

Abstract
Context
We previously presented evidence that TSH receptor (TSHR)-stimulating autoantibodies (TSAbs) bind to and activate TSHRs but do not bind to IGF1 receptors (IGF1Rs). Nevertheless, we showed that IGF1Rs were involved in thyroid eye disease (TED) pathogenesis because TSAbs activated crosstalk between TSHR and IGF1R. Teprotumumab, originally generated to inhibit IGF1 binding to IGF1R, was recently approved for the treatment of TED (Tepezza).
Objective
To investigate the role of TSHR/IGF1R crosstalk in teprotumumab treatment of TED.
Design
We used orbital fibroblasts from patients with TED (TEDOFs) and measured stimulated hyaluronan (HA) secretion as a measure of orbital fibroblast activation by TED immunoglobulins (TED-Igs) and monoclonal TSAb M22. We previously showed that M22, which does not bind to IGF1R, stimulated HA in a biphasic dose-response with the higher potency phase dependent on TSHR/IGF1R crosstalk and the lower potency phase independent of IGF1R. Stimulation by TED-Igs and M22 was measured in the absence or presence of teprotumumab biosimilar (Tepro) or K1-70, an antibody that inhibits TSHR.
Results
We show: (1) Tepro dose-dependently inhibits stimulation by TED-Igs; (2) Tepro does not bind to TSHRs; (3) Tepro inhibits IGF1R-dependent M22-induced HA production, which is mediated by TSHR/IGF1R crosstalk, but not IGF1R-independent M22 stimulation; and (4) β-arrestin 1 knockdown, which blocks TSHR/IGF1R crosstalk and prevents Tepro inhibition of HA production by M22 and by a pool of TED-Igs.
Conclusion
We conclude that Tepro inhibits HA production by TEDOFs by inhibiting TSHR/IGF1R crosstalk and suggest that inhibition of TSHR/IGF1R crosstalk is the mechanism of its action in treating TED.
Keywords: teprotumumab, thyroid eye disease (TED), TSH receptor (TSHR), insulin-like growth factor receptor 1 (IGF1R), crosstalk, TSHR stimulating antibody (TSAb), M22 (a monoclonal TSAb)
Tepezza (teprotumumab) is currently the sole drug therapy approved by the US Food and Drug Administration for the treatment of thyroid eye disease (TED) (1, 2). Teprotumumab (Tepro) was first generated as an IGF1 receptor (IGF1R) antagonist that inhibits IGF1 binding to IGF1R as a potential treatment of various cancers (3, 4). Based on initial data that showed that IGF1Rs were likely involved in TED pathogenesis (5), it was suggested that IGF1R-stimulating antibodies may bind to and activate IGF1Rs on cells in the orbits of Graves’ disease patients. However, although several laboratories around the world have attempted to identify and isolate IGF1R-stimulating antibodies from the blood of TED patients, none has been found (6). Nevertheless, several groups were able to show that some, but not all, antibodies that antagonize IGF1Rs (7) were able to inhibit stimulation by Graves’ autoantibodies, which activate the TSH receptor (TSHR), in orbital cells of patients with TED (for review, see (8)). Tepro has been shown to inhibit stimulation of cells derived from the blood of TED patients (fibrocytes) stimulated by TSH and a monoclonal TSHR-stimulating antibody (TSAb) M22 also (9, 10).
We proposed a mechanism not involving IGF1R-stimulating antibodies for the involvement of IGF1Rs in TED pathogenesis (for review, see (11)). Using cells in primary cultures derived from the orbits of TED patients undergoing decompression surgery (TEDOFs), we showed that IGF1Rs were activated by TSAbs that do not bind to nor directly activate IGF1Rs, but rather activate IGF1Rs via crosstalk between TSHRs and IGF1Rs (12). The phenomenon of activation of G protein-coupled receptors, like TSHR, activating receptor tyrosine kinases, like IGF1R, is a well-known mechanism for enhancing stimulation of multiple biologic responses (13, 14). This study similarly used Graves’ orbital fibroblasts (GOFs) in primary culture and measured secretion of hyaluronan (HA) to assess activation of TSHR/IGF1R crosstalk. We demonstrate that Tepro inhibits stimulation by patient-isolated TSAbs, immunoglobulins from patients with TED (TED-Igs), and by M22. In the case of M22, Tepro inhibits only the IGF1R-dependent component but not the IGF1R-independent component of TSHR activation. We conclude that inhibition of the component of M22 stimulation that is dependent on TSHR/IGF1R crosstalk is an integral part of Tepro function.
Materials and Methods
Materials
M22 (RRID: AB_2892140; http://antibodyregistry.org/AB_2892140), a human monoclonal TSAb, and K1-70 (RRID: AB_2637026; http://antibodyregistry.org/AB_2637026), a blocking type human monoclonal for TSHR, purchased from Kronus (Star, ID). DMEM, 1M HEPES buffer, and Hanks Balanced Salt Solution were obtained from Mediatech Inc. (Manassas, VA). Hydrocortisone, insulin, cholera toxin, 100x antibiotic-antimycotic solution, and human thyroglobulin ELISAs were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human epidermal growth factor was obtained from PeproTech (Rocky Hill, NJ). Fetal bovine serum (FBS), hygromycin B, 100x penicillin-streptomycin solution, L-glutamine, Ham’s F-12 nutrient mixture, trypsin-EDTA, and Alexa Fluor 647 Antibody Labeling Kit were obtained from Thermo Fisher Scientific (Carlsbad, CA). Accutase cell detachment solution was purchase from Innovative Cell Technologies (San Diego, CA). Bovine BSA V was obtained from MP Biomedicals (Santa Ana, CA). One megadalton HA was purchased from Lifecore Biomedical (Chaska, MN). Hyaluronic Acid Test Kits were purchased from Corgenix (Broomfield, CO). DharmaFECT transfection reagent and inhibitory RNA were purchased from Dharmacon (Lafayette, CO). Y-27632 dihydrochloride and teprotumumab biosimilar (Tepro, RRID: AB_2892149; http://antibodyregistry.org/AB_2892149) were purchased from Bio-Techne (Minneapolis, MN).
Cell Isolation and Tissue Culture
HEK-TSHR cells were generated as previously described (15). In short, human TSHR cDNA was inserted into pcDNA3.1(-)/hygromycin vector for high-level, constitutive expression. HEK-EM 293 cells were stably transfected with this plasmid and cultured at 37°C in a 5% CO2 humidified incubator using DMEM with FBS (10% vol/vol), penicillin (100 U/mL), streptomycin (100 µg/mL), and hygromycin (250 µg/mL).
Retro-orbital connective tissue was obtained from patients with severe TED who underwent orbital decompression surgery at the joint thyroid-eye-clinic of the Johannes Gutenberg University (JGU) Medical Center, Mainz, Germany. Patients were classified with phenotypically overt and clinically severe TED, according to the European Society of Endocrinology-endorsed 2021 guidelines of the European Group on Graves’ Orbitopathy Thyroid Association for TED management (16). Orbital decompression surgery was indicated because the anti-inflammatory immunomodulating medical treatment was either partially or not successful. Furthermore, all serum samples were collected from patients with Graves’ disease and associated severe and active orbitopathy who were off immunosuppressive treatment for at least 4 weeks. Time interval between collection of orbital tissue specimen and serum samples was 3 to 6 months. Orbital connective tissue and patient sera were stored at -80°C and -20°C, respectively.
The collection of patients’ tissues was approved by the Ethical Committee of the Medical Chamber of the State Rhineland-Palatinate, Germany, and by the institutional review board of the JGU Medical Center. Written, informed consent was received from all patients before collection of blood and tissue samples.
TEDOFs were isolated from tissue as previously described (17). Cells were passaged with trypsin/EDTA and cultured in DMEM with FBS (10% vol/vol), penicillin (100 U/mL), streptomycin (100 μg/mL), L-glutamine (2 mM), Ham’s F-12 nutrient mixture (25% vol/vol), hydrocortisone (25 ng/mL), epithelial growth factor (0.125 ng/mL), insulin (5 μg/mL), cholera toxin (11.7 nM), gentamicin (10 μg/mL), amphotericin B (250 ng/mL), and Y-27632 (5 μM). Cells were maintained at 37°C in a 7% CO2 humidified incubator.
Purification of TED-Igs From Patient Sera
Serum samples were obtained from patients with phenotypically overt and clinically severe TED at the joint thyroid-eye-clinic of the JGU Medical Center, Mainz, Germany. IgG antibodies were isolated from whole serum (TED-Igs) by thiophilic affinity chromatography as previously described (7). Aliquots of purified TED-Igs were pooled and used for experiments. For the Tepro dose-response, TED-Igs were pooled from 37 patients, and the concentration was 5.4 mg/mL, as measured by absorbance at 280 nm. In this experiment, the final concentrations were 4.8 mg/mL TED-Igs, 10% FBS, 10 mM HEPES, and 12 to 1000 ng/mL Tepro. For the β-arrestin 1 (ARRB1) knock down (KD) experiments, the IgG concentration of the resulting TED-Ig pool from 30 different patients was 2.1 mg/mL. TED-Igs were diluted 1:4 in complete media with a final concentration of 10% FBS, 10 mM HEPES, and 0.525 mg/mL TED-Igs.
Flow Cytometric Competitive Binding
Alexa-647 was covalently conjugated to M22 (Alexa-647-M22), using the Alexa Fluor 647 Antibody Labeling Kit according to the manufacturer’s directions. This batch of Alexa-647-M22 was validated in HEK parental cells, which do not express TSHR (ie, fluorescent intensity was not significantly different than cellular autofluorescence). For competitive binding experiments, HEK-TSHR cells were dissociated with Accutase and resuspended at a concentration of 1 × 105 cells per 100 µL volume. Cells were preincubated on ice with or without 2.5 µg unconjugated M22 or Tepro for 15 minutes, then incubated with added 0.5 µg of Alexa-647-M22 for 1 hour. Stained cell samples were analyzed by flow cytometry using a BD FACSAria II Cell Sorter (BD Biosciences) using a 70-μm nozzle and a sheath pressure of 65 psi. Each condition was done in duplicate, and data were from 3 independent experiments.
Measurement of Secreted HA
Following trypsinization, cell number was measured on a Vi-Cell XR Cell Counter (Beckman Coulter, Indianapolis, ID). TEDOFs were plated at confluence (4 × 105 cells/cm2) in 48-well plates and stimulated with M22 with or without K1-70 or Tepro in DMEM containing 10% FBS and 10 mM HEPES. Cells were incubated at 37°C, 7% CO2, for 5 days. Conditioned media were collected and stored at -20°C. HA concentration was measured by ELISA as previously described (18).
Gene Knockdown
TEDOFs were plated at a confluence (4 × 105 cells/cm2) in 48-well plates. After 1 day, cells were transfected with scrambled (nontargeting) or ARRB1 siRNA using DharmaFECT Transfection Reagent 1 according to the manufacturer’s directions. Following a 48-hour incubation with siRNA transfection medium, cells were stimulated with M22 with or without Tepro in DMEM containing 10% FBS and 10 mM HEPES. Three TEDOF strains were used in this experiment; mean ± SEM knockdown efficiency was 87.7 ± 5%. In cells treated with TED-Igs, mean ± SEM knockdown efficiency was 84.6 ± 7%. Experiments where down-regulation of ARRB1 mRNA was < 70% were excluded.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, CA; www.graphpad.com). Biological replicates from the same strains and passages of TEDOFs were normalized, then averaged together. The means from different strains were further averaged to calculate mean ± SEM. A minimum of 3 strains was used for each experiment.
Results
We measured secreted HA from TEDOFs stimulated by pooled TED-Igs in the presence of increasing doses of Tepro (Fig. 1). Tepro efficaciously inhibited TED-Ig-induced HA secretion in 3 TEDOF strains. Maximally effective doses of Tepro were 0.3 to 1.0 µg/mL. We next performed flow cytometry experiments to eliminate the possibility that Tepro inhibits TED-Ig stimulation by blocking their binding to TSHR. We fluorescently tagged M22, a monoclonal TSAb that activates the TSHR in TEDOFs (17), and measured binding of Alexa-647-M22 to HEK293 cells that stably express transfected TSHRs. Figure 2A shows representative histograms from the flow cytometry experiments. HEK-TSHR cells labeled with Alexa-647-M22 (control) exhibited a fluorescent peak that was 2 orders of magnitude higher than cellular autofluorescence (Fig. 2A, top panel). The peak shift was lost in the presence of excess unconjugated M22 (+M22; Fig. 2A, middle panel), that is, unconjugated M22 blocked binding of Alexa-647-M22. In contrast, excess unconjugated Tepro (+Tepro) did not inhibit the peak shift caused by Alexa-647-M22 (Fig. 2A, bottom panel). Figure 2B quantifies mean fluorescent intensities (MFIs) as a % of control for 6 independent measurements. MFI of control was 25-fold higher than cellular autofluorescence (Autofl), P < 0.001. The MFI of + M22 was not significantly different than Autofl. MFI for + Tepro was 30-fold higher than Autofl, P = 0.005, but not significantly different than control. Thus, inhibition by Tepro is not due to steric blocking of TED-Ig binding to TSHR.
Figure 1.

Tepro dose-dependently inhibits TED-Ig-induced HA secretion. Dose-response inhibition by Tepro was measured in TEDOFs (see Materials and Methods) stimulated with 4.9 mg/mL TED-Igs pooled from 37 patients. Secreted hyaluronan (HA) was measured in the conditioned media as described in Materials and Methods. Data represent the mean ± SD of biological duplicates in each strain.
Figure 2.

Tepro does not block M22 binding to TSHR. Tepro binding to TSH receptor (TSHR) was tested by determining whether Tepro, an anti-IGF1 receptor (anti-IGF1R) antibody, could inhibit binding of the monoclonal TSHR stimulating autoantibody (TSAb), M22, tagged with the fluorescent moiety Alexa-647 (Alexa-647-M22). Competitive binding between Alexa-647-M22 and untagged M22 or Tepro on HEK-TSHR cells, which were engineered to express TSHRs at high levels (see Materials and Methods), were tested by fluorescence activated cell sorting (FACS) analysis. Binding was performed by incubating cells with 1:5 ratio of Alexa-647-M22 to unlabeled M22 and Tepro. Competing ligands were added at 4°C 15 minutes before the addition of Alexa-647-M22 and subsequent incubation was for 1 hour at 4°C. (A) White histograms represent autofluorescence. Dark gray histograms show Alexa-647-M22 fluorescence intensity. Cells labeled with Alexa-647-M22 alone (control) exhibited an approximately 2-log peak shift (top panel). The peak shift on cells incubated with excess unconjugated M22 (+M22) was less than 1 order of magnitude. Excess unconjugated Tepro (+Tepro) did not affect the shift in peak (bottom panel). (B) Mean fluorescence intensity (MFI) was calculated as a % of control. Bars depict the average ± SEM of 3 experiments. MFI for autofluorescence (Autofl, white bar) was 4% of control (black bar), P < 0.0001). MFI for + M22 (gray bar) was 8% of control but not significantly different than Autofl. MFI for + Tepro (striped bar) was not significantly different than control.
Because we did not find evidence that Tepro blocks TSAb binding to TSHR, we sought to determine the possible role of TSHR/IGF1R receptor crosstalk in its mechanism of action. We had previously shown in TEDOFs that M22 stimulates TSHR within a putative signalosome containing IGF1R (19) in a biphasic manner. Lower M22 doses activate TSHR-IGF1R crosstalk and higher doses are necessary to stimulate IGF1R-independent TSHR signaling. Moreover, IGF1R antagonists, such as the IGF1R kinase inhibitor linsitinib and the IGF1R antagonist antibody 1H7 can inhibit lower, but not higher, M22 doses, whereas TSHR antagonists can inhibit both components of M22 stimulation (18). Taking advantage of this phenomenon, we measured the Tepro dose-response to low (0.25 nM) and high (1 nM) doses of M22 and compared it with that of K1-70, an inhibitory anti-TSHR antibody (20, 21). The results presented in Fig. 3 confirm that the TSHR antagonist K1-70 was completely efficacious against both low and high doses of M22. In contrast, Tepro fully inhibited low-dose M22 stimulation but only partially inhibited high doses of M22. Thus, Tepro inhibited the IGF1R-dependent but not the IGF1R-independent component of M22 stimulation in TEDOFs.
Figure 3.

Tepro fully inhibits IGF-1R-dependent (low-dose) M22 stimulation but only partially inhibits high dose M22 stimulation of hyaluronan secretion that is comprised of IGF1R-dependent and IGF1R-independent stimulating pathways. Dose-response inhibition by Tepro or K1-70 was measured in TEDOFs (see Materials and Methods) stimulated with 0.25 nM (M22low) or 1 nM (M22high) M22. K1-70 was fully efficacious against M22low (black circles) and M22high (black squares). Secreted hyaluronan (HA) was measured in the conditioned media as described in Materials and Methods. Data represent the mean ± SEM of 3 donor TEDOF strains plotted as percent HA levels of the 1 nM M22 response in the absence of antagonistic antibody.
To demonstrate the dependence of the Tepro effect on TSHR/IGF1R crosstalk, we performed a ARRB1 knockdown experiment in TEDOFs and assessed Tepro’s ability to inhibit M22-induced HA secretion. We showed previously that ARRB1 scaffolded TSHR and IGF1R within 40 nm of each other in a putative signalosome and that this proximity could be lost by knocking down ARRB1 (22). ARRB1 knockdown in TEDOFs prevented crosstalk between TSHR and IGF1R. Here, Fig. 4 shows that knockdown of ARRB1 had no significant effect on basal and 1 nM M22 stimulation of HA production but virtually completely prevented the inhibition caused by Tepro. Thus, ARRB1 1 knockdown prevented TSHR/IGF1R crosstalk and consequently inhibition of M22-induced HA secretion by Tepro in TEDOFs.
Figure 4.
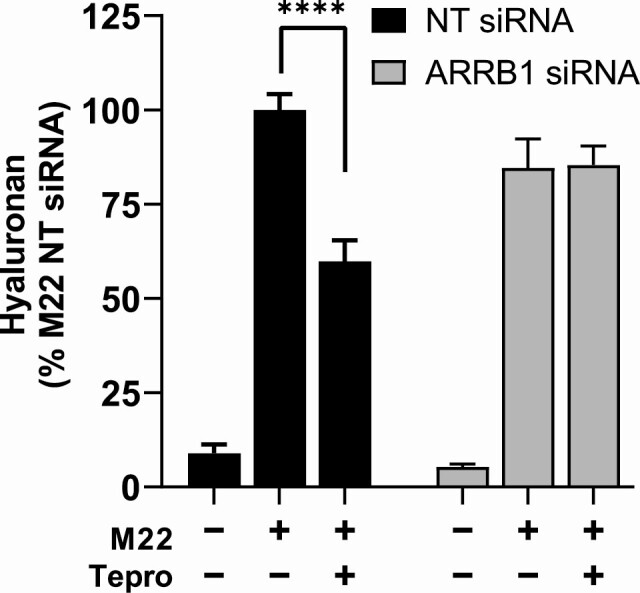
Tepro inhibition of M22 stimulation of hyaluronan production is dependent on TSHR/IGF1R crosstalk in TEDOFs. TEDOFs transfected with nontargeting (NT) or β-arrestin 1 (ARRB1) siRNA were stimulated with 1 nM M22 with or without 1 µg/mL Tepro. Cells treated with ARRB1 siRNA exhibited at least a 70% decrease in ARRB1 mRNA. Secreted hyaluronan (HA) was measured in the conditioned media as described in the Materials and Methods. Data were normalized to percent maximum of M22 in cells exposed to NT siRNA, and data points depict mean ± SEM computed from the averages of triplicate measurements in 3 different TEDOF strains. Under control conditions (black bars), basal HA secretion was 9% of M22 alone, P < 0.0001. Partial inhibition by Tepro lowered HA secretion to 60%, P < 0.0001. Following ARRB1 knockdown (gray bars), HA secretion was 85% for both M22 alone and M22 + Tepro.
The monoclonal TSAb M22 might not represent the mechanism of TEDOF stimulation by all polyclonal autoantibodies found in sera of TED patients. Therefore, our next objective was to determine whether TSHR/IGF1R crosstalk was necessary for Tepro inhibition of TED-Igs, a pool of polyclonal antibodies isolated from TED patients. HA stimulation by TED-Igs was seen in TEDOFs with or without ARRB1 (Fig. 5). In control cells, Tepro significantly inhibited TED-Ig-stimulated HA (Fig. 5). Following knockdown of ARRB1, the inhibitory effect of Tepro on TED-Ig-stimulated HA was lost.
Figure 5.
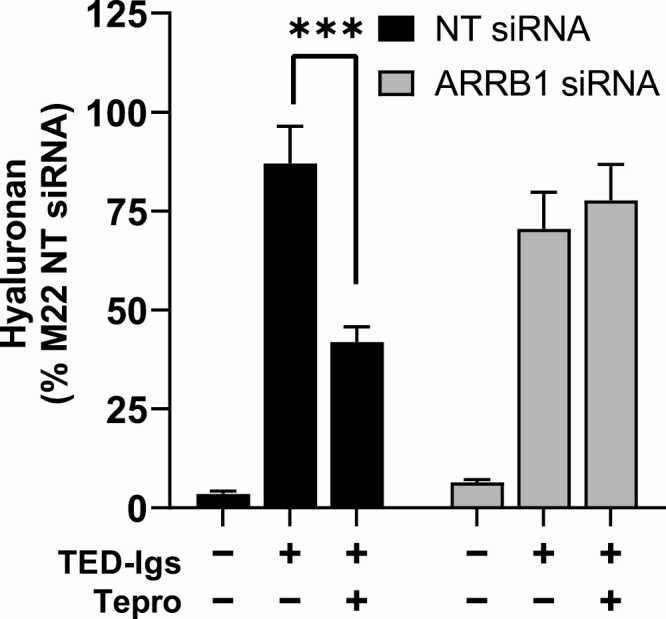
TSHR/IGF1R crosstalk is necessary for Tepro inhibition of GO antibodies. β-arrestin 1 (ARRB1) gene knockdown in TEDOFs was performed as described in the Fig. 4 legend. Cells were incubated with purified antibodies (TED-Igs) isolated from the sera of 30 patients with or without 1 µg/mL Tepro. Secreted hyaluronan (HA) was measured as described in Materials and Methods. Cells treated with ARRB1 siRNA exhibited at least a 70% decrease in ARRB1 mRNA. Data were normalized to percent maximum of M22 in cells exposed to nontargeting (NT) siRNA, and bars represent mean ± SEM computed from the averages of triplicate measurements in 3 different TEDOF strains. Under control conditions (black bars), TED-Ig stimulation of HA secretion was 87% of M22 and partially inhibited by Tepro, P = 0.0004. In cells without β-arrestin 1 (gray bars), TED-Igs stimulation was 70% and not significantly different with Tepro (78%).
Discussion
We previously proposed that IGF1R’s role in TED pathogenesis is through TSHR/IGF1R crosstalk and demonstrated that a subset of IGF1R inhibitory antibodies affect TSHR signaling by disrupting this crosstalk (7). Our hypothesis was that Tepro’s ability to treat TED was through a similar mechanism. To prove this, we first needed to confirm that Tepro did not block binding of M22 to TSHR. Because we had previously shown that TSHR and IGF1R are within 40 nm of each other in a signalosome (22), it was possible the receptors were close enough that binding of Tepro to IGF1R could sterically block binding of TSHR ligands. In our flow cytometry experiment, excess Tepro did not interfere with Alexa-647-M22 binding to TSHR (Fig. 2). This confirms that M22 and Tepro bind to different receptors. Because Tepro did not interfere with ligand binding to TSHR, inhibition of M22-stimulated signaling was likely to be through receptor crosstalk.
We used primary cultures of human cells isolated from the retroorbital tissue of TED patients to model the disease. The 2 doses of M22 used in TEDOFs could distinguish TSHR signaling that was IGF1R-dependent (low doses, high potency) or both IGF1R-dependent and IGF1R-independent (high doses, low potency). Comparing Tepro inhibition at these doses revealed a discrepancy in its effectiveness. Tepro was fully efficacious at low M22 doses, where TSHR predominantly signals through receptor crosstalk. At higher M22 doses—where TSHR signals through both IGF1R-dependent and IGF1R-independent pathways—Tepro inhibition was partial (Fig. 2). The likely explanation is that binding of Tepro to IGF1R only blocked TSHR/IGF1R crosstalk but had no effect on IGF1R-independent TSHR signaling that was induced by M22. We acknowledge there may be other effects of Tepro that contribute to its inhibition of TSAb stimulation, and these may involve noncanonical IGF1R pathways that have yet to be explored (23).
The ARRB1 knockdown experiment directly tested whether crosstalk was necessary for Tepro inhibition of M22. Our previous investigation revealed that activation of ARRB1/ERK signaling led to increased HA secretion from TEDOFs (24), and disruption of that pathway inhibited HA. We additionally showed that the association of TSHR and IGF1R within a putative signalosome was dependent on ARRB1 acting as a scaffold (22). Therefore, in the absence of ARRB1, TSHR predominantly signaled through IGF1R-independent pathways. In cells in which the level of ARRB1 was decreased, Tepro inhibition of M22 was lost (Fig. 4). Taken together, these data demonstrate that Tepro can only inhibit TSHR signaling when IGF1R and TSHR are close to each other, perhaps in a putative signalosome that would allow for TSHR/IGF1R crosstalk. Notably, TSHR/IGF1R crosstalk was necessary for Tepro inhibition of TED-Igs also (Fig. 5).
In conclusion, Tepro inhibits HA secretion by TEDOFs in vitro by inhibiting TSHR/IGF1R crosstalk not by inhibiting binding of TSAbs (or putative IGF1R-stimulating antibodies) to IGF1R. We propose that inhibition of TSHR/IGF1R crosstalk is the mechanism of action of Tepro in treating TED. This study and previous work (18) suggest that drugs that inhibit TSHR activation, such as K1-70 or small molecule TSHR antagonists (18), may be as effective as Tepro in treating TED, and possibly more, because they inhibit both IGF1R-dependent and IGF1R-independent pathways. We additionally suggest that combination treatments using IGF1R and TSHR antagonists may offset adverse effects because they could be used at lower doses and advocate future in vivo studies to test these ideas.
Acknowledgments
The authors thank Tanja Diana, PhD, MSc (Molecular Thyroid Laboratory), Heike Elflein, MD, and Katharina Ponto, MD (Department of Ophthalmology), Johannes Gutenberg University Medical Center, Mainz, Germany, who have generously prepared and provided TED patient tissue and TED serum samples for our experiments.
Funding : This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (Z01 DK011006).
Glossary
Abbreviations
- ARRB1
β-arrestin 1
- Autofl
autofluorescence
- FBS
fetal bovine serum
- HA
hyaluronan
- IGF1R
IGF1 receptor
- MFI
mean fluorescence intensity
- TED
thyroid eye disease
- TEDOF
orbital fibroblast from patients with thyroid eye disease
- TED-Ig
thyroid eye disease immunoglobulin
- Tepro
teprotumumab
- TSAb
TSH receptor-stimulating antibody
- TSHR
TSH receptor
Additional Information
Disclosure Summary : The authors at the National Institutes of Health have nothing to disclose. The Johannes Gutenberg University (JGU) Medical Center, Mainz, Germany, has received research-associated funding from the JGU Medical Faculty, AdvanceCor (Germany), Apitope (UK), Berlin-Chemie (Germany), Byondis (the Netherlands), Horizon Therapeutics (USA), Immunovant (USA), ISAR (Germany), Mediomics (USA), Merck (Germany), Novartis (USA), Quidel (USA), River Vision (USA), and Roche (Switzerland).
Data Availability
All data generated or analyzed during this study are included in this published article.
References
- 1. Douglas RS, Kahaly GJ, Patel A, et al. Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med. 2020;382(4):341-352. [DOI] [PubMed] [Google Scholar]
- 2. Kahaly GJ, Douglas RS, Holt RJ, Sile S, Smith TJ. Teprotumumab for patients with active thyroid eye disease: a pooled data analysis, subgroup analyses, and off-treatment follow-up results from two randomised, double-masked, placebo-controlled, multicentre trials. Lancet Diabetes Endocrinol. 2021;9(6):360-372. [DOI] [PubMed] [Google Scholar]
- 3. Laugwitz KL, Allgeier A, Offermanns S, et al. The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc Natl Acad Sci U S A. 1996;93(1):116-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pappo AS, Vassal G, Crowley JJ, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014;120(16):2448-2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith TJ, Hegedüs L, Douglas RS. Role of insulin-like growth factor-1 (IGF-1) pathway in the pathogenesis of Graves’ orbitopathy. Best Pract Res Clin Endocrinol Metab. 2012;26(3):291-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Minich WB, Dehina N, Welsink T, et al. Autoantibodies to the IGF1 receptor in Graves’ orbitopathy. J Clin Endocrinol Metab. 2013;98(2):752-760. [DOI] [PubMed] [Google Scholar]
- 7. Krieger CC, Place RF, Bevilacqua C, et al. TSH/IGF-1 receptor cross talk in Graves’ ophthalmopathy pathogenesis. J Clin Endocrinol Metab. 2016;101(6):2340-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krieger CC, Morgan SJ, Neumann S, Gershengorn MC. Thyroid stimulating hormone (TSH)/insulin-like growth factor 1 (IGF1) receptor cross-talk in human cells. Curr Opin Endocr Metab Res. 2018;2:29-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen H, Mester T, Raychaudhuri N, et al. Teprotumumab, an IGF-1R blocking monoclonal antibody inhibits TSH and IGF-1 action in fibrocytes. J Clin Endocrinol Metab. 2014;99(9): E1635-E1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen H, Shan SJ, Mester T, Wei YH, Douglas RS. TSH-mediated TNFα production in human fibrocytes is inhibited by teprotumumab, an IGF-1R antagonist. PLoS One. 2015;10(6):e0130322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krieger CC, Neumann S, Gershengorn MC. Is there evidence for IGF1R-stimulating abs in Graves’ orbitopathy pathogenesis? Int J Mol Sci. 2020;21(18):6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krieger CC, Neumann S, Marcus-Samuels B, Gershengorn MC. TSHR/IGF-1R cross-talk, not IGF-1R stimulating antibodies, mediates Graves’ ophthalmopathy pathogenesis. Thyroid. 2017;27(5):746-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pyne NJ, Pyne S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: out of the shadow? Trends Pharmacol Sci. 2011;32(8):443-450. [DOI] [PubMed] [Google Scholar]
- 14. Di Liberto V, Mudò G, Belluardo N. Crosstalk between receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCR) in the brain: focus on heteroreceptor complexes and related functional neurotrophic effects. Neuropharmacology. 2019;152:67-77. [DOI] [PubMed] [Google Scholar]
- 15. Neumann S, Kleinau G, Costanzi S, et al. A low-molecular-weight antagonist for the human thyrotropin receptor with therapeutic potential for hyperthyroidism. Endocrinology. 2008;149(12):5945-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bartalena L, Kahaly GJ, Baldeschi L, et al. ; EUGOGO . The 2021 European Group on Graves’ orbitopathy (EUGOGO) clinical practice guidelines for the medical management of Graves’ orbitopathy. Eur J Endocrinol. 2021;185(4):G43-G67. [DOI] [PubMed] [Google Scholar]
- 17. Krieger CC, Neumann S, Place RF, Marcus-Samuels B, Gershengorn MC. Bidirectional TSH and IGF-1 receptor cross talk mediates stimulation of hyaluronan secretion by Graves’ disease immunoglobins. J Clin Endocrinol Metab. 2015;100(3):1071-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Place RF, Krieger CC, Neumann S, Gershengorn MC. Inhibiting thyrotropin/insulin-like growth factor 1 receptor crosstalk to treat Graves’ ophthalmopathy: studies in orbital fibroblasts in vitro. Br J Pharmacol. 2017;174(4):328-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krieger CC, Gershengorn MC. A modified ELISA accurately measures secretion of high molecular weight hyaluronan (HA) by Graves’ disease orbital cells. Endocrinology. 2014;155(2):627-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Furmaniak J, Sanders J, Rees Smith B. Blocking type TSH receptor antibodies. Auto Immun Highlights. 2013;4(1):11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ryder M, Wentworth M, Algeciras-Schimnich A, et al. Blocking the thyrotropin receptor with K1-70 in a patient with follicular thyroid cancer, Graves’ disease, and Graves’ ophthalmopathy. Thyroid. 2021;31(10):1597-1602. [DOI] [PubMed] [Google Scholar]
- 22. Krieger CC, Boutin A, Jang D, et al. Arrestin-β-1 physically scaffolds TSH and IGF1 receptors to enable crosstalk. Endocrinology. 2019;160(6):1468-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janssen J. New insights from IGF-IR stimulating activity analyses: pathological considerations. Cells. 2020;9(4):862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krieger CC, Perry JD, Morgan SJ, Kahaly GJ, Gershengorn MC. TSH/IGF-1 receptor cross-talk rapidly activates extracellular signal-regulated kinases in multiple cell types. Endocrinology. 2017;158(10):3676-3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.