

Abstract
Background:
Innate immunity is mediated by a variety of cell types, including microglia, macrophages, and neutrophils, and serves as the immune system's first line of defense. There are numerous pathways involved in innate immunity, including the interferon (IFN) pathway, TRK pathway, mitogen-activated protein kinase (MAPK) pathway, Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, interleukin (IL) pathways, chemokine pathways (CCR5), GSK signaling, and Fas signaling.
Summary:
JAK/STAT is one of these important signaling pathways and this review focused on JAK/STAT signaling pathway only. The overactivation of microglia and astrocytes influences JAK/STAT's role in neuroinflammatory disease by initiating innate immunity, orchestrating adaptive immune mechanisms, and ultimately constraining inflammatory and immunological responses. The JAK/STAT signaling pathway is one of the critical factors that promotes neuroinflammation in neurodegenerative diseases.
Key message:
Given the importance of the JAK/STAT pathway in neurodegenerative disease, this review discussed the feasibility of targeting the JAK/STAT pathway as a neuroprotective therapy for neurodegenerative diseases in near future.
Keywords: JAK/STAT signaling, Neuroinflammation, Neurodegenerative disorders, Microglia cells, Aβ amyloid
Introduction
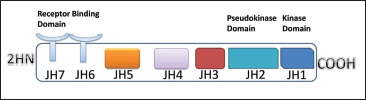
An inflammatory response within the brain or spinal cord is referred to as “neuroinflammation.” This inflammation is mediated by the production of cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers. These mediators are produced by resident CNS glia (microglia and astrocytes), endothelial cells, and peripherally derived immune cells. These neuroinflammatory responses affect the immune system, as well as the physiological, biochemical, and psychological systems. Moreover, the degree of neuroinflammation depends on the context, duration, and course of the primary stimulus or insult. For instance, inflammation can lead to the recruitment of immune cells followed by the JAK/STAT pathway, which is the predominant signaling pathway used by cytokines and is critical for initiating innate immunity, orchestrating adaptive immune mechanisms, and ultimately constraining inflammatory and immune responses. 1 2 3 More than 70 cytokines use this pathway. It is an important pathway in many cancers and neurological disorder. The Janus kinase (JAK) has four isoforms (JAK1, JAK2, JAK3, and TYK2) and STAT has seven isoforms (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6). 4 Structurally they share five domains, which are amino-terminal domain, a coiled-coil domain, a DNA-binding domain, SH2 domain, and a carboxy-terminal transactivation domain (Figure 1).
Figure 1. Schematic Structure of STAT Proteins.
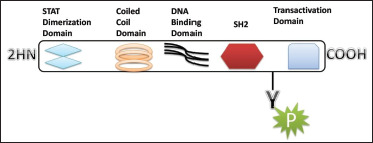
The STAT proteins were discovered as cytoplasmic transcription factors that mediate cellular responses to cytokines and growth factors 1 and 2. Once a ligand interacts with its receptor, STAT activation is induced by the phosphorylation of the key tyrosine residue in the STAT transactivation domain by growth factor receptors, JAKs (Figure 2), SRC family kinases, and other tyrosine kinases. This leads to numerous events including STAT–STAT dimerization through a reciprocal phospho-tyrosine (pTyr)-SH2 domain interaction, nuclear translocation, DNA binding, and the transcriptional induction of genes in the nucleus. Physiological negative regulators, such as suppressors of cytokine signaling (SOCS) and protein tyrosine phosphatases (PTPs), ultimately downregulate the active STAT signaling. However, cytokines, through activation of the JAK/STAT pathway, are of paramount importance in regulating the development, differentiation, and function of myeloid and lymphoid cells. Dysregulation of the JAK/STAT pathway, particularly by activating and polarizing myeloid cells and T cells to pathogenic phenotypes, has pathological implications for neuroinflammatory diseases such as Parkinson's and Alzheimer's. Thus, this review discusses the role of JAK/STAT pathways in neuroinflammatory disease and their contribution to pathogenic inflammation linked with their therapeutic management.
Figure 2. Schematic Structure of JAK Proteins.
Neuroinflammation
Role of JAK/STAT Pathway in Neuroinflammation
The JAK/STAT is a complex signaling pathway regulated by many cytokines and plays a dual role in cell proliferation and cell death. It acts as an activator for many cytokines which results in cell growth and death (Table 1). These activated cytokines cause inflammation in cells and are also responsible for cell death in the many regions of the brain. 5 The JAK/STAT-mediated activation of cytokines regulates the differentiation and function of myeloid and lymphoid cells. 6 Dysregulation of the JAK/STAT pathway has pathological implications for neuroinflammatory diseases, especially by triggering and polarizing myeloid cells and T cells to pathogenic phenotypes. 7
Table 1. The Role of STAT Isoform and Their Deficiency.
| Isoform of STATs | Activators | Function | Deficiency |
| STAT1(antiapoptotic) | Interferon (IFN) stimulation and supports immune | Function partly by controlling the growth and apoptosis of immune cells 4 | Leads to tumor |
| STAT2 | IFN-α and IFN-β | Promotes STAT3 activation 10 | Up regulation of interleukin-6 (IL-6) |
| STAT3 | These genes include those that encode p21WAF1/CIP2, cyclin D1, MYC, BCL-XL, BCL-2, vascular endothelial growth factor (VEGF), matrix metalloproteinase 1 (MMP1), MMP7, and MMP9, and survivin16,17,19,19, 21,22,23 | Essential for early embryonic development | That STAT3 functions are essential for IL-6-mediated
antiapoptotic responses
13
STAT3 for promoting malignant transformation and progression |
| STAT4 | Crucial mediator of IL-12 function that regulates the differentiation of TH1 cells and their inflammatory responses 5 | Associated with autoimmune diseases | |
| STAT5A | Important in mammary gland development and milk production | Defects in IL-2 receptor-α expression in T lymphocytes 12 | |
| STAT5B | Promoting cancer, including the induction of proproliferative and antiapoptotic genes | Aberrant STAT5 activation by BCR–ABL in CML | |
| STAT6 | IL-4 and IL-13 |
Activation of STATs occurs by the phosphorylation at the carboxy-terminal domain that contains serine/tyrosine residue. For the translocation of STAT from the cytosol to the nucleus, dimerization of STAT is required. Phosphorylation on tyrosine residue leads to dimerization of STAT molecules and that on serine residue enhances the transcriptional activation. 8 9 10 Previously STAT was known as a cytoplasmic transcription factor that facilitates the activation of growth factors and cytokines. The activity of STAT is regulated by phosphatase and JAK is regulated by the SOCS. Phosphorylation at the tyrosine residue plays an important role in the activation of STATs. The fundamental process such as cell growth, differentiation, apoptosis, development, inflammation, and immune response requires STAT. 11
The IFN-mediated STAT1 signaling regulates the immune function followed by controlling the growth and apoptosis of immune cells 12 (Table 1). STAT1 signaling controls the production of cytokines by T helper type 1 (TH1) cells, which alters immune function and inflammatory responses by changing the balance between TH1 and TH2 cells (Table 2). 13 Indeed, mice with STAT1 deficiency lose their ability to respond to IFN, making them more vulnerable to bacterial and viral infections 6 . Furthermore, malignant cells benefit from the loss of IFN responsiveness caused by STAT1 deficiency, which contributes to increased tumor development. 14 Although recent results indicate that STAT1 plays a more complex role in carcinogenesis, this finding indicates that the protein has a tumor-suppressive function. 15 Furthermore, STAT1 null mice with atherosclerosis-prone bone marrow transplantation have less foam cell formation and atherosclerosis, indicating that STAT1 plays a proatherogenic position. 16 Benefit of function mutations in the STAT1 gene, on the other hand, affect TH1 and TH17 cell responses and cause chronic mucocutaneous candidiasis because of STAT1 hyperactivation and defective nuclear dephosphorylation. 17
Table 2. The Inflammatory and Cellular Function of the JAK/STAT Pathway.
| Pathways | Cellular Function | Major Disease | Cytokines and Factor |
| STAT1 | Cell growth and apoptosis TH1 cell-specific cytokine
production Antimicrobial defense |
Atherosclerosis Infection Immune disorders |
IL-2, IL-6, IL-10, IFN-a, IFN-b, IFN-g, IL-27 |
| STAT2 | Mediation of IFN/IFN signal | Cancer Infection Immune disorders |
IFN-a, IFN-b |
| STAT3 | Cell proliferation and survival Inflammation Immune response Embryonic development Cell motility |
Cancer | LIF, IL-10, IL-6, IL-27, Growth hormone |
| STAT4 | TH1 cell differentiation Inflammatory responses Cell proliferation |
Experimental autoimmune encephalomyelitis (EAE) (multiple
sclerosis; MS) Systemic lupus erythematosus |
IL-12 |
| STAT5a | Cell proliferation and survival IL-2R expression in T
lymphocytes Mammary gland development Lactogenic signaling |
Cancer, chronic myelogenous Leukemia |
Prolactin, growth hormone, thrombopoietin |
| STAT5b | Cell proliferation and survival IL-2R expression in T
lymphocytes Sexual dimorphism of body growth rate NK cell cytolytic activity |
Cancer, chronic myelogenous leukemia | |
| STAT6 | Inflammatory and allergic immune response B cell and T cell proliferation |
Asthma, allergy | IL-4, IL-13 |
| JAK1 | Cellular proliferation, immune cell division, phosphorylation, and activation of transcription factors (STAT) | IL-2, IL-7, IL-9, IL-15, IL-4, IL-13, IL-6, IL-11, | |
| JAK2 | IL-3, IL-12, IL-13, IL-6, IL-11, IFN-gama, CT-1, | ||
| JAK3 | IL-2, IL-7, IL-9, IL-15, IL-4 | ||
| TyK2 | Dendritic cell differentiation, pro-inflammatory TH cell response, TH2 responses | Hyper IgE syndrome, Job's syndrome | IL-6, IL-11, IL-12, IL-13, CT-1, IFN-α, IFN-β, |
Role of Microglia in Neuroinflammation
The word “microglia” is associated with cells residing in the parenchyma of CNS and functionally differentiates as resident macrophage of CNS. The defensive role of microglia is shown in CNS pathologies like brain injury, cancer, age-associated neurodegenerative disorders, and stroke.18, 19 Although the role of microglia cells in healthy and unhealthy conditions is to establish synaptic pruning and maintain the inflammation, phagocytosis, and regeneration in CNS pathologies.20, 21 Therefore, the defensive role of microglia initiated with initiation of neuroinflammation coexist with neurodegenerative disease and other CNS pathologies. The pathological stimulus mediates microglia induction, allows effective function with site-specific migration of microglial cells with secretion pro-inflammatory cytokines and reactive oxygen and nitrogen species (ROS, NOS) engulfing and digesting dying cells, infectious agents, toxic protein aggregates, and phagocytosis cell debris, and also secretes anti-inflammatory cytokines and trophic factors for nerve tissue repair and regeneration.22, 23 Therefore, the functional activity of microglial classify in M1 “classical activated” and M2 “alternatively activated.” The M1 induces a pro-inflammatory response with activation of (tumor necrosis factor-α; TNF-α, IL-1β, IL-6, COX-2, ROS, and NO) and performs a neurotoxic role. Whereas M2 secretes anti-inflammatory response and induces neuroprotective role and induces phagocytosis to remove toxic protein aggregation associated with cellular debris. 24 The opposing role of microglial cells is associated with a crucial and successful immune response against the CNS pathologies. Thus, the imbalance in microglial role contributes for the development of neurological disorders. However, during brain injury, M2 is eventually replaced by M1 in later phase and initiates inflammation with impairment of neuronal recovery and regeneration. 25
Role of Astrocytes in Neuroinflammation
Astrocytes are resistant to endoplasmic stress (ER) and instead activate an inflammatory program involving activation of STAT3 in a JAK1-dependent fashion. The activation of astrocytes is ER-dependent and mediated by a series of activators. Furthermore, STAT3 signaling is dependent on PERK, a key component of the ER stress response. PERK inhibition prevents ER stress-induced activation of STAT3 and subsequent gene expression. 26 Additionally, ER-stress activated astrocytes induced a pro-inflammatory response and are shown to be critical for induction of EAE.27, 28 SOCS negatively regulate the immune response, primarily by interfering with the JAK/STAT pathway. However, the SOCS protein is an inhibitor of JAK/STAT signaling in EAE. The study reported in mice model shows that the conditional deletion of SOC3 in myeloid cells develops a critical, nonresolving form of EAE after MOG immunization. 2 The study observed upregulation of STAT3 with increased production of inflammatory cytokine/chemokines in CNS of mice model. Therefore, the activated JAK/STAT signaling hyperactivates the pro-inflammatory macrophage phenotype with elevation in the production of IL-6, IL-12, IL-23, Th1, and Th17 with pathogenic role in EAE. 29
Parkinson and Neuroinflammation
Parkinson’s disease is a movement disorder associated with loss of dopaminergic neurons in substantia nigra pars compact and aggregation of misfolded synuclein (SYN), followed by the common neurodegenerative disorder. 30 However, the regulation of the SYN gene is linked to mutation and leads to overexpression and aggregation of protein. 31 The secretion of oligomeric SYN induces toll-like-receptor-mediated activation of microglia and macrophages and leads to the production of pro-inflammatory mediators. 32 Emerging evidence reported in PD patients microgliosis in substantia nigra pars compact induce pro-inflammatory response with upregulation of major histocompatibility complex class II (MHCII) and intense the toxicity of SYN with critical role in PD pathophysiology.33, 34
Alzheimer’s and Neuroinflammation
Alzheimer's disease (AD) is a brain ailment that causes cell degradation and is the leading cause of dementia, which is defined by deterioration in thinking and independence in routine daily tasks. AD is regarded as a complex disease, with two major hypotheses offered as causes of the disease: the cholinergic and amyloid hypothesis. Currently, research is focusing on understanding AD pathophysiology by addressing multiple processes, including aberrant tau protein metabolism, amyloid, inflammatory response, and cholinergic and free radical damage. Neuroinflammation, along with Aβ or neurofibrillary tangles (NFT), is the third fundamental neuropathological characteristic in AD brains. Activated astrocytes and microglia are commonly observed around neurons and plaques. 35 Several pro-inflammatory cytokines or inflammatory markers were also shown to be overexpressed in AD brains.36, 37 This inflammatory response has been hypothesized as a response to the gradual accumulation of Aβ plaques and NFTs. Chronic or uncontrolled activation of these inflammatory pathways causes neuronal injury or death. Various clinical and preclinical approaches have been used to unravel the involvement of neuroinflammation in AD, including histopathological inspection, neuroimaging, and the identification of proteomic and other biomarker signatures in blood and cerebrospinal fluid (CSF). 38 Several studies have looked at cytokines such IL-1, IL-2, IL-6, IL-8, IL-10, IL-12, IL-18, IFN, TNF, and transforming growth factor (TGF) in the blood and CSF of AD patients. 39 40 41 A pooled research discovered overall elevations in IL-1, IL-6, IL-12, IL-18, TNF, and TGF levels in AD patients compared to healthy controls, as well as increases in APP expression and tau protein phosphorylation via the p38-MAPK pathway. 42 Despite this, oligodendrocytes are the primary generator of myelin in the central nervous system (CNS). The involvement of oligodendrocytes in the neuroinflammation of AD is still poorly understood. Oligodendrocytes were found to be capable of complementing manufacturing in an in vitro investigation, suggesting that they could contribute to the neuroinflammation process. Many extracellular signals, such as cytokines, growth factors, nucleotides, endothelins, or ephrins, can be sensed by astrocytes and can activate various intracellular signaling pathways, including the mitogen-activated protein kinase (MAPK), the nuclear factor B (NF-B), and the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways. The JAK/STAT3 pathway appears to be a central player in the induction of astrocyte reactivity. It is activated by a number of cytokines and growth factors that communicate via the gp130 receptor.
Multiple Sclerosis (MS) and Neuroinflammation
Multiple sclerosis (MS) is an inflammatory, demyelinating, and neurodegenerative disorder of the CNS that affects over two million people worldwide. 43 Early MS lesions are distinguished by focal infiltration of lymphocytes and monocytes into brain or spinal cord regions, breakdown of the blood–brain barrier (BBB), and varying degrees of demyelination, remyelination, and axonal loss. Magnetic resonance imaging (MRI) can detect inflammatory lesions, which are about ten times more common than periods of acute clinical worsening. 44 Within lesions, astrocytes have been shown to express the pro-inflammatory cytokine IL-17. As a result, while resident cells are anti-inflammatory in the physiologic state, they can switch to a pro-inflammatory phenotype in inflammatory conditions such as EAE and MS. The majority of evidence suggests that this involvement is merely a secondary event following leukocyte infiltration. However, some investigators proposed a primary involvement of residents, in particular, astrocytes and microglia in disease pathogenesis.
Huntington’s Disease (HD) and Neuroinflammation
Huntington's disease (HD) is characterized by the loss of neurons in the striatum and cortex, which results in motor dysfunction, cognitive decline, and psychiatric disorders. HD is a genetic disorder caused by trinucleotide (CAG) repeat expansion in the Huntingtin (HTT) gene on chromosome 4p16.3. Other metabolic pathways and mechanisms, in addition to impaired systems for dealing with abnormal proteins, may contribute to neurodegeneration and the progression of HD. Inflammation appears to play a role in HD pathogenesis. The current review summarizes the research on immune and/or inflammatory changes in HD. HD is associated with an increase in inflammatory mediators in both the CNS and the peripheral nervous system. As a result, some attempts have been made to slow the progression of HD by targeting the immune system. 45 At first glance, neuroinflammation appears to be beneficial to neuronal tissue because it promotes cell debris clearance, but the situation is far more complicated. Inflammatory mediators, for example, act on neurons as well as immune cells, potentially contributing to neuronal death. Neurodegeneration activates inflammatory mechanisms further, resulting in a vicious cycle of inflammation and neurodegeneration. Systemic inflammation, in addition to inflammatory processes in the CNS, may contribute to neuronal damage in HD. Increased inflammatory mediator production is caused by microglial activation and astrocytosis. IL-6, IL-8, TNF-α, monocyte chemoattractant protein-1 (MCP-1/CCL2), and IL-10 mRNA levels were significantly higher in the striatum of HD patients compared to controls. Furthermore, increased expression of IL-6, IL-8, and MMP-9 was observed in the cortex and cerebellum of HD patients.
Microglial Involvement in Parkinson Disease
The dysregulation of the immune system is associated with chronic inflammation in CNS and plays a major role in the pathogenesis of PD patients. 46 The activation of innate system induce an overexpression of SNCA1 gene and favor the pathogenic character. 47 However, microglia have a pro-inflammatory phenotype in PD and urge the invasion of macrophages, as reported in studies. 34 In addition, the reported data show that the overexpression of SYNCA1 induces microgliosis and activates MHCII complex with upregulation of NO and pro-inflammatory markers TNF-α. IL-1β 36 ensue the neuronal damage. However, the involvement of JAK/STAT pathways in activation and recruitment of pro-inflammatory marker in PD was repealed by the inhibition of JAK/STAT signaling. 48
Much as enhanced microglial activation leads to the development of PD, inhibition of microglial activation tends to be beneficial. Blocking the CD200-CD200R interaction, an inhibitory mechanism that suppresses microglial activation exacerbates microglial activation and dopamine (DA) neurodegeneration in the PD rat model. 49 Similarly, treatment with minocycline, an FDA-approved tetracycline derivative that inhibits microglial activation, prevented nigrostriatal DA neurodegeneration in the PD MPTP model. Mice with IFN-gamma deficiency, which promotes the polarization of pro-inflammatory macrophages by activation of the JAK/STAT pathway, showed attenuated MPTP-induced substantia nigra pars compacta DA neuron loss. 50 Genetic models also indicate the role of microglia in PD pathogenesis. Leucine-rich repeat kinase 2 (LRRK2) is the most frequently mutated gene in idiopathic and family PD. 51 Expression of LRRK2 is enhanced in immune cells, especially in B cells, monocytes, and DCs. 52 LRRK2 is an IFN-gamma-mediated gene and is involved in the development of ROS during the host defense. 53 While LRRK2 mutations have been shown to be associated with PD, their function, particularly in immune cells during neuroinflammation, is not well understood. The G2019S mutation in LRRK2 has been shown to worsen the induced neuroinflammation and DA neurodegeneration of α-SYN and has been mitigated by inhibition of LRRK2. 54 Recent studies have shown that the G2019S mutation of LRRK2 attenuates microglial motility by inhibiting focal adhesion kinase activity and, as a result, suppresses microglial response to stab-wound and laser-induced brain injury. 55 The genome-wide association study (GWAS) also showed a link between PD susceptibility and HLA-DR, a gene that encodes MHCII in humans. 56 Additional evidence further supports the role of MHCII in PD pathogenesis. In the mouse model of PD, the overexpression of 5-007-SYN contributed to an upregulation of MHCII in microglia. In addition, MHCII deletion blocked the activation of microglia, CD4+ T cell proliferation, and DA neurodegeneration. 57
Microglial Interaction with Aβ Amyloid
In AD, microglia interact with Aβ oligomers and fibrils via scavenger receptors (SCARA-1, MARCO, SCARB-1, CD36, and RAGE), G-protein coupled receptors (FPR2 and CMKLR1), toll-like receptors (TLR2, TLR4, TLR6, and TLR9), CD47, 61 integrin, and TERM2. 58 59 60 Moreover, early studies established that different species of Aβ aggregates could induce glial activation and the production of pro-inflammatory cytokines (including IL-1, IL-6, IL-8, and TNF), anti-inflammatory cytokines (including MCP1 and macrophage inflammatory protein 1), chemokines (including MCP1 and macrophage inflammatory protein 1), various cell adhesion molecules, NO, and ROS, all of which could lead to neuron death.61, 62 Smaller oligomers of Aβ have been found to be significantly more potent stimuli for inducing the microglial response and to be more lethal to neurons than larger oligomers135. Thus, these molecules activate the receptor-ligand-induced molecular pathways that cause microglial phenotypic changes.63, 64 For example, binding of Aβ fibrils to TLR2 induces IL-8 and TNF expression 137; activation of scavenger receptors in the presence of Aβ increases IL-1 and NO production via the NF-B, c-Jun N-terminal kinase (JNK), and MAPK pathways; 65 and recognition of Aβ by the CD36–CD47–61 integrin complex mediates phagocytosis. However, chronically activated microglia caused by Aβ generate pro-inflammatory mediators, which reduce phagocytosis ability and prolong neuroinflammation. 66 Aβ also stimulates astrocytes, potentially via the NF-B pathway. 67 Activated astrocytes degrade Aβ and promote microglia phagocytosis via ApoE lipidation. 68 However, active astrocytes create inflammatory mediators, which contribute to the neuroinflammation process. 69
Role of JAK/STAT Pathway in Brain
Brain cells are derived from neural stem cells (NSC) of neural progenitor cells and these stem cells also differentiate into neurons, astrocytes, or oligodendrocytes, and as the brain grows, the number of NSC decreases. For the differentiation and proliferation of brain cells from NSC, JAK/STAT pathway plays a major role. Two major regions of neurogenic cells in the brain are the subventricular zone (SVZ) of the olfactory bulbs and the dentate gyrus (DG) of the hippocampus. IL15 is a cytokine that localizes at SVZ and DG and causes the activation of STAT1, STAT3, STAT 5, and is regulated by leptin. Leptin is a JAK inhibitor and also regulates the neuro-proliferation in the DG of adult mice via activation of STAT3 and Akt. The embryonic cortical precursor cells activate the ciliary neurotrophic factor (CNTF) receptor leading to the activation of JAK1, STAT1, and STAT3 and causing the differentiation of these precursor cells into astrocytes. Cytokine and hormones lead to the proliferation of differentiation of NSC through the JAK/STAT pathway. Many studies reveal the role of STAT3 in glial differentiation. However, neurogenesis in some parts of the hippocampus is dependent on STAT3 activation, which explains the different gene expressions at embryonic and adult NSC.
JAK/STAT Pathway and Parkinson Disease
The JAK/STAT signaling prompts the production of cytokine followed by the activation of innate and adaptive response and eventually constraining the inflammatory and immune reaction. 70 The JAK/STAT-mediated cytokine activation have crucial importance in regulation, differentiation, and function of myeloid, a lymphoid cell. 71 Therefore, dysregulation of JAK/STAT pathways involve in neuroinflammatory disease by polarizing myeloid and T cells in pathogenic phenotype. 2 The JAK/STAT pathways are a therapeutic target for the treatment of autoimmune disease. Thus, Jakinibs FDA approved drug use to downregulate the JAK/STAT-induced cytokine expression in CSF and CNS. 72 However, the AAV2-SYN rat model studies reported that the SYN overexpression mediates the activation of JAK/STAT signaling and dysregulates the innate and adaptive immune response leading to neurodegeneration. Whereas, the AAV2-SYN treatment with Jaknibs is therapeutic. These results collectively record that repression of the JAK/STAT pathway disrupts the neuroinflammation and neurodegeneration circuitry characteristic of PD models.73
JAK/STAT Pathway and Alzheimer’s Disease (AD)
STAT3 is one of the key regulators of the pathways associated with astrogliosis, activated in response to cytokines, intercellular mediators, and growth factors. 74 Activation of JAK/STAT3 has been observed in astrocytes in many conditions and disease models such as stroke, AD, spinal cord injury, MS, and epilepsy. 74 STAT3 may increase neurodegeneration in a context-dependent manner which suggests that the many cellular pathways initiated by STAT3 signaling remain to be investigated. Alteration in the JAK2/STAT3 pathway may involve neurodegenerative diseases like AD independent of any inflammation process. Nicotinic acetylcholine receptors reduce Aβ neurotoxicity by activating JAK2/STAT3, but whether the neuroprotection requires STAT3 gene regulation is not known. Humanin and its derivatives abolish Aβ neurotoxicity by activating the JAK2/STAT3 pathway and maintaining cholinergic activity. 4
JAK/STAT Pathway and Multiple Sclerosis (MS)
The JAK/STAT pathway is important in the pathophysiology of MS, according to GWAS. STAT3, STAT4, and TYK2 genetic variants have been linked to MS susceptibility in a variety of populations. 7 Many JAK/STAT pathway-related genes, such as IL-2RA, IL-7, IL-7R, IL-12A, IL-12B, and HLA-DRB1 and HLA-A, have been found as MS risk factors in GWAS research. Overall, both genetic evidence and elevated levels of JAK/STAT-related cytokines show that the JAK/STAT pathway plays a significant role in the pathogenesis of MS, either directly or indirectly. Oligodendrocytes aid in the production of myelin in the CNS. One of the fundamental hallmarks of leukodystrophies and MS, two of the most common white matter illnesses, is the death of these cells and the resultant demyelination. STAT1 and/ or STAT3 activation by cytokines or JAK-STAT or STAT5 by glucocorticoid receptor enhances oligodendrocytes survival, which counteracted by SOCS3. Pharmacological manipulation of SOCS and STAT activity could lead to new and intriguing therapeutic options for demyelinating disorders. 4
JAK/STAT Pathway and Huntingtin’s Disease (HD)
Ben Haim et al. shows that inhibiting the JAK/STAT3 pathway increased the frequency of Huntingtin aggregates, a neuropathological hallmark of HD, in reactive astrocytes. 75 In HD, researchers looked at intracellular signaling networks in peripheral blood cells. The JAK/STAT pathway is the most typically activated signaling pathway downstream of cytokine receptors, coordinating cytokine-mediated gene expression and repression. Trager et al. (2013) used flow cytometry to look at the JAK/STAT pathway in monocytes from HD patients. While phosphorylated (p)STAT1 and pSTAT3 levels were unaffected at the start, pSTAT5 levels in HD gene carriers' monocytes were considerably higher than in control cells. Targeted activation of JAK/STAT signaling molecules with specific STAT1, STAT3, and STAT5 activators (IFN-, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF), respectively) found the same level of pathway activation in control and HD monocytes, indicating that the signaling cascade functions normally in HD monocytes. 45
STAT Protein Target for Drugs—(Therapeutic Approaches that Target STAT Proteins)
The first peptide inhibitor of a STAT protein was discovered more than a decade ago, and attempts to target STAT signaling for therapeutic purposes are still ongoing. As the evidenced number of studies relating to STAT3 shows, extensive drug research efforts are underway. In comparison, as shown by the few published papers, research attempts for inhibitory modulators of STAT1, STAT2, STAT4, STAT5, and STAT6 have been modest. STAT signaling and function are being studied using a variety of inhibitory techniques. The majority of these are peptides or peptidomimetics created by structure-based design, small molecules found through molecular modeling, computer or library scanning, or natural products that prevent STAT dimerization. In addition, inhibition of tyrosine kinases that phosphorylate STAT proteins or inducing phosphatases that dephosphorylate STAT proteins may also stop STAT dimerization, and signaling and other methods include the use of oligodeoxynucleotide (ODN) decoys as the basic STAT DNA-binding domain inhibitors and antisense oligodeoxynucleotide (ASO) inhibitors. As we describe above, STAT plays a major role in the pathophysiology of many diseases and can be the target for the drugs. The STAT-3 is mutated in many cancer and disease there are different types of inhibitor such as natural product, SiRNAs and oligodeoxynucleotide decoy in which some of them are under clinical trial at different phases. Thus, JAK/STAT pathway is a therapeutic target in autoimmune diseases and solid and liquid tumors that interrupt signaling downstream of multiple cytokines to various degrees, which may be a useful approach for PD, which is characterized by elevated levels of numerous cytokines in the periphery, CSF, and CNS.
It has been recently discovered that microRNA can play an important role in the regulation of brain immune response and can act as a regulator of these immunological reactions. The role of miR155 is well associated with MS, and many other miRNAs involve in the pathogenesis of the disease. Some miRNAs are found to be upregulated and many downregulated; the changes in the concentration of miRNA can increase the permeability (open a breach to peripheral immune cells) of peripheral immune cells through BBB by sacrificing the tight junction protein. This evidence was found in the MS patients with the regulation of the TLR. 76 In a study, in the treatment of TNF-α, IL-1β with LPS in monocytic cells elevates the expression of miR-155, miR-132, and miR-146 with downregulation of TLR signaling to inhibit the inflammation. Whereas, the suppression of CCAAT-enhancer-binding protein β (C/EBPβ) via overexpression of miRNA155 elevates the self-renewal capacity of neuronal stem cells with suppression of Msi, Hes1, and Bmi1 gene with role in proliferation of neuronal stem cells. 77 Suppression of these genes is mediated by the transcription factor C/EBPβ, which is also the target for miR155. Therefore, miR155 can be used as a potential therapeutic target. 77 However, the expression of miR124 is crucial to maintain the microglial cells in the quiescent state in MS and EAE. 78 Moreover, the activated microglial cells downregulate the expression of miR124 with upregulation of CCAAT-enhancer-binding-protein α (C/EBPα) and PU.1, 79 where PU.1 regulates the macrophage cell differentiation and proliferation which involve in phenotypic activation monocyte lineage. 80
Conclusion
Inflammation occurred in a minority of diseases and is caused by a variety of factors, including cytokine, signal transduction proteins, ROS, stress, toxin, bacteria, and viruses. In the current scenario, studies on the level of inflammation are being conducted all over the world in order to correlate disease with inflammation. However, inflammation is not the only factor that plays a significant role in the pathophysiology of the disease, but it is also associated with the factors that cause the disease. These neuroinflammatory processes are not primarily related to the pathogenesis of AD and PD. Furthermore, these diseases and their consequences are a global health burden, and targeting neuroinflammation may pave the way for the management and therapeutic intervention.
Acknowledgments
Mayank Jain is grateful to the University Grant Commission for providing him the Maulana Azad National Fellowship.
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: In compliance with the ICMJE uniform disclosure form, all authors declare that they have no known competing financial interests or personal relationships that could have appeared to influenced the work reported in this paper.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Mayank Jain https://orcid.org/0000-0002-5321-8340
https://orcid.org/0000-0002-5321-8340
Mukul Kumar Singh https://orcid.org/0000-0003-1074-4647
https://orcid.org/0000-0003-1074-4647
Authors’ Contribution
Substantial contributions to the conception or design of the work and the acquisition; drafting the work or revising it critically for important intellectual content.
Statement of Ethics
This study did not include any human subjects, so it is inapplicable. Therefore, no ethics committee approval was required for this study.
References
- 1.O'Shea JJ and Plenge R.. JAK STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity; April 20, 2012; 36(4): 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benveniste EN, Liu Y, McFarland BC, et al. Involvement of the Janus kinase/signal transducer and activator of transcription signaling pathway in multiple sclerosis and the animal model of experimental autoimmune encephalomyelitis. J Interferon Cytokine Res August 2014; 34(8): 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Shea JJ, Schwartz DM, Villarino AV, et al. The JAK/STAT pathway: Impact on human disease and therapeutic intervention. Ann Rev Med 2015; 66: 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolas CS, Amici M, Bortolotto ZA, et al. The role of JAK/STAT signaling within the CNS. JAK-STAT; January 01, 2013; 2(1): e22925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan Z, Gibson SA, Buckley JA, et al. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol , 2018. April; 189: 4–13. DOI: 10.1016/j.clim.2016.09.014. E pub 2016 October 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jatiani SS, Baker SJ, Silverman LR, et al. JAK/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer October 2010; 1(10): 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Gibson SA, Benveniste EN, et al. Opportunities for translation from the bench: Therapeutic intervention of the JAK/STAT pathway in neuroinflammatory diseases. Crit Rev Immunology 2015; 35(6): 505–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darnell Jr JE.STATs and gene regulation. Science 1997; 277(5332): 1630–1635. [DOI] [PubMed] [Google Scholar]
- 9.Bromberg J and Darnell JE Jr.. The role of STATs in transcriptional control and their impact on cellular function. Oncogene May 15, 2000; 19(21): 2468–2473. [DOI] [PubMed] [Google Scholar]
- 10.Stark GR, Darnell JE. The JAK/STAT pathway at twenty. Immunity; April 20, 2012; 36(4): 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miklossy G, Hilliard TS, Turkson J.. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov August 2013; 12(8): 611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sironi JJ, Ouchi T.. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem; February 6, 2004; 279(6): 4066–4074. [DOI] [PubMed] [Google Scholar]
- 13.Kaiko GE, Horvat JC, Beagley KW, et al. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology March 2008; 123(3): 326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castro F, Cardoso AP, Goncalves RM, et al. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol 2018; 9: 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koromilas AE, Sexl V.. The tumor suppressor function of STAT1 in breast cancer. JAK-STAT; April 1, 2013; 2(2): e23353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal S, Febbraio M, Podrez E, et al. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation; June 12, 2007; 115(23): 2939–2947. [DOI] [PubMed] [Google Scholar]
- 17.Zheng J, van de Veerdonk FL, Crossland KL, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol October 2015; 45(10): 2834–2846. [DOI] [PubMed] [Google Scholar]
- 18.Rock RB, Gekker G, Hu S, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev October 2004; 17(4): 942–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hickman S, Izzy S, Sen P, et al. Microglia in neurodegeneration. Nat Neurosci October 2018; 21(10): 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez-Nicola D and Perry VH.. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neuroscientist: Rev J Bringing Neurobiol, Neurol Psychiatry April 2015; 21(2): 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GP, Clark IA, Zinn R, et al. Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Memory October 2013; 105: 40–53. [DOI] [PubMed] [Google Scholar]
- 22.Nayak D, Roth TL, McGavern DB.. Microglia development and function. Ann Rev Immunol 2014; 32: 367–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraft AD, Harry GJ.. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health July 2011; 8(7): 2980–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco R and Fernandez-Suarez D.. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol August 2015; 131: 65–86. [DOI] [PubMed] [Google Scholar]
- 25.Donat CK, Scott G, Gentleman SM, et al. Microglial activation in traumatic brain injury. Front Aging Neurosci 2017; 9: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meares GP, Liu Y, Rajbhandari R, et al. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol October 2014; 34(20): 3911–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprenkle NT, Sims SG, Sanchez CL, et al. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegeneration; May 25, 2017; 12(1): 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Constantinescu CS, Farooqi N, O'Brien K, et al. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol October 2011; 164(4): 1079–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turner MD, Nedjai B, Hurst T, et al. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta November 2014; 1843(11): 2563–2582. [DOI] [PubMed] [Google Scholar]
- 30.Mhyre TR, Boyd JT, Hamill RW, et al. Parkinson's disease. Subcell Biochem 2012; 65: 389–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefanis L.Alpha-Synsclein in Parkinson's disease. Cold Spring Harb Perspect Med February 2012; 2(2): a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moussaud S, Jones DR, Moussaud-Lamodiere EL, et al. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol Neurodegeneration October 29, 2014; 9: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belloli S, Morari M, Murtaj V, et al. Translation imaging in Parkinson's disease: Focus on neuroinflammation. Front Aging Neurosci 2020; 12: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreira SA, Romero-Ramos M.. Microglia response during Parkinson's disease: Alpha-synuclein intervention. Front Cell Neurosci 2018; 12: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeTure MA, Dickson DW.. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegeneration; August 2, 2019; 14(1): 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang WY, Tan MS, Yu JT, et al. Role of proinflammatory cytokines released from microglia in Alzheimer's disease. Ann Translational Med June 2015; 3(10): 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JC, Han SH, Mook-Jung I.. Peripheral inflammatory biomarkers in Alzheimer's disease: A brief review. BMB Rep January 2020; 53(1): 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hampel H, Caraci F, Cuello AC, et al. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer's disease. Front Immunol 2020; 11: 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baird AL, Westwood S, Lovestone S.. Blood-based proteomic biomarkers of Alzheimer's disease pathology. Front Neurol 2015; 6: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayaprolu S, Higginbotham L, Bagchi P, et al. Systems-based proteomics to resolve the biology of Alzheimer's disease beyond amyloid and tau. Neuropsychopharmacology January 2021; 46(1): 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedrero-Prieto CM, Frontiñán-Rubio J, Alcaín FJ, et al. Biological significance of the protein changes occurring in the cerebrospinal fluid of alzheimer's disease patients: Getting clues from proteomic studies. Diagnostics (Basel, Switzerland: ) 2021; 11(9): 1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wyss-Coray T and Rogers J.. Inflammation in Alzheimer disease: A brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med January 2012; 2(1): a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutta R and Trapp BD.. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology 2007; 68(22 Suppl 3): S22–S31. [DOI] [PubMed] [Google Scholar]
- 44.Naegele M and Martin R.. The good and the bad of neuroinflammation in multiple sclerosis. Handbook Clinical Neurol 2014; 122: 59–87. [DOI] [PubMed] [Google Scholar]
- 45.Rocha NP, Ribeiro FM, Furr-Stimming E. et al. Neuroimmunology of Huntington's disease: Revisiting evidence from human studies. Mediators Inflamm 2016; 2016: 8653132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amor S, Puentes F, Baker D, et al. Inflammation in neurodegenerative diseases. Immunology February 2010; 129(2): 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen Reish HE, Standaert DG.. Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson’s disease. J Parkinson's Dis 2015; 5(1): 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiu H and Nicholson SE.. Biology and significance of the JAK/STAT signalling pathways. Growth Factors April 2012; 30(2): 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu CY, Wang X, Liu C. et al. Pharmacological targeting of microglial activation: New therapeutic approach. Front Cell Neurosci 2019; 13: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tikka T, Fiebich BL, Goldsteins G. et al. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci; April 15, 2015; 21(8): 2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schapansky J, Nardozzi JD, LaVoie MJ.. The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson's disease. Neuroscience August 27, 2015; 302: 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cook DA, Kannarkat GT, Cintron AF, et al. LRRK2 levels in immune cells are increased in Parkinson's disease. NPJ Parkinson's Dis 2017; 3: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardet A, Benita Y, Li C. et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol; November 1, 2010; 185(9): 5577–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cresto N, Gardier C, Gubinelli F, et al. The unlikely partnership between LRRK2 and alpha-synuclein in Parkinson's disease. Eur J Neurosci February 2019; 49(3): 339–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi I, Kim B, Byun JW. et al. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat Commun September 14, 2015; 6: 8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Drongelen V and Holoshitz J.. Human leukocyte antigen-disease associations in rheumatoid arthritis. Rheum Dis Clin North Am August 2017; 43(3): 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin HL, Santoro M, Mustafa S, et al. Evidence for a role of adaptive immune response in the disease pathogenesis of the MPTP mouse model of Parkinson's disease. Glia March 2016; 64(3): 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sung PS, Lin PY, Liu CH, et al. Neuroinflammation and neurogenesis in alzheimer's disease and potential therapeutic approaches. Int J of Mol Sci 2020; 21(701): 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu Y and Ye RD.. Microglial Abeta receptors in Alzheimer's disease. Cell Mol Neurobiol January 2015; 35(1): 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilkinson K and El Khoury J.. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimer's Dis 2012; 2012: 489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kinney JW, Bemiller SM, Murtishaw AS, et al. Inflammation as a central mechanism in Alzheimer's disease. Alzheimer's Dement 2018; 4: 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Domingues C, da Cruz ESOAB, and Henriques AG.. Impact of cytokines and chemokines on Alzheimer's disease neuropathological hallmarks. Curr Alzheimer Res 2017; 14(8): 870–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amar F, Sherman MA, Rush T, et al. The amyloid-beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signaling May 9, 2017; 10(478): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leng F and Edison P.. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol March 2021; 17(3): 157–172. [DOI] [PubMed] [Google Scholar]
- 65.Chang YH, Hsieh SL, Chen MC, et al. Lymphotoxin beta receptor induces interleukin 8 gene expression via NF-kappaB and AP-1 activation. Exp Cell Res; August 15, 2002; 278(2): 166–174. [DOI] [PubMed] [Google Scholar]
- 66.Miller TW, Isenberg JS, Shih HB, et al. Amyloid-beta inhibits No-cGMP signaling in a CD36- and CD47-dependent manner. PloS One; December 22, 2010; 5(12): e15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol December 2017; 7(12): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang Q, Lee CY, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron; June 12, 2008; 58(5): 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li K, Li J, Zheng J, Qin S.. Reactive astrocytes in neurodegenerative diseases. Aging Dis June 2019; 10(3): 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan Z, Gibson SA, Buckley JA, et al. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol April 2018; 189: 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seif F, Khoshmirsafa M, Aazami H, et al. The role of JAK/STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signaling; June 21, 2017; 15(1): 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerjee S, Biehl A, Gadina M, et al. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: Current and future prospects. Drugs April 2017; 77(5): 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qin H, Buckley JA, Li X, et al. Inhibition of the JAK/STAT pathway protects against alpha-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci; May 4, 2016; 36(18): 5144–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Villarino AV, Kanno Y, O'Shea JJ.. Mechanisms and consequences of JAK/STAT signaling in the immune system. Nat Immunol; March 22, 2017; 18(4): 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ben Haim L, Ceyzeriat K, Carrillo-de Sauvage MA, et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer's and Huntington's diseases. J Neurosci; February 11, 2015; 35(6): 2817–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Slota JA, Booth SA. MicroRNAs in neuroinflammation: Implications in disease pathogenesis, biomarker discovery and therapeutic applications. Noncoding RNA April 24, 2019; 5(2): 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Obora K, Onodera Y, Takehara T, et al. Inflammation-induced miRNA-155 inhibits self-renewal of neural stem cells via suppression of CCAAT/enhancer binding protein beta (C/EBPbeta) expression. Sci Rep February 27, 2017; 7: 43604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tahamtan A, Teymoori-Rad M, Nakstad B, et al. Anti-inflammatory microRNAs and their potential for inflammatory diseases treatment. Front Immunol 2018; 9: 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ponomarev ED, Veremeyko T, Barteneva N, et al. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med January 2011; 17(1): 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu YP, Thomas GD, Hedrick CC. 2014, Jeffrey M.. Hoeg award lecture: Transcriptional control of monocyte development. Arterioscler Thromb Vasc Biol September 2016; 36(9): 1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]