

Abstract
Hepatocellular carcinoma (HCC) is one of the most lethal human cancers. Liver transplantation has been an effective approach to treat liver cancer. However, significant numbers of patients with HCC experience cancer recurrence, and the selection of suitable candidates for liver transplant remains a challenge. We developed a model to predict the likelihood of HCC recurrence after liver transplantation based on transcriptome and whole‐exome sequencing analyses. We used a training cohort and a subsequent testing cohort based on liver transplantation performed before or after the first half of 2012. We found that the combination of transcriptome and mutation pathway analyses using a random forest machine learning correctly predicted HCC recurrence in 86.8% of the training set. The same algorithm yielded a correct prediction of HCC recurrence of 76.9% in the testing set. When the cohorts were combined, the prediction rate reached 84.4% in the leave‐one‐out cross‐validation analysis. When the transcriptome analysis was combined with Milan criteria using the k‐top scoring pairs (k‐TSP) method, the testing cohort prediction rate improved to 80.8%, whereas the training cohort and the combined cohort prediction rates were 79% and 84.4%, respectively. Application of the transcriptome/mutation pathways RF model on eight tumor nodules from 3 patients with HCC yielded 8/8 consistency, suggesting a robust prediction despite the heterogeneity of HCC. Conclusion: The genome prediction model may hold promise as an alternative in selecting patients with HCC for liver transplant.
We have determined algorithms which predict with high accuracy the possibility of a hepatocellular carcinoma (HCC) reappearing to a new transplanted liver, after the original HCC‐containing liver resection. The algorithm is based on genomic analyses of the HCC, predicated on transcriptome expression, and gene mutations in selected pathways.
Abbreviations
- AUC
area under the curve
- CDC
cell division cycle
- CoA
coenzyme A
- GO
Gene Ontology
- HCC
hepatocellular carcinoma
- k‐TSP
k‐top scoring pairs
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LDA
linear discriminant analysis
- LOOCV
leave‐one‐out cross‐validation
- RF
random forest
- RNA‐seq
RNA‐sequencing
- ROC
receiver operating characteristic
- SVM
support vector machine
Hepatocellular carcinoma (HCC) is the most frequent type of liver cancer and has an overall 5‐year survival of 18%,( 1 ) with only glioblastoma and pancreatic cancer having higher mortality.( 2 ) Currently, surgical intervention remains the most effective therapy. When HCC is localized and the liver function is adequate, tumor resection or cryoablation may be a treatment option. However, liver transplantation is the mainstay of successful treatment because it addresses both the tumor and the underlying liver disease, thereby eliminating the primary risk factor for additional tumors.
The first successful liver transplant was performed in 1967.( 3 ) Since then, the number of liver transplants applied to treat HCC has steadily increased. The Milan criteria were developed in 1996 to guide the selection of HCC patients by limiting transplantation to those individuals with HCC comprising a single lesion <5 cm in diameter or up to three tumor nodules but with no tumor nodule >3 cm in diameter.( 4 ) However, the Milan criteria were ultimately viewed by some as too restrictive, denying transplantation to a large number of potentially treatable patients with HCC. Several subsequent criteria were developed to address this,( 5 ) and the latest Extended Toronto criteria include patients with any size or number of tumors in the absence of systemic cancer‐related symptoms, extrahepatic disease, or poorly differentiated tumor.( 6 ) Posttransplant survival rates applying these criteria range from 65%‐85%.( 7 ) A major consideration in selection of transplant candidates is posttransplant HCC recurrence. Based on various studies, the HCC recurrence rate has been up to 20% among liver transplant patients, with a median recurrence time of 14 months following transplant. The median post‐recurrence survival time is only 12 months.( 8 , 9 ) Thus, a better prediction method of HCC recurrence is necessary to improve the clinical outcomes of patients with HCC.
Taking advantage of high‐throughput genomic technology, biomarkers can be detected and measured on a genome‐wide scale. For the transcriptomic study, gene expressions are able to be quantified by both large‐scale gene‐expression microarray and RNA‐sequencing (RNA‐seq) technology. These gene‐expression and microRNA expression techniques have been applied to HCC studies to detect new biomarkers, explore molecular mechanisms, and discover novel therapeutic targets.( 10 ) For DNA‐level analysis, genome‐wide somatic mutations and copy number variations can be detected by both single‐nucleotide polymorphism microarray and whole exome/genome sequencing. Investigators have used these cutting‐edge technologies to discover cancer‐driven mutations and to study HCC molecular profiles.( 11 , 12 , 13 )
Given the current need to rationally assess the likelihood of posttransplant tumor recurrence, and taking advantage of current genomic methodologies, we propose a prediction model in this report. We constructed and validated a prediction model based on the transcriptomic and exomic analyses on HCC samples to predict the likelihood of HCC recurrence following liver transplantation.
Materials and Methods
Clinical samples were obtained in accordance with the guidelines approved by the Institutional Review Board of the University of Pittsburgh. All methods were carried out in accordance with relevant guidelines and regulations. Informed consent exemptions were obtained from the University of Pittsburgh Institutional Review Board with # STUDY19070068.
Tissue Samples
The 128 tissue specimens in this study were obtained from the University of Pittsburgh Medical Center archived tissue deposit center in compliance with institutional regulatory guidelines. The clinical features of the samples, including etiology, pre‐transplant treatment, pathology grade, microvascular/macrovascular invasion, sizes and numbers of the tumors, immunosuppression, mammalian target of rapamycin inhibitor application, and status of follow‐up are listed in Table 1 and Supporting Table S1. Milan status was assessed based on the examination of the explanted liver. In some cases, tumors were found to be of size or number beyond Milan criteria for one of three reasons: First, some transplants were performed before the use of Milan criteria as a screening process. Second, clinical Milan status was based on radiologic assessment, and in some cases additional small tumors or variations in tumor size were discovered on analysis of the liver explant. Third, some patients with tumors beyond Milan criteria underwent living donor transplant outside the national match program. Cancer tissues from the explanted livers were identified through hematoxylin and eosin staining. The position of the cancer in the slide was matched with the tissue block and circled. The identified positions were then used to obtain needle cores from the cancer tissues, to simulate a clinical needle biopsy. Non‐liver and benign tissues some distance from the cancer were used as matched normal controls. The sample size was estimated by power analysis and specimen availability. The processes and protocols followed the guidelines approved by the Institutional Review Board of the University of Pittsburgh.
TABLE 1.
Clinical Features of the HCC Cohort
| Clinical Features | Category/Statistical Measurements | Training | Validation |
|---|---|---|---|
| Number of samples | Count | 38 | 26 |
| Latest recurrence status | Nonrecurrent | 23 | 18 |
| Recurrent | 15 | 8 | |
| Milan score | In | 16 (42.1%) | 20 (76.9%) |
| Out | 22 (57.9%) | 6 (23.1%) | |
| Tx age | Mean ± SD | 56.2 ± 8.9 | 61.9 ± 5.7 |
| Tx type | Orthotopic | 37 | 16 |
| Living related | 1 | 3 | |
| Living nonrelated | 0 | 7 | |
| Underlying disease | HBV | 5 (13.2%) | 0 (0.0%) |
| HCV | 17 (44.7%) | 9 (34.6%) | |
| Nodular regenerative hyperplasia | 1 (2.6%) | 0 (0.0%) | |
| Hemochromatosis | 2 (5.3%) | 0 (0.0%) | |
| NASH | 5 (13.2%) | 8 (30.8%) | |
| EtOH | 8 (21.1%) | 10 (38.5%) | |
| A1AT | 1 (2.6%) | 0 (0.0%) | |
| PBC | 1 (2.6%) | 1 (3.8%) | |
| NRH | 0 (0.0%) | 4 (15.4%) | |
| Orig. number of tumors | 1 | 10 | 9 |
| 2 | 8 | 6 | |
| 3 | 4 | 3 | |
| 4+ | 16 | 8 | |
| Orig. tumor sizes (cm) | [min, max] | [0.2, 21.0] | [0.3, 6.0] |
| Alive status at last follow‐up | Alive | 13 | 17 |
| Dead | 24 | 9 | |
| Unknown | 1 | 0 | |
| Pretransplant Rx | Y | 18 | 17 |
| N | 10 | 6 | |
| Unknown | 10 | 3 | |
| PreTx Rx type | RFA | 6 (15.8%) | 9 (34.6%) |
| Resection | 2 (5.3%) | 4 (15.4%) | |
| TACE | 13 (34.2%) | 14 (53.8%) | |
| Sorafenib | 0 (0.0%) | 5 (19.2%) | |
| None | 10 (26.3%) | 6 (23.1%) | |
| Immunosuppression | Tacrolimus | 18 (47.4%) | 25 (96.2%) |
| Mycophenolate | 11 (28.9%) | 25 (96.2%) | |
| Cyclosporine | 5 (13.2%) | 3 (11.5%) | |
| Everolimus | 3 (7.9%) | 14 (53.8%) | |
| Azathioprine | 0 | 4 (15.4%) | |
| mTOR inhibitor | Y | 3 | 13 |
| N | 28 | 12 | |
| Unknown | 7 | 1 | |
| Highest AFP level | [min, median, max] | [4, 40, 34,818] | [2.1, 34.05, 22,256] |
| HCC differentiation | Poor | 6 | 4 |
| Moderate | 21 | 17 | |
| Well | 11 | 5 | |
| Microvascular invasion | Yes | 23 | 14 |
| No | 13 | 12 | |
| Unknown | 2 | 0 | |
| Macrovascular invasion | Yes | 5 | 2 |
| No | 32 | 24 | |
| Unknown | 1 | 0 |
Abbreviations: A1AT, alpha‐1 antitrypsin deficiency; AFP, alpha‐fetoprotein; EtOH, ethanol; HBV, hepatitis B virus; HCV, hepatitis C virus; mTOR, mammalian target of rapamycin; N, no; NASH, nonalcoholic steatohepatitis; NRH, nodular regenerative hyperplasia; PBC, primary biliary cholangitis; RFA, radiofrequency ablation; Rx, prescription; TACE, transarterial chemoembolization; Tx, transplant; and Y , yes.
Inclusion and Exclusion Criteria
Samples were obtained from native liver explants at the time of liver transplantation on the basis of tissue availability. HCC represents a subset of liver transplant patients. Samples with sufficient quantity of RNA and DNA were selected for the study. For both training and testing data sets, patients were required to have clinical follow‐up for at least 3 years (Table 2 and Supporting Table S1). HCC recurrence was monitored by a combination of magnetic resonance imaging, computed tomography, ultrasound, alpha‐fetoprotein level, and clinical evaluation. Re‐emergence of HCC within 3 years after transplant is defined as recurrence, whereas absence of HCC detection in the same period is defined as non‐recurrence.
TABLE 2.
Clinical Features of Samples Collected From HCC Transplant Patients
| Cohort | Sample | Surgical Year | Recur Status | Milan | Months to Recur | Follow‐up (Months) |
|---|---|---|---|---|---|---|
| Training | Training 1 | 1988 | Non‐Recur | Out | NA | 72 |
| Training | Training 2 | 2009 | Non‐Recur | In | NA | 134 |
| Training | Training 3 | 2007 | Non‐Recur | Out | NA | 121.5 |
| Training | Training 4 | 2008 | Non‐Recur | In | NA | 136.9 |
| Training | Training 5 | 2008 | Non‐Recur | In | NA | 76.7 |
| Training | Training 6 | 2008 | Non‐Recur | Out | NA | 157.3 |
| Training | Training 7 | 2008 | Non‐Recur | Out | NA | 103 |
| Training | Training 8 | 2008 | Non‐Recur | In | NA | 104.2 |
| Training | Training 9 | 2008 | Non‐Recur | In | NA | 121.1 |
| Training | Training 10 | 2008 | Non‐Recur | In | NA | 113.1 |
| Training | Training 11 | 2009 | Non‐Recur | Out | NA | 110.4 |
| Training | Training 12 | 2009 | Non‐Recur | In | NA | 92.1 |
| Training | Training 13 | 2009 | Non‐Recur | In | NA | 127.8 |
| Training | Training 14 | 2009 | Non‐Recur | Out | NA | 116.2 |
| Training | Training 15 | 2009 | Non‐Recur | In | NA | 134.3 |
| Training | Training 16 | 2009 | Non‐Recur | In | NA | 92.2 |
| Training | Training 17 | 2009 | Non‐Recur | In | NA | 73.5 |
| Training | Training 18 | 2009 | Non‐Recur | In | NA | 137.2 |
| Training | Training 19 | 2009 | Non‐Recur | In | NA | 129.2 |
| Training | Training 20 | 2009 | Non‐Recur | In | NA | 145.8 |
| Training | Training 21 | 2009 | Non‐Recur | Out | NA | 142.3 |
| Training | Training 22 | 2012 | Non‐Recur | In | NA | 101.8 |
| Training | Training 23 | 1991 | Non‐Recur | Out | NA | 298.8 |
| Training | Training 24 | 1988 | Recur | Out | 26.3 | 31.1 |
| Training | Training 25 | 1989 | Recur | Out | 25.2 | 25.2 |
| Training | Training 26 | 1989 | Recur | Out | 5.8 | 5.8 |
| Training | Training 27 | 1990 | Recur | Out | 27.7 | 47.4 |
| Training | Training 28 | 1991 | Recur | Out | 9.1 | 9.1 |
| Training | Training 29 | 1992 | Recur | Out | 19.6 | 21.3 |
| Training | Training 30 | 2004 | Recur | Out | 25.5 | 29.1 |
| Training | Training 31 | 2007 | Recur | Out | 27.1 | 80.5 |
| Training | Training 32 | 2007 | Recur | Out | 10.9 | 13.6 |
| Training | Training 33 | 2007 | Recur | Out | 5.2 | 16.7 |
| Training | Training 34 | 2008 | Recur | Out | 15.2 | 79.1 |
| Training | Training 35 | 2012 | Recur | In | 35.4 | 43.2 |
| Training | Training 36 | 1988 | Recur | Out | 12.5 | 15.2 |
| Training | Training 37 | 1989 | Recur | Out | 6.6 | 6.6 |
| Training | Training 38 | 1990 | Recur | Out | 15.6 | 33.5 |
| Validation | Testing 1 | 2012 | Non‐Recur | In | NA | 55.6 |
| Validation | Testing 2 | 2015 | Non‐Recur | In | NA | 53.3 |
| Validation | Testing 3 | 2015 | Non‐Recur | In | NA | 49.7 |
| Validation | Testing 4 | 2015 | Non‐Recur | In | NA | 61 |
| Validation | Testing 5 | 2015 | Non‐Recur | In | NA | 61 |
| Validation | Testing 6 | 2014 | Non‐Recur | Out | NA | 48.7 |
| Validation | Testing 7 | 2015 | Non‐Recur | In | NA | 61 |
| Validation | Testing 8 | 2016 | Non‐Recur | In | NA | 56 |
| Validation | Testing 9 | 2016 | Non‐Recur | In | NA | 57.8 |
| Validation | Testing 10 | 2016 | Non‐Recur | In | NA | 37.9 |
| Validation | Testing 11 | 2016 | Non‐Recur | Out | NA | 37.3 |
| Validation | Testing 12 | 2016 | Non‐Recur | In | NA | 48.6 |
| Validation | Testing 13 | 2016 | Non‐Recur | In | NA | 57.4 |
| Validation | Testing 14 | 2016 | Non‐Recur | In | NA | 49.3 |
| Validation | Testing 15 | 2015 | Non‐Recur | In | NA | 65.8 |
| Validation | Testing 16 | 2016 | Non‐Recur | In | NA | 36.2 |
| Validation | Testing 17 | 2016 | Non‐Recur | In | NA | 51.5 |
| Validation | Testing 18 | 2016 | Non‐Recur | In | NA | 50.4 |
| Validation | Testing 19 | 2013 | Recur | In | 35.7 | 62.5 |
| Validation | Testing 20 | 2016 | Recur | Out | 7.5 | 38.6 |
| Validation | Testing 21 | 2016 | Recur | Out | 7.5 | 38.6 |
| Validation | Testing 22 | 2016 | Recur | Out | 7.5 | 38.6 |
| Validation | Testing 23 | 2016 | Recur | Out | 7.5 | 38.6 |
| Validation | Testing 24 | 2016 | Recur | In | 6.7 | 18.6 |
| Validation | Testing 25 | 2016 | Recur | In | 6.7 | 18.6 |
| Validation | Testing 26 | 2019 | Recur | In | 7.7 | 20.9 |
Abbreviations: NA, not available; and Recur, recurrence.
Transcriptome Sequencing
Paraffin was removed by incubating tissue cores with xylene overnight. RNA extraction and transcriptome sequencing procedures were described previously.( 14 , 15 , 16 , 17 , 18 ) Briefly, total RNA was extracted from tissue cores using the TRIzol method. DNase1 was used to degrade DNA, and a RIBO‐Zero Magnetic Kit (Epicentre, Madison, WI) was used to remove ribosomal RNA from the samples. RNA was reverse‐transcribed to complementary DNA, and a TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA) was used for library preparation. The procedure was guided by the manufacturer’s manual. The quality of the transcriptome library was analyzed with quantitative PCR using Illumina sequencing primers and quantified in an Agilent 2000 Bioanalyzer. The sequencing procedure followed the manual for paired‐end sequencing with 200 cycles as specified for the HiSeq 2500 or with 300 cycles as specified for the NextSeq550 platform by Illumina.
Exome Sequencing
Illumina TruSeq DNA Exome prep kit was used to prepare the exome library. Briefly, the extracted DNA (100 ng) was fragmented in Covaris sonicator to 200 bp length. This was followed by ends repairing, adenylation of 3’ ends, and adapter ligation. After clean‐up by magnetic beads, the DNA fragments were PCR‐amplified for eight cycles of 98°C for 20 seconds, 60°C for 20 seconds, and 72°C for 30 seconds. The amplified DNA was used to hybridize the probes, and the hybridized probes were captured by Streptavidin magnetic beads.
After repeating the probe hybridization and probe capturing, the enriched DNA fragments were amplified for eight cycles at 98°C for 10 seconds, 60°C for 35 seconds, and 72°C for 30 seconds. The libraries were then assessed for quality and quantity in an Agilent 2000 Bioanalyzer. The sequencing procedure followed the manual for paired‐end sequencing with 200 cycles, as specified for the HiSeq 2500 or with 300 cycles as specified for the NextSeq550 platform by Illumina.
Bioinformatics Analysis for Transcriptome Sequencing Data
The sequencing quality control was first performed on RNA‐seq data through FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ ). Adapter sequences and low‐quality reads were trimmed out by Trimmomatic.( 19 ) After preprocessing, surviving reads were aligned to human reference genome hg19 by aligner Hisat2.( 20 ) Gene fragments per kilobase per million reads (FPKM) were quantified by Cufflinks.( 21 ) All of the pipelines were run using default parameters.
Bioinformatics Analysis for Whole‐Exome Sequencing Data
DNA specimens from paired data (tumor and benign tissue for the same patient) were collected for whole‐exome sequencing (WES). Similar as for RNA‐seq data, each WES data point first went through a pipeline of quality control (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ ) and filtering. Reads that passed the quality control were then aligned to human reference genome hg19 by Burrows‐Wheeler Aligner mem function.( 22 ) Tool Picard (http://broadinstitute.github.io/picard/ ) was then applied to sort, index, and mark duplicates on the aligned reads. The Genome Analysis Toolkit( 23 ) analysis pipeline was then used to perform realignment and mutation calling. Eventually, paired samples (tumor and normal) were matched to call somatic mutations by GATK Mutect2.( 23 ) All pipelines were run by default parameters.
Prediction Model on Transcriptome Expression Profiles
Genomic and machine learning methods were introduced to predict the recurrence status of liver transplantation. These machine learning algorithms generally take in the genomic features (e.g., gene expression and mutation pathways across the samples) and generate a prediction probability per sample. For the transcriptome model, genome‐wide gene‐expression profiles were quantified across all tumor samples. FPKM values were first log2‐scaled. Several machine learning algorithms were applied to the transcriptome expression data, specifically, support vector machine (SVM),( 24 ) random forest (RF),( 25 , 26 ) linear discriminant analysis (LDA),( 27 ) logistic regression,( 28 ) and k‐top scoring pairs (k‐TSP).( 29 ) Quantile normalization across the training and testing cohorts was applied to correct the batch effect for the first four algorithms, whereas k‐TSP is a non‐parametric method in which quantile normalization is not required. For all of these methods, leave‐one‐out cross‐validation (LOOCV) was performed on the training cohort to evaluate the prediction algorithms and select the best parameters (the best top number of genes or paired genes). The best algorithm was then applied to the whole training cohort to train a model and apply to the testing cohort. Eventually, the training and testing cohorts were pooled together to generate the best model for prediction of recurrence of a new sample. All biostatistical analyses were performed by R programming and available R packages: “randomForest,” “MASS,” “e1071,” and “switchBox.”( 30 )
Prediction Model Integrating Transcriptome Expression and Gene Mutation
All machine learning algorithms applied to transcriptome analysis were used to integrate both RNA and DNA data. At the RNA level, gene expressions were used as features, which is similar to the model working only on transcriptome expression data. At the DNA level, somatic mutations were called on each tumor‐normal pair individually. Known Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with defined functional gene sets were collected from the public database.( 31 , 32 ) The total number of genes with somatic mutations were then calculated for each functional pathway and used as DNA‐level features.
For the machine learning models RF, SVM, LDA and logistic regression, both transcriptome expression at RNA level and pathway mutation at DNA level, were used as prediction features. RF regression prediction was used to predict a probability score for recurrence. This score ranges from 0 to 1, where a score >0.5 represents recurrence and score <0.5 predicts nonrecurrence. For the k‐TSP model, this was applied to transcriptome expression and gene‐mutation profiles individually. To combine the two ‐omics data sets, scores calculated from the transcriptome expression data and scores from the gene‐mutation data were weighted and summed for the prediction. The final score ranges from −1 to 1, where a positive score represents recurrence, and a negative score means nonrecurrence for binary prediction.
Similar to the model involving only transcriptome expression data, the model integrating both the RNA and DNA was first applied to the training cohort. The best parameters selected by LOOCV were used as the final model for the training cohort and then applied to the testing cohort for evaluation. In the final stage, both cohorts were pooled to provide a final prediction model on leave‐one‐out cross‐validation. All biostatistical analyses were performed by R programming and available R packages.( 30 )
Prediction Model Integrating Transcriptome Expression, Gene Mutation, and Milan Score
Similar to transcriptome expression and gene‐mutation integration, multiple machine learning models were used to integrate RNA expression, DNA mutation, and Milan score. For the k‐TSP model, it assigned a weight to the RNA score, DNA score, and Milan score (−1 for “in” and 1 for “out”). The final prediction score is the sum of all three weighted scores. For RF, SVM, LDA and logistic regression, the following were used as features contributing to prediction: gene expression, pathway mutation, and Milan score. RF generated a probability score ranging from 0 to 1, where a score higher than 0.5 is indicative of recurrence, and a score less than 0.5 predicts nonrecurrence.
Downstream Functional Pathway Analysis
When combining the training and testing data, the top 500 differentially expressed genes (DEGs) were selected by the ranking of P values. These genes were then used for functional pathway analysis. Four pathway databases were collected for the enrichment test: GO,( 31 ) KEGG,( 32 ) Reactome,( 33 ) and BioCarta.( 34 ) The top significant enriched pathways were selected by false discovery rate = 5%. Genes involved in selected pathways were used for network analysis. Clustering heatmap, pathway barplot, and network figure were generated by R programming (package ComplexHeatmap( 35 ) and ggplot( 36 ) and Cytoscape software( 37 ).
Statistical Analysis
All of the statistical analyses were performed by R programming. The receiver operating characteristic (ROC) curves and Kaplan‐Meier analyses were analyzed and plotted by R/Bioconductor packages survival (https://CRAN.R‐project.org/package=survival), pROC,( 38 ) ggfortify,( 39 ) and GGally (https://CRAN.R‐project.org/package=GGally).
Power calculation
In this study, the prediction model can generally reach 85% prediction accuracy (P = 0.85) with n = 64 samples. To achieve 95% confidence level (α = 0.05, Z = 1.96), the confidence interval was calculated as . That is, when we claim that the prediction accuracy is 85%, the corresponding confidence interval is [76.3%, 93.7%].
Results
Pre‐determination of Training Cohort and Testing Cohort
In previous studies we showed that alterations of genome and gene expression occurring in HCC are associated with aggressiveness of the cancer.( 40 , 41 ) However, it was unclear whether these changes contained predictive values for patients with HCC undergoing liver transplants. To explore this possibility, two cohorts based on the surgical timeline were constructed for transcriptome and exome sequencing analyses. The training cohort (38 samples) included HCC samples obtained from patients who had liver transplants from 1988 through the first half of 2012, while the testing cohort (26 samples) included HCC samples who received liver transplantation from the second half of 2012 up to 2016. The results of the transcriptome and exome analyses of the training cohort were combined to develop a classification algorithm as a training set (Fig. 1). The algorithm was then applied to predict the clinical outcomes of the samples from the testing set (second cohort).
FIG. 1.
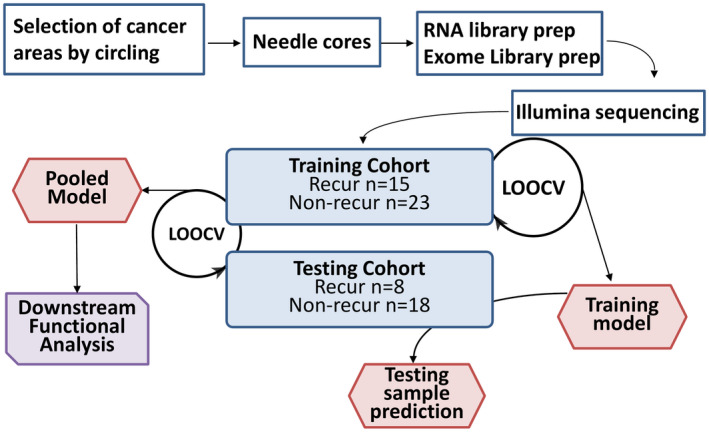
Flow chart of procedures for training and validation of genome prediction model. The procedure starts with the identification of cancer samples by the year of liver transplant surgery using 2012 as the demarcation. All samples before the first half of 2012 were used in the training set, whereas samples after the second half of 2012 were used as the testing set. The cancer areas and benign tissues from the non‐liver organ of the paraffin block were needle‐cored and used as “cancer” and “normal” tissues, respectively. All clinical information was blind to the researchers before the prediction.
Transcriptome Sequencing to Predict HCC Outcomes
The transcriptome analysis was performed using an RF( 25 , 26 ) model in which all genes were ranked based on differential expression between recurrence and nonrecurrence samples. The top 10 differentially expressed genes were first used to predict recurrence status of the samples in the training set using the LOOCV method. Subsequently, the top 20, 30, 40, 50, 100, 200, 500, or 1,000 differentially expressed genes were added to train the model and to examine whether the addition of genes improved the results. The final model was selected based on the best Youden index (sensitivity + specificity −1). As shown in Fig. 2A, 500 differentially expressed genes were found to produce the best results in predicting cancer recurrence for patients with HCC. The ROC curve yielded an area under the curve (AUC) of 0.87 and a P value of 2.8 × 10‐9. The LOOCV model based on 500 genes produced 84.2% accuracy, with a sensitivity of 80% and specificity of 87% (Supporting Table S2). When this algorithm was applied to the testing cohort, the AUC of the ROC was 0.806 with a P value of 0.00049 (Fig. 2B). The accuracy was 73.1%, with 87.5% sensitivity and 66.7% specificity (Supporting Table S2).
FIG. 2.
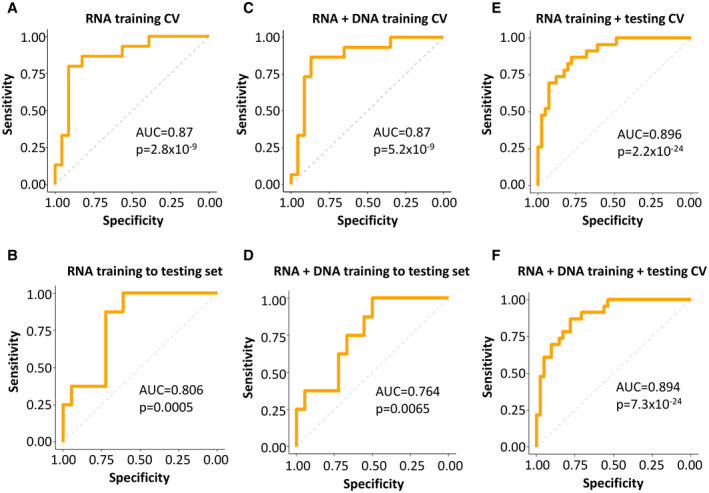
ROC analysis of genome prediction model. (A) Training set ROC based on top 500 differentially expressed genes between recurrence and nonrecurrence samples from the transcriptome sequencing using LOOCV strategy with RF method. (B) Testing set ROC based on the algorithm determined in the training set of (A). (C) Training set ROC based on transcriptome and exome sequencing results using RF method. (D) Testing set ROC based on the algorithm determined in the training set of (C). (E) ROC of pooled training and testing cohorts based on transcriptome sequencing using LOOCV strategy with RF method. (F) ROC of pooled training and testing cohorts based on transcriptome and exome sequencings using LOOCV strategy with RF method. Abbreviation: CV, cross validation.
Mutation Pathways Analysis to Enhance Prediction of HCC Outcomes
To examine whether genome mutations of HCC also have a role in predicting the clinical outcomes of the HCC transplant patients, we performed exome sequencing on the same HCC samples and their matched non‐liver benign tissue samples from both cohorts. Somatic mutations were identified by subtracting the single‐nucleotide variants in the cancer sample from the matched normal tissue of the same individual. A total of 30,090 somatic mutations were identified in 64 HCC samples of both cohorts, with an average 470 (15‐2,657) mutations per HCC sample (Supporting Table S3). These mutations were distributed among 6,977 pathways based on GO and KEGG. The difference of mutation numbers between the recurrence and nonrecurrence samples in each pathway was ranked through t tests. The pathway with the smallest P value was ranked at the top. The top 5 mutation pathways were then combined with the 500 genes from the transcriptome sequencing to examine whether the mutation status of the pathways improves the transcriptome prediction. This model was then added to the top 10, 15, 20, 25, or 30 pathways. The model with the best Youden index was selected through LOOCV. As shown in Fig. 2C,D, the combination of five mutation pathways and 500 differential expressed genes in the training set improved accuracy to 86.8% with a sensitivity of 86.7% and specificity of 87% (AUC = 0.87 and P = 5.2 × 10‐9). When this algorithm was applied to the testing set, the accuracy was 77% (AUC = 0.764 and P = 0.0065), with a sensitivity of 100% and specificity of 66.7% (Supporting Table S2).
When both training and testing set data were combined to create a prediction model based on the LOOCV method, the transcriptome model predicted 81.3% correctly (AUC = 0.896 and P = 2.2 × 10‐24; Fig. 2E), whereas the combination of mutation pathways and transcriptome generated a correct prediction of 84.4% (AUC = 0.894 and P = 7.3 × 10‐24) with a sensitivity of 78.3% and specificity of 87.8% (Fig. 2F and Supporting Table S2).
Using transcriptome analysis alone, survival analysis in the training set showed that 87% of the transplant patients predicted as nonrecurrent enjoyed recurrence‐free survival up to 298.8 months, while those patients predicted as recurrence had a 20% 3‐year recurrence‐free survival rate (P = 1.6 × 10‐6; Fig. 3A). When the same algorithm from the training set was applied to the testing set, the patients predicted as nonrecurrence had a 92.3% recurrence‐free survival up to 60 months, whereas the patients predicted as recurrence had only about 46% recurrence‐free survival in the same period (P = 0.01; Fig. 3B). The combination of transcriptome and mutation pathways analyses showed that the recurrence‐free survival rates reached 90.9% for patients predicted as nonrecurrence in the training set and 100% in the testing set, while the patients predicted as recurrence had recurrence‐free survival rates of 18.8% (P = 2.5 × 10‐7) in the training set and 42.9% (P = 0.002) in the testing set (Fig. 3C,D). These results suggest a minor improvement in the prediction of recurrence‐free survival when mutation pathways analysis was added to the prediction model. When both training and testing cohorts were combined, a similar mild improvement of survival prediction by combined transcriptome and mutation pathways model was shown: 87.8% patients predicted as nonrecurrence by the transcriptome/mutation pathways RF model experienced at least 3 years of recurrence‐free survival versus 85.4% by the transcriptome RF model, whereas only 21.7% patients predicted as recurrence by the transcriptome/mutation pathways RF model survived recurrence‐free for the similar period versus 26.1% for the transcriptome RF model (Fig. 3E,F).
FIG. 3.
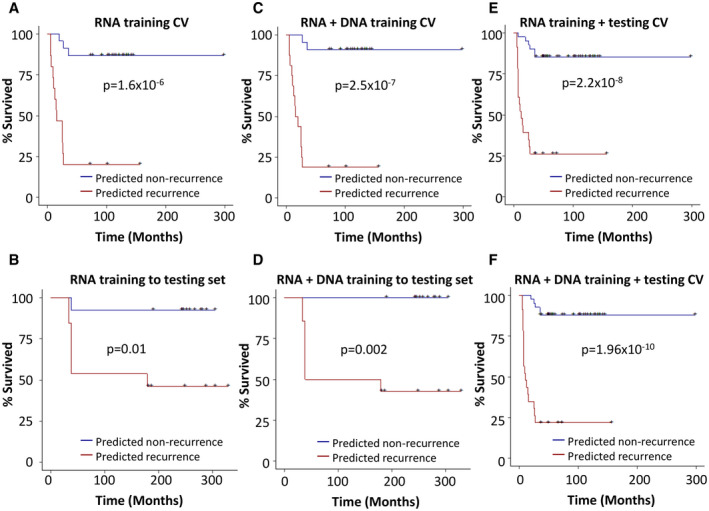
Kaplan‐Meier analysis of genome prediction model. (A) Training set Kaplan‐Meier analysis based on 500 differentially expressed genes from the transcriptome sequencing using LOOCV strategy with RF method. (B) Testing set Kaplan‐Meier analysis based on the algorithm determined in the training set of (A). (C) Training set Kaplan‐Meier analysis based on transcriptome and exome‐sequencing results using RF method. (D) Testing set Kaplan‐Meier analysis based on the algorithm determined in the training set of (C). (E) Kaplan‐Meier analysis of pooled training and testing cohorts based on transcriptome sequencing using LOOCV strategy with RF method. (F) Kaplan‐Meier analysis of pooled training and testing cohorts based on transcriptome and exome sequencings using LOOCV strategy with RF method.
Role of Milan Criteria in Predicting the Recurrence of HCC in the Transplant Patients
The Milan criteria constitute a radiology‐based parameter defined by the size and number of HCC tumor nodules. Based on Milan‐in (low risk of recurrence) and Milan‐out (high risk of recurrence) assessment, the prediction rate of recurrence for the entire cohort is 76.6%, with a sensitivity of 78.2% and specificity of 75.6%. To investigate whether the addition of Milan criteria improves the prediction rate of the genome prediction model, the transcriptome/mutation pathways model and Milan score were combined to create a transcriptome/mutation pathways/Milan RF model to predict the likelihood of HCC recurrence of the liver transplant patients. As shown in Supporting Figs. S1 and S2, even though the transcriptome/mutation pathways/Milan RF model offered significant improvement of the prediction rates over the Milan criteria, the addition of the Milan criteria did not improve the prediction rate of the transcriptome/mutation pathways RF model in the training analysis or training to testing analysis (Supporting Table S2).
To examine whether the other machine learning models were improved by Milan criteria, we analyzed the transcriptome sequence results through the k‐TSP method, a non‐parametric algorithm especially suitable for cross‐platform studies. The model provides a prediction score based on the k‐top scoring pairs, in which a positive value indicates recurrence, and a negative score predicts nonrecurrence. The k‐TSP model was applied to the training set for LOOCV with different numbers of top gene pairs (5, 7, 9, … 49), and the best model was selected by the highest Youden index. The transcriptome k‐TSP model alone yielded 79% accuracy in the training analysis, 73.1% in the testing analysis, and 79.7% in the combined training and testing analyses (Supporting Figs. S3 and S4, Supporting Table S2). The combination of Milan criteria and transcriptome sequencing produced a significant improvement over either analysis alone (Figs. 4 and 5): The Milan/transcriptome k‐TSP model generated a 79% prediction rate in the training analysis, 80.8% in the testing analysis, and 84.4% in the combined training and testing cross‐validation analysis. Interestingly, when the DNA mutation pathway analysis was combined with the Milan/transcriptome k‐TSP model, mixed results were obtained: The Milan/transcriptome/mutation pathways k‐TSP model improved the prediction to 89.5% in the training set and 87.5% in the combined training and testing set, but dropped the prediction rate to 73.1% in the testing analysis (Supporting Table S2, Supporting Figs. [Link], [Link], [Link]). These results suggest that Milan criteria may improve the prediction of the k‐TSP machine learning model, particularly the k‐TSP transcriptome analysis, when they are combined into an integrative prediction model.
FIG. 4.
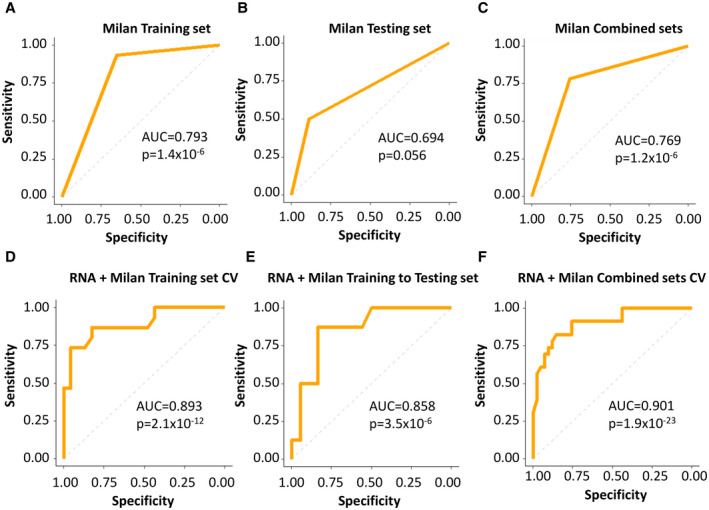
ROC analysis of Milan criteria with the genome prediction model. (A) ROC analysis based on Milan criteria in the training set. (B) ROC analysis based on Milan criteria in the testing set. (C) ROC analysis based on Milan criteria in the combined training and testing sets. (D) ROC analysis of the training set based on Milan/transcriptome k‐TSP prediction model using LOOCV. (E) ROC analysis of the testing set based on Milan/transcriptome k‐TSP prediction algorithm determined in (D). (F) ROC analysis of the combined training and testing sets based on Milan/transcriptome k‐TSP prediction model using LOOCV.
FIG. 5.
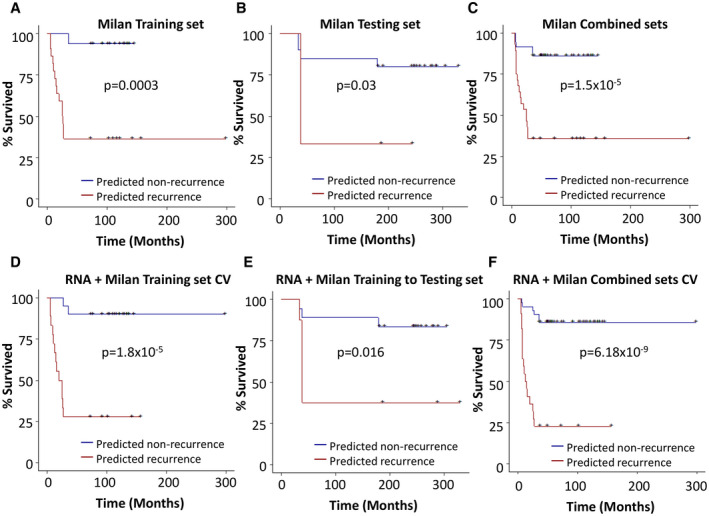
Kaplan‐Meier analysis of Milan criteria with the genome prediction model. (A) Kaplan‐Meier analysis based on Milan criteria in the training set (A), the testing set (B), and the combined training and testing sets (C). (D) Kaplan‐Meier analysis of the training set based on Milan/transcriptome k‐TSP prediction model using LOOCV. (E) Kaplan‐Meier analysis of the testing set based on Milan/transcriptome k‐TSP prediction algorithm determined in (D). (F) Kaplan‐Meier analysis of the combined training and testing sets based on Milan/transcriptome k‐TSP prediction model using LOOCV.
Survival analysis showed that 94% of patients with HCC with Milan “in” enjoyed a recurrence‐free survival of 3 years or more in the training set. However, the 3‐year recurrence‐free survival for Milan “in” patients decreased to 80% in the testing set and 86% in the combined data sets (Fig. 5A‐C). The Milan/transcriptome k‐TSP model showed a 90% 3‐year survival rate in the training set when patients were predicted as nonrecurrence (Fig. 5D). The testing validation analysis showed that 83% of patients with HCC predicted by Milan/transcriptome k‐TSP model as nonrecurrence survived up to 60 months without recurrence, compared with 37.5% patients predicted as recurrence survived similar periods without recurrence (P = 0.016; Fig. 5E). When both training and testing cohorts were combined, the cancer‐free survival improved to 85.7% for patients predicted as nonrecurrence, and 22.7% for patients as recurrence (P = 6.18 × 10‐9; Fig. 5F), very similar to the survival results produced by transcriptome/mutation pathways/Milan RF model in the same data set: 85.7% of patients with HCC with 3 years’ recurrence‐free survival when predicted as nonrecurrence, compared to 22.7% patients with 3 years or longer cancer‐free survival when predicted as recurrence (P = 6.54 × 10‐10). These compare favorably with Milan criteria alone: 86% 3‐year survival for Milan “in,” with 35.7% for Milan “out” (P = 1.5 × 10‐5; Supporting Fig. 2A‐F).
Next, the entire cohort was divided into low risk of recurrence (Milan‐in) and high risk of recurrence (Milan‐out) based on Milan criteria. The transcriptome/mutation pathways RF model was applied to predict outcomes. When Milan is “in,” the model predicted 88.9% correctly based on the transcriptome/mutation pathways RF model (Supporting Table S2). Interestingly, when Milan is “out,” the model had an accuracy of 82.1%, with 94.4% sensitivity and 60% specificity, including predicting 17 of 18 recurrent patients correctly (Supporting Table S2). These results suggest that the genome model may have a significant utility in predicting the clinical outcomes of patients outside the Milan criteria.
Impact of Heterogeneity of HCC
HCC may have significant heterogeneity in terms of genomic profile and differentiation even in the same individual.( 42 ) A tumor nodule may have different gene‐expression and mutation profiles from its nearby nodules. To investigate whether the genome prediction model is sufficiently robust to overcome the heterogeneous nature of HCC, we examined 3 individuals with multiple tumor nodules, including an individual (patient #V19) having four tumor nodules and 2 individuals (patients V#7 and #V21) having two tumor nodules each. These eight nodules are listed as individual samples in Table 3 and were predicted independently. As indicated in Table 3, the transcriptome/mutation pathways RF prediction model consistently produced scores indicating HCC recurrence from each of the four tumor nodules of patient #V19, matching the clinical outcome of the patient. The transcriptome/mutation pathways RF model correctly predicted nonrecurrence outcomes from two tumor nodules of patient #V7, whereas the same model predicted two tumor nodules of patient #V21 as recurrence outcomes, matching the real clinical results. Of the eight tumor nodules, the genome prediction yielded consistent prediction results for the multiple nodes collected from the same patients (8 of 8). Overall, the genome prediction model appears to be reasonably robust in predicting the clinical outcomes of HCC samples despite the heterogeneity of the cancers. A larger number of samples will of necessity need to be examined in a future study to further support this initial conclusion.
TABLE 3.
Multiple Cancer Nodule Predictions From Patients With HCC
| Patient | Recur Status | Milan | RF Probability Score* | Prediction Status | Months to Recur |
|---|---|---|---|---|---|
| #V19A | Recur | Out | 0.6275 | Recur | 7.5 |
| #V19B | Recur | Out | 0.7909 | Recur | 7.5 |
| #V19C | Recur | Out | 0.8400 | Recur | 7.5 |
| #V19D | Recur | Out | 0.7534 | Recur | 7.5 |
| #V7A | Non‐Recur | In | 0.1235 | Non‐Recur | NA |
| #V7B | Non‐Recur | In | 0.0120 | Non‐Recur | NA |
| #V21A | Recur | In | 0.6690 | Recur | 6.7 |
| #V21B | Recur | In | 0.9183 | Recur | 6.7 |
Score > 0.5 = likely recurrence, and score <0.5 = likely nonrecurrence.
Signaling Pathways Involved in the Genome Prediction Model
When the relative expression levels of the top 500 genes were used as parameters, most cancers with nonrecurrence outcomes appeared to aggregate together in a hierarchical clustering analysis (Fig. 6A) and principal component analysis (Supporting Fig. S6), separate from the samples with recurrence outcomes. A similar segregation of recurrence and nonrecurrence samples was achieved when using the top 43 pairs of genes from the k‐TSP model (Supporting Fig. S7A). At the DNA level, mutations in the dopamine binding pathway were dominant in samples from patients with HCC recurrence in the RF analysis (Fig. 6B), whereas mutations in the pathways of Syntaxin binding, Golgi associated vesicle biogenesis, and regulation of hormonal metabolic process were included in the k‐TSP model (Supporting Fig. S7B). Disruption of these pathways may impact the homeostasis and metabolism of the cancer cells, thereby affecting cancer survival. At the transcriptome level, 77 of 86 genes in the k‐TSP model overlapped with those from the RF models (Supporting Tables S4 and S5). The top 500 DEGs from the RF model and 43 pairs of genes from the k‐TSP model were applied for pathway enrichment analysis (Fig. 6, Supporting Figs. S7 and S8). Many tumorigenesis pathways were identified, such as pathways related to chromosome segregation, cell cycle, and DNA synthesis. Genes involved in DNA replication, chromosome segregation and mitosis, such as cyclin dependent kinase inhibitor 3, minichromosome maintenance 6 homologous recombination repair factor 8 (MCM8), minichromosome maintenance 8 homologous recombination repair factor 6 (MCM6), BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B), kinesin family member 23, and cell division cycle 6 (CDC6), dominated the pathway analyses (Fig. 6C; Supporting Figs. S7C and S8).
FIG. 6.
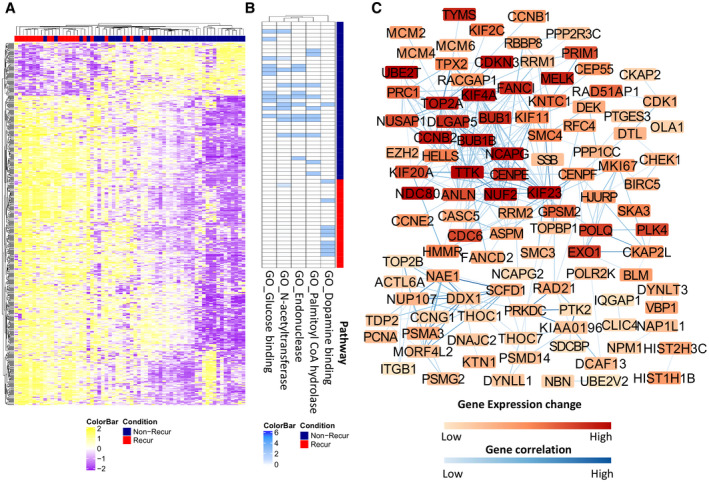
Transcriptomic alteration related to recurrence and mutation pathways of HCC samples. (A) Hierarchical clustering of HCC samples based on top 500 differential expression genes between nonrecurrence and recurrence HCC samples. (B) Heat map of five signaling pathways based on the differential mutation numbers in the pathways between nonrecurrence and recurrence samples. (C) Gene‐expression alterations and connections based on GO analysis.
Discussion
Liver transplantation is one of the main approaches to treat liver cancers and is particularly useful for patients with HCC with late‐stage cirrhosis. The Milan criteria have been useful for gauging patient suitability for liver transplant in the last 25 years. Although most patients inside the Milan criteria experience cancer‐free recovery from the liver transplant,( 43 ) these may be too restrictive and may preclude some eligible patients from liver transplant.( 43 ) The genome prediction model described in this report, whether in combination with Milan criteria or not, represents a potential alternative for the selection of HCC‐bearing liver transplant candidates. At least two potential clinical scenarios can be envisioned using this model: First, the Milan criteria are used as a first line of selection of patient candidates for the liver transplant. Patients with “Milan‐in” status are selected as viable candidates for liver transplant, while patients with “Milan‐out” status can be screened through the genome prediction model for transplant. Second, Milan criteria can be integrated into the genome prediction model to screen all HCC candidates for appropriateness of liver transplant. In either scenario, this model may represent an improvement on the Milan criteria alone.
Attempts to predict the likelihood of HCC recurrence after liver transplantation have been made in the past. Some prediction models used clinical features such as Milan score, maximal fludeoxyglucose uptake value, tumor size, tumor number, and pathology grading information( 44 , 45 ) as their base to predict the behavior of the cancer. One study by Kim et al.( 46 ) used microarray gene‐expression analysis to predict the outcomes of HCC recurrence for partial hepectomy patients of HBV‐related liver cancers. This study integrates both RNA/DNA sequencings and machine learning technologies to create a model to predict the recurrence of human liver cancers for liver transplant patients. The robust results from our analyses suggest that the genome analysis adds accuracy to select liver transplant candidates for this life‐saving procedure.
Overfitting is one of the potential pitfalls of molecular prediction models. To overcome potential overfitting issues, we preselected the HCC samples into two unconnected cohorts based on the year of transplant surgery. The testing cohort represents an ongoing prospective analysis. To increase the robustness of the analysis, most samples in one cohort (training) were analyzed through Illumina HiSeq2500, while another (testing) were analyzed through NextSeq550. Due to the differences of the platforms, the read lengths of the sequencing were also different: HiSeq2500 platform was limited to 100 bases per read, whereas NextSeq550DX was 150 bases. The sequencings were performed in different time frames (2015‐2017 for the training set, 2018‐2020 for the testing set). Despite the non‐connected nature of the cohorts, different sequencing platforms and different time frames, the variation in prediction accuracies between the two cohorts was consistently less than 10%, suggesting a good reproducibility of the model. The robustness of the genome prediction model is not limited to RF method. When we applied other machine learning methods such as k‐TSP, SVM, LDA, or logistics regression, similar results were obtained (Supporting Table S2).
A surprising finding in our analysis is that most of the frequent mutations of HCC such as tumor protein p53, catenin beta 1, and telomerase reverse transcriptase were not found to play important roles in predicting the behavior of HCC in liver transplant patients. Rather, mutations in dopamine signaling pathway such as dopamine receptors and G‐protein coupled receptors are frequent in patients with HCC who experienced recurrence after the liver transplant, whereas mutations in genes involved in glucose binding/metabolism such as hexokinase domain containing 1, glucose 6 phosphate dehydrogenase, and endonuclease such as ribonuclease A family member 2, X‐ray repair cross complementing 3, were more frequent in patients with HCC who were less likely to have cancer recurrence. The altered functions of these proteins may have an impact on the survival and metabolism of the cancer cells. In contrast, the transcriptome analysis shows that the most altered expression genes are those involving DNA synthesis (MCM8, MCM6, DNA topoisomerase II alpha, and CDC7), chromatin segregation (BUB1 and CDC6), and mitosis (NDC80 kinetochore complex component and protein phosphatase catalytic subunit gamma) (Fig. 6C, Supporting Fig. S7C). Copy number gain or overexpression of these genes has been previously reported in human cancers.( 16 , 47 , 48 ) These changes may facilitate DNA replication and growth of cancer cells. However, most of these genes were not mutated.
The relative irrelevance of the cancer driver mutations for predicting posttransplant recurrence is understandable. Tumor recurrence occurs after circulating HCC cells present at the time of transplantation traverse through the circulation, survive the turbulent flow environment of the cardiac valves, proceed through the pulmonary circulation without attaching to the lungs, and finally lodge themselves within the new liver.( 49 , 50 ) This may be a complicated process, and the pathways operating within the cells must allow them to withstand the immune and shear/stress forces likely to be encountered. The pathways enabling these capabilities are not well understood, and the findings from the current study are likely to provide useful information as to their nature. The mutation and transcriptome analyses appear to uncover two different facets of the cancer genome: a qualitative alteration without much change in expression levels and a quantitative change without the alteration of quality. Each change may have an impact on the cancer cells and contribute to recurrence and metastasis. Future dissection of these pathways may help to gain a better understanding of the cancer behavior.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1
Table S2
Table S3
Table S4
Table S5
Supported by the National Institutes of Health (1R56CA229262‐01 and UL1TR001857) and Pittsburgh Liver Research Center (NIH/NIDDK P30DK120531).
Contributor Information
George Michalopoulos, Email: luoj@upmc.edu.
Jian‐Hua Luo, Email: luoj@upmc.edu.
References
- 1. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7‐30. [DOI] [PubMed] [Google Scholar]
- 3. Starzl TE, Marchioro TL, Porter KA, Brettschneider L. Homotransplantation of the liver. Transplantation 1967;5:790‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roayaie S, Schwartz JD, Sung MW, Emre SH, Miller CM, Gondolesi GE, et al. Recurrence of hepatocellular carcinoma after liver transplant: patterns and prognosis. Liver Transpl 2004;10:534‐540. [DOI] [PubMed] [Google Scholar]
- 5. de'Angelis N, Landi F, Carra MC, Azoulay D. Managements of recurrent hepatocellular carcinoma after liver transplantation: a systematic review. World J Gastroenterol 2015;21:11185‐11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sapisochin G, Goldaracena N, Laurence JM, Dib M, Barbas A, Ghanekar A, et al. The extended Toronto criteria for liver transplantation in patients with hepatocellular carcinoma: a prospective validation study. Hepatology 2016;64:2077‐2088. [DOI] [PubMed] [Google Scholar]
- 7. Sapisochin G, Goldaracena N, Astete S, Laurence JM, Davidson D, Rafael E, et al. Benefit of treating hepatocellular carcinoma recurrence after liver transplantation and analysis of prognostic factors for survival in a large Euro‐American series. Ann Surg Oncol 2015;22:2286‐2294. [DOI] [PubMed] [Google Scholar]
- 8. Jain A, Reyes J, Kashyap R, Dodson SF, Demetris AJ, Ruppert K, et al. Long‐term survival after liver transplantation in 4,000 consecutive patients at a single center. Ann Surg 2000;232:490‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poynard T, Naveau S, Doffoel M, Boudjema K, Vanlemmens C, Mantion G, et al. Evaluation of efficacy of liver transplantation in alcoholic cirrhosis using matched and simulated controls: 5‐year survival. Multi‐centre group. J Hepatol 1999;30:1130‐1137. [DOI] [PubMed] [Google Scholar]
- 10. Boyault S, Rickman DS, de Reyniès A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007;45:42‐52. [DOI] [PubMed] [Google Scholar]
- 11. Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, et al. Whole‐genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 2016;48:500‐509. [DOI] [PubMed] [Google Scholar]
- 12. Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z, et al. Genome‐wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology 2011;54:1227‐1236. [DOI] [PubMed] [Google Scholar]
- 13. von Felden J, Villanueva A. Role of molecular biomarkers in liver transplantation for hepatocellular carcinoma. Liver Transpl 2020;26:823‐831. [DOI] [PubMed] [Google Scholar]
- 14. Luo J‐H, Yu YP, Cieply K, Lin F, Deflavia P, Dhir R, et al. Gene expression analysis of prostate cancers. Mol Carcinog 2002;33:25‐35. [DOI] [PubMed] [Google Scholar]
- 15. Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol 2004;22:2790‐2799. [DOI] [PubMed] [Google Scholar]
- 16. Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006;25:1090‐1098. [DOI] [PubMed] [Google Scholar]
- 17. Yu YP, Ding Y, Chen Z, Liu S, Michalopoulos A, Chen R, et al. Novel fusion transcripts associate with progressive prostate cancer. Am J Pathol 2014;184:2840‐2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo JH, Liu S, Tao J, Ren BG, Luo K, Chen ZH, et al. Pten‐NOLC1 fusion promotes cancers involving MET and EGFR signalings. Oncogene 2020;40(6):1064‐1076. 10.1038/s41388-020-01582-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 2019;37:907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cortes C, Vapnik V. Support‐vector networks. Mach Learn 1995;20:273‐297. [Google Scholar]
- 25. Amit Y, Geman D. Shape quantization and recognition with randomized trees. Neural Comput 1997;9:1545‐1588. 10.1162/neco.1997.9.7.1545 [DOI] [Google Scholar]
- 26. Bauer E, Kohavi R. An empirical comparison of voting classification algorithms: bagging, boosting, and variants. Mach Learn 1999;36:105‐139. [Google Scholar]
- 27. McLachlan GJ. Discriminant analysis and statistical pattern recognition. Appl Probab Stat 2004;1‐526. [Google Scholar]
- 28. Tolles J, Meurer WJ. Logistic regression: relating patient characteristics to outcomes. JAMA 2016;316:533‐534. [DOI] [PubMed] [Google Scholar]
- 29. Shi P, Ray S, Zhu Q, Kon MA. Top scoring pairs for feature selection in machine learning and applications to cancer outcome prediction. BMC Bioinformatics 2011;12:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Afsari B, Fertig EJ, Geman D, Marchionni L. switchBox: an R package for k‐Top scoring pairs classifier development. Bioinformatics 2015;31:273‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontology Consortium. Nat Genet 2000;25:25‐29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28:27‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, et al. The reactome pathway knowledgebase. Nucleic Acids Res 2020;48:D498‐D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016;2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847‐2849. [DOI] [PubMed] [Google Scholar]
- 36. Maag JLV. gganatogram: An R package for modular visualisation of anatograms and tissues based on ggplot2. F1000Res 2018;7:1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang Y, Horikoshi M, Li W. ggfortify: Unified Interface to Visualize Statistical Results of Popular R Packages. R J 2016;8:474‐485. [Google Scholar]
- 40. Luo J‐H, Ren B, Keryanov S, Tseng GC, Rao UNM, Monga SP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006;44:1012‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nalesnik MA, Tseng G, Ding Y, Xiang G‐S, Zheng Z‐L, Yu YanPing, et al. Gene deletions and amplifications in human hepatocellular carcinomas: correlation with hepatocyte growth regulation. Am J Pathol 2012;180:1495‐1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fransvea E, Paradiso A, Antonaci S, Giannelli G. HCC heterogeneity: molecular pathogenesis and clinical implications. Cell Oncol 2009;31:227‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pavel MC, Fuster J. Expansion of the hepatocellular carcinoma Milan criteria in liver transplantation: future directions. World J Gastroenterol 2018;24:3626‐3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marsh JW, Dvorchik I, Subotin M, Balan V, Rakela J, Popechitelev EP, et al. The prediction of risk of recurrence and time to recurrence of hepatocellular carcinoma after orthotopic liver transplantation: a pilot study. Hepatology 1997;26:444‐450. [DOI] [PubMed] [Google Scholar]
- 45. Chaiteerakij R, Zhang X, Addissie BD, Mohamed EA, Harmsen WS, Theobald PJ, et al. Combinations of biomarkers and Milan criteria for predicting hepatocellular carcinoma recurrence after liver transplantation. Liver Transpl 2015;21:599‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim JH, Sohn BH, Lee H‐S, Kim S‐B, Yoo JE, Park Y‐Y, et al. Genomic predictors for recurrence patterns of hepatocellular carcinoma: model derivation and validation. PLoS Med 2014;11:e1001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He D‐M, Ren B‐G, Liu S, Tan L‐Z, Cieply K, Tseng G, et al. Oncogenic activity of amplified miniature chromosome maintenance 8 in human malignancies. Oncogene 2017;36:3629‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010;10:59‐64. [DOI] [PubMed] [Google Scholar]
- 49. Rejniak KA. Circulating tumor cells: when a solid tumor meets a fluid microenvironment. Adv Exp Med Biol 2016;936:93‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krog BL, Henry MD. Biomechanics of the circulating tumor cell microenvironment. Adv Exp Med Biol 2018;1092:209‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1
Table S2
Table S3
Table S4
Table S5