

Abstract
Osteopontin (OPN) expression correlates with tumor progression in many cancers, including hepatocellular carcinoma (HCC); however, its role in the onset of HCC remains unclear. We hypothesized that increased hepatocyte‐derived OPN is a driver of hepatocarcinogenesis. Analysis of a tissue microarray of 366 human samples revealed a continuous increase in OPN expression during hepatocarcinogenesis. In patients with cirrhosis, a transcriptome‐based OPN correlation network was associated with HCC incidence along 10 years of follow‐up, together with messenger RNA (mRNA) signatures of carcinogenesis. After diethylnitrosamine (DEN) injection, mice with conditional overexpression of Opn in hepatocytes (Opn Hep transgenic [Tg]) showed increased tumor burden. Surprisingly, mice with conditional ablation of Opn in hepatocytes (Opn ΔHep) expressed a similar phenotype. The acute response to DEN was reduced in Opn ΔHep, which also showed more cancer stem/progenitor cells (CSCs, CD44+AFP+) at 5 months. CSCs from Opn Hep Tg mice expressed several mRNA signatures known to promote carcinogenesis, and mRNA signatures from Opn Hep Tg mice were associated with poor outcome in human HCC patients. Treatment with rOPN had little effect on CSCs, and their progression to HCC was similar in Opn −/− compared with wild‐type mice. Finally, ablation of Cd44, an OPN receptor, did not reduce tumor burden in Cd44 −/− Opn Hep Tg mice. Conclusions: Hepatocyte‐derived OPN acts as a tumor suppressor at physiological levels by controlling the acute response to DEN and the presence of CSCs, while induction of OPN is pro‐tumorigenic. This is primarily due to intracellular events rather that by the secretion of the protein and receptor activation.

Abbreviations
- Alb
Albumin
- CCl4
carbon tetrachloride
- Cd44 −/−
global Cd44 knockout mice
- Cd44 −/− OpnHep Tg
transgenic mice overexpressing Osteopontin in hepatocytes in Cd44 null background
- CSC
cancer stem/progenitor cell
- DE
differentially expressed
- DEN
diethylnitrosamine
- DN
dysplastic nodule
- ECM
extracellular matrix
- FDR
false discovery rate
- GO
Gene Ontology
- GSEA
gene‐set enrichment analysis
- H&E
hematoxilin and eosin
- IHC
immunohistochemistry
- IPA
Ingenuity Pathway Analysis
- mRNA
messenger RNA
- NT
nontumor
- Opn
Osteopontin gene or mRNA
- OPN
Osteopontin protein
- Opn−/−
global Osteopontin knockout mice
- Opn−/−Hep Tg
transgenic mice overexpressing Osteopontin in hepatocytes in Osteopontin null background
- OpnHep Tg
transgenic mice overexpressing Osteopontin in hepatocytes
- OpnΔHep
conditional Osteopontin knockout mice in hepatocytes
- PBS
phosphate buffered saline
- PI3K
Phosphoinositide 3‐kinase
- RNA‐seq
RNA sequencing
- rOPN
recombinant OPN
- STAT
signal transducer and activator of transcription
- TCA
tricarboxylic acid
- Tg
transgenic
- WT
wild‐type
Hepatocellular carcinoma (HCC) accounts for approximately 80% of liver cancers and is the fourth cause of cancer‐related death worldwide.( 1 ) In patients with advanced stage, immunotherapy shows superiority compared with Sorafenib( 2 ); however, its curative potential still needs to be demonstrated. When cancer is diagnosed early, patients benefit from liver resection, which increases the 5‐year overall survival up to 75%.( 3 ) Liver transplant is the only efficient therapeutic option, with recurrence rates of about 13% plus the additional advantage of eliminating the underlying liver disease,( 4 ) yet donor availability is limited. Hence, better understanding of the molecular mechanisms involved in the onset and progression of HCC is needed to develop novel therapies.
A major limitation for achieving progress in understanding hepatocellular carcinogenesis is the phenotypic and genetic heterogeneity of HCC, which shows the combined effect of a disrupted liver microenvironment and the unknown cellular origin.( 5 ) An emerging concept is that hepatocyte‐derived cancer stem/progenitor cells (CSCs) lead to HCC. This is supported by identification of these cells mostly in zone 3 from the liver of mice injected with diethylnitrosamine (DEN).( 6 , 7 ) These cells do not originate from liver progenitor cells.( 7 ) In vitro, genetic alteration of mature hepatocytes can retro‐differentiate them into CSCs.( 8 ) However, how hepatocyte retro‐differentiate into CSCs to give raise to HCC is not well understood, and the molecular mechanisms involved are unknown.
Osteopontin (OPN) is an extracellular matrix (ECM) protein that signals through integrins and CD44 to induce matrix remodeling and angiogenesis.( 9 , 10 ) Upon liver injury, OPN is secreted mostly by hepatocytes, macrophages, and stellate cells.( 11 ) In several cancers, elevated OPN expression is associated with tumor invasion, proliferation, and metastasis.( 9 ) In HCC, the increased concentration of OPN in plasma indicates that it could be a diagnostic and prognostic biomarker.( 12 ) The role of OPN in the progression of HCC has been investigated and suggests extracellular activation of CD44 and Integrin signaling.( 11 , 13 , 14 , 15 , 16 ) Increasing evidence points at a role for OPN signaling in the onset of HCC; however, its specific contribution is not well understood. Indeed, previous publications show discrepancy on the effect of global ablation of Opn in the DEN model of HCC,( 17 , 18 , 19 ) which could be explained by the cellular source of OPN or by different effects of intracellular compared with secreted OPN.
In this study, we hypothesized that hepatocyte‐derived OPN is a driver of hepatocarcinogenesis. To demonstrate this, we analyzed human data and used a mouse model of HCC with genetic manipulation of Opn expression in hepatocytes.
Materials and Methods
Human Samples
Subjects consisted of 153 patients who underwent liver transplantation at the University of Illinois at Chicago Hospital and Health Sciences System, from 2004 to 2012, and 8 healthy controls. All patients had available clinical data and archived liver tissue, which were obtained through the Tissue Biorepository. Microscopic evaluation of histological sections of liver tissues was performed by a liver pathologist for the diagnosis of cirrhosis, dysplastic nodules (DNs) and HCC, as detailed in the Supporting Material and Methods. Corresponding tissue blocks of cirrhotic liver explants were sampled in areas representing cirrhosis, DNs and HCC, such that the total number of specimens exceeded the number of explants. Forty‐five specimens had cirrhosis with HCC, 108 had cirrhosis without HCC, and 143 of the cases had DNs including 98 cases without HCC and 45 with HCC. Eight normal liver control samples were obtained from uninvolved, nondiseased liver tissues of benign resection specimens for hemangioma and focal nodular hyperplasia. The demographic and clinical features of all 153 subjects in the study population were obtained from the review of electronic medical records.
Mice
Wild‐type (WT) (Stock 000664), Albumin (Alb)‐Cre (Alb.Cre + ) (Stock 003574, B6.Cg‐Tg[Alb‐cre]21Mgn/J), Opn −/− (Stock 004936, B6.129S6[Cg]‐Spp1tm1Blh /J), and Cd44 −/− (Stock 005878, NOD.129[Cg]‐Cd44tm1Hbg /J) mice were purchased from the Jackson Laboratories (Bar Harbor, ME). Opnfl/fl mice were generated in our laboratory, creating the OpnloxP allele by inserting loxP sites to remove exons 4‐7. Opnfl/fl mice were bred with Alb.Cre + to generate hepatocyte‐specific knockout (Opn ΔHep) mice. Alb.Cre + mice were used as controls. Opn Hep transgenic (Tg) mice were donated by Dr. Satoshi Mochida (Saitama Medical University, Japan)( 20 ) and were crossbred with Opn −/− mice for ten generations to generate Opn −/− Hep Tg mice and with Cd44 −/− to generate Cd44 −/− Opn Hep Tg. All mice were in C57BL/6J background and lacked a liver phenotype in the absence of treatment.
Model of HCC
We used male mice in our study, as DEN induces less HCC in females.( 21 ) Mice were injected with 20 mg/kg of body weight of DEN (Sigma, St. Louis, MO) at 14 days of age and were sacrificed at 48 hours or after 5, 8 or 12 months.
CSC Isolation and Transplantation
CSCs were obtained as described.( 6 , 22 ) Primary hepatocytes were isolated 5 months after DEN injection by perfusing the livers with Liberase (Roche, Indianapolis, IN). Hepatocytes were sequentially strained through 100, 70, and 40 μm nylon sieves. The aggregate fraction, enriched in CSCs, was retained on top of the 70 and 40 μm sieves. The remainder cells were considered the nonaggregate fraction. To transplant CSCs, 4‐week‐old recipient mice were injected intraperitoneally (i.p.) twice with 50 mg/kg of retrorsine (Sigma) 2 weeks apart, to inhibit hepatocyte proliferation and facilitate the engraftment of transplanted hepatocytes.( 6 ) Four weeks later, mice were injected intrasplenically with 2 × 105 hepatocytes from the aggregate fraction of 5‐month‐old DEN‐injected WT mice, mixed with 8 × 105 hepatocytes from the nonaggregate fraction from the same mice, in a final volume of 100 μL of phosphate buffered saline (PBS). One week later, mice were injected i.p. with 0.5 mL/kg of CCl4 weekly for 3 weeks to induce fibrosis.
Results
OPN Expression Increases During the Progression of Chronic Liver Disease and Is Associated With Poor Outcome in Human HCC
First, using tissue microarrays, we analyzed the OPN protein expression in 366 samples from patients with normal liver, cirrhosis, dysplastic nodules, or HCC (Fig. 1A,B). In normal tissue, OPN protein expression in hepatocytes was modest. The intensity of the staining increased in cirrhosis (P < 10−3), was higher in dysplasia than in cirrhosis (P = 0.02), and was the highest in well‐established HCCs (P < 10−3) (Fig. 1A). This increase in OPN expression along with the progression of chronic liver disease was consistent regardless of etiology, as it was alike in hepatitis B virus and hepatitis C virus (HCV) (despite insignificant difference between cirrhosis and dysplasia in HCV; P = 0.15) (Supporting Fig. S1). Next, using publicly available data sets, we determined the clinical relevance of OPN messenger RNA (mRNA) levels in tumor tissues from patients with HCC after curative resection.( 23 , 24 ) Elevated OPN mRNA expression was significantly associated with lower overall survival and disease‐free survival as well as with tumor stage, tumor size, serum alanine aminotransferase, or vascular invasion in two independent data sets (n = 221 and n = 210, respectively) (Fig. 1C,D). In addition, OPN mRNA expression was significantly increased in the STEM subclass of HCCs, associated with the worst outcome in the 1,133‐HCC metadata set( 25 ) (Fig. 1E) and correlated with genes involved in inflammation, ECM remodeling, and cell proliferation (Supporting Fig. S2).
FIG. 1.
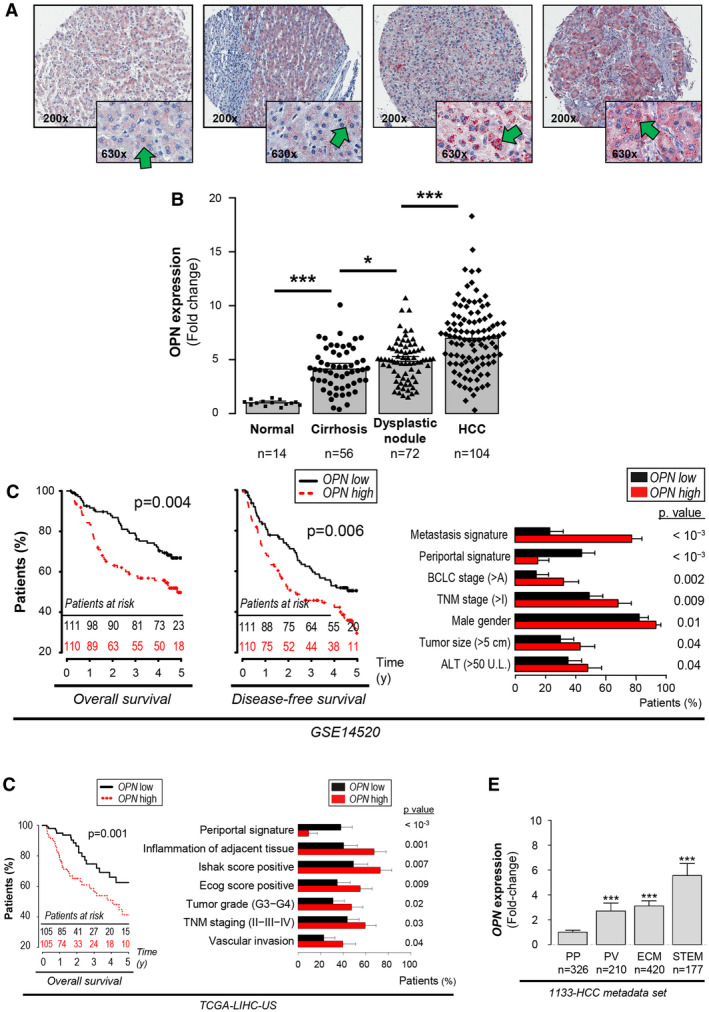
OPN expression increases with progression of chronic liver disease and is associated with poor differentiation and outcome in human HCC. Samples from 120 patients (n = 246 samples) in tissue microarrays were analyzed for OPN protein expression. (A) IHC for OPN (green arrows). (B) Quantification of OPN immunostaining by computer‐assisted morphometry analysis. (C,D) Kaplan‐Meier curves of overall survival and/or disease‐free survival and clinical data analysis of patients with HCC from two publicly available data sets based on the OPN mRNA expression, using the median as a threshold. (E) OPN mRNA expression in 1,133 patients with HCC according to the HCC subclasses indicating HCC progression and poor outcome (P = 5 × 10−14; 3 × 10−26; and 1 × 10−34).( 25 ) Data are expressed as fold change versus the PP subclass. *P < 0.05 and ***P < 0.001. Abbreviations: ALT, alanine aminotransferase; BCLC, Barcelona Clinic Liver Cancer; TGA‐LIHC, The Cancer Genome Atlas–Liver HCC; and TNM, tumor‐node‐metastasis.
OPN mRNA Expression in Early and Advanced Cirrhosis Is Associated With HCC Incidence and Signatures of Carcinogenesis
In nontumor (NT) tissue from patients with HCC after surgical resection,( 24 ) high OPN expression was associated with late recurrence (Fig. 2A), generally interpreted as de novo carcinogenesis.( 26 ) In patients with early cirrhosis,( 26 ) hierarchical clustering, based on the expression of genes correlating with OPN mRNA, classified individuals as OPN‐high and OPN‐low (Fig. 2B). OPN‐high genes were significantly enriched in Gene Ontology (GO) terms associated with cell cycle and ECM remodeling, whereas OPN‐low genes were enriched in GO terms associated with metabolic processes of healthy liver (Fig. 2D). Importantly, OPN‐high patients showed increased HCC incidence after 10 years of follow‐up (Fig. 2C). Gene‐set enrichment analysis (GSEA) revealed that these patients showed significant enrichment in signatures associated with ECM remodeling, integrin signaling, loss of P53 function, and the tumor field effect( 27 ) (Fig. 2E). Overall, these results identify OPN as a potential marker of early carcinogenesis and a robust marker of HCC progression and poor outcome.
FIG. 2.
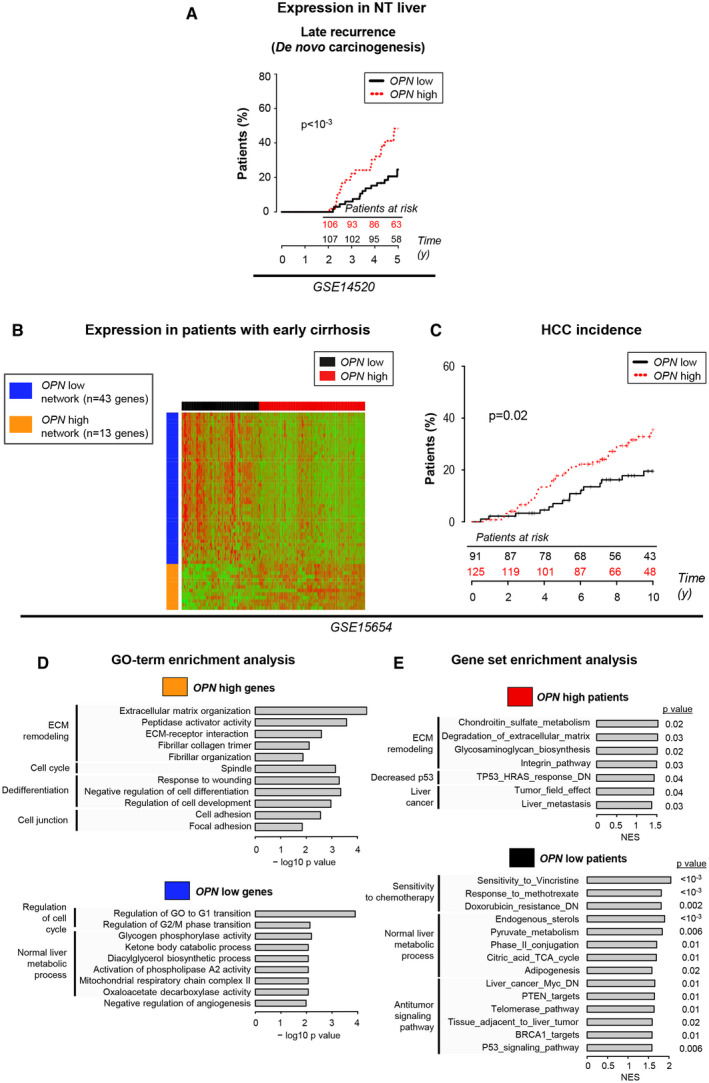
OPN mRNA expression in early and advanced cirrhosis correlates with HCC incidence and signatures of carcinogenesis. (A) Kaplan‐Meier curves of HCC late recurrence and clinical data analysis based on the OPN mRNA expression in NT tissue, using the median as a threshold. (B) In 216 patients with early‐stage liver cirrhosis,( 26 ) hierarchical clustering was performed based on the mRNA expression of genes positively (orange) or negatively (blue) correlated with OPN (abs[Pearson] > 0.3) and identified two groups of patients (OPN‐high [red] and OPN‐low [black]). (C) Kaplan‐Meier curves of HCC incidence of OPN‐high and OPN‐low patients. (D) GO‐term enrichment analysis of the OPN‐high and OPN‐low genes. (E) GSEA of the OPN‐high and OPN‐low patients. Abbreviation: NES, normalized enrichment score.
Overexpression as Well as Ablation of Opn in Hepatocytes Promote Carcinogenesis
To determine the effects of hepatocyte‐derived OPN in the development of HCC, we injected WT, Opn−/−, Opn Hep Tg, Opn −/− Hep Tg, Alb.Cre +, and Opn ΔHep mice with DEN and sacrificed them 12 months later. Overexpression of Opn in Opn Hep Tg and Opn −/− Hep Tg and ablation in Opn −/− and Opn ΔHep mice were validated by immunohistochemistry (IHC) (Fig. 3A and Supporting Fig. S3A). Opn Hep Tg showed an increase and Opn ΔHep a decrease in the concentration of OPN in serum (P = 0.0009 and P = 0.008), confirming the major contribution of hepatocytes to OPN secretion during HCC (Fig. 3B). There were more tumors in Opn Hep Tg (P = 0.01) and in Opn −/− Hep Tg (P = 0.04) compared with WT and Opn −/− mice, respectively (Fig. 3C,D and Supporting Fig. S3B,C). In Opn −/−, there was no difference in tumor burden compared with WT mice (Supporting Fig. S3C), confirming inconclusive results from earlier studies.( 17 , 18 , 19 ) Surprisingly, in Opn ΔHep mice the tumor burden increased compared with Alb.Cre +, as shown by increased number of tumors >3 mm (P = 0.01) and the liver‐to–body weight ratio (P = 0.02), despite no significant difference in the total number of tumors (Fig. 3C,E,F). A separate group of mice was sacrificed 8 months after DEN injection; yet, Opn Hep Tg mice only showed a modest increase in the number of tumors (Supporting Fig. S4), and very few Opn ΔHep mice showed tumors (1 in 5 mice, not shown). After pathological diagnosis of the hematoxylin and eosin (H&E) staining, HCC tumors were annotated for growth pattern matching human HCC (Supporting Fig. S5). HCCs from WT mice showed mostly trabecular and pseudoglandular patterns, known to be associated with well‐differentiated HCCs.( 28 ) All other groups of mice also showed a predominant trabecular pattern (with the exception of Opn −/−) together with a decrease in pseudoglandular and an increase in clear cell patterns (predominant in Opn −/− mice). The solid growth pattern, associated with poorly differentiated HCCs,( 28 , 29 ) was found in at least one case in Opn −/−, Opn ΔHep, and Opn Hep Tg, but not in WT or Alb.Cre + mice. However, this remained very infrequent, especially in Opn Hep Tg mice, where only one case presented that pattern. In summary, conditional ablation and overexpression of Opn in hepatocytes but not global ablation promotes liver carcinogenesis. This suggests that hepatocyte‐derived OPN at physiological levels acts as a tumor suppressor, while elevated OPN expression is pro‐tumorigenic.
FIG. 3.
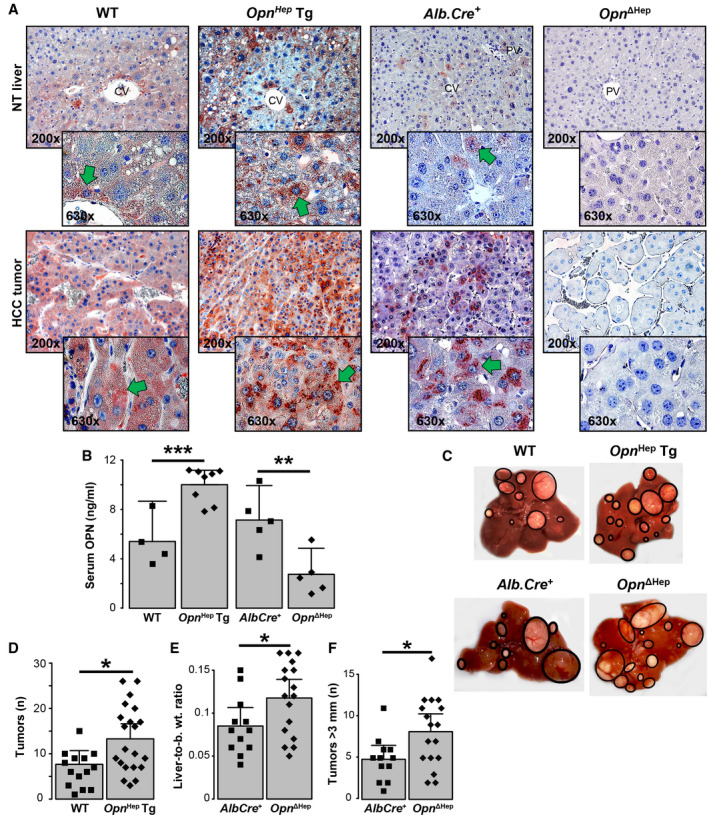
Overexpression and ablation of Opn in hepatocytes drives carcinogenesis in DEN‐injected mice. WT (n = 15), Opn Hep Tg (n = 21), Alb.Cre + (n = 12), and Opn ΔHep (n = 17) mice were injected DEN and sacrificed 12 months later. (A) IHC for OPN (green arrows). (B) Serum OPN in WT (n = 4), Opn Hep Tg (n = 8), Alb.Cre + (n = 5), and Opn ΔHep (n = 5) mice (mean ± SEM). (C) Representative gross appearance of the livers. (D) Number of macroscopic tumors per group (mean ± SEM), number of macroscopic tumors >3 mm per mouse liver (mean ± SEM) (E), and liver‐to–body weight ratio (mean ± SEM) (F). *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviation: b. wt., body weight.
Conditional Ablation of Opn in Hepatocytes Represses the Early Response to DEN and Drives the Emergence of CSCs
Previous studies suggest that the acute response to DEN (24 or 48 hours) could play a role protecting hepatocytes from HCC.( 30 , 31 ) We then performed RNA sequencing (RNA‐seq) in Opn ΔHep and Alb.Cre + as well as in Opn Hep Tg mice 48 hours after DEN or PBS injection. In Alb.Cre +, DEN induced a strong cellular response, with 1,347 differentially expressed (DE) genes (Fig. 4A), further characterized using the Ingenuity Pathway Analysis (IPA) platform (Fig. 4B,E). There was significant metabolic reprograming (activation of oxidative phosphorylation, tricarboxylic acid [TCA] cycle and lipid synthesis, reduced cholesterol, and unfolded protein response), activation of mammalian target of rapamycin, c‐JUN and p53 signaling, and decreased cell cycle. In Opn ΔHep mice, this response was considerably reduced (1,281 of 1,347 DE genes not affected by DEN) (Fig. 4A). The affected genes in common were linked to nuclear erythroid 2 p45‐related factor 2 (NRF2) signaling and xenobiotic metabolism (Fig. 4C), and glutathione metabolism was activated only in Opn ΔHep mice (Fig. 4D). In Opn Hep Tg mice, there was partial protection from the response to DEN (822 of 1,347 DE genes were not affected by DEN) (Fig. 4E and Supporting Fig. S6).
FIG. 4.
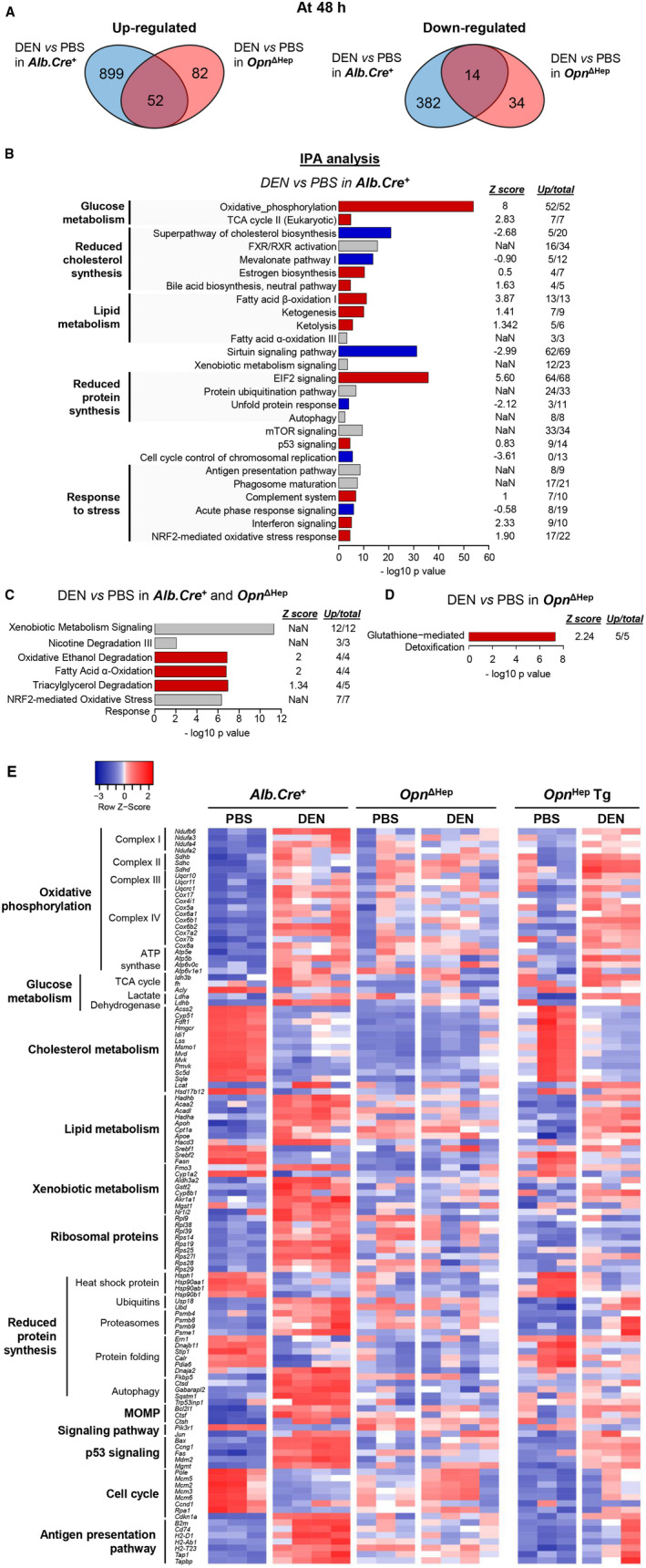
Ablation of Opn in hepatocytes decreases the early response to DEN. (A) Venn diagrams show the number of DE genes based on the RNA‐seq analysis of liver tissue from Alb.Cre + and Opn ΔHep mice injected with PBS (n = 3) or DEN (n = 4) and sacrificed at 48 hours. (B‐D) IPA analysis shows the signaling pathways changed by DEN in Alb.Cre +, Opn ΔHep mice or both. Red, blue, and gray represent positive, negative, or nonavailable Z scores, respectively, indicating positive or negative regulation of the pathway; “Up/total” refers to the number of up‐regulated genes among the DE genes within each signature (the rest of the genes are downregulated). (E) Heatmap shows the expression of genes of interest in all groups. Abbreviations: ATP, adenosine triphosphate; mTOR, mammalian target of rapamycin; and NaN, not a number (i.e., not available).
Five months after DEN injection, CSCs were detected in the aggregate fraction of primary hepatocytes by co‐expression of the progenitor markers CD44 and AFP.( 6 ) In our mice, we observed increased CD44+AFP+ cells in the aggregate fraction (Fig. 5A,B) compared with the non‐aggregate fraction (Supporting Fig. S7A). Aggregates of AFP+ cells also expressed hepatocyte nuclear factor 4, confirming that these were hepatocytes (Supporting Fig. S7B). There was no difference in the number of CSCs between WT and Opn Hep Tg mice, but it was significantly higher (P = 0.03) in Opn ΔHep versus Alb.Cre + (Fig. 5A‐C), suggesting that Opn ablation drives the emergence of CSCs after DEN injection.
FIG. 5.
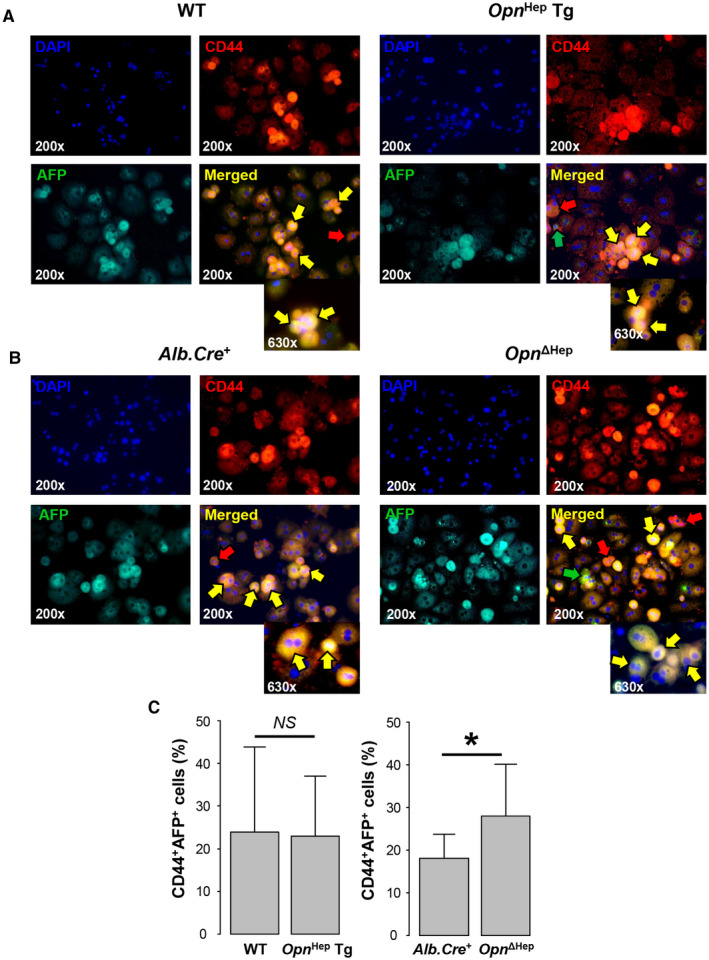
Ablation of Opn in hepatocytes drives the emergence of CSCs. (A,B) Immunofluorescence of CD44 (red) and AFP (green) (markers of CSCs) in the aggregate fraction of primary hepatocytes from WT, Opn Hep Tg, Alb.Cre +, and Opn ΔHep 5 months after DEN injection. Red arrow, CD44+AFP− cells; green arrow, CD44−AFP+ cells; and yellow arrow, CD44+AFP+ cells (CSCs). (C) Number of CSCs by group based on computer‐assisted morphometry analysis of the immunostaining of the aggregate fraction (n = 3/group, mean ± SEM). *P < 0.05. Abbreviation: NS, not significant.
Overexpression of Opn Induces mRNA Signatures of Carcinogenesis in CSCs and HCC Associated With Poor Outcome in Patients
Next, we performed RNA‐seq in the aggregate and non‐aggregate fractions of primary hepatocytes from 5‐month‐old DEN‐injected WT and Opn Hep Tg mice, as well as in the NT and HCC tissue from 12‐month‐old DEN‐injected WT, Opn −/−, and Opn Hep Tg mice. At 5 months, there was a significant difference in gene expression between the CSCs (enriched in the aggregates) and normal hepatocytes (non‐aggregates) in WT as well as in Opn Hep Tg mice, with little overlap between groups (4 of 448 genes) (Fig. 6A). The difference was analyzed using the IPA platform. In CSCs, overexpression of Opn induced ECM remodeling, inflammation, and activated signaling pathways associated with liver carcinogenesis, such as signal transducer and activator of transcription 3 (STAT3), HIPPO or phosphoinositide 3‐kinase (PI3K)/Akt, together with decreased p53 signaling (Fig. 6B). There was also a decrease in the complement system and coagulation (found in CSCs from human HCC( 32 )). Furthermore, overexpression of Opn induced a very different phenotype in normal hepatocytes (Supporting Fig. S8A). Inflammation was greatly reduced as well as signaling pathways associated with cancer (e.g., endothelial growth factor, NRF2, hepatocyte growth factor [HGF], Janus kinase 2/STAT), stem signatures, and signatures of other cancers. The similarities among CSCs and normal hepatocytes regarding Opn overexpression were a global decrease in normal liver metabolism, suggesting cell dedifferentiation, activation of PI3K/Akt, and HIPPO signaling together with decreased p53 signaling. At 12 months, NT tissue from Opn Hep Tg mice expressed signatures of cell cycle, inflammation, and activation of signaling pathways associated with carcinogenesis (STAT3, MYC, and Wnt), together with decreased healthy liver metabolic pathways and p53 signaling, based on GSEA (Supporting Fig. S8B). HCC tissue from the same mice showed signatures of ECM remodeling, inflammation, and poor outcome for patients with HCC. These mice also presented decreased signatures of healthy hepatocyte metabolism, likely reflecting de‐differentiation and negative association with HCC signatures of well‐differentiated HCC (Hoshida S3 and Chiang CTNNB1 subclasses( 33 , 34 )) (Fig. 6C). Similarly, HCCs from Opn −/− mice expressed mRNA signatures of highly proliferative and poorly differentiated tumors, including inflammation, ECM remodeling, and poorly differentiated HCC subclasses (Hoshida S1 and Chiang proliferation class) (Supporting Fig. S8C). Opn −/− mice also displayed decreased signatures of healthy liver metabolism and Hoshida’s S3 subclass.( 33 )
FIG. 6.
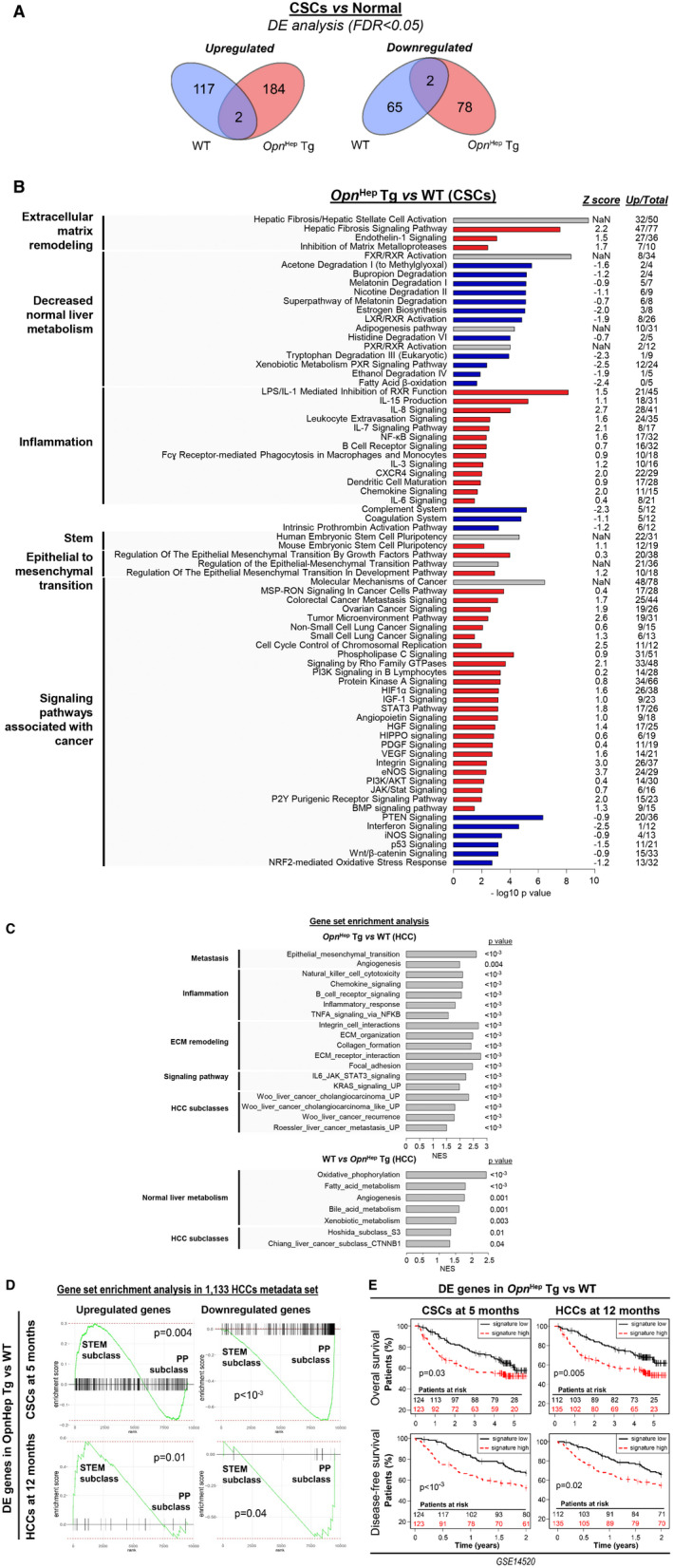
Overexpression of Opn induces mRNA signatures of carcinogenesis in CSCs and of HCC progression in HCC, associated with poor outcome in patients. (A) Venn diagrams show the number of DE genes between the aggregate (CSCs) and non‐aggregate (normal hepatocytes) fraction of primary hepatocytes from WT and Opn Hep Tg mice 5 months after DEN injection, based on RNA‐seq analysis (n = 4/group). (B) Enrichment analysis using the IPA platform. Black, white, and gray represent positive, negative, or non‐available Z scores, respectively, indicating positive or negative regulation of the pathway; “Up/total” refers to the number of up‐regulated genes among the DE genes within each signature (the rest of the genes are down‐regulated). (C) mRNA signatures enriched in Opn Hep Tg (top) or in WT (bottom) mice based on GSEA after RNA‐seq of HCC tissues from DEN‐injected mice (n = 4/group). GSEA of the association between DE genes in Opn Hep Tg versus WT mice and HCCs subclasses in 1,133‐HCC metadata set.( 25 ) (D) STEM and PP subclasses represent the ones with worth and best outcome respectively. (E) Survival analysis after patients clustering based on the gene expression of those gene sets in Roessler et al.( 24 )
Next, we analyzed the association of the mRNA signatures from mouse with clinical outcomes in human HCC patients. First, we extracted the DE genes in Opn Hep Tg versus WT at 5 months in CSCs and at 12 months in HCCs. Then, we analyzed the association of these gene sets with the STEM subclass of poorly differentiated HCCs in the 1,133 HCCs metadata set by GSEA (Fig. 6D). The genes up‐regulated in Opn Hep Tg in CSCs as well as in HCCs were positively enriched in the STEM subclass, characterized by the lowest survival rate among patients. Similarly, genes down‐regulated in Opn Hep Tg were negatively enriched in STEM. Finally, we used those gene sets to cluster patients with HCC from Roessler et al.( 24 ) Opn Hep Tg signatures from CSCs and HCCs were associated with decreased overall and disease‐free survival in these patients (Fig. 6E). Altogether, our RNA‐seq data suggest that Opn overexpression in CSCs promote carcinogenesis, whereas both Opn ablation and overexpression in HCCs enhance HCC progression.
Secreted OPN Has Limited Impact on CSCs Proliferation and Progression to HCC
Because Opn Hep Tg mice showed increased OPN secretion and the OPN/CD44 signaling axis participates in the progression of HCC, we examined whether this pathway could be involved in the progression of CSCs to HCC. To this end, first, we isolated the aggregate and non‐aggregate fractions from 5‐month‐old DEN‐injected WT mice and treated them with recombinant OPN (rOPN) for 24 hours (Fig. 7A). RNA‐seq revealed that rOPN had little effect on both cell populations, as there were no differentially DE genes after treatment (no genes with significant false discovery rate [FDR]). Second, we isolated CSCs from 5‐month‐old DEN‐injected WT mice and transplanted them into WT and Opn −/− mice pretreated with retrorsine (Fig. 7B). One week later, mice were treated with CCl4 for 3 weeks to induce fibrosis, which is required for CSCs to become pro‐carcinogenic.( 6 ) This also stimulated OPN secretion, with a strong reduction in Opn −/− mice, because in those mice only the transplanted hepatocytes carried the Opn gene. Magnetic resonance imaging at 5 and 8 months showed small (<2 mm) and large (>10 mm) tumors, respectively, in both groups (Supporting Fig. S9A). After 8 months, analysis of tumor burden revealed that most mice developed at least one tumor, but there was no difference between groups (Fig. 7C,D). The tumor tissue from Opn −/− mice expressed OPN, confirming that HCC arose from transplanted CSCs that originated from WT mice (Supporting Fig. S9B). Third, we investigated the role of OPN signaling through CD44 by injecting Cd44 −/− and Cd44 −/− Opn Hep Tg mice with DEN and sacrificing them after 12 months. Cd44 ablation was confirmed by IHC (Supporting Fig. S9C). Cd44 −/− Opn Hep Tg showed more tumors than Cd44 −/− mice (P = 0.007), and there was no difference compared with Opn Hep Tg mice (Fig. 7E,F). Altogether, these results suggest that secreted OPN plays a limited role in driving CSCs progression to HCC and that CD44 signaling is not involved.
FIG. 7.
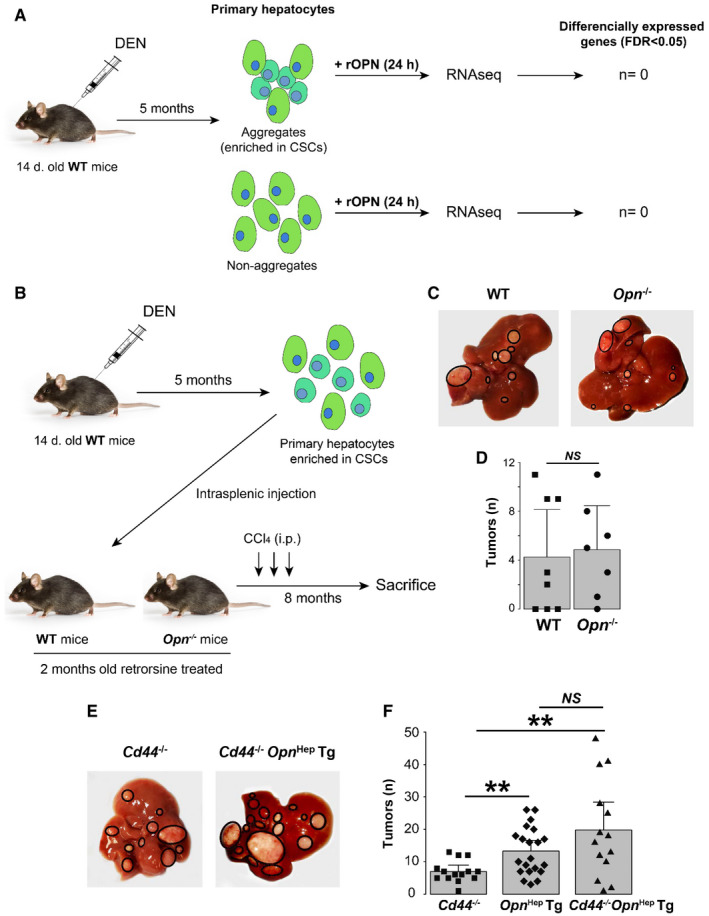
Secreted OPN has limited effect on CSC proliferation and progression to HCC. (A) Experimental approach to study the effect of rOPN on primary hepatocytes and CSCs (n = 3/group). (B‐D) Experimental approach to study the role of extracellular OPN on CSC progression to cancer, representative gross appearance of the livers, and number of tumors in both groups (mean ± SEM). (E) Representative gross appearance of livers from Cd44 −/− (n = 14) and Cd44 −/− Opn Hep Tg (n = 14) mice injected with DEN and sacrificed 12 months later. (F) Number of macroscopic tumors per mouse liver (mean ± SEM).
Discussion
The complexity of liver carcinogenesis can be explained by the involvement of multiple cell types and by the molecular diversity among patients. The mean number of mutations in HCC is quite elevated compared with other cancers (~40 per tumor)( 35 ); thus, induction of multiple oncogenes and reduction of several tumor suppressors are needed to trigger this cancer. Over the last 20 years, many oncogenes and tumor suppressors were identified. Among them, a significant number act both as oncogenes and tumor suppressors, depending on their level of expression. For example, ablation of c‐Met,( 36 ) inhibitor of nuclear factor kappa B kinase subunit beta (IKKβ),( 37 ) JNK,( 38 ) β‐catenin,( 39 ) or Shp2( 40 ) in hepatocytes induces more tumors, while overexpression also promotes carcinogenesis in mice. Based on our results, OPN appears to behave similarly. In addition, hepatocyte‐derived OPN has different effects during initiation and progression of liver cancer. At physiological levels, OPN is a tumor suppressor and is involved during the first stages of carcinogenesis by lessening the response to DEN and increasing CSCs in Opn ΔHep; however, it does not stimulate the progression of CSCs to HCC, as shown by the presence of more tumors at 12 months in Opn ΔHep but not earlier. When OPN is overexpressed, it is pro‐tumorigenic mostly by affecting the last stages of carcinogenesis, as shown by the number of CSCs not increasing in Opn Hep Tg mice but expressing signatures of carcinogenesis.
In this study, we first showed that the expression in hepatocytes increases along with progression of chronic liver disease to HCC. Although the increase in OPN in HCC has been reported,( 12 , 41 , 42 ) our data show intermediate expression in dysplastic nodules, suggesting a role for hepatocyte‐derived OPN in the onset of HCC. This was confirmed by showing significant association between OPN mRNA and incidence of HCC in patients with cirrhosis. Hence, we hypothesized that increased hepatocyte‐derived OPN is a driver of hepatocarcinogenesis, which was confirmed in mouse models of HCC. Because the pro‐tumorigenic effect of Opn overexpression occurred in three mouse genotypes (WT, Opn −/−, and Cd44 −/−), it indicates a solid effect of elevated hepatocyte‐derived OPN in carcinogenesis.
In Opn Hep Tg mice, the number of CSCs at 5 months did not increase, suggesting that high expression of OPN rather drives CSC progression to HCC. RNA‐seq revealed a profound change in their phenotype, shown by activation of signaling pathways involved in carcinogenesis such as STAT3,( 43 ) HIPPO/YAP/NOTCH( 44 ) and PI3K/Akt,( 29 ) as well as in HCC progression (hypoxia inducible factor 1 alpha subunit [HIF1α], HGF, vascular endothelial growth factor, and platelet‐derived growth factors) and decreased p53 signaling. In patients with cirrhosis expressing the OPN‐high gene network, p53 signaling also decreased, as well as in the NT liver from Opn Hep Tg mice at 12 months. P53 is a well‐known tumor suppressor, mutated in about 20% of human HCC.( 45 ) Mutations of TP53 and activation of MYC in hepatocytes induce poorly differentiated HCCs in a few months.( 29 ) However, how OPN overexpression represses P53 is unknown. Our initial hypothesis was the well‐described CD44/PI3K/Akt/MDM2 axis,( 31 ) but we ruled out this possibility experimentally by showing no involvement of CD44 in Opn Hep Tg mice. On the other hand, Opn overexpression induces a decrease in cancer‐associated signatures in normal hepatocytes. The distinct roles of OPN overexpression in CSCs and hepatocytes could partially explain some of the inconclusive results in this field. We then investigated the possibility that secreted OPN could drive CSCs progression to HCC; however, it was ruled out using three approaches (treatment with rOPN, CSC transplantation, and ablation of an OPN receptor). Thus, if secreted OPN is involved in liver carcinogenesis, its role is minimal and most of the effects observed in Opn Hep Tg mice occur intracellularly.
The only study showing more tumors in Opn −/− mice injected with DEN, proposed that intracellular OPN is a negative regulator of carcinogenesis.( 17 ) In mice fed alcohol, Opn ablation increases steatosis( 46 ) and iron deposition,( 47 ) while Opn overexpression in hepatocytes decreases steatosis.( 46 ) Likewise, in NASH‐induced HCC, Opn ablation worsens steatosis and increases tumorigenesis.( 48 ) Altogether, these data point to a physiological role of OPN in hepatocytes not previously known. Our study shows that at physiological levels, OPN acts as a tumor suppressor. Of note, Opn ΔHep also show ablation of Opn in cholangiocytes, but DEN only targets hepatocytes, because high levels of P450 are necessary to metabolize this pro‐carcinogenic chemical to induce DNA mutations.( 21 ) Moreover, there was no alteration in cholangiocytes or ductular reaction in Opn ΔHep mice.
Regarding the mechanisms involved, we hypothesized that it could be through regulation of the acute response to DEN, as suggested in recent studies.( 30 , 31 ) The acute response identified by RNA‐seq included metabolic reprograming and activation of previously reported p53 and cJun signaling,( 30 , 31 ) which was strongly repressed in Opn ΔHep mice. To date, the role of this cellular response is not fully understood but it could involve DNA repair.( 31 ) Although this could be a mechanism by which OPN drives liver carcinogenesis, we did not find a difference in genetic mutations in publicly available data from human HCC based on OPN expression (data not shown). A second possibility is that this response changes DNA methylation to protect cells from cancer( 49 ); however, we did not observe a change in global DNA methylation after DEN treatment in Opn ΔHep or Alb.Cre + (data not shown). Thus, the molecular mechanisms involved in the response to DEN remain elusive, but our findings suggest a central role for OPN in its regulation. This response likely protects from carcinogenesis, as we observed more CSCs at 5 months in Opn ΔHep mice.
The finding that global ablation of Opn does not induce more tumors points at a cell‐specific role of OPN in carcinogenesis with a potential pro‐tumorigenic effect in other cells that compensate when Opn is ablated in hepatocytes. Especially, the role of OPN in immune cells during carcinogenesis is worthy of future investigation.
The data confirm the role of OPN in driving the HCC phenotype, as already suggested in vitro and in orthotopic models.( 11 , 13 , 14 , 15 , 16 ) Here, we show in vivo that HCCs overexpressing OPN display a transcriptomic profile of proliferative HCC, associated with worst outcome.( 50 ) We confirm, in large cohorts of patients, that HCCs expressing high levels of OPN are associated with HCC subclasses of highly proliferative and poorly differentiated tumors with worst patient outcome. In addition, mRNA signatures from mice with Opn overexpression matched these patient subclasses. The mechanisms remain to be determined, but in vitro work suggests that OPN could cooperate with nuclear factor kappa B,( 13 ) HIF1α,( 15 ) matrix metalloproteinase 2,( 14 ) or vimentin.( 16 ) On the other hand, Opn ablation in HCC also triggers highly proliferative and poorly differentiated tumors in mice, consistent with the tumor suppressor role attributed to OPN at physiological levels.
In conclusion, this study identifies that OPN acts both as pro‐tumorigenic and as a tumor suppressor during liver carcinogenesis, mostly through intracellular mechanisms. Future studies are needed to better understand the physiological and pathological role of OPN in different cell types during chronic liver disease.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S51
Fig S52
Fig S6
Fig S7
Fig S81
Fig S82
Fig S91
Fig S92
Method S1
Acknowledgment
The authors thank Dr. Satoshi Mochida (Gastroenterology and Hepatology, Internal Medicine, Saitama Medical School, Saitama, Japan) for providing the Opn Hep Tg mice. They also thank Dr. Debanjan Dhar (Department of Pharmacology, University of California, San Diego, CA) for his help with the protocol for CSC enrichment and transplantation.
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases (DK099558), U.S. Department of Defense (CA170172), NIH/National Cancer Institute (CA233794), Cancer Prevention and Research Institute of Texas (RR180016), and the European commission (ERC‐AdG‐2014 HEPCIR).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T‐Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 3. Allaire M, Goumard C, Lim C, Le Cleach A, Wagner M, Scatton O. New frontiers in liver resection for hepatocellular carcinoma. JHEP Rep 2020;2:100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tan DJH, Wong C, Ng CH, Poh CW, Jain SR, Huang DQ, et al. A meta‐analysis on the rate of hepatocellular carcinoma recurrence after liver transplant and associations to etiology, alpha‐fetoprotein, income and ethnicity. J Clin Med 2021;10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer 2015;15:653‐667. [DOI] [PubMed] [Google Scholar]
- 6. He G, Dhar D, Nakagawa H, Font‐Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL‐6 signaling. Cell 2013;155:384‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Font‐Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015;162:766‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holczbauer Á, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, Raggi C, et al. Modeling pathogenesis of primary liver cancer in lineage‐specific mouse cell types. Gastroenterology 2013;145:221‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis‐promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med 2010;14:2037‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, et al. Osteopontin, an oxidant stress sensitive cytokine, up‐regulates collagen‐I via integrin alpha(V)beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology 2012;55:594‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song Z, Chen W, Athavale D, Ge X, Desert R, Das S, et al. Osteopontin takes center stage in chronic liver disease. Hepatology 2021;73:1594‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cabiati M, Gaggini M, Cesare MM, Caselli C, De Simone P, Filipponi F, et al. Osteopontin in hepatocellular carcinoma: a possible biomarker for diagnosis and follow‐up. Cytokine 2017;99:59‐65. [DOI] [PubMed] [Google Scholar]
- 13. Zhao J, Dong LI, Lu B, Wu G, Xu D, Chen J, et al. Down‐regulation of osteopontin suppresses growth and metastasis of hepatocellular carcinoma via induction of apoptosis. Gastroenterology 2008;135:956‐968. [DOI] [PubMed] [Google Scholar]
- 14. Sun B‐S, Dong Q‐Z, Ye Q‐H, Sun H‐J, Jia H‐L, Zhu X‐Q, et al. Lentiviral‐mediated miRNA against osteopontin suppresses tumor growth and metastasis of human hepatocellular carcinoma. Hepatology 2008;48:1834‐1842. [DOI] [PubMed] [Google Scholar]
- 15. Cao L, Fan X, Jing W, Liang Y, Chen R, Liu Y, et al. Osteopontin promotes a cancer stem cell‐like phenotype in hepatocellular carcinoma cells via an integrin‐NF‐kappaB‐HIF‐1alpha pathway. Oncotarget 2015;6:6627‐6640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong Q, Zhu X, Dai C, Zhang X, Gao X, Wei J, et al. Osteopontin promotes epithelial‐mesenchymal transition of hepatocellular carcinoma through regulating vimentin. Oncotarget 2016;7:12997‐13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fan X, He C, Jing W, Zhou X, Chen R, Cao L, et al. Intracellular Osteopontin inhibits toll‐like receptor signaling and impedes liver carcinogenesis. Cancer Res 2015;75:86‐97. [DOI] [PubMed] [Google Scholar]
- 18. Lee SH, Park JW, Woo SH, Go DM, Kwon HJ, Jang JJ, et al. Suppression of osteopontin inhibits chemically induced hepatic carcinogenesis by induction of apoptosis in mice. Oncotarget 2016;7:87219‐87231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu Y, Yang J, Xu DA, Gao X‐M, Zhang ZE, Hsu JL, et al. Disruption of tumour‐associated macrophage trafficking by the osteopontin‐induced colony‐stimulating factor‐1 signalling sensitises hepatocellular carcinoma to anti‐PD‐L1 blockade. Gut 2019;68:1653‐1666. [DOI] [PubMed] [Google Scholar]
- 20. Mochida S, Yoshimoto T, Mimura S, Inao M, Matsui A, Ohno A, et al. Transgenic mice expressing osteopontin in hepatocytes as a model of autoimmune hepatitis. Biochem Biophys Res Commun 2004;317:114‐120. [DOI] [PubMed] [Google Scholar]
- 21. Rao KV, Vesselinovitch SD. Age‐ and sex‐associated diethylnitrosamine dealkylation activity of the mouse liver and hepatocarcinogenesis. Cancer Res 1973;33:1625‐1627. [PubMed] [Google Scholar]
- 22. Zhang J, Han C, Ungerleider N, Chen W, Song K, Wang Y, et al. A Transforming growth factor‐beta and H19 signaling axis in tumor‐initiating hepatocytes that regulates hepatic carcinogenesis. Hepatology 2019;69:1549‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Research Network . Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327‐1341.e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roessler S, Jia H‐L, Budhu A, Forgues M, Ye Q‐H, Lee J‐S, et al. A unique metastasis gene signature enables prediction of tumor relapse in early‐stage hepatocellular carcinoma patients. Cancer Res 2010;70:10202‐10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Désert R, Rohart F, Canal F, Sicard M, Desille M, Renaud S, et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017;66:1502‐1518. [DOI] [PubMed] [Google Scholar]
- 26. Hoshida Y, Villanueva A, Sangiovanni A, Sole M, Hur C, Andersson KL, et al. Prognostic gene expression signature for patients with hepatitis C‐related early‐stage cirrhosis. Gastroenterology 2013;144:1024‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stearman RS, Dwyer‐Nield L, Grady MC, Malkinson AM, Geraci MW. A macrophage gene expression signature defines a field effect in the lung tumor microenvironment. Cancer Res 2008;68:34‐43. [DOI] [PubMed] [Google Scholar]
- 28. Schlageter M, Terracciano LM, D'Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol 2014;20:15955‐15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molina‐Sánchez P, Ruiz de Galarreta M, Yao MA, Lindblad KE, Bresnahan E, Bitterman E, et al. Cooperation between distinct cancer driver genes underlies intertumor heterogeneity in hepatocellular carcinoma. Gastroenterology 2020;159:2203‐2220.e2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bopp A, Wartlick F, Henninger C, Schwarz M, Kaina B, Fritz G. Rac1 promotes diethylnitrosamine (DEN)‐induced formation of liver tumors. Carcinogenesis 2015;36:378‐389. [DOI] [PubMed] [Google Scholar]
- 31. Dhar D, Antonucci L, Nakagawa H, Kim JY, Glitzner E, Caruso S, et al. Liver cancer initiation requires p53 inhibition by CD44‐enhanced growth factor signaling. Cancer Cell 2018;33:1061‐1077.e1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J, Liu K, Sheng Y, Zhang Q, Chen L, Qian H, et al. Enrichment and identification of differentially expressed genes in hepatocellular carcinoma stem‐like cells. Oncol Lett 2020;20:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet J‐P, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385‐7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779‐6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takami T, Kaposi‐Novak P, Uchida K, Gomez‐Quiroz LE, Conner EA, Factor VM, et al. Loss of hepatocyte growth factor/c‐Met signaling pathway accelerates early stages of N‐nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res 2007;67:9844‐9851. [DOI] [PubMed] [Google Scholar]
- 37. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005;121:977‐990. [DOI] [PubMed] [Google Scholar]
- 38. Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev 2011;25:634‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang XF, Tan X, Zeng G, Misse A, Singh S, Kim Y, et al. Conditional beta‐catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet‐derived growth factor receptor alpha/phosphoinositide 3‐kinase signaling. Hepatology 2010;52:954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Han T, Xiang D‐M, Sun W, Liu NA, Sun H‐L, Wen W, et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J Hepatol 2015;63:651‐660. [DOI] [PubMed] [Google Scholar]
- 41. Kim J, Ki SS, Lee SD, Han CJ, Kim YC, Park SH, et al. Elevated plasma osteopontin levels in patients with hepatocellular carcinoma. Am J Gastroenterol 2006;101:2051‐2059. [DOI] [PubMed] [Google Scholar]
- 42. Sun T, Li P, Sun D, Bu Q, Li G. Prognostic value of osteopontin in patients with hepatocellular carcinoma: a systematic review and meta‐analysis. Medicine (Baltimore) 2018;97:e12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF‐kappaB inhibits tumor promotion and progression by preventing oxidative stress‐driven STAT3 activation. Cancer Cell 2010;17:286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patel SH, Camargo FD, Yimlamai D. Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 2017;152:533‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ge X, Leung TM, Arriazu E, Lu Y, Urtasun R, Christensen B, et al. Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor‐alpha and prevents early alcohol‐induced liver injury in mice. Hepatology 2014;59:1600‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Magdaleno F, Ge X, Fey H, Lu Y, Gaskell H, Blajszczak CC, et al. Osteopontin deletion drives hematopoietic stem cell mobilization to the liver and increases hepatic iron contributing to alcoholic liver disease. Hepatol Commun 2018;2:84‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nardo AD, Grün NG, Zeyda M, Dumanic M, Oberhuber G, Rivelles E, et al. Impact of osteopontin on the development of non‐alcoholic liver disease and related hepatocellular carcinoma. Liver Int 2020;40:1620‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ito Y, Nakajima K, Masubuchi Y, Kikuchi S, Saito F, Akahori Y, et al. Expression characteristics of genes hypermethylated and downregulated in rat liver specific to nongenotoxic hepatocarcinogens. Toxicol Sci 2019;169:122‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Desert R, Nieto N, Musso O. Dimensions of hepatocellular carcinoma phenotypic diversity. World J Gastroenterol 2018;24:4536‐4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S51
Fig S52
Fig S6
Fig S7
Fig S81
Fig S82
Fig S91
Fig S92
Method S1