

Abstract
The application of hepatitis B virus (HBV)–T‐cell receptor (TCR) T‐cell immunotherapy in patients with HBV‐related hepatocellular carcinoma (HBV‐HCC) has been apathetic, as the expression of HBV antigens by both normal HBV‐infected hepatocytes and HCC cells with HBV‐DNA integration increases the risk of on‐target off‐tumor severe liver inflammatory events. To increase the safety of this immunotherapeutic approach, we developed messenger RNA (mRNA) HBV‐TCR‐redirected T cells that—due to the transient nature of mRNA—are functionally short lived and can be infused in escalating doses. The safety of this approach and its clinical potential against primary HBV‐HCC have never been analyzed in human trials; thus, we studied the clinical and immunological parameters of 8 patients with chronic HBV infection and diffuse nonoperable HBV‐HCC treated at weekly intervals with escalating doses (1 × 104, 1 × 105, 1 × 106, and 5 × 106 TCR+ T cells/kg body weight) of T cells modified with HBV‐TCR encoding mRNA. The treatment was well tolerated with no severe systemic inflammatory events, cytokine storm, or neurotoxicity observed in any of these patients throughout treatment. Instead, we observed a destruction of the tumor lesion or a prolonged stable disease in 3 of 8 patients. Importantly, the patients without clinically relevant reductions of HCC did not display any detectable peripheral blood immunological alterations. In contrast, signs of transient localized liver inflammation, activation of the T‐cell compartment, and/or elevations of serum chemokine (C‐X‐C motif) ligand (CXCL) 9 and CXCL10 levels were detected in patients with long‐term clinical benefit. Conclusion: We show that despite the reduced in vivo half‐life (3‐4 days), adoptive transfer of mRNA HBV‐TCR T cells into patients with HBV‐HCC show long‐term clinical benefit that was associated with transient immunological alterations.
Abbreviations
- AFP
alpha‐fetoprotein
- ALT
alanine aminotransferase
- CAR
chimeric antigen receptor
- CHB
chronic HBV infection
- CRP
C‐reactive protein
- CT
computed tomography
- CXCL
chemokine (C‐X‐C motif) ligand
- HBV
hepatitis B virus
- HBV‐HCC
HBV‐related HCC
- HBV‐TCR T cell
T cell engineered with HBV‐specific T‐cell receptors
- HBV‐Env
HBV envelope
- HBsAg
HBV surface antigen
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigen
- IL
interleukin
- LMR
lymphocyte/monocyte ratio
- mRNA
messenger RNA
- NLR
neutrophil/lymphocyte ratio
- PBMC
peripheral blood mononuclear cell
- pgRNA
pre‐genomic RNA
- PLR
platelet/lymphocyte ratio
- TBil
total bilirubin
- TCR
T‐cell receptor
Hepatitis B–related hepatocellular carcinoma (HBV‐HCC) remains a global issue with increasing cases particularly in Asia.( 1 ) At present, approved systemic therapies are favorable for early stages of the disease, while effective therapy options for advanced HBV‐HCC cases are limited to liver transplantations or the use of immune checkpoint inhibitors alone or in conjunction with anti‐angiogenics, which has varying response rates and survival benefits.( 2 ) We have pioneered a personalized T‐cell therapy that targets HBV antigens expressed on HBV‐HCC cells using T cells engineered with HBV‐specific T cell receptors (HBV‐TCR T cells).( 3 , 4 , 5 ) These engineered HBV‐TCR T cells have demonstrated the ability to recognize both HBV‐infected hepatocytes and HCC cells expressing HBV antigens. Importantly, we showed that short HBV–DNA integrations in HBV‐HCC cells can generate functional CD8 T‐cell epitopes capable of activating HBV‐TCR T cells even in the absence of whole HBV antigens detected through antibody‐based assays.( 5 ) Furthermore, more than 90% of HBV‐HCCs have DNA integrations that are typically truncated fragments of the cognate open reading frame,( 5 , 6 , 7 ) suggesting the possibility of treating a large quantity of patients with HBV‐HCC. However, an important drawback of such a strategy is that the HBV T‐cell epitopes derived from the processing of the whole or truncated forms of HBV antigens within infected or HBV‐HCC cells are identical and may lead to off‐target effects. Of note, there are no experimental indications to support whether HBV‐TCR T cells could preferentially target either normal HBV‐infected hepatocytes or HCC cells with HBV–DNA integrations. To address the safety concerns related to the possible targeting of large numbers of normal HBV‐infected hepatocytes, we test the efficacy of HBV‐TCR T cells in patients with HBV‐HCC relapses after liver transplant.( 5 , 8 ) In these patients, tumor and liver‐graft human leukocyte antigen (HLA) haplotypes are mismatched, unlike in patients with primary HBV‐HCC. This specific patient‐selection criterion minimizes the occurrence of off‐tumor inflammatory events by allowing the engineered TCR T cells to be redirected toward epitopes presented by HLA molecules only on the tumor cells but not on the normal hepatocytes, hence allowing us to demonstrate the ability of our therapy in selected patients to target HCC cells with HBV‐DNA integration in vivo.
In addition, to translate this therapy into patients with primary HBV‐HCC, we developed a strategy of HBV‐TCR T‐cell production based on the transient expression of HBV TCRs obtained through messenger RNA (mRNA) electroporation. This approach has two advantages. First, multiple infusions of functionally short‐lived mRNA HBV‐TCR T cells could provide better antitumor effects. Using a murine xenograft model, it was recently demonstrated that the continuous activation of adoptively transferred chimeric antigen receptor (CAR) T cells by signaling through the stably expressed CARs induced exhaustion that requires transient cessation of CAR signaling in order to restore functionality.( 9 ) This implies that the adoptive transfer of TCR T cells engineered to stably express the exogenous TCR—and hence proliferate and engraft in the patient—might only provide an antitumor function that eventually wanes with the development of exhaustion. Instead, an approach that relies on multiple infusions of short‐lived mRNA‐electroporated TCR T cells would in effect refresh this effector T‐cell population and potentially enhance the antitumor efficacy. Second, short‐lived HBV‐TCR T cells have a better safety profile that should not perpetuate on‐target off‐tumor adverse events if it occurs. We showed that multiple adoptive transfers of such short‐lived (3‐4 days) mRNA HBV TCRs into HBV‐infected chimeric mice engrafted with human hepatocytes triggered only a transient and reversible liver inflammation,( 10 ) so it is reasonable for us to presume that multiple infusions of optimal quantity of short‐lived mRNA HBV‐TCR T cells in patients with chronic HBV (CHB) with primary HBV‐HCC could be tolerated.
From a different perspective, the induction of immune‐related events after mRNA HBV‐TCR T‐cell immunotherapy might also be indicative of a less‐compromised immune system rather than a mere undesirable adverse event. This can have important implications in the establishment and maintenance of the cancer immunity cycle that can perpetuate a long‐term antitumor effect. For example, patients with non‐small cell lung cancer who experienced treatment‐related adverse events following therapy with anti–programmed death 1 (PD1) checkpoint inhibitor (Pembrolizumab; KEYNOTE‐001 trial) had an increased objective response rate, progression‐free survival, and overall survival compared with patients who did not.( 11 ) Similarly, a meta‐analysis of clinical trials with another anti‐PD1 checkpoint inhibitor (Nivolumab) also found a positive correlation of the incidence rate of immune‐related adverse events with the objective response rate of patients with advanced solid tumors.( 12 ) It would then be important to understand whether immunological alterations can be induced by the transiently functional mRNA HBV‐TCR T cells in patients with primary HBV‐HCC and its association with antitumor response.
To address these issues, we first tested the safety of escalating doses of mRNA HBV‐TCR T‐cell immunotherapy in patients with CHB with diffused unresectable HCC. We also longitudinally monitored the biochemical, radiological, and immunological alterations to better understand the potential efficacy of HBV‐TCR T‐cell immunotherapy in these patients.
Materials and Methods
Patients
Patients with primary HBV‐HCC (n = 8) were recruited at the Fifth Medical Center of Chinese PLA General Hospital, Beijing, China, for an open‐label, phase 1 dose‐escalation study (NCT03899415). None of the patients had a decompensated disease and were at least Child‐Pugh grade B at the start of therapy. All patients had a history of chronic HBV infection and received nucleotide analog treatment. In addition to patients B003 and B006, all other patients received Sorafenib before TCR T‐cell therapy. Patients B001, B002, B003, B004, B005, B007, and B008 also received Sorafenib treatment in combination with TCR T‐cell infusion. None of the patients received immunotherapy before this study. This study was approved by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital and was conducted according to the principles of the Declaration of Helsinki with informed consent obtained from all patients involved.
HBV‐TCR T cells
All of the patients were HLA‐typed to determine whether they were expressing either HLA‐A0201 or HLA‐Cw0801. HBV envelope (HBV‐Env)–specific TCRs restricted by the respective HLA molecules were then selected accordingly.
Production of HBV‐specific TCR T cells for infusion is similar to that described previously.( 5 ) In brief, peripheral blood mononuclear cells (PBMCs) from the patient were isolated by Ficoll density gradient centrifugation, followed by activation for 8 days with 600 IU/mL of GMP‐grade interleukin (IL) 2 (Miltenyi, Germany) and 50 ng/mL of GMP‐grade muromonab‐CD3 (Miltenyi, Germany) in cell therapy–grade AIM‐V (Invitrogen, Carlsbad, CA) supplemented with 5% CTS Serum Replacement (Invitrogen, Carlsbad, CA). Activated T cells were then electroporated using the AgilePulse Max system (BTX, Holliston, MA) according to the manufacturer’s recommended protocol.
Quality Control of Engineered HBV‐Specific T Cells
Electroporation efficiency of TCR‐redirected T cells was characterized for every infusion in all patients. Electroporation efficiency was quantified by staining with the appropriate TCR‐Vβ antibodies (Beckman Coulter, Pasadena, CA).
Infusion Protocol
All patients started the experimental therapy with a planned dose‐escalation phase, in which increasing numbers of TCR T cells (104 to 5 × 106 TCR+ T cells / kg) were infused every week (total of four infusions). This was followed by a monitoring period of at least 1 month before infusions of high numbers (maximum 5 × 106 TCR+ T cells/ kg) of TCR T cells were given when indicated. Serum alanine aminotransferase (ALT) and total bilirubin (TBil) levels were monitored regularly. Infusions were temporarily suspended, as determined by the primary physician if patients experienced clinically significant adverse events and the doses were reduced for subsequent infusions if applicable.
Immunophenotyping of Activated and Proliferating T Cells
To detect activated and proliferating T cells, the following antibodies (abs) were used: anti‐CD3‐APC‐Cy7 (clone SK7), anti‐CD8‐BV510 (clone SK1), anti‐Ki67‐AF488 (clone ki67), and anti‐CD39‐APC (clone A1). All abs were purchased from BioLegend (San Diego, CA). PBMCs were labeled with the previously mentioned Abs on ice for 30 minutes and then thoroughly washed and fixed for further analysis by flow cytometry.
Serum Cytokine/Chemokine Analysis
Serum levels of IL‐6, IL‐10, IL‐18, interferon gamma (IFN‐γ), tumor necrosis factor alpha (TNF‐α), chemokine (C‐X‐C motif) ligand (CXCL) 9, and CXCL10 were evaluated using a Luminex‐procartaplex 7 plex kit (PPX‐07; eBioscience, Waltham, MA) following the manufacturer’s instructions.
Hematological, Biochemical, and Imaging Analysis of Treated Patients
All hematological, biochemical, and imaging analysis was performed by the Fifth Medical Center of PLA General Hospital in accordance with their standard operating procedures for each assay requested.
Results
mRNA HBV‐TCR T‐Cell Immunotherapy of Patients With CHB With Primary HBV‐HCC Did Not Cause Frequent Liver Inflammatory Events
We recruited 8 patients with CHB with diffused nonoperable HBV‐HCC. The HLA class I haplotype of all patients was defined, and HBV‐Env‐specific TCRs restricted by either HLA‐A0201 or HLA‐Cw0801 were used to engineer the respective mRNA HBV‐TCR T cells (Table 1). All of the patients were scheduled to receive four escalating doses of mRNA HBV‐TCR T cells (104 to 5 × 106 TCR+ T cells/kg) infused weekly (Fig. 1A). Quality control experiments aimed at evaluating the mRNA electroporation efficiency (by antibody staining of the appropriate TCR‐Vβ chain) were performed for every production cycle of mRNA HBV‐TCR T cells (Fig. 1B). Electroporation efficiency varies for each patient and typically ranges between 30% and 60% of live cells recovered after electroporation (Fig. 1B).
TABLE 1.
Summary of Treatment‐Induced Immunological Alterations and the Pretreatment Levels of HBV, Tumor, and Inflammatory Markers Detected in the Peripheral Blood Compartment of All Treated Patients
| Patient No. | HBV‐TCRs | Treatment‐Induced Immunological Alterations | Pretreatment Levels | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adverse Events (Y/N) | T‐cell Activation Response | Serum chemokine Response | Radiological Response | HBV Virological Response | HBsAg (IU/mL) | ALT (IU/L) | CRP (mg/L) | PLR | NLR | LMR | ||
| B001 | HBV‐Env HLA‐A0201 restricted | Y | + | − | + | <LLOD | 202 | 26 | 30.2 | 153.89 | 2.53 | 5.57 |
| B008 | HBV‐Env HLA‐A0201 restricted | N | + | + | + | <LLOD | 303.8 | 14 | 3.5 | 81.52 | 2.44 | 4.72 |
| B005 | HBV‐Env HLA‐A0201 restricted | N | − | + | ±* | <LLOD | 942.6 | 26 | 2.4 | 83.33 | 1.31 | 7.38 |
| B002 | HBV‐Env HLA‐A0201 restricted | Y | − | − | ± | + | 1444 | 32 | 1.6 | 30.59 | 2.01 | 4.05 |
| B003 | HBV‐Env HLA‐A0201 restricted | N | − | − | − | <LLOD | 929.9 | 40 | 5.9 | 81.91 | 2.90 | 4.70 |
| B004 | HBV‐Env HLA‐Cw0801 restricted | N | − | − | − | <LLOD | 210.9 | 48 | 48.6 | 326.42 | 4.87 | 2.52 |
| B006 | HBV‐Env HLA‐A0201 restricted | N | − | − | − | <LLOD | 1549 | 20 | 9.3 | 166.07 | 3.24 | 3.86 |
| B007 | HBV‐Env HLA‐A0201 restricted | N | − | − | − | <LLOD | 231.5 | 28 | 126.8 | 313.64 | 7.15 | 2.20 |
Abbreviations: LLOD, lower limit of detection; N, no; Y, yes.
Both target lesions remain stable during therapy.
FIG. 1.
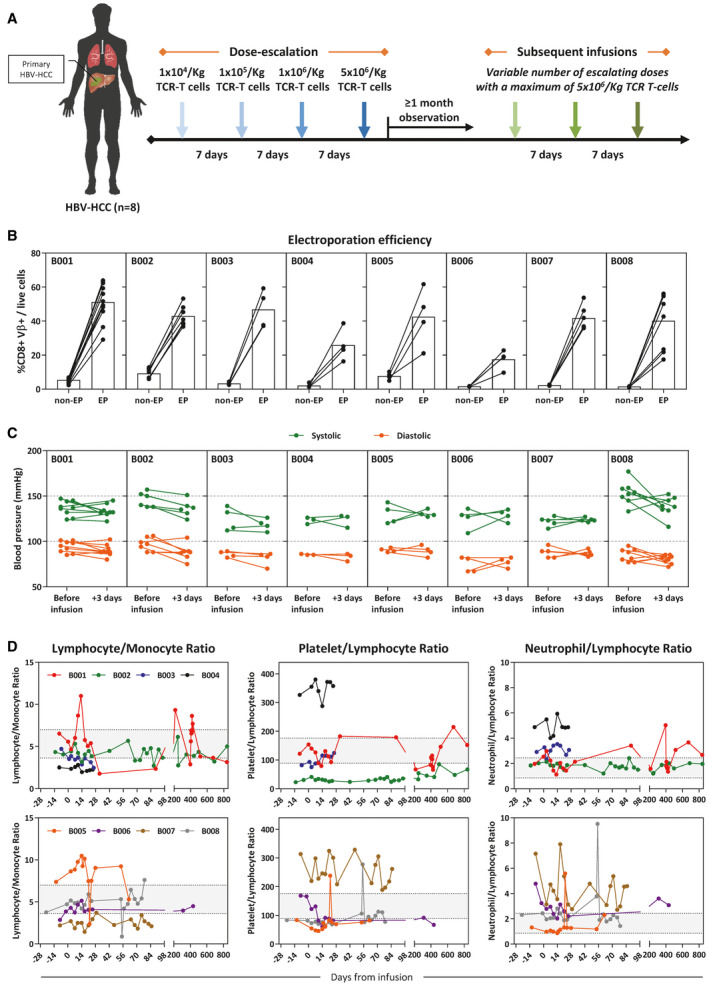
HBV‐specific TCR T‐cell therapy of patients with CHB with primary HBV‐HCC did not induce an acute systemic inflammatory reaction. (A) Schematic representation of the infusion protocol. Patients with CHB primary HBV‐HCC (n = 8) were given four infusions of autologous mRNA electroporated HBV‐specific TCR T cells at increasing doses (1 × 104 to 5 × 106 CD8+Vβ+ T cells/Kg) 1 week apart. Subsequent infusions were given with variable number of escalating doses, with a maximum of 5 × 106/kg TCR T cells. (B) CD8+Vβ+ T‐cell frequency of all TCR T‐cell productions were quantified. The frequency before and after HBV‐TCR mRNA electroporation is shown. (C) Systolic and diastolic blood pressure of all treated patients before and 3 days after each infusion. (D) Differential blood counts were performed for all patients during the course of therapy, and markers of systemic inflammation are shown. The shaded regions demarcate the reference ranges of each parameter derived from the mean ± SD observed in healthy individuals.( 16 ) Abbreviation: EP, electroporation.
None of the treated patients experienced an immediate systemic adverse event after any infusion, even at the maximum dose of 5 × 106 TCR+ T cells/kg. Vital signs of all patients remained stable after every infusion (Fig. 1C), and markers of systemic inflammation (lymphocyte/monocyte ratio [LMR]; platelet/lymphocyte ratio [PLR]; and neutrophil/lymphocyte ratio [NLR]) fluctuated without any dose‐dependent patterns (Fig. 1D). These observations indicate that infusions of mRNA HBV‐TCR T cells into patients with CHB and primary HBV‐HCC were generally well tolerated and did not induce an acute systemic inflammatory reaction.
Focusing on liver inflammatory events, 2 of 8 patients developed liver inflammation upon receiving a low dose of HBV‐TCR T cells (5 × 104 to 105 TCR+ T cells/kg) (Fig. 2A). B001 tolerated the first dose of 104 TCR+ T cells/kg without changes in any clinical/virological parameters. In contrast, a second dose of 105 TCR+ T cells/kg induced a large and rapid elevation of serum ALT and TBil levels evident 3 days after the infusion (Fig. 2A,B), accompanied with nausea and the development of jaundice for about 25 days. Despite the transient expression of HBV‐TCR by the engineered T cells, serum ALT, and TBil levels progressively increased, peaking at about 16 days after the second infusion (Fig. 2A,B). Clinical symptoms and liver function gradually improved without clinical intervention, and a full recovery was made about 80 days from the onset of inflammation. On the other hand, patient 2 (B002) tolerated the first dose and only had a mild and gradual increase of serum ALT levels after receiving the second infusion of 5 × 104 TCR+ T cells/kg without accompanying elevations of TBil (Fig. 2A,B). Serum ALT levels returned to baseline in about 35 days from the second infusion. In both patients, serum C‐reactive protein (CRP) levels remained stable throughout the liver inflammatory event (Fig. 2B). Interestingly, when both patients resumed therapy thereafter, subsequent infusions at even higher doses (maximum of 5 × 106 TCR+ T cells/kg) did not cause any observable liver inflammation (Fig. 2B). Based on these observations, the occurrence of the localized liver inflammation appears to be dose‐independent (Fig. 2), and the pretreatment serum levels of hepatitis B surface antigen (HBsAg), ALT, and CRP also do not predict the induction of liver inflammation (Table 1). At the moment, a clear cause for these adverse events remains to be determined.
FIG. 2.
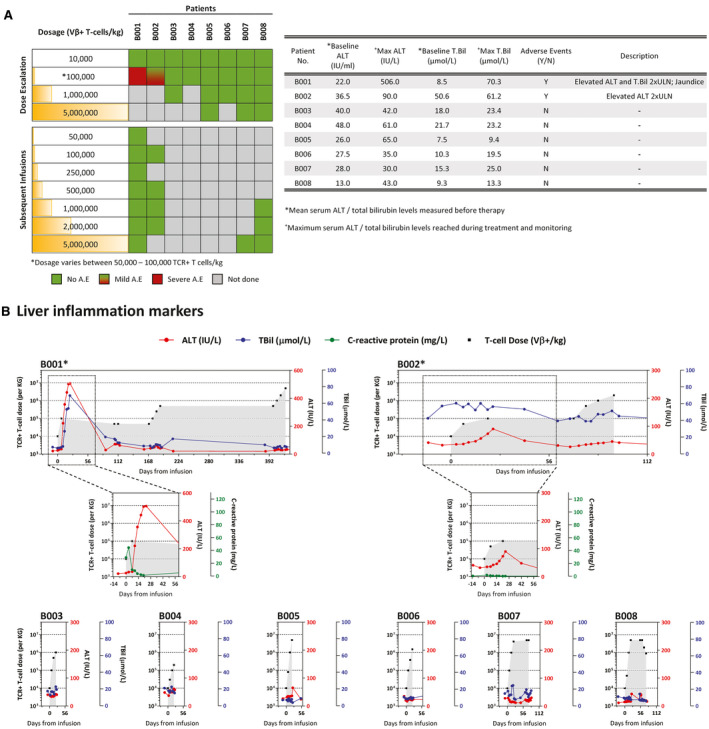
HBV‐specific TCR T‐cell therapy can cause a self‐limiting and reversible liver‐specific adverse event in some patients. (A) Occurrence of adverse events in relation to the dose of HBV‐specific TCR T cells infused into the patient (left). Table summarizes the adverse events that occurred during the course of the therapy, and the liver‐specific parameters recorded at baseline and the maximum levels achieved during treatment. (B) Longitudinal levels of liver inflammation markers ALT (red) and TBil (blue) of all patients treated with HBV‐specific TCR T cells. CRP levels (green) of patients with documented adverse events are shown in the respective inserts. The number of HBV‐specific TCR T cells infused are indicated in gray. *Patients with reported adverse events.
In all, these results demonstrate that infusions of as little as 6 million HBV‐specific T cells (frequency in total T cells approximately 0.000003%) can trigger a localized liver inflammation in some patients without systemic involvement. Liver damage, however, was well tolerated and fully reversible despite the advanced pathological liver condition present in the patients.
mRNA HBV‐TCR T‐Cell Infusions Induce Immunological Alterations in the Peripheral Blood
We next analyzed whether mRNA HBV‐TCR T‐cell infusions can induce detectable immunological alterations in the peripheral blood of the patients. The frequencies of activated and proliferating CD8 and CD4 T cells (Ki67+ CD39+/CD8 or CD4 T cells) were quantified before and after every TCR T‐cell infusion to determine whether an activation of the T‐cell compartment could be induced. Serum from peripheral blood was also collected at regular intervals and the levels of IFN‐γ, TNF‐α, IL‐6, IL‐18, IL‐10, CXCL9, and CXCL10 were quantified. Only CXCL9 and CXCL10 exhibited significant fluctuations after HBV‐TCR T‐cell infusions, while the other measured analytes remain stable in all patients.
Immune‐related alterations were induced in 3 of 8 HBV‐TCR T cell–treated patients (Fig. 3). A transient increase in Ki67+ CD39+ CD8 and CD4 T‐cell frequencies, indicative of an activated T‐cell compartment, was observed in patient B008 without the induction of liver inflammation (Fig. 3A). These changes peaked as early as 3 days after infusion and subsided in the next 4 days (Fig. 3A). Activation of peripheral blood T cells was also accompanied with the transient elevation of serum CXCL9 and CXCL10 levels (Fig. 3B). This increase was more rapid, peaking 1 day after mRNA HBV‐TCR T‐cell infusion and subsiding 2 days later. Both immunological alterations were observed only after high‐dose HBV‐TCR T‐cell infusions (5 × 106 TCR+ T cells/kg) and not at the lower doses (Fig. 3). In a similar fashion, induction of serum CXCL9 and CXCL10 was also observed in patient B005, albeit at lower levels and without detectable liver inflammation or activation of the T‐cell compartment (Fig. 3).
FIG. 3.
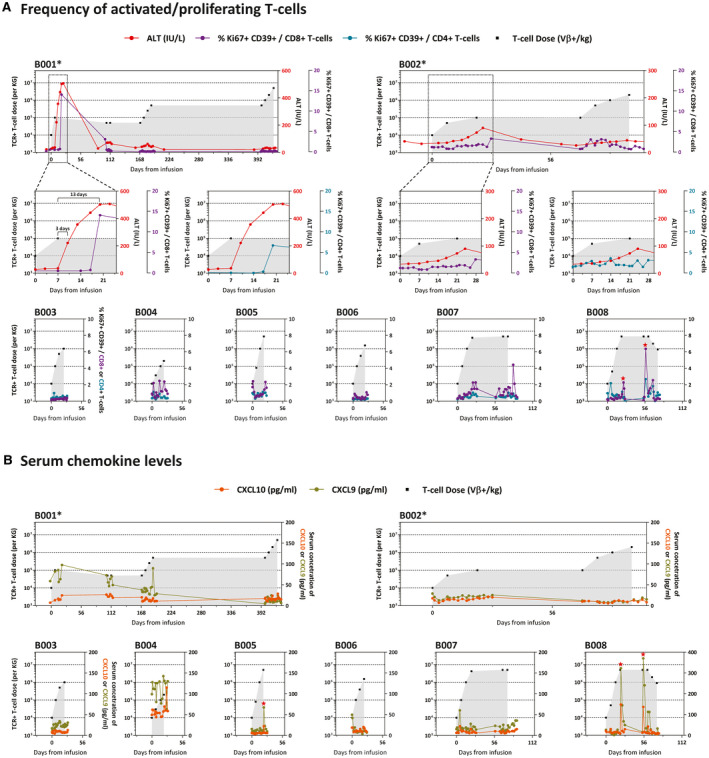
HBV‐specific TCR T‐cell therapy induced observable immunological alterations in the peripheral blood. (A) Longitudinal frequencies of peripheral blood activated and proliferating (Ki67+ CD39+) CD8 and CD4 T cells. (B) Serum concentrations of CXCL9 and CXCL10 of all patients treated with HBV‐specific TCR T cells. Serum ALT levels and the number of HBV‐specific TCR T cells infused are indicated as before. *Patients with reported adverse events.
In patient B001 who developed severe liver inflammation, there was a temporal association between the induction of liver inflammation and the increase in the frequencies of activated T cells in the peripheral blood (Fig. 3A). The kinetics, however, were slightly different. Ki67+ CD39+ CD8 and CD4 T‐cell frequencies were only elevated 10 days after the development of liver inflammation before the frequencies returned to baseline levels upon resolution (Fig. 3A). Serum levels of CXCL9 were elevated in the first 28 days of treatment when liver inflammation occurred, and gradually declined with time (Fig. 3B). Unfortunately, the serum sampling time points for B001 did not allow us to track the levels of both chemokines with high resolution during liver inflammation. Both T‐cell activation and serum chemokine changes were no longer observed after subsequent higher‐dose infusions in which liver inflammation was not triggered (Fig. 3). Unlike patient B001, the mild liver inflammation induced in B002 was not associated with a concomitant increase in the frequency of activated T cells or CXCL9 and CXCL10 levels in the peripheral blood (Fig. 3).
Taken together, we demonstrate that mRNA HBV‐TCR T‐cell treatment of patients with primary HBV‐HCC can induce detectable immune‐related alterations with differing magnitude in the peripheral blood with or without the occurrence of liver inflammation.
Immunological Alterations Induced by mRNA HBV‐TCR T‐Cell Therapy Is Positively Associated With Treatment Response
The association between immune‐related alterations induced by mRNA HBV‐TCR T‐cell therapy and anti‐tumor response was then evaluated by monitoring the tumor growth during treatment. In addition, because the treatment targets HBV antigens, HBV‐related parameters could also be used to monitor treatment response. These patients with chronic HBV‐HCC are under nucleoside analogue (NA) therapy for HBV infection; hence, their serum HBV‐DNA load is well suppressed and not useful for monitoring purposes. However, serum HBV pre‐genomic RNA (pgRNA) have been shown to be more readily detected in NA‐treated patients with CHB, and also have good correlation with HBV‐DNA load in untreated patients with CHB or during acute infection.( 13 ) Hence, to supplement the radiological evaluation, we also quantified the serum levels of HBV pgRNA, which were above the limit of detection only in patient B002.
The severe liver inflammation, together with an activation of the T‐cell compartment and subsequent elevation of CXCL9 after HBV‐TCR T‐cell infusion in patient B001, was temporally associated with tumor destruction (Fig. 4A). One hundred and sixty‐five days after treatment commencement and after the liver inflammation has subsided, we observed a dramatic necrosis of the liver tumor through contrast computed tomography (CT) imaging (Fig. 4A). Follow‐up CT imaging at day 425 showed a clear reduction of the tumor size that was maintained until day 520, over 3 months after the last infusion. In patients in whom immune‐related alterations were less pronounced, like in patient B008, in whom T‐cell activation and elevation of serum chemokines were observed, or in patient B005, in whom only serum chemokines were elevated, both without detectable liver inflammation, the total tumor load remained relatively stable with some liver lesion decreasing in size (Fig. 4B,C). Instead, in all other patients in whom immune‐related alterations were undetectable, tumor load continued to increase (Fig. 4B,C). Interestingly, even though radiological imaging did not detect reduction of the tumor load of patient B002 after treatment despite the mild liver inflammation, analysis of the serum HBV‐pgRNA levels showed a clear decline following treatment, which was indicative of treatment response (Fig. 4D).
FIG. 4.
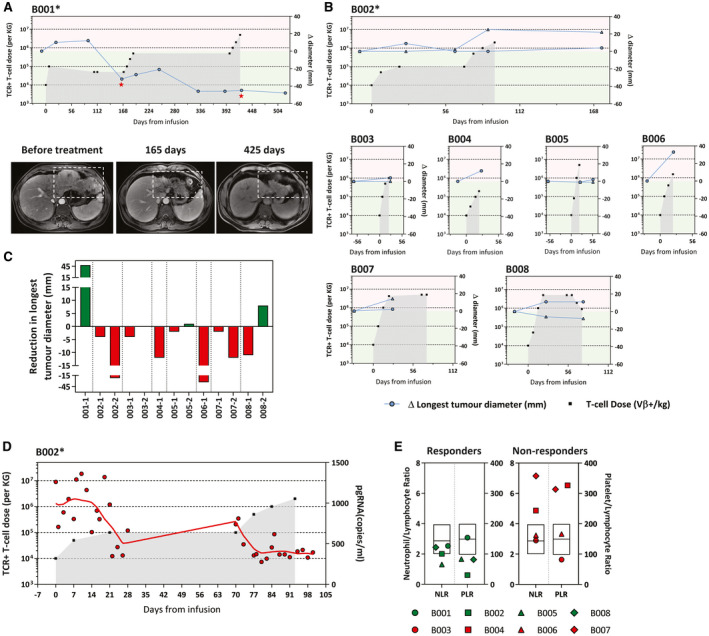
Immunological alterations in the peripheral blood correlate with observable antitumor response. The longest diameter of the target lesions in the liver was monitored throughout the treatment. The number of HBV‐specific TCR T cells infused are indicated as before. *Patients with reported adverse events. (A) Changes in the longest diameter (compared with baseline) of the liver target lesion in patient B001. Representative CT images of the liver target lesion at different time points are shown and indicated in the graph. (B) Changes in the longest diameter (compared with baseline) of the target lesion in the liver of patients B002‐B008. Each line represents a single liver target lesion. (C) Summary of the reduction in the longest diameter (compared with baseline) of individual liver target lesions in all treated patients analyzed from radiological imaging performed before treatment and within 3 months of the last infusion. (D) Levels of serum HBV pgRNA were analyzed in all patients. Only patient B002 (shown here) had detectable serum HBV pgRNA at baseline and was followed throughout treatment. (E) Baseline pre‐infusion neutrophil/lymphocyte and platelet/lymphocyte ratio of patients who exhibited immunological alterations after receiving HBV‐specific TCR T cells (responders) and those who do not (nonresponders). Box plot overlay shows the median and interquartile range of both parameters, as observed in the total population analyzed in Sangro et al.( 14 )
Recently, pretreatment levels of systemic inflammation markers have been shown to have a negative association with the survival outcome of Nivolumab monotherapy.( 14 ) To further support our observations, we segregated the treated patients based on their pretreatment levels of several inflammatory biomarkers (PLR, NLR, LMR, CRP, and ALT) and the HCC‐associated antigen alpha‐fetoprotein (AFP). Consistent with their findings, all of the treatment responders (B001, B002, B005, and B008) had lower pretreatment NLR and PLR levels compared with the nonresponders, whereas other inflammatory markers like ALT, CRP, LMR, and AFP showed no discernible association (Table 1; Fig. 4E).
These data demonstrate the positive association between the induction of immune‐related events after HBV‐TCR T‐cell immunotherapy of patients with primary HBV‐HCC and treatment response (Table 1). A better treatment response was also observed when stronger immune‐related alterations were induced, suggesting that such alterations are prognostic of the antitumor effects of HBV‐TCR T‐cell immunotherapy.
Discussion
Immune therapy with mRNA HBV‐TCR T cells of patients with HBV‐HCC with HCC recurrence following liver transplantation have shown good safety profile without occurrences of adverse events.( 5 ) However, in patients with primary HBV‐HCC, HBV‐TCR T‐cell killing of infected and transformed hepatocytes can no longer be segregated by the expression of mutually exclusive HLA molecules, leading to a concern of on‐target off‐tumor toxicity. Here we report evidence regarding the safety profile of short‐lived mRNA HBV‐TCR T‐cell therapy of patients with CHB with primary diffused nonoperable HBV‐HCC.
Multiple infusions of escalating numbers of short‐lived mRNA HBV‐TCR T cells was safe with no occurrences of severe cytokine storms, neurotoxicity, or systemic inflammation detected in any of the patients throughout treatment. Nevertheless, some patients developed liver inflammation that was self‐limiting and fully reversible, even with their existing liver pathology. However, while it is not entirely surprising that the hepatitis following HBV‐TCR T‐cell infusion was occurring in 2 of 8 patients, it noteworthy that we did not detect any correlation among the triggering of liver inflammation, the quantity of adoptively transferred mRNA HBV‐TCR T cells (specific for HBV‐Env), or the pretreatment levels of HBsAg in the serum (Table 1). For example, the high levels of ALT were induced in patient B001 by the adoptive transfer of only 100,000 TCR+ T cells/kg (approximately 6 million cells in total). Given that the total number of lymphocytes in a healthy human is approximately 1‐2 × 1012, the final frequency of HBV‐TCR T cells infused in this patient is approximately 3 of 100 million T cells. It appears completely implausible that such low quantities of T cells could be able to sustain a direct lysis of hepatocytes, causing altered levels of ALT and TBil that lasted for approximately 80 days observed in this patient. HBV‐specific T cells capable of proliferating and cytokine production (thus not completely exhausted) can be detected in so‐called immune‐tolerant patients with CHB characterized by normal levels of ALT. Even though the frequency of these T cells has not been directly quantified ex vivo in these patients, the fact that we can expand HBV‐specific T cells from 0.5 × 106 PBMCs indicated that HBV‐specific T cells circulating in these patients are at frequencies superior to the one achieved in patient B001, and yet with no occurrences of hepatitis in such patients.
We think that the only possible explanation of the ability of the limited quantity of HBV‐TCR T cells to trigger a prolonged inflammatory event is the fact that the immunological environment of the liver in a patient with advanced liver disease differs from that of a patient with CHB. In particular, we have demonstrated that an enrichment of pro‐inflammatory myeloid cells is present in patients with advanced liver disease.( 15 ) Therefore, it seems plausible to hypothesize that the quantity of such cells and their ability to respond to minimal quantity of cytokines or damage signals induced directly by the lysis of few HBV‐expressing hepatocytes by HBV‐TCR T cells is responsible for driving the prolonged liver inflammation detected in some patients. Further support of this hypothesis can also be derived by the lack of any correlation between the induction of liver inflammation and the pretreatment serum HBsAg quantity (Table 1). Even though the serum HBsAg quantity is only a generic proxy of the number of hepatocytes expressing HBsAg, as high amounts of HBsAg can be released by few hepatocytes, we should theoretically expect that patients with higher quantities of serum HBsAg might be at higher risk of liver inflammation due to the increased availability of the T‐cell trigger. This was also not observed in our cohort. On the other hand, our observations support the idea that the liver environment composition, and not the number of available targets nor the dose of HBV‐TCR T cells infused, drives the triggering of the inflammatory events. In addition, subsequent infusions of higher numbers of HBV‐TCR T cells in patient B001 after resolution of the first episode of hepatitis did not result in any peripheral signs of liver damage, suggestive of the induction of unknown regulatory mechanisms within the liver environment. Hence, while mRNA HBV‐TCR T‐cell therapy in patients with primary HBV‐HCC can be well tolerated, the inability to detect a relation between liver inflammation triggering and dose of HBV‐TCR T‐cell or HBV virological parameters highlights the importance of carefully dosing the quantity of adoptively transferred mRNA HBV‐TCR T cells and supports the implementation of a cautious approach to mitigate potential liver inflammation occurring unpredictably even at low doses.
The immune‐related changes induced by the infusion of short‐lived mRNA HBV‐TCR T cells have also provided important insights. In a previous analysis of patients with post‐liver‐transplant recurrence of HBV‐HCC who were treated with mRNA HBV‐TCR T cells, we observed an increased frequency of activated T cells in the peripheral blood within 5 days after the T‐cell infusions without triggering hepatitis.( 5 , 8 ) Such changes were detected even in the presence of the immunosuppressive drugs Tacrolimus and Mycophenolate Mofetil, which suppress T‐cell function to prevent liver graft rejection. This elevated frequency was also only seen in patients who exhibited antitumor response either through radiological imaging or from the reduction of serum tumor markers AFP, providing initial evidence of the association between the antitumor effects and the activation of the T‐cell compartment. In the current work, the increase in T‐cell activation following mRNA HBV‐TCR T‐cell infusions was still associated with treatment response, and the alterations could occur in association with liver inflammation or be independent of acute hepatitis without the presence of confounding immunosuppressive drugs. Importantly, we expanded the analysis to include the detection of serum CXCL9 and CXCL10 in the peripheral blood. Because CXCL9 and CXCL10 are not produced by T cells directly and are instead secreted by other cells secondary to T‐cell activation, the increase of these chemokines is suggestive of a successful antigen‐specific stimulation of the mRNA HBV‐TCR T cells through engagement with HBV‐HCC cells. The rapid kinetics of the chemokine elevations was also consistent with the transient expression of the HBV‐TCR on the adoptively transferred mRNA HBV‐TCR T cells. Although both chemokines increase in response to the HBV‐TCR T‐cell infusions in some patients, elevations of CXCL9 levels were generally higher and could represent a better prognostic marker of efficacy than CXCL10. This would have to be investigated in a larger cohort of mRNA HBV‐TCR T cell–treated patients.
Combining the incidence of all immune‐related events (liver inflammation, T‐cell activation, and serum chemokine increase), only patients with detectable immunological alterations had subsequent treatment response, whereas those without any detectable immune‐related changes after treatment had a progressive disease. It was also interesting that the treatment responders had a lower baseline systemic inflammation, as evidenced by the pretreatment NLR and PLR values compared to the patients with no treatment response. Although speculative, a lower inflammatory state could allow for the activation of the patient’s immune system following treatment, leading to tumor destruction, whereas a persistently inflamed situation would prevent the successful induction of antitumor immune response, possibly through multiple immunoregulatory processes that try to limit inflammation. Collectively, all of the evidence supports an alternate interpretation of immunological events, including adverse events, following mRNA HBV‐TCR T‐cell therapy of patients with CHB with HBV‐HCC, in whom such inductions are prognostic of successful treatment.
The limitation of this study is its small sample size; therefore, future trials with a larger cohort of patients with CHB with HBV‐HCC will be required to validate the association of treatment‐induced immunological alterations and antitumor response. Nevertheless, the data presented here show that despite the reduced in vivo half‐life (3‐4 days), repetitive infusions of mRNA HBV‐TCR T cells into patients with CHB with HCC can trigger long‐term clinical benefits that were associated with transient immunological alterations, further supporting the potential of such an immunotherapeutic strategy in the treatment of advanced HCC.
Supported by the Singapore Translational Research Investigator Award, Singapore Ministry of Health's National Medical Research Council (MOH‐000019), Innovative Research Team in the National Natural Science Foundation of China (81721002), Capital Clinical Diagnosis and Treatment Technology Research and Demonstration Application Project in China (Z20110000552), and National Science and Technology Major Project (2018ZX10301202).
Potential conflict of interest: Nothing to report.
Contributor Information
Antonio Bertoletti, Email: antonio@duke-nus.edu.sg.
Fu‐sheng Wang, Email: fswang302@163.com.
References
Author names in bold designate shared co‐first authorship.
- 1. Liu Z, Jiang Y, Yuan H, Fang Q, Cai N, Suo C, et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol 2019;70:674‐683. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6. [DOI] [PubMed] [Google Scholar]
- 3. Koh S, Shimasaki N, Suwanarusk R, Ho ZZ, Chia A, Banu N, et al. A practical approach to immunotherapy of hepatocellular carcinoma using T cells redirected against hepatitis B virus. Mol Ther Nucleic Acids 2013;2:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Qasim W, Brunetto M, Gehring AJ, Xue S‐A, Schurich A, Khakpoor A, et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol 2015;62:486‐491. [DOI] [PubMed] [Google Scholar]
- 5. Tan AT, Yang N, Lee Krishnamoorthy T, Oei V, Chua A, Zhao X, et al. Use of expression profiles of HBV‐DNA integrated into genomes of hepatocellular carcinoma cells to select T cells for immunotherapy. Gastroenterology 2019;156:1862‐1876.e1869. [DOI] [PubMed] [Google Scholar]
- 6. Chiu Y‐T, Wong JKL, Choi S‐W, Sze KMF, Ho DWH, Chan L‐K, et al. Novel pre‐mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J Hepatol 2016;64:1256‐1264. [DOI] [PubMed] [Google Scholar]
- 7. Furuta M, Tanaka H, Shiraishi Y, Uchida T, Imamura M, Fujimoto A, et al. Characterization of HBV integration patterns and timing in liver cancer and HBV‐infected livers. Oncotarget 2018;9:25075‐25088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hafezi M, Lin M, Chia A, Chua A, Ho ZZ, Fam R, et al. Immunosuppressive drug resistant armored TCR T cells for immune‐therapy of HCC in liver transplant patients. Hepatology 2021;74:200‐213. [DOI] [PubMed] [Google Scholar]
- 9. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR‐T cells through epigenetic remodeling. Science 2021;372:eaba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kah J, Koh S, Volz T, Ceccarello E, Allweiss L, Lütgehetmann M, et al. Lymphocytes transiently expressing virus‐specific T cell receptors reduce hepatitis B virus infection. J Clin Invest 2017;127:3177‐3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lisberg A, Tucker DA, Goldman JW, Wolf B, Carroll J, Hardy A, et al. Treatment‐related adverse events predict improved clinical outcome in NSCLC patients on KEYNOTE‐001 at a single center. Cancer Immunol Res 2018;6:288‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xing P, Zhang F, Wang G, Xu YU, Li C, Wang S, et al. Incidence rates of immune‐related adverse events and their correlation with response in advanced solid tumours treated with NIVO or NIVO+IPI: a systematic review and meta‐analysis. J Immunother Cancer 2019;7:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Butler EK, Gersch J, McNamara A, Luk K‐C, Holzmayer V, de Medina M, et al. Hepatitis B virus serum DNA and RNA levels in nucleos(t)ide analog‐treated or untreated patients during chronic and acute infection. Hepatology 2018;68:2106‐2117. [DOI] [PubMed] [Google Scholar]
- 14. Sangro B, Melero I, Wadhawan S, Finn RS, Abou‐Alfa GK, Cheng A‐L, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab‐treated patients with advanced hepatocellular carcinoma. J Hepatol 2020;73:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan‐Garcia A, Lai F, Yeong JPS, Irac SE, Ng PY, Msallam R, et al. Liver fibrosis and CD206(+) macrophage accumulation are suppressed by anti‐GM‐CSF therapy. JHEP Rep 2020;2:100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee JS, Kim NY, Na SH, Youn YH, Shin CS. Reference values of neutrophil‐lymphocyte ratio, lymphocyte‐monocyte ratio, platelet‐lymphocyte ratio, and mean platelet volume in healthy adults in South Korea. Medicine (Baltimore) 2018;97:e11138. [DOI] [PMC free article] [PubMed] [Google Scholar]