

Abstract
The critical functions of the human liver are coordinated through the interactions of hepatic parenchymal and non‐parenchymal cells. Recent advances in single‐cell transcriptional approaches have enabled an examination of the human liver with unprecedented resolution. However, dissociation‐related cell perturbation can limit the ability to fully capture the human liver’s parenchymal cell fraction, which limits the ability to comprehensively profile this organ. Here, we report the transcriptional landscape of 73,295 cells from the human liver using matched single‐cell RNA sequencing (scRNA‐seq) and single‐nucleus RNA sequencing (snRNA‐seq). The addition of snRNA‐seq enabled the characterization of interzonal hepatocytes at a single‐cell resolution, revealed the presence of rare subtypes of liver mesenchymal cells, and facilitated the detection of cholangiocyte progenitors that had only been observed during in vitro differentiation experiments. However, T and B lymphocytes and natural killer cells were only distinguishable using scRNA‐seq, highlighting the importance of applying both technologies to obtain a complete map of tissue‐resident cell types. We validated the distinct spatial distribution of the hepatocyte, cholangiocyte, and mesenchymal cell populations by an independent spatial transcriptomics data set and immunohistochemistry. Conclusion: Our study provides a systematic comparison of the transcriptomes captured by scRNA‐seq and snRNA‐seq and delivers a high‐resolution map of the parenchymal cell populations in the healthy human liver.

Abbreviations
- CST
CHAPS with salts and Tris
- CV
central venous
- CYP3A4
cytochrome P450 3A4
- FB
fibroblast
- GSEA
gene‐set enrichment analysis
- HSC
hepatic stellate cell
- IL
interleukin
- KRT
keratin 1
- LSEC
liver sinusoidal endothelial cell
- MAML
mastermind like transcriptional coactivator
- NK
natural killer
- NST
Nonidet P40 with salts and Tri
- PDE
phosphodiesterase
- RBP
recombination signal binding protein
- scRNA‐seq
single‐cell RNA sequencing
- snRNA‐seq
single‐nucleus RNA sequencing
- SOX9
SRY (sex determining region Y)‐box 9
- TAGLN
transgelin
- TST
Tween‐20 with salts and Tris
- VSMC
vascular smooth muscle cell
The liver is an essential organ responsible for critical functions including lipid and glucose metabolism, protein synthesis, bile secretion, and immune functions. Single‐cell RNA sequencing (scRNA‐seq) technologies enable the analysis of the transcriptome of individual cells and have provided important insights regarding the development,( 1 ) physiology,( 2 , 3 ) and pathology( 4 , 5 , 6 ) of the human liver. These studies have shed light into previously inaccessible aspects of human liver physiology such as hepatic lobular zonation, cell to cell interactions, and immune cell phenotype and heterogeneity.
Previously, we examined the cellular complexity of the human liver by scRNA‐seq and identified 20 distinct cell clusters including two distinct populations of liver‐resident macrophages with immunoregulatory and inflammatory properties.( 2 ) An observation from this work was that enzymatic and mechanical dissociation of the human liver tissue significantly affected the composition of the liver map, in that hepatocytes were sensitive to dissociation‐induced damage and cholangiocytes and liver mesenchymal cells such as hepatic stellate cells (HSCs) were not well‐released by our dissociation technique. For example, cholangiocytes (i.e., parenchymal cells that form the bile duct and are expected to make up 3%‐5% of all liver cells)( 7 ) consist of only 199 (0.64%) cells of our 8,444 cell scRNA‐seq map. Capturing cholangiocyte heterogeneity is key to understanding the pathogenesis of cholangiopathies, such as primary sclerosing cholangitis, for which there are no curative therapeutic interventions.( 8 )
Single‐nucleus RNA sequencing (snRNA‐seq) is an approach that bypasses the cell dissociation step required for scRNA‐seq by using detergents to release nuclei from intact cells. SnRNA‐seq data are also compatible with snap‐frozen samples that may be available from tissue archives. Recently, Slyper et al.( 9 ) assessed three different nuclei isolation protocols for snRNA‐seq from frozen tissues that each used different detergent‐based buffers: Nonidet P40 with salts and Tris (NST), Tween‐20 with salts and Tris (TST), and CHAPS with salts and Tris (CST). Here, we carried out matched snRNA‐seq using these three protocols and scRNA‐seq using our published experimental and analysis workflow( 2 ) on four healthy human liver samples (Fig. 1A). Using multiple protocols on the same samples enables us to evaluate the ability of these three snRNA‐seq protocols to reduce dissociation‐related effects compared with scRNA‐seq protocols, contrast the expression profiles of cells measured with each protocol, and develop an approach to integrate the results into a single map that is more comprehensive than what is achieved with any individual method.
FIG. 1.
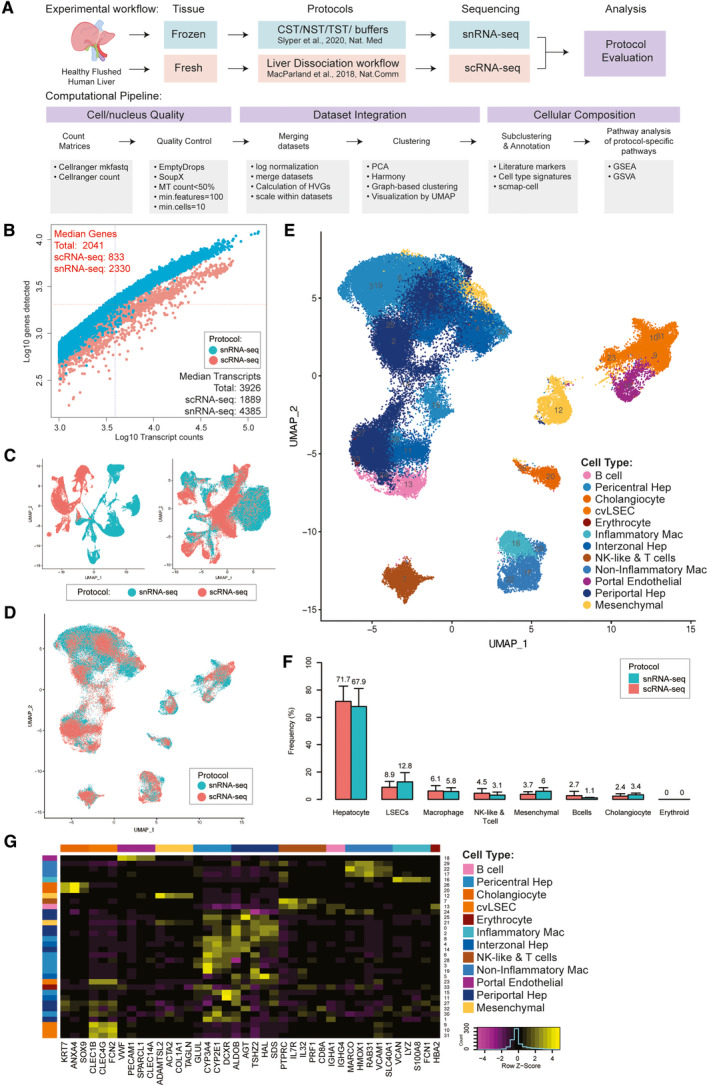
Technical differences between scRNA‐seq and snRNA‐seq in profiling cells from the healthy human liver. (A) Overview of single‐cell and single‐nucleus isolation, data set integration, and analysis workflows. (B) Sensitivity of each approach as measured by the number of genes and transcripts identified in each cell/nucleus. (C) UMAP projection of cells derived from scRNA‐seq and snRNA‐seq merged (i) and then scaled individually before merging (ii). (D) UMAP projection of cells from scRNA‐seq (pink) and snRNA‐seq (blue) individually scaled before merging and then integrated using harmony. (E) UMAP plot showing the assigned identity for each cluster after scaling individually, merging and integrating. (F) Frequency of each major cell population in their source data set; error bars indicate 95% confidence intervals across samples. (G) Heatmap showing scaled mean expression of known marker genes in each cluster. Abbreviations: cvLSECS, central venous LSECs; GSVA, gene‐set variation analysis; MT, proportion of RNA content derived from the mitochondiral genome; PCA, Principal Component Analysis; and UMAP, Uniform Manifold Approximation and Projection.
Our work highlights cell‐type composition differences between snRNA‐seq and scRNA‐seq technologies as applied to human liver, and reveals cholangiocyte and liver mesenchymal cell subpopulations specific to the snRNA‐seq data, previously not identified in single‐cell transcriptomic studies. Combining results from both technologies creates a rich data set for the interpretation of human liver biology, and the identification of key cell‐type defining marker genes across both technologies (Supporting Table S1).
Results
Examining the Quality of Liver Mapping via scRNA‐seq Versus snRNA‐seq
Single‐cell and single‐nucleus transcriptomes were generated from four healthy human livers from neurologically deceased donors that were undergoing transplantation into living recipients. In total, 29,432 single cells were captured from fresh liver dissociates and 43,863 single nuclei were captured from matched snap‐frozen tissue and sequenced using the 10× Chromium platform. These data underwent identical quality control processing, and the matched samples were systematically compared (Fig. 1A).
SnRNA‐seq captured a greater diversity of genes than scRNA‐seq (Fig. 1B). These differences are largely due to the high proportion of unique molecular identifiers in scRNA‐seq data that are derived from transcripts encoding ribosomal proteins and genes encoded in the mitochondrial genome, which are not present in snRNA‐seq data (Supporting Table S2). Minimal differences were observed between different detergents used to extract nuclei (Supporting Table S3). Furthermore, snRNA‐seq contained a significantly higher proportion of ambient RNA than scRNA‐seq for most samples, estimated by SoupX (Supporting Fig. S1).
Integrating scRNA‐seq and snRNA‐seq Maps
Using a typical computational processing pipeline, scRNA‐seq and snRNA‐seq data do not cluster together (Fig. 1C; Supporting Fig. S2), due to the systematic differences between RNA found in the nucleus versus the cytoplasm of cells. Additional technical confounding effects may be introduced during tissue processing and cell handling. The scRNA‐seq samples were derived from fresh tissue that was enzymatically and mechanically dissociated, which may introduce stress responses in cells. In contrast, the snRNA‐seq samples were extracted from flash‐frozen tissue, which should be less impacted by dissociation‐related stresses. In addition, we see significant batch effects between individual donors when using the same sequencing technology, particularly in hepatocytes (Supporting Fig. S2). This may be related to environmental influences on liver metabolism and is consistent with our previously reported scRNA‐seq liver map.( 2 )
However, if samples are normalized and scaled individually before merging, cells and nuclei broadly cluster by cell type rather than by transcriptome mapping technology (Fig. 1C). However, significant differences between technologies are still evident, and the application of Harmony,( 10 ) a commonly used single‐cell data integration method, overcomes this and enables integration and co‐clustering of scRNA‐seq and snRNA‐seq data (Fig. 1D).
Systematic Differences in Nuclear and Whole‐Cell Transcriptomes
The necessity to scale data sets individually to enable scRNA‐seq and snRNA‐seq to be integrated demonstrates that there are significant systematic gene‐expression differences between data generated by these technologies. Examining these systematically differentially expressed genes revealed that both gene function and gene length were strongly associated with expression as measured by scRNA‐seq or snRNA‐seq (Supporting Figs. S3 and S4). Genes encoded by the mitochondrial genome and nuclear genes that encode mitochondrial proteins are more highly expressed in scRNA‐seq (Supporting Fig. S3). Similarly, mRNA encoding ribosome‐related proteins are more than four‐fold more highly expressed in scRNA‐seq than in snRNA‐seq. This is expected, as mitochondria and ribosomes are prevalent in the cytoplasm in liver cells, especially hepatocytes, and cytoplasm is mostly missing from snRNA‐seq material input. In contrast, other “housekeeping” protein‐coding genes( 11 ) were equally expressed in snRNA‐seq and scRNA‐seq. In agreement with results from other tissues,( 12 , 13 ) long noncoding RNAs were 0.7‐fold more highly expressed in snRNA‐seq than scRNA‐seq; however, this is not significantly different from other non‐housekeeping protein‐coding genes, which were 0.8‐fold more highly expressed in snRNA‐seq (Supporting Fig. S3).
Aside from gene function, overall gene length had the strongest correlation with relative expression in snRNA‐seq versus scRNA‐seq (Supporting Fig. S4). Longer genes (>37 kb) were more highly expressed in snRNA‐seq, and short genes (<15 kb) were more highly expressed in scRNA‐seq (Supporting Fig. S4). This was true even after excluding ribosomal and mitochondrial‐related genes.
Cells of the Human Liver as Revealed by scRNA‐seq Versus snRNA‐seq
After integration (see Methods), the data were clustered and annotated using known markers( 2 ); the resulting map revealed all major known hepatic cell types (Fig. 1E‐G). These cell types were represented in both scRNA‐seq and snRNA‐seq, as well as in all samples (Supporting Figs. S5 and S6) but were captured at different frequencies. In particular, scRNA‐seq captured a higher proportion of immune cells with 7% of all cells sequenced identified as lymphocytes and 9% identified as macrophages, compared to snRNA‐seq with 3% lymphocytes and 5% macrophages. In contrast, snRNA‐seq captured 50% more cholangiocytes and hepatic mesenchymal cells than scRNA‐seq (Fig. 1F; Supporting Table S4). We obtained similar percentages of liver sinusoidal endothelial cells (LSECs) and endothelial cells with both methods. However, these frequencies depend on the detergent used for the snRNA‐seq. CST and NST extracted a higher frequency of LSECs (25% and 20%, respectively) and higher frequencies of mesenchymal cells (7% in CST and 12% in NST), but lower frequencies of hepatocytes.
Hepatocytes
Hepatocytes are the main parenchymal cell of the hepatic lobule and are responsible for most liver function. They exhibit functional zonation from the pericentral vein to the periportal region. Recent work has demonstrated the importance of interzonal hepatocytes for liver homeostasis and regeneration,( 14 ) but they have been difficult to identify due to a paucity of known markers.( 2 , 3 ) Subclustering our hepatocyte cluster revealed six distinct clusters (Fig. 2A,B) sourced from both scRNA‐seq and snRNA‐seq (Fig. 2C), and across all samples (Fig. 2D). Correlating these clusters with zonated expression from microdissected regions of the liver lobules in mice( 15 ) enabled the annotation of three of these clusters as containing either pericentral or periportal hepatocytes (Fig. 2E). These annotations were confirmed using known pericentral marker genes (cytochrome P450 3A4 [CYP3A4], alcohol dehydrogenase 4 [ADH4], glutamate‐ammonia ligase [GLUL], and butyrylcholinesterase [BCHE]) and periportal marker genes (histidine ammonia‐lyase [HAL], carbamoyl‐phosphate synthase 1 [CPS1], and 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1 [HMGCS1]) (Fig. 2F). Of the remaining three subclusters, one was most strongly correlated with mouse layer 7; another was most strongly correlated with mouse layer 5, suggesting these two clusters (IZ1 and IZ2) represent human interzonal hepatocytes (Fig. 2E). We validated the putative interzonal hepatocyte identity by comparing it to bulk RNA‐seq derived from human microdissected liver lobule regions, where these clusters correlated most strongly with interzonal layer 4 (Supporting Fig. S7).( 5 ) Markers identified from these two clusters (histidine triad nucleotide binding protein 1 [HINT1], cytochrome C oxidase subunit 7C [COX7C], apolipoprotein C1 [APOC1], fatty acid binding protein 1, liver [FABP1], metallothionein 2A [MT2A], metallothionein 1G [MT1G], and ubiquinone oxidoreductase subunit B1 [NDUFB1]) were validated using spatial transcriptomics and immunohistochemistry, where they exhibit clearly distinct expression patterns from either periportal or pericentral markers but remain ambiguous (Fig. 2G‐I; Supporting Figs. S8‐S12).
FIG. 2.
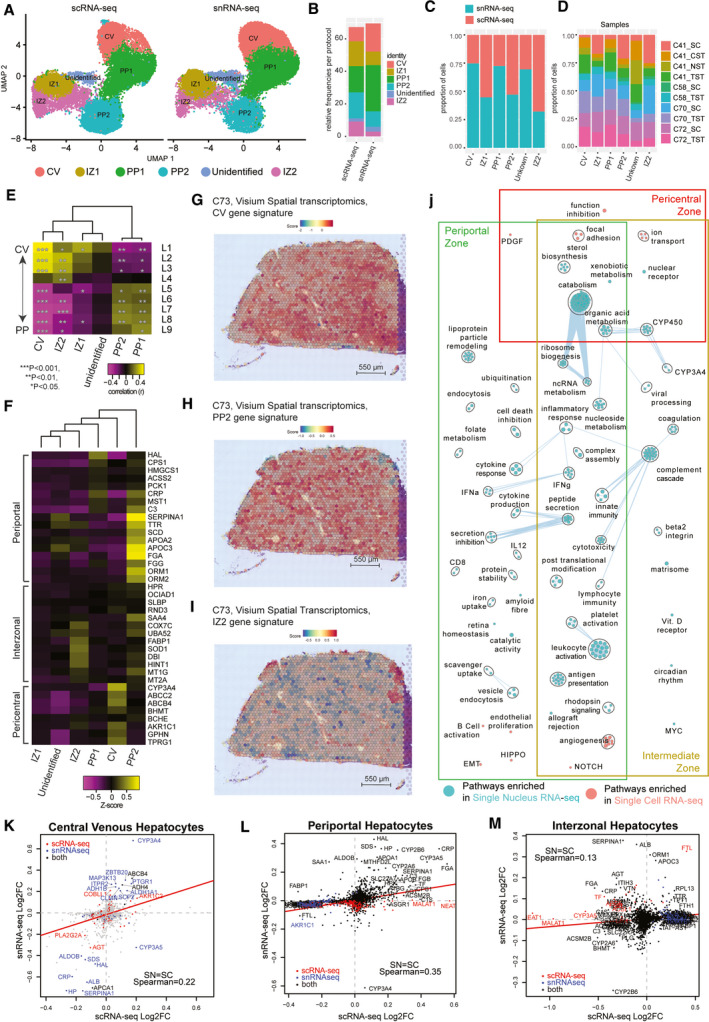
Hepatocyte populations in sample‐matched scRNA‐seq and snRNA‐seq data are spatially resolved by spatial transcriptomics. (A) UMAP plot with the six major populations of hepatocytes split by protocol. (B) Stacked bar plot indicating the frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of hepatocytes by protocol (C) or by donor sample (D) in the combined data set. (E) Correlation of human hepatocyte clusters to known mouse liver sinusoid layers calculated using Spearman correlation. ***P < 0.001, ** P < 0.01, *P < 0.05. (F) Expression of known marker genes in hepatocyte subpopulations in the combined data set. Gene signature scores of the top 30 marker genes in clusters CV (G), PP2 (H), and IZ2 (I) across the spatial transcriptomics spots of a healthy human liver cryosection. (J) Pathway enrichment analysis examining which cellular pathways are better represented by snRNA‐seq (cyan) and scRNA‐seq (pink) in the central venous, periportal, and interzonal hepatocyte populations. Circles (nodes) represent pathways, sized by the number of genes included in that pathway. Related pathways, indicated by light blue lines, are grouped into a theme (black circle) and labeled. Intra‐pathway and inter‐pathway relationships are shown in light blue and represent the number of genes shared between each pathway. Log2FC of significant genes (q‐value < 0.05) within either scRNA‐seq (red) or snRNA‐seq (blue) or both (black) for CV hepatocytes (K) (cluster CV), PP clusters (L) (clusters PP1 and PP2), and IZ clusters (M) (clusters IZ1 and IZ2). Abbreviations: IZ, interzonal; and PP, periportal.
The final cluster of albumin (ALB)–expressing hepatocytes expressed periportal hepatocyte markers like serpin family A member 1 (SERPINA1), transthyretin (TTR), apolipoprotein A1 (APOA1), and apolipoprotein C3 (APOC3). However, this cluster did not correlate strongly with the periportal mouse sinusoid regions, nor did it express many other periportal marker genes like HAL, CPS1, and HMGCS1. Furthermore, the expression profile correlated most strongly with the interzonal layer 4 of the human sinusoid, but did not correlate with any mouse zonation layer (top differentially expressed genes (DE): ALB, SERPINA1, APOA1, haptoglobin [HP], ferritin light chain [FTL], APOC3, serum amyloid A1 [SAA1], stabilin 1 [STAB1], TTR, beta‐2‐microglobulin [B2M], and LIF receptor subunit alpha [LIFR]). These results suggest that this cluster may represent a human‐specific interzonal hepatocyte cluster, but further work will be required to confirm this identity.
Hepatocytes as a whole were captured equally well in scRNA‐seq and snRNA‐seq (Supporting Table S2); however, central venous (CV1) hepatocytes were almost twice as frequent (17% vs. 9%; P < 10−30) in snRNA‐seq than scRNA‐seq (Fig. 2B,C; Supporting Tables S3 and S5). In contrast, interzonal hepatocytes were most frequent in scRNA‐seq (15% and 9% vs. 8% and 3%; P < 10−15). Despite these differences, we observe almost all hepatocyte‐related pathways exhibiting elevated expression in snRNA‐seq‐derived hepatocytes (Fig. 2J). Several significant genes identified in CV1 hepatocytes by snRNA‐seq were not identified by scRNA‐seq (Fig. 2K), whereas the gene‐expression patterns for periportal hepatocytes correlated significantly across both technologies (Fig. 2L). The greatest discordance exists for the interzonal hepatocytes in which many of the genes up‐regulated in snRNA‐seq are down‐regulated in scRNA‐seq (Fig. 2M). This may reflect poor viability or disrupted cellular state within hepatocytes due to the dissociation( 16 ) required for scRNA‐seq, which is not present when examining hepatocytes with snRNA‐seq. Thus, snRNA‐seq may provide a better characterization of hepatocytes than scRNA‐seq, despite similar capture rates.
Cholangiocytes
Cholangiocytes are epithelial cells that line the bile ducts and generate 30% of the total bile volume.( 17 ) Our previous attempt to characterize these cells using exclusively scRNA‐seq identified only a single population encompassing 1.4% (199 of 8,444) of the cells expressing cholangiocyte markers (epithelial cell adhesion molecule [EPCAM], SRY (sex determining region Y)‐box 9 [SOX9], and keratin 1 [KRT1]).( 2 )
In this study, we found that snRNA‐seq captured a higher proportion of KRT7, SOX9, and annexin A4 (ANXA4) expressing cholangiocytes (3.4% vs. 2.4%), resulting in a total of 448 cholangiocyte‐like cells (Fig. 1G; Supporting Table S2). Subclustering this KRT7+ SOX9+ population revealed six transcriptionally distinct subpopulations (labeled Chol‐1 to Chol‐6) (Fig. 3A). We identified three asialoglycoprotein receptor 1–positive (ASGR1+) hepatocyte‐like clusters, two typical cholangiocyte‐like clusters (KRT7, KRT18, and solute carrier family 4 member 2 [SLC4A2] high), and a cluster of progenitors, 82% of which were derived from snRNA‐seq (Fig. 3B‐F; Supporting Table S6). These clusters were not specific to a particular sample or donor, indicating that they were not the result of batch effects or donor‐specific variation (Fig. 3D). Rather, these clusters formed a branching trajectory extending from bipotent progenitors to both hepatocyte and cholangiocyte cell fates, as computed using both Slingshot( 18 ) and diffusion maps (Fig. 3G; Supporting Fig. S13).
FIG. 3.
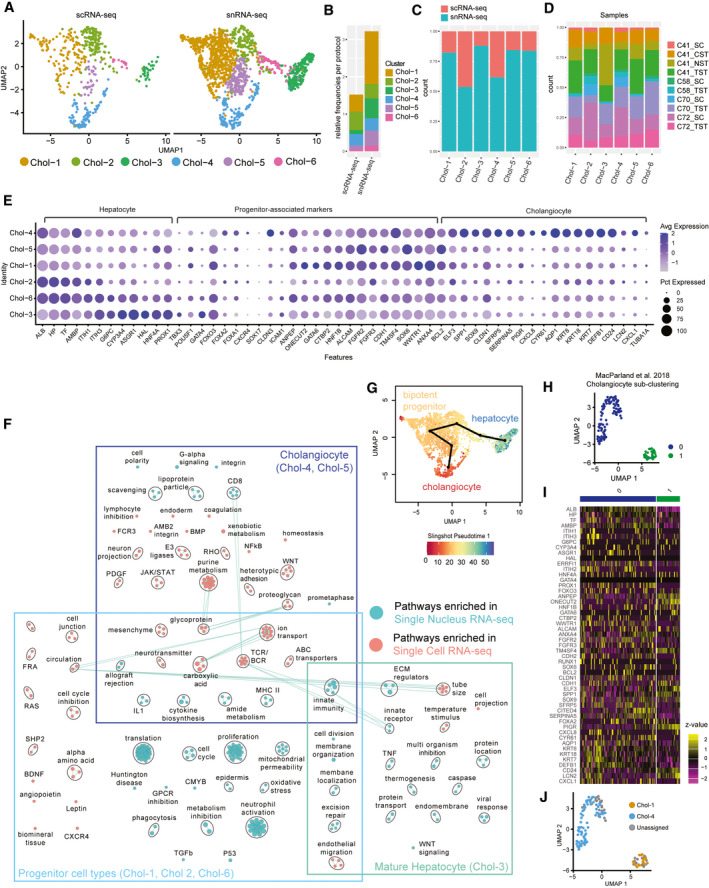
Cholangiocyte‐associated cells as revealed by snRNA‐seq. (A) UMAP plot with the six major populations of cholangiocytes and progenitor cells identified in the combined data set split by protocol. (B) Frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of each population by protocol (C) and by sample (D) in the combined data set. (E) Expression of known cholangiocyte, progenitor, and hepatocyte marker genes in each population. The size of the circle indicates the percentage of cells in each population expressing each gene. (F) Pairwise pathway analysis comparing cholangiocyte‐like, progenitor‐like, and mature hepatocyte‐like cells from snRNA‐seq (cyan) to those from scRNA‐seq (pink). Circles (nodes) represent pathways, sized by the number of genes included in that pathway. Related pathways, indicated by light blue lines, are grouped into a theme (black circle) and labeled. Due to low cell number, similar clusters were combined for pathway analysis (see Supporting Fig. S15) for more details. (G) Color value in UMAP plot indicates distance along the cholangiocyte to bipotent progenitor cells to hepatocyte pseudo‐time trajectory as inferred by Slingshot.( 18 ) (H) UMAP of subclustered cholangiocyte‐like populations from an independent scRNA‐seq healthy human liver data set.( 2 ) (I) Heatmap depicting the relative expression of cholangiocyte subpopulation‐associated marker genes in cholangiocyte‐like cells.( 2 ) (J) Projection of cell‐type annotations from the combined scRNA‐seq and snRNA‐seq cholangiocyte data set onto scRNA‐seq data from MacParland et al.( 2 ) using scmap‐cell.( 23 )
At the most differentiated end of the cholangiocyte branch was the Chol‐4 population, which contained mature cholangiocytes expressing differentiated cholangiocyte‐associated markers (Top DE: aquaporin 1 [AQP1], KRT8, KRT18, KRT7, defensin beta 1 [DEFB1], CD24, polymeric immunoglobulin receptor [PIGR], and ANXA4) (Fig. 3E; Supporting Fig. S14A), many of which are highly specific to human bile ducts (Supporting Fig. S15A).( 19 ) Furthermore, classical cholangiocyte pathways, such as cell polarity, ion transport, ABC transporters and many immune pathways, were enriched among the genes up‐regulated in this cluster (Fig. 3F; Supporting Fig. S16). Cells of this cluster were derived in equal measure from scRNA‐seq and snRNA‐seq, and the significantly up‐regulated markers, including AQP1, secreted phosphoprotein 1 [SPP1] and DEFB1, were relatively consistent across both technologies (Supporting Fig. S17). Thus, mature cholangiocytes are well characterized by either snRNA‐seq or scRNA‐seq.
The less‐differentiated cholangiocyte population (Chol‐5) was specific to snRNA‐seq (177 nuclei vs. 33 cells). Cholangiocyte identity was confirmed by the expression of key transcription factors (hepatocyte nuclear factor‐1‐beta [HNF1B], one cut homeobox 1 [ONECUT1], and SOX9) and the enrichment of WNT signaling, ABC, and ion transporters( 20 ) (Supporting Fig. S16). We determined this cluster contained specifically small cholangiocytes on the basis of B‐cell leukemia/lymphoma 2 (BCL2) expression (Fig. 3E; Supporting Fig. S14A), which is not expressed by large cholangiocytes.( 20 ) This was confirmed by noting bile‐duct restricted expression of BCL2 by immunohistochemistry( 19 ) (Supporting Fig. S15B). We note high expression of many stem‐ness markers( 21 ) in this cluster, which is consistent with previous reports of a less‐differentiated phenotype of these cholangiocytes( 20 ) (Fig. 3E; Supporting Fig. S14A,B). Markers of this cluster were identified using snRNA‐seq (CYP3A5, PPARG coactivator 1 alpha [PPARGC1A], fragile histidine triad diadenosine triphosphatase [FHIT], flavin containing dimethylaniline monooxygenase 5 [FMO5], tryptophan 2,3‐dioxygenase [TDO2], and SOX6); however, these were not recapitulated in scRNA‐seq, subsequently reinforcing that this population can only be characterized using snRNA‐seq (Supporting Fig. S17).
At the far end of the opposite branch was Chol‐3, a population of central venous hepatocyte‐like cells, expressing high levels of CYP3A4, glypican 6 (GPC6), and aldehyde oxidase 1 (AOX1) (Supporting Figs. S9 and S14C). These cells clustered together with cholangiocytes because of their high expression of many bile metabolism genes (i.e., ATP‐binding cassette subfamily A member 8 [ABCA8], ABCA6, hydroxyacid oxidase 1 [HAO1], ABCA1, and SLC27A2), indicating involvement in biliary function. Interestingly, these cells express hepatocyte nuclear factor 4 (HNF4a), which is first expressed in progenitor hepatocytes and is a central regulator of hepatocyte differentiation.( 22 )
Two additional hepatocyte‐biased clusters, Chol‐2 and Chol‐6, expressed some typical cholangiocyte markers (KRT7, SOX9, and CD24) but also expressed early hepatocyte lineage‐defining transcription factors (HNF4a, forkhead box A2 [FOXA2], prospero homeobox 1 [PROX1], ONECUT1, and ONECUT2), suggesting an intermediate or progenitor phenotype (Fig. 3E; Supporting Figs. S14B‐E and S18). The highly expressed genes of both Chol‐6 and Chol‐2 related to progenitor‐associated markers, such as FOXA2, fibroblast growth factor receptor 3 (FGFR3), hairy and enhancer of split‐1 [HES1], and jagged canonical notch ligand 1 (JAG1) (Fig. 3E; Supporting Fig. S14B,D). The key difference between the clusters is the expression of proliferation‐related genes in Chol‐6, indicating that these cells were proliferative and non‐proliferative hepatic progenitors.
At the root of these lineage‐biased branches was a large cluster of bipotent progenitors (Chol‐1), containing predominantly sequenced single nuclei (621 nuclei vs. 134 cells) (Extended Table 6; Fig. 3C). This cluster did not express mature cholangiocyte markers, but rather a collection of many stem‐like and progenitor markers, including pou class 5 homeobox 1 (POU5F1), FOXO2, runt‐related transcription factor 2 (RUNX2), SOX6, CD133, alanyl aminopeptidase, membrane (ANPEP), and SOX9 (Supporting Fig. S14B‐D). These characteristics of this cluster are consistent with bipotent progenitors that have previously been observed only in vitro (Supporting Fig. S14D).( 3 ) Furthermore, using pathway analysis, we identify the NOTCH2 signaling pathway (notch receptor 2 [NOTCH2], recombination signal binding protein for immunoglobulin kappa J region [RBPJ], mastermind like transcriptional coactivator 2 [MAML2], MAML3, MAML4) as a key feature of these progenitor cells. Endoderm, cell cycle, and cell division pathways were enriched in this population relative to the other clusters supporting a stem‐like state (Supporting Figs. S16 and S18). Marker genes of Chol‐1 (i.e., FOXO3, GATA binding protein 6 [GATA6], FGFR3, and ANPEP) were localized to bile ducts in both our spatial transcriptomics data and publicly available immunohistochemistry data, suggesting that these may be the progenitor niche (Supporting Figs. S19 and S20).( 19 )
Using the combined map presented here as a reference, we were able to use the automatic annotation tool scmap‐cell( 23 ) to identify and label two distinct subsets of cholangiocytes that were unable to be detected in our previous map (Fig. 3H). We identified cells from both mature cholangiocytes (Chol‐4) and a small number of bipotent progenitors (Chol‐1), which could only be identified using the snRNA‐seq data, demonstrating the utility of our combined scRNA‐seq and snRNA‐seq liver map (Fig. 3I,J).
Liver Mesenchymal Cells
Various mesenchymal cell populations such as HSCs, fibroblasts (FBs), and vascular smooth muscle cells (VSMCs) have been proposed as the source of pathogenic myofibroblasts, that promote fibrosis following liver injury through the production of extracellular matrix proteins.( 24 ) To design ideal antifibrotic therapies that specifically target myofibroblasts in disease, the baseline cellular functions of these mesenchymal cells at homeostasis must be described. Previous single‐cell studies that have focused on the heterogeneity in the mesenchyme, specifically HSCs during disease or injury, typically make use of additional cell‐handling steps to enrich for these cells.( 4 , 25 ) Subclustering our mesenchymal cell cluster revealed seven distinct populations (Mes1‐7) present in the healthy liver, of which only Mes1, 2, and 4 contained more than 10 cells captured with scRNA‐seq, and only Mes4 was approximately equally captured by both methodologies and in all samples (Fig. 4A‐D; Supporting Table S7). Overall, mesenchymal cells consisted of 2.5% of single nuclei, more than double their 1% of single cells captured (Fig. 4B). Using snRNA‐seq, we reveal the transcriptome of VSMCs and FBs present in healthy livers at low frequencies (Fig. 4).
FIG. 4.
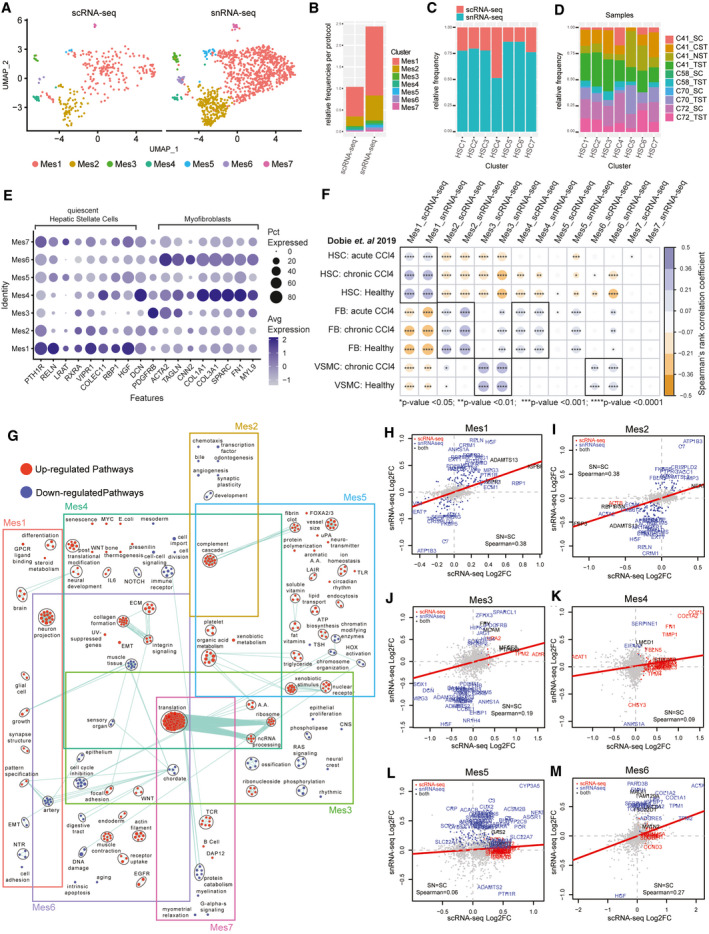
Identification of HSCs, FBs, and VSMCs in the healthy human liver through scRNA‐seq and snRNA‐seq. (A) UMAP plots with the seven major clusters of mesenchymal cells in the combined data set, split by protocol. (B) Stacked bar plot indicating the frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of each population by protocol (C) and by sample (D) in the combined data set. (E) Dot plot indicating the relative expression of known quiescent HSCs and myofibroblast‐specific marker genes in each population. The size of the circle indicates the percentage of cells in each cluster expressing each gene. (F) Spearman correlation of transcriptional gene signatures from mouse HSCs, FBs and VSMCs, sourced from healthy and carbon tetrachloride–induced acute and chronically fibrotic livers (Dobie et al. 2019) to the human mesenchymal cells clusters from this study.( 29 ) ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. (G) Pathway enrichment analysis examining what are the up‐regulated (red) and down‐regulated (cyan) biological pathways in each mesenchymal cell cluster in the combined scRNA‐seq and snRNA‐seq data set. Circles (nodes) represent pathways, sized by the number of genes included in that pathway. Related pathways, indicated by light blue lines, are grouped into a theme (black circle) and labeled. (H)‐(M) Log2FC of significant genes (q‐value < 0.05) within either scRNA‐seq (red) or snRNA‐seq (blue) or both (black) for each cluster within the mesenchymal cell data set.
Under physiological conditions, HSCs maintain a quiescent and non‐proliferative phenotype and are localized between the layers of LSECs and hepatocytes in the space of Disse.( 2 , 26 ) Activated HSCs are thought to be the primary contributor to the myofibroblast pool in liver fibrosis and are characterized by the loss of retinol‐bearing lipid droplets, the development of a contractile phenotype, and the production of collagens and extracellular matrix remodeling proteins.( 26 , 27 , 28 ) Mes1, 5, and 7 expressed HSC‐associated retinol storage genes (Fig. 4E), while the rest expressed myofibroblast‐associated genes. To further determine the cellular identity of these clusters, we carried out a cross‐species correlation analysis using previously published transcriptomic signatures of fluorescence‐activated cell–sorted HSCs, FBs, and VSMCs from healthy and carbon tetrachloride–induced fibrotic mouse livers.( 29 ) The only mesenchymal cluster to correlate strongly with the healthy and fibrotic HSC signatures was Mes1. Seventy percent of mesenchymal nuclei consisted of quiescent HSCs (Mes‐1 and Mes‐5) specializing in vitamin A storage and metabolism, with high expression of RBP1, lecithin retinol acyltransferase (LRAT), and phosphodiesterase 3B (PDE3B) (Fig. 4F; Supporting Fig. S21A).( 30 ) Mes1 specifically expressed hepatocyte growth factor (HGF), which is a key growth factor in liver regeneration and hepatocyte differentiation and growth, and displayed enrichment for neuronal pathways as expected.( 31 ) Mes5 expressed antigen presentation genes and showed an enrichment of pathways associated with both fat and water‐soluble vitamins, lipid transport, and organic acid metabolism (Fig. 4G; Supporting Fig. S21A). The presence of lipid metabolism and fibrin clot pathways and inflammation‐associated genes indicate that these may represent activated HSCs (Fig. 4G; Supporting Fig. S21A‐C). Mes7 is the smallest cluster of cells that express RBP1, HGF, LRAT and Decorin (DCN), yet the presence of many lymphocyte‐associated transcripts suggests that they are likely to be doublets.
Mes2 and Mes4 transcriptomes from both scRNA‐seq and snRNA‐seq correlated well with mouse FB signatures, whereas Mes3 and Mes6 correlated with VSMC signatures and not activated HSCs from the chronically fibrotic mouse liver (Fig. 4F). These observations held true for both scRNA‐seq and snRNA‐seq. Mes2 cells were the second‐most‐frequent population in the data set (24% of nuclei). They expressed more lipid‐processing genes in comparison to Mes4 (AOX1, PDE3D, and PDE4D), as well as an enrichment of the complement cascade pathways but lacked typical HSC markers and strong myofibroblast markers (Fig. 4F). Furthermore, this cluster also showed expression of cytokine and growth factor receptors, and angiogenic and proliferative factors (i.e., transforming growth factor beta 1 [TGFB1], transforming growth factor receptor type 2 [TGFBR2]), indicating that they may be liver FBs (Supporting Fig. S21D,D).
The other three non‐HSC Mes clusters consisted of only 10% of mesenchymal cell nuclei (112 of 1,069), and contained heterogeneous subsets of tissue remodeling cell types. Myofibroblasts typically arise following liver damage, in response to cytokines, local damage, growth factors, and fibrogenic signals.( 26 , 32 ) In contrast to Mes2, Mes4 had enriched pathways associated with inflammatory signaling, collagen production and matrix remodeling, and expressed high levels of fibrosis and myofibroblast‐associated genes (Supporting Fig. S21B,D). Both Mes4 and Mes6 expressed several activation‐associated genes [ACTA2, secreted protein acidic and cysteine rich (SPARC), transgelin (TAGLN), collagen type 1 alpha 1 chain (COL1A1), and TIMP metallopeptidase inhibitor 1 (TIMP1)], and enriched pathways in both clusters included collagen formation, extracellular matrix, and integrin signaling (Fig. 4E,F; Supporting Fig. S21B). Furthermore, Mes4 was enriched in senescence‐related pathways and inflammatory genes (interleukin‐32 [IL‐32], colony stimulating factor 1 [CSF1], TNF superfamily member 10 [TNFSF10], C‐C motif chemokine ligand 2 [CCL2], and IL6ST), indicating a more activated FB phenotype (Supporting Fig. S21C). Mes6 expressed VSMC markers like calponin 2 (CNN2), myosin light chain 9 (MYL9), TAGLN and ACTA2, high levels of fibrogenic growth factors, and was enriched in the contractile phenotype, suggesting a fibrogenic state of VSMCs (Fig. 4G; Supporting Fig. S21D). Mes3 nuclei also express ACTA2, MYL9 and TAGLN, but not collagen or matrix remodelers or quiescent HSC genes. Instead, this cluster is enriched for genes that support angiogenesis and local cell proliferation (apoptosis), suggesting a less‐activated VSMC state (Supporting Fig. S21D,E).
Spatial transcriptomics independently confirmed HSC gene expression (Mes1 and 5) to be higher and dispersed throughout liver tissue, whereas VSMCs (Mes3 and 6) and FBs (Mes2 and 4) were primarily located in periportal regions, as seen previously( 29 ) (Supporting Fig. S22). As most of these clusters were primarily identified in snRNA‐seq data, most of the differentially expressed genes in the snRNA‐seq data were not identified as significant in the scRNA‐seq samples (Fig. 4H‐M), reinforcing the value of a combined approach in capturing liver mesenchymal cell heterogeneity.
Liver Endothelial Cells
The endothelium of the liver vasculature is made up of LSECs and vascular endothelial cells. LSEC populations were annotated using previously defined markers (Figs. 1G and 5A‐E).( 2 ) Similar to hepatocytes, central venous LSECs were more frequently captured with snRNA‐seq (7.52%) than scRNA‐seq (5.2%) (Fig. 5B‐D), whereas periportal LSECs and portal endothelial cells were present in similar frequencies using either technology (ppLSECs: 1.15% in snRNA‐seq and 1.32% in scRNA‐seq; PortalEndo: 0.76% and 0.63%, respectively) (Supporting Table S8). Markers and associated pathways of these populations were generally consistent across both technologies. Although gene‐expression fold changes were typically smaller in snRNA‐seq (Fig. 5F‐I), this was particularly true for portal markers: von Willebrand factor (VWF), C‐type lectin domain containing 14A (CLEC14A), inhibitor of DNA binding 1 (ID1), SPARC like 1 (SPARCL1), and connective tissue growth factor (CTGF) (Fig. 5E). The central venous LSECs and portal endothelial‐associated marker genes and gene signatures are distributed peri‐centrally and peri‐portally, respectively (Supporting Fig. S23). However, the expression of marker genes for periportal LSECs and the periportal LSEC gene signature was not well captured using the 10X Genomics Visium spatial gene‐expression platform.
FIG. 5.

Analysis of LSECs in the combined scRNA‐seq and snRNA‐seq data set. (a) UMAP plots with the three major endothelial cell populations in the combined data set split by protocol. (B) Frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of each population by protocol (C) and by sample (D) in the combined data set. (E) Dot plot indicating the relative expression of known LSEC marker genes in each population by protocol. The size of the circle indicates the percentage of cells in each population expressing each gene. (F) Pathway enrichment analysis examining which cellular pathways are better represented by snRNA‐seq (cyan) and scRNA‐seq (pink) in each of the LSEC subpopulations. Circles (nodes) represent pathways, sized by the number of genes included in that pathway. Related pathways, indicated by light blue lines, are grouped into a theme (black circle) and labeled. (G),(I) Log2FC of significant genes (q‐value < 0.05) within either scRNA‐seq (red) or snRNA‐seq (blue) or both (black) for each cluster within the LSEC data set.
Intrahepatic Monocytes/Macrophages
We previously characterized two populations of intrahepatic macrophages with distinct immunoregulatory and inflammatory properties.( 2 ) We identified both of these populations in scRNA‐seq and snRNA‐seq; however, we note a higher overall proportion of macrophages in snRNA‐seq (7.4% of nuclei vs. 4.1% of cells) and a lower relative proportion of inflammatory macrophages in snRNA‐seq (46% vs. 59%) (Fig. 6A‐D; Supporting Table S9). Although snRNA‐seq was more efficient at capturing non‐inflammatory macrophages and their associated marker genes (Fig. 6B,E), several marker genes for these populations are present in both snRNA‐seq and scRNA‐seq maps (CD68, protein tyrosine phosphatase receptor type C [PTPRC], and macrophage receptor with collagenous structure [MARCO]). Meanwhile, the markers used to describe inflammatory macrophages (lysozyme [LYZ], S100A8, and S100A9) were better represented by scRNA‐seq (Fig. 6F). Immune‐associated pathways (interferon gamma [IFNγ], leukocyte activation, phagocytosis, and bacterial response) were more highly expressed in snRNA‐seq rather than scRNA‐seq (Fig. 6G), suggesting that macrophages may be dissociation‐sensitive. Spatial transcriptomics of healthy liver tissue indicated that the non‐inflammatory macrophage genes and gene signature were present in the periportal regions, whereas the inflammatory macrophage genes and gene signature were expressed closer to the central vein (Supporting Fig. S24).
FIG. 6.

Analysis of liver‐resident macrophages in the combined scRNA‐seq and snRNA‐seq data set. (A) UMAP plots depicting the clustering of inflammatory and non‐inflammatory macrophages in the combined data set split by protocol. (B) Stacked bar plot indicating the frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of each population by protocol (C) and by sample (D) in the combined data set. (E) Dot plot indicating the relative expression of known inflammatory and non‐inflammatory macrophage marker genes in each cluster by protocol. The size of the circle indicates the percentage of cells in each population expressing each gene. (F) Log2FC of significant genes (5% false discovery rate) within either scRNA‐seq (red) or snRNA‐seq (blue) or both (black) for each cluster within the macrophage populations; nonsignificant shown in gray. (G) Pairwise pathway enrichment analysis comparing snRNA‐seq to scRNA‐seq in each macrophage subpopulation. Pathways enriched in snRNA‐sq are labeled in cyan, and pathways enriched in scRNA‐seq are indicated in pink. Circles (nodes) represent pathways, sized by the number of genes included in that pathway. Related pathways, indicated by light blue lines, are grouped into a theme (black circle) and labeled. Abbreviation: Macs, macrophages.
Intrahepatic Lymphocyte Populations
We previously observed that lymphocytes are well‐detected after hepatic tissue disruption.( 2 ) Major lymphocyte populations were captured by each protocol (Fig. 7A) and in each sample (Fig. 7B); lymphocytes comprise 5.2% of the scRNA‐seq data set (1,524 of 29,432) but make up only 1.8% of the snRNA‐seq data set (804 of 43,863). All lymphocyte subpopulations were captured at higher frequency by scRNA‐seq (Fig. 7C,D). B‐cell populations in particular make up only 0.2% of the snRNA‐seq data set, but are enriched almost 2 times more in scRNA‐seq data (0.5%) (Supporting Table S10). In our examination of marker genes for each cluster present across both protocols (Fig. 7E), IL7R and S100A4 serve as the best markers for resident memory T cells. The top marker genes for γδ T cells and natural killer (NK) have significant overlap. Unfortunately, B‐cell receptor and T‐cell receptor genes were not well‐captured by snRNA‐seq (Supporting Fig. S25). As such, scRNA‐seq captures transcripts that provide the resolution for differentiating distinct lymphocyte populations.
FIG. 7.
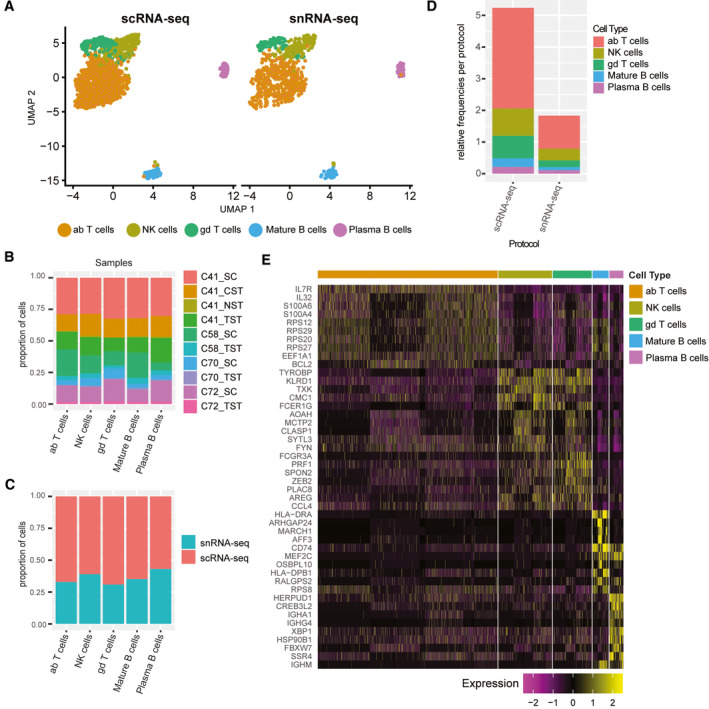
Liver‐resident lymphocytes are enriched in scRNA‐seq data sets. (A) UMAP plots depicting the clustering of various lymphocyte subpopulations in the combined data set split by protocol. (B) Frequency of each population in either scRNA‐seq or snRNA‐seq data sets. Distribution of each population by protocol (C) and by sample (D) in the combined data set. (E) Heat map showing the most significantly up‐regulated genes per cluster. Abbreviations: ab, alpha‐beta; and gd, gamma‐delta.
Discussion
Comparing scRNA‐seq and snRNA‐seq protocols applied to four samples of healthy human liver revealed several significant differences. Both techniques produced high‐quality data that could elucidate the major cell classifications in the human liver. However, cell‐type frequencies are distorted in scRNA‐seq, primarily due to the resiliency of immune cells to tissue dissociation compared with parenchymal cells. These differences affect the sensitivity of each method to delineate subtypes of the respective cell types. SnRNA‐seq enables the detection of many additional cell subtypes of cholangiocytes and mesenchymal cells, which are difficult to distinguish using scRNA‐seq. For example, the transcriptional profile of small‐duct cholangiocytes provided in this data set may act as a reference for assessing how cholangiocytes in small‐duct cholangio‐pathologies, like primary biliary cholangitis (PBC), might differ transcriptionally to those in the healthy liver. Furthermore, this platform could potentially allow for distinguishing differences in the cellular landscape of poorly understood autoimmune liver diseases, such as differentiating between PBC and primary sclerosing cholangitis, which is a disease primarily affecting large bile ducts.
A complete characterization of the intrahepatic immune landscape is also crucial to understanding the pathogenesis of liver disease. Despite the advantages of nuclei profiling, many important markers of immune cells were completely absent from snRNA‐seq data. For instance, none of the T‐cell or B‐cell receptor components were detected in our snRNA‐seq samples. Thus, we recommend that studies investigating intrahepatic immune populations use scRNA‐seq.
The ability of snRNA‐seq to overcome the limitations associated with tissue dissociation and to capture parenchymal cells in high resolution opens an avenue for a detailed examination of the interplay between parenchymal cells and non‐parenchymal cells in health and disease. Additionally, the ability to perform snRNA‐seq in frozen tissues can enable the examination of biobanked samples with single‐cell resolution. Taken together, we have shown that single‐cell and single‐nucleus RNA‐seq generate high‐quality data from normal liver samples, with snRNA‐seq allowing for better examination of rare liver stromal cells including FBs, HSCs and VSMCs, and parenchymal cells such as cholangiocytes. This combined data set enables a complex examination of parenchymal cell complexity and provides a foundation for single‐cell liver disease studies.
Materials and Methods
Preparation of Fresh Tissue Homogenates and Nuclei From Snap‐Frozen Human Liver Tissue
Human liver tissue from the caudate lobe was obtained from neurologically deceased donor liver acceptable for liver transplantation. Samples were collected with institutional ethics approval from the University Health Network (REB# 14‐7425‐AE). A 3‐mm‐cubed fragment of tissue was preserved for snRNA‐seq by snap freezing in liquid nitrogen. Within a 3‐mm‐cubed segment of tissue, we would expect two to three lobules would be assessed, and the presence of all cell clusters in all populations in our analysis suggests the relative homogeneity of liver cell compositions between individuals. Single‐cell suspensions of fresh human liver were generated as previously described( 2 ) the two‐step collagenase perfusion protocol (https://doi.org/10.17504/protocols.io.m9sc96e). Samples with paired snap‐frozen tissues (frozen in liquid nitrogen at the time of dissociation within 1 hour of caudate lobe removal from the donor organ) were selected for scRNA‐seq and snRNA‐seq. Nuclei were extracted from snap‐frozen tissues within 2 years of snap freezing. Single‐nucleus extraction from frozen tissue was performed as previously described.( 9 ) The full description of processing is found in the extended methods.
Sample Processing, Complementary DNA Library Preparation, Sequencing, and Data Processing
Single cell samples were prepared as outlined by the 10x Genomics Single‐Cell 3′ v2 and 3’ v3 Reagent Kit user guides and as described previously.( 2 ) Single‐cell data were processed using 10x Cell Ranger software version 3.01, mapping reads to the GRCh38 human genome. Single‐nucleus data were processed using Cell Ranger version 3, and reads were mapped to a modified transcriptome based on GRCh38, which included intronic regions to ensure quantification of reads derived from immature, unspliced messenger RNA (mRNA) present in the nucleus. The full description of 10x sample and data processing is found in the extended methods.
Data Integration and Clustering
The data were integrated using default parameters of Harmony,( 10 ) then clustered using Seurat’s SNN‐Louvain clustering algorithm.( 33 ) The data were clustered using 30 sets of parameters, and the most consistent clusterings were identified using apcluster( 34 ) on the cluster–cluster distance matrix calculated using the Variation of Information criterion.( 35 ) The full description of data integration and clustering can be found in the extended methods.
Gene‐Type and Gene‐Length Biases Between scRNA‐seq and snRNA‐seq Data
A total of 3,804 housekeeping genes were obtained from the literature.( 11 ) Long noncoding RNAs and protein coding gene lists were obtained from Ensembl. Nuclear‐encoded mitochondrial proteins were obtained from MitoCarta3.0.( 36 ) Ribosomal genes were obtained from the Ribosomal Protein Gene database.( 37 ) Transcript GC content, microRNA binding sites, transcript length, untranslated region lengths, and intron length were obtained from Ensembl Biomart. Log‐fold changes of mean expression across all cell types in scRNA‐seq and snRNA‐seq were calculated across all samples.
Pathway Enrichment, Correlation, and Trajectory Inference Analysis
Pathway enrichment analysis was performed as previously described( 2 ) with the addition of a dissociation signature to the pathway gene‐set database.( 16 ) Slingshot (v1.8.0) was used to infer the pseudo‐time based on the Harmony embedding matrix of cells. Lineages were calculated using the Slingshot Uniform Manifold Approximation and Projection (UMAP) embedding protocol.( 18 ) Diffusion maps( 38 ) (destiny, v3.1.1) were computed with both the raw counts matrix and the principal component analysis loadings. Spearman’s rank correlation coefficient was calculated on each pair of outputs of these analyses and plotted using corrplot (v0.84). The full description of this analysis is found in the extended methods.
Validation of Zonated Gene Signatures Using Spatial Transcriptomics
Healthy human liver tissue was embedded in optimal cutting temperature, frozen, and cryosectioned with 16‐um thickness at −10ºC (cryostar NX70 HOMP). Sections were placed on a chilled Visium Tissue Optimization Slide (10x Genomics) and processed following the Visium Spatial Gene Expression User Guide. Tissue was permeabilized for 12 minutes, based on an initial optimizations trial and libraries were prepared according to the Visium Spatial Gene Expression User Guide. Samples were sequenced on a NovaSeq 6000.
Visium Spatial Transcriptomics
The Visium spatial transcriptomic data were sequenced to a depth of 167,400,637 reads, a saturation of 77%. These reads were mapped to the GRCh38 human genome and expression was quantified with the spaceranger‐1.1.0. Further processing and visualization were performed with Seurat (version 3.2.1). The full description of Visium data processing is found in the extended methods.
Validation of zonated protein expression via the Human Protein Atlas
Immunostaining images were obtained from the Human Protein Atlas (https://www.proteinatlas.org).( 19 ) Lobule annotation was confirmed by a liver pathologist (C. Thoeni).
Supporting information
Supplementary Material
Table S6
Table S11
Table S12
Acknowledgment
This project has been made possible in part by grant number CZF2019‐002429 from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation. This research was supported in part by the University of Toronto’s Medicine by Design initiative, which receives funding from the Canada First Research Excellence Fund (CFREF) to SAM, GDB and IDM; by the NRNB (U.S. National Institutes of Health, grant P41 GM103504) to GDB and by the Toronto General and Western Hospital Foundation. JA has received graduate fellowships from NSERC (CGS‐M) and an Ontario Graduate Scholarship. CTP has received postdoctoral funds from the Canadian Network on Hepatitis C (CanHepC) and PSC Partners Canada. CanHepC is funded by a joint initiative of the Canadian Institutes of Health Research (CIHR) (NHC‐142832) and the Public Health Agency of Canada (PHAC).
Supported by the Chan Zuckerberg Initiative (CZF2019‐002429).
Potential conflict of interest: Dr. Slyper consults for Genentech. Dr. Regev owns stock and holds intellectual property rights with Celsius and Immunitas. Dr. Regev owns stock in Roche and Neogene.
Contributor Information
Ian D. McGilvray, Email: Ian.Mcgilvray@uhn.ca.
Gary D. Bader, Email: gary.bader@utoronto.ca.
Sonya A. MacParland, Email: s.macparland@utoronto.ca.
Data Availability Statement
Raw data and raw count matrices, and final annotations are available in GEO under accession: GSE185477; count‐matrices and fully processed data are available via figshare (https://figshare.com/projects/Human_Liver_SC_vs_SN_paper/98981) and Dropbox (https://www.dropbox.com/sh/sso15ehqmrrh6mk/AACKHOsSlZW0_Zy9cbCkOmMfa?dl=0). Code used in the analysis is available on github: https://github.com/tallulandrews/Liver_sc_sn_paper_scripts. Spatial transcriptomics data visualization tool is available at https://macparlandlab.shinyapps.io/healthylivermapspatialgui/
References
Author names in bold designate shared co‐first authorship.
- 1. Segal JM, Kent D, Wesche DJ, Ng SS, Serra M, Oulès B, et al. Single cell analysis of human foetal liver captures the transcriptional profile of hepatobiliary hybrid progenitors. Nat Commun 2019;10:3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacParland SA, Liu JC, Ma X‐Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramachandran P, Dobie R, Wilson‐Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature 2019;575:512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Massalha H, Bahar Halpern K, Abu‐Gazala S, Jana T, Massasa EE, Moor AE, et al. A single cell atlas of the human liver tumor microenvironment. Mol Syst Biol 2020;16:e9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma A, Seow JJW, Dutertre C‐A, Pai R, Blériot C, Mishra A, et al. Onco‐fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 2020;183:377‐394.e21. [DOI] [PubMed] [Google Scholar]
- 7. Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019;16:269‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med 2016;375:1161‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single‐cell and single‐nucleus RNA‐Seq toolbox for fresh and frozen human tumors. Nat Med 2020;26:792‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single‐cell data with Harmony. Nat Methods 2019;16:1289‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet 2013;29:569‐574. [DOI] [PubMed] [Google Scholar]
- 12. Selewa A, Dohn R, Eckart H, Lozano S, Xie B, Gauchat E, et al. Systematic comparison of high‐throughput single‐cell and single‐nucleus transcriptomes during cardiomyocyte differentiation. Sci Rep 2020;10:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single‐nucleus over single‐cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 2019;30:23‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei Y, Wang YG, Jia Y, Li L, Yoon J, Zhang S, et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 2021;371:eabb1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halpern KB, Shenhav R, Matcovitch‐Natan O, Tóth B, Lemze D, Golan M, et al. Single‐cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017;542:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson‐Maduro J, Baron CS, et al. Single‐cell sequencing reveals dissociation‐induced gene expression in tissue subpopulations. Nat Methods 2017;14:935‐936. [DOI] [PubMed] [Google Scholar]
- 17. Banales J‐M, Prieto J, Medina J‐F. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol 2006;12:3496‐3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: cell lineage and pseudotime inference for single‐cell transcriptomics. BMC Genom 2018;19:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue‐based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 20. Tabibian JH, Masyuk AI, Masyuk TV, O’Hara SP, LaRusso NF. Physiology of cholangiocytes. Compr Physiol 2013;3:541‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai J, Weiss ML, Rao MS. In search of “stemness.” Exp Hematol 2004;32:585‐598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonzo JA, Ferry CH, Matsubara T, Kim J‐H, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J Biol Chem 2012;287:7345‐7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kiselev VY, Yiu A, Hemberg M. scmap: projection of single‐cell RNA‐seq data across data sets. Nat Methods 2018;15:359‐362. [DOI] [PubMed] [Google Scholar]
- 24. Iwaisako K, Jiang C, Zhang M, Cong M, Moore‐Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA 2014;111:E3297‐E3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori‐Bachash M, Hornstein S, et al. Acute liver failure is regulated by MYC‐ and microbiome‐dependent programs. Nat Med 2020;26:1899‐1911. [DOI] [PubMed] [Google Scholar]
- 26. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 27. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275:2247‐2250. [DOI] [PubMed] [Google Scholar]
- 28. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobie R, Wilson‐Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, et al. Single‐cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep 2019;29:1832‐1847.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiskirchen R, Tacke F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg Nutr 2014;3:344‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwiecinski M, Noetel A, Elfimova N, Trebicka J, Schievenbusch S, Strack I, et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA‐29 induction. PLoS One 2011;6:e24568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feldstein AE, Papouchado BG, Angulo P, Sanderson S, Adams L, Gores GJ. Hepatic stellate cells and fibrosis progression in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2005;3:384‐389. [DOI] [PubMed] [Google Scholar]
- 33. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 2015;33:495‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frey BJ, Dueck D. Clustering by passing messages between data points. Science 2007;315:972‐976. [DOI] [PubMed] [Google Scholar]
- 35. Meilă M. Comparing clusterings by the variation of information. In: Schölkopf B, Warmuth MK, eds. Learning Theory and Kernel Machines. Berlin, Heidelberg: Springer, Berlin Heidelberg; 2003:173‐187. [Google Scholar]
- 36. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res 2021;49:D1541‐D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakao A, Yoshihama M, Kenmochi N. RPG: the Ribosomal Protein Gene database. Nucleic Acids Res 2004;32:D168‐D170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, Buettner F. destiny: diffusion maps for large‐scale single‐cell data in R. Bioinformatics 2016;32:1241‐1243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S6
Table S11
Table S12
Data Availability Statement
Raw data and raw count matrices, and final annotations are available in GEO under accession: GSE185477; count‐matrices and fully processed data are available via figshare (https://figshare.com/projects/Human_Liver_SC_vs_SN_paper/98981) and Dropbox (https://www.dropbox.com/sh/sso15ehqmrrh6mk/AACKHOsSlZW0_Zy9cbCkOmMfa?dl=0). Code used in the analysis is available on github: https://github.com/tallulandrews/Liver_sc_sn_paper_scripts. Spatial transcriptomics data visualization tool is available at https://macparlandlab.shinyapps.io/healthylivermapspatialgui/