

Abstract
Recent genome-wide association studies have identified 78 loci associated with Parkinson’s disease susceptibility but the underlying mechanisms remain largely unclear. To identify likely causal variants for disease risk, we fine-mapped these Parkinson’s-associated loci using four different fine-mapping methods. We then integrated multi-assay cell type–specific epigenomic profiles to pinpoint the likely mechanism of action of each variant, allowing us to identify Consensus single nucleotide polymorphism (SNPs) that disrupt LRRK2 and FCGR2A regulatory elements in microglia, an MBNL2 enhancer in oligodendrocytes, and a DYRK1A enhancer in neurons. This genome-wide functional fine-mapping investigation of Parkinson’s disease substantially advances our understanding of the causal mechanisms underlying this complex disease while avoiding focus on spurious, non-causal mechanisms. Together, these results provide a robust, comprehensive list of the likely causal variants, genes and cell-types underlying Parkinson’s disease risk as demonstrated by consistently greater enrichment of our fine-mapped SNPs relative to lead GWAS SNPs across independent functional impact annotations. In addition, our approach prioritized an average of 3/85 variants per locus as putatively causal, making downstream experimental studies both more tractable and more likely to yield disease-relevant, actionable results. Large-scale studies comparing individuals with Parkinson’s disease to age-matched controls have identified many regions of the genome associated with the disease. However, there is widespread correlation between different parts of the genome, making it difficult to tell which genetic variants cause Parkinson’s and which are simply co-inherited with causal variants. We therefore applied a suite of statistical models to identify the most likely causal genetic variants (i.e. fine-mapping). We then linked these genetic variants with epigenomic and gene expression signatures across a wide variety of tissues and cell types to identify how these variants cause disease. Therefore, this study provides a comprehensive and robust list of cellular and molecular mechanisms that may serve as targets in the development of more effective Parkinson’s therapeutics.
Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease, occurring in 2–3% of individuals 65 years of age or older (1). Many efforts have been made to better understand the biological mechanisms underpinning this disease in hopes of developing more effective treatments. Genome-wide association studies (GWAS) have offered insights into the molecular etiology of this debilitating disease (2–4). The largest PD GWAS to date recently identified 90 independent PD-associated variants distributed across 78 loci (3), more than doubling the number of previously known PD-risk signals (2). However, for any given locus, the lead or tag single nucleotide polymorphism (SNP) may merely be correlated with the causal SNP(s) due to linkage disequilibrium (LD), thus limiting our ability to interpret the functional consequences of genetic variation through which they affect complex disease risk (e.g. PD) (5–7).
Fine-mapping is a methodology that aims to prioritize putative causal variants (8–10). The necessity of fine-mapping has been effectively demonstrated across a number of conditions, including diabetes (11,12), rheumatoid arthritis (13,14) and Alzheimer’s disease (15). Statistical fine-mapping tools, such as approximate Bayes factor ABF (16) FINEMAP (17,18) (as in a previous PD fine-mapping study (19)), do not utilize any additional functional data and instead infer causal SNPs based on GWAS summary statistics and LD structure alone. Alternatively, functional fine-mapping tools integrate information genomic annotations, epigenetic data and quantitative trait loci (QTL) to improve the accuracy of causal SNP predictions. One such tool, PolyFun (20) further improves upon functional fine-mapping by automatically upweighting the most informative annotations during a training phase, while downweighting less relevant ones. All existing fine-mapping tools have their respective strengths and weaknesses, and consequently the 95% probability credible set (CS95%) SNPs they predict can differ substantially (8,20,21). Despite this, there is considerable overlap between subsets of their predictions (8,20).
In order to more robustly identify causal SNPs underlying PD, we utilized a consensus (i.e. ensemble) approach that integrates predictions from multiple statistical and functional fine-mapping tools. We previously developed echolocatoR (21), an open-access R package that enables automated end-to-end fine-mapping across many loci, using multiple fine-mapping tools in a single R function (Fig. 1). Using echolocatoR, we fine-mapped the majority of the PD loci using four different statistical and functional fine-mapping tools, which reduced the average number of candidate SNPs per locus from 4981 (85 of which are genome-wide significant) to just 3. The resulting fine-mapped SNPs, especially the multi-tool Consensus SNPs, were greatly enriched for functional annotations compared with the lead GWAS SNPs. Furthermore, we identified many Consensus SNPs that are within cell type–specific regulatory regions (e.g. active microglia enhancers), providing comprehensive and novel insights into PD molecular etiology.
Figure 1.
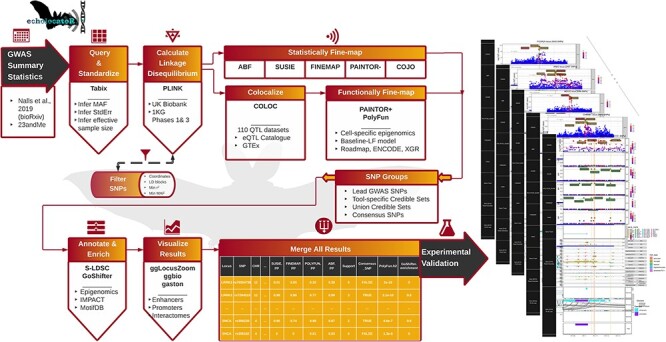
Outline of the study workflow. echolocatoR (21) enables end-to-end fine-mapping using the following workflow: (1) Nalls et al. (3) GWAS summary statistics imported, (2) locus-specific subsets are queried and standardized, (3) LD matrices are extracted from a European-ancestry subset of UK Biobank (UKB) (20,69,70), or the 1000 Genomes Project (1KG), (4) statistical and functional fine-mapping are conducted across multiple tools, (5) SNPs are categorized, (6) enrichment for tissue- and cell type-specific epigenomics, S-LDSC heritability, predictions of functional impact from several machine learning models (IMPACT, Basenji, DeepSEA), (7) all results are merged into a single table with one row per SNP, (8) high-resolution multi-track plots are generated for each locus.
Results
Fine-mapping of PD GWAS loci
Loci that fall within the HLA or Tau/17q21.31 regions were excluded due to the known complexity of their LD architectures (22). We present here high-confidence fine-mapping results for 74/78 loci using four complementary fine-mapping tools: ABF (16), FINEMAP (17,18), SuSiE (23) and PolyFun+SuSiE (20,23). Several key SNP groups were compared here: 1) GWAS lead: the SNP with the smallest P-value in each PD GWAS locus (3) after filtering steps (i.e. removing SNPs with MAF < 0.05 or are absent in the LD reference panel, and considering all SNPs within ±1 Mb of the original pre-filtered GWAS lead SNP), 2) [fine-mapping tool name] CS95%: tool-specific 95% credible sets, generally defined as having ≥95% posterior probability (PP) of being causal for the phenotype (i.e. PD), 3) UCS: Union Credible Set SNPs defined as the union of all tool-specific CS95% and 4) Consensus: SNPs in the CS95% of at least two fine-mapping tools.
Using our multi-tool fine-mapping strategy, we identified UCS and Consensus SNPs in 100% of these 74 loci. In total, there were 598 UCS SNPs (8.1 per locus on average) and 190 Consensus SNPs (2.6 per locus on average). In 69/74 (93.2%) loci, the lead SNP was in the respective UCS. However, the lead SNP was a Consensus SNP in only 29/74 (39.2%) of loci, highlighting the need for fine-mapping. The proportion of loci that each tool was able to identify at least one CS95% SNP varied considerably (Fig. 2). For example, ABF (a relatively simple fine-mapping model that can only assume one causal SNP/locus) was only able to identify a CS95% in eight loci, whereas SuSiE (which can assume multiple causal SNPs) produced CS95% in all loci. We also note that the SNP-wise posterior probabilities (PP) of PolyFun+SuSiE and SuSiE are expectedly much more highly correlated with one another than those of ABF or FINEMAP (Supplementary Material, Fig. S1g).
Figure 2.
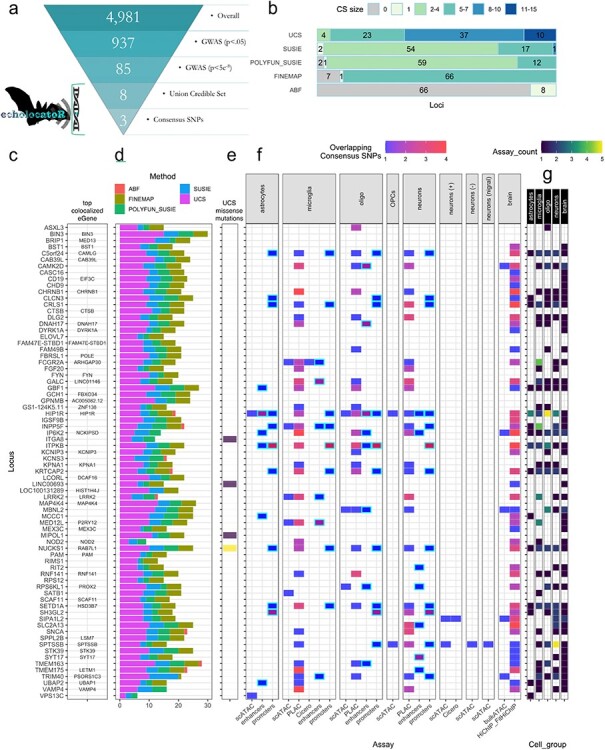
Summary of all fine-mapped loci. (a) The average number of potentially causal SNPs per PD-associated GWAS locus was reduced from 4981 SNPs (85 of which are genome-wide significant) to just three consensus SNPs. (b) The number of loci in which each fine-mapping tool identified a 95% credible set (CS95%) of a given size. These counts are also shown for the union credible set (UCS) of all CS95% together. (c) Rows are names of each locus as designated in the Nalls et al. (3) GWAS, while ‘top co-localized eGene’ displays the gene that showed the highest co-localization probability across all tested eGenes in all eQTL Catalogue datasets (after filtering spurious RP11-genes). (d) The number of SNPs in each tool-specific CS95%, as well as UCS size. (e) The number of UCS SNPs that were missense mutations. (f) The number of Consensus SNPs that fell within different cell type–specific epigenomic annotations. Nott et al. (24) data include enhancers or promoters called from peaks across multiple assays as well as overlap with PLAC-seq co-accessibility anchors (PLAC). Data from Corces et al. (25) include single-cell ATAC-seq peaks (scATAC), co-accessibility anchors called from the scATAC-seq data using the tool Cicero (Cicero), as well as peaks called from ATAC-seq in bulk brain tissue (bulkATAC) and co-accessibility anchors from HiChIP-FitHiChIP in bulk brain. (OPCs = oligodendrocyte progenitor cells, neurons (+) = excitatory neurons, neurons (−) = inhibitory neurons, neurons (nigra) = dopaminergic neurons from the substantia nigra. Enhancers and promoters are highlighted in cyan outlines. (g) The number of assays in which at least one consensus SNP overlapped with the annotation, aggregated by cell type (or bulk brain). This highlights the most relevant cell type(s) in each locus.
When checking for overlap with cell type–specific epigenomic annotations (24,25), we found that 66/74 loci (89.2%) had at least one overlapping UCS SNP and 54/74 loci (73%) loci had at least one overlapping Consensus SNP (Fig. 2f). We also checked for exclusivity among brain cell type–specific epigenomic signatures (astrocytes, microglia, neurons and oligodendrocytes in any assay) and found that Consensus SNPs overlapped with annotations in only one cell type in 14 loci, two cell types in 12 loci, three cell types in 10 loci and four cell types in 9 loci. UCS SNPs overlapped with enhancers in 42 loci and promoters in 20 loci, while Consensus SNPs overlapped with enhancers in 21 loci and promoters in 11 loci. Of all brain cell types investigated here, Consensus SNPs most frequently fell within epigenomic peaks present in microglia (n = 141), followed by those in neurons/neuronal subtypes (n = 122), and oligodendrocytes/OPCs (n = 102). These peaks were not necessarily mutually exclusive and do not preclude shared epigenomic signals in cell types not investigated here. See the Functional enrichment section below for quantitative enrichment analysis results.
Mismatch between the population compositions of the LD reference panel and the original GWAS, as well as insufficient LD reference panel size, can have drastic effects on fine-mapping results (26). We therefore repeated the fine-mapping pipeline on all PD loci using the European ancestry subset of 1000 Genomes Phase 3 (1KG; n = 503 samples) as the LD reference panel instead of UKB (n = 337 000 samples). Due to differences between 1KG and UKB, not all SNPs were present in both panels. Thus, only 62/74 (83.8%) of loci had the same lead SNP between panels. We next compared fine-mapping UCS using LD from each panel and found that all loci had at least one shared UCS SNP between panels. The multi-tool mean PP was highly correlated between LD panels (Spearman rho = 0.65, P < 2.2 x 10−16), though see Supplementary Material, Fig. S1a-g for plots of inter-tool variation. In 46/74 (62.2%) of loci, at least one Consensus SNP overlapped between LD panel results. Despite this, when we specifically checked the loci that we show in multi-track plots (LRRK2, Fig. 3a; MBNL2, Fig. 3b; DYRK1A, Fig. 4a; FCGR2A, Fig. 4b), we found that all Consensus SNPs using the UKB panel were also Consensus SNPs using the 1KG panel, affirming that the Consensus SNPs in these example loci are robust and reproducible.
Figure 3.
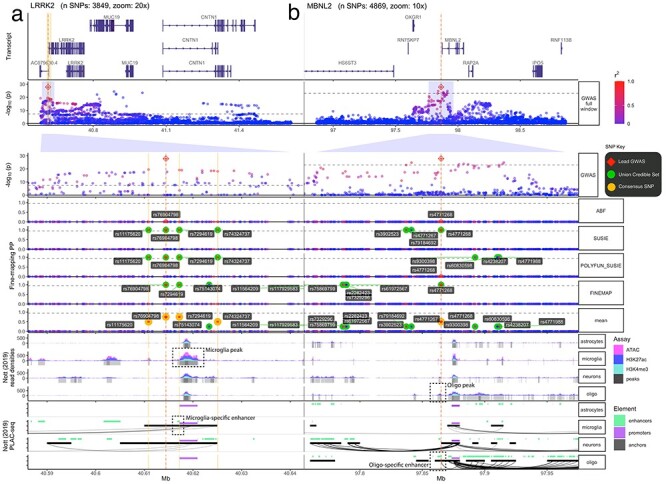
Fine-mapping of LRRK2 and MBNL2 loci. Multi-track plots within two PD-associated loci. (a) LRRK2 contains Consensus SNPs that are common in the population and overlap with microglia-specific epigenomic peaks (indicated by green arrows). Specifically, rs7294619 (which is not the lead SNP) falls within a microglia-specific enhancer, implicating a cell type-specific mechanism of PD risk. (b) MBNL2 contains a single consensus SNP (rs4771268), which happens to also be the lead GWAS SNP that overlaps with oligodendrocyte-specific epigenomic peaks (indicated by purple arrows). It also falls within an enhancer, with direct interactions (as indicated by PLAC-seq arches) with the MBNL2 promoter. The following tracks are shown (track labels in gray boxes on the left, sub-track labels in white boxes on the right): GWAS: –log10(P-value) from the Nalls et al. (3) PD GWAS zoomed out to show the entire locus. Below is the same GWAS data but zoomed into 10x to better show the fine-mapped SNPs (red diamond = lead GWAS SNP, green label = UCS SNPs, gold label = consensus SNPs). ABF, SUSIE, POLYFUN_SUSIE, FINEMAP: Fine-mapping results from four different tools, with the posterior probability (PP) that each SNP is causal as the y-axis (red diamond = lead GWAS SNP, green label = tool-specific CS95% SNPs, green circles = UCS SNPs). Mean: Per-SNP PP averaged across all fine-mapped tools. Gene track: Gene model of one of the LRRK2 transcripts (other transcripts not shown for simplicity). Nott (2019) Read Densities: Histograms of cell type-specific assays (oligo = oligodendrocytes). Called peaks are indicated by gray bars. Nott (2019) PLAC-seq: PLAC-seq interactome data as well as enhancers and promoters called by the original authors.
Figure 4.
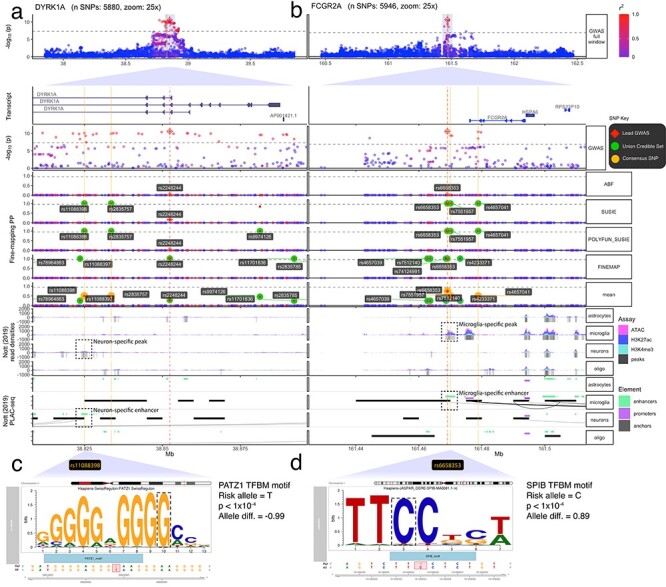
Fine-mapping of DYRK1A and FCGR2A loci. Multi-track plots within two PD-associated loci. (a) The DYRK1A locus contains common consensus SNPs that exclusively overlap with neuron-specific epigenomic peaks (indicated by blue arrows). Specifically rs11088398 (which is not the lead SNP) falls within a neuron-specific enhancer that has direct interactions with an upstream DYRK1A promoter. (c) motifbreakR results indicating that rs11088398 strongly disrupts a PATZ1 TFBM, which remains highly significant even after accounting for background effects and multiple testing correction (P < 1 x 10−4). (b) The FCGR2A locus contains three consensus SNPs, all of which exclusively overlap with microglia-specific epigenomic peaks (indicated by green arrows). (d) Specifically, rs665835 falls within a microglia-specific enhancer that is predicted to strongly disrupt binding in an SPIB TFBM (P < 1 x 10−4). It also falls within an enhancer, with direct interactions (as indicated by PLAC-seq arches) with the MBNL2 promoter. Within subplots (a) and (b), the following tracks are shown (track labels in gray boxes on the left, sub-track labels in white boxes on the right):GWAS: –log10(P-value) from the Nalls et al. (3) PD GWAS zoomed out to show the entire locus. Below is the same GWAS data but zoomed into 10x to better show the fine-mapped SNPs (red diamond = lead GWAS SNP, green label = UCS SNPs, gold label = consensus SNPs). ABF, SUSIE, POLYFUN_SUSIE, FINEMAP: Fine-mapping results from four different tools, with the PP that each SNP is causal as the y-axis. (red diamond = lead GWAS SNP, green label = tool-specific CS95% SNPs, green circles = UCS SNPs). ***Mean: Per-SNP PP averaged across all fine-mapped tools. Gene Track: Gene model of one of the LRRK2 transcripts (other transcripts not shown for simplicity). Nott (2019) Read Densities: Histograms of cell type-specific assays (oligo = oligodendrocytes). Called peaks are indicated by gray bars. Nott (2019) PLAC-seq: PLAC-seq interactome data as well as enhancers and promoters called by the original authors.
To assess reproducibility, we also compared our FINEMAP results with those from a recent study that performed statistical fine-mapping (using FINEMAP) on the same PD GWAS meta-analysis dataset but using a TOPMed (n = 16 257 samples) as the LD reference panel (19) (Supplementary Material, Fig. S1h-m). When comparing our UCS SNPs to a subset of the FINEMAP results provided by Grenn and colleagues (19) (42 CS95% SNPs across 18 loci), we found 10 overlapping SNPs, 6 of which were Consensus SNPs and 5 of which were also in our FINEMAP CS95%, across 9 loci (HIP1R, KRTCAP2, SH3GL2, SLC2A13, FAM47E-STBD1, TMEM175, TMEM163, CRHR1 and KCNS3). This limited concordance (10/42 SNPs = 23.8%) likely stems from several key methodological differences, including different LD panels and the fact that we specify a maximum of five causal SNPs for all loci, whereas Grenn et al. (27) first estimated the number of independent causal signals in each locus using the stepwise model selection procedure in GCTA-COJO. Despite this, we observed moderate correlation between the SNP-wise FINEMAP posterior inclusion probabilities (PIP) from Grenn et al. and those of our FINEMAP analyses (Spearman rho = 0.68, P-value < 2.2 x 10−16, n = 6391 overlapping SNPs; Supplementary Material, Fig. S1k), as well as with mean. PP in UCS SNPs only (Spearman rho = 0.68, P-value < 2.2 x 10−16, n = 10). This suggests that despite substantial methodological differences, there is moderate concordance in the pattern of results between studies.
Examples of fine-mapped loci: LRRK2, MBNL2, DYRK1A and FCGR2A
To exemplify the utility of our fine-mapping approach, here we highlight results from four loci and further provide a web app for results in all loci: https://rajlab.shinyapps.io/Fine_Mapping_Shiny. These were selected based on several criterion: 1) Explainable: At least one Consensus SNP fell within a regulatory element identified by the cell type–specific epigenomic signatures. 2) Interpretable: Consensus SNPs implicated a single cell type, as opposed to more complex loci that implicated multiple cell types or had very widespread LD architectures. 3) Visible: Consensus SNPs could be visualized within a window size that was not so wide as to prevent readers from seeing the peak-specific details in a static plot.
Rare mutations within protein-coding domains of the LRRK2 gene are frequently found in familial PD (28,29). However, less is known about potential common causal variants at this locus (MAF > 1%). Here, we identified four common Consensus SNPs (MAF > 10%) within the LRRK2 locus, of which only rs7294619 (not the lead PD GWAS SNP) is within a cell type–specific (microglia) enhancer (Fig. 3a). This confirms early independent reports of rs7294619 as a PD risk factor in smaller subpopulations (30). That said, another Consensus SNP (rs11175620) also falls within a microglia-specific peak, though this region did not meet the criterion for a regulatory element in the original publication. Deeper epigenomic sequencing of this region in microglia will be needed to resolve whether rs11175620 could also be a causal variant in this locus. eQTL co-localization tests further implicate LRRK2 as the most functionally relevant gene in this locus in both microglia (coloc PP.H4 = 0.65, n = 93 individuals) (31) and peripheral monocytes, a closely related myeloid cell type, in multiple more well-powered datasets (BLUEPRINT: PP.H4 = 0.999, n = 197; Fairfax_2014: PP.H4 = 0.968, n = 424; Quach_2016: PP.H4 = 0.991, n = 200; Supplementary Material, Fig. S2) (32–34). Specifically, the PD risk allele of rs7294619 (C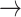 T) significantly increases only LRRK2 expression in microglia (P = 1.27x10−6, beta = 0.263) (31). The involvement of LRRK2 in microglia dysregulation aligns with substantial previous research implicating this gene in systemic and central nervous system (CNS) inflammation aspects of PD (28,29,35) and other inflammatory diseases (36–38).
T) significantly increases only LRRK2 expression in microglia (P = 1.27x10−6, beta = 0.263) (31). The involvement of LRRK2 in microglia dysregulation aligns with substantial previous research implicating this gene in systemic and central nervous system (CNS) inflammation aspects of PD (28,29,35) and other inflammatory diseases (36–38).
Within the MBNL2 locus, we identified 11 UCS SNPs and just one Consensus SNP (rs4771268), which was also the lead GWAS SNP. rs4771268 overlaps with an enhancer exclusively active in oligodendrocytes, lending further support to recent studies that have indicated a more important role of oligodendrocytes in PD than previously suspected (24,39). This oligodendrocyte-specific enhancer interacts with a downstream promoter of the gene MBNL2 (see Proximity Ligation-Assisted ChIP (PLAC) -seq interactome tracks in Fig. 3b), supporting the hypothesis that MBNL2 is likely to be the causal gene within this locus (though the eQTL co-localization tests could not identify any eGene for this region). We also conducted transcription factor binding motif (TFBM) analyses of the LRRK2 and MBNL2 loci using motifbreakR (40) (Supplementary Material, Fig. S3a-c), although the proposed TFBMs were less consistent across motif databases and we therefore advise readers to interpret these with caution.
At the DYRK1A locus, we identified two Consensus SNPs (rs11088398 and rs2835757, neither of which were the lead GWAS SNP) but only rs11088398 falls within a neuron-specific enhancer and has direct interactions with the upstream promoter of DYRK1A, as shown by multiple interactome assays (Fig. 4a). Previously, Grenn et al. (19) used FINEMAP to nominate rs2248244 (the lead SNP) and rs11701722 as putative causal variants. Despite the aforementioned methodological differences, rs2248244 also appeared in our UCS. However, in contrast to rs11088398, rs2248244 does not fall within any brain cell type–specific epigenomic peak or regulatory annotation in the datasets explored here. In further support of this observation, rs11088398 had considerably greater machine learning model-derived functional impact scores (max IMPACT score = 0.862, max Basenji H3K4ME3 Brain score = 3.64 x 10−5), than those of rs2248244 (max IMPACT score = 0.512, max Basenji H3K4ME3 Brain score = 8.32 x 10−8). While multiple variants within this locus may influence PD risk via different mechanisms, we find the functionally mechanistic explanation for rs11088398 particularly compelling. Furthermore, analysis of this region using motifbreakR (40), which searches a comprehensive database of positional weight matrices (PWM) and applies significance-based background correction, revealed that rs11088398 is within a PATZ1 TFBM, as well as KLF5 and MAZ TFBM to a lesser extent (P < 4.15x10−4, allele diff. > −0.785), and that the PD risk allele (G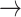 T) is predicted to strongly decrease binding affinity relative to the reference allele (P < 1x10−10, allele diff. = −0.990; Fig. 4c). This association remained highly significant even after background correction and stringent Bonferroni multiple-testing correction (q < 1x10−10). PATZ1 plays an important role in embryonic development and neurogenesis (e.g. in midbrain) and is expressed in neurons (and not glia) (Supplementary Material, Fig. S3d) and its downregulation is associated with premature senescence in mouse models, cell cultures and human brain tissue (41,42). Lastly, DYRK1A is the top eQTL-nominated gene from our co-localization analysis, which aligns with prior eQTL-based gene nominations in this locus (19). It is expressed in all brain cell types, but more so neurons than glia (Supplementary Material, Fig. S3e).
T) is predicted to strongly decrease binding affinity relative to the reference allele (P < 1x10−10, allele diff. = −0.990; Fig. 4c). This association remained highly significant even after background correction and stringent Bonferroni multiple-testing correction (q < 1x10−10). PATZ1 plays an important role in embryonic development and neurogenesis (e.g. in midbrain) and is expressed in neurons (and not glia) (Supplementary Material, Fig. S3d) and its downregulation is associated with premature senescence in mouse models, cell cultures and human brain tissue (41,42). Lastly, DYRK1A is the top eQTL-nominated gene from our co-localization analysis, which aligns with prior eQTL-based gene nominations in this locus (19). It is expressed in all brain cell types, but more so neurons than glia (Supplementary Material, Fig. S3e).
Lastly, the gene Fc Fragment of IgG Receptor IIa (FCGR2A) codes for an IgGFc receptor protein expressed on the surface of immune response cells and is important for phagocytosis and debris clearing (43). Our fine-mapping analyses revealed three Consensus SNPs, two of which (rs6658353 and rs7551957) fell within a microglia-specific enhancer (to the exclusion of other cell type–specific epigenomic peaks) in the FCGR2A locus. While the Consensus SNP did fall within multiple interactome assay anchors (PLAC-seq, Cicero, HiChIP-FitHiChIP) (24,25), the data did not associate it with a specific gene. FCGR2A was nevertheless the closest to the enhancer that the Consensus SNPs were located in. Furthermore, the Consensus SNP rs6658353 falls within an SPI1 (a well-established transcriptional regulator in microglia) TFBM, and its PD risk allele (G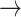 C) greatly disrupts its binding (Fig. 4d; P < 1 x 10−4, allele diff. = 0.89). FCGR2A itself is very highly expressed in both microglia and macrophages (Supplementary Material, Fig. S3f), as is SPI1 (Supplementary Material, Fig. S3g).
C) greatly disrupts its binding (Fig. 4d; P < 1 x 10−4, allele diff. = 0.89). FCGR2A itself is very highly expressed in both microglia and macrophages (Supplementary Material, Fig. S3f), as is SPI1 (Supplementary Material, Fig. S3g).
Functional enrichment
To efficiently verify the functionality of the fine-mapped SNPs, we employed an functional enrichment validation strategy across a diverse set of functional annotations (i.e. validation datasets), including 1) heritability enrichment scores derived from the L2-regularized S-LDSC regression (44–46) step in PolyFun (20), 2) probability scores from IMPACT, an elastic net logistic regression model that integrates 503 cell type–specific epigenomic annotations to predict each variant’s functional impact in the context of a particular tissue and cell type, primarily through the disruption of TFBM (47), 3) per-variant P-values from tests of genotypes impact on gene expression from the survey of regulatory elements (SuRE) massively parallel reporter assay (MPRA) (48), as well as 4) predictions from deep learning models, Basenji (49) and DeepSEA (50), trained on blood-, brain- or non–tissue-specific annotations (51).
Specifically, we tested two hypotheses within each validation dataset (H1 and H2). H1: fine-mapped (e.g. UCS, Consensus) SNPs have greater functional impact and thus are more likely to impact disease risk, than GWAS lead SNPs. H1 was tested using pairwise Wilcoxon rank-sum tests on per-Locus means of each SNP group. H2: fine-mapped SNPs more frequently have greater functional impact than randomly selected SNPs than do GWAS lead SNPs. H2 was tested using 10 000 boot-strapped iterations to compare functional annotations from each resampled SNP group to those of randomly sampled SNPs. In addition to the four main SNP groups defined above, we also compared SNP groups called without any functional fine-mapping (i.e. PolyFun+SuSIE) to avoid circularity, as some of the validation datasets (i.e. IMPACT, Basenji and DeepSEA) used annotations that were also used in the PolyFun baseline model. These additional SNP groups were 5) UCS (-PolyFun) and 6) Consensus (-PolyFun). All P-values listed below are post-adjustment, unless otherwise specified. While an in-depth analysis of inter-tool performance is beyond the scope of this study, we do provide extended functional enrichment validation results comparing tool-specific CS95% (Supplementary Material, Figs S4 and S5). See Methods and Supplementary Methods for extended details on these analyses.
Relative to GWAS lead SNPs, UCS SNPs had significantly greater h2 enrichment scores (H1 P = 3.2x10−6, H2 P = 6.2x10−88, mean = 1.46; Fig. 5a), IMPACT probability scores (H1 P = 0.0053, H2 P = 3.7x10−238, mean = 0.69; Fig. 5b) and functional impact probability in all 28 deep learning model-tissue-assay combinations (H1 P < 0.05; H2 P < 2.7 x 10−303, mean = 0.0056; Fig. 5d). The significant gain in predicted functionality remained true for UCS (-PolyFun) SNPs: h2 enrichment (H1 P = 1 x 10−5, H2 P = 7.3 x 10−132, mean = 1.37), IMPACT probability scores (H1 P = 0.046, H2 P = 2.2 x 10−308, mean = 0.68) and functional impact probability in all 28 deep learning model-tissue-assay combinations (H1 adj. P < 0.05; H2 norm. P = 2.7 x 10−303, mean = 0.0056; Fig. 5d). Likewise, Consensus SNPs also showed significantly greater functional impact than GWAS lead SNPs: h2 enrichment (H1 P = 0.00052, H2 P = 9.3x10−239, mean = 1.68), IMPACT probability scores (H1 P = 0.00031, H2 P = 2.2 x 10−308, mean = 0.74) and functional impact probability in 18/28 deep learning model-tissue-assay combinations (H1 P < 0.05; H2 P = 2.7 x 10−303, mean = 0.0053; Fig. 5d). While the H1 test of SuRE MPRA did not show significant differences between SNP groups, the bootstrapping test revealed that there was indeed significantly greater impact on gene expression in UCS, UCS (-PolyFun) and Consensus SNPs (H2 P = 2.7 x 10−303). This is perhaps due to the ability of the bootstrapping approach to approximate the true underlying distribution, thus better guarding against sampling error. While Consensus SNPs were not significantly greater than UCS or UCS (-PolyFun) in any of the H1 or H2 tests, they still have fewer SNPs on average (~3 per locus) making them better suited for follow-up wet laboratory experiments.
Figure 5.
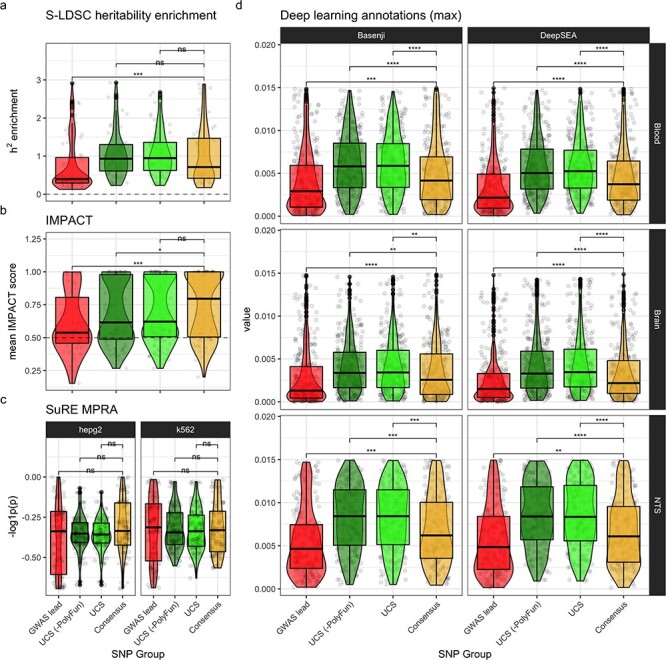
Functional enrichment. Functional enrichment validation was performed by statistically comparing each SNP group. For each validation (panel), per-locus mean values were computed for each SNP group (each represented by a point). Significance labels are from Holm-adjusted P-values of pairwise Wilcoxon rank-sum tests. Significance key: ns: P > 0.05, *: P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001, ****: P ≤ 0.0001. (a) Mean PolyFun-implemented (20) S-LDSC (44–46) heritability (h2) enrichment per locus per SNP group. (b) For each locus, the annotation with the highest mean IMPACT score across all SNPs in that locus was selected (to reduce noise from less relevant annotations). (c) SuRE MPRA results. Negative log10-transformed mean P-values from Wilcoxon rank-sum tests comparing gene expression changes between reference and alternative allele genotypes. Cell lines (i.e. HepG2 and K562) are separated into columns. (d) Deep learning model predictions of the epigenomic impact that mutating each genomic position would have. Tissue-specific models (i.e. Blood and Brain) were trained on datasets from only that tissue. Non-tissue-specific (NTS) models were also trained using all tissues together. For the purposes of readability, we collapse values across assays and remove outliers above the 75% percentile (see Supplementary Material, Fig. S4 for full view).
Discussion
Using a suite of complementary fine-mapping tools and validating using multiple independent lines of evidence, we identified 1–4 high-confidence consensus SNPs in most known PD-associated loci. These Consensus SNPs have a high probability of being causal in PD, often through the disruption of regulatory elements. Co-localization tests and brain cell type–specific chromatin accessibility and histone modifications allowed us to explore the locus-specific mechanisms by which the Consensus SNPs exert their effects. We showed that PD risk alleles in Consensus SNPs likely alter the functioning of microglia-specific enhancers in the loci LRRK2 (Fig. 3a) and FCGR2A (Fig. 4b,d), specifically through disrupting an SPIB-binding motif in the latter. We also identified Consensus SNPs affecting an oligodendrocyte-specific MBNL2 enhancer (Fig. 3b) and a neuron-specific DYRK1A enhancer (Fig. 3a), the latter of which is mediated through altered binding with PATZ1. Additionally, in two loci (KCNIP3 and TRIM40), our Consensus SNPs were missense mutations, both of which were found to contain missense mutations (2 and 6, respectively) and the former of which was significant in a Bonferroni-corrected rare coding variant burden analysis (3).
The fine-mapped SNPs identified in this study consistently demonstrate higher functional relevance than GWAS lead SNPs according to multiple lines of evidence, including h2 enrichment, predictions from a suite of high-accuracy machine learning models, cell type–specific epigenomic assays and high-throughput variant-editing experimental assays. In particular, Consensus SNPs have the advantages of high functional enrichment, high coverage across all loci (100%) and relatively small set sizes (1–4 SNPs per locus), making them ideal candidates for further experimental validation. This remains true even after multiple testing correction, rigorous bootstrapped testing regimes and removing any functionally informed fine-mapping results during UCS identification to avoid circularity in our validation strategy. Together, these multiple lines of evidence support our hypotheses that high-quality fine-mapped SNPs are much more likely to be causal for PD than simply selecting the SNPs with the smallest P-values, or random SNPs.
The present study has several limitations and could further be improved upon in the future by addressing the following: 1) our LD reference panels did not come from the same participants as the original PD GWAS studies, which reduces the accuracy of fine-mapping (26); 2) not all fine-mapped variants are mediated via cell type–specific enhancers/promoters, and thus future studies would benefit from exploring other mechanisms not explored here such as splicing (e.g. in MAPT) (52,53); 3) the PD GWAS was conducted using almost entirely genotyping data (as opposed to whole-genome sequencing), which can introduce substantial bias and miss rare variants as a consequence of imputation procedures; 4) the functional genomic annotations used in this study are far from a complete representation of all PD-relevant tissues, brain regions (e.g. substantia nigra), cell types and assay modalities across varying PD stages and clinical subtypes and 5) functional experiments would further validate these results. It is also possible that our consensus fine-mapping procedure missed certain causal SNPs (false negatives). However, this is difficult to resolve with certainty as we can currently only infer causality through functional and phenotypic impacts. Therefore, the best assessment of false-negative rates comes from simulation studies presented in the original publications of each fine-mapping tool, where accuracy and precision can be directly estimated. Despite these challenges, we hope these results will continue to be a resource for future studies with better powered, more diverse datasets across a variety of tissues, cell types and physiological conditions.
In summary, we have fine-mapped almost all known PD-associated loci using a suite of complementary fine-mapping methods and identified putative mechanisms of actions through which they increase risk of PD, including cell type and regulatory element type. Furthermore, our consensus fine-mapping strategy, implemented in echolocatoR (21), can easily be applied to any other GWAS/QTL summary statistics, opening up many opportunities for rapid and robust identification of causal genetic variants. This sets the stage to further our understanding of PD through uncovering potential shared genomic mechanisms underlying both PD and other neurological diseases, such as Alzheimer’s disease. Lastly, all SNP-wise fine-mapping results, LD matrices, merged annotations and plots from this study have been made available through a dedicated web application (https://rajlab.shinyapps.io/Fine_Mapping_Shiny), opening many opportunities to explore each locus in greater depth and, importantly, to validate putative causal SNPs through experimental validation (e.g. CRISPR-cas9 editing in patient-derived iPSC models).
Materials and Methods
All variant-level annotations, tools and analyses used in our pipeline were integrated into echolocatoR (21), either directly or via APIs, and run in R v3.6.3. See Supplementary Methods for a more detailed description of each dataset.
GWAS
Full genome-wide GWAS summary statistics from Nalls and colleagues (3) were provided by the authors and by 23andMe. If unspecified, we identified the lead GWAS SNP as the one with the lowest corrected P-value within that locus. Then, for each locus, we gathered all SNPs within 2-Mb windows (i.e. ± 1 Mb flanking the lead GWAS SNP) and filtered out SNPs with a minor allele frequency (MAF) < 0.05. We focused on common variants in order to maximize the relevance of these results to a larger proportion of the PD population.
Fine-mapping
Statistical fine-mapping was performed on each locus separately with ABF (16), FINEMAP (16–18) and SuSiE (23). Functional fine-mapping was performed using PolyFun+SuSiE (20,23), which computes SNP-wise heritability-derived prior probabilities using an L2-regularized extension of stratified LD SCore (S-LDSC) regression (44–46). For PolyFun+SuSiE, we used the default UK Biobank baseline model composed of 187 binarized epigenomic and genic annotations (54). In all subsequent analyses presented here, loci that fall within the HLA region or the Tau/17q21.31 region were excluded due to the particularly complex LD architectures (22).
While the specifics of each fine-mapping model differ from tool-to-tool, they are united by several key features: 1) they are Bayesian models; 2) they provide the PP that each SNP is a causal SNP, on a scale from 0 to 1 and 3) they provide CS of SNPs that have been identified as having a high PP of being causal, which we have set at a threshold of PP ≥ 0.95 for all tools (see Supplementary Methods for details). We only included tools that met the following criteria: 1) can take into account LD and 2) can operate using only summary statistics, which are more widely accessible and perform comparably to models using individual-level genotype data (5). By running all of these tools on each PD locus, we reduced the average number of candidate SNPs per locus from ~ 5000 to 8 (Fig. 2). For all fine-mapping models, we set the (maximum) number of causal SNPs to five, except for ABF, which can only assume a single causal SNP.
As ABF can only assume a single causal SNP, and thus cannot model scenarios where this is more than one true causal SNP, or for that matter, scenarios where two or more SNPs are indistinguishable due to perfect LD. SNPs with PP ≥ 0.95 were considered part of the ABF credible set and added to the overall cross-tool Credible Set. However, we decided to exclude this method when identifying Consensus SNPs because it almost exclusively returned the lead SNP, suggesting this simpler algorithm was not offering any additional information beyond which SNP had the lowest GWAS P-value.
LD
LD correlation matrices (in units of r) were acquired for each locus from the UK Biobank (UKB) reference panel, pre-calculated by Weissbrod et al. (20). We also acquired LD from the 1000 Genome Project (1KG), both Phases 1 and 3 (55), by downloading the relevant variant call files (VCF) from the 1KG file transfer protocol (FTP) server using Tabix (56) and then using the R package snpStats (57) set to the default parameters to calculate all pairwise r values. For both the UKB and 1KG reference panels, individuals of non-European ancestry were removed to best match the populations in the PD GWAS data. Any SNPs that could not be identified within the LD reference were necessarily removed from subsequent analyses.
eQTL
echolocatoR (21) uses another R package developed by our laboratory, catalogueR, to automatically query all 110 eQTL datasets (from 20 studies) in the eQTL Catalogue (58) via an API. This comprised the majority of the eQTL datasets used in this study as they have all been uniformly re-analyzed using the exact same pipeline with standardized formatting. This includes data from the Genotype-Tissue Expression (GTEx) project V8 (59), including 13 brain regions: amygdala, anterior cingulate cortex (BA24), caudate, cerebellar hemisphere, cerebellum, cortex, frontal cortex (BA9), hippocampus, hypothalamus, nucleus accumbens, putamen, spinal cord (cervical c-1) and substantia nigra. We also obtained genotype gene expression data from the Multi-Ethnic Study of Atherosclerosis (MESA) (60), which contains eQTL data in monocytes from multiple subpopulations: 233 African American (AFA), 578 European (CAU) and 352 Hispanic (HIS) individuals.
Co-localization
A GWAS-QTL locus pair was considered ‘co-localized’ if the following criterion were met, where the PP that the signals are associated with their respective traits and shared is PP.H4 and the PP that the signals are associated with their respective traits but not shared is PP.H3:
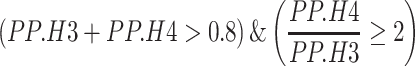 |
This provides a more robust means of identifying whether the signal in a GWAS dataset can be considered the same as the signal within a QTL dataset, as opposed to simply looking at the number of overlapping significant QTLs that can be confounded by factors such as LD (61).
Transcription factor binding motifs
We ran motifbreakR (40) on select Consensus SNPs, which 1) identifies whether a given variant falls within binding motifs or one or more transcription factors (TFBM) and 2) calculates how much each allele of that variant impacts motif binding. Motifs are compiled by a supporting package, MotifDb, which contains a comprehensive database of positional weight matrices (PWM) (n = 9933) from 14 TFBM databases gathered from multiple organisms (n = 16 species) and assay types (including ChIP-seq). For each variant, motifbreakR first queries the PWM databases to identify any motifs that the variant may fall within and returns metrics assessing how much each allele of that variant disrupts (or enhances) binding in the putative TFBM(s). Particularly important metrics include pct_ref/_alt: the proportion of maximum binding affinity of the motif to a given TF (0 to 1 scale) afforded by the variant’s reference/alternative alleles (respectively), and allele diff.: the difference in the proportion of binding affinity between the reference (pct_ref) and alternative (pct_alt) alleles (−1 to 1 scale). Next, motifbreakR applies a robust background correction regime to compute P-values. This is a computationally expensive but nevertheless important step as it helps guard against false positives, a substantial concern as many motifs have similar sequences and/or promiscuously bind to multiple transcription factors. Lastly, we applied the Benjamini-Hochberg procedure to compute false discovery rate (FDR) to account for querying multiple variants (62).
Functional enrichment
S-LDSC heritability, IMPACT, SuRE MPRA and deep learning annotations
For all S-LDSC heritability scores, IMPACT scores, SuRE MPRA P-values and deep learning epigenomic predictions (collectively referred to as validation annotations), we calculated the mean of the respective values per locus per SNP group (GWAS lead, UCS (-PolyFun), UCS, Consensus). Mean was used as opposed to max values, to avoid bias due to differences in SNP group sizes. We evaluated independence between SNP groups using a series of pairwise Wilcoxon rank-sum tests with multiple-testing adjusted P-values (Holm-Bonferroni method) (Fig. 5).
To ensure the robustness of these results, we also employed a bootstrapping hypothesis testing procedure, analogous to that proposed by Tibshirani and Efron (63). For 10 000 iterations in each SNP group, repeated separately for each validation annotation, we sampled (with replacement) 20 per-locus validation annotation means of the SNP group of interest and used a Wilcoxon rank-sum test (coin::wilcox_test in R) to determine whether their validation annotation values significantly differed from that of 20 randomly sampled SNPs (Random). Importantly, our Random samples were taken only from the 2-Mb windows defining our PD loci (as opposed to the whole genome), as an appropriate background to which compare the SNP groups. Resampling 10 000 times ensures that these results are not merely due to chance. Finally, we used a generalized linear model (using the R stats::glm function) to test whether the normalized test statistic distribution (z-value) of each fine-mapped SNP group (UCS (-PolyFun), UCS, Consensus (-PolyFun), Consensus) was significantly different from the GWAS lead z-value distribution. This bootstrapping procedure can easily be replicated using a single echolocatoR function, VALIDATION.bootstrap (21).
Lastly, we repeated enrichment tests for each SNP group against each combination of bulk brain tissue and cell type–specific epigenomic peaks, regulatory elements and interactomes (Supplementary Material, Fig. S6).
Cell type–specific epigenomic annotations
We conducted a series of negative binomial enrichment tests, with the XGR::xGRviaGenomicAnno function in R (64), using the following annotations: 1) cell type–specific epigenomic peaks (scATAC, ATAC, H3K27ac, H3K4me3), 2) cell type–specific regulatory regions (enhancers, promoters), 3) cell type–specific interactome anchors (PLAC, cicero), 4) bulk brain epigenomic peaks (ATAC) and 5) bulk brain interactome anchors (HiChIP_FitHiChIP) (24,25). We conducted these tests separately for each combination of assay/regulatory element and cell type. Furthermore, enrichment tests were repeated separately for each SNP group. All SNPs within any 2-Mb locus were used as the background. These same epigenomic datasets were used in summary plots (Fig. 2) and track plots (Figs 3 and 4). Annotations for missense variants were gathered from biomaRt (65,66) and HaploReg (67).
Active promoters and enhancers from Nott et al (24) were defined as follows. H3K4me3 and H3K27ac ChIP-seq data were collected for each purified cell type. Then, within each cell type, peaks were called using MACS2 (68). Active promoters were defined as the intersection between H3K4me3 peaks and H3K27ac peaks that were within 2 kb of the nearest transcription start site (TSS). Active enhancers were defined as H3K27ac peaks that were not within H3K4me3 peaks.
Data and Code Availability
A significant subset of the PD GWAS summary statistics can be found in the Supplementary Materials of the original publication (3). For full summary statistics, please contact the respective authors of that publication. eQTL Catalogue data are freely accessible through the main website (https://www.ebi.ac.uk/eqtl) or through the catalogueR software (https://github.com/RajLabMSSM/catalogueR).
All results of this study (fine-mapping, co-localization, plots) are accessible through the echolocatoR Fine-mapping Portal (21), which the authors have created to easily share and visualize the results of this study and others by our laboratory (https://rajlab.shinyapps.io/Fine_Mapping_Shiny).
R scripts containing all of the analyses conducted in this study are also made freely available on GitHub: https://github.com/RajLabMSSM/Fine_Mapping
Code for the echolocatoR Fine-mapping Portal (21) is also available on GitHub: https://github.com/RajLabMSSM/Fine_Mapping_Shiny
echolocatoR and catalogueR (21) are an open-source R packages that can be installed through the following GitHub repositories:
Supplementary Material
Acknowledgements
We would like to thank Kuan-lin Huang, Jack Humphrey, Ricardo Vialle, Elisa Navarro, Giulietta Riboldi, Gloriia Novikova, Cecilia Lindgren and Teresa Ferreira for their valuable feedback and suggestions. We would also like to thank Omer Weissbrod, Christopher Glass, Alexi Nott and Kaur Alasoo for their guidance with data and/or tool integration. This work was supported in part through the computational and data resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. We thank the research participants and employees of 23andMe who contributed to the PD GWAS.
A note from the first author (BMS):
This study is dedicated to Robert Neil Cronin, my grandfather. He is the reason I decided to pursue this field as a career, has continued to be my source of motivation, especially when challenges arose, and has kept this work grounded in its core mission of helping others. Without him, this study as it exists today would not have been possible. For all of this, and so much more, I am eternally grateful.
Conflict of Interest Statement. None declared.
Contributor Information
Brian M Schilder, Nash Family Department of Neuroscience & Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Ronald M. Loeb Center for Alzheimer’s disease, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Estelle and Daniel Maggin Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Towfique Raj, Nash Family Department of Neuroscience & Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Ronald M. Loeb Center for Alzheimer’s disease, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA; Estelle and Daniel Maggin Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Funding
Michael J. Fox Foundation (Grants #14899 and #16743 to T.R.) and US National Institutes of Health (NIH NIA R01-AG054005 and NINDS R01-NS116006 to T.R.). The author B.M.S. was additionally supported by the UK Dementia Research Institute that receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK.
References
- 1. Poewe, W., Seppi, K., Tanner, C.M., Halliday, G.M., Brundin, P., Volkmann, J., Schrag, A.-E. and Lang, A.E. (2017) Parkinson disease. Nat. Rev. Dis. Primers, 3, 17013. [DOI] [PubMed] [Google Scholar]
- 2. Chang, D., Nalls, M.A., Hallgrímsdóttir, I.B., Hunkapiller, J., van der Brug, M., Cai, F., International Parkinson’s Disease Genomics Consortium, 23andMe Research Team, Kerchner, G.A., Ayalon, G. et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet., 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nalls, M.A., Blauwendraat, C., Vallerga, C.L., Heilbron, K., Bandres-Ciga, S., Chang, D., Tan, M., Kia, D.A., Noyce, A.J., Xue, A. et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol., 18, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blauwendraat, C., Nalls, M.A. and Singleton, A.B. (2019) The genetic architecture of Parkinson’s disease. Lancet Neurol., 19, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pasaniuc, B. and Price, A.L. (2017) Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet., 18, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang, J., Weedon, M.N., Purcell, S., Lettre, G., Estrada, K., Willer, C.J., Smith, A.V., Ingelsson, E., O’Connell, J.R., Mangino, M. et al. (2011) Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet., 19, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pritchard, J.K. and Przeworski, M. (2001) Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet., 69, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Broekema, R.V., Bakker, O.B. and Jonkers, I.H. (2020) A practical view of fine-mapping and gene prioritization in the post-genome-wide association era. Open Biol., 10, 190221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hutchinson, A., Asimit, J. and Wallace, C. (2020) Fine-mapping genetic associations. Hum. Mol. Genet., 29, R81–R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaid, D.J., Chen, W. and Larson, N.B. (2018) From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet., 19, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaulton, K.J., Ferreira, T., Lee, Y., Raimondo, A., Mägi, R., Reschen, M.E., Mahajan, A., Locke, A., Rayner, N.W., Robertson, N. et al. (2015) Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet., 47, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mahajan, A., Taliun, D., Thurner, M., Robertson, N.R., Torres, J.M., Rayner, N.W., Payne, A.J., Steinthorsdottir, V., Scott, R.A., Grarup, N. et al. (2018) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet., 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kichaev, G. and Pasaniuc, B. (2015) Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet., 97, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westra, H.-J., Martínez-Bonet, M., Onengut-Gumuscu, S., Lee, A., Luo, Y., Teslovich, N., Worthington, J., Martin, J., Huizinga, T., Klareskog, L. et al. (2018) Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet., 50, 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartzentruber, J., Cooper, S., Liu, J.Z., Barrio-Hernandez, I., Bello, E., Kumasaka, N., Young, A.M.H., Franklin, R.J.M., Johnson, T., Estrada, K. et al. (2021) Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet., 53, 585–586. [DOI] [PubMed] [Google Scholar]
- 16. Wakefield, J. (2007) A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet., 81, 208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benner, C., Spencer, C.C.A., Havulinna, A.S., Salomaa, V., Ripatti, S. and Pirinen, M. (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics, 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benner, C., Havulinna, A.S., Salomaa, V., Ripatti, S. and Pirinen, M. (2018) Refining fine-mapping: effect sizes and regional heritability. bioRxiv, 318618. [Google Scholar]
- 19. Grenn, F.P., Kim, J.J., Makarious, M.B., Iwaki, H., Illarionova, A., Brolin, K., Kluss, J.H., Schumacher-Schuh, A.F., Leonard, H., Faghri, F. et al. (2020) The Parkinson’s disease genome-wide association study locus browser. Mov. Disord., 35, 2056–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weissbrod, O., Hormozdiari, F., Benner, C., Cui, R., Ulirsch, J., Gazal, S., Schoech, A.P., van de Geijn, B., Reshef, Y., Márquez-Luna, C. et al. (2020) Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet., 52, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schilder, B.M., Humphrey, J. and Raj, T. (2021) echolocatoR: an automated end-to-end statistical and functional genomic fine-mapping pipeline. Bioinformatics, btab658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boettger, L.M., Handsaker, R.E., Zody, M.C. and McCarroll, S.A. (2012) Structural haplotypes and recent evolution of the human 17q21.31 region. Nat. Genet., 44, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, G., Sarkar, A., Carbonetto, P. and Stephens, M. (2020) A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol., 82, 1273–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nott, A., Holtman, I.R., Coufal, N.G., Schlachetzki, J.C.M., Yu, M., Hu, R., Han, C.Z., Pena, M., Xiao, J., Wu, Y. et al. (2019) Brain cell type-specific enhancer-promoter interactome maps and disease risk association. Science, 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corces, M.R., Shcherbina, A., Kundu, S., Gloudemans, M.J., Frésard, L., Granja, J.M., Louie, B.H., Eulalio, T., Shams, S., Bagdatli, S.T. et al. (2020) Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson's diseases. Nat. Genet., 52, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benner, C., Havulinna, A.S., Järvelin, M.-R., Salomaa, V., Ripatti, S. and Pirinen, M. (2017) Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Hum. Genet., 101, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang, J., Lee, S.H., Goddard, M.E. and Visscher, P.M. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cook, D.A., Kannarkat, G.T., Cintron, A.F., Butkovich, L.M., Fraser, K.B., Chang, J., Grigoryan, N., Factor, S.A., West, A.B., Boss, J.M. et al. (2017) LRRK2 levels in immune cells are increased in Parkinson’s disease. npj Parkinson’s Disease, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kluss, J.H., Mamais, A. and Cookson, M.R. (2019) LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans., 47, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mata, I.F., Checkoway, H., Hutter, C.M., Samii, A., Roberts, J.W., Kim, H.M., Agarwal, P., Alvarez, V., Ribacoba, R., Pastor, P. et al. (2012) Common variation in the LRRK2 gene is a risk factor for Parkinson’s disease. Mov. Disord., 27, 1822–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Young, A.M.H., Kumasaka, N., Calvert, F., Hammond, T.R., Knights, A., Panousis, N., Schwartzentruber, J., Liu, J., Kundu, K., Segel, M. et al. (2021) A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat. Genet., 53, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quach, H., Rotival, M., Pothlichet, J., Loh, Y.H.E., Dannemann, M., Zidane, N., Laval, G., Patin, E., Harmant, C., Lopez, M. et al. (2016) Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell, 167, 643, e17–e656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fairfax, B.P., Humburg, P., Makino, S., Naranbhai, V., Wong, D., Lau, E., Jostins, L., Plant, K., Andrews, R., McGee, C. et al. (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science, 343, 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen, L., Ge, B., Casale, F.P., Vasquez, L., Kwan, T., Garrido-Martín, D., Watt, S., Yan, Y., Kundu, K., Ecker, S. et al. (2016) Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell, 167, 1398, e24–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alessi, D.R. and Sammler, E. (2018) LRRK2 kinase in Parkinson’s disease. Science, 360, 36–37. [DOI] [PubMed] [Google Scholar]
- 36. Wallings, R.L. and Tansey, M.G. (2019) LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans., 47, 1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hui, K.Y., Fernandez-Hernandez, H., Hu, J., Schaffner, A., Pankratz, N., Hsu, N.-Y., Chuang, L.-S., Carmi, S., Villaverde, N., Li, X. et al. (2018) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson's disease. Sci. Transl. Med., 2, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Witoelar, A., Jansen, I.E., Wang, Y., Desikan, R.S., Gibbs, J.R., Blauwendraat, C., Thompson, W.K., Hernandez, D.G., Djurovic, S., Schork, A.J. et al. (2017) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol., 74, 780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bryois, J., Eating Disorders Working Group of the Psychiatric Genomics Consortium, Skene, N.G., Hansen, T.F., Kogelman, L.J.A., Watson, H.J., Liu, Z., Brueggeman, L., Breen, G., Bulik, C.M. et al. (2020) Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet., 52, 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coetzee, S.G., Coetzee, G.A. and Hazelett, D.J. (2015) motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics, 31, 3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fedele, M., Crescenzi, E. and Cerchia, L. (2017) The POZ/BTB and AT-hook containing zinc finger 1 (PATZ1) transcription regulator: physiological functions and disease involvement. Int. J. Mol. Sci., 12, 2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glaab, E. and Schneider, R. (2015) Comparative pathway and network analysis of brain transcriptome changes during adult aging and in Parkinson’s disease. Neurobiol. Dis., 74, 1–13. [DOI] [PubMed] [Google Scholar]
- 43. Indik, Z.K., Park, J.G., Hunter, S. and Schreiber, A.D. (1995) The molecular dissection of Fc gamma receptor mediated phagocytosis. Blood, 86, 4389–4399. [PubMed] [Google Scholar]
- 44. Gazal, S., Finucane, H.K., Furlotte, N.A., Loh, P.-R., Palamara, P.F., Liu, X., Schoech, A., Bulik-Sullivan, B., Neale, B.M., Gusev, A. et al. (2017) Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nat. Genet., 49, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finucane, H.K., Consortium, R.G., Bulik-Sullivan, B., Gusev, A., Trynka, G., Reshef, Y., Loh, P.-R., Anttila, V., Xu, H., Zang, C. et al. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet., 47, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bulik-Sullivan, B.K., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Loh, P.R., Finucane, H.K., Ripke, S., Yang, J., Patterson, N., Daly, M.J., Price, A.L. and Neale, B.M. (2015) LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amariuta, T., Luo, Y., Gazal, S., Davenport, E.E., van de Geijn, B., Ishigaki, K., Westra, H.-J., Teslovich, N., Okada, Y., Yamamoto, K. et al. (2019) IMPACT: genomic annotation of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. Am. J. Hum. Genet., 104, 879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Arensbergen, J., Pagie, L., FitzPatrick, V.D., de Haas, M., Baltissen, M.P., Comoglio, F., van der Weide, R.H., Teunissen, H., Võsa, U., Franke, L. et al. (2019) High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet., 51, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kelley, D.R., Reshef, Y.A., Bileschi, M., Belanger, D., McLean, C.Y. and Snoek, J. (2018) Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res., 28, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou, J. and Troyanskaya, O.G. (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dey, K.K., Van de Geijn, B., Kim, S.S., Hormozdiari, F., Kelley, D.R. and Price, A.L. (2020) Evaluating the informativeness of deep learning annotations for human complex diseases. Nat. Commun., 11, 4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Raj, T., Li, Y.I., Wong, G., Humphrey, J., Wang, M., Ramdhani, S., Wang, Y.-C., Ng, B., Gupta, I., Haroutunian, V. et al. (2018) Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet., 50, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li, Y.I., Wong, G., Humphrey, J. and Raj, T. (2019) Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun., 10, 994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gazal, S., Loh, P.-R., Finucane, H.K., Ganna, A., Schoech, A., Sunyaev, S. and Price, A.L. (2018) Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat. Genet., 50, 1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li, H. (2011) Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics, 27, 718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Clayton, D. (2020) snpStats: SnpMatrix and XSnpMatrix classes and methods. Bioconductor.
- 58. Kerimov, N., Hayhurst, J.D., Peikova, K., Manning, J.R., Walter, P., Kolberg, L., Samoviča, M., Sakthivel, M.P., Kuzmin, I., Trevanion, S.J. et al. (2021) A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet., 53, 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mogil, L.S., Andaleon, A., Badalamenti, A., Dickinson, S.P., Guo, X., Rotter, J.I., Craig Johnson, W., Im, H.K., Liu, Y. and Wheeler, H.E. (2018) Genetic architecture of gene expression traits across diverse populations. PLoS Genet., 14, e1007586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Giambartolomei, C., Vukcevic, D., Schadt, E.E., Franke, L., Hingorani, A.D., Wallace, C. and Plagnol, V. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet., 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., 57, 289–300. [Google Scholar]
- 63. Tibshirani, R.J. and Efron, B. (1993) An introduction to the bootstrap. Monogr. Stat. Appl. Prob., 57, 1–436. [Google Scholar]
- 64. Fang, H., Knezevic, B., Burnham, K.L. and Knight, J.C. (2016) XGR software for enhanced interpretation of genomic summary data, illustrated by application to immunological traits. Genome Med., 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Durinck, S., Moreau, Y., Kasprzyk, A., Davis, S., De Moor, B., Brazma, A. and Huber, W. (2005) BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics, 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- 66. Durinck, S., Spellman, P.T., Birney, E. and Huber, W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc., 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhbannikov, I.Y., Arbeev, K., Ukraintseva, S. and Yashin, A.I. (2017) haploR: an R package for querying web-based annotation tools. F1000Research, 6, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Feng, J., Liu, T., Qin, B., Zhang, Y. and Liu, X.S. (2012) Identifying ChIP-seq enrichment using MACS. Nat. Protoc., 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L.T., Sharp, K., Motyer, A., Vukcevic, D., Delaneau, O., O’Connell, J. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sudlow, C., Gallacher, J., Allen, N., Beral, V., Burton, P., Danesh, J., Downey, P., Elliott, P., Green, J., Landray, M. et al. (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Habib, N., Li, Y., Heidenreich, M., Swiech, L., Avraham-Davidi, I., Trombetta, J.J., Hession, C., Zhang, F. and Regev, A. (2016) Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science, 353, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tirosh, I., Izar, B., Prakadan, S.M., Wadsworth, M.H., 2nd, Treacy, D., Trombetta, J.J., Rotem, A., Rodman, C., Lian, C., Murphy, G. et al. (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tasic, B., Menon, V., Nguyen, T.N., Kim, T.K., Jarsky, T., Yao, Z., Levi, B., Gray, L.T., Sorensen, S.A., Dolbeare, T. et al. (2016) Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci., 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A significant subset of the PD GWAS summary statistics can be found in the Supplementary Materials of the original publication (3). For full summary statistics, please contact the respective authors of that publication. eQTL Catalogue data are freely accessible through the main website (https://www.ebi.ac.uk/eqtl) or through the catalogueR software (https://github.com/RajLabMSSM/catalogueR).
All results of this study (fine-mapping, co-localization, plots) are accessible through the echolocatoR Fine-mapping Portal (21), which the authors have created to easily share and visualize the results of this study and others by our laboratory (https://rajlab.shinyapps.io/Fine_Mapping_Shiny).
R scripts containing all of the analyses conducted in this study are also made freely available on GitHub: https://github.com/RajLabMSSM/Fine_Mapping
Code for the echolocatoR Fine-mapping Portal (21) is also available on GitHub: https://github.com/RajLabMSSM/Fine_Mapping_Shiny
echolocatoR and catalogueR (21) are an open-source R packages that can be installed through the following GitHub repositories: