locates into the cytoplasm [9]. Despite the fact that the functional role of FAM83H remains to be elucidated, it has been demonstrated with a mouse model that the mutation mechanism causing ADHCAI is neomorphic [10].
In this study, we recruited a four-generation Colombian family with hypocalcified AI and sequenced the FAM83H gene to identify a disease-causing mutation. Cephalometric analyses were performed to characterize whether there is a common craniofacial skeletal feature among the affected family members in the family.
2. Materials and Methods
We recruited an extended Colombian AI family. The study protocol was reviewed and approved by the institutional review board of the Pontificia Universidad Javeriana and the Seoul National University Dental Hospital. Informed consent was obtained from all subjects participating in this study with an understanding of the nature of the study.
The family history was investigated to build the pedigree. Clinical examinations were performed for the participating family members to identify and characterize the dental phenotype. Panoramic and lateral cephalogram radiographs were taken from the participating family members. After tracing the cephalograms with Dolphin® software version 11.9 (Dolphin Imaging & Management Solutions, Chatsworth, CA, USA), the measurement standards were analyzed to establish the skeletal and facial phenotype.
Saliva samples were collected for the mutational analysis using Oragene® DNA saliva collection kits (DNA Genotek Inc., Ottawa, ON, Canada). Genomic DNA was isolated from saliva samples with the conventional salting-out method and the quantity and quality were measured. Candidate gene sequencing for the FAM83H gene was performed with a DNA sample of the proband as previously described [5]. The segregation of the identified mutation within the family was investigated by Sanger sequencing. Sanger DNA sequencing was performed using ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) at the DNA sequencing center (Macrogen, Seoul, Korea).
3. Results
In this study, we recruited a four-generation Colombian AI family (Figure 1). A pedigree analysis strongly suggested an autosomal dominant inheritance pattern. No family member reported other remarkable past medical history, including genetic abnormalities or systemic diseases. Radiographically, the erupted teeth presented thin enamel coverage by breakdown but the unerupted developing teeth showed enamel with normal thickness but reduced radiopacity, being similar to the dentin. The affected enamel was very weak and easily worn, leaving brown-discolored, rough surfaces, suggesting hypocalcified AI. Therefore, mutational screening for the FAM83H gene was performed as a candidate gene approach.
Figure 1.
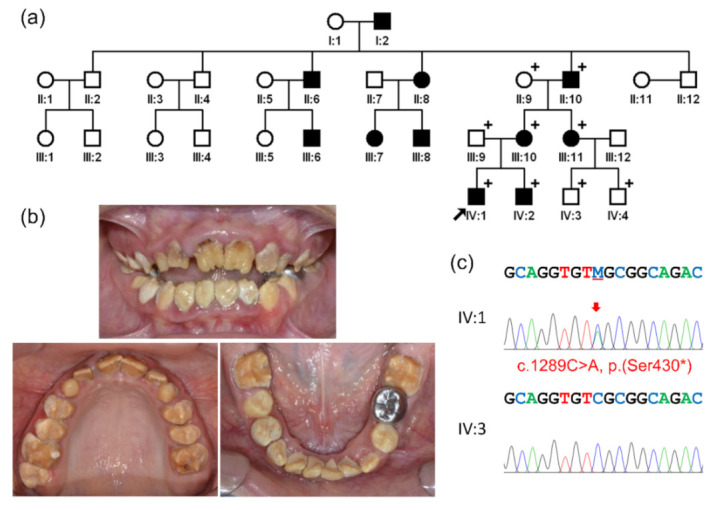
Pedigree, clinical photos and sequencing chromatograms: (a) pedigree of the family. Affected family members are indicated by black-filled symbols and the participating members are indicated with a plus sign (+) at the upper right corner of the symbols. The black arrow indicates the proband. (b) Clinical photos of the proband at age 10 years 6 months. A brown-discolored, rough enamel surface can be seen in all teeth. Heavy calculus deposits and an anterior open bite can be observed. (c) Sequencing chromatograms are shown for the proband (IV:1) and an unaffected cousin (IV:3). The mutation is indicated by the red arrow (M: C and A).
Sanger sequencing of the exons and exon/intron boundaries of all five exons of FAM83H revealed a nonsense mutation in the last exon. The mutation was a transversion change of cytosine to adenine (NM_198488.5:c.1289C>A), changing serine (TCG) to an amber stop codon (TAG) at amino acid position 430 (NP_940890.4:p.(Ser430 *)). The mutation would produce a truncated protein of 429 amino acids instead of 1179 wild-type FAM83H. Perfect segregation of the mutation and the disease phenotype in the family was confirmed. This is the first FAM83H mutation identified in the Colombian population.
Clinically, the proband (IV:1) presented an anterior open bite, unlike his younger brother (IV:2), who showed a deep bite. The cephalometric analysis of proband revealed skeletal Class III malocclusion and skeletal open bite. The other affected individuals (III:11 and IV:2) presented skeletal Class I malocclusion as well as straight profile.
4. Discussion
The mutation identified in this study was initially reported in genotype-phenotype studies involving 71 AI families and the affected ADHCAI family was African-American (personal communication with Dr. John Timothy Wright) [11]. Later, a sporadic case was reported in a European study of a cohort of 101 unrelated patients from France, Germany, and Morocco [12]. The family in this study is Mestizo Colombian, suggesting that this mutation is a mutational hotspot in the FAM83H gene.
The FAM83H gene consists of five exons. The translation start site is located in exon 2 and the last exon encodes most of the amino acids (933 out of 1179 amino acids). The FAM83H mutations causing ADHCAI clustered in the anterior half of exon 5. Therefore, the truncated proteins without a specific sequence in the exon 5, lose their normal cytoplasmic localization but translocate to the nucleus and hamper normal enamel calcification process [7].
The open bite tendency has been identified in AI patients regardless of their molecular etiology [13,14]. Some, but not all, ADHCAI families also show an association of the open bite or Angle’s Class III malocclusion [5,6]. The proband in this study had an anterior open bite. To find the association between the malocclusion and the identified FAM83H mutation, cephalometric analyses were conducted on several affected individuals (III:11, IV:1, and IV:2). As a result, the anterior open bite was not consistent among the affected individuals, despite the fact that their skeletal characteristics did not differ considerably (Figure 2, Table 1). Slightly increased overjet was identified in a younger brother of the proband. Therefore, it appears that the anterior open bite is not associated with the FAM83H mutation itself, at least in this family.
Figure 2.
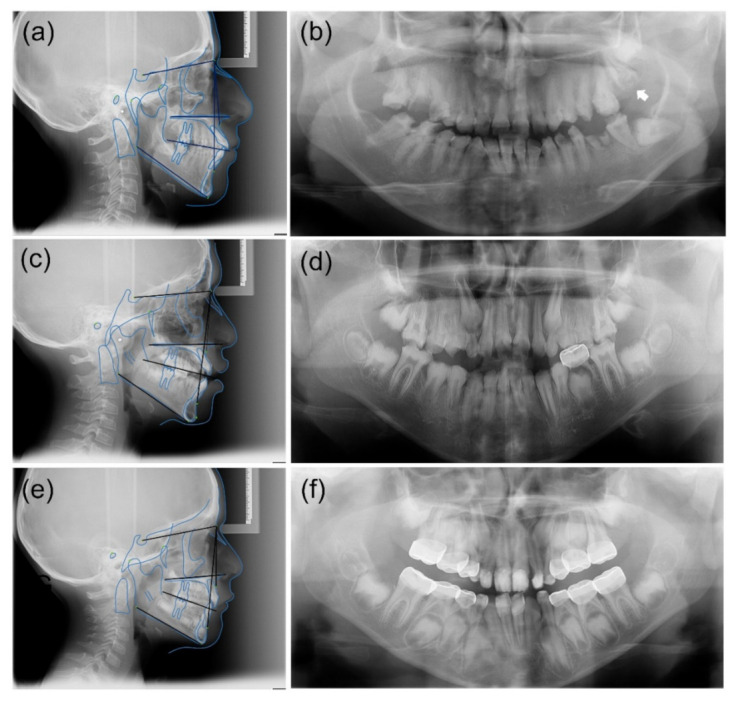
Cephalometric and panoramic radiographs of the affected individuals. (a,b) Cephalometric and panoramic radiographs of an aunt of the proband (III:11) at age 29 years 10 months. Impactions of the mandibular left third molar and maxillary left second and third molars were identified. Crown resorption (white arrow) of the impacted maxillary left second molar was found to have occurred. The anterior overbite appears to be within the normal range. (c,d) Cephalometric and panoramic radiographs of the proband (IV:1) at age 10 years 6 months. Hypocalcified enamel with reduced radiodensity is clearly observable in the developing second and third molars. The proband has an anterior open bite. (e,f) Cephalometric and panoramic radiographs of a younger brother of the proband (IV:2) at age 8 years 8 months. Multiple restorations were performed with a stainless-steel crown and resin composite. Anterior teeth tended toward mild to moderately deep overbite.
Table 1.
Cephalometric Measurements of the Affected Individuals.
| Measurements | Normal Values | III:11 | IV:1 | IV:2 |
|---|---|---|---|---|
| SNA | 82° ± 2° | 84° | 82° | 83° |
| SNB | 80° ± 2° | 80° | 75° | 77° |
| ANB | 2° ± 2° | 3° | 7° | 6° |
| A to N perp | −2.0 ± 3.7 mm | 2 mm | 7 mm | −7 mm |
| B to N perp | −6.9 ± 4.3 mm | −11 mm | −15 mm | −13 mm |
| A-N | 0.4 ± 1 mm | 1 mm | 4 mm | 3 mm |
| Pg-N | −1.8 ± 4.5 mm | −6 mm | −11 mm | −9 mm |
| N-A-Pg | 4° ± 6.4° | 7° | 8° | 6° |
| SN-PM | 32° | 38° | 40° | 32° |
| U1-SN | 103.9° ± 5.5° | 112° | 111.5° | 99.5° |
| L1-PM | 95° ± 7° | 93° | 101.2° | 84.2° |
| PP-PM | 25° | 35° | 33° | 32° |
Abbreviations: S = Sella; N = Nasion; A = A-point; B = B-point; Pg = Pogonion; Nperp = Nasion perpendicular line; PM = mandibular plane; U1 = axis of the upper incisor; L1 = axis of the upper incisor; and PP = palatal plane.
Tooth impaction is most frequently related to the enamel renal syndrome (ERS, OMIM #204690) [15,16,17]. This is caused by recessive FAM20A mutations, characterized by hypoplastic AI, pulp stones, delayed or failed eruptions of secondary dentition, gingival overgrowth, and nephrocalcinosis. In addition to tooth impaction caused by FAM20A mutations, impaction and/or crown resorption have been reported in the other non-syndromic AI patients [7,18]. Specifically, hypoplastic and hypocalcified AI are more frequently linked to this phenotype compared to hypomature AI. Tooth eruption could be less efficient with a rough enamel surface than a smooth and well-mineralized enamel surface. Furthermore, a rough and less-mineralized enamel surface could be prone to resorption when impaction occurs. In this study, impaction and crown resorption of the left maxillary second molar were noted in an affected individual (III:11). Routine follow-up and early intervention, such as surgical window opening, could prevent extraction caused by impaction and crown resorption.
In conclusion, in this report, we present a recurrent FAM83H mutation as a candidate gene approach, for the first time, in an extended ADHCAI Colombian family. Impaction and crown resorption were observed in an affected individual, supporting the previous finding. The proband had an anterior open bite, but it was not correlated with the disease in this family. This finding will improve our understanding of the genotype–phenotype relationship in ADHCAI caused by the FAM83H mutation.
Acknowledgments
We are grateful to all family members who participated in this study.
Author Contributions
C.A., M.A.A., Y.L. and D.A.G. conducted sample collection, experiments, and data analyses, and critically revised manuscript; S.G. and P.M. contributed to clinical evaluation, data analysis, and interpretation, and critically revised manuscript; L.O. and J.-W.K. contributed to conception, design, data acquisition, analysis, and interpretation, and drafted and critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2018R1A5A2024418 and NRF-2020R1A2C2100543) and by Pontifical Xavierian University (Bogota, Colombia) through the grant No. 3922.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of the Pontifical Xavierian University (CIEFOUJ 003 (2011) and 18 February 2011) and the Seoul National University Dental Hospital (CRI05003G and 10 December 2020).
Informed Consent Statement
Informed consent was obtained from all individual participants included in the study.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Witkop C.J., Jr., Sauk J.J., Jr. Heritable defects of enamel. In: Stewart R.E., Prescott G.H., editors. Oral Facial Genetics. C.V. Mosby Co.; St. Louis, MO, USA: 1976. pp. 151–226. [Google Scholar]
- 2.Kim Y.J., Kang J., Seymen F., Koruyucu M., Zhang H., Kasimoglu Y., Bayram M., Tuna-Ince E.B., Bayrak S., Tuloglu N., et al. Alteration of Exon Definition Causes Amelogenesis Imperfecta. J. Dent. Res. 2020;99:410–418. doi: 10.1177/0022034520901708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith C.E.L., Poulter J.A., Antanaviciute A., Kirkham J., Brookes S.J., Inglehearn C.F., Mighell A.J. Amelogenesis Imperfecta; Genes, Proteins, and Pathways. Front. Physiol. 2017;8:435. doi: 10.3389/fphys.2017.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witkop C.J., Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. Med. 1988;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- 5.Kim J.-W., Lee S.-K., Lee Z.H., Park J.-C., Lee K.-E., Lee M.-H., Park J.-T., Seo B.-M., Hu J.C.-C., Simmer J.P. FAM83H Mutations in Families with Autosomal-Dominant Hypocalcified Amelogenesis Imperfecta. Am. J. Hum. Genet. 2008;82:489–494. doi: 10.1016/j.ajhg.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright J., Frazier-Bowers S., Simmons D., Alexander K., Crawford P., Han S., Hart P., Hart T. Phenotypic Variation in FAM83H-associated Amelogenesis Imperfecta. J. Dent. Res. 2009;88:356–360. doi: 10.1177/0022034509333822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S., Zhang H., Hu C., Liu J., Chadha S., Kim J., Simmer J., Hu J. FAM83H and Autosomal Dominant Hypocalcified Amelogenesis Imperfecta. J. Dent. Res. 2020;100:293–301. doi: 10.1177/0022034520962731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S., Hu Y., Yang J., Smith C.E., Richardson A.S., Yamakoshi Y., Lee Y., Seymen F., Koruyucu M., Gencay K., et al. FAM83H null mice support a neomorphic mechanism for human ADHCAI. Mol. Genet. Genom. Med. 2015;4:46–67. doi: 10.1002/mgg3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S.-K., Lee K.-E., Jeong T.-S., Hwang Y.-H., Kim S., Hu J.-C., Simmer J., Kim J.-W. FAM83H Mutations Cause ADHCAI and Alter Intracellular Protein Localization. J. Dent. Res. 2010;90:377–381. doi: 10.1177/0022034510389177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang S., Hu Y., Smith C.E., Yang J., Zeng C., Kim J., Hu J.C., Simmer J.P. The Enamel Phenotype in Homozygous FAM83H Truncation Mice. Mol. Genet. Genom. Med. 2019;7:e724. doi: 10.1002/mgg3.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright J.T., Torain M., Long K., Seow K., Crawford P., Aldred M.J., Hart P.S., Hart T.C. Amelogenesis Imperfecta: Genotype-Phenotype Studies in 71 Families. Cells Tissues Organs. 2011;194:279–283. doi: 10.1159/000324339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasad M.K., Geoffroy V., Vicaire S., Jost B., Dumas M., Le Gras S., Switala M., Gasse B., Laugel-Haushalter V., Paschaki M., et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 2015;53:98–110. doi: 10.1136/jmedgenet-2015-103302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowley R., Hill F., Winter G. An investigation of the association between anterior open-bite and amelogenesis imperfecta. Am. J. Orthod. 1982;81:229–235. doi: 10.1016/0002-9416(82)90055-0. [DOI] [PubMed] [Google Scholar]
- 14.Ravassipour D.B., Powell C.M., Phillips C.L., Hart P.S., Hart T.C., Boyd C., Wright J.T. Variation in dental and skeletal open bite malocclusion in humans with amelogenesis imperfecta. Arch. Oral Biol. 2005;50:611–623. doi: 10.1016/j.archoralbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Cho S.H., Seymen F., Lee K.-E., Lee S.-K., Kweon Y.-S., Kim K.J., Jung S.-E., Song S.J., Yildirim M., Bayram M., et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum. Mutat. 2011;33:91–94. doi: 10.1002/humu.21621. [DOI] [PubMed] [Google Scholar]
- 16.O’Sullivan J., Bitu C.C., Daly S.B., Urquhart J., Barron M., Bhaskar S.S., Martelli-Júnior H., Neto P.E.D.S., Mansilla M.A., Murray J.C., et al. Whole-Exome Sequencing Identifies FAM20A Mutations as a Cause of Amelogenesis Imperfecta and Gingival Hyperplasia Syndrome. Am. J. Hum. Genet. 2011;88:616–620. doi: 10.1016/j.ajhg.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S.-K., Aref P., Hu Y., Milkovich R.N., Simmer J.P., El-Khateeb M., Daggag H., Baqain Z.H., Hu J.C.-C. FAM20A Mutations Can Cause Enamel-Renal Syndrome (ERS) PLoS Genet. 2013;9:e1003302. doi: 10.1371/journal.pgen.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee K.-E., Lee S.-K., Jung S.-E., Song S.J., Cho S.H., Lee Z.H., Kim J.-W. A novel mutation in the AMELX gene and multiple crown resorptions. Eur. J. Oral Sci. 2011;119:324–328. doi: 10.1111/j.1600-0722.2011.00858.x. [DOI] [PubMed] [Google Scholar]