

Abstract
The development and application of trimetallic nanoparticles continues to accelerate rapidly as a result of advances in materials design, synthetic control, and reaction characterization. Following the technological successes of multicomponent materials in automotive exhausts and photovoltaics, synergistic effects are now accessible through the careful preparation of multielement particles, presenting exciting opportunities in the field of catalysis. In this review, we explore the methods currently used in the design, synthesis, analysis, and application of trimetallic nanoparticles across both the experimental and computational realms and provide a critical perspective on the emergent field of trimetallic nanocatalysts. Trimetallic nanoparticles are typically supported on high-surface-area metal oxides for catalytic applications, synthesized via preparative conditions that are comparable to those applied for mono- and bimetallic nanoparticles. However, controlled elemental segregation and subsequent characterization remain challenging because of the heterogeneous nature of the systems. The multielement composition exhibits beneficial synergy for important oxidation, dehydrogenation, and hydrogenation reactions; in some cases, this is realized through higher selectivity, while activity improvements are also observed. However, challenges related to identifying and harnessing influential characteristics for maximum productivity remain. Computation provides support for the experimental endeavors, for example in electrocatalysis, and a clear need is identified for the marriage of simulation, with respect to both combinatorial element screening and optimal reaction design, to experiment in order to maximize productivity from this nascent field. Clear challenges remain with respect to identifying, making, and applying trimetallic catalysts efficiently, but the foundations are now visible, and the outlook is strong for this exciting chemical field.
1. Introduction
Nanoparticles (NPs) have been utilized for millennia, as evidenced by their discovery in cave paintings1 their presence in the Lycurgus cup from the fourth century2 and in glassware dating back to the late Bronze Age.3 A notable modern focus in nanotechnology emerged in the late 20th century,2 where the application of NPs has broadened; considerable research has also been performed since this period in the development of nanotubes, -wires, -fibers, porous materials, quantum dots, and dendrimers.4−11 Because of the large surface-to-volume ratio of NPs compared with bulk materials, NPs are attractive, but not limited to, the catalysis industry. NPs are also notable across varied industries such as transport, food, textiles, and personal care products. For example, the catalytic converters present in the exhaust systems of motor vehicles are a widely known application of multimetal NPs. Comprising Pd/Pt, Rh, CeO2, and Al2O3, catalytic converters are crucial in reducing and controlling the release of toxic gases and pollutants.12 Self-cleaning surfaces based on lanthanum doped titania13 and indium-doped tin oxide for photovoltaic thin films in solar panels14 are other applications of multicomponent materials.
Specific focus on trimetallic nanoparticles (TMNPs) in catalysis is an emerging area, one developed from the benefits observed upon addition of a second metal to NP catalysts, such as Pd to Au NPs supported on a high-surface-area metal oxide.15,16 In depth studies of bimetallic catalysts have been explored; therefore, the fundamental understanding of these systems can be utilized in the design, synthesis, characterization, and application of trimetallic catalysts. Bimetallic catalysts are well-known to exhibit different properties from their monometallic counterparts. Synergistic effects between the two metals are often responsible for the increased activity, selectivity, and stability. Ferrando et al. discussed several crucial considerations when mixing two metals,17 which include (i) the relative bond strength between two metals, (ii) the surface energy, (iii) relative atomic sizes, (iv) geometric effects, and (v) electronic effects. Sankar et al. presented a thorough and critical review of bimetallic catalysts, providing an overview of the recent developments in material design, synthesis methods, characterization, and catalytic applications.18
Differences in electronic and geometric properties are among the most common explanations why bimetallic catalysts often show increased catalytic performances. Xie et al. demonstrated the positive influences of adding a second metal to Ni.19 They reported an electronic effect that in this system weakened the bonding of CO, suppressing methanation for aqueous-phase reforming of ethylene glycol. Additionally, the formation of a SnNi bimetallic alloy was shown through X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) and was suggested as a center for H2O activation where surface hydroxyls are formed on Sn, facilitating higher H2 selectivity.19 Ferretti et al. used XPS to suggest a partial electron transfer when a binding energy shift for the Rh 3d5/2 peak was observed upon Sn addition relative to monometallic Rh. The successful application of a SnRh/SiO2 catalyst to the hydrogenation of crotonaldehyde was reported.20 The addition of a third metal adds to the complexity of the system, but key principles learned and understood from bimetallic systems can be directly applied to aid the synthesis of trimetallic catalysts. In particular, advanced characterization techniques are relevant to both bi- and trimetallic systems, such as high-resolution electron microscopy, including high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and X-ray adsorption techniques such as X-ray absorption fine structure (XAFS). Significantly, the distinction of the different elements present in a solid solution, core@shell, or segregated mixed system should be explored when possible to provide a better understanding of the complex reaction site. For example, distinguishing the concentration of elements present on the surface of a bimetallic supported catalyst was reported by Rioux and Vannice through chemisorption studies.21 Chemisorption of CO, N2O, and H2 was used to probe the surface concentration of carbon-supported Pt–Cu catalysts in comparison with their bulk compositional values and to differentiate observations in the rate of isopropyl alcohol dehydrogenation. Zhu et al.(22) applied a multitude of characterization techniques, including XPS, high-resolution transmission electron microscopy (HRTEM), and high-sensitivity low-energy ion scattering (LEIS) spectroscopy to differentiate between structural variations. LEIS spectroscopy was employed to investigate the surface atomic composition of the catalysts, including bimetallic NiCo and RuNiCo TMNPs, upon exposure to different heat treatments. Tarditi and Cornaglia23 also utilized LEIS spectroscopy to investigate surface segregation of novel PdAgCu ternary alloys. It is known that the chemical and physical properties of the bulk composition vary from those of metallic alloy surfaces. Such techniques are encouraged to determine the role of metal alloys at the surface as well as their bulk compositions. Realizing the full benefits of using trimetallic compositions as catalysts requires advanced characterization concurrent with building on the successful application of these complex materials.
Over 130 articles related to TMNP catalysts were published in 2020 (Web of Science; search “trimetallic catalyst”), with an upward trend in publications over the past decade (Figure 1) that is far greater than the general growth in scientific publications.24 Consequently, TMNPs are considered to be a key component in the advancement of catalyst-based processes. However, fundamental studies on TMNP catalysts remain elusive, and much work will be required to truly advance this field. Despite this, TMNPs have been successfully applied as catalysts to alcohol oxidation25,26 and dehydrogenation27,28 reactions, where increased catalytic activity over mono- and bimetallic counterparts has been reported. Therefore, TMNPs can be considered highly promising, as the addition of a third metal often synergistically enhances the catalytic performance. However, the catalytic activity is highly dependent on the composition, morphology, and dimensions, and thus, it is necessary to apply multiple characterization techniques to understand the nature of TMNPs. In this respect, computational approaches have become a vital component in the development of TMNP catalysts, particularly in defining the expected stability of a given composition.29−31 As the growth of reported bi- and trimetallic materials has increased, so too has the number of terms used to describe these systems, principally because of their complexity. Terms such as solid solution, core–shell, intermetallic solution, ordered alloy, and segregated systems are abundant and often used in relation to the intended composition or when suitable characterization techniques can resolve the elemental composition and structure of a nanoparticle. To further complicate matters, this structure may not be applicable to all nanoparticles, so a larger pool of candidates must be considered. We have attempted to summarize the variety of compositional descriptors in Figure 2, and these terms are then used throughout the review and should be referenced when distinguishing the systems commonly reported. These include core@shell-types, such as M1@M2@M3/support, and solid solutions or alloys, such as M1M2M3/support, where M represents a metal. Furthermore, where appropriate, the nature of the nanoparticle structure can be described through the use of a descriptor to notate order, e.g., o-M1M2M3. Where this is not used, a random structure can be assumed, e.g., M1M2M3. Defining these clearly and consistently across the variety of the fields of which TMNPs are part should ideally be standardized to avoid confusion.
Figure 1.
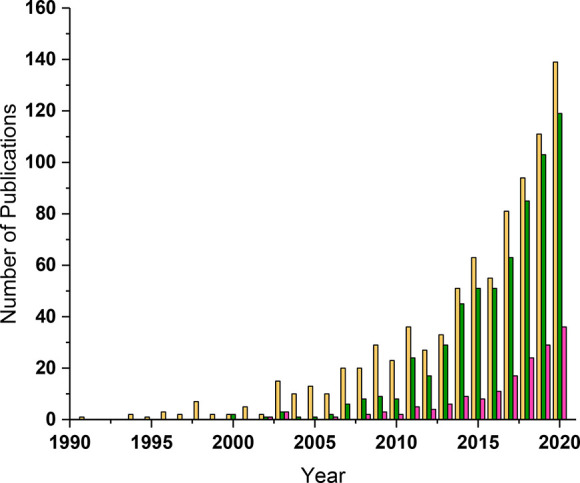
Number of publications based on a Web of Science (February 2021) search for “trimetallic catalyst” (tan), “trimetallic nanoparticles” (green), and “trimetallic nanoparticles” with “applications” (pink) and limited from 1990 to 2020.
Figure 2.

Diagrammatical representations of different compositional descriptors used in multicomponent nanomaterials (left to right) noting ordered structures (o-) where appropriate: bimetallic core@shell (M1@M2), trimetallic inner-core@core@shell (M1@M2@M3), core@random-shell (M1@M2M3), random alloy (M1M2), ordered-core@shell (o-M1M2@M3), core@ordered-shell (M1@o-M2M3), ordered alloy (o-M1M2), random-core@shell (M1M2@M3), core@shell where M2 is distributed across the particle (M1M2@M2M3, often described as an AB@AC structure), ordered alloy with M3 randomly distributed (o-M1M2-M3), random trimetallic solid solution (M1M2M3), and ordered trimetallic alloy or intermetallic solution (o-M1M2M3). Where a support is used, the nomenclature will be given as M1M2M3/support (e.g., PtPdAu/TiO2).
Understanding the operation and benefit of TMNPs as catalytic active sites is challenging because of the additional degree of freedom of ternary metal nanoparticles. Synergy is often broadly used to describe the benefit of additional metals; however, this term is not as clear as it could be in terms of molecular transformations through electron transfer on supported nanoparticle surfaces. Bond breaking and making are crucial processes in catalysis, and the d-band model has been an impactful development to describe how the electronic properties of a material influence bond formation at the surface. In the d-band model, the strength of a surface–adsorbate interaction is determined by the degree of electronic occupation in the hybridized d−σ antibonding states that form between the surface d and adsorbate σ orbitals.32 The degree of occupation in the surface d states is element- and/or composition-dependent, and higher occupancy leads to greater occupancy of the surface–adsorbate d−σ antibonding states, which results in weaker surface–adsorbate interactions. Thus, the center of the d states (or the d-band center for short), which signifies the occupancy of the surface d states, has become synonymous with the strength of any surface–adsorbate interactions. In the case of TMNPs, such considerations are highly important because of the potential tunability of the position of the d-band center by compositional variation.33
This critical review attempts to provide insight and understanding of TMNP material descriptions, preparation methods, and catalytic activities, particularly emphasizing the positive influence that TMNPs are envisaged to have on the future of catalyst-based industries.
2. Properties of Trimetallic Nanoparticles
When nanoparticles are being synthesized for catalytic applications, there are several factors to consider, including size, shape, morphology, structure, composition, phases, supports, and defects. To date, a plethora of studies involving mono- and bimetallic nanoparticles (BNMPs) have taken these factors into account in the careful design of systems for catalytic applications.18,34−37 For trimetallic nanoparticles, the same fundamental principles can be applied. These considerations become even more complex because of the increased degrees of freedom; however, the greater flexibility afforded with respect to tuning these properties can greatly improve catalytic performance. Where possible, we have attempted to relate the various catalytic processes discussed according to the rate of product formation or turnover frequency (TOF) to better compare catalyst compositions. This is particularly key when attempting to gauge the efficacy of a TMNP catalyst compared to their mono- or bimetallic counterparts. Often this is a challenge because of the lack of complementary specific activity data given in the literature. In this case we have used the available data such as catalyst mass or volume, supported metal loading (mass or moles), flow rate, or gas hourly space velocity and calculated the activity as moles of product per unit catalyst mass per hour or as TOF (moles of product per unit time as a function of moles of metal present on the catalyst). Ideally, productivity values should be calculated at moderate conversion percentages on the basis of active site density, as the productivity is insensitive when taken at high conversions. However, the values should offer the reader a way to initially assess the influence of metal composition. Therefore, the productivity values are presented in tables in the following sections for comparison where appropriate. We submit that through these metrics we can compare catalysts for a given process, but care must be taken because it is rare to possess activity rate data for a range of compositions of BMNPs and TMNPs within one study. Therefore, we take this opportunity to make the point that activity rate data should be provided routinely, normalized to metal loading as a function of time, to better understand the improvements that are often claimed for TMNP catalysts.
2.1. Key Properties of TMNPs to Consider
The composition of a metal nanoparticle is an important consideration that includes the identities of the species present in the nanoparticle in addition to the ratio of these species. In a trimetallic nanoparticle, the metal ratio A:B:C can have drastic implications for the morphology, structure, and performance of a catalyst, which have been explored through both synthetic and computational studies. The key properties of composition and morphology will be introduced here.
As an example, Figure 3 illustrates the importance of composition, specifically the ratio of the three metals on the resultant catalytic activity.16 Hutchings and co-workers developed a contour plot of the rate of hydrogen peroxide productivity as a function of the Au:Pd:Pt metal ratio. The most active catalysts were those with a small amount of Pt relative to an Au:Pd weight ratio of 1:1 (Au:Pd:Pt = 2.4:2.4:0.2). Where the Pd or Pt concentration was higher, the hydrogenation rate was significantly increased, reducing the overall peroxide yield. To generate this plot, many experiments were carried out with various compositional ratios, and the results show that a small change in composition can have a significant impact on the overall reaction rate. Presently, this complexity is a key issue in the preparation and application of TMNPs.
Figure 3.

Contour diagram of catalytic activity of AuPdPt/CeO2 catalysts, showing how the rate of H2O2 production (molH2O2 kgcat–1 h–1) depends on the Au:Pd:Pt ratio. Adapted with permission from ref (16). Copyright 2014 Wiley-VCH.
The influence of the metal nanoparticle (MNP) oxidation state on the catalytic performance can be significant. Metals within a nanoparticle system are not necessarily of neutral charge, which may impact the outcome of reactions because of the altered electronic interactions between the metal and the substrates. For example, different oxidation states change the redox properties of the nanoparticle, which in turn may enhance or suppress its performance.38 To illustrate this phenomenon, we use a monometallic example from the work of Hutchings et al.(39) on the mechanism of acetylene hydrochlorination to vinyl chloride over single-site Au/C catalysts. These catalytic sites were investigated using in situ XAFS and XPS. According to analysis of these measurements, the active form of gold under the reaction conditions was not Au0 but rather Au cation sites, specifically, Au1+ or Au3+. The results indicated the catalytic activity was strongly correlated with the Au1+:Au3+ ratio, with Au1+ showing high catalytic activity and the Au0 and Au3+ sites exhibiting little catalytic activity. Interestingly, the catalyst shows a 1 h induction period of modest activity, suggesting that the active Au1+ form of the catalyst is formed under the reaction conditions. In a multimetallic system, the oxidation state of just one of the three metals may impact the catalytic performance.40,41 For example, Sial et al. noted that in their PtCoFe catalysts, a high ratio of Pt0 to oxidized Pt2+ vastly improved the electrocatalytic performance for ethanol oxidation and oxygen reduction.42 Analysis of XPS data revealed that the other metals were present as Co2+ and Fe0, and in addition to the strong structural character of this composition, these factors facilitated the enhanced catalytic activity observed.
Morphology is another crucial factor to consider when assessing catalytic performance. Many examples of size- and shape-dependent catalytic performances have been reported, including recent literature reviews that describe the influences of size, shape, and support on CO oxidation reactions.43−46 In the case of TMNPs, the addition of a third metal can be more complicated but can result in greater flexibility and control. Metal NP preparation methods influence the different shape formations and the various ways atoms can pack together.6,47 Darby et al. explored this concept through computational modeling of bimetallic Au clusters in which one Au atom was replaced by a Cu atom.48 Their calculations indicated that replacement of one Au atom could lead to significant structural changes for clusters of 14, 16, and 55 atoms compared with the monometallic counterparts. Figure 4 illustrates various model particle shapes such as (a) cubic, (b) tetrahedral, (c) octahedral, (d) truncated octahedral, and (e) spherical.43
Figure 4.
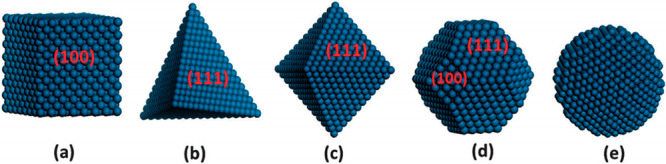
Examples of metal NPs with different morphologies: (a) cube with (100) facets, (b) tetrahedron with (111) facets, (c) octahedron with (111) facets, (d) cuboctahedron with (100) and (111) facets, and (e) a spherical shape. Adapted with permission from ref (43). Copyright 2016 Royal Society of Chemistry.
Miller indices (hkl) are often used to categorize surfaces of NPs, and for a metal catalyst with a face-centered-cubic (FCC) lattice, the (100), (111), and (110) planes are the main surfaces of a shape-controlled NP.43 The different surfaces, with different coordination numbers (CNs), influence the adsorption, dissociation, coupling, and interaction of intermediate adsorbates in a catalytic event. Selectivity is also sensitive to the arrangement of atoms and exposed facets on the surface.49 For example, the difference between the adsorption energies of the Pt(111) and Pt(100) surfaces can be traced to their intrinsic structural differences and the difference in binding energies, with density functional theory (DFT) studies showing that the binding energy of NHx species on the Pt(100) surface is greater than that on Pt(111) surface by ca. 0.7 eV.50 This difference between the adsorption energies of adsorbates on the two surfaces probably results from the difference in coordination numbers of Pt atoms on the Pt(111) and Pt(100) facets. A shape-dependent catalytic reaction for the hydrogenation of benzene over Pt NPs was reported by Somorjai et al., who synthesized cubic and cuboctahedral NPs shapes using tetradecyltrimethylammonium bromide as a capping agent.51 The cubic particles consisted of Pt(100) facets (ca. 12.3 nm diameter), whereas the cuboctahedral particles contained both Pt(100) and Pt(111) facets (ca. 13.5 nm diameter). The hydrogenation of benzene over Pt NPs was reported to be highly dependent on the shape; it was concluded that the Pt(111) surface of the cuboctahedral NPs resulted in cyclohexene production, as both possible products (cyclohexane and cyclohexene) formed on the cuboctahedral nanoparticles whereas only cyclohexane was produced on the cubic nanoparticles. On Pt(111), the weak adsorption energy of cyclohexene makes desorption of cyclohexene facile, whereas the selectivity for cyclohexane on Pt(100) was considered to be a result of the high adsorption energy and the relatively strong binding of cyclohexene, encouraging further hydrogenation. Hence, the cuboctahedral Pt nanoparticle catalysts, containing both (111) and (100) exposed facets, resulted in both cyclohexene and cyclohexane formation.
Wang et al. demonstrated a shape-controlled synthesis of trimetallic PtPdCu nanocrystals applied as electrocatalysts for the methanol oxidation reaction (MOR).47 The different surfactants used in the coreduction synthesis, cetyltrimethylammonium bromide (CTAB) and cetyltrimethylammonium chloride (CTAC), resulted in the unique formation of nanocubes and nanodendrites, respectively. According to Xu et al., the stabilization of the Pd(100) facets was due to binding of the Br– ions from CTAB, promoting the formation of Pd nanocubes. When CTAB was replaced with CTAC, the chloride ions did not efficiently stabilize and promote the formation of (100) surfaces of Pd.52 Similar reasoning was considered to account for the different nanocrystal formations reported by Wang et al.(47) The materials characterized by TEM displayed the sizes and morphologies of the PtPdCu porous nanocubes and nanodendrites. The lattice fringe measurements shown in HRTEM images can be assigned to the (200) crystal planes of PtPdCu alloy in the nanocubes (Figure 5b) and the (111) and (200) crystal planes of PtPdCu alloy in the nanodendrites (Figure 5d). Alloy formation was also observed in a shift in the XRD pattern and a positive binding energy shift in the Pd 3d5/2 peak in the XPS data, providing evidence of the PtPdCu alloy compared with monometallic Pd. HAADF-STEM images with elemental mapping also confirmed alloy formation but additionally indicated a Pt-rich surface in the nanocubes. The PtPdCu nanocrystals exhibited higher electrochemical activity for MOR in comparison with a commercial Pt/C catalyst. The mass activity and specific activity of the catalysts were reported to be in the order of PtPdCu nanodendrites > PtPdCu porous nanocubes > commercial Pt/C. This trend could be related to the higher electrochemically active surface area (ECSA) of the PtPdCu nanodendrites (75 m2 gPt–1) in comparison with the PtPdCu nanocubes (50 m2 gPt–1) and commercial Pt/C catalyst (34 m2 gPt–1). The PtPdCu nanocrystals have a higher ECSA because of their porous morphology compared with the monometallic Pt/C catalyst, resulting in more active sites. Therefore, the PtPdCu nanocrystals exhibit higher activity due to the combination of the porous morphology and the synergistic effects from adding Pd and Cu atoms. The addition of Cu atoms may modify the electronic state of Pt atoms, resulting in a weaker Pt–CO bond during MOR, decreasing catalytic poisoning by CO.47
Figure 5.

(a) TEM image of PtPdCu porous nanocubes. (b) HRTEM image of PtPdCu porous nanocubes. (c) TEM image of PtPdCu porous nanodendrites. (d) HRTEM image of PtPdCu porous nanodendrites. Adapted from ref (47). Copyright 2019 American Chemical Society.
In the field of nanoparticle catalysis, surface defect sites have long been known to exhibit enhanced catalytic performance. Such surface defects include atomic steps, kinks, and edges.53 These defect sites contain a high concentration of low-coordinate atoms, which may facilitate catalysis of certain processes. For example, the cleavage of N2 readily occurs on coordination-unsaturated Ru sites.54 The enhancement of these catalytic sites in bond-breaking steps can be attributed to local electronic or geometric effects.55,56 Because of the localized change in electronic and geometric structure, defect sites are more susceptible to act as nucleation sites, allowing for site-selective addition of the third metal around the sites.
Li et al.(26) reported a defect-dominated shape recovery phenomenon of nanocrystals. Fine-tuned geometric and electronic structures were rationally designed by means of a step-induced growth mechanism. Two key requirements were reported for the synthesis of new trimetallic o-M1M2@M3 structures by shape recovery: the metals must have the same crystal structure and similar atomic radii. This preparative concept involves chemical etching of an Pt3Ni octahedral crystal to form a defect-rich concave structure.26 Other defect-rich trimetallic structures obtained using etching methods have been reported. These include nanoframes, which are hollow and retain only the atoms on the edges of the facets. Nanoframes possess a high surface area and many low-coordinate defects that are known to be highly catalytically active. This structural motif along with the trimetallic composition has been shown to further improve catalytic performance.57−62
Perhaps one of the most compelling properties of multimetallic NPs is the many structures they can manifest. TMNPs with varied morphologies, such as wires,63−67 dumbbells,68 hollow interiors,5,69−71 nanotubes,72,73 and dendrimers,74−76 have been reported, exemplifying the flexible outcomes available through variation of the synthesis conditions and metal choice. Interestingly, the different metals within NPs can be distributed in many ways on and within the nanoparticle. Therefore, with respect to TMNPs, many structures can be investigated, such as random alloys and core–shell structures, similar to those seen in bimetallic systems.
Alloyed TMNPs can exhibit an essentially random distribution of the three metals within the nanoparticle, with no clear structural segregation present. For example, Wang et al. prepared NiAuPd/C samples in which the metal nanoparticles were composed of random alloys, and the catalysts were applied to the dehydrogenation of formic acid.77 The catalysts were prepared by simultaneous reduction of all three metal precursors in aqueous solution in the presence of an active carbon support suspended within the solution. The Ni:Au:Pd molar ratio was 40:15:45. HRTEM images illustrated that the diameter of the nanoparticles was in the range of 16–35 nm (Figure 6). Furthermore, images from energy-dispersive X-ray (EDX) analysis (Figure 6) confirmed the randomly distributed and homogeneous structure of the three metals within the pseudospherical nanoparticles and the lack of a core@shell structure. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements confirmed that the overall metal loading was 9.91% and the Ni:Au:Pd ratio was 36:18:46.
Figure 6.
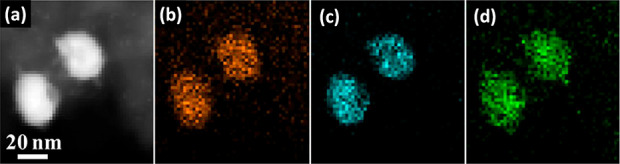
(a) HAADF-STEM image of Ni40Au15Pd45/C and (b–d) the corresponding EDX elemental maps of (b) Ni, (c) Au, and (d) Pd. Adapted with permission from ref (77). Copyright 2014 Elsevier.
Using a different design approach, Fu et al.(78) prepared unsupported, randomly alloyed PtAuCo TMNPs as catalysts for the dehydrogenation of ammonia borane. First, Au@Co core@shell bimetallic nanoparticles were prepared, and these were added to a solution of Pt precursor, digested with 1,2-hexadecanediol, and heated to 493 K. The approach led to TMNPs with a mean diameter of 4–5 nm in addition to smaller 1–2 nm particles, which are considerably smaller than most other reported TMNPs. The synthetic Pt:Au:Co molar ratio was 6.3:1:1. Analysis of EDX spectra confirmed that the metals were randomly distributed throughout the spherical particles. The design strategy is illustrated in Figure 7, along with electron microscopy images and a selected-area electron diffraction (SAED) pattern of such alloy catalysts.
Figure 7.

(left) Synthesis scheme to produce PtAuCo alloy catalysts. (right) Resultant morphology and structure of the unsupported Pt76Au12Co12 alloy TMNPs from electron microscopy analysis: (a, b) TEM images taken at different magnifications; (b1–b4) HRTEM images recorded from the different regions in (b); (c) selected-area electron diffraction (SAED) pattern. Adapted from ref (78). Copyright 2020 American Chemical Society.
Because of the additional degree of freedom when three metals are utilized, more variation is expected in the NP structures. Core@shell materials as M1@M2M3 were prepared by Kang et al. as shown in Figure 2).79Via a one-pot synthesis method, trimetallic Au@PdPt core–shell NPs were successfully synthesized with fine control over the nucleation and growth kinetics through manipulation of the reducing agents.79 In this example, an octahedral Au core and a highly crystalline PdPt alloy shell were prepared, which exhibited excellent catalytic activity and stability for the electrooxidation of methanol in acidic media. The enhanced catalytic activity was attributed to their optimized binding affinity for adsorbates due to the improved charge transfer between the core and the shell of the NPs. Computational modeling has been used extensively to understand how the geometric and chemical structure vary with AuPdPt trimetallic composition. Zhang et al.(80) observed charge transfer from Pd atoms to Au and Pt in Au-rich compositions, leaving the latter negatively charged; the Au present is therefore postulated to be able to activate molecular oxygen by electron donation.
There has been further extensive computational work using global optimization to investigate how the composition and nuclearity affect the chemical distribution and geometric structure for AuPdPt across a range of specific compositions and nuclearities, using both molecular mechanics and electronic structure simulation. There is a general consensus that Pt favors segregation to the interior of the nanoparticles while Au is strongly segregated to the surface for global optimizations under vacuum conditions, which is in line with thermodynamic expectations considering elemental cohesive and surface energies.81−84 In dynamical simulations for AuPdPt NPs encapsulated in carbon nanotubes (CNTs), Wei et al. showed that multishell structures are formed with Au accumulating near the core and Pt distributed near the nanotube wall, which highlights how thermodynamic stability can be influenced by the environment or support (as discussed in section 2.2).85
In several cases, AuPdPt and AgPdPt have been compared during simulation work, as both Au and Ag are in group 11. Wu et al. identified a preference for decahedral structures in both compositions but a greater preference for icosahedral structures for AuPdPt than for AgPdPt at a nuclearity of 75 atoms. With respect to composition, Ag was observed to alloy more than Au. Au prefers homogeneous bonding (i.e., to other Au atoms),86 which was further confirmed by Du et al. for nuclearities up to 147 atoms.87
Further work on AgPdPt has confirmed observations that Pt tends to occupy a position at the core of these trimetallic NPs, with Ag at the surface (similar to Au).88 Khanal et al. produced an interesting study looking at the growth of AgPdPt nanoparticles using step-based synthesis, which is often overlooked in the search for thermodynamic global minima.89 Monte Carlo simulations showed that Pt grown on AgPd nanoalloys forms three-dimensional (3D) islands at the beginning of the deposition process (Figure 8). Akbarzadeh et al. also looked at the AgPd@Pt configuration with molecular dynamics simulations and identified a higher melting temperature when phase separation is realized in this core@shell manner. When the Pd concentration was increased and the Ag concentration reduced, the melting temperature rose, which is in line with elemental properties but also associated with reduced strain on the system when Pd atoms are at the core of the NP.90
Figure 8.
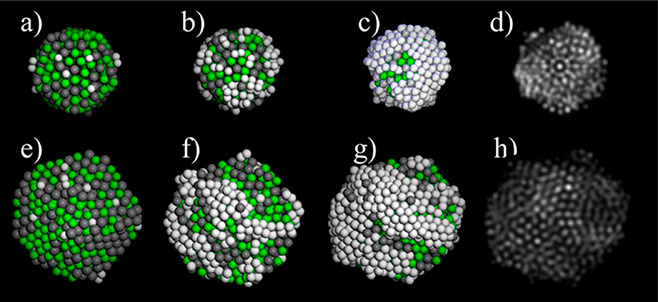
Monte Carlo simulations showed that Pt grown on AgPd nanoalloys forms 3D islands at the beginning of the deposition process. Ag0.5Pd0.5 icosahedra (top row) and decahedra (bottom row): (a, e) initial stage of Pt (light-gray spheres) nucleation on Ag0.5Pd0.5; formation of Pt deposits on AgPd at (b, f) Θ = 0.5 monolayer (ML) and (c, g) Θ = 1 ML; (d, h) STEM simulations of configurations at Θ = 1 ML. Adapted with permission from ref (89). Copyright 2013 Royal Society of Chemistry.
Considering further such layered NP structures, Au@Co@Fe triple-layer core@shell NPs (Au:Co:Fe molar ratio = 6:6:88) were synthesized via a one-step preparation method using poly(vinylpyrrolidinone) (PVP) as a stabilizing agent.91 As the reducing agent was exposed to the metal precursors at the same time, the resultant triple-layer core@shell structure is based on the reduction potentials of the metal cations as a measure of their ability to undergo reduction (EFe(III)/Fe(II)° = +0.77 V vs the standard hydrogen electrode (SHE); EFe(II)/Fe = −0.44 V vs SHE; ECo(II)/Co° = −0.28 V vs SHE; EAu(III)/Au = +0.93 V vs SHE). The Au core can be formed relatively quickly because of its higher reduction potential and can serve as an in situ seed for the catalytic reduction leading to the formation of the outer layers. This is an example of a successive reduction process for the formation of triple-layer core@shell trimetallic catalysts, as illustrated by HAADF-STEM imaging and electron energy loss spectroscopy (EELS) elemental maps revealing a Au core, a Co-rich interlayer, and an Fe-rich shell (Figure 9).
Figure 9.
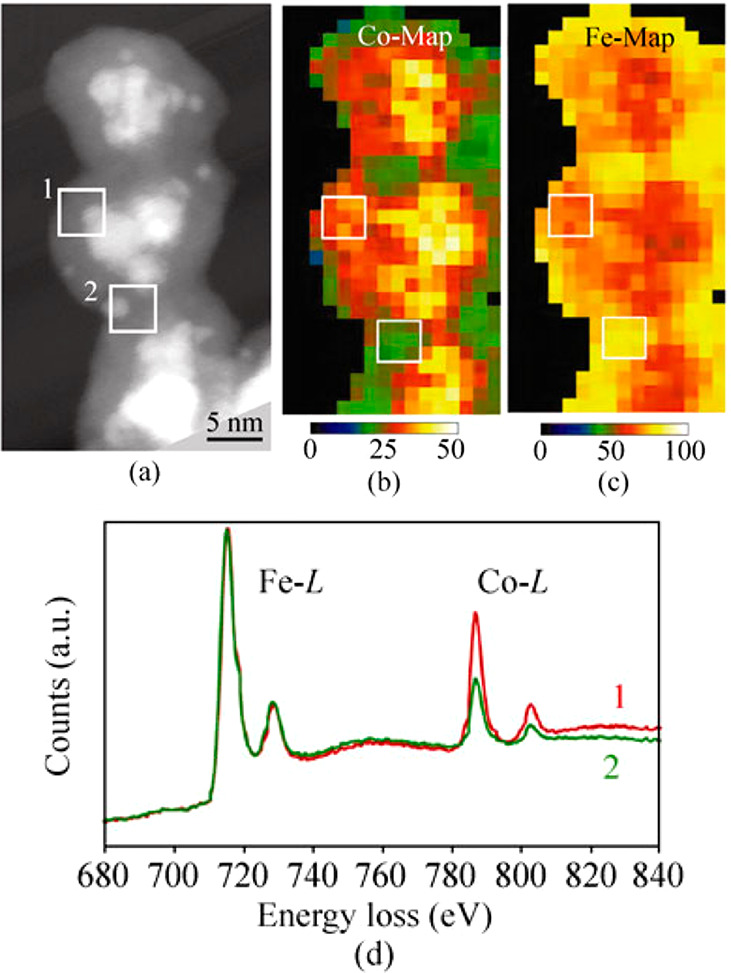
(a) HAADF-STEM image of AuCoFe triple-layer core@shell NPs, (b, c) EELS elemental maps for (b) Co and (c) Fe, and (d) EELS spectra at the areas indicated in (a). Adapted with permission from ref (91). Copyright 2011 Tsinghua University Press and Springer.
Combinations of group 11 elements are popular in TMNPs because of the aspiration to replace expensive Au with cheaper Ag and Cu without significantly changing the valence electronic structure, and these have been extensively investigated using computational methods. For example, utilizing Ag, Au, and Cu, Yildirim and Arslan92 showed that Ag and Au segregate to the surface of 38- and 55-atom NPs while Cu segregates to the core, in agreement with the higher surface energy of Cu. Segregation varies slightly for smaller systems (N < 10), where Cu and Ag take higher-coordination sites and Au the lower-coordination exposed sites.93 The preference of Au for surface sites has also been observed for CuAuPt alloys with nuclearities up to 147 atoms, with Pt and Cu preferring positions in the core; this observation is driven by the stronger cohesion energies and high surface energies of Pt and Cu.94,95 Taran considered how composition affects the melting of 55-atom Cu–Au–Pt trimetallic nanoalloys using molecular dynamics; Au was observed to be more mobile than Cu and Pt, which would favor Au diffusion to the surface of the aforementioned larger particles.96 For larger models that considered the specific structure of CuPt alloys on a Au core with molecular dynamics (MD) simulations, increasing concentrations of Pt led to greater stability, which is due to the greater cohesive energy of this species.97,98 Mattei et al.(99) considered AuPtPd systems, comparing MD and Metropolis Monte Carlo (MMC) simulations with laboratory experiments; the experiments showed Pd-rich shells and Au/Pt cores, and this observation was validated by modeling of the growth dynamics, with Pd structures forming quicker than Au/Pt and enveloping these particles during coalescence.
In some cases, subnano clusters have been considered, such as the four-atom AlTiNi clusters modeled by Koh et al.,100 where complex multiscale global optimization was applied. The interest in this system is motivated by the jellium behavior observed for small Al clusters; in this case, the stability of the clusters increases with the number of nickel atoms. Doping of 13-atom NiAg clusters with Cd and Cr was investigated by Datta using DFT calculations.101 The undoped NiAg clusters form a core@shell segregated system with smaller Ni in the core; doping was observed to enhance the mixing of the system, with Cd dopants being more effective than Cr. Taran et al. performed global optimization simulations on PtNi@Ag core@shell structures, which are of interest for electrocatalytic applications,102 and observed that, similar to Datta’s work, Ni atoms occupy core regions close to Pt atoms in the icosahedral structures; however, the core Pt atoms are under high stress, which is reduced by replacement with Ni atoms.
Overall, it is clear that the composition of TMNPs can give rise to morphological diversity and interesting structural features across a variety of sizes, which may then be exploited in catalytic processes. The broad research efforts in both synthetic and computational studies not only speak to this but also highlight the opportunity to develop this field further in novel ways.
2.2. Influence of the Support
Commonly, NP catalysts are synthesized with a support, which can improve their overall performance. Although unsupported colloidal trimetallic catalysts have shown promise,103−106 there are also many advantages of using a support. The advantages include improved dispersion of NPs, enhanced surface area, and increased stability against deactivation processes such as agglomeration.
However, it is important to consider that the support is not necessarily inert and may participate in the reaction. In the 1970s, the term “strong metal–support interaction” (SMSI) was introduced by Tauster to explain the unusual adsorption behavior of H2 over noble metal catalysts supported on TiO2.107,108 The metal–support interaction (MSI) is therefore a crucial consideration, as are the many influential factors on MSI,109 and it is desirable to understand these influences because tuning the MSI may facilitate optimization of catalytic properties through catalyst design.110−114
The number of catalyst supports available is large, and when selecting a support there are a number of factors one must consider, including mechanical and thermal stability, surface area, accessibility of active sites, porosity, and nanostructure. The support may also provide beneficial characteristics toward the reaction, such as defect sites, charge transfer ability, spillover, and additional functionality to the NPs.109 The development of a wide variety of novel supports has resulted in a huge diversification of supported heterogeneous catalysts, and a number of these novel supports have been employed in trimetallic catalysis.115 Graphene is a very promising option with regard to stability and is particularly favorable for electrocatalysis because of its unique charge transfer properties.116 Graphene is commonly in the form of few-layer graphene rather than the single-layer graphene to increase the stability,117 and graphene and graphene oxide (GO) have shown encouraging results as catalyst supports for a diverse selection of reactions.118−123 CNTs have incredible stability due to their nanostructure, which also allows exceptional distribution of NPs and small average particle sizes.28,124−126
There is currently limited information available for direct comparison of supports for trimetallic catalysts because of the recent development of the TMNP domain; however, reports have emerged that illustrate the importance of choosing the appropriate support and method of preparation when working with trimetallic catalysts, such as the use of different carbon support types for CO hydrogenation over alkali-promoted CoRhMo catalysts.112,113 These choices can drastically impact the overall catalytic performance.124,125,127−132 These aspects of TMNP catalyst design will be examined in greater detail in the following sections on catalyst preparation and testing.
3. Preparation of Trimetallic Catalysts
A variety of methods are used to synthesize NP catalysts, including impregnation, sol immobilization, coprecipitation, and microwave-assisted methods. Not only have these techniques been used to synthesize supported monometallic catalysts, but they have also provided stable and active multimetallic catalysts. The addition of a third metal can result in complications over the control of the NP composition but can offer greater tunability. Herein, we shall explore these methods to demonstrate how they can be successfully applied to the synthesis of heterogeneous TMNP catalysts.
3.1. Impregnation Preparations
Impregnation is one of the more traditional methods for the synthesis of heterogeneous catalysts. An excess of solution is used in wet impregnation, where a dissolved metal component is present. Wet impregnation is a relatively simple method to deposit metal NPs onto a support. Activation, through calcination or reduction in a high temperature furnace, is preceded by a drying step. Wet impregnation has been successfully applied to prepare various TMNPs as catalysts.133,134 Examples of supported TMNPs prepared via this technique include NiMoW supported on SBA-15135 and PdNiAg NPs supported on carbon.27 An analogue to this technique is incipient wetness impregnation (IWI), which uses a volume of metal precursors in solution equal to (or just below) the pore volume of the support, followed by a drying step; the catalyst is then typically activated by heat treatment under either a reductive, oxidative, or inert atmosphere as a final step.136 Other various TMNPs have been synthesized by impregnation: for example, a pore-filling coimpregnation method was applied by Nakaya et al. in preparing Pt3(Fe0.75M0.25)/SiO2, where M = Co, Ni, Cu, Zn, Ga, In, Sn, or Pb.137 Supported trimetallic AuPdPt alloys were synthesized by a conventional wet impregnation method by He et al.(133) Enhanced catalytic performances were observed in the solvent-free oxidation of benzyl alcohol over these AuPdPt catalysts, in part as a result of the reduction in selectivity for the byproduct toluene. However, the catalytic activity was much lower than over TMNPs prepared by alternative methods. The lower activity observed was thought to be due to the significant increase in mean particle diameter when such an impregnation technique was used compared with the colloid-derived material.
Generally, impregnation methods are known to produce a greater particle size and particle size distribution. Lopez-Sanchez et al.(138) reported the particle size distribution of supported AuPd metal NPs prepared by both sol immobilization and an impregnation route. The synthesized particles ranged in diameter between 4 and 7 nm in the catalyst prepared by sol immobilization, and a much wider particle diameter distribution of 2–14 nm was observed in the latter route. Bahruji et al. also observed that Pd/ZnO catalysts synthesized by impregnation produced larger diameters of metal NPs after heating in H2 at 673 K (ca. 8.8 nm) in comparison with those prepared by sol immobilization (ca. 4.9 nm).139
3.2. Colloidal Preparations
In an attempt to eliminate the larger particle sizes observed with some impregnation methods (i.e., those over 8 nm in diameter), facile chemical reduction preparation methods are often utilized, with various terminology including, but not limited to, coreduction, sol immobilization, and one-pot synthesis. Although coreduction methods demonstrate greater control of particle size and particle size distribution in multimetallic systems, compositional fluctuations with respect to neighboring particles are often observed, which needs further investigation.133,138 As trimetallic catalysts attract greater interest, colloidal preparation methods have been successfully reported and applied to a range of catalytic reactions such as glycerol oxidation,25 methanol oxidation,118 and hydrolytic dehydrogenation of ammonia borane.91 Colloidal nanoalloys can be prepared by adding an excess of stabilizing agent and reducing agent to the metal precursor(s). These stabilizing agents are often donor ligands, polymers, and surfactants, such as PVP, poly(vinyl alcohol) (PVA), and CTAC, which also control the growth of the initially formed nanoclusters.139−142 Protective agents are often deemed as essential through either electrostatic stabilization or steric stabilization of the colloids, as demonstrated by Nam et al. in the shape-controlled synthesis of intermetallic PdSn nanocrystals.143
Conversely, studies have been conducted to show successful activity and stability under surfactant-free conditions using a facile chemical reduction preparation method. Wang et al. demonstrated the synthesis of a well-dispersed random alloyed trimetallic catalyst composed of NiAuPd NPs deposited on carbon black in the absence of a stabilizing agent.77 Yurderi et al. also synthesized a stabilizer free PdNiAg/C catalyst that showed high activity for the dehydrogenation of formic acid, and maintained activity after the forth reusability test, without a stabilizing agent.27 These studies successfully demonstrated the synthesis of stable TMNPs as catalysts that resist agglomeration under surfactant-free conditions; however, the use of protecting agents is more important for unsupported colloids, as these NPs are unstable with respect to agglomeration.144
Often, a major drawback of the use of TMNPs in catalysis is the complexity that potentially arises from combining three metal precursors, all with different reduction potentials. The multielement composition makes the growth kinetics and structural characteristics complex and difficult to control, which will affect the overall catalytic performance. Kang et al.(79) developed a facile one-pot aqueous synthesis method to obtain AuPdPt TMNPs with a controlled nanostructure by using simultaneous reduction of all three metal precursors in a 1:1:1 molar ratio with separate reducing agents (hydrazine and ascorbic acid) in the presence of CTAC. Their method resulted in an octahedral Au core with a single-crystalline shell of dendritic Pd–Pt alloy.
In order to study the formation mechanism, coreduction of the Au and Pd precursors with CTAC and no additional reagents was performed and yielded core@shell Au@Pd {111}-faceted octahedral nanoparticles with an average edge length of 61.0 ± 1.3 nm.79 When the synthesis was repeated with the addition of the Pt precursor, spherical Au@PdPt nanoparticles were formed with a polycrystalline shell having an average edge length of 43.3 ± 5.1 nm. Thus, in this case the addition of Pt influenced the metal and surfactant interactions. When just hydrazine was added to the mixture of metal precursors and CTAC, polyhedral Au monometallic nanoparticles formed in a variety of shapes, and Pd or Pt were not visible via EDX analysis of the final product; when only ascorbic acid was added, dendritic Pd–Pt nanoparticles were formed. The observations confirmed that both ascorbic acid and hydrazine were essential to produce the well-defined shape of the octahedral core. Further investigations with UV–vis spectroscopy revealed a peak at 285 nm, which was attributed to ligand-to-metal charge transfer (LMCT) between the Pd precursor and CTAC. The Pd–CTAC complex is essential in promoting the structure direction of the nanoparticles. If CTAC is substituted with other surfactants, the octahedral shape of the Au core is not obtained. Although the method outlined clearly works very well for Au@PdPt nanoparticles, it does not appear to be applicable to other trimetallic systems, as altering the type or amount of reducing agents, the surfactant, or the molar ratio of metal precursors has been shown to affect the composition, structure, and morphology of the nanoparticles, which has a negative effect on the catalytic performance.79
The successful generation of the Auoct@PdPt core@shell NPs with well-defined octahedral Au core and a dendritic PdPt shell was achieved exclusively when both ascorbic acid and hydrazine were employed in the synthesis as reducing agents. The Auoct@PdPt particles had an average edge length of 61.0 ± 1.3 nm and shell thickness of 13.6 ± 1.6 nm. When ascorbic acid was solely used as a reductant in the synthesis, dendritic PdPt alloy NPs were produced instead of ternary AuPdPt NPs. Spherical Au cores were formed without the presence of ascorbic acid and hydrazine, indicating the influence of the reducing agents. EDX elemental mapping confirmed the core–shell structure, and the Pd–Pt shell was further observed via XRD analysis. ICP-AES estimated the Au:Pd:Pt atomic ratio to be 55:25:20.79 The effect of the reaction time was investigated by studying the NPs after a range of reaction times. After 1 min, octahedral NPs formed, and after 10 min small dendrites were observed on the NP surface. These protrusions were more abundant after 20 min. Further increasing the reaction time continued to increase the growth of the branches on the NP surface, and after 150 min the well-defined Auoct@PdPt core–shell TMNPs were formed.79
The effect of the reducing agent on the nucleation and growth kinetics was also investigated. When only CTAC was used in combination with the metal precursors, spherically shaped Au@PdPt NPs were formed with an average particle diameter of 43.3 ± 5.1 nm. The Au core was spherical in shape with an average diameter of 27.8 ± 3.9 nm, and the Pd–Pt shell was dendritic in structure with an average thickness of 7.6 ± 1.3 nm. The Pd–Pt shell was observed to be polycrystalline by SAED, suggesting that the shape of the Au core may influence the crystallinity of the shell. The Auoct@PdPt NPs could be produced only when both ascorbic acid and hydrazine were used simultaneously as reducing agents. When only ascorbic acid was used, bimetallic dendritic Pd–Pt NPs were produced. EDX showed Pd–Pt alloying with no Au present, even though Au has the highest reduction potential of the three metals; the Pd:Pt atomic ratio was 65:35 as determined by ICP-AES. When hydrazine was used exclusively as the reducing agent, polyhedral monometallic Au NPs were produced in a variety of shapes, and EDX showed that no Pd or Pt was present. The formation mechanism of the octahedral Au core was found to be influenced also by the metal precursors present, as when just HAuCl4 and K2PdCl4 were reduced by hydrazine, homogeneous octahedral Au NPs were synthesized, but the polyhedral Au NPs were produced when the K2PtCl6 precursor was also included. UV–vis adsorption spectra showed evidence of a peak corresponding to LMCT between the Pd precursor and CTAC, indicating that this interaction is a crucial component for producing the well-defined octahedral shape. Replacing CTAC with a different surfactant that does not contain ammonium resulted in an absence of the octahedral shape. When Pt is present, it preferentially binds to hydrazine ahead of Pd because of the stronger binding affinity. Ascorbic acid is therefore additionally required to direct the formation of the Pd–CTAC complex. The equimolar combination of the metal precursors was also crucial to form the well-defined Auoct@PdPt NPs; when Au was in excess, the octahedral shape of the Au cores could not be directed. Similarly, when Pt was in excess, its inhibiting effect on Pd–CTAC complex formation could not be eliminated by ascorbic acid. Au@PdPt NPs did form when Pd was in excess, but the shapes of the cores were inhomogeneous. Using the appropriate amounts of both reducing agents was therefore crucial to both the morphology and the material yield.79
Other trimetallic systems that have been synthesized by a one-pot method include Cu/Au/Pt,106,145 PtAuRu,146 Ag@PdAu,147 and (Pd,Co)@Pt.148 These examples appear to have been prepared following a significant amount of trial and error to identify the optimum conditions for reduction, and the “one-pot method” does not appear to be universally applicable.
A technique that is complementary to colloidal methods is the use of polyols (e.g., the diol ethylene glycol) to synthesize metal NPs through chemical reduction of metal salts. These liquid organic compounds can act as both the solvent and the reducing agent. At elevated temperatures, ethylene glycol can reduce metal cations to their metallic state and therefore encourage the growth of nanostructures. Advantages of using polyols include facile formation of crystallized materials and the ability to coordinate to metal particle surfaces to minimize aggregation. Such polyol compounds often have high viscosities, favoring diffusion-controlled particle growth that results in controlled structures and morphologies.149 The polyol method has been successfully applied to prepare TMNPs as catalysts, such as PdRuNi supported on graphene oxide for the dehydrocoupling of dimethylamine borane121 and PdCoPt/C for electrocatalytic oxygen reduction reactions.148 Adaptations of the polyol method have been used to synthesize (Pd,Co)@Pt NPs supported on Ketjenblack carbon by sonochemical reactions of Pt(acac)2, Pd(acac)2, and Co(acac)2 in ethylene glycol; with an elemental composition of Pd50Co20Pt30, the resultant trimetallic catalysts showed superior activity and stability, due to synergistic effects, for electrocatalytic oxygen reduction reactions compared with commercial Pt/C.148
Microwave (MW)-assisted polyol reduction methods have also been reported for the successful preparation of TMNPs, such as PdRuNi/GO,121 used as a catalyst for dehydrocoupling of dimethylamine borane, and PtRuFe supported on N-doped graphene, used in methanol oxidation and oxygen reduction reactions.150 Further examples of trimetallic catalyst preparation by MW irradiation methods have recently emerged. Typically, MW-assisted material synthesis is a highly controllable and rapid method to synthesize nanoparticles. The main benefit of MW techniques is that the internal heating provided by MW irradiation is much faster than that in conventional chemical reactions heated in an oil bath or oven. Furthermore, modern microwave reactors provide a high level of control over reaction conditions such as temperature, pressure, and heating rate, creating a very stable reaction environment. Depending on the method, MW irradiation may be applied to all of the metal precursors at once,121,151 or one metal precursor may be reduced to form a core NP first, after which the remaining precursors are added before MW synthesis is carried out.152−154
3.3. Obtaining Multimetallic Nanoparticles via Galvanic Replacement
Galvanic replacement or displacement has emerged as one of the more commonly used methods for synthesizing both supported and unsupported multimetallic nanoparticles.155−162 The method can be used to apply a third metal to a bimetallic NP system, resulting in core@shell or pseudo-core@shell structures as well as “nanorings”.163
The galvanic replacement reaction (GRR) occurs between the zero-valent surface metal atoms of the nanoparticle and metal precursors in solution. It is driven by the difference in reduction potentials between the surface metal atoms and the cations in solution, resulting in oxidation of the surface atom via reduction of the cation to its neutral state. As such, the process is sensitive to the chemistry of the metallic species at the surface and the cation in solution.164 Galvanic replacement demonstrates a flexible design approach in which the morphology and electronic composition of the active sites of the TMNP can be finely and independently tuned.165−168Equation 1 summarizes the overall reaction occurring at the surface sites:
| 1 |
The process effectively replaces n surface atoms of metal M2 with m atoms of metal M3, resulting in an M1M2@M1M3 pseudo-core@shell structure. The reaction is highly selective, as only one element on the surface is replaced. The process is also strictly a surface modification, as the third metal does not enter the interior of the nanoparticle, and therefore, the inherent morphology of the nanoparticle is not changed.165 Since the replacement reaction is a gradual process, the degree of replacement can be controlled by varying the reaction time, hence affording control over the stoichiometry of metal M3.169
Sahoo et al.(170) used the sacrificial oxidation of cobalt nanoparticles that were initially prepared from a CoCl2 precursor, reduced with NaBH4, and capped with sodium citrate to prepare TMNP catalysts. Initially, the reaction takes place under N2 to prevent oxidation of Co. After reduction is complete, a deaerated solution of HAuCl4 is transferred to the Co nanoparticle solution in the absence of oxygen. A solution containing PVP is then added as a stabilizing agent, and the sacrificial oxidation of Co0 to Co2+ spontaneously reduces the Au3+ ions to Au0 as Au nanoparticles are formed and Co2+ re-enters the solution. NaBH4 is then added to the solution of Au nanoparticles and Co2+ ions, which rereduces Co2+ ions to deposit a layer of Co0 around the Au nanoparticles. When this reduction is complete, a K2PdCl4 precursor solution is added to the mixture and stirred for 6 h at room temperature and then for a further 2 h at 323 K. Again, the precious metal, in this case Pd, is reduced from Pd2+ to Pd0 while Co0 is oxidized to Co2+. This solution is deaerated, and the process is then repeated using the RuCl3·H2O precursor to obtain triple core@shell Au@Pd@Ru nanoparticles.170 The disadvantage of the galvanic displacement method is that the final average nanoparticle diameter is large, at 110 nm, which means that a lot of the metal atoms are in the bulk and therefore unavailable for catalytic processes on the surface of the nanoparticle. Nevertheless, the Au@Pd@Ru nanoparticles were evaluated as catalysts for dye degradation and wastewater treatment and were promising compared with mono- and bimetallic analogues.
Miyazaki et al. reported the use of GRR to add a third metal (M) to PdZn/SiO2 such that Pd:Zn:M = 1:1:0.25.165 First, PdZn/SiO2 was synthesized by pore-filling coimpregnation of aqueous Pd(NO3)2 and Zn(NO3)2·6H2O on dried silica gel (specific surface area of 470 m2 g–1) to obtain a 3% loading of PdZn. The resulting product was reduced and stored under argon prior to the introduction of an aqueous solution of the third metal precursor. HRTEM images indicated that the diameters of the PdZn/SiO2 BMNPs were between 2 and 6 nm with a mean of ca. 3 nm. HRTEM images of a single PdZn nanoparticle, with the CsCl-type crystal structure, showed lattice fringes with a spacing of 0.217 nm. This spacing corresponds to the PdZn(010) planes, calculated to be 0.219 nm, and is indicative of an intermetallic bimetallic alloy of PdZn. Following addition of Pb via galvanic replacement, TEM images of PdZn@Pb0.25 showed a mean diameter of 3.3 nm (Figure 10). Minimal changes in the morphology of the nanoparticle were reported following galvanic replacement, which is as expected. The spacing of the (010) plane lattice fringes was reported to be 0.224 nm, similar to that of the PdZn(010) planes previously calculated. A fast Fourier transform of the HRTEM image confirmed the P4/mmm space group oriented along the (010) plane, comparable to analysis of a PdZn single crystal. EDX mapping of the HRTEM image confirmed the pseudo-core@shell structure, wherein Pd was homogeneously dispersed throughout the nanoparticle, Zn was found exclusively within the core, and Pb was found exclusively in the shell (M1M2@M1M3 structure). In this galvanic replacement reaction, Zn in the surface is replaced with Pb, whereas Pd remains in place. This is the case because Zn has a lower oxidation potential than Pd and is capable of reducing Pb2+ to Pb, as well as many other late transition and p-block metals. (Zn2+ + 2e– → Zn, −0.76 V vs NHE). This catalyst composition was compared to the Lindlar catalyst (Pd–Pb/CaCO3) in the hydrogenation of phenylacetylene to styrene, with the galvanic replacement prepared catalyst, comprised of PdZn@Pb0.25, exhibiting a high activity and importantly an enhanced R1/R2 ratio, indicating a reduced overhydrogenation rate of styrene.
Figure 10.
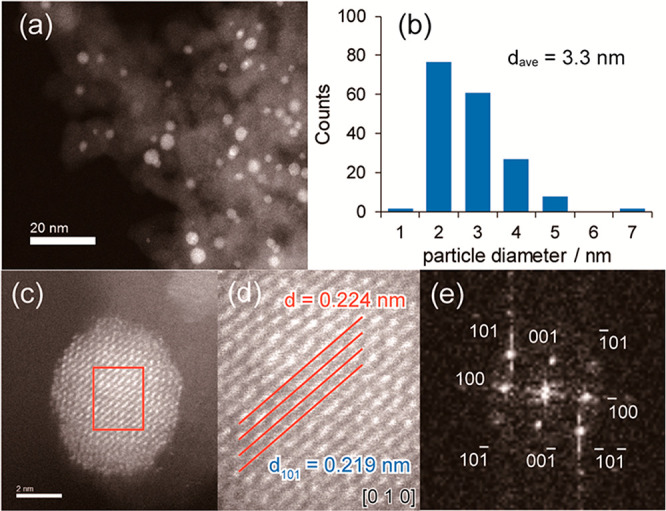
(a) TEM image of PdZn@Pb0.25 TMNPs. (b) Size distribution of the TMNPs. (c) HRTEM image of a single nanoparticle. (d) Further magnification of the region denoted by the red square in (c), highlighting the lattice fringes on the (010) plane. (e) Fast Fourier transform of the single nanoparticle. Adapted from ref (165). Copyright 2019 American Chemical Society.
Galvanic replacement has been extensively investigated by Skrabalak and co-workers as a way to access multicomponent materials and catalysts, including heterostructures that may offer unique opportunities in catalysis. As an extension or development of the work discussed above, the formation of heterostructures from Pd nanocubes or AgPd heterodimers has been reported,171 along with structures from intermetallic PdCu nanoparticles.172 These architecturally complex, multicomponent materials possess unique structural motifs and junctions, for example in the formation of a PdCu–Au Janus particle172 However, when Pt was added to the same intermetallic PdCu nanoparticles, no Janus particles were formed. The difference was ascribed to the surface mobility of Au versus Pt, as Au has a higher surface mobility and resulted in the segregated domains.
The use of TMNPs or nanostructures as templates for quaternary or quintenary NPs via GRR appears to be rare. However, the preparation of tetrametallic AgAuPtPd nanotubes from multidomain AgAuPd nanotubes has been investigated.173 This four-component structure was formed via a sequential GRR starting with Ag nanowires, to which Au was added with CTAB to form multidomain nanotubes. Pd (with CTAB) was added to this structure, and finally, Pt (with CTAB) was added to form the nanotubes. The material was imaged with HAADF-STEM, which revealed that the metals were well-mixed and were effective at catalytic reduction of 4-nitrophenol with NaBH4.
3.4. Preparations of Specialized Trimetallic Nanostructures
Novel approaches toward trimetallic systems have been reported in which defect sites on a bimetallic system have acted as seeds for controlled addition of a third metal.26 Such a method has paved the way for design of trimetallic catalysts at the atomic level. One example used Pt3Ni nanocrystals with a concave octahedral morphology containing many surface defect sites, and deposition of Ni was carried out to form Pt3Ni@Ni, which was of regular octahedral shape. Overall, several trimetallic catalysts of the form Pt3Ni@M8 (M = Au, Ag, Cu, Rh) were prepared. As the high-energy defect sites are less thermodynamically favored, nanoparticles often possess relatively few natural defect sites; this issue can be overcome for the outlined purposes by using chemical etching to introduce many additional defect sites.174 In the example reported by Wu et al., the recovered nanocrystals consisted of many surface sites of segregated Pt, with step edges and step terraces in contrast to the eight (111) facets expected in a regular octahedron.26 Proof-of-concept experiments with a PVP (Mw = 8000)-capped Ni shell confirmed that growth of the shell was seeded by these defect sites, reforming a regular octahedron with eight Ni(111) facets. Finally, catalysts were prepared using other metals (M) to form Pt3Ni@Mx core@shell TMNPs. The catalysts containing 0.5 atom % Pt were evaluated for the Suzuki–Miyaura reaction and reduction of nitrobenzene using formic acid as a hydrogen source. The stoichiometry of M was controlled by varying the concentration of M precursor and was set at x = 0.5, 2, or 8. Pt3Ni@M8 represented a perfect octahedron, whereas a low M stoichiometry such as Pt3Ni@M0.5 was still of concave morphology. XPS data confirmed the addition of the third metal on the defect-rich concave region of the nanoparticles. TEM images confirmed particle diameters of 10–13 nm for all of the catalysts that were synthesized. Catalysts containing Au (0.5 atom %) as the third metal had significantly enhanced reaction rates compared with a commercial example and for the reduction of nitrobenzene, where with other metals (Cu, Rh, and Ag) the rate was lower.
Etching methods have also been used for the synthesis of trimetallic nanoframes, as reported by Yin et al.(175) First, trimetallic PtRuCu nanoparticles were synthesized via hydrothermal methods using K2PtCl6, CuCl2·2H2O, and RuCl3·xH2O with oleic acid and oleylamine capping (Figure 11). The mixture was heated to 473 K with stirring and then heated in a Teflon-lined stainless-steel autoclave. The PtRuCu TMNPs were dispersed in a 1:1 mixture of cyclohexane and DMF. The etching agent, BF4NO, was added with stirring. The mixture was centrifuged and washed with DMF. The nanoframes were dispersed in 3:1 DMF/acetone, and finally, carbon black (Vulcan XC-72) was dispersed into the mixture followed by sonication. The catalysts were activated in a N2-saturated HClO4 aqueous solution.
Figure 11.
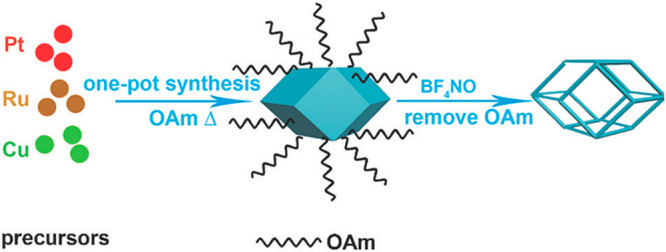
Schematic displaying the formation of PtRuCu nanoframes via formation of the nanoparticle and subsequent etching to form the hollow frame. Adapted with permission from ref (175). Copyright 2020 Royal Society of Chemistry.
The resulting frames had a rhombic dodecahedral shape and were 40–80 nm in diameter. The three metals within the frame were reported to be alloyed and evenly distributed, and the Pt:Ru:Cu ratio was 40.7:0.3:59. The low Ru content was understood to be due to a mismatch of the Ru lattice structure with those of Cu and Pt; Ru adopts a hexagonal closed-packed (HCP) structure, whereas Cu and Pt adopt FCC structures.176
Another specialized nanostructure, the one-dimensional nanowire, was reported by Zhu et al.(177) and was synthesized with a sacrificial template composed of a tellurium nanowire. These materials were evaluated as electrocatalysts for ethanol electrooxidation. The Pd–Pt–Au nanowire was prepared by hydrothermal synthesis from an aqueous suspension of Te nanowires that was charged with H2PdCl4, H2PtCl6, and HAuCl4. Bimetallic samples, PdPt and PdAu, were also prepared. Trimetallic formulations included Pd:Pt:Au molar ratios of 5:5:1 (resulting in an atomic formulation of Au18Pt6Pd76), 4:16:1 (resulting in Au17Pt24Pd59), and 2:8:1 (resulting in Au34Pt18Pd48). The compositional differences observed in the final materials were attributed to the galvanic replacement reaction rates (HAuCl4 > H2PdCl4 > H2PtCl6). The nanowires were shown by EDX mapping to contain an evenly distributed and fully alloyed configuration, with no remaining Te apparent. The detailed mechanism of formation of the nanowires is still currently under investigation; however, in comparison with the commercial E-TEK Pd/C electrocatalyst, the Au17Pt24Pd59 nanowires were shown to have greater activity for ethanol electrooxidation in alkaline media.
Hydrothermal synthesis is another important method that has been used for the preparation of TMNPs. The technique involves heating a solution of dissolved metal precursors in an autoclave under autogenous pressure and can be an effective method to produce highly ordered structures in a facile manner. Examples of trimetallic nanoparticles prepared this way include PdNiAl NPs,178 NiFeCo NPs,179 PtAgCu@PtCu core@shell concave nanooctahedrons as catalysts for formic acid oxidation,180 (111)-terminated PtPdRh nanotruncated octahedrons for conversion of ethanol to CO2,181 and CoNiCu hydroxyphosphate nanosheets as electrocatalysts for the oxygen evolution reaction.182
An interesting approach to the formation of highly ordered arrays of multimetal nanoparticles was developed by Mirkin and co-workers through a tip-directed deposition technique.183,184 Here a metal or multimetallic precursor solution containing a block copolymer could be deposited on a suitable substrate such as Si via a technique termed scanning probe block copolymer lithography.185 This technique results in a high degree of deposition control, with particles spaced at regular intervals, and nanoparticles with a diameter of ca. 10 nm are formed following annealing at 500 °C. Formulations of nanoparticles included AuPd, PtNi, PdNi, PtCo, PdCo, and CoNi BMNPs and also AuAgPd TMNPs. These arrays were evaluated as catalysts for the conversion of 4-nitrophenol to 4-aminophenol with NaBH4. As an extension of this technique, the formation of quaternary and quinary nanoparticles that can be prepared as heterostructured nanoparticles was subsequently reported.184 The diversity of preparation techniques available to synthesize TMNPs as NPs deposited on a surface or as trimetallic nanoframes highlights the degree of control available to tailor materials to potential catalytic processes. In the next section we evaluate the catalytic applications of many of the materials discussed in this section and explore the benefits of TMNPs in important processes.
4. Catalytic Applications
There are several examples of the uses of trimetallic nanoparticles in catalysis, including oxidations, reductions, hydrogenations, decomposition reactions, and electrochemical reactions. The following section will include applications of specific compositions that have received particular attention in the literature. The majority of examples in the literature are for transition metals in groups 8–11. The exact role played by the addition of a third metal in their catalytic performance can be complicated, but it is apparent that TMNP-based catalysts can exhibit increased catalytic activity compared with their mono- and bimetallic counterparts. Here we explore the potential causes of this enhancement with different compositions of TMNPs. These enhancements often appear through side-reaction suppression via dilution of an active surface or subtle alteration of the active site through sterics or an electronic change of an active metal center upon addition of third metal.
4.1. Hydrogenation
Hydrogenation encompasses a wide variety of reactions that involve reduction of a molecule via the addition of hydrogen.186 Without a catalyst, hydrogenation would require high temperatures and pressures, so an appropriate catalyst is essential to ensure appropriate selectivity and feasible reaction conditions. Common examples of heterogeneous catalysts used in hydrogenation include ruthenium,187,188 Raney nickel,189 and the palladium-based Lindlar’s catalyst.190 More recently, a number of trimetallic examples have been reported that can further address the issues related to selectivity, and a selection are discussed in further detail in this section.
4.1.1. Hydrogenation of CO to Higher Alcohols
Mixtures of higher alcohols have the potential to be used to blend with gasoline to improve its octane number and reduce the output of harmful emissions. Although these higher alcohols can be obtained from biomass-derived synthesis gas (syngas), the product is typically contaminated with H2S, which is a strong poison for many catalysts. Hence, two essential properties of catalysts for the synthesis of higher alcohols from biomass-derived syngas are (i) high activity for the water gas shift (WGS) reaction and (ii) superior sulfur resistance. MoS2 catalysts fulfill these criteria but have low selectivity for higher alcohols, instead producing methanol, hydrocarbons, and CO2. The addition of Rh shifts the activity toward oxygenates, and further addition of Co increases C1 to C2 homologation, encouraging the production of ethanol as a dominant product. Table 1 summarizes the influence of the support on the activities of TMNP catalyst examples discussed here, when applied to this reaction.
Table 1. Summary of a Selection of TMNP Catalysts Applied to CO Hydrogenation to Higher Alcohols.
| catalyst | catalyst type | reaction conditions | % CO conversion (selectivity for higher alcohols) | ref |
|---|---|---|---|---|
| Rh–Mo–K/MWCNT | alkali-promoted BMNP | T = 593 K; p = 8.3 MPa; GHSVa = 3.6 m3 (STP)/(h kg cat) over 24 h; H2/CO molar ratio = 1 | 40.1 (24.6) | (124) |
| 4.5 wt % Co–Rh–Mo–K/MWCNT | alkali-promoted TMNP | 45.2 (31.4) | ||
| 6.0 wt % Co–Rh–Mo–K/MWCNT | 48.9 (27.8) | |||
| 4.5 wt % Co–Rh–Mo–K/AC | 31.2 (18.8) | |||
| 6.0 wt % Co–Rh–Mo–K/AC | 35.3 (15.9) | |||
| 4.5 wt % Co–Rh–Mo–K/AC-Darco (microporous) | alkali-promoted TMNP | T = 603 K; p = 8.3 MPa; GHSVa = 3.6 m3 (STP)/(h kg cat) over 48 h; H2/CO molar ratio = 2 | 35.6 (11.7) | (125) |
| 4.5 wt % Co–Rh–Mo–K/AC-RX3 Extra (microporous) | 39.6 (12.1) | |||
| 4.5 wt % Co–Rh–Mo–K/AC-Fluid Coke (mesoporous) | 41.8 (12.7) | |||
| 4.5 wt % Co–Rh–Mo–K/AC-CGP Super (mesoporous) | 44.5 (13.1) | |||
| 4.5 wt % Co–Rh–Mo–K/MWCNTs | 52.4 (16.8) |
Gas hourly space velocity.
The activities of alkali-promoted trimetallic Co–Rh–Mo sulfide catalysts for higher alcohol synthesis (C2 and higher) from syngas were assessed.124 When used as a support, activated carbon (AC) does not favor hydrocarbon formation and was reported to be resistant to changes in pH, high temperatures, and pressures. However, AC still suffers from coking, as it has micropores (<2 nm pore diameter) that are easily blocked, leading to transport limitations. Multiwalled carbon nanotubes (MWCNTs) have properties similar to those of AC, albeit with a meso/macroporous structure that circumvents the limitations of transport and metal dispersion. To synthesize the MWCNT-supported catalysts, the supports were pretreated with HNO3, and then metals were added using an incipient wetness impregnation method through the addition of first a K-promoter (9 wt %) followed by the metal precursors. A range of reaction conditions were tested, with higher temperatures and pressures leading to increased CO conversion and hydrocarbon formation rate, decreasing methanol selectivity, and increasing higher alcohol selectivity (593 K and 8.28 MPa were optimal).
Bimetallic Rh–Mo catalysts were also compared for higher alcohol synthesis (C2 and higher) from syngas, but the CO uptake increased when Co was added, up to 4.5 wt %, to form the trimetallic catalyst.124 The Co–Mo–S phase is considered to be the active site for higher alcohol synthesis, and the selectivity for higher alcohols over the 4.5 wt % Co–Rh–Mo–K/MWCNT catalyst was 31.4 wt %, compared with 24.6 wt % over Rh–Mo–K/MWCNT. When the Co content was increased above 4.5 wt %, an increase in large Co9S8 particles was observed that decreased the surface area and hence the metal dispersion of the Co–Mo–S phase. The selectivity for higher alcohols over catalysts containing 6 wt % Co supported on either AC or MWCNTs was subsequently reduced. Further analysis of the catalysts by temperature-programmed reduction (TPR) revealed that the addition of Co caused the reduction temperature to decrease, showing that addition of Co enhances the reducibility of the other metal species present at lower temperatures, and as the Co content was increased, this effect intensified. The highest CO conversion (48.9%) was observed over the 6 wt % Co–Rh–Mo-K/MWCNT catalyst.124 The catalyst with the lower Co content (4.5 wt % Co–Rh–Mo–K/MWCNT) showed a lower CO conversion (45.2%). The results imply that Co addition increases the number of active sites, as an improved CO conversion was observed, but when the Co concentration is increased beyond a certain level, the methanation activity increases as large Co9S8 sites form.
In a later study, the same group assessed the influence of the porosity of the carbon support on the performance of the alkali-modified trimetallic Co–Rh–Mo catalysts for CO hydrogenation to higher alcohols, testing four different commercial AC supports.125 Two of the AC supports had microporous characteristics (AC-Darco and AC-RX3 Extra), while the remaining two AC supports were mesoporous (AC-Fluid Coke and AC-CGP Super). The supports were compared to commercial MWCNTs, which are also mesoporous. In reflection of the group’s previous work, a 4.5 wt % Co–Rh–Mo–K catalyst was studied on each support.124 TEM studies showed that the TMNPs present were well-dispersed on the mesoporous AC and MWCNT supports with small average particle diameters (3–6 nm on mesoporous AC and 1–3 nm on MWCNTs). Conversely, TMNPs agglomerated on the microporous AC supports, resulting in lower dispersion. The different dispersions were reflected in the measured CO uptake on each catalyst; the highest CO uptake was observed on the MWCNT-supported catalyst, followed by the mesoporous-AC-supported catalysts, and the microporous-AC-supported catalysts had the lowest CO uptake. The CO hydrogenation activity followed the same trend due to pore structure of the catalyst supports. The uniform pore size of the MWCNT support led to the best TMNP dispersion, which resulted in the highest number of active sites, giving the highest activity. The MWCNT-supported catalyst also had the highest selectivity for higher alcohols, 16.8%, compared with 13.1% and 12.7% for the two mesoporous-AC-supported catalysts and 12.1% and 11.7% for the two microporous-AC-supported catalysts. The observations suggest that the Brunauer–Emmett–Teller (BET) surface area and pore volume of the supports are not the important features of these catalysts but rather that the textural properties of pore size and percent mesoporosity have the most significant effects on the activity and selectivity.
4.1.2. Fischer–Tropsch Synthesis
Fischer–Tropsch synthesis (FTS) refers to the hydrogenation of carbon monoxide to form hydrocarbons (HCs). This reaction is of environmental importance, as it is a potential route for non-petroleum-based fuel production from waste products (CO and H2/H2O). The routes of alkene and alkane production are given in eqs 2 and 3, respectively:
| 2 |
| 3 |
First developed in the 1920s by Franz Fischer and Hans Tropsch, Fe-based catalysts were initially favored, although later Co-based catalysts were found to perform well for the hydrogenation of CO to produce a mixture of hydrocarbons and oxygenates.191 Vannice studied different transition metals supported on alumina for their specific activities and product distributions for CO hydrogenation and found Ru/Al2O3 to be the most active catalyst, yielding the highest molecular weight of products.192 The wide variety of products possible via these routes leads to complicated product mixtures, and therefore, a highly selective catalyst is desirable. The implication of this is that the development of a high-performing trimetallic catalyst could be vastly beneficial.
Badoga et al. studied the effects of four different promoters—Mn, Mg, Co, and Ni—on the performance of a KCuFe catalyst supported on mesoporous Al2O3 for FTS.193 The catalysts were synthesized by a sequential incipient wetness impregnation method. First Fe (equivalent to 25 wt %) was added to the Al2O3, and after drying Cu (equivalent to 0.5 wt %) was added. Then the catalyst was dried again and calcined at 400 °C for 4 h. K was then added (1 wt %), and after drying and calcination the KCuFe/Al2O3 catalyst was obtained. To this the various promoters could be added by impregnation, and for comparison, an unpromoted monometallic Fe/Al2O3 catalyst was also synthesized.
The catalysts were reduced for 16 h at 623 K prior to the reaction and tested for FTS at 523 K and 2 MPa with a space velocity of 2000 h–1. The unpromoted monometallic Fe/Al2O3 catalyst showed a high CO conversion of 95% but also a high selectivity for CO2 (46.9%). Of all the hydrocarbon products produced, the selectivities were 34.1% for CH4, 11.1% for C2–C4 products, and 54.8% for C5+ HCs. The production ratio of olefins to paraffins (O/P) was 0.35. Over the KCuFe/Al2O3 catalyst, the CO conversion remained high at 94%. However, the C5+ selectivity increased in the hydrocarbon product distribution to 77.0%, and the CH4 selectivity decreased to 12.0%. The O/P ratio was significantly increased to 0.71, and the CO2 selectivity decreased to 44.0%.
When Co was added as a promoter, the CO conversion remained comparable (93%), but the CH4 selectivity increased to 16.8% and subsequently the O/P ratio dropped to 0.25. TPR and X-ray absorption near-edge structure (XANES) analyses of this catalyst showed relatively low ease of reduction of Fe compared with other promoted KCuFe/Al2O3 catalysts. When Mg was added as a promoter, the CO conversion was unaffected (93%), but the O/P ratio decreased with respect to KCuFe/Al2O3 to 0.60. The CH4 selectivity increased from 12.0% to 14.8%, and the C5+ HC selectivity decreased from 77.0% to 74.0%. The CO2 selectivity was similar for KCuFe/Al2O3 and the Co- and Mg-promoted equivalents, suggesting that these two promoters are not effective in inhibiting the WGS reaction.
Analysis of the Ni–KCuFe/Al2O3 catalyst with TPR and XANES suggested that the extent of Fe reduction was high, but the testing data revealed this catalyst to have the lowest CO conversion (77%) and the highest CH4 selectivity (29.3%) among all of the bi- and trimetallic catalysts tested. This was thought to be due to the H2 spillover mechanism being active on Ni. The Ni–KCuFe/Al2O3 catalyst did show a reduction in CO2 selectivity to 40.7%, which suggests that the WGS reaction was suppressed. However, in turn this lowered the H2 availability and may account for the decrease in CO conversion. The Mn–KCuFe/Al2O3 catalyst was the only promoted catalyst to exhibit an increase in CO conversion versus the unpromoted catalyst to 95%. Both the CO2 selectivity (39.4%) and CH4 selectivity (10.0%) decreased, whereas the selectivity for the desirable C5+ HCs increased to 81.0%. Meanwhile, the C2–C4 HC selectivity dropped to 9.0%, and the O/P ratio increased to 0.85.
XPS analysis of the Mn–KCuFe/Al2O3 catalyst showed that it had the highest surface concentration of Fe, whereas the Mg–KCuFe/Al2O3 catalyst had the lowest surface concentration of Fe (almost 40% lower than that of Mn–KCuFe/Al2O3), which helps to rationalize the varying performances of these catalysts. TPR and XANES analyses of the Mn–KCuFe/Al2O3 sample also showed that the ease of Fe reduction on this catalyst and the electron-donating nature of Mn lead to strong electronic interactions between Mn and Fe.
To summarize this work, Mn–KCuFe/Al2O3 outperforms all of the other catalysts tested with respect to C5+ HC selectivity, CO conversion, and minimization of undesired products (CO2, CH4, and C2–C4 HCs). This was reported to be due to increased Fe dispersion and reducibility.193
Golestan et al. also explored the promoting nature of Mn for FTS using unsupported FeCoMn catalysts.194 The catalysts were synthesized by a hydrothermal method in which the precursors Fe(NO3)3·9H2O, Co(NO3)3·6H2O, and Mn(NO3)2·4H2O, the capping agent CTAB, and the precipitating agent urea were stirred together for 1 h, after which the mixture was placed in an autoclave and heated to 180 °C for 18 h. The product was washed, collected by centrifugation, dried, and calcined (500 °C, 4 h). The catalysts were synthesized with a range of Mn loadings (wt %), and once the highest-performing catalyst from this series had been identified, the Fe:Co molar ratio was varied to study the impact of this. The catalysts were tested for FTS under the conditions identified as optimum through further study (563 K, 0.2 MPa, and a space velocity of 3000 h–1 with a H2:CO molar ratio of 1:1).
The desired products for this study were light olefins (C2–C4), and suppression of CH4 production and an improvement in the ratio of olefins produced to methane and paraffins (O/(M+P)) were also intended. It was found that increasing the concentration of Mn improved the selectivity for C2–C4 olefins and decreased the selectivity for CH4; however, the C5+ HC selectivity was unaffected. The catalyst containing 16 wt % Mn was the highest-performing with respect to O/(M+P). It was also found that as the Mn loading increased, the surface area, pore size, and pore volume of the catalysts all increased too. The effect of varying the Fe:Co molar ratio was next investigated using a catalyst with 16 wt % Mn. The Fe:Co content was varied from 68 wt % Co and 16 wt % Fe to 16 wt % Co and 68 wt % Fe. The highest-performing catalyst regarding light olefin production was 68%Fe–16%Co–16%Mn. However, although the increasing Fe content and decreasing Co content improved the O/(M+P) ratio, the catalysts with higher Co content had higher activity for syngas conversion.
XRD analysis of the used 68%Fe–16%Co–16%Mn catalyst showed the presence of oxidic and carbide phases, which are active phases for FTS. TPR studies confirmed that the addition of Co reduces the level of Fe2O3, which is reduced to FeO and Fe and subsequently forms iron carbides, including Fe5C2, which is especially important in FTS. N2 adsorption experiments found the BET surface area of the 68%Fe–16%Co–16%Mn catalyst to be 75 m2/g, which is significantly higher than that of any of the other catalysts synthesized and may be a contributing factor to its improved performance.194 The same group also studied FeCoMn catalysts supported on MgO, which appeared to improve the activity of the catalyst, although the selectivity for the desired C2–C4 olefins was reduced.195
Razmara et al. compared the performance of Co2–Ni–Mn/SiO2 catalysts prepared by three different methods for the Fischer–Tropsch reaction.196 In addition to the Co2–Ni–Mn/SiO2 catalysts prepared by impregnation and coprecipitation, a third sample was prepared by the thermal decomposition of a novel inorganic precursor, [Ni(H2O)5Co(dipic)2]·2H2O + [Mn(H2O)5Co(dipic)2]/SiO2, where dipic denotes the ligand pyridine-2,6-dicarboxylic acid. Thermogravimetric analysis results showed that the precursor decomposed to the metal oxides NiO4 and MnCo2O4 at 427 °C, so in order to ensure full decomposition, the precursor was calcined at 500 °C for 4 h in static air. The calcination process was shown to significantly increase the measured BET surface area and pore volume in the nanocatalyst compared with the precursor. This was reflected in the morphology observed by scanning electron microscopy (SEM), where the catalyst particle size is visibly much smaller than the precursor.
The three catalysts produced by thermal decomposition, coprecipitation, and impregnation were tested for the Fischer–Tropsch reaction, conducted at 553–633 K and atmospheric pressure with a H2:CO molar ratio of 1:1 (gas hourly space velocity (GHSV) = 3600 h–1). It was found that over all three samples, as the reaction temperature was increased, the activity for CO conversion also increased, but this also corresponded to an increase in methane selectivity. At all of the temperatures tested, the catalyst produced by thermal decomposition was the highest-performing with respect to both CO conversion and hydrocarbon selectivity. At 633 K, the CO conversions for the thermal decomposition, coprecipitation, and impregnation catalysts were 68.7, 58.1, and 43.2% respectively. This directly correlates with the Debye–Scherrer calculations from XRD, which gave crystallite sizes of 12.5, 21.3, and 29.0 nm for the thermal decomposition, coprecipitation, and impregnation catalysts, respectively. This is also in agreement with the observations from SEM and surface area measurements and suggests that the smaller crystallite size of the thermal decomposition catalyst exposes a larger amount of surface area, thereby improving the catalyst activity. All three catalysts showed stable performance over 8 h of time on-line, but after that there was a decrease in CO conversion over the 30 h reaction, with final CO conversion values of 61, 47, and 33% for the thermal decomposition, coprecipitation, and impregnation catalysts, respectively. In addition to its relative superior performance, other advantages of the thermal decomposition method were noted as being the accurate metal loading on the support, a homogeneous metal distribution, and the formation of a strong metal–support interaction.196
Zhai et al. used zinc- and sodium-modulated Fe5C2 catalysts synthesized by coprecipitation to investigate the formation of C5+ alkenes via FTS.197 In Fe-based catalysts, Zn may often be used to inhibit the reduction of FeO to increase CO adsorption, hence increasing the formation of long-chain hydrocarbons. However, the WGS reaction remained prevalent, and the methane productivity was high. Potassium is commonly used to increase the hydrogenation barrier of CHx species, subduing methane production and increasing the selectivity for other products, including alkenes. By using sodium instead, the authors hoped to show a similar effect.
The catalysts tested were denoted as Fe–Zn–xNa, where x is the concentration of Na (3.4, 0.81, 0.36, or 0.0%) and the concentrations of Fe and Zn were equal. Furthermore, a catalyst containing no Zn and 1.2% Na (Fe–1.2Na) was also tested, along with a version of this catalyst that was washed to remove the Na, leaving a Na-free Fe catalyst. XRD characterization of the catalysts showed similar diffraction patterns for all of the Fe–Zn–Na catalysts, with the ZnO and ZnFe2O4 phases dominating, illustrating the strong Zn and Fe interactions in these samples. The most intense ZnO diffraction pattern was observed in the catalyst with the highest concentration of Na. In the catalysts in which Zn was absent, the crystallite size increased from the range 8.4–13.0 nm to 15.4–19.6 nm. This was supported by observations from the CO uptake survey, which suggested a larger exposed surface for the catalysts containing Zn.
The catalysts were tested for FTS at 340 °C and 2.0 MPa with a 6:16:2:1 H2/CO/CO2/Ar synthesis gas mixture. The catalyst found to have the highest syngas conversion was Fe–Zn–0.36Na, followed by Fe–Zn–0.81Na, Fe–Zn–3.4Na, and Fe–1.2Na, and finally the monometallic Fe catalyst had the lowest conversion. The two catalysts containing no Zn performed much worse than the Zn-containing catalysts, as Zn acts as a structural promoter leading to smaller Fe crystals with more exposed surface, which corresponds to a greater activity with respect to the mass of Fe. Figure 12 compares the performance of tri-, bi-, and monometallic catalysts.
Figure 12.
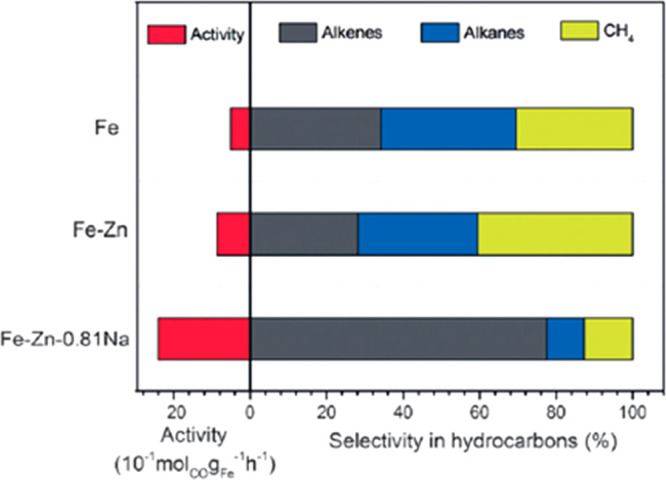
Activities and product distributions on Fe–Zn–0.81Na, Fe–Zn, and Fe catalysts. Reaction conditions: catalyst (20 mg), 340 °C, 2.0 MPa, syngas (6:16:2:1 CO/H2:/CO2/Ar, 20 mL min–1). Reproduced with permission from ref (197). Copyright 2016 Wiley-VCH.
It is common for Fe-based catalysts to produce large amounts of CO2 in FTS, and this was indeed observed over the Fe–Zn catalyst (33.1%). However, for the Na-containing catalysts the CO2 selectivity was reduced to as low as 21.9% (Fe–1.2Na) and 22.5% for the best-performing trimetallic catalyst (Fe–Zn–3.4Na). Fe–Zn–0.81Na also showed the lowest selectivity for CH4 among all of the catalysts tested. Increasing the Na content suppressed the alkane formation and increased the alkene production. Fe–Zn–3.4Na had the greatest production of C5+ alkenes (46.3% selectivity). Fe–1.2Na and Fe–Zn–0.81Na, the two catalysts with the most comparable Na contents, showed similar product distributions, but Fe–1.2Na had much lower syngas conversion. This indicates that Na plays an important role in determining the product distribution. DFT studies were used to show that Na is an electronic promoter, donating electrons to Fe to increase the CO activation ability and decrease the rate of C=C bond hydrogenation by promoting desorption of the alkene.197
There are other synthetic fuels that can be produced from the hydrogenation of CO, such as ethanol. Liu et al. studied the effects of the addition of Fe and Mn to Rh/SiO2 catalysts for the production of C2+ oxygenates and short chain hydrocarbons from syngas.198 The reactant mixture had a CO:H2 ratio of 1:1. The performances of mono-, bi-, and trimetallic combinations of the three metals were compared. To produce monometallic 5 wt % Rh/SiO2, 0.41 wt % Fe/SiO2, and 0.27 wt % Mn/SiO2 catalysts for comparison purposes, an incipient wetness impregnation method was used. The metal precursors were RhCl3, Fe(NO3)3, and Mn(NO3)2, respectively. To produce the bimetallic and trimetallic catalysts, controlled surface reaction (CSR) processes were used. For RhFe/SiO2 or RhMn/SiO2 catalysts, the reduced Rh/SiO2 catalyst was held in a Schlenk flask under an inert atmosphere, and (cyclohexadiene)iron tricarbonyl or (cyclohexane)manganese tricarbonyl dissolved in n-pentane was added. After 2 h of stirring, the excess pentane was evaporated, and the catalyst was then reduced. For RhFeMn/SiO2 or RhMnFe/SiO2 catalysts, the third metal was added to the reduced RhFe/SiO2 or RhMn/SiO2 catalysts via a Schlenk line using the same process as described for the production of bimetallic catalysts. The catalysts are denoted as Rh–xFe–yMn, where x and y are the nominal Fe:Rh and Mn:Rh molar ratios, respectively. The Rh loading was consistent in all of the catalysts at 5 wt %.
UV–vis spectroscopy was used to characterize the catalysts, and it was found that neither Fe nor Mn interacted with the silica support and that the Rh–Fe and Rh–Mn interactions were much more significant. The extent of metal uptake from CSR was also assessed; for the trimetallic catalysts, when Fe is deposited before Mn, the percentage of Mn adsorbed decreased as the total metal loading increased. That is to say, deposition of the Mn precursor is limited by the number of Rh sites available after Fe deposition. For example, in the Rh–0.05Fe–0.10Mn catalyst, the Mn uptake was measured as 63%. When the iron content was tripled in the Rh–0.15Fe–0.10Mn catalyst, the Mn uptake decreased to 48%. A further increase in iron loading to Rh–0.30Fe–0.10Mn saw an uptake of Mn of only 20%. Likewise, when Mn was deposited first in the trimetallic catalyst, the Fe loading was found to be lower than anticipated. This suggests that in the trimetallic system Fe and Mn act independently over the Rh surface, apart from when the loading of the first metal is very low, leaving enough Rh sites available for the deposition of the second metal.
The catalysts were tested for CO conversion at 523 K and 4.0 MPa with a CO:H2 ratio of 1:1. The CO conversion was kept close to 1% for all of the tests in order to study the product selectivity over each catalyst in detail. Over the monometallic Rh/SiO2 catalyst, the major products were CH4 and acetaldehyde. The Fe/SiO2 catalyst was much less active than Rh/SiO2 and primarily produced C2+ hydrocarbons, while the Mn/SiO2 catalyst was completely inactive.
The bimetallic RhFe/SiO2 catalysts were synthesized with Rh:Fe ratios of 0.05, 0.15, and 0.30. It was found that the selectivity for ethanol increased up to the Rh–0.15Fe catalyst and the selectivity for acetaldehyde decreased, and the Rh–0.15Fe catalyst had the highest selectivity for total oxygenates among all of the mono- and bimetallic catalysts tested. In fact, the Rh–0.30Fe catalyst had the highest TOF for total oxygenate production; however, as the TOF for CH4 production also increased, the overall selectivity for oxygenates decreased. It appears that the promoting role of Fe facilitates acetaldehyde hydrogenation: as the loading of Fe is increased, the TOF for acetaldehyde production decreases and the TOF for ethanol production increases.
The bimetallic RhMn/SiO2 catalysts had Rh:Mn ratios of 0.05, 0.10, and 0.15. The Rh–0.10Mn catalyst showed the highest selectivity for ethanol. This catalyst had less of a promotional effect than Fe, but Rh–Mn interactions still resulted in increased selectivity for oxygenates compared with the monometallic catalysts. The Rh–Mn catalysts are much more selective for C2+ hydrocarbons (2 times more than Rh/SiO2 and 4 times more than RhFe/SiO2) and showed a larger decrease in selectivity for CO2 and CH4 compared with Rh/SiO2 and RhFe/SiO2. Rh–0.10Mn had the highest selectivity for total oxygenates (28.4%), and the TOF for CO consumption was 4 times higher compared with Rh/SiO2—the highest of any of the catalysts tested in this study. The promotional effect observed for the Rh–Mn interactions is related to the increase in C2+ hydrocarbon production and CO consumption.
The synergistic effect between both the Fe and Mn promoters and Rh can be examined by testing the trimetallic catalysts. First, the amount of Fe was kept fixed while the level of Mn was varied. For the series Rh–0.05Fe–yMn (y = 0.05, 0.10, 0.15), the methane and CO2 production levels were reduced, and the C2+ hydrocarbon and ethanol production levels were improved. Rh–0.05Fe–0.10Mn was found to have the best selectivity for total oxygenates (36.8%). The C2 oxygenate formation rate and CO consumption rate increased as the Mn loading increased. In general, this series of catalysts was found to favor production of alkenes over alkanes, which was the same trend as observed over the bimetallic Rh–Mn catalysts. For the series Rh–0.15Fe–yMn, selectivity patterns similar to those for the Rh–0.05Fe–yMn catalysts were observed, with Rh–0.15Fe–0.10Mn showing the highest selectivities for ethanol, C2 oxygenates, and total oxygenates. This catalyst was also the most selective for oxygenates among all of the catalysts prepared and had the lowest methane selectivity. However, its CO consumption rate was low. Oxygenates accounted for 44.8% of the products produced by this catalyst (2 times more than over Rh/SiO2). Conversely, Rh–0.30–yMn showed a different pattern of selectivity compared with the previously tested trimetallic catalysts. For this series it was found that changing the Mn loading did not substantially change the reactivity of the catalyst, as Fe was in excess first, limiting Rh–Mn interactions. Therefore, in general, the Rh–0.30–yMn catalysts followed the same trends in selectivity as observed for the Rh–xFe catalysts.
The effect of the order of secondary metal deposition on the catalytic performance was also investigated (Figure 13). Rh–0.15Fe–0.05Mn and Rh–0.05Mn–0.15Fe were found to perform similarly. However, the Rh–0.15Fe–0.10 Mn and Rh–0.10Mn–0.15Fe catalysts and the Rh–0.15Fe–0.15Mn and Rh–0.15Mn–0.15Fe catalysts showed more differences in reactivity for CO hydrogenation. When Fe was deposited before Mn, the total production of oxygenates was larger. When Mn was deposited before Fe, the ethanol selectivity was lowered, and the C2+ hydrocarbon selectivity was increased. This is the same pattern as observed for the Rh–xFe and Rh–yMn catalysts, respectively, and shows that the effect of the precursor deposited first is more important to the overall performance of the catalyst compared with the precursor deposited last. Observations made in an analysis of the uptake of the second precursor showed this to be less than 100%. However, this does not apply for the catalysts with very low loadings of Mn (i.e., Rh–0.15Fe–0.05Mn and Rh–0.05Mn–xFe).198
Figure 13.

Active sites for CO hydrogenation on Rh–Mn and Rh–Fe active sites and product distributions for various Rh–xFe–yMn catalysts tested for syngas conversion. Reproduced from ref (198). Copyright 2017 American Chemical Society.
4.1.3. Hydrogenation of CO2 to Methanol
The use of CO2 to produce platform chemicals is emerging as an important process to reduce harmful greenhouse gas emissions. Of these valorization products, methanol has been identified as a valuable option because it can be used as a solvent or as a chemical feedstock. Methanol can also be produced as a renewable fuel, provided that sustainable H2 is available for the hydrogenation of CO2.199 Ramirez et al. investigated the hydrogenation of CO2 under atmospheric pressure over PdCuZn/SiC trimetallic catalysts with various palladium metal loadings (Table 2).200 The bimetallic combinations PdCu/SiC, PdZn/SiC, and CuZn/SiC were also compared for their catalytic performance. The methanol synthesis reaction (eq 4) is in competition with the reverse water gas shift (RWGS) reaction (eq 5):
| 4 |
| 5 |
Table 2. Summary of Catalysts Discussed Here for the Hydrogenation of CO2 to Methanol.
| catalyst | catalyst type | reaction conditions | TOF;a activity;b selectivity | ref |
|---|---|---|---|---|
| Cu/ZnO/Al2O3 (CZA) | commercial | T = 513 K; p = 2 MPa; H2:CO2 = 3; GHSV = 6000 h–1 | activity = 3.17; selectivity for MeOH = 35% | (205) |
| CuZn/SiC | BMNP | T = 523 K; p = 0.1 MPa; CO2:H2 = 1:9 v/v | activity = 0.03; selectivity for MeOH = 5% | (200) |
| PdZn/SiC | activity = 0.23; selectivity for MeOH = 21% | |||
| PdCu/SiC | activity = 0; selectivity for MeOH = 0% | |||
| 12.5PdCuZn/SiC | TMNP | activity = 0.07; selectivity for MeOH = 23% | ||
| 25PdCuZn/SiC | activity = 0.14; selectivity for MeOH = 40% | |||
| 37.5PdCuZn/SiC | activity = 0.30; selectivity for MeOH = 50% | |||
| Ni–Ga | BMNP | T = 533 K; p = 1 MPa; H2:CO2 = 3 | TOF = 2880; activity = 1.2; selectivity for MeOH + DME = 43% (DME selectivity < 1%) | (203) |
| Au–Ni–Ga | TMNP | TOF = 11160; activity = 1.1; selectivity for MeOH + DME = 51% (DME selectivity < 1%) | ||
| Cu–Ni–Ga | TOF = 5040; activity = 1.8; selectivity for MeOH + DME = 56% (DME selectivity < 1%) | |||
| Co–Ni–Ga | TOF = 1800; activity = 1.3; selectivity for MeOH + DME = 55% (DME selectivity < 1%) |
TOF (h–1);
Specific activity toward MeOH formation (molMeOH kgcat–1 h–1).
These catalysts were prepared by an incipient wetness impregnation method and were shown to have Type II–IV isotherms, correlating to macro- and mesoporous materials that were unchanged from the β-SiC support. However, the surface area, pore volume, and average pore radius were all reduced following the addition of the metal nanoparticles. For the bimetallic CuZn/SiC catalyst, the pores were fully blocked as a result of the larger size of the nanoparticles; however, in the palladium-containing bi- and trimetallic nanoparticles, the particle size was smaller, so the pores in the support were only partially blocked, reducing the average pore size. In general, it was observed that the particle diameter decreased as the palladium content increased, as confirmed through studying XRD patterns and applying the Scherrer equation to estimate the particle sizes. The average particle sizes for the trimetallic catalysts containing 25 mol % and 37.5 mol % Pd were smaller than those for the bimetallic PdZn/SiC catalyst. TEM studies showed a Gaussian particle size distribution for all of the catalysts; however, the smallest average particle diameter was for the trimetallic catalyst containing the highest Pd content (37.5 mol %). Through TPR experiments, the reduction peak representing the formation of Cu0 was observed to occur at lower temperatures for the trimetallic catalysts than for the CuZn/SiC catalyst. The TPR profiles also showed a peak at around 673 K for the trimetallic catalyst, corresponding to PdZn alloy formation. As the CO2 hydrogenation reaction temperature was increased, the CO formation rate increased. The highest rate for the RWGS reaction was measured over the PdZn/SiC catalyst, followed by the CuZn/SiC catalyst. Maximum methanol formation at atmospheric pressure was generated in the temperature range 498–548 K. The methanol formation rate increased with the palladium content of the TMNP catalysts. Although the Cu-containing bimetallic catalysts performed poorly, the trimetallic catalysts were more active, which highlights the potential of the synergistic effects among the three metals to achieve high selectivity and activity. It must be noted, however, that the methanol production by these catalysts at atmospheric pressure is much less than has been obtained at thermodynamic equilibrium.
The combination of Pd, Zn, and Cu is thought to be effective for CO2 hydrogenation because of the formation of a PdZn alloy, which is considered to be an active site for methanol synthesis. Conversely, metallic Pd0 is present in the active surface at which the RWGS reaction occurs. The addition of Cu prevents the formation of metallic Pd0 and hence decreases the rate of CO formation and increases the selectivity for methanol.200 Furthermore, as reported by Zhao et al.,201 DFT calculations on PdCuAu TMNPs with various compositions indicated that the average bond distance of PdaCubAuc clusters, (where a:b:c is the compositional ratio), decreases with increasing Cu content. Hydrogen adsorption is favored by Pd > Cu > Au, highlighting the role of the Pd atoms in these trimetallic catalyst compositions. Pd3Cu2Au2 exhibited the highest H2 adsorption energy and was deemed to be the most suitable for H–H activation among the studied catalysts.201 When Cu and Au are mixed with Pt instead of Pd, the TMNPs are active toward the WGS reaction. Xue et al.(202) showed Au@o-AuCuPt to be most stable among the TMNP catalysts examined for the WGS reaction, with greater stability than respective binary alloys. The increased electronic activity was explained in terms of the d-band center of −3.05 eV being closer to the Fermi level. Both reactants (CO and H2O) bind strongly to the catalyst surface, and exhaustive mechanism analysis has shown that the formic acid mechanism has a lower barrier (3.09 eV) than the redox (4.84 eV) and carboxyl (3.15 eV) routes. The elementary reaction energy barrier of CO and desorption of chemisorbed CO2 is large, which was attributed to the strong adsorbate interaction with Pt.
Metal-promoted NiGa/SiO2 intermetallic catalysts, prepared via incipient wetness impregnation, have also been reported for CO2 hydrogenation.203 The activity order for CO2 hydrogenation to methanol (1.0 MPa, 473–543 K) as a function of TOF was AuNiGa > CuNiGa > NiGa > CoNiGa. The supported TMNP AuNiGa/SiO2 exhibited the highest TOF in this study (>3 s–1 at 533 K), which was a 4-fold improvement over o-NiGa/SiO2. The promotional effect of Au weakened the interaction between the catalyst surface and adsorbates binding through oxygen or carbon and was considered to be due to electronic modification of adding Au to NiGa. The active center for the high activity over AuNiGa is thought to be Au decorated on Ni3Ga, as supported by CO chemisorption, XRD, and STEM-EDX analyses. CuNiGa outperformed the other catalysts in terms of specific activity toward methanol; however, AuNiGa was more active. The addition of Cu is thought to form a Cu–Ga phase, affecting the selectivity.203 Further work will be needed to understand the intrinsic activities of the phases and the correlation to changes in catalytic performance. Other Ga-based catalysts have been prepared and tested for CO2 hydrogenation to methanol. With an optimized composition, Ni2FeGa catalysts were prepared by incipient wetness impregnation with an optimal reduction temperature of 823 K, resulting in alloy formation of the three metals and no evidence of sintering at this temperature. The introduction of Ga into NiFe NPs resulted in higher methanol selectivity through a promotional effect;204 however, the as-prepared Ni2FeGa/SiO2 did not outperform the commercial Cu/ZnO/Al2O3 (CZA) catalyst that is commonly used for methanol synthesis. Trimetallic alloyed systems could be desirable to deviate away from problems associated with CZA, such as water formation from the RWGS reaction, which promotes Cu sintering. Therefore, further investigation into suppressing the RWGS reaction and combining the favorable formation of a PdZn alloy has the potential to optimize CO2 hydrogenation to methanol.
4.1.4. Direct Synthesis of Hydrogen Peroxide
TMNPs have recently been applied to the direct formation of hydrogen peroxide from H2 and O2. Industrially, H2O2 is produced by the anthraquinone method, and in 2014 the global annual production was 3 million metric tonnes, most of which was used for fine chemical synthesis and bleaching of paper and textiles.206 The anthraquinone method is indirect and involves hydrogenation and oxidation of an alkylanthraquinone precursor in organic solvents; an extraction step is also required to recover the H2O2. The multistep method generates significant waste and comes with high transport costs.206 Direct H2O2 synthesis is a potential alternative production route, but a major challenge is obtaining a high selectivity over water and preventing further hydrogenation of H2O2 to water.
Recent studies have investigated the use of supported Pd-based catalysts for the direct formation of H2O2;207−210 however, these materials require promoters (typically acid and halide) to obtain high selectivity.211 Hutchings et al. used multimetallic NPs for the direct formation of H2O2.212 The research team determined that the addition of Au to the supported Pd catalysts resulted in high H2O2 selectivity, eliminating the need for promoters. However, significant overhydrogenation of H2O2 to water was still observed. To further improve the yield of H2O2, the addition of Pt to form a TMNP-based catalyst was investigated.
The study by Hutchings et al. set out to determine an optimal Au:Pd:Pt stoichiometry when supported on CeO2.16 The TMNPs were prepared using an impregnation method, where the total metal content was fixed at 5 wt % and mono- and bimetallic analogues were also prepared. For catalytic testing, the total pressure of the reactor was 4 MPa, and the temperature was kept at 275 K. Each experiment was run for 30 min. The catalytic activity of each trimetallic composition can be seen in Figure 3, given in the form of a contour map. In general, the catalysts consisted of equal parts Pd and Au with a small quantity of Pt added. The best catalyst would exhibit a high H2O2 productivity rate (PR) with a low rate of H2O2 hydrogenation to water. Of the monometallic analogues supported on CeO2, Pd showed the highest PR of 97 molH2O2 kgcat–1 h–1. Supported monometallic Pt and Au achieved PRs of 8 and 1 molH2O2 kgcat–1 h–1, respectively. The PRs for the hydrogenation of H2O2 to water observed over Pd, Pt, and Au were 329, 126, and 118 molH2O2 kgcat–1 h–1, respectively. For each of these catalysts, PRproduct/PRhydrog is less than 1, which is undesirable. Bimetallic Au50Pd50/CeO2 exhibited a PR of 68 molH2O2 kgcat–1 h–1; however, the hydrogenation PR was 145 molH2O2 kgcat–1 h–1, again resulting in a PRproduct/PRhydrog lower than 1. Of the trimetallic compositions consisting of equal-weight quantities of Pd and Au with a small (<10%) weight fraction of Pt, the PR was generally over 100 molH2O2 kgcat–1 h–1. The highest H2O2 PRproduct of 170 molH2O2 kgcat–1 h–1 was observed with Au48Pd48Pt4/CeO2. The corresponding hydrogenation PRhydrog was 145 molH2O2 kgcat–1 h–1, giving a PRproduct/PRhydrog ratio greater than 1. Compositions with more and less Pt saw a decline in PRproduct, but the PRproduct/PRhydrog ratio was ca. 1.1–1.5.
A further composition was investigated whereby a portion of the Pd was replaced with Pt: Au50Pd46Pt4/CeO2.16 The catalyst showed a lower H2O2 PR of 86 molH2O2 kgcat–1 h–1; interestingly, however, this composition highly suppressed the hydrogenation reaction, with a hydrogenation PR of just 11 molH2O2 kgcat–1 h–1. This resulted in a PRproduct/PRhydrog ratio of almost 8, or a selectivity of 88%. Two other regions on the compositional contour map also showed high productivity of H2O2, most notably an almost-entirely Pd composition with some Pt and Au added and a region that was predominantly Pd but with more Pt and Au added. However, catalysts prepared within these compositional regions also showed high hydrogenation and decomposition PRs. Only in the region of nearly equal Au and Pd with a minor concentration of Pt were simultaneous promotion of H2O2 production and suppression of hydrogenation and decomposition observed. Thus, Au50Pd46Pt4/CeO2 was concluded to be the optimal composition for the direct synthesis of H2O2. As the optimal catalyst had a lower H2O2 productivity than the other trimetallic formations yet yielded the highest selectivity, this outcome once again demonstrates the trade-off between total activity and high selectivity that is often observed within the field of catalysis.
Following the impregnation synthesis of each catalyst, a 673 K heat treatment in air was carried out. XPS data showed that following heat treatment the Pd:Au molar ratio on the surface of the TMNPs was far greater than the theoretically expected value.16 Au50Pd46Pt4/CeO2 was expected to have a Pd:Au molar ratio of 1.7; however, experimental data proved the surface ratio to be 165, nearly 100 times greater, which is indicative of a core@shell structure with a Pd shell. Interestingly, Au50Pd50/CeO2 saw a far smaller change in the Pd:Au molar ratio from 1.9 to 7.1. This leads to the possibility that the presence of Pt induces a drastic reorganization of the surface and reconfiguration of the structure of the nanoparticle. As with many other TMNP systems, the electronic induction between the three metals was thought to drive the enhanced catalytic performance toward the production of H2O2. The influence of the support on the efficacy of Pd–Pt–Au mixtures can be compared with supporting research on TiO2 by the same group.213 Again, the inclusion of a low concentration of Pt in the AuPd NPs resulted in the highest PRproduct/PRhydrog ratio of 1.14 in this study, over 2.45%Au–2.45%Pd–0.05%Pt/TiO2, and increasing this Pt concentration decreased the PR ratio (Table 3). Analysis of the catalysts prepared by impregnation indicated that the distribution of NP diameters narrowed with the inclusion of Pt. There was a significant quantity of smaller Pd-rich NPs, and the Pd concentration in the nanoalloys decreased as the particle diameter increased. Interestingly, the inclusion of Pt suppresses the core@shell motif common with AuPd BMNPs supported on TiO2.
Table 3. Summary of Mono-, Bi- and Trimetallic NP Catalysts for the Direct Synthesis of H2O2 from H2 and O2.
| catalyst | catalyst type (supported) | H2O2 formation rate/molH2O2 h–1 kgcat–1 (H2 selectivity/%)a | rate of overhydrogenation (molH2O2 h–1 kgcat–1) | PRproduct/PRhydrog | ref |
|---|---|---|---|---|---|
| 5%Au/C | MMNP | 1.0 | NDb | – | (212) |
| 2.5%Au–2.5%Pd/C | BMNP | 110 (80) | 28 | 3.92 | |
| 5%Pd/C | MMNP | 55 (34) | 107 | 0.51 | |
| 5%Au/Al2O3 | MMNP | 2.6 | NDb | – | |
| 2.5%Au–2.5%Pd/Al2O3 | BMNP | 15 (14) | 92 | 0.16 | |
| 5%Pd/Al2O3 | MMNP | 9.0 | NDb | – | |
| 5%Au/TiO2 | MMNP | 7.0 | NDb | – | |
| 2.5%Au–2.5%Pd/TiO2 | BMNP | 64 (70) | 27 | 2.37 | |
| 5%Pd/TiO2 | MMNP | 30 (21) | 113 | 0.26 | |
| 2.5%Au–2.5%Pd/TiO2 (1:1.9 mol % Au:Pd) | BMNP | 64 | 235 | 0.27 | (213) |
| 2.5%Au–2.5%Pt/TiO2 (1:1 mol % Au:Pd) | BMNP | 65 | 103 | 0.63 | |
| 2.5%Pd–2.5%Pt/TiO2 (1.9:1 mol % Au:Pd) | BMNP | 124 | 129 | 0.96 | |
| 2.45%Au–2.45%Pd–0.05%Pt/TiO2 (1:1.9:0.025 mol % Au:Pd:Pt) | TMNP | 154 | 135 | 1.14 | |
| 2.45%Au–2.45%Pd–0.1%Pt/TiO2 (1:1.9:0.05 mol % Au:Pd:Pt) | TMNP | 159 | 283 | 0.56 | |
| 2.40%Au–2.40%Pd–0.20%Pt/TiO2 (1:1.9:0.10 mol % Au:Pd:Pt) | TMNP | 156 | 318 | 0.49 | |
| 2.28%Au–2.28%Pd–0.45%Pt/TiO2 (1:1.9:0.20 mol % Au:Pd:Pt) | TMNP | 106 | 263 | 0.40 | |
| 0.20%Au–4.60%Pd–0.20%Pt/TiO2 (1:43:1 mol % Au:Pd:Pt) | TMNP | 184 | 382 | 0.48 | |
| Pd/CeO2 (5 wt % Pd) | MMNP | 97 | 329 | 0.29 | (16) |
| Pt/CeO2 (5 wt % Pt) | MMNP | 8 | 126 | 0.063 | |
| Au/CeO2 (5 wt % Au) | MMNP | 1 | 118 | 0.0084 | |
| Au50Pd50/CeO2 (5 wt % M) | BMNP | 68 | 145 | 0.47 | |
| Au48Pd48Pt4/CeO2 (5 wt % M) | TMNP | 170 | 145 | 1.17 | |
| Au50Pd46Pt4/CeO2 (5 wt % M) | TMNP | 86 | 11 | 7.81 | |
| PdAg/C | BMNP | 70 | 29 | 2.41 | (216) |
Reaction conditions: T = 275 K; p = 4 MPa; H2:O2 ratio = 1:2.
ND = not determinable at such low yields.
There have been several other reports of direct H2O2 synthesis using MNP catalysts.214,215 In general, the PRproduct over BMNPs was reported to be lower than that over the TMNP formulations, typically in the 50–100 molH2O2 kgcat–1 h–1 region, with selectivities of <75%. The Au50Pd46Pt4/CeO2 catalyst has a PR of 86 molH2O2 kgcat–1 h–1, similar to the PRproduct of 70 molH2O2 kgcat–1 h–1 for the bimetallic PdAg/C catalyst reported by Gu et al.;216 however, the selectivity was 71%, which was lower than the 88% selectivity obtained with Au50Pd46Pt4/CeO2.
Aside from earlier publications by the Hutchings team, which are summarized in Table 3 (e.g., trimetallic ruthenium-containing PdRuAu/TiO2 catalysts with activity and selectivities similar to those for the AuPdPt formulations),217 to our knowledge there are no other reports of TMNPs for this reaction. Thus, the use of TMNPs for the direct synthesis of H2O2 is a new and promising area of investigation.
4.1.5. Selective Hydrogenation of Alkynes to Alkenes
TMNPs have also been investigated for application in the selective hydrogenation of phenylacetylene to styrene. Industrially, styrene is an important monomer unit used for the production of polystyrene, 22 million tonnes of which was produced in 2015.218 For this reaction, the catalyst must simultaneously inhibit overhydrogenation to ethylbenzene.165
Miyazaki et al. reported several TMNPs based on o-PdZn/SiO2 in which a third metal (M = Cu, Rh, Sn, Au, Pb, or Bi) was added via galvanic replacement, and the catalysts were compared on the basis of their catalytic activities (Figure 14).165 In the context of the reaction, it is important that reaction rate R1 (the rate of formation of styrene) is higher than R2 (the rate of formation of ethylbenzene) to ensure selectivity for styrene over ethylbenzene. The compositions with the highest reaction rate were M = Au, Sn, Rh, and Cu, but these compositions exhibited a greater propensity for overhydrogenation to form ethylbenzene. Furthermore, supported PdZn favors the formation of ethylbenzene over styrene. The inclusion of Pb or Bi, however, favored the formation of styrene, with Pb producing the highest yield of styrene (Figure 14a). The authors suggested that the product preference for styrene is due to steric hindrance because the large atomic radii of Pb and Bi atoms suppress further hydrogenation of the C=C moiety. This observation was supported by previous work by this group whereby overhydrogenation of internal C=C bonds was suppressed as a result of steric repulsion in reactions where large Bi atoms were present on the surface.219 The addition of Pb to the PdZn surface causes different surface structures to form according to DFT calculations; these substituted surfaces appear to increase the length of the Pd–C bond in ethylene over that of acetylene. To demonstrate the steric hindrance effect, other compositions were prepared such that Pbx with x = 0.1–0.4. The R1/R2 ratio was reported to increase with increasing quantities of Pb, but the rates of both hydrogenation reactions were diminished.
Figure 14.

(a) Catalytic performance data for PdZn with several third metals added via galvanic replacement. (b) Conversion of phenylacetylene as a function of reaction time under the reaction conditions, treated at 673 K. Data for an unsupported PdZn0.75Pb0.25 alloy are provided for comparison. Corresponding TOF values are given in brackets. Adapted from ref (165). Copyright 2019 American Chemical Society.
The TOF over the PdZn@Pb0.25/SiO2 catalyst was 450 min–1, in comparison with 273 min–1 over PdZn. The TOF over the commercial catalyst for the same reaction, PdPb/CaCO3 (Lindlar catalyst) was 429 min–1. Upon heating to 673 K, however, the TOF of PdZn@Pb0.25/SiO2 diminished to 141 min–1. The diminished TOF was reportedly due to a phase change that resulted in the loss of the AB@AC structure and the formation of a random PdZnPb alloy. The partial hydrogenation to styrene demonstrates the trade-off between reaction rate and selectivity, as the compositions with the highest conversion rates had the lowest selectivity for the desired product; however, the superior TOF, selectivity, and yield over the PdZn@Pb0.25/SiO2 catalyst suggest that the benefits of high selectivity could outweigh the disadvantage of a lower conversion rate.
In summary, trimetallic catalysts have been successfully applied to a variety of hydrogenation reactions, but because of the recent nature of these discoveries, the number of publications on this subject remains relatively low. The work that has been published to date highlights the importance of continued research into the area and of the balance between selectivity and reaction rate in designing catalysts. The progression to trimetallic catalysts for use in hydrogenation reactions has been shown to improve catalytic performance compared with bimetallic catalysts, particularly for hydrogenation of carbon monoxide and carbon dioxide. Often the third metal can enhance the selectivity through reduction of overhydrogenation, such as in the direct synthesis of H2O2 or alkyne hydrogenation.
4.2. Dehydrogenation Reactions
Dehydrogenation reactions involve the removal or liberation of hydrogen from a molecule. At present, many commercial dehydrogenation reactions are practiced. The dehydrogenations of molecules such as formic acid220−223 and ammonia borane224−226 are of high importance, as are dehydrogenations of alkanes to alkenes227−229 or alkynes.230−232 In finding cleaner and more sustainable energy sources, dehydrogenation reactions have become important in the realization of hydrogen storage for energy, including the specific examples of harvesting stored hydrogen from simple molecules such as formic acid and ammonia borane. Similar to hydrogenation reactions, heterogeneous catalysts (often Pt- or Pd-based) are important for obtaining high activities and selectivities under milder conditions.233−235 The following section details recent examples of the use of TMNPs in various dehydrogenation reactions.
4.2.1. Dehydrogenation of Formic Acid to Hydrogen and Carbon Dioxide
The dehydrogenation of formic acid (FA) to give H2 and CO2 has been widely studied because of the interest in use of FA for hydrogen storage and potential energy generation.236,237 There have been extensive studies investigating modifications of the commercially used Pd/C catalyst for this dehydrogenation reaction.238 As a reference point, Hu et al. characterized and evaluated a commercial Pd/C catalyst and reported a TOF of >500 h–1 at 333 K in aqueous solution.239 Multiple supports have also been investigated, and high TOFs have been observed over catalysts using CeO2, N-functionalized porous carbon (N-MSC-30), and reduced graphene oxide, for example.240,241
Enhanced catalytic performance for formic acid dehydrogenation has been observed through the use of multiple metals in bimetallic and trimetallic systems. For example, the addition of Ag to Pd/C showed a significant improvement in activity; as an example, Zhang et al.(242) prepared a AgPd/C catalyst via a coreduction method, and the Ag42Pd58 alloy catalyst exhibited a TOF of 382 h–1 at 323 K. The enhancement in catalytic performance compared with the as-synthesized monometallic counterparts was ascribed to the charge transfer between Ag and the active Pd center due to the net difference in work function.238
Yurderi et al. prepared trimetallic PdNiAg NPs supported on activated carbon via wet impregnation followed by simultaneous reduction in the absence of stabilizers.27 The dehydrogenation of FA was carried out over a series of mono-, bi-, and trimetallic NP catalysts, and the highest activity was observed over the catalyst composed of Pd0.58Ni0.18Ag0.24 at 323 K (TOF = 85 h–1), with a high selectivity of ca. 100% (Figure 15a). The supported TMNPs were reported to be well-dispersed, with typical particle diameters of 2.5–8.4 nm, and alloyed, as supported by powder XRD, TEM, STEM-EDX mapping, and HAADF-STEM elemental mapping (Figure 15b,c). In the same study, the TMNPs provided higher catalytic activity than the monometallic counterparts prepared by the same method.27 The high activities were explained by the combination of (i) the composition of the trimetallic catalyst, (ii) low activation energies, and (iii) the synergistic effects from a change in the electronic density of Pd resulting from PdNiAg alloy formation. Additionally, the TMNP catalyst showed significant stability, maintaining activity (>94%) after four reusability tests. From ICP-OES data, the PdNiAg/C catalyst exhibited high stability against leaching, as no leaching was observed.27
Figure 15.

(a) Volumes of H2 gas generated as functions of reaction time from the dehydrogenation of FA over MMNP (Pd/C, Ag/C, Ni/C), BMNP (Pd0.55 Ni0.45/C, Pd0.52 Ag0.48/C, Ni0.58Ag0.42/C), and TMNP (Pd0.74Ni0.12Ag0.14/C, Pd 0.42Ni0.36Ag0.22/C, Pd0.40Ni0.16Ag0.44/C, Pd0.18Ni0.38Ag0.44/C) catalysts. Reaction conditions: [metal] = 2.85 mM and [FA] = [SF] = 0.175 M in 10.0 mL of aqueous solution at 323 K. (b) HRTEM images of PdNiAg/C. (c) Elemental distribution of components in the PdNiAg NPs obtained by line-scan analysis using STEM-EDX along the white arrow in the HAADF-STEM image of PdNiAg/C given in the inset. Adapted with permission from ref (27). Copyright 2014 Elsevier.
Wang et al. synthesized NiAuPd/C catalysts by a coreduction method in the absence of a surfactant and applied the catalysts to the dehydrogenation of FA.77 In comparison with the study reported by Yurderi et al.,27 larger TMNP diameters were formed via this method (16–35 nm), and an initial TOF of 12.4 h–1 at 298 K over Ni0.40Au0.15Pd0.45/C was reported, representing an increase in performance compared with the as-synthesized mono- and bimetallic counterparts. The enhanced activity may be ascribed to the synergistic effects that arise through the modification of the electronic structure of the NPs, which lead to different interactions between the catalytic surface and FA. In the example considered here, monometallic Pd/C showed low activity and is known to be susceptible to deactivation through CO poisoning.239,243 Additionally, comparison of the diffraction peaks for NiAuPd NPs to monometallic Au showed a shift to a lower diffraction angle for TMNPs, indicating an increase in the crystal lattice spacing from the formation of a NiAuPd alloy. The study also noted a strong correlation between composition and activity, with the volume of the evolved gas (1 h reaction time) correlated positively with the increase in Ni content to an optimized ratio. As a result, Wang et al. concluded the best composition to be Ni0.40Au0.15Pd0.45/C.77 The catalytic activity as a function of composition (Figure 16) highlights the need to optimize the respective metal concentrations of the active site.
Figure 16.

Illustration of the catalytic activity as a function of composition of TMNPs through measurements of gas generation from the dehydrogenation of FA (0.5 M, 10.0 mL) catalyzed by (a) Nix(Au0.25Pd0.75)1.0–x/C with different x values and (b) Ni0.40(AuyPd1.0–y)0.60/C with different y values at 298 K under ambient atmosphere (nmetal/nFA = 0.02, reaction time = 1 h). Adapted with permission from ref (77). Copyright 2014 Elsevier.
Fang et al. used DFT calculations to investigate Au(core)Pd(shell)Pt(cluster) TMNPs for oxidation of CO and FA. CO adsorption is rare on “two Pd atoms between two Pt clusters mixed-hollow sites”, termed D-sites, which remain free to catalyze the electrooxidation of FA, leading to the high catalytic activity. The optimum Pt coverage on Pd was θPt ≈ 0.5, thus maximizing the number of mixed-hollow D-sites and promoting the catalytic reactivity. The catalytic activity was observed to decrease with increasing thickness of the Pd overlayer, specifically when the thickness was greater than two monolayers. The authors proposed that the Au core can be replaced by less expensive Ag, since Au is recognized as providing structure morphology and not an electronic contribution;244 such a concept could be potentially extended to other low-cost, stable transition metals. Duan et al.(245) used computational methods to investigate nine possible decomposition routes of HCOOH on analogous Au(core)Pd(shell)Pt(cluster) TMNPs, considering a variety of different adsorption sites, as shown in Figure 17.
Figure 17.

Optimized structures of (A) HCOO, (B) HCOOHu, (C) HCOOHd, (D) COOHu, and (E) COOHd adsorbed on different sites in the center area of Au@2 ML Pd@0.5 ML Pt NPs. A key is provided at the top left. Adapted with permission from ref (245). Copyright 2013 The PCCP Owner Societies.
CO, which may poison the catalyst, was repelled by the Pt/Pd edge site, as illustrated in Figure 18. The repulsion of this poisonous species contributed to the high electrocatalytic performance by effectively leaving the site vacant for the CO2 pathway, which itself was simultaneously enhanced in the edge area. H and O atoms were shown to prefer to adsorb on the FCC and HCP sites in the center area of the Pt/Pd surface on Au@2 ML Pd@0.5 ML Pt NPs (core@shell@shell configuration).
Figure 18.

Schematic adsorption areas at (A) the center of a Pd facet and (B) the edge of a Pd/Pt moiety for various adsorption sites (i.e., bridge, FCC, HCP, and top from left to right) and optimized structures of CO adsorbed on Au@2 ML Pd@0.5 ML Pt NPs. A key is provided in the top left corner. Adapted with permission from ref (245). Copyright 2013 The PCCP Owner Societies.
Wu et al. also investigated dehydrogenation of formic acid using TMNPs, with the intent of generating H2in situ for the hydrogenation of nitrobenzene to aniline.26 In the chemical industry, aniline is an important intermediate246 for dyes,247 agricultural chemicals,248 and pharmaceuticals.249 These catalysts were synthesized using the defect-dominated shape recovery method to fine-tune the stoichiometry of the nanoparticle. Among the Pt3Ni-based catalysts and stoichiometries prepared, Pt3Ni@Au0.5 was reported to have the highest activity and selectivity toward the formation of aniline, with a yield of 97% and a TOF of 124 h–1, which is reportedly a 10-fold increase over bimetallic Pt3Ni, for which the TOF was calculated to be 12 h–1. An increase in gold content resulted in a loss of activity, with the TOF of Pt3Ni@Au2 diminishing to 84 h–1. The activity further diminished to merely 6 h–1 with the fully octahedral and defect-free nanoparticle Pt3Ni@Au8.
A loss of activity with increasing metal coverage was also observed when Ag, Cu, and Rh were used as the third metal for the same systems, which supports the postulate that defect sites exhibit catalytic properties, as increasing metal coverage corresponds to a loss of defect sites; however, the identity of the third metal remains important. The activities of catalysts where the third metal was Ag, Cu, or Rh were far lower than for Au as the third metal. The electronic properties of Pt3Ni@Aux were investigated; reported electronic coupling within the structure indicated electron transfer from Au to Pt,250 thereby effectively reducing Pt(II) within the nanoparticle to a lower and more catalytically active oxidation state. Conversely, Au and Ni are slightly oxidized. The TOF of 124 h–1 obtained for the Pt3Ni@Au0.5 composition was considerably greater than the TOF of 85 h–1 obtained with the PdNiAg/C catalyst reported by Yurderi et al.,27 which may in part be due to the 20 K higher temperature used by Wu et al. or the considerably smaller nanoparticle size.
Using a unique support, Mori et al.(251) prepared PdCuCr TMNPs within a macroreticular basic resin. The effect of the slightly basic resin was to assist with O–H cleavage, aiding the dehydrogenation of FA. At 348 K, this catalyst exhibited a high TOF of 830 h–1, which was substantially higher than those over bimetallic PdCu and PdCr catalysts and monometallic Pd. The addition of Cr was thought to stabilize the PdCu NPs against agglomeration and to enrich the Pd center with electrons, which are beneficial synergistic effects that achieve higher catalytic performances.
Khan252 demonstrated that TMNPs with the same elemental composition created by different synthetic methods can lead to different outcomes with regard to both particle morphology and catalytic activity. Using three different orders of metal introduction, Khan synthesized different unsupported core@shell TMNP structures consisting of Pd, Ag, and Fe. The ratio of the metals in each experiment was fixed at 1:1:1, and each TMNP had an M1M2@M3 structure. These catalysts were then applied to the degradation of formic acid to carbon dioxide and hydrogen gas.
First, unsupported PdAg@Fe TMNPs were synthesized using a seedless ligand-capped reduction method with CTAB as the capping agent and hydrazine as the reducing agent. In contrast, FePd@Ag TMNPs were synthesized through stepwise addition of the metals. The Fe precursor was reduced with hydrazine and CTAB, forming Fe0 MNPs. An aqueous Pd2+ solution was added to the Fe MNP suspension along with additional CTAB, depositing Pd onto the surface via a displacement reaction. The FePd BMNPs were isolated from the reaction mixture and dispersed in deionized water; repeating the procedure with an aqueous Ag+ solution in the FePd BMNP suspension then yielded FePd@Ag TMNPs. Finally, FeAg@Pd TMNPs were synthesized by simultaneously reducing Ag+ and Pd2+ onto Fe0 MNPs, and the final product was washed with copious amounts of deionized water to remove any CTAB. The different reduction potentials of the metal salt precursors were used to explain how these materials formed through the formation of a galvanic cell replacement reaction.252
Interestingly, the activity profiles for the dehydrogenation of formic acid over this series of catalysts were different. FeAg@Pd, with kobs = 0.83 h–1 (TOF = 75 molH2 molcat–1 h–1), had the highest activity, whereas over FePd@Ag and PdAg@Fe the observed kobs values were calculated to be 0.54 and 0.33 h–1, respectively (Table 4).252 Over FeAg@Pd, the calculated kobs corresponded to the formation of 88 mL of the (CO2 + H2) product in 120 min; in contrast, over monometallic Fe and Ag catalysts, 3 mL of the product was formed in 120 min. The activation energy over the FeAg@Pd catalyst was 51 kJ mol–1, compared with 60 and 66 kJ mol–1 over the FePd@Ag and PdAg@Fe catalysts, respectively. Although these rates were considerably lower than previous reports, these reactions were carried out at 303 K, whereas the reactions were carried out at >323 K as reported by other groups (Table 4). Overall, the report demonstrates how different preparation methods for TMNPs with the same composition can have a direct and stark impact on the catalytic outcome. This can be due to the different morphologies, sizes, and nanoparticle structures that can manifest even with the same elemental composition, depending on the method used.
Table 4. Summary of Catalysts for Dehydrogenation of Formic Acid.
| catalyst | catalyst type | reaction conditions | performance | ref |
|---|---|---|---|---|
| AgPd/C | supported BMNP | 323 K | TOF = 382 h–1 | (242) |
| Pd/C | spported MMNP | TOF = 75 h–1; volume of H2 generated after 140 min = 63 mL | (27) | |
| Ni/C | supported MMNP | TOF = 4 h–1; volume of H2 generated after 140 min = 4 mL | ||
| Ag/C | supported MMNP | TOF = 2 h–1; volume of H2 generated after 140 min = 2 mL | ||
| Pd0.58Ni0.18Ag0.24/C | supported TMNP | TOF = 85 molH2 molcat–1 h–1; volume of H2 generated after 140 min = 76 mL | ||
| Pd/C | supported MMNP | T = 298 K; p = 0.1 MPa | TOF = 8.3 h–1; formic acid conversion after 600 min = 39% | (77) |
| Ni/C | supported MMNP | formic acid conversion after 600 min = 2% | ||
| Au/C | supported MMNP | formic acid conversion after 600 min = 0% | ||
| Ni0.4Pd0.6/C | supported BMNP | TOF = 8.4 h–1; formic acid conversion after 600 min = 52% | ||
| Au0.25Pd0.75/C | supported BMNP | TOF = 10.1 h–1; formic acid conversion after 600 min = 42% | ||
| Ni0.4Au0.6/C | supported BMNP | formic acid conversion after 600 min = 1% | ||
| Ni0.4Au0.15Pd0.45/C | supported TMNP | TOF = 12.4 h–1; formic acid conversion after 600 min = 73% | ||
| physical mixture of Ni/C, Au/C, and Pd/C (Ni:Au:Pd = 0.4:0.15:0.45) | mixture of MMNPs | TOF = 3.7 h–1; formic acid conversion after 600 min = 22% | ||
| PdAg@Fe | core@shell TMNP | 303 K | kobs = 0.328 h–1 (TOF = 30 molH2 molcat–1 h–1) | (252) |
| FePd@Ag | core@shell TMNP | kobs = 0.540 h–1 (TOF = 49 molH2 molcat–1 h–1) | ||
| FeAg@Pd | core@shell TMNP | kobs = 0.828 h–1; CO2 + H2 generated after 120 min = 88 mL; TOF = 75 molH2 molcat–1 h–1 | ||
| PdCuCr | TMNP constructed in situ | 348 K; HCOOH/HCOONa = 9:1 aqueous solution | H2 production rate = 174 000 mL h–1 gPd–1; TOF = 830 h–1 | (251) |
The emerging reports of TMNPs as catalysts for the dehydrogenation of FA have in part been inspired by the improved catalytic performances observed with bimetallic catalysts for this reaction. In combination with other enhancements, such as using different supports, further improvements can be achieved by trimetallic systems that will improve outcomes compared with the commercial Pd/C catalyst. The optimized electronic character of the NPs is commonly ascribed to the enhanced activity, and this is where optimizing the metal composition can be exploited with TMNPs.
4.2.2. Dehydrogenation of Alkanes to Alkenes
When technology for the dehydrogenation of alkanes to alkenes was first developed in the late 1930s, chromia–alumina catalysts were used. These were mainly employed to produce butenes from butane, which could then be upgraded to more highly valued octenes and octanes. In the 1960s, Bloch identified that platinum-based catalysts could be used as highly active and stable catalysts to selectively dehydrogenate heavy linear alkanes to internal monoalkenes.253 As of 2014, the global production of propene from the dehydrogenation of propane was 5 million tons, and this is set to increase with at least 12 new propane dehydrogenation plants planned to be commisioned.254 There are two patented industrial processes for the dehydrogenation of propane, called the Oleflex process and the Catofin process, which use Pt–Sn/Al2O3 and Cr2O3/Al2O3 catalysts, respectively.
To improve on the Oleflex commercial formulation, the support influence of pure alumina, pure zirconia, and different percentage mixtures of zirconia and alumina (χZr–Al, where χ represents the mass percentage of ZrO2) on PtSnIn-based trimetallic catalysts was assessed.128 The supports were prepared by precipitation/coprecipitation, and the metals were then added by sequential impregnation.
The performance of the catalysts for propane dehydrogenation was assessed by measuring the propene selectivity, the initial and final propane conversion, and the decline in propane conversion over a 2.5 h reaction cycle. The propane conversions, both initial and final, decreased in the order PtSnIn/08Zr–Al > PtSnIn/04Zr–Al > PtSnIn/16Zr–Al > PtSnIn/Al2O3 ≫ PtSnIn/ZrO2. The results of these catalytic studies have been collated in Table 5 for comparison. The PtSnIn/08Zr–Al catalyst also had the highest propene selectivity (>98%). Furthermore, the PtSnIn/08Zr–Al catalyst was proven to be very stable, showing only a 3% decrease in initial conversion over five 2.5 h reaction cycles.
Table 5. Comparison of Catalysts Containing Pt, Sn, and In for the Dehydrogenation of Propane to Propene.
| conversion/%
(selectivity/%) |
|||||
|---|---|---|---|---|---|
| catalyst | catalyst type | reaction conditions | initial | ≥150 min | ref |
| PtSn/In-Al2O3 | supported TMNP | T = 873 K; p = 0.1 MPa; H2:C3H8:Ar molar ratio = 7:8:35; WHSV = 3.3 h–1; mcat = 0.3 g | 47.0 (96.6) | 36.0 (97.1) | (128) |
| PtSnIn/ZrO2 | 22.0 (96.1) | 15.0 (95.9) | |||
| PtSnIn/04Zr–Al2O3 | 54.8 (98.0) | 47.6 (98.8) | |||
| PtSnIn/08Zr–Al2O3 | 57.7 (98.3) | 51.7 (98.7) | |||
| PtSnIn/16Zr–Al2O3 | 51.2 (97.2) | 45.8 (98.6) | |||
| PtSn/In-Al2O3 | supported TMNP | T = 873 K; H2:C3H8:Ar molar ratio = 7:8:35; WHSV = 3.3 h–1; mcat = 0.3 g | 62.0 (96.4) | 37.0 (97.3)a | (127) |
| PtSnIn/0.6Ca–Al2O3 | 62.0 (96.8) | 54.0 (97.6)a | |||
| PtSnIn/1.2Ca–Al2O3 | 60.0 (97.0) | 57.5 (97.8)a | |||
| PtSnIn/1.5Ca–Al2O3 | 62.0 (97.6) | 58.0 (97.8)a | |||
| PtSnIn/1.8Ca–Al2O3 | 54.5 (96.6) | 49.0 (96.25)a | |||
| PtSnIn/2.4Ca–Al2O3 | 50.0 (96.5) | 34.5 (96.25)a | |||
| In/Al2O3 | supported MMNP | T = 893 K; H2:C3H8:Ar molar ratio = 7:8:35; GHSV = 1500 h–1; mcat = 0.3 g | 10.0 (44.0) | 9.5 (58.5)b | (255) |
| Pt/Al2O3 | 29.5 (55.5) | 15.5 (67.0)b | |||
| PtSn/Al2O3 | supported BMNP | 37.5 (83.0) | 23.5 (86.0)b | ||
| PtIn/Al2O3 | 53.0 (93.5) | 37.0 (92.0)b | |||
| PtSn/In-Al2O3 | supported TMNP | 58.0 (92.5) | 48.0 (93.5)b | ||
After 22 h.
After 2.75 h.
Further examination revealed that the poor performance of the pure-zirconia-supported catalyst for propane dehydrogenation was probably due to its negligible surface acidity and low surface area. The ZrO2 was also found to obscure some of the Pt particles through a strong metal–support interaction, which resulted in a zirconia suboxide species decorating the surface of the Pt and obscuring its active sites. The PtSnIn/χZr–Al catalysts, however, performed well because of the moderate surface acidity arising from the high percentage of weak and medium-strength acid sites. These catalysts also have the smallest particle size and most homogeneous dispersion of metal. The χZr–Al mixture can promote the reduction of Pt species and stabilize the Sn oxidation states, which helps to maintain the catalyst stability. Assessment of the catalysts through C3H6 TPD showed that the reduction temperature of propene decreased in the order PtSnIn/ZrO2 > PtSnIn/Al2O3 > PtSnIn/08Zr–Al. The reduction temperatures show that the combination of ZrO2 and Al2O3 weakens the propene–support interaction compared with the other supports, and hence, the product is more easily removed from the catalyst before it reacts further to give higher-order dehydrogenation products, ensuring high selectivity.
In an earlier study, the same group investigated doping of γ-Al2O3-supported PtSnIn catalysts with calcium.127 As propane dehydrogenation is an endothermic reaction, high temperatures are usually employed, leading to rapid catalyst deactivation and poor selectivity due to the formation of coke and cracking. Ca has been shown to improve the stability of the catalyst and metal NP dispersion in bimetallic PtSn catalysts, resulting in improved performance. Long et al. prepared a range of PtSnIn/χCa–Al catalysts (where χ represents the mass percentage of Ca) by sequential impregnation. The initial conversions of propane over PtSnIn supported on 0.0Ca–Al, 0.6Ca–Al, 1.2Ca–Al, and 1.5Ca–Al were similar at ca. 60%; however, the final conversions were 36.7%, 54.0%, 57.0%, and 58.9%, respectively, indicating how the surface concentration of Ca affects the efficacy of the catalyst as a function of reaction time. The most stable catalyst was PtSnIn/1.5Ca–Al, which exhibited just a 1.2% decline in propane conversion over a 22 h reaction cycle; however, as the Ca content was increased above 1.5 wt %, the propane conversion and catalyst efficacy decreased. The PtSnIn/1.8Ca–Al catalyst showed an initial conversion of 54.6% and a final conversion of 49.5% after a 22 h reaction cycle, and the PtSnIn/2.4Ca–Al catalyst exhibited an initial conversion of 50.0% and a final conversion of 34.5%. Furthermore, over both of these catalysts the selectivity for propene was below 97%, whereas the catalysts with <1.5 wt % Ca had a propene selectivity of over 97.5%.
This study suggests that among all of the Ca-doped catalysts tested, the ideal catalyst for propane dehydrogenation was PtSnIn/1.5Ca–Al, which showed the least decline in propane conversion during time on stream and a high selectivity of over 98.0%. For this catalyst, the initial propane conversion decreased from 60.1% to 58.9% after 22 h on stream, which is a minimal decrease of 1.2%. After 100 h, the propane conversion had decreased by a further 24.9–34.0%, but this represents much higher stability than catalysts with other Ca concentrations. The reason given for the improved performance is that Ca introduces a bifunctional effect by providing acidic active sites on the support in addition to the metal active sites in the nanoparticles. These two kinds of active sites work synergistically and as such exist in an optimum ratio. The alkaline nature of Ca means that it neutralizes the otherwise strongly acidic support sites on the alumina, reducing negative effects caused by isomerization and coking that are prevalent on strongly acidic sites; however, if the concentration of Ca is too high, the optimum ratio of metal sites to acidic support sites is lost.
Comparison of these two studies by Long et al.(127,128) (Table 5) shows that the PtSnIn/1.5Ca–Al catalyst displayed the best activity and selectivity, with an initial propane conversion of 60.1%, which decreased by 0.8% to 59.3% after 2.5 h. This is a higher conversion and smaller decrease than observed over the PtSnIn/08Zr–Al catalyst, which had an initial propane conversion of 57.7% that decreased by 6.0% to 51.7% after 2.5 h on stream. However, the PtSnIn/08Zr–Al catalyst was more selective for the desired product, propene, with an initial selectivity of 98.3% that increased to 98.6% after 2.5 h; comparatively, over the PtSnIn/1.5Ca–Al catalyst the initial propene selectivity of 96.6% increased to 97.0% after 2.5 h.
The addition of a third metal in propane dehydrogenation catalysts has been shown to have beneficial effects. Long et al.(255) tested trimetallic PtSnIn catalysts, alongside monometallic Pt and bimetallic PtSn, supported on γ-Al2O3. Again, an impregnation method was used to produce the catalyst. The study showed that the addition of a third metal, indium, to a bimetallic PtSn catalyst resulted in improved catalytic performance and stability because the In helps to neutralize the strongly acidic sites on the γ-Al2O3 surface, enabling higher selectivity toward propene. After a 53 h reaction cycle, the final propane conversion was >41.0% and the selectivity remained stable at >96% at 873 K. The dilution effect caused by adding an additional metal also facilitated a reduction of the particle dimensions, with the average diameter of the PtSnIn TMNPs being 10.8 nm, compared with 17.9 nm for monometallic Pt/Al2O3 and 12.1 nm for bimetallic PtSn/Al2O3.
To compare monometallic, bimetallic, and trimetallic catalysts for the propane dehydrogenation reaction, an experiment was run over 2.75 h with monometallic In/Al2O3 and Pt/Al2O3 catalysts, bimetallic PtSn/Al2O3 and PtIn/Al2O3 catalysts, and trimetallic PtSn/In-Al2O3. Under the reaction conditions of 893 K and GHSV = 1500 h–1, the results outlined in Tables 5 and 6 were obtained. These results highlight the advantages of a TMNP catalyst for the propane dehydrogenation reaction, as the propane conversion decreased less than with the BMNP catalysts during the experiment and the propene selectivity was high, modestly improving to 93.5% during the test.
Table 6. Comparison of the Performances of Different Catalysts for Propane Dehydrogenation at 2.75 h Time on Linea,255.
| catalyst | XI (%) | Xf (%) | ±X (%) | SI (%) | Sf (%) | ±S (%) |
|---|---|---|---|---|---|---|
| In/Al2O3 | 10.0 | 9.5 | –0.5 | 44.0 | 58.5 | +14.5 |
| Pt/Al2O3 | 29.5 | 15.5 | –14.0 | 55.5 | 67.0 | +11.5 |
| PtSn/Al2O3 | 37.5 | 23.5 | –14.0 | 83.0 | 86.0 | +3.0 |
| PtIn/Al2O3 | 53.0 | 37.0 | –16.0 | 93.5 | 92.0 | –1.5 |
| PtSn/In-Al2O3 | 58.0 | 48.0 | –10.0 | 92.5 | 93.5 | +1.0 |
Xi and Xf are the initial and final propane conversions, respectively, and ±X is the change in conversion over the reaction cycle. Si and Sf are the initial and final propene selectivities respectively, and ±S is the change in selectivity over the reaction cycle.
4.2.3. Dehydrogenation of Ammonia Borane
Ammonia borane is considered to be a suitable material for hydrogen storage because of its high hydrogen storage capacity, air stability, nonflammability, and low toxicity.256,257 The key driver for this technology is to efficiently dehydrogenate ammonia borane and release molecular hydrogen; consequently, many catalyst formulations have been evaluated, including those with TMNPs.
Randomly alloyed PtAuCo catalysts prepared by Fu et al. were tested for the dehydrogenation of ammonia borane (Figure 19).78 Several ratios of PtAuCo were synthesized and tested: Pt76Au12Co12, Pt76Au11Co13, Pt74Au21Co5, Pt80Au16Co4, Pt78Au6Co16, and Pt56Au4Co40. The performance was compared to that of Pt85Au15 and Pt86Co14 bimetallic alloys, core@shell Au@Co, and monometallic Pt, Au, and Co. The TOF (moles of hydrogen formed per mole of metal per hour) obtained for each catalyst is displayed in Table 7, and these values are compared with those obtained over other catalyst formulations discussed here.
Figure 19.

(a) Plots of H2 generation vs time and (b) the corresponding TOFs (molH2 molM–1 min–1) for the ammonia borane hydrolysis reaction at 298 K catalyzed by trimetallic Pt–Au–Co catalysts with different compositions. (c, d) TEM images of Pt56Au4Co40 obtained after refluxing for 1 h and (e, f) the corresponding EDX data indicating the segregation of elements present due to high Co concentration, causing a reduction of catalytic activity. Adapted from ref (78). Copyright 2020 American Chemical Society.
Table 7. Catalytic Performance of a Variety of Supported and Unsupported TMNP, BMNP, and MMNP Formulations for the Ammonia Borane Dehydrogenation Reaction.
| catalyst | catalyst type | reaction temperature (K) | TOF (h–1) | ref |
|---|---|---|---|---|
| Pt76Au12Co12 | TMNP alloy | 298 | 27000 | (78) |
| Pt76Au11Co13 | 23460 | |||
| Pt74Au21Co5 | 15900 | |||
| Pt80Au16Co4 | 15540 | |||
| Pt78Au6Co16 | 6000 | |||
| Pt56Au4Co40 | 5640 | |||
| Pt85Au15 | BMNP alloy | 8220 | ||
| Pt86Co14 | 3960 | |||
| Au@Co | BMNP core@shell | 840 | ||
| Pt | MMNP | 240 | ||
| Au | 180 | |||
| Co | 120 | |||
| Pt (black) | MMNP | 298 | 840 | (258) |
| Ru@Co | BMNP core@shell | 298 | 20640 | (259) |
| AuCo/CN | supported BMNP | 303 | 2880 | (260) |
| PtCo | BMNP | 293 | 18180 | (261) |
| PtCu | BMNP | 298 | 6480 | (262) |
| Cu40Co60/BN | supported BMNP | 298 | 480 | (263) |
| PtCoCu@SiO2 | protected TMNP | 303 | 16380 | (264) |
| Cu72Co18Mo10 | TMNP | 298 | 7140 | (265) |
| CuNiCo/MOF | supported TMNP | 298 | 4200 | (266) |
| RuCuNi/CNTs | supported TMNP | 298 | 18669 | (28) |
The TOFs obtained over the catalysts prepared by Fu et al.(78) varied greatly according to the stoichiometry of the three metals; however, the Pt76Au12Co12 composition proved to be superior to the other catalysts in the study, with a TOF of 27 000 h–1. By comparison, the most active BMNP formulation within this study was Pt85Au15, with a TOF of 8220 h–1, which is less than one-third of the reported TOF of Pt76Au12Co12. A separate study by Cao et al.(259) found that unsupported Ru@Co core@shell NPs are a more active BMNP formulation than Pt85Au15, with a TOF of 20 640 min–1; however, this turnover is still outperformed by the Pt76Au12Co12 composition. The performance of the Pt, Au, and Co MMNPs was relatively poor, with TOFs of 120–240 h–1, which are 2 orders of magnitude lower than those of the most active catalysts.
Analysis of XPS, TEM, and EDX characterizations of the PtAuCo catalysts indicated that as a result of a phase and size mismatch, a high concentration of Co caused segregation of the metals, as can be seen in TEM images of Pt56Au4Co40 (Figure 19), and this was correlated to a decrease in catalytic performance.78 Nanoparticle segregation occurred at high concentrations of Co, as can be seen for the high-cobalt composition (Pt14Au6Co80), where the majority of the MNP population were monometallic Co, with segregated Au/Pt islands. Similarly, Pt78Au6Co16 exhibited a small amount of Co segregation. The PtAuCo catalysts were assessed for their stability using two methods: first, another equivalent of ammonia borane solution was added following the cessation of H2 formation; second, the catalyst was recovered by centrifugation, washed with deionized water, and redispersed in a fresh ammonia borane solution. With both methods, the catalysts showed significant degradation of performance after five runs. The TOF of the Pt76Au12Co12 catalyst fell to 15 120 h–1 by the fifth run with the first method. The second method resulted in less degradation, with the TOF falling to 21 900 h–1 by the fifth experiment. The bimetallic Pt85Au15 composition exhibited a similar decline in TOF, falling from 8220 to 3900 h–1 by the fifth run with the first method. With the second method, the activity decreased modestly, to 5580 h–1. The second method proved to be superior to facilitate reusability, as the Pt76Au12Co12 retained 81% activity after five runs compared with 56% activity with the first method. The loss of activity was attributed to the production of BO2– ions in the reaction, which increased the viscosity of the solution and slowed the diffusion, adsorption, and desorption processes.267
As Pt is the main component of many highly active catalysts for the dehydrogenation of ammonia borane, Pt likely provides the main active sites for Pt76Au12Co12. XPS measurements indicated that the Pt in the nanoalloys was slightly oxidized. The electronegativity of Pt is 2.20, whereas that of Au is 2.54 and that of Co is 1.70. The presence of Au results in a positive shift of the binding energy (BE), whereas Co results in a negative shift (Pt 4f BE = 71.34 eV for Pt76Au12Co12, 71.52 eV for Pt85Au15, and 71.25 eV for Pt86Co14). The data suggested that the intermediate binding energy of the Pt76Au12Co12 composition provided the optimal interaction energy between the catalyst and the active intermediates in the ammonia borane dehydrogenation reaction.
Other TMNP catalysts, which combine different metals than those already discussed, have been investigated for the dehydrogenation of ammonia borane. Highly dispersed, alloyed RuCuNi NPs were supported on carbon nanotubes (40–60 nm diameter) via a chemical reduction method and tested for ammonia borane dehydrogenation.28 Ru/CNTs, Cu/CNTs, Ni/CNTs, bimetallic RuCu/CNTs and RuNi/CNTs, and trimetallic RuCuNi/CNTs were prepared and then tested at room temperature. The resulting catalytic activity order was RuCuNi/CNTs > RuNi/CNTs > RuCu/CNTs > Ru/CNTs > Ni/CNTs > Cu/CNTs. The importance of alloy formation was investigated by Xiong et al. through comparison of RuCuNi/CNTs with physical mixtures of the as-prepared monometallic Ru/CNTs, Cu/CNTs, and Ni/CNTs, which showed inferior catalytic activity compared with the alloyed trimetallic catalyst. Overall, the study showed strong synergistic effects among the three alloyed metals, as manifested by the reported increase in catalytic activity for this reaction; it is also to be noted that the RuCuNi/CNTs catalysts exhibited excellent catalytic activity in recycling tests, where 61.9% of the initial activity was retained after five cycles.28
Sen et al. reported superior catalytic activity for the dehydrocoupling of dimethylamine borane over trimetallic PdRuNi NPs supported on graphene oxide.121 With a TOF of 737 h–1, this was reported as one of the leading catalytic activities in this field, along with RuPtNi/GO (TOF = 727 h–1)268 and RuCo/f-MWCNT (TOF = 775 h–1).269 In contrast, TOFs of 38 and 272 h–1 were reported for the dehydrocoupling of dimethylamine borane over Pd/GO and PdNi/GO, respectively.270,271 The superior catalytic performance observed was ascribed to the synergy of the three alloyed metals along with the small diameter (ca. 3.8 nm), high monodispersity, and high metallic ratio. The prepared trimetallic nanocomposites showed excellent catalytic performance and can be considered as a competitive alternative to other catalysts to obtain hydrogen gas from the dehydrocoupling of dimethylamine borane.
The use of TMNP catalysts for dehydrogenation reactions reflects the emerging use of TMNP catalysts in general. Publications involving the use of TMNPs for dehydrogenation reactions have appeared only within the last 10 years, alluding to the novelty of such catalytic systems. From the examples detailed in this section and collated in Table 7, the addition of a third metal has proven beneficial in many systems, particularly the dehydrogenation of formic acid and ammonia borane as well as the generation of alkenes and alkynes. In many cases, the addition of a third metal significantly enhanced the catalytic activity and improved the selectivity for the desired product through electronic modification and/or morphology control of the NPs.
4.3. Catalytic Oxidation Reactions over TMNPs
Catalytic oxidation reactions play crucial roles in the contribution of green and sustainable processes and in the production of key intermediates and chemicals. Oxidations often lead to low selectivity and can be difficult to control, and therefore, selective oxidation requires careful design of catalysts to overcome this challenge.272 Au- and Pd-based catalysts are among the more commonly used materials in catalytic oxidations of alcohols and polyols.273−275 The continual research into finding more active, selective, and stable catalysts is apparent, and alloying of other transition metals with noble metals is desirable to enhance the catalytic performance and also reduce the cost.276 Herein we discuss the impact of a variety of reports of oxidation reactions over TMNP catalysts.
4.3.1. Oxidation of Glucose and Alcohols
Oxidation of abundant chemicals such as glucose can generate important intermediates that are used in the food and pharma industries; however, glucose oxidation is typically achieved via enzymatic catalysis, and therefore, high catalytic activity is demanded of heterogeneous catalysts for them to be considered as a replacement technology.
Polymer-protected Au70Pt20Ag10 NPs exhibited high catalytic performance for the aerobic oxidation of glucose, with a high average activity of 20 090 molglucose h–1 molM–1 at 2 h (3.8 times higher than that of monometallic Au NPs) under operating conditions of 333 K and 0.1 MPa O2.277 The Au70Pt20Ag10 catalyst was reported to comprise small-diameter particles (ca. 1.5 nm) formed by a chemical reduction method and protected by PVP. It was concluded that in addition to the small NP size, electronic charge transfer from adjacent elements resulted in higher catalytic activities in comparison with those of mono- and bimetallic NPs. With the addition of Ag as the third metal, this species could donate electrons to Au and Pt (the ionization energies of Ag, Au, Pt are 7.58, 9.22, and 9.02 eV, respectively) (Figure 20). Compared with the bimetallic system, where there is only one route for charge transfer, the TMNP system had two further charge transfer routes: Ag → Au and Ag → Pt. The synergistic effect of the combination of the three metals was used to explain the increased activity.277 To support the synergistic postulate, XPS measurements and DFT calculations indicated the presence of negatively charged Au atoms in such trimetallic alloys that were formed through electron transfer from Ag. The negatively charged Au atoms were thought to be the crucial active sites in glucose oxidation. For the study outlined, it is also noted that 70% of the initial activity was maintained after four cycles, giving a total of 8 h for the long-term activity of the aerobic oxidation of glucose.
Figure 20.

(a) Illustration of potential electronic charge transfer routes in PVP-protected Au/Ag and Au/Pt/Ag NPs. (b) Metal time yield (MTY, in units of moles of glucose per hour per mole of metal) over TMNPs with different compositions, highlighting the advantages of an optimized metal ratio according to improved charge transfer. Adapted from ref (277). Copyright 2011 American Chemical Society.
Subsequently, Zhang et al. reported the use of AuPdPt TMNPs in a colloidal suspension for the aerobic oxidation of glucose.80 TMNPs with a variety of Au:Pt:Pd atomic ratios were synthesized, and the particles were compared with monometallic Pd, Pt, and Au and bimetallic Pt75Pd25, Au60Pt40, and Au60Pd40 materials. On the basis of TEM images, the reported diameters of these nanoparticles ranged from 1.3 ± 0.5 nm (Au60Pt10Pd30) to 2.1 ± 0.6 nm (Au70Pt20Pd10). The reported catalytic activities for the aerobic oxidation of glucose over the various species indicate that an improvement in catalytic activity can be achieved via addition of a second metal. The Au60Pt40 bimetallic nanoparticles exhibited a 4-fold increase in the conversion rate in comparison with their monometallic counterparts. Upon the addition of a third metal, such as in the case of Au60Pt30Pd10, a further increase in the rate was achieved in comparison with the bimetallic compositions; however, the data in Figure 21b highlight how variance in the Au:Pt:Pd ratio can drastically influence the catalytic activity. Higher concentrations of Au resulted in a reduced rate, with Au90Pt5Pd5 having a rate similar to that for monometallic Au, corresponding to a 4-fold decrease in activity compared with Au60Pt30Pd10. Figure 21c illustrates that variations in the stoichiometry of Pt and Pd, with the concentration of Au fixed at 60%, had an impact on the catalytic performance. The optimal stoichiometry was reported as Au60Pt10Pd30, with a glucose oxidation rate of 26 430 molglucose h–1 molM–1. The activity of this composition is more than 5 times greater than those of the corresponding monometallic NP catalysts and more than 15% greater than for the bimetallic compositions such as Au60Pt40.
Figure 21.

(a) Catalytic activities of Pd, Pt, Au, Pt75Pd25, Au60Pt40, Au60Pd40, and Au60Pt30Pd10. The corresponding mean diameters of the nanoparticles are shown. (b) Catalytic activities of AuPtPd nanoparticles with Au fractions of 60, 70, 80, and 90%. (c) Catalytic activities of AuPtPd nanoparticles with 60% Au. Adapted with permission from ref (80). Copyright 2013 Elsevier.
The increase in activity in the highlighted work was again concluded to originate from electronic charge transfer between elements, which is due to their different electronegativities. In this case, Pd is the most electropositive metal and donates electron density to neighboring Au and Pt atoms. DFT calculations confirmed the Mulliken charges on a simulated Au37Pt12Pd6 nanoparticle to be −0.035 for Au, −0.024 for Pt, and +0.168 for Pd. The induced (slight) negative charge of the Au atoms could activate O2 molecules by donation of electronic charge into the antibonding orbital of the O atom to form a superoxo-type radical; this highly active oxygen species then would promote the glucose oxidation reaction, which in turn is facilitated by the Pt and Pd sites.80 Zhao et al. investigated various TMNP AuPdPt compositions with DFT calculations and observed that Au took exposed positions (lower CN) with Pd in the center (higher CN).278Figure 22a illustrates the calculated average interatomic distances of the cluster according to the composition. Interestingly, longer O–O bonds appear on clusters with shorter average metal–metal distances as the cluster composition changes. Subsequent calculations indicated that high Pt and low Au content yielded a greater oxygen binding energy and shorter bonds, while the largest O2 bond elongation and lowest binding energies were observed for Au2Pd2Pt3 and Au2PdPt4. Mixing was preferred over segregation of bi- and monometallic phases, and O2 favored binding in the order Pt > Pd > Au, as depicted in Figure 22b. The observations were attributed to lower vertical ionization potentials and electron affinities of clusters with higher Pd content. The adsorption energy increased with decreasing Au content, and this was approximately correlated with the electron transfer from the metal cluster to O, which itself aligned with the highlighted experiment.278
Figure 22.

Ternary contour maps of (a) calculated average Au–Pd, Au–Pt, and Pd–Pt distances (in Å) for AuxPdyPtz (x + y + z = 7) clusters and (b) calculated O–O distances (in Å) in AuxPdyPtz–O2 (x + y + z = 7) clusters as functions of the atomic composition. Adapted from ref (278). Copyright 2017 American Chemical Society.
Selective oxidation of glycerol can yield a variety of useful commercial products and intermediates in organic synthesis, including glyceric acid (GLYA), dihydroxyacetone (DHA), and tartronic acid.279,280 With regard to glycerol oxidation, it is crucial to tune the selectivity, overcome deactivation caused by strong adsorption of acids and ketones, and avoid overoxidation.281 Kimura et al. pioneered a commercial Pt/C catalyst with added bismuth.282 The DHA selectivity increased from 10% to 80% when Bi was added to the parent Pt/C catalyst, and it was proposed that surface submonolayers of bismuth acted as site blockers on Pt(111), controlling the glycerol orientation.
Earlier studies used basic conditions for glycerol oxidation, but base-free conditions have recently been developed due to the use of base additives forming impurities.281 Additional studies have been conducted to further enhance the commercial Pt/C and BiPt/C catalysts (where C is activated carbon). Villa et al. modified AuPt/C with bismuth (0.1–1 wt %) and compared the resulting catalysts to the commercial catalysts (Pt/C and BiPt/C) in the base-free oxidation of glycerol.283 Adding 1 wt % Bi to AuPt/C had no significant effect on the activity (TOF = 585 h–1) compared to nonmodified AuPt/C (541 h–1). Conversely, a higher TOF (773 h–1) was observed with the addition of only 0.1 wt % Bi. In comparison, commercial Pt/C and BiPt/C exhibited activities of 586 and 914 h–1, respectively (353 K, pO2 = 0.3 MPa, base-free). High amounts of Bi were thought to block the active sites, reducing the promotional effect of Bi addition, as seen in previous studies using addition of 1–3 wt % Bi to a bimetallic AuPd catalyst.284
In terms of selectivity, the addition of Bi directed the oxidation of the secondary alcohol function, yielding DHA as the main product. In the absence of Bi, glyceric acid was the main product, as oxidation of the primary alcohol group was favored; however, the selectivity for DHA depended on the amount of Bi, with 1 wt % Bi decreasing the DHA selectivity. With a low Bi loading, stable DHA selectivity (63–65%) with conversion was obtained, in contrast to a drastic drop in selectivity over commercial BiPt/C, where at 30% and 80% conversion the selectivities were 53% and 35%, respectively. Analysis of XPS measurements revealed that Bi did not modify the electronic properties of Au and Pt; therefore, the group proposed that the change in selectivity was due to a change in the geometric nature of the NPs.284
Recycling tests were performed to compare 0.1 wt % Bi-modified AuPt/C with commercial BiPt/C (4 h reaction, catalyst filtered and reused after each run without any further treatment). Interestingly, 0.1 wt % BiAuPt/C showed better resistance to deactivation, as Au enhanced the stability by limiting the leaching of Bi, whereas BiPt/C resulted in high Bi leaching, as confirmed by ICP measurements.283
Other bimetallic systems have shown considerable activities toward glycerol oxidation, with a mixture of selectivities for GLYA or DHA, including PdPt/TiO2,25 AuPt/NiO,285 PtBi/MWCNTs, and PtSb/MWCNTs.286 Kondrat et al. prepared AuPdPt TMNPs supported on TiO2 by sol immobilization and reported enhanced catalytic performance for base-free glycerol oxidation (373 K, pO2 = 0.3 MPa, 0.5 h).25 Bimetallic PdPt/TiO2 showed high selectivity for C3 products. However, the addition of Au as the third metal increased the catalytic activity: the TOF increased from 210 h–1 over PdPt/TiO2 to 378 h–1 over the trimetallic AuPdPt/TiO2 catalyst. Notably, monometallic Pt NPs were reported to prevent toluene formation in glycerol oxidation. After the first reusability test, the particle diameter was determined to be ca. 2.6 nm by TEM. The catalyst showed small amounts of leaching and agglomeration after the second reuse, and the average particle diameter increased to 4.3 nm, indicating instability after two runs.
Using sol immobilization preparation, the same group also supported AuPdPt NPs on activated carbon, and the catalytic activity for the solvent-free oxidation of benzyl alcohol was investigated.133 Detailed aberration-corrected STEM-EDX analysis confirmed the formation of ternary alloys but also indicated compositional differences between particles for the trimetallic catalysts. The fluctuation in composition varied systematically with NP size, as larger particles were Pd-rich whereas smaller particles were Pd-deficient—an example of size-dependent composition. Ostwald ripening could also be a factor in the Pd-rich particles.287 In comparison, the compositional variation was not observed in the AuPt bimetallic system. Oxidations were carried out for 2 h at 393 K and pO2 = 1.0 MPa. Over a 1 wt % Au0.45Pd0.45Pt0.1/C trimetallic catalyst, a TOF of 31 900 h–1 was reported, with 80.2% selectivity for benzaldehyde. In contrast, over monometallic Pt/C, a TOF of 2910 h–1 was recorded, but a high selectivity of 90.7% for benzaldehyde. Over bimetallic Au0.5Pt0.5/C, a TOF of 8450 h–1 was reported, with 84.9% selectivity for benzaldehyde. Addition of low levels of Pt to the AuPd system resulted in higher selectivity by suppression of toluene formation, which was thought to be due to the enhanced relative stability of platinum hydride in comparison with palladium hydride.272
4.3.2. Oxidation of Carbon Monoxide to Carbon Dioxide
Carbon monoxide is a well-known toxic gas and a pollutant with various environmental and health-related problems. Catalytic CO oxidation is an important research area because of its applicability in several processes, such as methanol synthesis, the water gas shift reaction, and automotive exhaust controls.288−290 Haruta et al. demonstrated that supported Au NPs exhibited high catalytic activity when the diameter of the Au NPs was less than 5 nm.291 The excellent affinity of Au to selectively bind with CO enhances the catalytic activity, and CO oxidation is thought to take place at the interface between the Au metal NP and an oxide support, such as ZnO, TiO2, or CeO2.45,292 Pt metal catalysts are also highly active for CO oxidation,293 but other non-noble metals such as Fe, Co, Ni, and Cu have received recent focus due to the motivation to reduce cost.294,295 High activity, selectivity, and stability are constant challenges in heterogeneous catalysis, so it is crucial to design catalysts with highly active sites, for example, to render them useful in applications. Adding a second or third metal can not only increase the activity, selectivity, and stability through synergistic effects but also reduce the price, for example by reducing the weight percent of costly platinum-group metals by addition of other metals such as Fe and Cu.
There is limited literature on the use of TMNPs as catalysts for CO oxidation, but successful reports concern mono- and bimetallic catalysts.293,296−298 Yang et al.(276) synthesized PtNiCo alloyed NPs via a polyol method with hexadecanediol as the reducing agent, and three different support materials were investigated: carbon black, silica, and titania. The CO conversion rates (in terms of Pt-specific mass activity of the NPs supported on carbon black) followed the trend PtNiCo/C > PtCo/C > PtNi/C. Pt was the active site, but the combination of Pt, Ni, and Co in the alloy led to enhanced activity and stability in comparison with the mono- and bimetallic counterparts. Ni was responsible for the enhanced stability, while the synergy of adding Co promoted the activity. The formation of PtNiCo trimetallic alloy sites promoted oxygen activation on the surface, facilitating the adsorption and activation of CO and O2. Overall, the study highlighted the importance of oxygen-activation sites on the nanoalloy trimetallic surface and the tunability of active sites related to support–nanoalloy interactions.
Tripathi et al. studied a 55-atom Pt31Ni12Co12 FCC catalyst using DFT modeling approaches.299 The system exhibited limited CO poisoning, which is advantageous, but was inferior to PtNi for CO oxidation activity. The occupancy of d states and the position of the d-band center were used to determine optimal alloying metals with Pt and revealed that lowering the back-donation ratio was key to reduce the CO binding energy. There remains a trade-off between activity and reduction in CO poisoning, and a balance can be struck by analysis of the occupancy of d states and the position of the d-band center, which can be harnessed in further work in this field.
Zhu et al. tested highly mixed PtPdRh FCC (111) and (100) slabs with a major focus on C–C splitting and CO oxidation.181 They found that alloy systems were better for C–C bond cleavage over monometallic equivalents, indicating synergy. Pd in the Pt–Pd–Rh nanocrystals was shown to promote activity and durability by offering oxidative hydroxyl groups that facilitated the oxidation of adsorbates. For (100), Pd-rich compositions are favorable for C–C cleavage; for (111), the highest reactivity was observed with a 1:1:1 composition, which was considered suitable for CO oxidation as well. The CO oxidation capability increased with increasing Pt content in ternary nanotruncated octahedrons as a result of a lower energy barrier, while Rh improved the selectivity for CO2.
4.3.3. Partial Oxidation of Methane to Methanol
The direct formation of methanol from methane remains a promising process to generate an important chemical with the potential to be used to form fuels or resins, for example. Methane reserves have been estimated to exceed 2 × 1014 m3.300 Currently, the dominant method of converting methane to methanol is a two-step process. The first step involves the conversion of methane to syngas (CO + H2), and this step is highly energy-intensive. The second step converts syngas selectively to methanol over a methanol-selective catalyst,301 which is highly selective, but again the high energy demand and harsh conditions of the process leave much room for improvement. The direct or one-pass conversion of methane to methanol, for example, is characterized by high selectivity; however, the low conversion of methane provides the main challenge.
Previous studies have used H2O2 as the oxidizing agent;302−305 however, the use of concentrated H2O2 is associated with its own challenges, including storage and transport. Therefore, the catalyst must both catalyze the direct synthesis of H2O2 and the oxidation of methane. Previous reports have detailed a supported BMNP formulation, AuPd/TiO2, to catalyze the oxidation of methane with H2O2 in water at 323 K.306,307
With the addition of copper, Ab Rahim et al.(308) expanded the AuPd/TiO2 system to a trimetallic CuAuPd/TiO2 system in order to investigate the effect of a third metal on the catalysis. The effects on the H2O2 production, methanol production, and methanol selectivity were investigated. Catalysts with various stoichiometries were prepared using the IWI method. In a process relying on in situ generation of H2O2, the addition of Cu actually led to a sharp decrease in catalytic performance in methanol production in comparison with the AuPd/TiO2 system. Using 2.5 wt % Au and 2.5 wt % Pd on TiO2 resulted in an activity of 0.9 molCH4converted kgcat–1 h–1 with a methanol selectivity of 68.2%; the activity using CuAuPd/TiO2 (2.5 wt % loading of each metal) was 0.1 molCH4converted kgcat–1 h–1 with a methanol selectivity of 81.8%. The TOF of the trimetallic material further diminished with decreasing Cu loading, and no activity was recorded over 2.5 wt % Cu/TiO2.
When H2O2 was added to the reaction mixture, far greater activity was recorded, and the TMNP system proved to be superior. AuPd/TiO2 showed an activity of 1.9 molCH4converted kgcat–1 h–1 but only 49.3% methanol selectivity, and other bimetallic combinations gave low activities and selectivities toward methanol. The addition of 2.5 wt % Cu boosted the activity to 2.2 molCH4converted kgcat–1 h–1, but further compromised the methanol selectivity to 27.8%. However, when 1.0 wt % Cu was used, a slightly higher activity of 2.7 molCH4converted kgcat–1 h–1 was recorded, with a methanol selectivity of 82.7%.
Three possible explanations were proposed for the increase in catalytic performance with the addition of Cu. First, the redox properties of the AuPd sites could have been altered with the addition of Cu through electronic inductive effects. Second, the Cu could have blocked or neutralized active sites for undesirable H2O2 decomposition pathways. Third, the Cu itself could have lowered the rate of H2O2 decomposition.
XPS was used to analyze the CuAuPd/TiO2 (1.0 wt %, 2.5 wt %, and 2.5 wt %, respectively) following calcination at 673 K, and Cu+ and Cu0 were present, but Cu2+ was not. Pd was present as both Pd2+ and Pd0, with an atomic ratio of 6.14:1. Little change in the Pd/Cu ratio was observed, indicating that the formation of Au@Pd with Cu highly dispersed across the TiO2 surface is more likely. The mean diameter of the nanoparticles was 1.2 nm, as measured with TEM imaging; however, EDX studies of such particles with greater resolution would be required to positively support the conclusion of a core@shell structure as postulated on the basis of the XPS measurements, particularly with respect to the small NP size.
With regard to catalytic oxidation reactions, reports on TMNPs as catalysts are limited, and ternary metal oxides as catalysts dominate.309−311 From the stated examples, however, the benefits that alloying a third metal into a binary system can provide to catalytic performance are evident.
4.4. Electrochemical Reactions
TMNPs have recently garnered much attention as catalysts in many electrochemical oxidations and reductions. With the ongoing environmental cost related to high usage of nonrenewable fossil fuels, the search for renewable, efficient, sustainable, and green energy sources is of high importance. Electrochemical oxidation of methanol,312−320 ethanol,321−326 and other simple oxygen-containing organic compounds327 to give carbon dioxide and water is a potential candidate to meet this demand.
4.4.1. Electrochemical Oxidation of Alcohols
Methanol possesses a high energy density, and the products of the methanol oxidation reaction are CO2 and H2O, both of which can be recycled and reused in other processes. Methanol is also easy to store and transport in large quantities. The current commercial catalyst for the direct methanol fuel cell is Pt/C,328 but the kinetics for the oxidation reaction of methanol are slow, and intermediates are formed that poison the catalyst, such as CO, formaldehyde, and formic acid.329 The addition of a second metal, notably Ru, increases the catalytic performance considerably as a result of electronic and geometric effects.175 Addition of Ru adsorbs •OH at a lower potential than Pt, so Ru–OHads is formed more readily than Pt–OHads and oxidizes COads to CO2, liberating the catalytic site of the strongly adsorbed CO molecule that would poison a Pt-only catalyst. Cu has also been reported as a second metal, since the Pt–Cu interaction shifts the d band of Pt, reducing the strength of adsorption of CO on Pt.330,331
Yin et al. reported the use of trimetallic PtRuCu nanoframes for the electrochemical oxidation of methanol (Figures 11 and 23).175 The PtRuCu nanoframes aim to combine the promotional effects of the addition of Ru and Cu to the Pt-only catalyst, resulting in a catalyst with activity superior to that of bimetallic formulations. Figure 23b illustrates that the commercial Pt/C catalyst exhibits an electrocatalytic performance normalized to catalyst mass of 0.32 A mgPt–1, whereas the PtRuCu/C nanoframe catalysts were 3 times more active, with an activity of 0.99 A mgPt–1. The unetched PtRuCu/C TMNP catalyst had an activity of 0.36 A mgPt–1, which was marginally greater than Pt/C (Table 8), demonstrating the increase in catalytic performance that can be achieved from high exposure of low-coordinate metal sites.
Figure 23.

(a) Cyclic voltammograms obtained for the methanol oxidation reaction in N2-saturated aqueous HClO4 over PtRuCu nanoframes and carbon-supported PtRuCu and Pt NPs. (b) Mass activities of the catalysts. (c) Specific activities, with peak current density on the left and mass activity on the right. (d) Durability comparison following 800 CV cycles. Adapted with permission from ref (175). Copyright 2020 Chinese Chemical Society, Institute of Chemistry of the Chinese Academy of Sciences, and Royal Society of Chemistry.
Table 8. Summary of Catalysts Used for the Electrochemical Oxidation of Methanol.
| catalyst | catalyst type | performancea | ref |
|---|---|---|---|
| PtRuCu/C | supported trimetallic nanoframe | MA = 0.99; SA = 2.17; decay after 800 cycles = 21% | (175) |
| supported TMNP | MA = 0.36; SA = 1.22; decay after 800 cycles = 48% | ||
| PtCu/C | supported BMNP | MA = ca. 0.5 | |
| Pt/C | supported MMNP | MA = 0.32; SA = 0.63; decay after 800 cycles = 77% | |
| Auoct@PdPt | colloidal core@shell TMNP | MA = 1.50; SA = 2.19 | (79) |
| Pt2Au1Sn1/CNT | supported TMNP | MA = 0.50 (acidic) | (126) |
| MA = 1.70 (basic) |
MA is the mass activity in units of A mgmetal-1, and SA is the specific activity in units of mA cm-2.
Furthermore, Figure 23c illustrates that the specific activity of the PtRuCu/C nanoframes was 2.17 mA cm–2, which is 3.4 times greater than that over Pt/C (0.63 mA cm–2). To demonstrate that a modest quantity of Ru could improve the catalytic performance, PtCu/C samples were also prepared. The mass activity of the PtCu/C catalysts was ca. 0.5 A mgPt–1, half that of the trimetallic formulation. Moreover, the specific activity of the Ru-free sample was one-third of that for the trimetallic sample. Studies with Cu-free samples showed similar results, strongly confirming the synergistic interaction between the three metals within the system.
In further work from the same study, Yin et al. reported degradation of the catalytic performance following 800 cyclic voltammetry (CV) cycles (Figure 23d). The PtRuCu/C nanoframe exhibited a 21% decay in activity. By comparison, acetic acid-capped PtRuCu/C samples showed a 39% decay, TMNP samples a 48% decay, and the conventional Pt/C catalyst a 77% decay. All of the trimetallic formulations showed stability superior to that of the conventional Pt/C catalyst. The observations support the importance of Ru and Cu in lowering the strength of adsorption of intermediates that poison the active sites, thus making desorption of these intermediates more favorable. Analysis following the durability study suggested that the PtRuCu/C nanoframes retained their shape and integrity.
The PtRuCu/C nanoframe samples capped with oleylamine proved to be superior to the commercial Pt/C catalyst with regard to both activity and stability, highlighting the benefits of adding additional metals. In this case, the combination of metals used in the study served to improve the durability of the catalyst by increasing the rate of desorption of catalytic poisons; meanwhile, the defect-rich nanoframe structure facilitated greater catalytic performance in the oxidation of methanol.
Kang et al. explored a facile one-pot synthesis method to produce highly controlled Au@PdPt TMNPs with a well-defined octahedral Au core and a highly crystalline dendritic Pd–Pt shell (labeled as Auoct@PdPt).79 The catalysts showed excellent performance for methanol electrooxidation, originating from optimal binding of adsorbate molecules due to improved charge transfer between the core and the shell. Although the morphologies of BMNPs have been widely studied to tune the catalytic performance, in TMNPs this has been comparatively unexplored. The addition of a third metal increases the number of degrees of freedom with respect to the structure–composition–property relationships. In order to control the nucleation and growth kinetics, preformed NP seeds or structure-directing templates are commonly used, but the method developed by Kang et al. instead uses dual reducing agents (namely, ascorbic acid and hydrazine) to coreduce the metal precursors. The single-step process is therefore a simpler approach for synthesizing multimetallic nanoparticles with a desirable structure.79
The facile one-pot method involved first mixing the metal precursors (HAuCl4, K2PdCl4, and K2PtCl6) in an equimolar ratio with CTAC as a stabilizing agent; ascorbic acid was then added, and the mixture was gently shaken. The second reducing agent, hydrazine, was then added, and the mixture was sealed and heated to 95 °C in an oven for 150 min. The resulting NPs were collected via centrifugation. SEM and TEM were used to show the high yield (>90%) of octahedral-shaped NPs (Figure 24a). The prepared NPs showed excellent catalytic activity and stability for the electrooxidation of methanol in acidic media. The mass activity and current density of the Auoct@PdPt NPs were ca. 1.5 A mgPt–1 and 2.19 mA cm–2 (Figure 24c,d and Table 8), which compare favorably to those for the nanoframes prepared by Yin et al.(175) The enhanced catalytic activity was attributed to the optimized binding affinity of these trimetallic catalysts for the adsorbates due to the improved charge transfer between the core and the shell of the NPs. This conjecture was supported by analysis of the XPS data, as the d-band centers of the Au@PdPt NP catalysts shifted downward compared with that of the PdPt alloy NP catalyst.
Figure 24.

Characterization of the Au@PdPt TMNP electrooxidation catalyst. (a) HAADF-STEM image and cross-sectional compositional line profiles of an Auoct@PdPt NP. The scale bar indicates 20 nm. (b) HAADF-STEM-EDX elemental mapping images of an Auoct@PdPt NP. (c) CVs of Auoct@PdPt NPs, Ausph@PdPt NPs, dendritic Pd–Pt alloy NPs, and Pt/C catalysts in 0.1 M HClO4 + 0.5 M methanol. Scan rate = 50 mV s–1. (d) Chronoamperometry curves for the various catalysts in 0.1 M HClO4 + 0.5 M methanol at 0.6 V vs Ag/AgCl. Adapted from ref (79). Copyright 2013 American Chemical Society.
Singh et al. tested PtAuSn trimetallic catalysts in combination with a range of carbon-based supports for methanol electrooxidation.126 Activated carbon is a widely used catalyst support, and it is popular because of its high surface area, which provides good particle dispersion. However, activated carbon is also highly microporous, and this can cause metal nanoparticles to become trapped and inaccessible, which eliminates their catalytic activity. Alternative carbon-based supports, such as CNTs and carbon nanofibers (CNFs), are now emerging. Mono-, bi-, and trimetallic platinum-based catalysts (20% w/w) on different carbon supports were tested for methanol electrooxidation. The CNTs and CNFs have to be pretreated with acid to increase their hydrophilicity in order for the metal(s) to be deposited onto the support. The pretreatment develops functionality on the surface that can be confirmed through the use of Fourier transform infrared spectroscopy (FTIR) and XPS, showing evidence of −OH, C–O, C=O, and C(=O)–O functionalities.
The subsequent evaluation of Pt2Au1Sn1/CNT revealed that this catalyst can be considered among the best catalysts for methanol electrooxidation.126 The catalyst was measured to have methanol oxidation mass activities of ca. 0.5 A mgPt–1 in acidic media and 1.7 A mgPt–1 under basic conditions. Although Pt2Au1Sn1/CNF, prepared in the same study, had very reasonable metal dispersion, TEM and XRD analyses indicated that the particle diameter on average was 3.8 ± 1.5 nm (median = 3.7 nm) and that the particle size distribution (PSD) was broad, with only ca. 40% of the particles in this 3–4 nm range. Conversely, the Pt2Au1Sn1/CNT catalyst had a smaller average metal particle diameter of 3.3 ± 1.8 nm (median = 2.6 nm), and ca. 75% of the particles had diameters of ≤3 nm. The measurement of particle size also showed that the particle size increased slightly with increasing Au content; as the fraction of Au in the metal particles can be reduced in trimetallic catalysts, hence removing its effect on particle size growth, it is advantageous to consider using the trimetallic catalyst over a bimetallic combination.126 The unique structure of CNTs appeared to encourage a narrow PSD and small particle diameter of Pt, Au, and Sn TMNP alloys, which make the active sites highly accessible to the reactants, although the reasons for this require further work.
Evidence of an electronic effect caused by the presence of Au was uncovered from analysis of the XPS measurements. In the Pt/AC catalyst, 53% of the platinum was in the oxidized state (as a combination of Pt2+ and Pt4+); as the Au content was increased to form the Pt1Au4/C catalyst, the percentage of oxidized platinum was 72%, and in both the trimetallic Pt2Au1Sn1/C and Pt2Au1Sn1/CNT catalysts, the percentage of oxidized platinum was ca. 45%. These measurements suggest that the presence of gold in the catalyst enhances the catalytic oxidizing potential of Pt.126
Methanol is highly toxic, and therefore, ethanol is considered as an alternative candidate for fuel cell applications. However, the 12-electron reaction for complete transformation is challenging because of the C–C bond cleavage required.332 Despite this challenge, bioderived ethanol has a strong case as a renewable resource for energy applications,333 particularly as ethanol has a greater theoretical energy density than methanol (8.0 vs 6.1 kWh kg–1, respectively).334
Zhu et al., reported on the electrochemical lower-alcohol oxidation reaction over TMNP materials.177 For the electrochemical oxidation of ethanol (EOR), previous studies had already indicated that Pd is an effective catalyst under basic conditions.335,336 As shown elsewhere within this review, the AuxPtyPdz formulation has received much attention in the fast-developing field of trimetallic catalysts; however, as opposed to nanoparticles or nanoframes, one-dimensional nanowires were used in the study reviewed here.177
The effect of the composition of the trimetallic nanowires was investigated using CV. As the quantity of Pt was increased, the ethanol oxidation peak potential and the onset of the peak potential were negatively shifted, and the peak current increased. Thus, among the three trimetallic compositions tested, the composition with the most Pt (Au17Pt24Pd59) had the highest catalytic activity. Figure 25 illustrates that the peak current density is highest for the Au17Pt24Pd59 composition, where the loading of noble metal was 35.7 μg cm–2 for each catalyst. Moreover, Figure 25c shows that the peak current density and catalytic activity of Au17Pt24Pd59 are superior to those of the bimetallic formulations Pt11Pd89 and Au27Pd73 as well as the commercial Pd/C catalyst.
Figure 25.

Cyclic voltammetry measurements and current density–time curves in the ethanol electrooxidation reaction. (a) CVs of the three trimetallic catalysts on electrodes under basic conditions. (b) Current density–time curves for the three trimetallic catalysts in a 0.5 M NaOH + 1 M ethanol solution at 0.2 V. (c) CVs of bimetallic Pt11Pd89 and Au27Pd73 as well as Au17Pt24Pd59 and commercial E-TEK Pd/C electrodes. (d) Current density–time curves of the bimetallic compositions, Au17Pt24Pd59, and commercial E-TEK Pd/C electrodes at 0.2 V. Adapted with permission from ref (177). Copyright 2012 Royal Society of Chemistry.
A computational study of Pt–AuSnOx by Dai et al.(337) showed that Pt-based catalysts in which an oxygen-containing species decorates a Pt surface can greatly enhance the ethanol oxidation reaction. Bader charge analysis revealed that in the Pt–AuO catalyst, the small negatively charged O in AuO has a higher-energy O p band and less negative charge to stabilize H and thus was found to be more active than the other screened oxygen-containing species. The high segregation energy of Pt–SnO and the strong oxygen adsorption energy provide durability by preventing dissolution of the Pt–oxygen-containing species into the subsurface or bulk of Pt. Combining the two materials into a trimetallic Pt–AuSnOx species increased the mass activity by a factor of 9.7 compared with that of pure Pt while maintaining the desired stability.
It was proposed that in the ethanol oxidation reaction using a AuxPtyPdz catalyst, a CH3CO intermediate forms on the Pd surface, and its reaction with adsorbed OH is the rate-determining step because of the low concentration of OH.338 The addition of Pt and Au activate the surface, forming OH through electronic synergistic effects. The highlighted study noted that the catalytic performance of the AuxPtyPdz nanowires could be further improved with finer optimizations and adjustments of the Au:Pd:Pt ratio.
Dai et al. computationally investigated Pt3RhM (M = Fe, Co, Ni, Cu, Ga, In, Sn, Pb) surface models built with the FCC structure and (111) facets using a 2:1:1 composition and four-layer depth in their models for ethanol dehydrogenation. Descriptors developed for selectivity and reactivity were based on the binding energies for O + H2O and O + C, respectively; Rh and M were identified to control intermediate adsorption and thus the barrier in the rate-determining step. Furthermore, M influences H2O adsorption and tunes the d-band center, which is believed to be responsible for modulating the reactivity. Group IIIA and IVA metals increase the selectivity and preference for CO2; this was most impactful for Pt3RhSn/C, which exhibited 67- and 7-fold increases in specific activity and mass activity, respectively, when referenced against a commercial Pt/C catalyst.339 Further work investigated Pt6SnAg nanorods, which showed enhanced EOR activity and stability with a homogeneous Pt–Sn–Ag surface configuration.340 The reactivity enhancement was attributed to a structural effect of Sn–Ag pairs formed on the Pt surface, and the homogeneity of the catalyst was highlighted as key to the reactivity.340
4.4.2. Oxygen Reduction Reaction
The oxygen reduction reaction (ORR) is of high interest in electrochemistry and has been heavily studied due to its importance in fuel cell technology.341 Pt NPs supported on carbon are commonly used as an ORR electrocatalyst, which is characterized by adequate catalytic activity and durability.342,343 Research into enhancing the Pt catalyst has shown that introducing additional metals can improve the catalytic performance further as a result of synergistic effects. Tang et al.(344) synthesized AuxPty electrocatalysts deposited on fluorinated tin oxide and carbon disk substrates. Through the addition of Au, a higher resistance to poisoning in an acidic environment was achieved, with a significant improvement in catalytic performance in an alkaline electrolyte for the ORR. Through the electronic interactions between Pt and Au in alloy particles, the differences in catalytic performance were rationalized. Compared with monometallic Pt electrocatalysts, the overpotential for the reduction was decreased by the bimetallic NPs, leading to an increased cell voltage. Ternary PdxPtyNiz alloy catalysts were synthesized by a polyol reduction method and were tested for the ORR in proton-exchange-membrane fuel cells. With a narrow particle diameter distribution of ca. 5 nm, the Pt-based mass activity of PdxPtyNiz was double that of commercial Pt; in addition, the long-term durability testing of the trimetallic PdxPtyNiz alloy within a 200 h operation time was comparable to that of a commercial Pt catalyst.345 Furthermore, Mazumder et al.(346) highlighted that ternary Pd/PtFe NPs had high durability over 10 000 cycles without loss of the core@shell structure. The use of multiple metals can decrease the total Pt loading in the catalytic system, making such catalysts more attractive for commercial applications with regard to cost and efficiency. A core@shell model with the notion of a thin Pt shell and a core incorporating less expensive and more abundant metals has been considered to improve Pt catalysts; Mazumder et al.(346) demonstrated a unique synthesis approach for preparing structured Pd@FePt NPs with a Pd-rich core and a FePt-rich shell. The ORR under acidic conditions was reported to be dependent on the thickness of the FePt shell, where the highest activity and durability were achieved with a shell thickness of <1 nm. The high activity was thought to be due to the change of the electronic structure of Pt upon alloying, but compared to the ORR over a commercial Pt catalyst, the activity was not drastically improved. In terms of the ORR in the study of Mazumder et al., the authors summarized that the interfacial interactions between the FePt shell and the Pd core resulted in higher activity, as upon careful alterations of shell thickness correlating activities were achieved.
Other groups have also focused on incorporating multiple metals to modify Pt NP catalysts, concluding that the added synergy, including altering the Pt electronic structure, improves the catalytic performance. Au@FePt3 NPs with well-defined surfaces were compared against a commercial Pt catalyst and bimetallic FePt3.347 The trimetallic Au@FePt3 catalyst showed superior durability and activity due to the tailored morphology, composition, and synergy. The initial ORR activity, normalized to the catalyst mass over Au@FePt3, was reported to be 5 times higher than that over Pt/C. Figure 26indicates the mass activity before and after 60 000 potential cycles over Pt/C, FePt3/C, and Au@FePt3/C catalysts, highlighting the durability of the ternary catalyst. TEM characterizations before and after the stability studies were also performed and indicated a considerable change in the size of the Pt and FePt3 NPs that was proposed to be due to Ostwald ripening.348 In comparison, Au@FePt3 NPs were of a similar size and shape before and after the potential cycling, indicating a higher relative durability.
Figure 26.
Mass activities of the Pt/C, FePt3/C, and Au@FePt3/C catalysts before and after 60 000 potential cycles between 0.6 and 1.1 V vs RHE in oxygen-saturated 0.1 M HClO4 electrolyte at 20 °C with a sweep rate of 50 mV/s. Adapted from ref (347). Copyright 2010 American Chemical Society.
The preparation of TMNP catalysts without Pt, comprising PdCoAu NP alloys, were prepared via a microemulsion method and supported on carbon black.349 Voltammetric and valence-band spectral measurements indicated a change in electronic structure properties of Pd caused by alloying with Co and Au. The PdCoAu/C system exhibited a decreased density of states at the Fermi level of Pd through a shift of the Pd d-band center. As a result, the formation of Oads or OHads was inhibited on the Pd surface, leading to enhanced performance in the ORR. The activity at +0.8 V was 0.15 mA cm–2 for the as-synthesized Pd/C monometallic catalysts compared with 1.32 mA cm–2 over the heat-treated trimetallic PdCoAu/C catalyst.
Modern DFT techniques have become increasingly significant in recent studies of TMNP catalysts for the ORR. Ternary nanoparticles based on Pt–Au–M (M = Cr, Mn, Co, Cu, Zn) were explored by Jennings et al. as electrocatalysts for the ORR.350 Alloying with Au provided stability under harsh operating conditions at the cathode, and Cr-, Co-, and Cu-rich cores were shown to lead to an icosahedral structure due to the small lattice constants that was determined to be highly catalytically active because of the interplay between strain and ligand effects. Compressive strain exerted on the Au subsurface by the 3d transition metal core led to a favorable weakening of the Pt–OH binding energy, which subsequently resulted in an overall enhancement of the reaction kinetics.350
Noh et al. investigated alternative Ni@Cu@Pt nanoparticles for the ORR, again using DFT, and observed that there is a strong compressive strain on Pt, leading to the high ORR activity among the nanoparticles according to the d-band center energy model.351 At a diameter of ca. 3 nm, the electrochemical stability of the TMNPs exceeded that of the monometallic Pt NPs.352 An oxygen monolayer coverage exceeding 0.34 induced Cu segregation to the core of the Ni@Cu@Pt TMNPs, implying high stability under reaction conditions between 0.99 and 1.10 V, where up to 0.33 monolayer of O will be available.352 Similarly, Flores-Rojas et al. studied 44-atom octahedral CoNi bimetallic NPs wrapped in Pt with a ConNi6–nPt38 (0 ≤ n ≤ 6) atomic composition.353 The ORR activity was described in terms of O and OH binding energies, which were lower for the trimetallic NPs than for Pt44, suggesting an overall higher activity relative to the monometallic Pt catalyst.353 In a nicely complementary study, these findings were then confirmed experimentally with two comparable nanoparticle compositions, Co30Ni70–20Pt/C and Co70Ni30–20Pt/C, which exhibited 400% and 300% increases in specific activity, respectively, with respect to Pt/C and overall a 1.5-fold higher mass activity than a commercially available Pt/C catalyst.353 Cruz-Martínez et al.(354) investigated the electrocatalytic activity of M6@Pd30Pt8 (M = Co, Ni, Cu) toward the ORR and observed similarity to Pt44; an activity trend of Pt44 > binary M@Pd38 > Pd44 was reported. The O and OH adsorption energies were again evaluated as descriptors of the electrocatalytic activity, and the trend in activity based on compositional variation was shown to be M6@Pd30Pt8 > M6@Pd38 > Pd44,354 supporting the inference that the benefits of TMNPs over mono- and bimetallic formulations for this reaction are significant.
Oxygen is activated on Ni@PdPt; calculations indicated that M atoms (M = Ni, Co, Cu) have lower energies in the core in ternary compositions, which changes for bimetallic nanoparticles in an oxidizing environment. Kim et al.(355) performed computational analysis on PtxCuyPdz alloy catalysts for the ORR. They reported that the strength of the bond between surface Pt and adsorbates reaches a maximum when the Pt:Pd ratio reaches 50 atom % and that Pd and Au improve the durability of the (111) surface in an acidic and oxidizing environment while maintaining the catalytic reactivity. The enhanced catalyst stability is crucial for achieving the durability target for proton-exchange-membrane fuel cells set by the U.S. Department of Energy.356
In Pd13−nNin@Pt systems, it appears that reactant adsorption is strongly affected by the coordination number of the shell atoms; the adsorption energy and d-band center were found to be inversely correlated with the activity, as shown in Figure 27. In general, the most active catalysts among those investigated were composed of NiPt; therefore, it was proposed that surface Pd impedes this reaction.357 For Pt@PdNi compositions, 0.8% compressive strain was observed in the structure compared with Pt3Pd, which aligned with EXAFS observations. The adsorption energy decrease was attributed to negative d-band shifts following the structural compression, which was further supported by a report that OH adsorption was 0.13 eV weaker than on the binary system.358
Figure 27.

Pt d-band center and adsorption energies of O2 and O atoms in the Pd13–nNin@Pt42 (n = 0, 1, 12, 13) NPs. Adapted with permission from ref (357). Copyright 2017 Elsevier.
Pd–Pt based FCC TMNPs have been investigated as potential catalysts for scavenging of reactive oxygen species.359,360 Wang et al.(360) used experimental and DFT studies to investigate idealized PtPdRh FCC fragments with fixed structure and mixed compositions and showed that the O atoms are located further away from each other on the trimetallic structure compared with PtPd, which translated into remarkable scavenging efficiency for reactive oxygen species. Mu et al.(359) carried out an experimental study of nanozyme oxidation with additional computational modeling of a small PtPdMo FCC system (treated as a random alloy). The TMNPs in this study were reported to be attractive to electrons, thus capturing oxygen and nitrogen species on Mo, as confirmed by electrostatic potential and electron localization function analysis. Doping of PtPd with Mo caused lattice deformation and increased exposure of the (200) facets with highly active sites.
Han et al.(361) investigated o-CoFeW TMNP catalytic materials for the oxygen evolution reaction (OER) (Figure 28), an important step of the water splitting reaction that can allow for clean and renewable energy production and utilization. o-CoFeW clusters were investigated by DFT using basin-hopping Monte Carlo for structural ordering. In the presence of Fe, the individual Co CN increases, suggesting that Co has a bias toward the core and Fe toward the surface of the o-CoFeW nanoclusters. The ternary CoFeW cluster structures result in desired intermediary adsorption energies of reactants, which were otherwise too strong (o-WFe) or too weak (o-WCo) in the binary metallic clusters that were examined. However, a degradation of catalytic activity was observed, which on the basis of experimental XPS data was attributed to the formation of an oxyhydroxide surface layer. o-CoFeW exhibited an overpotential of 192 mV at a current density of 10 mA cm–2 when loaded on gold foam as the working electrode (Figure 28), which places it among the most active oxygen evolution reaction catalysts reported to date.361
Figure 28.

Schematic illustration of the synthesis of trimetallic clusters. (a) Space-filling representation of the structure of polyoxometalates (POMs). (b) Self-assembly of POMs and ethylenediamine-grafted C60 (EDA-C60) driven by electrostatic interactions. (c) Schematic structure of the POMs/EDA-C60 composites after freeze-drying. (d) Trimetallic clusters on a carbonic support. O, gray; Co/Fe, violet; W, red; P, yellow. (e) OER polarization curves of BNMP and TMNP catalysts loaded on gold foam (scan rate of 5 mV s–1 without iR correction). (f) Current density vs time for 3-CoFeW loaded on gold foam for 30 h at a constant applied potential of 1.46 V vs RHE for the OER. Adapted from ref (361). Copyright 2018 American Chemical Society.
In summary, the overall outcome of the studies of the OER and ORR utilizing polymetallic electrocatalysts with well-defined surfaces or cluster shapes and composition can be a path to optimal catalyst design. A plethora of computational and experimental techniques can effectively be used together for screening and benchmarking of catalytic materials on the basis of a combination of electronic, morphological, and steric effects that translate into synergy for better catalytic performance. For example, enhancements in catalytic activity or durability can be achieved through interplay between two contrasting weak–strong interactions of intermediates with atomic species present in a TMNP, morphological changes, and/or better exposure of active sites. Well-balanced composition can help reach intermediate properties that allow better catalytic activity while increasing durability, and novel TMNP catalysts often manifest properties far superior to those of well-established commercially available materials.
4.5. Further Applications of TMNPs and Beyond Ternary Compositions
This section sets out further examples of TMNP-based catalysts that are not strictly applicable to a preceding section. Here TMNP catalysts have been applied to hydrodesulfurization (HDS) and dry reforming of methane (DRM). In both cases, achieving high activities is discussed with respect to the optimal catalyst formulation or preparative conditions of these examples, with the benefits of a ternary composition made clear for the outlined applications.
HDS is an industrial chemical process in which sulfur is removed during oil refining. Sulfur is the third most common element in crude oil and may account for up to 7.9% per gram.362 Reduction of sulfur-based emissions from the use of fuels is a key aspect of the current energy sector. Sulfur is also a common poison for precious metal catalysts, which are typically used further along in the refining process. Metal catalysts commonly involved in HDS are based on Ni, Co, Mo, and W,363 and in particular, sulfided versions of these catalysts have been found to be most active. The activity of sulfided catalysts is problematic because it can cause unwanted sulfur contamination; thus, over recent years an effort has been made to produce non-sulfided catalysts that are as active as their sulfided counterparts.364 A potential solution to the outlined challenge could lie in the application of trimetallic catalysts.
In a study reported by Mendoza-Nieto et al.,129 trimetallic NiMoW catalysts were tested for HDS of petroleum fractions, specifically dibenzothiophene and 4,6-dimethyldibenzothiophene. The catalysts were supported on either γ-Al2O3 or mesoporous SBA-15, which had been prepared with or without the addition of citric acid. The NiMoW/γ-Al2O3 catalyst was unaffected by the addition of citric acid during its preparation, and the metals were well-dispersed for the oxide and sulfided states regardless of how they were prepared. Analysis of TPR profiles suggested that a large fraction of Mo6+ and W6+ species with tetrahedral coordination were present, providing evidence of a strong metal–support interaction; consequently, the catalyst was not reducible at temperatures as high as 873–1273 K. Conversely, NiMoW/SBA-15 possessed a weak metal–support interaction, suggesting that agglomerated NiMoO4 and WO3 species were formed when the catalyst was prepared without the addition of citric acid. Through the addition of citric acid during preparation, the metal dispersion was increased, and the metals became more easily reduced and sulfided. Additionally, the presence of citric acid decreased the degree of stacking of the Mo(W)S2 active phase. The final citric acid-prepared NiMoW/SBA-15 catalyst had the highest activity for HDS of dibenzothiophene and 4,6-dimethyldibenzothiophene, as it was >2 times more active than NiMoW/γ-Al2O3.129
Mozhaev et al. assessed the impact of nickel addition to sulfided CoMo/Al2O3 catalysts for HDS of dibenzothiophene.363 The Mo loading was constant in each catalyst (10 wt %), and the Co and Ni contents were varied from 1.3 to 3.8 wt % and from 0.8 to 3.8 wt %, respectively. The catalysts were prepared by incipient wetness impregnation using decamolybdodicobaltate heteropolyacid (Co2Mo10HPA) and cobalt or nickel citrate. The catalysts were sulfided prior to the reaction, and catalytic testing took place in a fixed-bed reactor at 543 K and 3.0 MPa.
Increasing the Co:Mo ratio was found to decrease the TOF of the Cox–Co2Mo10/Al2O3 catalysts. The trimetallic Nix–Co2Mo10/Al2O3 catalysts had higher TOF values than the bimetallic CoMo-based equivalents. These changes in activity were thought to be due to the formation of either mixed NiCoMoS active sites or, more likely, the coexistence of NiMoS and CoMoS active sites. However, it was crucial to have the correct ratio of metals: increasing the Co content in Cox–Co2Mo10/Al2O3 decreased the Co content in the CoMoS phase, causing an increase in the Co fraction in cobalt sulfide. Increasing the Ni content in Nix–Co2Mo10/Al2O3, however, reduced the Ni content in the NiCoMoS phase while the Co percentage remained constant. At a (Co + Ni)/(Co + Ni + Mo) atomic ratio of 0.33, the dibenzothiophene HDS activity was observed to be at its maximum. The rate constant observed for the trimetallic sulfide catalyst was higher than that of the CoMoS/Al2O3 catalysts with the same metal content, and when the Ni loading was further increased to a Ni/(Ni + Co + Mo) molar ratio of 0.34, the TOF decreased because of a reduction in the number of mixed active sites.363
Liu et al.(365) used rare-earth metals in a trimetallic catalyst for the hydrocracking of jatropha oil to produce green diesel consisting of C11–C20 straight-chain alkanes. The catalysts were prepared by a novel powder metallurgy (PM) technique, which was advantageous when more common preparation techniques were problematic because of the significant effect of pH on the activity. In this preparation technique, γ-Al2O3 powder is mixed with the metal oxide precursor powders (5 wt % NiO, 15 wt % MoO3, and 1 wt % La2O3), pressed into a cylindrical shape, and then heat-treated at 973 K for 3 h. The preparation method is simple and inexpensive. For comparison, the same catalyst was also synthesized by a wet impregnation method followed by heat treatment at 400 °C. The catalysts were then tested for hydrocracking of jatropha oil in a fixed-bed reactor under the reaction conditions of 370 °C, 3.0 MPa, LHSV = 0.9 h–1, and H2/feed ratio = 1000/1.
Surface area analysis on the catalyst through nitrogen adsorption revealed that the pore diameter at the surface of the catalyst was smaller than the diameter inside the pore. Both the bare support and the catalyst prepared by impregnation showed a Type IV isotherm with an H4-type hysteresis loop, indicative of a mesoporous material. The PM catalyst showed a Type IV isotherm, with an H3-type hysteresis loop, suggesting that the preparation method affects the textural properties. Analysis of XRD patterns showed that there were no crystalline phases of MoO3 or LaO3 present, suggesting that these oxides are highly dispersed or amorphous. In the PM catalyst, which had been heated to 973 K, NiO phases were detected. The impregnation catalyst, which had been heated at 673 K, showed much less intense peaks, indicating that higher temperatures result in crystalline phases. TEM images showed very different results for the two catalysts, with the impregnation catalyst showing a rough surface with agglomerated metal species whereas the PM catalyst had a much smoother surface with a more homogeneous dispersion of metals.365
The catalyst prepared by PM had a higher selectivity for C11–C20 products. However, the jatropha oil conversion over the catalyst prepared by impregnation was higher, but more short-chain hydrocarbons and alkanes longer than 20 carbons were formed, which are less useful. Increasing the liquid hourly space velocity (LHSV) was found to decrease the conversion and significantly reduced the selectivity for C11–C20 alkanes because of a reduction in the contact time.365
Dry reforming of methane is of great industrial interest because it produces syngas (H2 and CO), which can be used for other applications, such as Fischer–Tropsch synthesis.366,367 Ni-based catalysts are among the more common materials used for DRM but often suffer from deactivation due to carbon deposition.368−370 Therefore, various strategies, such as alloying Ni with other metals, offer a solution to prevent sintering. The addition of Fe to form NiFe alloys has been reported to enhance carbon resistance and activity;371 however, the stability of the catalysts was a concern because of Fe segregation during the process of DRM. The NiFe alloy catalysts are sensitive to dealloying during the process and subsequently sinter.372 Theofanidis et al. reported the synthesis of a NiFePd alloy catalyst via incipient wetness impregnation373 that had enhanced stability and activity achieved through the addition of Pd to FeNi/MgAl2O4, wherein an FeNi core and a trimetallic shell composed of a FeNiPd alloy were formed. With an optimal Ni:Pd molar ratio of 75:1, DFT calculations and catalytic performance studies indicated that the addition of Pd to FeNi reduced the tendency of Fe to segregate and controlled the carbon formation, resulting in a high activity of 24.8 mmol s–1 gmetals–1 after 21 h on stream (1023 K, 0.1 MPa). With use of other non-noble-metal additives, Jin et al. introduced Cu into NiFe alloy-based catalysts supported on MgAl2O4via coprecipitation to suppress Fe segregation and NP sintering.374 The effect of different Cu loadings was evaluated with DRM, and the subsequent catalytic performance was determined. The initial methane conversion as a function of time on stream followed the trend Ni3Cu1-MA > Ni3Fe1Cu0.5-MA > Ni3Fe1Cu1-MA ≈ Ni3Fe1-MA > Ni3Fe1Cu0.5-MA. Although Ni3Cu1-MA showed high activity initially, it severely deactivated over time; after 20 h, the methane conversion followed the trend Ni3Fe1Cu0.5-MA > Ni3Fe1-MA > Ni3Fe1Cu1-MA ≈ Ni3Cu1-MA > Ni3Fe1Cu1.5-MA. The trend at 20 h indicates that adding small amounts of Cu to form a trimetallic system enhanced activity and stability, which was correlated to an alloy structure formation according to EXAFS analysis. Excessive Cu addition, however, led to lower activity because Cu obscured the Ni active sites.
It is important to note that multimetal systems are not limited to trimetallic ones. As mentioned previously, incorporating multiple metals into a catalytic system increases the number of degrees of freedom, complicating preparative control and subsequent material characterization. High-entropy alloys (HEAs) are an example of systems that use multiple metals and are notably more complex than trimetallic systems. Yeh et al. first proposed HEAs as materials containing five or more elements in near equiatomic percentages.375 The entropic contribution to the total free energy overcomes the enthalpic contributions as the number of elements increases in an alloy, stabilizing solid solutions.375,376 HEAs have shown enhanced properties, including increased mechanical strength and stability and enhanced corrosion and oxidation resistance.376−378 There is appeal in the flexibility of designing systems with multiple metals, but the control and rationale behind these complex structures remain a challenge. Although HEAs are a relatively new field, they have been seen in a range of applications such as aerospace engineering, biomimetic materials, hydrogen storage materials, and electromagnetic shielding materials.376,378
In line with the purpose of this review, attention should be placed on their catalytic applications. Understanding is limited because of the premature nature of HEAs, and most efforts in this field have been placed on enhanced mechanical properties. However, reports have shown the application of HEAs in electrocatalytic systems, such as the hydrogen evolution reaction (HER),379,380 the ORR,379,381 and ammonia decomposition.382 Successful synthesis methods that have been developed for HEAs include a carbothermal shock method,383 fast-moving bed pyrolysis,379 and ball milling.384
Multielemental alloy nanoparticles (MEA-NPs) can also be used to describe HEA systems. Yao et al. reported a computer-aided study to illustrate their promise for catalyst discovery, with vast compositional space.385 Alloy phase characterization in MEA-NPs was reported using EDX elemental maps, HAAD-STEM, and FT-EXAFS (Figure 29). Bimetallic Ru–Ni NPs show phase separation due to their immiscibility, which may be attributed to the Hume–Rother principle.386 However, when the number of elements is increased, a uniform distribution with a homogeneous alloy structure can be observed (Figure 29b,c). The alloy structure is also supported by FT-EXAFS measurements, as a slight shift was observed in the radial distance of Ru and Ni compared with their corresponding metallic bonds. Furthermore, in Figure 29f, a modest difference in bond lengths among the different elements present suggests that short-range ordering of the NPs exists.
Figure 29.

(A–C) Elemental maps of (A) bimetallic Ru–Ni NPs, (B) Ru-4 (RuRhCoNi), and (C) Ru-5 (RuRhCoNiIr) supported on carbon nanofibers. (D–F) FT-EXAFS spectra of (D) Ru in Ru-5, (E) Ni in Ru-5, and (F) Ru, Rh, Co, Ni, and Ir in Ru-5. (G–I) HAADF-STEM images of Ru, Ru-4, and Ru-5 MEA-NPs, respectively. From ref (385). CC BY-NC 4.0.
Comparable to bimetallic and trimetallic catalysts, additional metals such as those found in HEAs can be beneficial as a result of synergistic effects. Facile synthesis methods are more commonly reported for bimetallic and trimetallic NPs in comparison with HEAs. Enhancing the relationship between theory-driven and experimental synthesis is crucial in the design, including the ability to prescreen a multitude of compositions. DFT, molecular dynamics, and Monte Carlo methods can aid the prediction of alloy formation and examine structural stability.385 To mass-produce HEAs efficiently, new synthesis strategies with low cost along with high catalytic performances should be thoroughly investigated to meet industrial needs and regulations.
Trimetallic systems already show challenges in terms of full characterization, but a multitude of studies, as presented in this review, have shown their catalytic enhancements through synergistic effects. HEAs require even more in-depth analysis, and to make progress, state-of-the-art characterization systems such as EXAFS, HAADF-STEM, and LEIS spectroscopy will be required to fully understand the properties of these multimetal systems.
5. Conclusions and Future Prospects
Reports on the preparation and use of TMNP catalysts have rapidly increased, and in this review these recent advances have been comprehensively discussed. Improvements in catalyst design in this field benefit from a combined computational and experimental approach because of the complexity of the supported and unsupported ternary nanoparticles. However, at this stage there are predominantly reports detailing novel preparation and particle compositions along with comparative catalyst testing studies of mono-, bi-, and trimetallic NPs. The elemental compositions of those discussed in the review often share common elements in combination, such as Pt, Au, Ni, Pd, Ag, Cu, Zn and/or Mo, Fe, and Co, other compositions include Rh, Ru, In, or Sn. Of these, the most successful or most often reported/applied compositions are often based on one or two of the following metals in combination or all three: Pd, Pt, and/or Au. This is the case because they readily form trimetallic alloys. These compositions have been applied as catalysts for both hydrogenation and oxidation reactions. Therefore, as a guide for future researchers hoping to use TMNPs, we offer that those preparations could involve two of these metals as a starting point.
Computational studies of TMNPs are increasing in parallel; however, there are only a handful of reports using a combined approach. The influence of a second or third metal on catalytic activity is often presented as a synergistic relationship and is strongly dependent on the nanoparticle composition, as evidenced with AuPdPt-based catalysts for the direct synthesis of hydrogen peroxide or the electrooxidation of ethanol. Despite major progress in studying TMNP compositions, there are great challenges, such as characterizing and understanding the particle makeup and inconsistencies from particle to particle. Furthermore, several reports detail a compromise between selectivity and activity, that is, some TMNPs may offer lower overall activity but higher selectivity, thus making them superior overall. The improved selectivity can be observed through steric hindrance, suppression of the formation of byproducts, electronic effects promoting the formation of product over a byproduct, or inhibition of unwanted side reactions by the third metal.
We have presented a mixture of articles, some focusing on materials and design whereas others directly compare catalytic activities of mono-, bi-, and trimetallic systems. Our aim in this review has been to bridge these aspects and give an overview of how each factor plays a role, particularly with respect to efforts to add non-noble metals to mono- or bimetallic NPs; this approach has proven to be more effective and economical, driving commercial attention. Advanced characterization, such as operando studies, may pave the way to a clear relationship of precise composition and structure with catalytic performance. Here computation can provide an essential insight in the design of catalysts and optimize influential factors on the reactivity, such as the d-band center, which themselves play a role in catalytic performance. However, challenges remain in exploring the full compositional spectrum and modeling reactive processes on realistic nanoparticles. When appropriate complementary characterization using bulk and surface spectroscopy, for example XPS, LEIS spectroscopy, surface-enhanced Raman spectroscopy, EXAFS and XANES, X-ray diffraction, high-resolution electron microscopy, NMR spectroscopy, and chemisorption should be used to discriminate and locate the metals within the NPs. The key information that we consider for researchers hoping to understand these systems starts with the composition and morphology of the nanoparticles, the chemical or electronic nature of the surface, support influences (if applicable), and finally detailed data on the mechanism of a given process. These key elements combined should provide sufficient information to relate the influence of a third metal to the experimental data. Following the techniques discussed by Ferrando et al.(17) on characterizing nanoalloys or BMNPs, we expect advances in resolution and modeling of ternary particles to greatly improve in the next 10 years, although effort is still necessary to unite computational modeling with materials and experimental design, particularly for supported systems. However, computational studies can already provide a wealth of information on potential for metals to alloy, phase composition, and surface electronic trends. On the basis of the results surveyed in this review, there is a wide scope for further research, principally in fundamental studies and development of TMNPs as catalysts, as the evident benefits will be applicable to other applications.
Acknowledgments
The authors thank the EPSRC for funding (EP/S030468/1). A.J.L. acknowledges funding by the UKRI Future Leaders Fellowship Program (MR/T018372/1).
Biographies
James. W. M. Crawley obtained his M.Chem. from the University of Southampton in 2019. He is currently undertaking his Ph.D. in the Hutchings’ research group in the Cardiff Catalysis Institute at Cardiff University, investigating novel trimetallic nanoparticles as catalysts for the hydrogenation of CO2 to methanol.
Isla E. Gow is a Ph.D. student in the Hutchings’ research group in the Cardiff Catalysis Institute at Cardiff University, investigating new trimetallic nanoparticles as catalysts for CO2 hydrogenation to methanol. She received her M.Chem. from Cardiff University in 2019.
Naomi Lawes obtained her M.Chem. with a Year in Industry from Cardiff University in 2019. Currently, she is doing her Ph.D. in the Hutchings’ group, researching novel trimetallic nanoparticles as catalysts for CO2 hydrogenation to methanol.
Igor Kowalec is a Ph.D. student in the Cardiff Catalysis Institute at Cardiff University, conducting computational research on CO2 hydrogenation to methanol using Pd-based catalysts under the supervision of Prof. Sir C. Richard A. Catlow and Dr. Andrew J. Logsdail. He completed his M.Chem. at Cardiff University in 2019.
Lara Kabalan obtained her Ph.D. in Physical Chemistry of Condensed Matter from the University of Bordeaux 1 in France, after which she moved to Pennsylvania State University as a postdoctoral researcher and then to University College London, Qatar Environmental Research Institute, and the University of Strathclyde before joining Cardiff University in 2020 as a postdoctoral research associate in the group of Prof. Richard Catlow and Dr. Andrew Logsdail. Her research interest lies in computational studies of homogeneous and heterogeneous catalysis.
C. Richard A. Catlow is Professor of Chemistry at Cardiff University and University College London (UCL). He studied chemistry at Oxford University, where he was later a research fellow. He moved to UCL as a Lecturer in Chemistry and subsequently to Keele University as Professor of Physical Chemistry—a joint appointment with the SERC Daresbury Laboratory. He was for several years Wolfson Professor and Director of the Davy Faraday Laboratory at the Royal Institution of Great Britain before returning to UCL as Head of Chemistry and then Dean of Mathematical and Physical Sciences. He was elected a Fellow of the Royal Society in 2004 and of the German National Science Academy (Leopoldina) in 2020. He has been awarded the Solid State Chemistry, Liversidge, and Faraday Awards of the Royal Society of Chemistry. He was elected Foreign Secretary of the Royal Society in 2016 and received a knighthood in 2020.
Andrew J. Logsdail received his Ph.D. in Chemistry from the University of Birmingham in 2012 under the supervision of Prof. Roy L. Johnston. He was a postdoctoral research associate with Prof. C. Richard A. Catlow, FRS, in the Department of Chemistry at University College London from 2012 to 2014 and then held a Ramsay Research Fellowship at the same institution from 2014 to 2016. He was appointed to a University Research Fellowship at the School of Chemistry of Cardiff University in 2016, promoted to Lecturer in Computational and Catalytic Chemistry in 2019, and awarded a UKRI Future Leaders Fellowship in 2020 (MR/T018372/1). His research expertise is in the development and application of computational software for modeling of catalytic materials and reactions.
Stuart H. Taylor completed his Ph.D. at the University of Liverpool in 1994 and took up an academic position at Cardiff University School of Chemistry in 1997. He was promoted to Professor in 2013 and currently is the Director of Research for the School. He has more than 29 years of experience in heterogeneous catalysis research and has published over 320 papers and patents. He has wide ranging expertise in experimental studies of catalysis, exploiting preparation techniques for fundamental catalyst understanding and design. His work impacts on chemicals, fuels, sustainability, energy, and the environment. He has given many invited presentations at international meetings and receives support from a range of funding bodies and industry. He collaborates extensively with many universities and industry both nationally and internationally.
Nicholas F. Dummer is a Research Fellow at Cardiff University. He obtained his Ph.D. under the supervision of Graham Hutchings in 2005 at Cardiff University. In 2012 he was appointed as a Special Assistant Professor in Wataru Ueda’s group in the Catalysis Research Center of Hokkaido University in Japan. Following this in 2013 he was awarded a Senior Research Fellowship at Wollongong University in Australia. In 2015 he returned to Cardiff University and is presently a MaxNet Research Fellow and local coordinator for the Max Planck Centre in Cardiff University on the Fundamentals of Heterogeneous Catalysis. He has co-authored over 65 articles on heterogeneous catalysts and their applications.
Graham J. Hutchings is Regius Professor of Chemistry at Cardiff University. He studied chemistry at University College London. His early career was with ICI and AECI Ltd., where he became interested in gold catalysis. In 1984 he moved to academia and has held chairs at the Universities of Witwatersrand, Liverpool, and Cardiff. He was elected Fellow of the Royal Society in 2009. He was awarded the Davy Medal of the Royal Society in 2013, the ENI Award for Advanced Environmental Solutions in 2017, the RSC Faraday Lectureship Prize and a CBE in 2018, and the Michel Boudart Award in 2021.
The authors declare no competing financial interest.
References
- Chalmin E.; Menu M.; Vignaud C. Analysis of Rock Art Painting and Technology of Palaeolithic Painters. Meas. Sci. Technol. 2003, 14, 1590–1597. 10.1088/0957-0233/14/9/310. [DOI] [Google Scholar]
- Mohan Bhagyaraj S.; Oluwafemi O. S.. Nanotechnology: The Science of the Invisible. In Synthesis of Inorganic Nanomaterials; Elsevier, 2018; pp 1–18. 10.1016/B978-0-08-101975-7.00001-4. [DOI] [Google Scholar]
- Brun N.; Mazerolles L.; Pernot M. Microstructure of Opaque Red Glass Containing Copper. J. Mater. Sci. Lett. 1991, 10, 1418–1420. 10.1007/BF00735696. [DOI] [Google Scholar]
- Khawaji M.; Chadwick D. Selective Catalytic Oxidation over Au-Pd/Titanate Nanotubes and the Influence of the Catalyst Preparation Method on the Activity. Catal. Today 2019, 334, 122–130. 10.1016/j.cattod.2018.11.080. [DOI] [Google Scholar]
- Sun Y.; Mayers B.; Xia Y. Metal Nanostructures with Hollow Interiors. Adv. Mater. 2003, 15, 641–646. 10.1002/adma.200301639. [DOI] [Google Scholar]
- Wiley B.; Sun Y.; Mayers B.; Xia Y. Shape-Controlled Synthesis of Metal Nanostructures: The Case of Silver. Chem. - Eur. J. 2005, 11, 454–463. 10.1002/chem.200400927. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Guerra N.; Moreno-López L.; Serrano-Ruiz J. C.; Valverde J. L.; de Lucas-Consuegra A. Gas Phase Electrocatalytic Conversion of CO2 to Syn-Fuels on Cu Based Catalysts-Electrodes. Appl. Catal., B 2016, 188, 272–282. 10.1016/j.apcatb.2016.02.010. [DOI] [Google Scholar]
- Wang T.; Ren D.; Huo Z.; Song Z.; Jin F.; Chen M.; Chen L. A Nanoporous Nickel Catalyst for Selective Hydrogenation of Carbonates into Formic Acid in Water. Green Chem. 2017, 19, 716–721. 10.1039/C6GC02866G. [DOI] [Google Scholar]
- Byrappa K.; Ohara S.; Adschiri T. Nanoparticles Synthesis Using Supercritical Fluid Technology – towards Biomedical Applications. Adv. Drug Delivery Rev. 2008, 60, 299–327. 10.1016/j.addr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Yamamoto K.; Imaoka T.; Tanabe M.; Kambe T. New Horizon of Nanoparticle and Cluster Catalysis with Dendrimers. Chem. Rev. 2020, 120, 1397–1437. 10.1021/acs.chemrev.9b00188. [DOI] [PubMed] [Google Scholar]
- Zhong J.; Yang X.; Wu Z.; Liang B.; Huang Y.; Zhang T. State of the Art and Perspectives in Heterogeneous Catalysis of CO2 Hydrogenation to Methanol. Chemical Society Reviews. 2020, 49, 1385–1413. 10.1039/C9CS00614A. [DOI] [PubMed] [Google Scholar]
- Kašpar J.; Fornasiero P.; Hickey N. Automotive Catalytic Converters: Current Status and Some Perspectives. Catal. Today 2003, 77, 419–449. 10.1016/S0920-5861(02)00384-X. [DOI] [Google Scholar]
- Saif M.; El-Molla S. A.; Aboul-Fotouh S. M. K.; Ibrahim M. M.; Ismail L. F. M.; Dahn D. C. Nanostructured Gd3+-TiO2 Surfaces for Self-Cleaning Application. J. Mol. Struct. 2014, 1067, 120–126. 10.1016/j.molstruc.2014.03.024. [DOI] [Google Scholar]
- Martínez M. A.; Herrero J.; Gutiérrez M. T. Optimisation of Indium Tin Oxide Thin Films for Photovoltaic Applications. Thin Solid Films 1995, 269, 80–84. 10.1016/0040-6090(95)06873-2. [DOI] [Google Scholar]
- Pawelec B.; Venezia A. M.; La Parola V.; Cano-Serrano E.; Campos-Martin J. M.; Fierro J. L. G. AuPd Alloy Formation in Au-Pd/Al2O3 Catalysts and Its Role on Aromatics Hydrogenation. Appl. Surf. Sci. 2005, 242, 380–391. 10.1016/j.apsusc.2004.09.004. [DOI] [Google Scholar]
- Edwards J. K.; Pritchard J.; Lu L.; Piccinini M.; Shaw G.; Carley A. F.; Morgan D. J.; Kiely C. J.; Hutchings G. J. The Direct Synthesis of Hydrogen Peroxide Using Platinum-Promoted Gold-Palladium Catalysts. Angew. Chem., Int. Ed. 2014, 53, 2381–2384. 10.1002/anie.201308067. [DOI] [PubMed] [Google Scholar]
- Ferrando R.; Jellinek J.; Johnston R. L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. 10.1021/cr040090g. [DOI] [PubMed] [Google Scholar]
- Sankar M.; Dimitratos N.; Miedziak P. J.; Wells P. P.; Kiely C. J.; Hutchings G. J. Designing Bimetallic Catalysts for a Green and Sustainable Future. Chem. Soc. Rev. 2012, 41, 8099–8139. 10.1039/c2cs35296f. [DOI] [PubMed] [Google Scholar]
- Xie F.; Chu X.; Hu H.; Qiao M.; Yan S.; Zhu Y.; He H.; Fan K.; Li H.; Zong B.; Zhang X. Characterization and Catalytic Properties of Sn-Modified Rapidly Quenched Skeletal Ni Catalysts in Aqueous-Phase Reforming of Ethylene Glycol. J. Catal. 2006, 241, 211–220. 10.1016/j.jcat.2006.05.001. [DOI] [Google Scholar]
- Reyes P.; Aguirre M. C.; Fierro J. L. G.; Santori G.; Ferretti O. Hydrogenation of Crotonaldehyde on Rh-Sn/SiO2 Catalysts Prepared by Reaction of Tetrabutyltin on Prereduced Rh/SiO2 Precursors. J. Mol. Catal. A: Chem. 2002, 184, 431–441. 10.1016/S1381-1169(02)00031-6. [DOI] [Google Scholar]
- Rioux R. M.; Vannice M. A. Dehydrogenation of Isopropyl Alcohol on Carbon-Supported Pt and Cu–Pt Catalysts. J. Catal. 2005, 233, 147–165. 10.1016/j.jcat.2005.04.020. [DOI] [Google Scholar]
- Zhu L.; Zhang H.; Zhong L.; Zheng J.; Yu C.; Zhang N.; Chen B. H. RuNiCo-Based Nanocatalysts with Different Nanostructures for Naphthalene Selective Hydrogenation. Fuel 2018, 216, 208–217. 10.1016/j.fuel.2017.12.023. [DOI] [Google Scholar]
- Tarditi A. M.; Cornaglia L. M. Novel PdAgCu Ternary Alloy as Promising Materials for Hydrogen Separation Membranes: Synthesis and Characterization. Surf. Sci. 2011, 605, 62–71. 10.1016/j.susc.2010.10.001. [DOI] [Google Scholar]
- White K.Publications Output: U.S. Trends and International Comparisons. National Science Foundation; December 17, 2019. https://ncses.nsf.gov/pubs/nsb20206/executive-summary (accessed 2021-03-11).
- Kondrat S. A.; Miedziak P. J.; Douthwaite M.; Brett G. L.; Davies T. E.; Morgan D. J.; Edwards J. K.; Knight D. W.; Kiely C. J.; Taylor S. H.; Hutchings G. J. Base-Free Oxidation of Glycerol Using Titania-Supported Trimetallic Au-Pd-Pt Nanoparticles. ChemSusChem 2014, 7, 1326–1334. 10.1002/cssc.201300834. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Wang D.; Chen X.; Zhou G.; Yu R.; Li Y. Defect-Dominated Shape Recovery of Nanocrystals: A New Strategy for Trimetallic Catalysts. J. Am. Chem. Soc. 2013, 135, 12220–12223. 10.1021/ja4068063. [DOI] [PubMed] [Google Scholar]
- Yurderi M.; Bulut A.; Zahmakiran M.; Kaya M. Carbon Supported Trimetallic PdNiAg Nanoparticles as Highly Active, Selective and Reusable Catalyst in the Formic Acid Decomposition. Appl. Catal., B 2014, 160–161, 514–524. 10.1016/j.apcatb.2014.06.004. [DOI] [Google Scholar]
- Xiong X.; Zhou L.; Yu G.; Yang K.; Ye M.; Xia Q. Synthesis and Catalytic Performance of a Novel RuCuNi/CNTs Nanocomposite in Hydrolytic Dehydrogenation of Ammonia Borane. Int. J. Hydrogen Energy 2015, 40, 15521–15528. 10.1016/j.ijhydene.2015.09.095. [DOI] [Google Scholar]
- Wen Y. H.; Li L.; Zhao T.; Huang R. Solid-Liquid Coexistence in Trimetallic Heterostructured Nanoparticle Catalysts: Insights from Molecular Dynamics Simulations. ACS Appl. Nano Mater. 2020, 3, 12369–12378. 10.1021/acsanm.0c02862. [DOI] [Google Scholar]
- Strasser P.; Fan Q.; Devenney M.; Weinberg W. H.; Liu P.; Nørskov J. K. High Throughput Experimental and Theoretical Predictive Screening of Materials - A Comparative Study of Search Strategies for New Fuel Cell Anode Catalysts. J. Phys. Chem. B 2003, 107, 11013–11021. 10.1021/jp030508z. [DOI] [Google Scholar]
- Cao L.; Zhao Z.; Liu Z.; Gao W.; Dai S.; Gha J.; Xue W.; Sun H.; Duan X.; Pan X.; Mueller T.; Huang Y. Differential Surface Elemental Distribution Leads to Significantly Enhanced Stability of PtNi-Based ORR Catalysts. Matter 2019, 1, 1567–1580. 10.1016/j.matt.2019.07.015. [DOI] [Google Scholar]
- Hammer B.; Norskov J. K. Why Gold Is the Noblest of All the Metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Norskov J. K.; Abild-Pedersen F.; Studt F.; Bligaard T. Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 937–943. 10.1073/pnas.1006652108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Zhang Y. W.; Tao F. Shape Control of Bimetallic Nanocatalysts through Well-Designed Colloidal Chemistry Approaches. Chem. Soc. Rev. 2012, 41, 8050–8065. 10.1039/c2cs35184f. [DOI] [PubMed] [Google Scholar]
- Jeong H. E.; Kim S.; Seo M. G.; Lee D. W.; Lee K. Y. Catalytic Activity of Pd Octahedrons/SiO2 for the Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen. J. Mol. Catal. A: Chem. 2016, 420, 88–95. 10.1016/j.molcata.2016.03.043. [DOI] [Google Scholar]
- Menegazzo F.; Signoretto M.; Ghedini E.; Strukul G. Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen. Catalysts 2019, 9, 251. 10.3390/catal9030251. [DOI] [Google Scholar]
- Kim D.; Resasco J.; Yu Y.; Asiri A. M.; Yang P. Synergistic Geometric and Electronic Effects for Electrochemical Reduction of Carbon Dioxide Using Gold-Copper Bimetallic Nanoparticles. Nat. Commun. 2014, 5, 4948 10.1038/ncomms5948. [DOI] [PubMed] [Google Scholar]
- Cuenya B. R. Synthesis and Catalytic Properties of Metal Nanoparticles: Size, Shape, Support, Composition, and Oxidation State Effects. Thin Solid Films 2010, 518, 3127–3150. 10.1016/j.tsf.2010.01.018. [DOI] [Google Scholar]
- Malta G.; Kondrat S. A.; Freakley S. J.; Davies C. J.; Lu L.; Dawson S.; Thetford A.; Gibson E. K.; Morgan D. J.; Jones W.; Wells P. P.; Johnston P.; Catlow C. R. A.; Kiely C. J.; Hutchings G. J. Identification of Single-Site Gold Catalysis in Acetylene Hydrochlorination. Science 2017, 355, 1399–1403. 10.1126/science.aal3439. [DOI] [PubMed] [Google Scholar]
- Abazari R.; Heshmatpour F.; Balalaie S. Pt/Pd/Fe Trimetallic Nanoparticle Produced via Reverse Micelle Technique: Synthesis, Characterization, and Its Use as an Efficient Catalyst for Reductive Hydrodehalogenation of Aryl and Aliphatic Halides under Mild Conditions. ACS Catal. 2013, 3, 139–149. 10.1021/cs300507a. [DOI] [Google Scholar]
- Khalid M.; Honorato A. M. B.; Tremiliosi Filho G.; Varela H. Trifunctional Catalytic Activities of Trimetallic FeCoNi Alloy Nanoparticles Embedded in a Carbon Shell for Efficient Overall Water Splitting. J. Mater. Chem. A 2020, 8, 9021–9031. 10.1039/C9TA13637A. [DOI] [Google Scholar]
- Sial M. A. Z. G.; Lin H.; Zulfiqar M.; Ullah S.; Ni B.; Wang X. Trimetallic PtCoFe Alloy Monolayer Superlattices as Bifunctional Oxygen-Reduction and Ethanol-Oxidation Electrocatalysts. Small 2017, 13, 1700250. 10.1002/smll.201700250. [DOI] [PubMed] [Google Scholar]
- Cao S.; Tao F. F.; Tang Y.; Li Y.; Yu J. Size- and Shape-Dependent Catalytic Performances of Oxidation and Reduction Reactions on Nanocatalysts. Chem. Soc. Rev. 2016, 45, 4747–4765. 10.1039/C6CS00094K. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ji S.; Liu Y.; Cao X.; Tian S.; Chen Y.; Niu Z.; Li Y. Well-Defined Materials for Heterogeneous Catalysis: From Nanoparticles to Isolated Single-Atom Sites. Chem. Rev. 2020, 120, 623–682. 10.1021/acs.chemrev.9b00311. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Murayama T.; Taketoshi A.; Haruta M. Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes. Chem. Rev. 2020, 120, 464–525. 10.1021/acs.chemrev.9b00551. [DOI] [PubMed] [Google Scholar]
- Sankar M.; He Q.; Engel R. V.; Sainna M. A.; Logsdail A. J.; Roldan A.; Willock D. J.; Agarwal N.; Kiely C. J.; Hutchings G. J. Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts. Chem. Rev. 2020, 120, 3890–3938. 10.1021/acs.chemrev.9b00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Zhang Y.; Shi R.; Wang Z. Shape-Controlled Synthesis of Trimetallic PtPdCu Nanocrystals and Their Electrocatalytic Properties. ACS Appl. Energy Mater. 2019, 2, 2515–2523. 10.1021/acsaem.8b02075. [DOI] [Google Scholar]
- Darby S.; Mortimer-Jones T. V.; Johnston R. L.; Roberts C. Theoretical Study of Cu-Au Nanoalloy Clusters Using a Genetic Algorithm. J. Chem. Phys. 2002, 116, 1536–1550. 10.1063/1.1429658. [DOI] [Google Scholar]
- Xia Y.; Xiong Y.; Lim B.; Skrabalak S. E. Shape-Controlled Synthesis of Metal Nanocrystals: Simple Chemistry Meets Complex Physics?. Angew. Chem., Int. Ed. 2009, 48, 60–103. 10.1002/anie.200802248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novell-Leruth G.; Valcárcel A.; Clotet A.; Ricart J. M.; Pérez-Ramírez J. DFT Characterization of Adsorbed NHx Species on Pt(100) and Pt(111) Surfaces. J. Phys. Chem. B 2005, 109, 18061–18069. 10.1021/jp051682l. [DOI] [PubMed] [Google Scholar]
- Bratlie K. M.; Lee H.; Komvopoulos K.; Yang P.; Somorjai G. A. Platinum Nanoparticle Shape Effects on Benzene Hydrogenation Selectivity. Nano Lett. 2007, 7, 3097–3101. 10.1021/nl0716000. [DOI] [PubMed] [Google Scholar]
- Niu W.; Li Z.-Y.; Shi L.; Liu X.; Li H.; Han S.; Chen J.; Xu G. Seed-Mediated Growth of Nearly Monodisperse Palladium Nanocubes with Controllable Sizes. Cryst. Growth Des. 2008, 8, 4440–4444. 10.1021/cg8002433. [DOI] [Google Scholar]
- Quan Z.; Wang Y.; Fang J. High-Index Faceted Noble Metal Nanocrystals. Acc. Chem. Res. 2013, 46, 191–202. 10.1021/ar200293n. [DOI] [PubMed] [Google Scholar]
- Dahl S.; Logadottir A.; Egeberg R. C.; Larsen J. H.; Chorkendorff I.; Törnqvist E.; Nørskov J. K. Role of Steps in N2 Activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814–1817. 10.1103/PhysRevLett.83.1814. [DOI] [Google Scholar]
- Liu Z.-P.; Hu P. General Rules for Predicting Where a Catalytic Reaction Should Occur on Metal Surfaces: A Density Functional Theory Study of C–H and C–O Bond Breaking/Making on Flat, Stepped, and Kinked Metal Surfaces. J. Am. Chem. Soc. 2003, 125, 1958–1967. 10.1021/ja0207551. [DOI] [PubMed] [Google Scholar]
- Stamenkovic V.; Mun B. S.; Mayrhofer K. J. J.; Ross P. N.; Markovic N. M.; Rossmeisl J.; Greeley J.; Nørskov J. K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem., Int. Ed. 2006, 45, 2897–2901. 10.1002/anie.200504386. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Wang D.; Zhou G.; Yu R.; Chen C.; Li Y. Sophisticated Construction of Au Islands on Pt-Ni: An Ideal Trimetallic Nanoframe Catalyst. J. Am. Chem. Soc. 2014, 136, 11594–11597. 10.1021/ja5058532. [DOI] [PubMed] [Google Scholar]
- Pei J.; Mao J.; Liang X.; Chen C.; Peng Q.; Wang D.; Li Y. Ir–Cu Nanoframes: One-Pot Synthesis and Efficient Electrocatalysts for Oxygen Evolution Reaction. Chem. Commun. 2016, 52, 3793–3796. 10.1039/C6CC00552G. [DOI] [PubMed] [Google Scholar]
- Gao D.; Yang S.; Xi L.; Risch M.; Song L.; Lv Y.; Li C.; Li C.; Chen G. External and Internal Interface-Controlled Trimetallic PtCuNi Nanoframes with High Defect Density for Enhanced Electrooxidation of Liquid Fuels. Chem. Mater. 2020, 32, 1581–1594. 10.1021/acs.chemmater.9b04789. [DOI] [Google Scholar]
- Nosheen F.; Anwar T.; Siddique A.; Hussain N. Noble Metal Based Alloy Nanoframes: Syntheses and Applications in Fuel Cells. Front. Chem. 2019, 7, 456. 10.3389/fchem.2019.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ruditskiy A.; Xia Y. Rational Design and Synthesis of Noble-Metal Nanoframes for Catalytic and Photonic Applications. Natl. Sci. Rev. 2016, 3, 520–533. 10.1093/nsr/nww062. [DOI] [Google Scholar]
- McEachran M.; Keogh D.; Pietrobon B.; Cathcart N.; Gourevich I.; Coombs N.; Kitaev V. Ultrathin Gold Nanoframes through Surfactant-Free Templating of Faceted Pentagonal Silver Nanoparticles. J. Am. Chem. Soc. 2011, 133, 8066–8069. 10.1021/ja111642d. [DOI] [PubMed] [Google Scholar]
- Li K.; Li X.; Huang H.; Luo L.; Li X.; Yan X.; Ma C.; Si R.; Yang J.; Zeng J. One-Nanometer-Thick PtNiRh Trimetallic Nanowires with Enhanced Oxygen Reduction Electrocatalysis in Acid Media: Integrating Multiple Advantages into One Catalyst. J. Am. Chem. Soc. 2018, 140, 16159–16167. 10.1021/jacs.8b08836. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ren G.; Wang M.; Zhang Z.; Liang Y.; Wu S.; Shen J. Facile Synthesis of Trimetallic PtAuCu Alloy Nanowires as High–Performance Electrocatalysts for Methanol Oxidation Reaction. J. Alloys Compd. 2019, 780, 504–511. 10.1016/j.jallcom.2018.12.016. [DOI] [Google Scholar]
- Jin L.; Xu H.; Chen C.; Song T.; Wang C.; Wang Y.; Shang H.; Du Y. Uniform PdCu Coated Te Nanowires as Efficient Catalysts for Electrooxidation of Ethylene Glycol. J. Colloid Interface Sci. 2019, 540, 265–271. 10.1016/j.jcis.2019.01.019. [DOI] [PubMed] [Google Scholar]
- Wang P.; Zhang Y.; Shi R.; Wang Z. Trimetallic PtPdCu Nanowires as an Electrocatalyst for Methanol and Formic Acid Oxidation. New J. Chem. 2018, 42, 19083–19089. 10.1039/C8NJ04723E. [DOI] [Google Scholar]
- Sun Y.; Li Y.; Qin Y.; Wang L.; Guo S. Interfacial Engineering in PtNiCo/NiCoS Nanowires for Enhanced Electrocatalysis and Electroanalysis. Chem. - A Eur. J. 2020, 26, 4032–4038. 10.1002/chem.201904473. [DOI] [PubMed] [Google Scholar]
- George C.; Genovese A.; Casu A.; Prato M.; Povia M.; Manna L.; Montanari T. CO Oxidation on Colloidal Au0.80Pd0.20-FexOy Dumbbell Nanocrystals. Nano Lett. 2013, 13, 752–757. 10.1021/nl304448p. [DOI] [PubMed] [Google Scholar]
- Chen H. Y.; Niu H. J.; Han Z.; Feng J. J.; Huang H.; Wang A. J. Simple Fabrication of Trimetallic Platinum-Nickel-Cobalt Hollow Alloyed 3D Multipods for Highly Boosted Hydrogen Evolution Reaction. J. Colloid Interface Sci. 2020, 570, 205–211. 10.1016/j.jcis.2020.02.090. [DOI] [PubMed] [Google Scholar]
- Zeng L.; Li X.; Chen S.; Wen J.; Huang W.; Chen A. Unique Hollow Ni-Fe@MoS2 Nanocubes with Boosted Electrocatalytic Activity for N2 Reduction to NH3. J. Mater. Chem. A 2020, 8, 7339–7349. 10.1039/C9TA13336D. [DOI] [Google Scholar]
- Wang Y.; Zhan W.; Chen Z.; Chen J.; Li X.; Li Y. Advanced 3D Hollow-Out ZnZrO@C Combined with Hierarchical Zeolite for Highly Active and Selective CO Hydrogenation to Aromatics. ACS Catal. 2020, 10, 7177–7187. 10.1021/acscatal.0c01418. [DOI] [Google Scholar]
- Aasen D.; Clark M. P.; Ivey D. G. Investigation of Transition Metal-Based (Mn, Co, Ni, Fe) Trimetallic Oxide Nanoparticles on N-Doped Carbon Nanotubes as Bifunctional Catalysts for Zn-Air Batteries. J. Electrochem. Soc. 2020, 167, 040503 10.1149/1945-7111/ab7094. [DOI] [Google Scholar]
- Karthikeyan B.; Loganathan B. Rapid Green Synthetic Protocol for Novel Trimetallic Nanoparticles. J. Nanoparticles 2013, 2013, 1–8. 10.1155/2013/168916. [DOI] [Google Scholar]
- Huan T. N.; Shinde D. V.; Kim S.; Han S.-H.; Artero V.; Chung H. Forest of Pt–Au–Ag Tri-Metallic Nanodendrites as an Efficient Electrocatalyst for Methanol Oxidation Reaction. RSC Adv. 2015, 5, 6940–6944. 10.1039/C4RA10205C. [DOI] [Google Scholar]
- Eid K.; Malgras V.; He P.; Wang K.; Aldalbahi A.; Alshehri S. M.; Yamauchi Y.; Wang L. One-Step Synthesis of Trimetallic Pt-Pd-Ru Nanodendrites as Highly Active Electrocatalysts. RSC Adv. 2015, 5, 31147–31152. 10.1039/C5RA01981H. [DOI] [Google Scholar]
- Sriphathoorat R.; Wang K.; Shen P. K. Trimetallic Hollow Pt-Ni-Co Nanodendrites as Efficient Anodic Electrocatalysts. ACS Appl. Energy Mater. 2019, 2, 961–965. 10.1021/acsaem.8b01741. [DOI] [Google Scholar]
- Wang Z.-L.; Ping Y.; Yan J.-M.; Wang H.-L.; Jiang Q. Hydrogen Generation from Formic Acid Decomposition at Room Temperature Using a NiAuPd Alloy Nanocatalyst. Int. J. Hydrogen Energy 2014, 39, 4850–4856. 10.1016/j.ijhydene.2013.12.148. [DOI] [Google Scholar]
- Fu L.-L.; Zhang D.-F.; Yang Z.; Chen T.-W.; Zhai J. PtAuCo Trimetallic Nanoalloys as Highly Efficient Catalysts toward Dehydrogenation of Ammonia Borane. ACS Sustainable Chem. Eng. 2020, 8, 3734–3742. 10.1021/acssuschemeng.9b06865. [DOI] [Google Scholar]
- Kang S. W.; Lee Y. W.; Park Y.; Choi B. S.; Hong J. W.; Park K. H.; Han S. W. One-Pot Synthesis of Trimetallic Au@PdPt Core-Shell Nanoparticles with High Catalytic Performance. ACS Nano 2013, 7, 7945–7955. 10.1021/nn403027j. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lu L.; Cao Y.; Du S.; Cheng Z.; Zhang S. Fabrication of Catalytically Active Au/Pt/Pd Trimetallic Nanoparticles by Rapid Injection of NaBH4. Mater. Res. Bull. 2014, 49, 393–398. 10.1016/j.materresbull.2013.09.025. [DOI] [Google Scholar]
- Fan T.-E.; Liu T.-D.; Zheng J.-W.; Shao G.-F.; Wen Y.-H. Structural Optimization of Pt–Pd–Au Trimetallic Nanoparticles by Discrete Particle Swarm Algorithms. J. Mater. Sci. 2015, 50, 3308–3319. 10.1007/s10853-015-8880-9. [DOI] [Google Scholar]
- Wu G. H.; Liu Q. M.; Wu X. Geometrical and Energetic Properties in 38-Atom Trimetallic AuPdPt Clusters. Chem. Phys. Lett. 2015, 620, 92–97. 10.1016/j.cplett.2014.12.022. [DOI] [Google Scholar]
- Zhao Z.; Li M.; Cheng D.; Zhu J. Understanding the Structural Properties and Thermal Stabilities of Au-Pd-Pt Trimetallic Clusters. Chem. Phys. 2014, 441, 152–158. 10.1016/j.chemphys.2014.07.016. [DOI] [Google Scholar]
- Wu X.; Dong Y. Theoretical Studies of Structures and Energies of Pd, Au-Pd, and Au-Pd-Pt Clusters. New J. Chem. 2014, 38, 4893–4900. 10.1039/C4NJ00984C. [DOI] [Google Scholar]
- Wei H.; Wei S.; Zhu X.; Lu X. Investigation of Structural, Thermal, and Dynamical Properties of Pd-Au-Pt Ternary Metal Nanoparticles Confined in Carbon Nanotubes Based on MD Simulation. J. Phys. Chem. C 2017, 121, 12911–12920. 10.1021/acs.jpcc.7b02434. [DOI] [Google Scholar]
- Wu X.; Liu Q.; Sun Y.; Wu G. Structural Characterization of Metal Dopants (M = Ag or Au) in Trimetallic M-Pd-Pt Clusters. RSC Adv. 2015, 5, 51142–51148. 10.1039/C5RA06457K. [DOI] [Google Scholar]
- Du R.-B.; Xu Y.-Q.; Wu X.; Liu T. Geometrical Structures of Trimetallic Ag–Pd–Pt and Au–Pd–Pt Clusters up to 147 Atoms. Struct. Chem. 2019, 30, 637–645. 10.1007/s11224-018-1211-4. [DOI] [Google Scholar]
- Taran S.; Garip A. K.; Arslan H. A Theoretical Study on Chemical Ordering of 38-Atom Trimetallic Pd–Ag–Pt Nanoalloys. Chin. Phys. B 2020, 29, 077801 10.1088/1674-1056/ab99b4. [DOI] [Google Scholar]
- Khanal S.; Bhattarai N.; Velázquez-Salazar J. J.; Bahena D.; Soldano G.; Ponce A.; Mariscal M. M.; Mejía-Rosales S.; José-Yacamán M. Trimetallic Nanostructures: The Case of AgPd-Pt Multiply Twinned Nanoparticles. Nanoscale 2013, 5, 12456–12463. 10.1039/c3nr03831a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarzadeh H.; Abbaspour M.; Mehrjouei E.; Kamrani M. Stability Control of AgPd@Pt Trimetallic Nanoparticles via Ag-Pd Core Structure and Composition: A Molecular Dynamics Study. Ind. Eng. Chem. Res. 2018, 57, 6236–6245. 10.1021/acs.iecr.8b00447. [DOI] [Google Scholar]
- Aranishi K.; Jiang H.-L.; Akita T.; Haruta M.; Xu Q. One-Step Synthesis of Magnetically Recyclable Au/Co/Fe Triple-Layered Core-Shell Nanoparticles as Highly Efficient Catalysts for the Hydrolytic Dehydrogenation of Ammonia Borane. Nano Res. 2011, 4, 1233–1241. 10.1007/s12274-011-0174-1. [DOI] [Google Scholar]
- Yildirim H.; Arslan H. Size and Composition Effect on Structural Properties and Melting Behaviors of Cu–Ag–Au Ternary Nanoalloys. Int. J. Mod. Phys. C 2020, 31, 2050078. 10.1142/S0129183120500783. [DOI] [Google Scholar]
- Zhao S.; Ren Y.; Yao K.; Tian X.; Wang J. Density Functional Study of Ternary AuxAgyCuz and AuxAgyCuz+ Clusters (x + y + z = 5, 6). Mol. Phys. 2019, 117, 474–488. 10.1080/00268976.2018.1524158. [DOI] [Google Scholar]
- Taran S.; Garip A. K.; Arslan H. Chemical Ordering Effect on Structural Stability of Trimetallic Cu-Au-Pt Nanoalloys. Phys. Scr. 2020, 95, 085404 10.1088/1402-4896/aba3ab. [DOI] [Google Scholar]
- Wu G.; Sun Y.; Wu X.; Chen R.; Wang Y. Large Scale Structural Optimization of Trimetallic Cu–Au–Pt Clusters up to 147 Atoms. Chem. Phys. Lett. 2017, 686, 103–110. 10.1016/j.cplett.2017.08.049. [DOI] [Google Scholar]
- Taran S. Composition Effect on Melting Behaviors of Cu-Au-Pt Trimetallic Nanoalloys. Comput. Theor. Chem. 2019, 1166, 112576. 10.1016/j.comptc.2019.112576. [DOI] [Google Scholar]
- Huang R.; Shao G. F.; Wen Y. H.; Sun S. G. Tunable Thermodynamic Stability of Au-CuPt Core-Shell Trimetallic Nanoparticles by Controlling the Alloy Composition: Insights from Atomistic Simulations. Phys. Chem. Chem. Phys. 2014, 16, 22754–22761. 10.1039/C4CP02930E. [DOI] [PubMed] [Google Scholar]
- Schebarchov D.; Wales D. J. Quasi-Combinatorial Energy Landscapes for Nanoalloy Structure Optimisation. Phys. Chem. Chem. Phys. 2015, 17, 28331–28338. 10.1039/C5CP01198A. [DOI] [PubMed] [Google Scholar]
- Mattei J. G.; Grammatikopoulos P.; Zhao J.; Singh V.; Vernieres J.; Steinhauer S.; Porkovich A.; Danielson E.; Nordlund K.; Djurabekova F.; Sowwan M. Gas-Phase Synthesis of Trimetallic Nanoparticles. Chem. Mater. 2019, 31, 2151–2163. 10.1021/acs.chemmater.9b00129. [DOI] [Google Scholar]
- Koh P. W.; Yoon T. L.; Lim T. L.; Chang Y. H. R. The Generation of Ground-state Structures and Electronic Properties of Ternary AlkTilNim Clusters (k + l + m = 4) from a Two-stage Density Functional Theory Global Searching Approach. Int. J. Quantum Chem. 2019, 119, e25884 10.1002/qua.25884. [DOI] [Google Scholar]
- Datta S. Ultra-Small Cd@NiAg and Cr@NiAg Nano-Clusters with Enhanced Mixing. AIP Adv. 2019, 9, 115316. 10.1063/1.5124718. [DOI] [Google Scholar]
- Taran S.; Garip A. K.; Arslan H. Investigation of the Chemical Ordering and Structural Properties of the Trimetallic (PtNi)@Ag Nanoalloys. J. Clust. Sci. 2021, 32, 199–208. 10.1007/s10876-020-01778-8. [DOI] [Google Scholar]
- Henglein A. Colloidal Palladium Nanoparticles: Reduction of Pd(II) by H2; PdCoreAuShellAgShell Particles. J. Phys. Chem. B 2000, 104, 6683–6685. 10.1021/jp000746j. [DOI] [Google Scholar]
- Wang H.-L.; Yan J.-M.; Wang Z.-L.; Jiang Q. One-Step Synthesis of Cu@FeNi Core–Shell Nanoparticles: Highly Active Catalyst for Hydrolytic Dehydrogenation of Ammonia Borane. Int. J. Hydrogen Energy 2012, 37, 10229–10235. 10.1016/j.ijhydene.2012.04.007. [DOI] [Google Scholar]
- Loganathan B.; Chandraboss V. L.; Senthilvelan S.; Karthikeyan B. Tailored Rh Surface Facilitates, Enhancement of Raman Scattering in Trimetallic AuPt Core/Rh Shell Composites: Experimental and Theoretical Evidences. Phys. E Low-dimensional Syst. Nanostructures 2016, 75, 223–234. 10.1016/j.physe.2015.09.006. [DOI] [Google Scholar]
- Wu P.; Ding P.; Ye X.; Li L.; He X.; Wang K. One-Pot Synthesized Cu/Au/Pt Trimetallic Nanoparticles as a Novel Enzyme Mimic for Biosensing Applications. RSC Adv. 2019, 9, 14982–14989. 10.1039/C9RA00603F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauster S. J.; Fung S. C.; Garten R. L. Strong Metal–Support Interactions. Group 8 Noble Metals Supported on Titanium Dioxide. J. Am. Chem. Soc. 1978, 100, 170–175. 10.1021/ja00469a029. [DOI] [Google Scholar]
- Tauster S. J. Strong Metal-Support Interactions. Acc. Chem. Res. 1987, 20, 389–394. 10.1021/ar00143a001. [DOI] [Google Scholar]
- van Deelen T. W.; Hernández Mejía C.; de Jong K. P. Control of Metal-Support Interactions in Heterogeneous Catalysts to Enhance Activity and Selectivity. Nat. Catal. 2019, 2, 955–970. 10.1038/s41929-019-0364-x. [DOI] [Google Scholar]
- Hong J.; Wang B.; Xiao G.; Wang N.; Zhang Y.; Khodakov A. Y.; Li J. Tuning the Metal-Support Interaction and Enhancing the Stability of Titania-Supported Cobalt Fischer–Tropsch Catalysts via Carbon Nitride Coating. ACS Catal. 2020, 10, 5554–5566. 10.1021/acscatal.0c01121. [DOI] [Google Scholar]
- Zhao Y.; Chen D.; Liu J.; He D.; Cao X.; Han C.; Lu J.; Luo Y. Tuning the Metal-Support Interaction on Chromium-Based Catalysts for Catalytically Eliminate Methyl Mercaptan: Anchored Active Chromium Species through Surface Hydroxyl Groups. Chem. Eng. J. 2020, 389, 124384. 10.1016/j.cej.2020.124384. [DOI] [Google Scholar]
- Kumar R.; Kumar K.; Pant K. K.; Choudary N. V. Tuning the Metal-Support Interaction of Methane Tri-Reforming Catalysts for Industrial Flue Gas Utilization. Int. J. Hydrogen Energy 2020, 45, 1911–1929. 10.1016/j.ijhydene.2019.11.111. [DOI] [Google Scholar]
- Li M.; van Veen A. C. Tuning the Catalytic Performance of Ni-Catalysed Dry Reforming of Methane and Carbon Deposition via Ni-CeO2 Interaction. Appl. Catal., B 2018, 237, 641–648. 10.1016/j.apcatb.2018.06.032. [DOI] [Google Scholar]
- Xiao Z.; Zhang X.; Hou F.; Wu C.; Wang L.; Li G. Tuning Metal-Support Interaction and Oxygen Vacancies of Ceria Supported Nickel Catalysts by Tb Doping for n-Dodecane Steam Reforming. Appl. Surf. Sci. 2020, 503, 144319. 10.1016/j.apsusc.2019.144319. [DOI] [Google Scholar]
- Yang Q.; Xu Q.; Jiang H.-L. Metal–Organic Frameworks Meet Metal Nanoparticles: Synergistic Effect for Enhanced Catalysis. Chem. Soc. Rev. 2017, 46, 4774–4808. 10.1039/C6CS00724D. [DOI] [PubMed] [Google Scholar]
- Sarno M.; Ponticorvo E.; Scarpa D. Active and Stable Graphene Supporting Trimetallic Alloy-Based Electrocatalyst for Hydrogen Evolution by Seawater Splitting. Electrochem. commun. 2020, 111, 106647. 10.1016/j.elecom.2019.106647. [DOI] [Google Scholar]
- Machado B. F.; Serp P. Graphene-Based Materials for Catalysis. Catal. Sci. Technol. 2012, 2, 54–75. 10.1039/C1CY00361E. [DOI] [Google Scholar]
- Zhao J.; Li H.; Liu Z.; Hu W.; Zhao C.; Shi D. An Advanced Electrocatalyst with Exceptional Eletrocatalytic Activity via Ultrafine Pt-Based Trimetallic Nanoparticles on Pristine Graphene. Carbon N. Y. 2015, 87, 116–127. 10.1016/j.carbon.2015.01.038. [DOI] [Google Scholar]
- Shi W.; Sun X.; Ding R.; Ying D.; Huang Y.; Huang Y.; Tan C.; Jia Z.; Liu E. Trimetallic NiCoMo/Graphene Multifunctional Electrocatalysts with Moderate Structural/Electronic Effects for Highly Efficient Alkaline Urea Oxidation Reaction. Chem. Commun. 2020, 56, 6503–6506. 10.1039/D0CC02132F. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Liu L.; Yang J.; Zhang Y.; Wan Z.; Zhou L. Pd-Ru-Bi Nanoalloys Modified Three-Dimensional Reduced Graphene Oxide/MOF-199 Composites as a Highly Efficient Electrocatalyst for Ethylene Glycol Electrooxidation. Appl. Surf. Sci. 2019, 492, 617–625. 10.1016/j.apsusc.2019.06.228. [DOI] [Google Scholar]
- Sen B.; Demirkan B.; Şavk A.; Karahan Gülbay S.; Sen F. Trimetallic PdRuNi Nanocomposites Decorated on Graphene Oxide: A Superior Catalyst for the Hydrogen Evolution Reaction. Int. J. Hydrogen Energy 2018, 43, 17984–17992. 10.1016/j.ijhydene.2018.07.122. [DOI] [Google Scholar]
- Huang L.; Han Y.; Dong S. Highly-Branched Mesoporous Au-Pd-Pt Trimetallic Nanoflowers Blooming on Reduced Graphene Oxide as an Oxygen Reduction Electrocatalyst. Chem. Commun. 2016, 52, 8659–8662. 10.1039/C6CC03073D. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Wang X.; Chen C.; An C.; Xu Y.; Huang Y.; Zhang Q.; Wang Y.; Jiao L.; Yuan H. Facile Synthesis of Cu@CoNi Core-Shell Nanoparticles Composites for the Catalytic Hydrolysis of Ammonia Borane. Int. J. Hydrogen Energy 2015, 40, 12253–12261. 10.1016/j.ijhydene.2015.07.067. [DOI] [Google Scholar]
- Surisetty V. R.; Dalai A. K.; Kozinski J. Alkali-Promoted Trimetallic Co-Rh-Mo Sulfide Catalysts for Higher Alcohols Synthesis from Synthesis Gas: Comparison of MWCNT and Activated Carbon Supports. Ind. Eng. Chem. Res. 2010, 49, 6956–6963. 10.1021/ie100427j. [DOI] [Google Scholar]
- Surisetty V. R.; Dalai A. K.; Kozinski J. Influence of Porous Characteristics of the Carbon Support on Alkali-Modified Trimetallic Co-Rh-Mo Sulfided Catalysts for Higher Alcohols Synthesis from Synthesis Gas. Appl. Catal., A 2011, 393, 50–58. 10.1016/j.apcata.2010.11.026. [DOI] [Google Scholar]
- Singh B.; Murad L.; Laffir F.; Dickinson C.; Dempsey E. Pt Based Nanocomposites (Mono/Bi/Tri-Metallic) Decorated Using Different Carbon Supports for Methanol Electro-Oxidation in Acidic and Basic Media. Nanoscale 2011, 3, 3334–3349. 10.1039/c1nr10273g. [DOI] [PubMed] [Google Scholar]
- Long L.-L.; Lang W.-Z.; Liu X.; Hu C.-L.; Chu L.-F.; Guo Y.-J. Improved Catalytic Stability of PtSnIn/xCa–Al Catalysts for Propane Dehydrogenation to Propylene. Chem. Eng. J. 2014, 257, 209–217. 10.1016/j.cej.2014.07.044. [DOI] [Google Scholar]
- Long L. L.; Xia K.; Lang W. Z.; Shen L. L.; Yang Q.; Yan X.; Guo Y. J. The Comparison and Optimization of Zirconia, Alumina, and Zirconia-Alumina Supported PtSnIn Trimetallic Catalysts for Propane Dehydrogenation Reaction. J. Ind. Eng. Chem. 2017, 51, 271–280. 10.1016/j.jiec.2017.03.012. [DOI] [Google Scholar]
- Mendoza-Nieto J. A.; Robles-Méndez F.; Klimova T. E. Support Effect on the Catalytic Performance of Trimetallic NiMoW Catalysts Prepared with Citric Acid in HDS of Dibenzothiophenes. Catal. Today 2015, 250, 47–59. 10.1016/j.cattod.2014.05.002. [DOI] [Google Scholar]
- Comignani V.; Sieben J. M.; Sanchez M. D.; Duarte M. M. E. Influence of Carbon Support Properties on the Electrocatalytic Activity of PtRuCu Nanoparticles for Methanol and Ethanol Oxidation. Int. J. Hydrogen Energy 2017, 42, 24785–24796. 10.1016/j.ijhydene.2017.08.079. [DOI] [Google Scholar]
- Anashkin Y. V.; Ishutenko D. I.; Maximov V. V.; Pimerzin A. A.; Kogan V. M.; Nikulshin P. A. Effect of Carrier Properties on the Activity of Supported KCoMoS Catalysts in the Synthesis of Alcohol from Syngas. React. Kinet. Mech. Catal. 2019, 127, 301–314. 10.1007/s11144-019-01580-2. [DOI] [Google Scholar]
- Boldushevskii R. E.; Koklyukhin A. S.; Mozhaev A. V.; Minaev P. P.; Guseva A. I.; Nikul’shin P. A. Inhibiting Effect of Quinoline on the Hydroconversion of Dibenzothiophene and Naphthalene on Trimetallic NiCoMoS Catalysts Supported on Al2O3, SiO2, and SBA-15. Russ. J. Appl. Chem. 2019, 92, 1789–1796. 10.1134/S1070427219120216. [DOI] [Google Scholar]
- He Q.; Miedziak P. J.; Kesavan L.; Dimitratos N.; Sankar M.; Lopez-Sanchez J. A.; Forde M. M.; Edwards J. K.; Knight D. W.; Taylor S. H.; Kiely C. J.; Hutchings G. J. Switching-off Toluene Formation in the Solvent-Free Oxidation of Benzyl Alcohol Using Supported Trimetallic Au–Pd–Pt Nanoparticles. Faraday Discuss. 2013, 162, 365. 10.1039/c2fd20153d. [DOI] [PubMed] [Google Scholar]
- Jahel A. N.; Moizan-Baslé V.; Chizallet C.; Raybaud P.; Olivier-Fourcade J.; Jumas J. C.; Avenier P.; Lacombe S. Effect of Indium Doping of γ-Alumina on the Stabilization of PtSn Alloyed Clusters Prepared by Surface Organostannic Chemistry. J. Phys. Chem. C 2012, 116, 10073–10083. 10.1021/jp301282r. [DOI] [Google Scholar]
- Mendoza-Nieto J. A.; Vera-Vallejo O.; Escobar-Alarcón L.; Solís-Casados D. A.; Klimova T. Development of New Trimetallic NiMoW Catalysts Supported on SBA-15 for Deep Hydrodesulfurization. Fuel 2013, 110, 268–277. 10.1016/j.fuel.2012.07.057. [DOI] [Google Scholar]
- Deraz N. M. The Comparative Jurisprudence of Catalysts Preparation Methods: II. Deposition-Precipitation and Adsorption Methods. J. Ind. Env. Chem. 2018, 2, 1–3. [Google Scholar]
- Nakaya Y.; Miyazaki M.; Yamazoe S.; Shimizu K.-I.; Furukawa S. Active, Selective, and Durable Catalyst for Alkane Dehydrogenation Based on a Well-Designed Trimetallic Alloy. ACS Catal. 2020, 10, 5163–5172. 10.1021/acscatal.0c00151. [DOI] [Google Scholar]
- Lopez-Sanchez J. A.; Dimitratos N.; Miedziak P.; Ntainjua E.; Edwards J. K.; Morgan D.; Carley A. F.; Tiruvalam R.; Kiely C. J.; Hutchings G. J. Au-Pd Supported Nanocrystals Prepared by a Sol Immobilisation Technique as Catalysts for Selective Chemical Synthesis. Phys. Chem. Chem. Phys. 2008, 10, 1921–1930. 10.1039/b719345a. [DOI] [PubMed] [Google Scholar]
- Bahruji H.; Bowker M.; Hutchings G.; Dimitratos N.; Wells P.; Gibson E.; Jones W.; Brookes C.; Morgan D.; Lalev G. Pd/ZnO Catalysts for Direct CO2 hydrogenation to Methanol. J. Catal. 2016, 343, 133–146. 10.1016/j.jcat.2016.03.017. [DOI] [Google Scholar]
- Bönnemann H.; Richards R. M. Nanoscopic Metal Particles – Synthetic Methods and Potential Applications. Eur. J. Inorg. Chem. 2001, 2001, 2455–2480. . [DOI] [Google Scholar]
- Collier P. J.; Iggo J. A.; Whyman R. Preparation and Characterisation of Solvent-Stabilised Nanoparticulate Platinum and Palladium and Their Catalytic Behaviour towards the Enantioselective Hydrogenation of Ethyl Pyruvate. J. Mol. Catal. A: Chem. 1999, 146, 149–157. 10.1016/S1381-1169(99)00084-9. [DOI] [Google Scholar]
- Jia C.-J.; Schüth F. Colloidal Metal Nanoparticles as a Component of Designed Catalyst. Phys. Chem. Chem. Phys. 2011, 13, 2457. 10.1039/c0cp02680h. [DOI] [PubMed] [Google Scholar]
- Chou N. H.; Schaak R. E. Shape-Controlled Conversion of β-Sn Nanocrystals into Intermetallic M-Sn (M = Fe, Co, Ni, Pd) Nanocrystals. J. Am. Chem. Soc. 2007, 129, 7339–7345. 10.1021/ja069032y. [DOI] [PubMed] [Google Scholar]
- Serpell C. J.; Cookson J.; Ozkaya D.; Beer P. D. Core@shell Bimetallic Nanoparticle Synthesis via Anion Coordination. Nat. Chem. 2011, 3, 478–483. 10.1038/nchem.1030. [DOI] [PubMed] [Google Scholar]
- Ye X.; He X.; Lei Y.; Tang J.; Yu Y.; Shi H.; Wang K. One-Pot Synthesized Cu/Au/Pt Trimetallic Nanoparticles with Enhanced Catalytic and Plasmonic Properties as a Universal Platform for Biosensing and Cancer Theranostics. Chem. Commun. 2019, 55, 2321–2324. 10.1039/C8CC10127B. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Xu H.; Wang J.; Li S.; Xiong Z.; Yan B.; Wang C.; Du Y. Visible Light Enhanced Electrochemical Detection of Caffeic Acid with Waxberry-like PtAuRu Nanoparticles Modified GCE. Sens. Actuators, B. 2018, 272, 135–138. 10.1016/j.snb.2018.05.160. [DOI] [Google Scholar]
- Pandey P. C.; Pandey G. One-Pot Two-Step Rapid Synthesis of 3-Aminopropyltrimethoxysilane-Mediated Highly Catalytic Ag@(PdAu) Trimetallic Nanoparticles. Catal. Sci. Technol. 2016, 6, 3911–3917. 10.1039/C5CY02040A. [DOI] [Google Scholar]
- Matin M. A.; Jang J. H.; Kwon Y. U. One-Pot Sonication-Assisted Polyol Synthesis of Trimetallic Core-Shell (Pd,Co)@Pt Nanoparticles for Enhanced Electrocatalysis. Int. J. Hydrogen Energy 2014, 39, 3710–3718. 10.1016/j.ijhydene.2013.12.137. [DOI] [Google Scholar]
- Fievet F.; Ammar-Merah S.; Brayner R.; Chau F.; Giraud M.; Mammeri F.; Peron J.; Piquemal J. Y.; Sicard L.; Viau G. The Polyol Process: A Unique Method for Easy Access to Metal Nanoparticles with Tailored Sizes, Shapes and Compositions. Chem. Soc. Rev. 2018, 47, 5187–5233. 10.1039/C7CS00777A. [DOI] [PubMed] [Google Scholar]
- Rethinasabapathy M.; Kang S. M.; Haldorai Y.; Jankiraman M.; Jonna N.; Choe S. R.; Huh Y. S.; Natesan B. Ternary PtRuFe Nanoparticles Supported N-Doped Graphene as an Efficient Bifunctional Catalyst for Methanol Oxidation and Oxygen Reduction Reactions. Int. J. Hydrogen Energy 2017, 42, 30738–30749. 10.1016/j.ijhydene.2017.10.121. [DOI] [Google Scholar]
- Gao P.; Cai Y.; Wang F.; Zhu H.; Pu M. Pt-Based Trimetallic Nanocrystals with High Proportions of M (M = Fe, Ni) Metals for Catalyzing Oxygen Reduction Reaction. Int. J. Hydrogen Energy 2020, 45, 16039–16048. 10.1016/j.ijhydene.2020.03.152. [DOI] [Google Scholar]
- Karthikeyan B.; Loganathan B. Strategic Green Synthesis and Characterization of Au/Pt/Ag Trimetallic Nanocomposites. Mater. Lett. 2012, 85, 53–56. 10.1016/j.matlet.2012.06.070. [DOI] [Google Scholar]
- Yadav N.; Jaiswal A. K.; Dey K. K.; Yadav V. B.; Nath G.; Srivastava A. K.; Yadav R. R. Trimetallic Au/Pt/Ag Based Nanofluid for Enhanced Antibacterial Response. Mater. Chem. Phys. 2018, 218, 10–17. 10.1016/j.matchemphys.2018.07.016. [DOI] [Google Scholar]
- Loganathan B.; Karthikeyan B. Au Core Pd/Pt Shell in Trimetallic Au/Pd/Pt Colloidal Nanocomposites - Physicochemical Characterization Study. Colloids Surf., A 2013, 436, 944–952. 10.1016/j.colsurfa.2013.08.035. [DOI] [Google Scholar]
- Chen S.; Reggiano G.; Thota S.; Zhao J. Au–Cu–Ag Nanorods Synthesized by Seed-Mediated Coreduction and Their Optical Properties. Part. Part. Syst. Charact. 2017, 34, 1600384. 10.1002/ppsc.201600384. [DOI] [Google Scholar]
- Li Q.; Xu P.; Zhang B.; Wu G.; Zhao H.; Fu E.; Wang H. L. Self-Supported Pt Nanoclusters via Galvanic Replacement from Cu2O Nanocubes as Efficient Electrocatalysts. Nanoscale 2013, 5, 7397–7402. 10.1039/c3nr02243a. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen S.; Reggiano G.; Thota S.; Wang Y.; Kerns P.; Suib S. L.; Zhao J. Au-Cu-M (M = Pt, Pd, Ag) Nanorods with Enhanced Catalytic Efficiency by Galvanic Replacement Reaction. Chem. Commun. 2019, 55, 1249–1252. 10.1039/C8CC08083F. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Yeo B. C.; Han S. S.; Lee T. J.; Bhang S. H.; Kim W. S.; Yu T. Facile Aqueous-Phase Synthesis of Ag–Cu–Pt–Pd Quadrometallic Nanoparticles. Nano Convergence 2019, 6, 38. 10.1186/s40580-019-0208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Deng Z.; Li J.; Liu H.; Wang Y.; Fu X. Efficient Electrolytic Water Splitting with FeCoPt Trimetallic Cubic Nanocatalysts. IOP Conf. Ser. Mater. Sci. Eng. 2019, 592, 012043 10.1088/1757-899X/592/1/012043. [DOI] [Google Scholar]
- Song Y.; Chen S. Trimetallic Ag@AuPt Neapolitan Nanoparticles. Nanoscale 2013, 5, 7284–7289. 10.1039/c3nr02269b. [DOI] [PubMed] [Google Scholar]
- Papaderakis A.; Mintsouli I.; Georgieva J.; Sotiropoulos S. Electrocatalysts Prepared by Galvanic Replacement. Catalysts 2017, 7, 80. 10.3390/catal7030080. [DOI] [Google Scholar]
- Li B.; Chan S. H. PtFeNi Tri-Metallic Alloy Nanoparticles as Electrocatalyst for Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells with Ultra-Low Pt Loading. Int. J. Hydrogen Energy 2013, 38, 3338–3345. 10.1016/j.ijhydene.2013.01.049. [DOI] [Google Scholar]
- da Silva A. G. M.; Rodrigues T. S.; Haigh S. J.; Camargo P. H. C. Galvanic Replacement Reaction: Recent Developments for Engineering Metal Nanostructures towards Catalytic Applications. Chem. Commun. 2017, 53, 7135–7148. 10.1039/C7CC02352A. [DOI] [PubMed] [Google Scholar]
- Lowe J. M.; Coridan R. H. Mechanistic Control of a Galvanic Replacement Reaction on Cuprous Oxide. Nanoscale Adv. 2019, 1, 1343–1350. 10.1039/C8NA00396C. [DOI] [Google Scholar]
- Miyazaki M.; Furukawa S.; Takayama T.; Yamazoe S.; Komatsu T. Surface Modification of PdZn Nanoparticles via Galvanic Replacement for the Selective Hydrogenation of Terminal Alkynes. ACS Appl. Nano Mater. 2019, 2, 3307–3314. 10.1021/acsanm.9b00761. [DOI] [Google Scholar]
- You D. J.; Jin S.; Lee K. H.; Pak C.; Choi K. H.; Chang H. Improvement of Activity for Oxygen Reduction Reaction by Decoration of Ir on PdCu/C Catalyst. Catal. Today 2012, 185, 138–142. 10.1016/j.cattod.2011.08.024. [DOI] [Google Scholar]
- Yang J.; Zhou W.; Cheng C. H.; Lee J. Y.; Liu Z. Pt-Decorated PdFe Nanoparticles as Methanol-Tolerant Oxygen Reduction Electrocatalyst. ACS Appl. Mater. Interfaces 2010, 2, 119–126. 10.1021/am900623e. [DOI] [PubMed] [Google Scholar]
- Merkoçi F.; Patarroyo J.; Russo L.; Piella J.; Genç A.; Arbiol J.; Bastús N. G.; Puntes V. Understanding Galvanic Replacement Reactions: The Case of Pt and Ag. Mater. Today Adv. 2020, 5, 100037. 10.1016/j.mtadv.2019.100037. [DOI] [Google Scholar]
- Wang D. Y.; Chou H. L.; Lin Y. C.; Lai F. J.; Chen C. H.; Lee J. F.; Hwang B. J.; Chen C. C. Simple Replacement Reaction for the Preparation of Ternary Fe1–xPtRux Nanocrystals with Superior Catalytic Activity in Methanol Oxidation Reaction. J. Am. Chem. Soc. 2012, 134, 10011–10020. 10.1021/ja3010754. [DOI] [PubMed] [Google Scholar]
- Sahoo A.; Tripathy S. K.; Dehury N.; Patra S. A Porous Trimetallic Au@Pd@Ru Nanoparticle System: Synthesis, Characterisation and Efficient Dye Degradation and Removal. J. Mater. Chem. A 2015, 3, 19376–19383. 10.1039/C5TA03959B. [DOI] [Google Scholar]
- Chen A. N.; McClain S. M.; House S. D.; Yang J. C.; Skrabalak S. E. Mechanistic Study of Galvanic Replacement of Chemically Heterogeneous Templates. Chem. Mater. 2019, 31, 1344–1351. 10.1021/acs.chemmater.8b04630. [DOI] [Google Scholar]
- Chen A. N.; Endres E. J.; Ashberry H. M.; Bueno S. L. A.; Chen Y.; Skrabalak S. E. Galvanic Replacement of Intermetallic Nanocrystals as a Route toward Complex Heterostructures. Nanoscale 2021, 13, 2618–2625. 10.1039/D0NR08255D. [DOI] [PubMed] [Google Scholar]
- Patarroyo Rengifo J. I.Exploring Synthetic Strategies for the Production of Complex Inorganic Nanoparticles. Doctoral Thesis, Universitat Autonoma de Barcelona, Bellaterra, Spain, 2018. [Google Scholar]
- Wu Y.; Wang D.; Niu Z.; Chen P.; Zhou G.; Li Y. A Strategy for Designing a Concave Pt-Ni Alloy through Controllable Chemical Etching. Angew. Chem., Int. Ed. 2012, 51, 12524–12528. 10.1002/anie.201207491. [DOI] [PubMed] [Google Scholar]
- Yin H.-J.; Zhang Z.-P.; Guo Y.; Yuan K.; Zhang Y.-W. Highly-Selective Synthesis of Trimetallic PtRuCu Nanoframes as Robust Catalysts for the Methanol Oxidation Reaction. Mater. Chem. Front. 2020, 4, 1985–1992. 10.1039/D0QM00033G. [DOI] [Google Scholar]
- Kumara L. S. R.; Sakata O.; Kohara S.; Yang A.; Song C.; Kusada K.; Kobayashi H.; Kitagawa H. Origin of the Catalytic Activity of Face-Centered-Cubic Ruthenium Nanoparticles Determined from an Atomic-Scale Structure. Phys. Chem. Chem. Phys. 2016, 18, 30622–30629. 10.1039/C6CP04088H. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Guo S.; Dong S. Facile Synthesis of Trimetallic AuPtPd Alloy Nanowires and Their Catalysis for Ethanol Electrooxidation. J. Mater. Chem. 2012, 22, 14851–14855. 10.1039/c2jm32663a. [DOI] [Google Scholar]
- Ding K.; Li Y.; Zhao Y.; Zhao J.; Chen Y.; Wang Q. Preparation of a Trimetallic Pd1NiyAl0.5 Composite Nanoparticle by a Hydrothermal Method for Ethanol Oxidation Reaction. Int. J. Electrochem. Sci. 2015, 10, 8844–8857. [Google Scholar]
- Annalakshmi M.; Balasubramanian P.; Chen S.-M.; Chen T.-W. One Pot Synthesis of Nanospheres-like Trimetallic NiFeCo Nanoalloy: A Superior Electrocatalyst for Electrochemical Sensing of Hydrazine in Water Bodies. Sensors Actuators B Chem. 2019, 296, 126620. 10.1016/j.snb.2019.05.097. [DOI] [Google Scholar]
- Fu G. T.; Xia B. Y.; Ma R. G.; Chen Y.; Tang Y. W.; Lee J. M. Trimetallic PtAgCu@PtCu Core@shell Concave Nanooctahedrons with Enhanced Activity for Formic Acid Oxidation Reaction. Nano Energy 2015, 12, 824–832. 10.1016/j.nanoen.2015.01.041. [DOI] [Google Scholar]
- Zhu W.; Ke J.; Wang S.-B.; Ren J.; Wang H.-H.; Zhou Z.-Y.; Si R.; Zhang Y.-W.; Yan C.-H. Shaping Single-Crystalline Trimetallic Pt–Pd–Rh Nanocrystals toward High-Efficiency C–C Splitting of Ethanol in Conversion to CO2. ACS Catal. 2015, 5, 1995–2008. 10.1021/cs5018419. [DOI] [Google Scholar]
- Zhang Y.; Qu T.; Bi F.; Hao P.; Li M.; Chen S.; Guo X.; Xie M.; Guo X. Trimetallic (Co/Ni/Cu) Hydroxyphosphate Nanosheet Array as Efficient and Durable Electrocatalyst for Oxygen Evolution Reaction. ACS Sustainable Chem. Eng. 2018, 6, 16859–16866. 10.1021/acssuschemeng.8b04180. [DOI] [Google Scholar]
- Chen P.-C.; Liu G.; Zhou Y.; Brown K. A.; Chernyak N.; Hedrick J. L.; He S.; Xie Z.; Lin Q.-Y.; Dravid V. P.; O’Neill-Slawecki S. A.; Mirkin C. A. Tip-Directed Synthesis of Multimetallic Nanoparticles. J. Am. Chem. Soc. 2015, 137, 9167–9173. 10.1021/jacs.5b05139. [DOI] [PubMed] [Google Scholar]
- Chen P. C.; Liu X.; Hedrick J. L.; Xie Z.; Wang S.; Lin Q. Y.; Hersam M. C.; Dravid V. P.; Mirkin C. A. Polyelemental Nanoparticle Libraries. Science 2016, 352, 1565–1569. 10.1126/science.aaf8402. [DOI] [PubMed] [Google Scholar]
- Chai J.; Huo F.; Zheng Z.; Giam L. R.; Shim W.; Mirkin C. A. Scanning Probe Block Copolymer Lithography. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20202–20206. 10.1073/pnas.1014892107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.; Chen J.-S. Synthetic Porous Materials Applied in Hydrogenation Reactions. Microporous Mesoporous Mater. 2017, 237, 246–259. 10.1016/j.micromeso.2016.09.039. [DOI] [Google Scholar]
- Yan Z. P.; Lin L.; Liu S. Synthesis of γ-Valerolactone by Hydrogenation of Biomass- DerivedLevulinic Acid over Ru/C Catalyst. Energy Fuels 2009, 23, 3853–3858. 10.1021/ef900259h. [DOI] [Google Scholar]
- Maegawa T.; Akashi A.; Yaguchi K.; Iwasaki Y.; Shigetsura M.; Monguchi Y.; Sajiki H. Efficient and Practical Arene Hydrogenation by Heterogeneous Catalysts under Mild Conditions.. Chem. - Eur. J. 2009, 15, 6953–6963. 10.1002/chem.200900361. [DOI] [PubMed] [Google Scholar]
- Gallezot P.; Cerino P. J.; Blanc B.; Flèche G.; Fuertes P. Glucose Hydrogenation on Promoted Raney-Nickel Catalysts. J. Catal. 1994, 146, 93–102. 10.1016/0021-9517(94)90012-4. [DOI] [Google Scholar]
- Lindlar H.; Dubuis R. Palladium Catalyst for Partial Reduction of Acetylenes. Org. Synth. 1966, 46, 89. 10.15227/orgsyn.046.0089. [DOI] [Google Scholar]
- Schulz H. Short History and Present Trends of Fischer–Tropsch Synthesis. Appl. Catal., A 1999, 186, 3–12. 10.1016/S0926-860X(99)00160-X. [DOI] [Google Scholar]
- Vannice M. A. The Catalytic Synthesis of Hydrocarbons from H2 CO Mixtures over the Group VIII Metals.I. The Specific Activities and Product Distributions of Supported Metals. J. Catal. 1975, 37, 449–461. 10.1016/0021-9517(75)90181-5. [DOI] [Google Scholar]
- Badoga S.; Kamath G.; Dalai A. Effects of Promoters (Mn, Mg, Co and Ni) on the Fischer–Tropsch Activity and Selectivity of KCuFe/Mesoporous-Alumina Catalyst. Appl. Catal., A 2020, 607, 117861. 10.1016/j.apcata.2020.117861. [DOI] [Google Scholar]
- Golestan S.; Mirzaei A. A.; Atashi H. Fischer–Tropsch Synthesis over an Iron–Cobalt–Manganese (Ternary) Nanocatalyst Prepared by Hydrothermal Procedure: Effects of Nanocatalyst Composition and Operational Conditions. Int. J. Hydrogen Energy 2017, 42, 9816–9830. 10.1016/j.ijhydene.2017.01.162. [DOI] [Google Scholar]
- Arsalanfar M.; Mirzaei A. A.; Bozorgzadeh H. R.; Atashi H. Effect of Process Conditions on the Surface Reaction Rates and Catalytic Performance of MgO Supported Fe–Co–Mn Catalyst for CO Hydrogenation. J. Ind. Eng. Chem. 2012, 18, 2092–2102. 10.1016/j.jiec.2012.06.003. [DOI] [Google Scholar]
- Razmara Z.; Rezvani A. R.; Saravani H. Fischer–Tropsch Reaction over a Co2-Ni-Mn/SiO2 Nanocatalyst Prepared by Thermal Decomposition of a New Precursor. Chem. Pap. 2016 714 2017, 71, 849–856. 10.1007/s11696-016-0100-9. [DOI] [Google Scholar]
- Zhai P.; Xu C.; Gao R.; Liu X.; Li M.; Li W.; Fu X.; Jia C.; Xie J.; Zhao M.; Wang X.; Li Y. W.; Zhang Q.; Wen X. D.; Ma D. Highly Tunable Selectivity for Syngas-Derived Alkenes over Zinc and Sodium-Modulated Fe5C2 Catalyst. Angew. Chem., Int. Ed. 2016, 55, 9902–9907. 10.1002/anie.201603556. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Göeltl F.; Ro I.; Ball M. R.; Sener C.; Aragão I. B.; Zanchet D.; Huber G. W.; Mavrikakis M.; Dumesic J. A. Synthesis Gas Conversion over Rh-Based Catalysts Promoted by Fe and Mn. ACS Catal. 2017, 7, 4550–4563. 10.1021/acscatal.7b01381. [DOI] [Google Scholar]
- Bowker M. Methanol Synthesis from CO2 Hydrogenation. ChemCatChem. 2019, 11, 4238–4246. 10.1002/cctc.201900401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Ramírez J.; Díaz J. A.; Sánchez P.; Dorado F. Optimization of the Pd/Cu Ratio in Pd-Cu-Zn/SiC Catalysts for the CO2 Hydrogenation to Methanol at Atmospheric Pressure. J. CO2 Util. 2017, 22, 71–80. 10.1016/j.jcou.2017.09.012. [DOI] [Google Scholar]
- Zhao S.; Zhao Z.; Yao K.; Liu H. Density Functional Study of PdaCubAuc (a + b + c = 7) Clusters: Geometry, Electronic and H2 Physisorption Properties. Comput. Theor. Chem. 2020, 1178, 112783. 10.1016/j.comptc.2020.112783. [DOI] [Google Scholar]
- Xue J. L.; Fang L.; Luo W.; Meng Y.; Chen T.; Xia S. J.; Ni Z. M. Density Functional Study of Water Gas Shift Reaction Catalyzed by Cu-Pt-Au Ternary Alloy. Ranliao Huaxue Xuebao/J. Fuel Chem. Technol. 2019, 47, 688–696. 10.1016/S1872-5813(19)30030-1. [DOI] [Google Scholar]
- Duyar M. S.; Gallo A.; Snider J. L.; Jaramillo T. F. Low-Pressure Methanol Synthesis from CO2 over Metal-Promoted Ni-Ga Intermetallic Catalysts. J. CO2 Util. 2020, 39, 101151. 10.1016/j.jcou.2020.03.001. [DOI] [Google Scholar]
- Smitshuysen T. E. L.; Nielsen M. R.; Pruessmann T.; Zimina A.; Sheppard T. L.; Grunwaldt J.; Chorkendorff I.; Damsgaard C. D. Optimizing Ni–Fe–Ga Alloys into Ni2FeGa for the Hydrogenation of CO2 into Methanol. ChemCatChem. 2020, 12, 3265–3273. 10.1002/cctc.202000174. [DOI] [Google Scholar]
- Pori M.; Arčon I.; Dasireddy V. D. B. C.; Likozar B.; Orel Z. C.; Marinšek M. Photo-Chemically-Deposited and Industrial Cu/ZnO/Al2O3 Catalyst Material Surface Structures During CO2 Hydrogenation to Methanol: EXAFS, XANES and XPS Analyses of Phases after Oxidation, Reduction, and Reaction. Catal. Lett. 2021, 151, 3114–3134. 10.1007/s10562-021-03556-1. [DOI] [Google Scholar]
- Campos-Martin J. M.; Blanco-Brieva G.; Fierro J. L. G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem., Int. Ed. 2006, 45, 6962–6984. 10.1002/anie.200503779. [DOI] [PubMed] [Google Scholar]
- Lunsford J. H. The Direct Formation of H2O2 from H2 and O2 over Palladium Catalysts. J. Catal. 2003, 216, 455–460. 10.1016/S0021-9517(02)00070-2. [DOI] [Google Scholar]
- Krishnan V. V.; Dokoutchaev A. G.; Thompson M. E. Direct Production of Hydrogen Peroxide with Palladium Supported on Phosphate Viologen Phosphonate Catalysts. J. Catal. 2000, 196, 366–374. 10.1006/jcat.2000.3036. [DOI] [Google Scholar]
- Freakley S. J.; He Q.; Harrhy J. H.; Lu L.; Crole D. A.; Morgan D. J.; Ntainjua E. N.; Edwards J. K.; Carley A. F.; Borisevich A. Y.; Kiely C. J.; Hutchings G. J. Palladium-Tin Catalysts for the Direct Synthesis of H2O2 with High Selectivity. Science 2016, 351, 965–968. 10.1126/science.aad5705. [DOI] [PubMed] [Google Scholar]
- Landon P.; Collier P. J.; Papworth A. J.; Kiely C. J.; Hutchings G. J. Direct Formation of Hydrogen Peroxide from H2/O2 Using a Gold Catalyst. Chem. Commun. 2002, 18, 2058–2059. 10.1039/b205248m. [DOI] [PubMed] [Google Scholar]
- Choudhary V. R.; Gaikwad A. G.; Sansare S. D. Activation of Supported Pd Metal Catalysts for Selective Oxidation of Hydrogen to Hydrogen Peroxide. Catal. Lett. 2002, 83, 235–239. 10.1023/A:1021066904862. [DOI] [Google Scholar]
- Edwards J. K.; Carley A. F.; Herzing A. A.; Kiely C. J.; Hutchings G. J. Direct Synthesis of Hydrogen Peroxide from H2 and O2 Using Supported Au-Pd Catalysts. Faraday Discuss. 2008, 138, 225–239. 10.1039/B705915A. [DOI] [PubMed] [Google Scholar]
- Edwards J. K.; Pritchard J.; Miedziak P. J.; Piccinini M.; Carley A. F.; He Q.; Kiely C. J.; Hutchings G. J. The Direct Synthesis of Hydrogen Peroxide Using Platinum Promoted Gold–Palladium Catalysts. Catal. Sci. Technol. 2014, 4, 3244–3250. 10.1039/C4CY00496E. [DOI] [PubMed] [Google Scholar]
- Ranganathan S.; Sieber V. Recent Advances in the Direct Synthesis of Hydrogen Peroxide Using Chemical Catalysis—A Review. Catalysts 2018, 8, 379. 10.3390/catal8090379. [DOI] [Google Scholar]
- Menegazzo F.; Signoretto M.; Ghedini E.; Strukul G. Looking for the “Dream Catalyst” for Hydrogen Peroxide Production from Hydrogen and Oxygen. Catalysts 2019, 9, 251. 10.3390/catal9030251. [DOI] [Google Scholar]
- Gu J.; Wang S.; He Z.; Han Y.; Zhang J. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Activated-Carbon-Supported Pd-Ag Alloy Catalysts. Catal. Sci. Technol. 2016, 6, 809–817. 10.1039/C5CY00813A. [DOI] [Google Scholar]
- Ntainjua E. N.; Freakley S. J.; Hutchings G. J. Direct Synthesis of Hydrogen Peroxide Using Ruthenium Catalysts. Top. Catal. 2012, 55, 718–722. 10.1007/s11244-012-9866-3. [DOI] [Google Scholar]
- Satterthwaite K.Plastics Based on Styrene. In Brydson’s Plastics Materials; Elsevier, 2017; pp 311–328. 10.1016/B978-0-323-35824-8.00012-8. [DOI] [Google Scholar]
- Miyazaki M.; Furukawa S.; Komatsu T. Regio- and Chemoselective Hydrogenation of Dienes to Monoenes Governed by a Well-Structured Bimetallic Surface. J. Am. Chem. Soc. 2017, 139, 18231–18239. 10.1021/jacs.7b08792. [DOI] [PubMed] [Google Scholar]
- Jeon H.-j.; Chung Y.-M. Hydrogen Production from Formic Acid Dehydrogenation over Pd/C Catalysts: Effect of Metal and Support Properties on the Catalytic Performance. Appl. Catal., B 2017, 210, 212–222. 10.1016/j.apcatb.2017.03.070. [DOI] [Google Scholar]
- Li J.; Zhu Q. L.; Xu Q. Dehydrogenation of Formic Acid by Heterogeneous Catalysts. Chimia 2015, 69, 348–352. 10.2533/chimia.2015.348. [DOI] [PubMed] [Google Scholar]
- Li S.; Zhou Y.; Kang X.; Liu D.; Gu L.; Zhang Q.; Yan J.; Jiang Q. A Simple and Effective Principle for a Rational Design of Heterogeneous Catalysts for Dehydrogenation of Formic Acid. Adv. Mater. 2019, 31, 1806781. 10.1002/adma.201806781. [DOI] [PubMed] [Google Scholar]
- Li Z.; Xu Q. Metal-Nanoparticle-Catalyzed Hydrogen Generation from Formic Acid. Acc. Chem. Res. 2017, 50, 1449–1458. 10.1021/acs.accounts.7b00132. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Kam L.; Trerise R.; Williams T. J. Ruthenium-Catalyzed Ammonia Borane Dehydrogenation: Mechanism and Utility. Acc. Chem. Res. 2017, 50, 86–95. 10.1021/acs.accounts.6b00482. [DOI] [PubMed] [Google Scholar]
- Denney M. C.; Pons V.; Hebden T. J.; Heinekey D. M.; Goldberg K. I. Efficient Catalysis of Ammonia Borane Dehydrogenation. J. Am. Chem. Soc. 2006, 128, 12048–12049. 10.1021/ja062419g. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Kam L.; Williams T. J. Dehydrogenation of Ammonia Borane through the Third Equivalent of Hydrogen. Dalton Trans. 2016, 45, 7672–7677. 10.1039/C6DT00604C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk M. J.; Crabtree R. H. Selective Catalytic Dehydrogenation of Alkanes to Alkenes. J. Am. Chem. Soc. 1987, 109, 8025–8032. 10.1021/ja00260a013. [DOI] [Google Scholar]
- Sterligov O. D.; Olfer’eva T. G.; Kononov N. F. Dehydrogenation of Alkanes and Alkenes at Low Pressures. Russ. Chem. Rev. 1967, 36, 498–511. 10.1070/RC1967v036n07ABEH001659. [DOI] [Google Scholar]
- James O. O.; Mandal S.; Alele N.; Chowdhury B.; Maity S. Lower Alkanes Dehydrogenation: Strategies and Reaction Routes to Corresponding Alkenes. Fuel Process. Technol. 2016, 149, 239–255. 10.1016/j.fuproc.2016.04.016. [DOI] [Google Scholar]
- Flick D. W.; Huff M. C. Acetylene Formation during the Catalytic Oxidative Dehydrogenation of Ethane over Pt-Coated Monoliths at Short Contact Times. Catal. Lett. 1997, 47, 91–97. 10.1023/A:1019088501248. [DOI] [Google Scholar]
- Håkonsen S. F.; Silberova B.; Holmen A. Oxidative Dehydrogenation of Ethane on Pt–Sn Impregnated Monoliths. Top. Catal. 2007, 45, 61–67. 10.1007/s11244-007-0241-8. [DOI] [Google Scholar]
- Cum G.; Gallo R.; Ipsale S.; Spadaro A. Selective Synthesis of Alkynes by Catalytic Dehydrogenation of Alkenes over Polymer-Supported Palladium Acetate in the Liquid Phase. J. Chem. Soc., Chem. Commun. 1985, 22, 1571. 10.1039/c39850001571. [DOI] [Google Scholar]
- Taubmann S.; Alt H. G. Heterogeneous Catalysts for the Dehydrogenation of Saturated Hydrocarbons. J. Mol. Catal. A: Chem. 2008, 287, 102–109. 10.1016/j.molcata.2008.02.023. [DOI] [Google Scholar]
- Delbecq F.; Loffreda D.; Sautet P. Heterogeneous Catalytic Hydrogenation: Is Double Bond/Surface Coordination Necessary?. J. Phys. Chem. Lett. 2010, 1, 323–326. 10.1021/jz900159q. [DOI] [Google Scholar]
- Shuikin N. I.; Timofeeva E. A.; Dobrynina T. P. Heterogeneous Catalytic Dehydrogenation of Isopentane as a Function of Temperature. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1958, 7, 702–705. 10.1007/BF00917279. [DOI] [Google Scholar]
- Johnson T. C.; Morris D. J.; Wills M. Hydrogen Generation from Formic Acid and Alcohols Using Homogeneous Catalysts. Chem. Soc. Rev. 2010, 39, 81–88. 10.1039/B904495G. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Kobayashi T.; Suenobu T. Unusually Large Tunneling Effect on Highly Efficient Generation of Hydrogen and Hydrogen Isotopes in pH-Selective Decomposition of Formic Acid Catalyzed by a Heterodinuclear Iridium-Ruthenium Complex in Water. J. Am. Chem. Soc. 2010, 132, 1496–1497. 10.1021/ja910349w. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wu W.; Jiang Z.; Fang T. A Review on Liquid-Phase Heterogeneous Dehydrogenation of Formic Acid: Recent Advances and Perspectives. Chem. Pap. 2018, 72, 2121–2135. 10.1007/s11696-018-0469-8. [DOI] [Google Scholar]
- Hu C.; Pulleri J. K.; Ting S. W.; Chan K. Y. Activity of Pd/C for Hydrogen Generation in Aqueous Formic Acid Solution. Int. J. Hydrogen Energy 2014, 39, 381–390. 10.1016/j.ijhydene.2013.10.067. [DOI] [Google Scholar]
- Akbayrak S.; Tonbul Y.; Özkar S. Nanoceria Supported Palladium(0) Nanoparticles: Superb Catalyst in Dehydrogenation of Formic Acid at Room Temperature. Appl. Catal., B 2017, 206, 384–392. 10.1016/j.apcatb.2017.01.063. [DOI] [Google Scholar]
- Li Z.; Yang X.; Tsumori N.; Liu Z.; Himeda Y.; Autrey T.; Xu Q. Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid. ACS Catal. 2017, 7, 2720–2724. 10.1021/acscatal.7b00053. [DOI] [Google Scholar]
- Zhang S.; Metin Ö.; Su D.; Sun S. Monodisperse AgPd Alloy Nanoparticles and Their Superior Catalysis for the Dehydrogenation of Formic Acid. Angew. Chem., Int. Ed. 2013, 52, 3681–3684. 10.1002/anie.201300276. [DOI] [PubMed] [Google Scholar]
- Jones S.; Kolpin A.; Tsang S. C. E. Modification of Pd for Formic Acid Decomposition by Support Grafted Functional Groups. Catal. Struct. React. 2015, 1, 19–24. 10.1179/2055075814Y.0000000004. [DOI] [Google Scholar]
- Fang P. P.; Duan S.; Lin X. D.; Anema J. R.; Li J. F.; Buriez O.; Ding Y.; Fan F. R.; Wu D. Y.; Ren B.; Wang Z. L.; Amatore C.; Tian Z. Q. Tailoring Au-Core Pd-Shell Pt-Cluster Nanoparticles for Enhanced Electrocatalytic Activity. Chem. Sci. 2011, 2, 531–539. 10.1039/C0SC00489H. [DOI] [Google Scholar]
- Duan S.; Ji Y. F.; Fang P. P.; Chen Y. X.; Xu X.; Luo Y.; Tian Z. Q. Density Functional Theory Study on the Adsorption and Decomposition of the Formic Acid Catalyzed by Highly Active Mushroom-like Au@Pd@Pt Tri-Metallic Nanoparticles. Phys. Chem. Chem. Phys. 2013, 15, 4625–4633. 10.1039/c3cp44053b. [DOI] [PubMed] [Google Scholar]
- Gómez S.; Torres C.; Gárcia Fierro J. L. G.; Apesteguía C. R.; Reyes P. Hydrogenation of Nitrobenzene on Au/ZrO2 Catalysts. J. Chil. Chem. Soc. 2012, 57, 1194–1198. 10.4067/S0717-97072012000200029. [DOI] [Google Scholar]
- Downing R. S.; Kunkeler P. J.; Van Bekkum H. Catalytic Syntheses of Aromatic Amines. Catal. Today 1997, 37, 121–136. 10.1016/S0920-5861(97)00005-9. [DOI] [Google Scholar]
- Nickson T. E. A Unique Application of the Sulfide Reduction Useful for the Preparation of Isomerically Pure Aromatic Nitro Compounds and Anilines. J. Org. Chem. 1986, 51, 3903–3904. 10.1021/jo00370a031. [DOI] [Google Scholar]
- Dale D. J.; Dunn P. J.; Golightly C.; Hughes M. L.; Levett P. C.; Pearce A. K.; Searle P. M.; Ward G.; Wood A. S. The Chemical Development of the Commercial Route to Sildenafil: A Case History. Org. Process Res. Dev. 2000, 4, 17–22. 10.1021/op9900683. [DOI] [Google Scholar]
- Yang J.; Ying J. Y. Nanocomposites of Ag2S and Noble Metals. Angew. Chem., Int. Ed. 2011, 50, 4637–4643. 10.1002/anie.201101213. [DOI] [PubMed] [Google Scholar]
- Mori K.; Naka K.; Masuda S.; Miyawaki K.; Yamashita H. Palladium Copper Chromium Ternary Nanoparticles Constructed In Situ within a Basic Resin: Enhanced Activity in the Dehydrogenation of Formic Acid. ChemCatChem 2017, 9, 3456–3462. 10.1002/cctc.201700595. [DOI] [Google Scholar]
- Khan Z. Trimetallic Nanoparticles: Synthesis, Characterization and Catalytic Degradation of Formic Acid for Hydrogen Generation. Int. J. Hydrogen Energy 2019, 44, 11503–11513. 10.1016/j.ijhydene.2019.03.122. [DOI] [Google Scholar]
- Bhasin M. M.; McCain J. H.; Vora B. V.; Imai T.; Pujadó P. R. Dehydrogenation and Oxydehydrogenation of Paraffins to Olefins. Appl. Catal., A 2001, 221, 397–419. 10.1016/S0926-860X(01)00816-X. [DOI] [Google Scholar]
- Amghizar I.; Vandewalle L. A.; Van Geem K. M.; Marin G. B. New Trends in Olefin Production. Engineering 2017, 3, 171–178. 10.1016/J.ENG.2017.02.006. [DOI] [Google Scholar]
- Liu X.; Lang W. Z.; Long L. L.; Hu C. L.; Chu L. F.; Guo Y. J. Improved Catalytic Performance in Propane Dehydrogenation of PtSn/γ-Al2O3 Catalysts by Doping Indium. Chem. Eng. J. 2014, 247, 183–192. 10.1016/j.cej.2014.02.084. [DOI] [Google Scholar]
- Peng B.; Chen J. Ammonia Borane as an Efficient and Lightweight Hydrogen Storage Medium. Energy Environ. Sci. 2008, 1, 479–483. 10.1039/b809243p. [DOI] [Google Scholar]
- Akbayrak S.; Özkar S. Ammonia Borane as Hydrogen Storage Materials. Int. J. Hydrogen Energy 2018, 43, 18592–18606. 10.1016/j.ijhydene.2018.02.190. [DOI] [Google Scholar]
- Chandra M.; Xu Q. A High-Performance Hydrogen Generation System: Transition Metal-Catalyzed Dissociation and Hydrolysis of Ammonia–Borane. J. Power Sources 2006, 156, 190–194. 10.1016/j.jpowsour.2005.05.043. [DOI] [Google Scholar]
- Cao N.; Su J.; Luo W.; Cheng G. Graphene Supported Ru@Co Core–Shell Nanoparticles as Efficient Catalysts for Hydrogen Generation from Hydrolysis of Ammonia Borane and Methylamine Borane. Catal. Commun. 2014, 43, 47–51. 10.1016/j.catcom.2013.09.003. [DOI] [Google Scholar]
- Guo L. T.; Cai Y. Y.; Ge J. M.; Zhang Y. N.; Gong L. H.; Li X. H.; Wang K. X.; Ren Q. Z.; Su J.; Chen J. S. Multifunctional Au-Co@CN Nanocatalyst for Highly Efficient Hydrolysis of Ammonia Borane. ACS Catal. 2015, 5, 388–392. 10.1021/cs501692n. [DOI] [Google Scholar]
- Wang Q.; Fu F.; Yang S.; Martinez Moro M.; Ramirez M. d. l. A.; Moya S.; Salmon L.; Ruiz J.; Astruc D. Dramatic Synergy in CoPt Nanocatalysts Stabilized by “Click” Dendrimers for Evolution of Hydrogen from Hydrolysis of Ammonia Borane. ACS Catal. 2019, 9, 1110–1119. 10.1021/acscatal.8b04498. [DOI] [Google Scholar]
- Zhou Q.; Qi L.; Yang H.; Xu C. Hierarchical Nanoporous Platinum–Copper Alloy Nanoflowers as Highly Active Catalysts for the Hydrolytic Dehydrogenation of Ammonia Borane. J. Colloid Interface Sci. 2018, 513, 258–265. 10.1016/j.jcis.2017.11.040. [DOI] [PubMed] [Google Scholar]
- Yang X.; Li Q.; Li L.; Lin J.; Yang X.; Yu C.; Liu Z.; Fang Y.; Huang Y.; Tang C. CuCo Binary Metal Nanoparticles Supported on Boron Nitride Nanofibers as Highly Efficient Catalysts for Hydrogen Generation from Hydrolysis of Ammonia Borane. J. Power Sources 2019, 431, 135–143. 10.1016/j.jpowsour.2019.05.038. [DOI] [Google Scholar]
- Ge Y.; Shah Z. H.; Lin X. J.; Lu R.; Liao Z.; Zhang S. Highly Efficient Pt Decorated CoCu Bimetallic Nanoparticles Protected in Silica for Hydrogen Production from Ammonia-Borane. ACS Sustainable Chem. Eng. 2017, 5, 1675–1684. 10.1021/acssuschemeng.6b02430. [DOI] [Google Scholar]
- Yao Q.; Yang K.; Hong X.; Chen X.; Lu Z. H. Base-Promoted Hydrolytic Dehydrogenation of Ammonia Borane Catalyzed by Noble-Metal-Free Nanoparticles. Catal. Sci. Technol. 2018, 8, 870–877. 10.1039/C7CY02365K. [DOI] [Google Scholar]
- Liang Z.; Xiao X.; Yu X.; Huang X.; Jiang Y.; Fan X.; Chen L. Non-Noble Trimetallic Cu-Ni-Co Nanoparticles Supported on Metal-Organic Frameworks as Highly Efficient Catalysts for Hydrolysis of Ammonia Borane. J. Alloys Compd. 2018, 741, 501–508. 10.1016/j.jallcom.2017.12.151. [DOI] [Google Scholar]
- Zhdanov V.; Note P. The Effect of Viscosity on the Rate of Diffusion-Limited Association of Nanoparticles. J. Chem. Phys. 2015, 143, 166102. 10.1063/1.4934948. [DOI] [PubMed] [Google Scholar]
- Sen B.; Kuzu S.; Demir E.; Onal Okyay T.; Sen F. Hydrogen Liberation from the Dehydrocoupling of Dimethylamine–Borane at Room Temperature by Using Novel and Highly Monodispersed RuPtNi Nanocatalysts Decorated with Graphene Oxide. Int. J. Hydrogen Energy 2017, 42, 23299–23306. 10.1016/j.ijhydene.2017.04.213. [DOI] [Google Scholar]
- Sen B.; Kuzu S.; Demir E.; Akocak S.; Sen F. Highly Monodisperse RuCo Nanoparticles Decorated on Functionalized Multiwalled Carbon Nanotube with the Highest Observed Catalytic Activity in the Dehydrogenation of Dimethylamine–Borane. Int. J. Hydrogen Energy 2017, 42, 23292–23298. 10.1016/j.ijhydene.2017.06.032. [DOI] [Google Scholar]
- Şen B.; Aygün A.; Okyay T. O.; Şavk A.; Kartop R.; Şen F. Monodisperse Palladium Nanoparticles Assembled on Graphene Oxide with the High Catalytic Activity and Reusability in the Dehydrogenation of Dimethylamine-Borane. Int. J. Hydrogen Energy 2018, 43, 20176–20182. 10.1016/j.ijhydene.2018.03.175. [DOI] [Google Scholar]
- Sen B.; Kuzu S.; Demir E.; Akocak S.; Sen F. Monodisperse Palladium–Nickel Alloy Nanoparticles Assembled on Graphene Oxide with the High Catalytic Activity and Reusability in the Dehydrogenation of Dimethylamine–Borane. Int. J. Hydrogen Energy 2017, 42, 23276–23283. 10.1016/j.ijhydene.2017.05.113. [DOI] [Google Scholar]
- Hutchings G. J. Selective Oxidation Using Supported Gold Bimetallic and Trimetallic Nanoparticles. Catal. Today 2014, 238, 69–73. 10.1016/j.cattod.2014.01.033. [DOI] [Google Scholar]
- Haider P.; Kimmerle B.; Krumeich F.; Kleist W.; Grunwaldt J. D.; Baiker A. Gold-Catalyzed Aerobic Oxidation of Benzyl Alcohol: Effect of Gold Particle Size on Activity and Selectivity in Different Solvents. Catal. Lett. 2008, 125, 169–176. 10.1007/s10562-008-9567-5. [DOI] [Google Scholar]
- Abad A.; Concepción P.; Corma A.; García H. A Collaborative Effect between Gold and a Support Induces the Selective Oxidation of Alcohols. Angew. Chem., Int. Ed. 2005, 44, 4066–4069. 10.1002/anie.200500382. [DOI] [PubMed] [Google Scholar]
- Sankar M.; Dimitratos N.; Knight D. W.; Carley A. F.; Tiruvalam R.; Kiely C. J.; Thomas D.; Hutchings G. J. Oxidation of Glycerol to Glycolate by Using Supported Gold and Palladium Nanoparticles. ChemSusChem 2009, 2, 1145–1151. 10.1002/cssc.200900133. [DOI] [PubMed] [Google Scholar]
- Yang L.; Shan S.; Loukrakpam R.; Petkov V.; Ren Y.; Wanjala B. N.; Engelhard M. H.; Luo J.; Yin J.; Chen Y.; Zhong C. J. Role of Support-Nanoalloy Interactions in the Atomic-Scale Structural and Chemical Ordering for Tuning Catalytic Sites. J. Am. Chem. Soc. 2012, 134, 15048–15060. 10.1021/ja3060035. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Okumura M.; Toshima N. Stable Dispersions of PVP-Protected Au/Pt/Ag Trimetallic Nanoparticles as Highly Active Colloidal Catalysts for Aerobic Glucose Oxidation. J. Phys. Chem. C 2011, 115, 14883–14891. 10.1021/jp203457f. [DOI] [Google Scholar]
- Zhao S.; Zhao B.; Tian X.; Ren Y.; Yao K.; Wang J.; Liu J.; Ren Y. Density Functional Study of Trimetallic AuxPdyPtz (x + y + z = 7) Clusters and Their Interactions with the O2 Molecule. J. Phys. Chem. A 2017, 121, 5226–5236. 10.1021/acs.jpca.7b04411. [DOI] [PubMed] [Google Scholar]
- Behr A.; Eilting J.; Irawadi K.; Leschinski J.; Lindner F. Improved Utilisation of Renewable Resources: New Important Derivatives of Glycerol. Green Chem. 2008, 10, 13–30. 10.1039/B710561D. [DOI] [Google Scholar]
- Katryniok B.; Kimura H.; Skrzyńska E.; Girardon J. S.; Fongarland P.; Capron M.; Ducoulombier R.; Mimura N.; Paul S.; Dumeignil F. Selective Catalytic Oxidation of Glycerol: Perspectives for High Value Chemicals. Green Chem. 2011, 13, 1960–1979. 10.1039/c1gc15320j. [DOI] [Google Scholar]
- Yang L.; Li X.; Chen P.; Hou Z. Selective Oxidation of Glycerol in a Base-Free Aqueous Solution: A Short Review. Chin. J. Catal. 2019, 40, 1020–1034. 10.1016/S1872-2067(19)63301-2. [DOI] [Google Scholar]
- Kimura H.; Tsuto K.; Wakisaka T.; Kazumi Y.; Inaya Y. Selective Oxidation of Glycerol on a Platinum-Bismuth Catalyst. Appl. Catal., A 1993, 96, 217–228. 10.1016/0926-860X(90)80011-3. [DOI] [Google Scholar]
- Villa A.; Campisi S.; Chan-Thaw C. E.; Motta D.; Wang D.; Prati L. Bismuth Modified Au-Pt Bimetallic Catalysts for Dihydroxyacetone Production. Catal. Today 2015, 249, 103–108. 10.1016/j.cattod.2014.12.012. [DOI] [Google Scholar]
- Villa A.; Wang D.; Veith G. M.; Prati L. Bismuth as a Modifier of Au-Pd Catalyst: Enhancing Selectivity in Alcohol Oxidation by Suppressing Parallel Reaction. J. Catal. 2012, 292, 73–80. 10.1016/j.jcat.2012.04.021. [DOI] [Google Scholar]
- Villa A.; Campisi S.; Mohammed K. M. H.; Dimitratos N.; Vindigni F.; Manzoli M.; Jones W.; Bowker M.; Hutchings G. J.; Prati L. Tailoring the Selectivity of Glycerol Oxidation by Tuning the Acid-Base Properties of Au Catalysts. Catal. Sci. Technol. 2015, 5, 1126–1132. 10.1039/C4CY01246A. [DOI] [Google Scholar]
- Nie R.; Liang D.; Shen L.; Gao J.; Chen P.; Hou Z. Selective Oxidation of Glycerol with Oxygen in Base-Free Solution over MWCNTs Supported PtSb Alloy Nanoparticles. Appl. Catal., B 2012, 127, 212–220. 10.1016/j.apcatb.2012.08.026. [DOI] [Google Scholar]
- Alloyeau D.; Prévot G.; Le Bouar Y.; Oikawa T.; Langlois C.; Loiseau A.; Ricolleau C. Ostwald Ripening in Nanoalloys: When Thermodynamics Drives a Size-Dependent Particle Composition. Phys. Rev. Lett. 2010, 105, 255901. 10.1103/PhysRevLett.105.255901. [DOI] [PubMed] [Google Scholar]
- Andreeva D.; Idakiev V.; Tabakova T.; Ilieva L.; Falaras P.; Bourlinos A.; Travlos A. Low-Temperature Water-Gas Shift Reaction over Au/CeO2 Catalysts. Catal. Today 2002, 72, 51–57. 10.1016/S0920-5861(01)00477-1. [DOI] [Google Scholar]
- Kummer J. T. Use of Noble Metals in Automobile Exhaust Catalysts. J. Phys. Chem. 1986, 90, 4747–4752. 10.1021/j100411a008. [DOI] [Google Scholar]
- Wang L.; Yang L.; Zhang Y.; Ding W.; Chen S.; Fang W.; Yang Y. Promoting Effect of an Aluminum Emulsion on Catalytic Performance of Cu-Based Catalysts for Methanol Synthesis from Syngas. Fuel Process. Technol. 2010, 91, 723–728. 10.1016/j.fuproc.2010.02.003. [DOI] [Google Scholar]
- Haruta M.; Kobayashi T.; Sano H.; Yamada N. Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature Far Below 0 °C. Chem. Lett. 1987, 16, 405–408. 10.1246/cl.1987.405. [DOI] [Google Scholar]
- Boccuzzi F.; Chiorino A.; Tsubota S.; Haruta M. FTIR Study of Carbon Monoxide Oxidation and Scrambling at Room Temperature over Gold Supported on ZnO and TiO2. 2. J. Phys. Chem. 1996, 100, 3625–3631. 10.1021/jp952259n. [DOI] [Google Scholar]
- Dey S.; Dhal G. C. Property and Structure of Various Platinum Catalysts for Low-Temperature Carbon Monoxide Oxidations. Mater. Today Chem. 2020, 16, 100228. 10.1016/j.mtchem.2019.100228. [DOI] [Google Scholar]
- Biabani-Ravandi A.; Rezaei M.; Fattah Z. Study of Fe-Co Mixed Metal Oxide Nanoparticles in the Catalytic Low-Temperature CO Oxidation. Process Saf. Environ. Prot. 2013, 91, 489–494. 10.1016/j.psep.2012.10.015. [DOI] [Google Scholar]
- Reddy B. M.; Rao K. N.; Bharali P. Copper Promoted Cobalt and Nickel Catalysts Supported on Ceria-Alumina Mixed Oxide: Structural Characterization and CO Oxidation Activity. Ind. Eng. Chem. Res. 2009, 48, 8478–8486. 10.1021/ie900755b. [DOI] [Google Scholar]
- Jing P.; Gong X.; Liu B.; Zhang J. Recent Advances in Synergistic Effect Promoted Catalysts for Preferential Oxidation of Carbon Monoxide. Catal. Sci. Technol. 2020, 10, 919–934. 10.1039/C9CY02073J. [DOI] [Google Scholar]
- Shen J.; Zhu Y.; Hu Y.; Li C. Atomically Dispersed Gold-Supported Catalysts: Preparation and Potential for Low-Temperature CO Oxidation. Mater. Today Nano 2018, 4, 54–69. 10.1016/j.mtnano.2018.12.004. [DOI] [Google Scholar]
- Soliman N. K. Factors Affecting CO Oxidation Reaction over Nanosized Materials: A Review. J. Mater. Res. Technol. 2019, 8, 2395–2407. 10.1016/j.jmrt.2018.12.012. [DOI] [Google Scholar]
- Tripathi A.; Hareesh C.; Sinthika S.; Andersson G.; Thapa R. CO Oxidation on Pt Based Binary and Ternary Alloy Nanocatalysts: Reaction Pathways and Electronic Descriptor. Appl. Surf. Sci. 2020, 528, 146964. 10.1016/j.apsusc.2020.146964. [DOI] [Google Scholar]
- Holz F.; Richter P. M.; Egging R. A Global Perspective on the Future of Natural Gas: Resources, Trade, and Climate Constraints. Rev. Environ. Econ. Policy 2015, 9, 85–106. 10.1093/reep/reu016. [DOI] [Google Scholar]
- Khirsariya P.; Mewada R. K. Single Step Oxidation of Methane to Methanol–Towards Better Understanding. Procedia Eng. 2013, 51, 409–415. 10.1016/j.proeng.2013.01.057. [DOI] [Google Scholar]
- Rahman A. K. M. L.; Kumashiro M.; Ishihara T. Direct Synthesis of Formic Acid by Partial Oxidation of Methane on H-ZSM-5 Solid Acid Catalyst. Catal. Commun. 2011, 12, 1198–1200. 10.1016/j.catcom.2011.04.001. [DOI] [Google Scholar]
- Sorokin A. B.; Kudrik E. V.; Bouchu D. Bio-Inspired Oxidation of Methane in Water Catalyzed by N-Bridged Diiron Phthalocyanine Complex. Chem. Commun. 2008, 22, 2562. 10.1039/b804405h. [DOI] [PubMed] [Google Scholar]
- Ab Rahim M. H.; Forde M. M.; Hammond C.; Jenkins R. L.; Dimitratos N.; Lopez-Sanchez J. A.; Carley A. F.; Taylor S. H.; Willock D. J.; Hutchings G. J. Systematic Study of the Oxidation of Methane Using Supported Gold Palladium Nanoparticles Under Mild Aqueous Conditions. Top. Catal. 2013, 56, 1843–1857. 10.1007/s11244-013-0121-3. [DOI] [Google Scholar]
- Mizuno N.; Seki Y.; Nishiyama Y.; Kiyoto I.; Misono M. Aqueous Phase Oxidation of Methane with Hydrogen Peroxide Catalyzed by Di-Iron-Substituted Silicotungstate. J. Catal. 1999, 184, 550–552. 10.1006/jcat.1999.2455. [DOI] [Google Scholar]
- Williams C.; Carter J. H.; Dummer N. F.; Chow Y. K.; Morgan D. J.; Yacob S.; Serna P.; Willock D. J.; Meyer R. J.; Taylor S. H.; Hutchings G. J. Selective Oxidation of Methane to Methanol Using Supported AuPd Catalysts Prepared by Stabilizer-Free Sol-Immobilization. ACS Catal. 2018, 8, 2567–2576. 10.1021/acscatal.7b04417. [DOI] [Google Scholar]
- Ab Rahim M. H.; Forde M. M.; Jenkins R. L.; Hammond C.; He Q.; Dimitratos N.; Lopez-Sanchez J. A.; Carley A. F.; Taylor S. H.; Willock D. J.; Murphy D. M.; Kiely C. J.; Hutchings G. J. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold-Palladium Alloy Nanoparticles. Angew. Chem., Int. Ed. 2013, 52, 1280–1284. 10.1002/anie.201207717. [DOI] [PubMed] [Google Scholar]
- Ab Rahim M. H.; Armstrong R. D.; Hammond C.; Dimitratos N.; Freakley S. J.; Forde M. M.; Morgan D. J.; Lalev G.; Jenkins R. L.; Lopez-Sanchez J. A.; Taylor S. H.; Hutchings G. J. Low Temperature Selective Oxidation of Methane to Methanol Using Titania Supported Gold Palladium Copper Catalysts. Catal. Sci. Technol. 2016, 6, 3410–3418. 10.1039/C5CY01586C. [DOI] [Google Scholar]
- Choi K. H.; Lee D. H.; Kim H. S.; Yoon Y. C.; Park C. S.; Kim Y. H. Reaction Characteristics of Precious-Metal-Free Ternary Mn-Cu-M (M = Ce, Co, Cr, and Fe) Oxide Catalysts for Low-Temperature CO Oxidation. Ind. Eng. Chem. Res. 2016, 55, 4443–4450. 10.1021/acs.iecr.5b04985. [DOI] [Google Scholar]
- Liu Z.; Wu Z.; Peng X.; Binder A.; Chai S.; Dai S. Origin of Active Oxygen in a Ternary CuOx/Co3O4–CeO2 Catalyst for CO Oxidation. J. Phys. Chem. C 2014, 118, 27870–27877. 10.1021/jp508487x. [DOI] [Google Scholar]
- Binder A. J.; Toops T. J.; Unocic R. R.; Parks J. E.; Dai S. Low-Temperature CO Oxidation over a Ternary Oxide Catalyst with High Resistance to Hydrocarbon Inhibition. Angew. Chem. 2015, 127, 13461–13465. 10.1002/ange.201506093. [DOI] [PubMed] [Google Scholar]
- Bhunia K.; Khilari S.; Pradhan D. Trimetallic PtAuNi Alloy Nanoparticles as an Efficient Electrocatalyst for the Methanol Electrooxidation Reaction. Dalton Trans. 2017, 46, 15558–15566. 10.1039/C7DT02608K. [DOI] [PubMed] [Google Scholar]
- Tang M.; Luo S.; Wang K.; Du H.; Sriphathoorat R.; Shen P. Simultaneous Formation of Trimetallic Pt-Ni-Cu Excavated Rhombic Dodecahedrons with Enhanced Catalytic Performance for the Methanol Oxidation Reaction. Nano Res. 2018, 11, 4786–4795. 10.1007/s12274-018-2063-3. [DOI] [Google Scholar]
- Thongthai K.; Pakawanit P.; Chanlek N.; Kim J. H.; Ananta S.; Srisombat L. Ag/Au/Pt Trimetallic Nanoparticles with Defects: Preparation, Characterization, and Electrocatalytic Activity in Methanol Oxidation. Nanotechnology 2017, 28, 375602. 10.1088/1361-6528/aa846b. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhang F.; Chan K. Y. Preparation of Pt-Ru-Co Trimetallic Nanoparticles and Their Electrocatalytic Properties. Catal. Commun. 2004, 5, 749–753. 10.1016/j.catcom.2004.09.010. [DOI] [Google Scholar]
- Lan J.; Li C.; Liu T.; Yuan Q. One-Step Synthesis of Porous PtNiCu Trimetallic Nanoalloy with Enhanced Electrocatalytic Performance toward Methanol Oxidation. J. Saudi Chem. Soc. 2019, 23, 43–51. 10.1016/j.jscs.2018.04.002. [DOI] [Google Scholar]
- Wang W.; Wang R.; Wang H.; Ji S.; Key J.; Li X.; Lei Z. An Advantageous Method for Methanol Oxidation: Design and Fabrication of a Nanoporous PtRuNi Trimetallic Electrocatalyst. J. Power Sources 2011, 196, 9346–9351. 10.1016/j.jpowsour.2011.06.101. [DOI] [Google Scholar]
- Yousaf A. B.; Afsar N.-U.; Zeb A.; Imran M.; Yuan C.-Z. Cubic PdPtRu Trimetallic Nanocomposite: Electrocatalytic Application as Anode Material for Direct Methanol Fuel Cell. Int. J. Electrochem. Sci. 2015, 10, 10292–10306. [Google Scholar]
- Poh C. K.; Tian Z.; Gao J.; Liu Z.; Lin J.; Feng Y. P.; Su F. Nanostructured Trimetallic Pt/FeRuC, Pt/NiRuC, and Pt/CoRuC Catalysts for Methanol Electrooxidation. J. Mater. Chem. 2012, 22, 13643. 10.1039/c2jm31956j. [DOI] [Google Scholar]
- Nie M.; Du S.; Li Q.; Hummel M.; Gu Z.; Lu S. Tungsten Carbide as Supports for Trimetallic AuPdPt Electrocatalysts for Methanol Oxidation. J. Electrochem. Soc. 2020, 167, 044510 10.1149/1945-7111/ab754d. [DOI] [Google Scholar]
- Liu H.; Li J.; Wang L.; Tang Y.; Xia B. Y.; Chen Y. Trimetallic PtRhNi Alloy Nanoassemblies as Highly Active Electrocatalyst for Ethanol Electrooxidation. Nano Res. 2017, 10, 3324–3332. 10.1007/s12274-017-1545-z. [DOI] [Google Scholar]
- Bhunia K.; Khilari S.; Pradhan D. Monodispersed PtPdNi Trimetallic Nanoparticles-Integrated Reduced Graphene Oxide Hybrid Platform for Direct Alcohol Fuel Cell. ACS Sustainable Chem. Eng. 2018, 6, 7769–7778. 10.1021/acssuschemeng.8b00721. [DOI] [Google Scholar]
- Sudachom N.; Warakulwit C.; Prapainainar C.; Witoon T.; Prapainainar P. One Step NaBH4 Reduction of Pt-Ru-Ni Catalysts on Different Types of Carbon Supports for Direct Ethanol Fuel Cells: Synthesis and Characterization. Ranliao Huaxue Xuebao/J. Fuel Chem. Technol. 2017, 45, 596–607. 10.1016/S1872-5813(17)30031-2. [DOI] [Google Scholar]
- Mao J.; Chen W.; He D.; Wan J.; Pei J.; Dong J.; Wang Y.; An P.; Jin Z.; Xing W.; Tang H.; Zhuang Z.; Liang X.; Huang Y.; Zhou G.; Wang L.; Wang D.; Li Y. Design of Ultrathin Pt-Mo-Ni Nanowire Catalysts for Ethanol Electrooxidation. Sci. Adv. 2017, 3, e1603068 10.1126/sciadv.1603068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W. J.; Li W. Z.; Song S. Q.; Zhou Z. H.; Jiang L. H.; Sun G. Q.; Xin Q.; Poulianitis K.; Kontou S.; Tsiakaras P. Bi- and Tri-Metallic Pt-Based Anode Catalysts for Direct Ethanol Fuel Cells. J. Power Sources 2004, 131, 217–223. 10.1016/j.jpowsour.2003.12.040. [DOI] [Google Scholar]
- Figueiredo M. C.; Sorsa O.; Arán-Ais R. M.; Doan N.; Feliu J. M.; Kallio T. Trimetallic Catalyst Based on PtRu Modified by Irreversible Adsorption of Sb for Direct Ethanol Fuel Cells. J. Catal. 2015, 329, 69–77. 10.1016/j.jcat.2015.04.032. [DOI] [Google Scholar]
- Ulas B.; Caglar A.; Kivrak A.; Aktas N.; Kivrak H. Tailoring the Metallic Composition of Pd, Pt, and Au Containing Novel Trimetallic Catalysts to Achieve Enhanced Formic Acid Electrooxidation Activity. Ionics (Kiel) 2020, 26, 3109–3121. 10.1007/s11581-020-03444-5. [DOI] [Google Scholar]
- Jiang Q. L.; Peng Z. D.; Xie X. F.; Du K.; Hu G. R.; Liu Y. X. Preparation of High Active Pt/C Cathode Electrocatalyst for Direct Methanol Fuel Cell by Citrate-Stabilized Method. Trans. Nonferrous Met. Soc. China (Engl. Ed.) 2011, 21, 127–132. 10.1016/S1003-6326(11)60688-2. [DOI] [Google Scholar]
- Housmans T. H. M.; Wonders A. H.; Koper M. T. M. Structure Sensitivity of Methanol Electrooxidation Pathways on Platinum: An on-Line Electrochemical Mass Spectrometry Study. J. Phys. Chem. B 2006, 110, 10021–10031. 10.1021/jp055949s. [DOI] [PubMed] [Google Scholar]
- Papadimitriou S.; Armyanov S.; Valova E.; Hubin A.; Steenhaut O.; Pavlidou E.; Kokkinidis G.; Sotiropoulos S. Methanol Oxidation at Pt–Cu, Pt–Ni, and Pt–Co Electrode Coatings Prepared by a Galvanic Replacement Process. J. Phys. Chem. C 2010, 114, 5217–5223. 10.1021/jp911568g. [DOI] [Google Scholar]
- Zhao Y.; Liu J.; Liu C.; Wang F.; Song Y. Amorphous CuPt Alloy Nanotubes Induced by Na2S2O3 as Efficient Catalysts for the Methanol Oxidation Reaction. ACS Catal. 2016, 6, 4127–4134. 10.1021/acscatal.6b00540. [DOI] [Google Scholar]
- Song S.; Zhou W.; Liang Z.; Cai R.; Sun G.; Xin Q.; Stergiopoulos V.; Tsiakaras P. The Effect of Methanol and Ethanol Cross-over on the Performance of PtRu/C-Based Anode DAFCs. Appl. Catal., B 2005, 55, 65–72. 10.1016/j.apcatb.2004.05.017. [DOI] [Google Scholar]
- Wachsmann U.; Tolmasquim M. T. Wind Power in Brazil—Transition Using German Experience. Renew. Energy 2003, 28, 1029–1038. 10.1016/S0960-1481(02)00212-4. [DOI] [Google Scholar]
- Zhou W. J.; Song S. Q.; Li W. Z.; Zhou Z. H.; Sun G. Q.; Xin Q.; Douvartzides S.; Tsiakaras P. Direct Ethanol Fuel Cells Based on PtSn Anodes: The Effect of Sn Content on the Fuel Cell Performance. J. Power Sources 2005, 140, 50–58. 10.1016/j.jpowsour.2004.08.003. [DOI] [Google Scholar]
- Monyoncho E. A.; Woo T. K.; Baranova E. A. Ethanol Electrooxidation Reaction in Alkaline Media for Direct Ethanol Fuel Cells. Electrochemistry (Cambridge, U. K.) 2018, 15, 1–57. 10.1039/9781788013895-00001. [DOI] [Google Scholar]
- Obradović M. D.; Stančić Z. M.; Lačnjevac U. Č.; Radmilović V. V.; Gavrilović-Wohlmuther A.; Radmilović V. R.; Gojković S. L. Electrochemical Oxidation of Ethanol on Palladium-Nickel Nanocatalyst in Alkaline Media. Appl. Catal., B 2016, 189, 110–118. 10.1016/j.apcatb.2016.02.039. [DOI] [Google Scholar]
- Dai S.; Huang T. H.; Chien P. C.; Lin C. A.; Liu C. W.; Lee S. W.; Wang J. H.; Wang K. W.; Pan X. Optimization of Pt–Oxygen-Containing Species Anodes for Ethanol Oxidation Reaction: High Performance of Pt-AuSnOx Electrocatalyst. J. Phys. Chem. Lett. 2020, 11, 2846–2853. 10.1021/acs.jpclett.0c00213. [DOI] [PubMed] [Google Scholar]
- Liang Z. X.; Zhao T. S.; Xu J. B.; Zhu L. D. Mechanism Study of the Ethanol Oxidation Reaction on Palladium in Alkaline Media. Electrochim. Acta 2009, 54, 2203–2208. 10.1016/j.electacta.2008.10.034. [DOI] [Google Scholar]
- Dai L. X.; Wang X. Y.; Yang S. S.; Zhang T.; Ren P. J.; Ye J. Y.; Nan B.; Wen X. D.; Zhou Z. Y.; Si R.; Yan C. H.; Zhang Y. W. Intrinsic Composition and Electronic Effects of Multicomponent Platinum Nanocatalysts with High Activity and Selectivity for Ethanol Oxidation Reaction. J. Mater. Chem. A 2018, 6, 11270–11280. 10.1039/C8TA03320J. [DOI] [Google Scholar]
- Dai S.; Huang T. H.; Yan X.; Yang C. Y.; Chen T. Y.; Wang J. H.; Pan X.; Wang K. W. Promotion of Ternary Pt-Sn-Ag Catalysts toward Ethanol Oxidation Reaction: Revealing Electronic and Structural Effects of Additive Metals. ACS Energy Lett. 2018, 3, 2550–2557. 10.1021/acsenergylett.8b01632. [DOI] [Google Scholar]
- Sui S.; Wang X.; Zhou X.; Su Y.; Riffat S.; Liu C. A Comprehensive Review of Pt Electrocatalysts for the Oxygen Reduction Reaction: Nanostructure, Activity, Mechanism and Carbon Support in PEM Fuel Cells. J. Mater. Chem. A 2017, 5, 1808–1825. 10.1039/C6TA08580F. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Marković N. M.; Schmidt T. J.; Stamenković V.; Ross P. N. Oxygen Reduction Reaction on Pt and Pt Bimetallic Surfaces: A Selective Review. Fuel Cells 2001, 1, 105–116. . [DOI] [Google Scholar]
- Tang W.; Jayaraman S.; Jaramillo T. F.; Stucky G. D.; McFarland E. W. Electrocatalytic Activity of Gold-Platinum Clusters for Low Temperature Fuel Cell Applications. J. Phys. Chem. C 2009, 113, 5014–5024. 10.1021/jp8089209. [DOI] [Google Scholar]
- Zhao J.; Jarvis K.; Ferreira P.; Manthiram A. Performance and Stability of Pd-Pt-Ni Nanoalloy Electrocatalysts in Proton Exchange Membrane Fuel Cells. J. Power Sources 2011, 196, 4515–4523. 10.1016/j.jpowsour.2011.01.026. [DOI] [Google Scholar]
- Mazumder V.; Chi M.; More K. L.; Sun S. Core/Shell Pd/FePt Nanoparticles as an Active and Durable Catalyst for the Oxygen Reduction Reaction. J. Am. Chem. Soc. 2010, 132, 7848–7849. 10.1021/ja1024436. [DOI] [PubMed] [Google Scholar]
- Wang C.; van der Vliet D.; More K. L.; Zaluzec N. J.; Peng S.; Sun S.; Daimon H.; Wang G.; Greeley J.; Pearson J.; Paulikas A. P.; Karapetrov G.; Strmcnik D.; Markovic N. M.; Stamenkovic V. R. Multimetallic Au/FePt3 Nanoparticles as Highly Durable Electrocatalyst. Nano Lett. 2011, 11, 919–926. 10.1021/nl102369k. [DOI] [PubMed] [Google Scholar]
- Ferreira P. J.; la O’ G. J.; Shao-Horn Y.; Morgan D.; Makharia R.; Kocha S.; Gasteiger H. A. Instability of Pt/C Electrocatalysts in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2005, 152, A2256. 10.1149/1.2050347. [DOI] [Google Scholar]
- Venkateswara Rao C.; Viswanathan B. Microemulsion Synthesis and Electrocatalytic Properties of Carbon-Supported Pd-Co-Au Alloy Nanoparticles. J. Colloid Interface Sci. 2012, 367, 337–341. 10.1016/j.jcis.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Jennings P. C.; Lysgaard S.; Hansen H. A.; Vegge T. Decoupling Strain and Ligand Effects in Ternary Nanoparticles for Improved ORR Electrocatalysis. Phys. Chem. Chem. Phys. 2016, 18, 24737–24745. 10.1039/C6CP04194A. [DOI] [PubMed] [Google Scholar]
- Noh S. H.; Han B.; Ohsaka T. First-Principles Computational Study of Highly Stable and Active Ternary PtCuNi Nanocatalyst for Oxygen Reduction Reaction. Nano Res. 2015, 8, 3394–3403. 10.1007/s12274-015-0839-2. [DOI] [Google Scholar]
- Noh S. H.; Han B.; Ohsaka T. First-Principles Computational Study of Highly Stable and Active Ternary PtCuNi Nanocatalyst for Oxygen Reduction Reaction. Nano Res. 2015, 8, 3394–3403. 10.1007/s12274-015-0839-2. [DOI] [Google Scholar]
- Flores-Rojas E.; Cruz-Martínez H.; Rojas-Chávez H.; Tellez-Cruz M. M.; Reyes-Rodríguez J. L.; Cabañas-Moreno J. G.; Calaminici P.; Solorza-Feria O. A Combined DFT and Experimental Investigation of Pt-Wrapped CoNi Nanoparticles for the Oxygen Reduction Reaction. Electrocatalysis 2018, 9, 662–672. 10.1007/s12678-018-0474-2. [DOI] [Google Scholar]
- Cruz-Martínez H.; Tellez-Cruz M. M.; Solorza-Feria O.; Calaminici P.; Medina D. I. Catalytic Activity Trends from Pure Pd Nanoclusters to M@PdPt (M = Co, Ni, and Cu) Core-Shell Nanoclusters for the Oxygen Reduction Reaction: A First-Principles Analysis. Int. J. Hydrogen Energy 2020, 45, 13738–13745. 10.1016/j.ijhydene.2019.08.245. [DOI] [Google Scholar]
- Kim J.-S.; Lee B.-J. Durability Screening of Pt Ternary Alloy (111) Surfaces for Oxygen Reduction Reaction Using Density Functional Theory. Surf. Interfaces 2020, 18, 100440. 10.1016/j.surfin.2020.100440. [DOI] [Google Scholar]
- The Department of Energy Hydrogen and Fuel Cells Program Plan; U.S. Department of Energy, 2011; http://hydrogen.energy.gov/pdfs/program_plan2011.pdf. (accessed 2021-03-11). [Google Scholar]
- Li S.; Yang Y.; Huang S. O2 Adsorption and Dissociation on the Pd13–nNin@Pt42 (n = 0, 1, 12, and 13) Tri-Metallic Nanoparticles: A DFT Study. Appl. Surf. Sci. 2017, 410, 291–298. 10.1016/j.apsusc.2017.03.083. [DOI] [Google Scholar]
- Kong F.; Liu S.; Li J.; Du L.; Banis M. N.; Zhang L.; Chen G.; Doyle-Davis K.; Liang J.; Wang S.; Zhao F.; Li R.; Du C.; Yin G.; Zhao Z.; Sun X. Trimetallic Pt–Pd–Ni Octahedral Nanocages with Subnanometer Thick-Wall towards High Oxygen Reduction Reaction. Nano Energy 2019, 64, 103890. 10.1016/j.nanoen.2019.103890. [DOI] [Google Scholar]
- Mu X.; Wang J.; Li Y.; Xu F.; Long W.; Ouyang L.; Liu H.; Jing Y.; Wang J.; Dai H.; Liu Q.; Sun Y.; Liu C.; Zhang X.-D. Redox Trimetallic Nanozyme with Neutral Environment Preference for Brain Injury. ACS Nano 2019, 13, 1870–1884. 10.1021/acsnano.8b08045. [DOI] [PubMed] [Google Scholar]
- Wang J. Y.; Mu X.; Li Y.; Xu F.; Long W.; Yang J.; Bian P.; Chen J.; Ouyang L.; Liu H.; Jing Y.; Wang J.; Liu L.; Dai H.; Sun Y.; Liu C.; Zhang X. D. Hollow PtPdRh Nanocubes with Enhanced Catalytic Activities for In Vivo Clearance of Radiation-Induced ROS via Surface-Mediated Bond Breaking. Small 2018, 14, 1703736. 10.1002/smll.201703736. [DOI] [PubMed] [Google Scholar]
- Han X. B.; Tang X. Y.; Lin Y.; Gracia-Espino E.; Liu S. G.; Liang H. W.; Hu G. Z.; Zhao X. J.; Liao H. G.; Tan Y. Z.; Wagberg T.; Xie S. Y.; Zheng L. S. Ultrasmall Abundant Metal-Based Clusters as Oxygen-Evolving Catalysts. J. Am. Chem. Soc. 2019, 141, 232–239. 10.1021/jacs.8b09076. [DOI] [PubMed] [Google Scholar]
- Doukeh R.; Bombos M.; Trifoi A.; Pasare M.; Banu I.; Bolocan I. Dimethyldisulphide Hydrodesulphurization on NiCoMo/Al2O3 Catalyst. Rev. Chim. 2017, 68, 1496–1500. 10.37358/RC.17.7.5703. [DOI] [Google Scholar]
- Mozhaev A. V.; Nikulshin P. A.; Pimerzin A. A.; Maslakov K. I.; Pimerzin A. A. Investigation of Co-Promotion Effect in NiCoMoS/Al2O3 Catalysts Based on Co2Mo10-Heteropolyacid and Nickel Citrate. Catal. Today 2016, 271, 80–90. 10.1016/j.cattod.2015.11.002. [DOI] [Google Scholar]
- Kordouli E.; Sygellou L.; Kordulis C.; Bourikas K.; Lycourghiotis A. Probing the Synergistic Ratio of the NiMo/γ-Al2O3 Reduced Catalysts for the Transformation of Natural Triglycerides into Green Diesel. Appl. Catal., B 2017, 209, 12–22. 10.1016/j.apcatb.2017.02.045. [DOI] [Google Scholar]
- Liu J.; Yoda S.; He J.; Deng L.; Fan K.; Rong L. A Novel Method of Preparing Ni–Mo–La/γ-Alumina Catalysts for Hydrocracking. Chem. Lett. 2014, 43, 310–312. 10.1246/cl.130962. [DOI] [Google Scholar]
- Nair M. M.; Kaliaguine S.; Kleitz F. Nanocast LaNiO3 Perovskites as Precursors for the Preparation of Coke-Resistant Dry Reforming Catalysts. ACS Catal. 2014, 4, 3837–3846. 10.1021/cs500918c. [DOI] [Google Scholar]
- Peña M. A.; Gómez J. P.; Fierro J. L. G. New Catalytic Routes for Syngas and Hydrogen Production. Appl. Catal., A 1996, 144, 7–57. 10.1016/0926-860X(96)00108-1. [DOI] [Google Scholar]
- Chen P.; Zhang H.-B.; Lin G.-D.; Tsai K.-R. Development of Coking-Resistant Ni-Based Catalyst for Partial Oxidation and CO2-Reforming of Methane to Syngas. Appl. Catal., A 1998, 166, 343–350. 10.1016/S0926-860X(97)00291-3. [DOI] [Google Scholar]
- Kim J. H.; Suh D. J.; Park T. J.; Kim K. L. Effect of Metal Particle Size on Coking during CO2 Reforming of CH4 over Ni-Alumina Aerogel Catalysts. Appl. Catal., A 2000, 197, 191–200. 10.1016/S0926-860X(99)00487-1. [DOI] [Google Scholar]
- Bitter J. H.; Seshan K.; Lercher J. A. Deactivation and Coke Accumulation during CO2/CH4 Reforming over Pt Catalysts. J. Catal. 1999, 183, 336–343. 10.1006/jcat.1999.2402. [DOI] [Google Scholar]
- Kim S. M.; Abdala P. M.; Margossian T.; Hosseini D.; Foppa L.; Armutlulu A.; Van Beek W.; Comas-Vives A.; Copéret C.; Müller C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949. 10.1021/jacs.6b11487. [DOI] [PubMed] [Google Scholar]
- Theofanidis S. A.; Galvita V. V.; Poelman H.; Marin G. B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. 10.1021/acscatal.5b00357. [DOI] [Google Scholar]
- Theofanidis S. A.; Galvita V. V.; Sabbe M.; Poelman H.; Detavernier C.; Marin G. B. Controlling the Stability of a Fe–Ni Reforming Catalyst: Structural Organization of the Active Components. Appl. Catal., B 2017, 209, 405–416. 10.1016/j.apcatb.2017.03.025. [DOI] [Google Scholar]
- Jin F.; Fu Y.; Kong W.; Wang J.; Cai F.; Yuan C.; Pan B.; Zhang J.; Sun Y. Stable Trimetallic NiFeCu Catalysts with High Carbon Resistance for Dry Reforming of Methane. ChemPlusChem 2020, 85, 1120–1128. 10.1002/cplu.202000217. [DOI] [PubMed] [Google Scholar]
- Yeh J. W.; Chen S. K.; Lin S. J.; Gan J. Y.; Chin T. S.; Shun T. T.; Tsau C. H.; Chang S. Y. Nanostructured High-Entropy Alloys with Multiple Principal Elements: Novel Alloy Design Concepts and Outcomes. Adv. Eng. Mater. 2004, 6, 299–303. 10.1002/adem.200300567. [DOI] [Google Scholar]
- Tsai M.-H.; Yeh J.-W. High-Entropy Alloys: A Critical Review. Mater. Res. Lett. 2014, 2, 107–123. 10.1080/21663831.2014.912690. [DOI] [Google Scholar]
- George E. P.; Raabe D.; Ritchie R. O. High-Entropy Alloys. Nat. Rev. Mater. 2019, 4, 515–534. 10.1038/s41578-019-0121-4. [DOI] [Google Scholar]
- Li K.; Chen W. Recent Progress in High-Entropy Alloys for Catalysts: Synthesis, Applications, and Prospects. Mater. Today Energy 2021, 20, 100638. 10.1016/j.mtener.2021.100638. [DOI] [Google Scholar]
- Batchelor T. A. A.; Pedersen J. K.; Winther S. H.; Castelli I. E.; Jacobsen K. W.; Rossmeisl J. High-Entropy Alloys as a Discovery Platform for Electrocatalysis. Joule 2019, 3, 834–845. 10.1016/j.joule.2018.12.015. [DOI] [Google Scholar]
- Zhang G.; Ming K.; Kang J.; Huang Q.; Zhang Z.; Zheng X.; Bi X. High Entropy Alloy as a Highly Active and Stable Electrocatalyst for Hydrogen Evolution Reaction. Electrochim. Acta 2018, 279, 19–23. 10.1016/j.electacta.2018.05.035. [DOI] [Google Scholar]
- Qiu H. J.; Fang G.; Wen Y.; Liu P.; Xie G.; Liu X.; Sun S. Nanoporous High-Entropy Alloys for Highly Stable and Efficient Catalysts. J. Mater. Chem. A 2019, 7, 6499–6506. 10.1039/C9TA00505F. [DOI] [Google Scholar]
- Xie P.; Yao Y.; Huang Z.; Liu Z.; Zhang J.; Li T.; Wang G.; Shahbazian-Yassar R.; Hu L.; Wang C. Highly Efficient Decomposition of Ammonia Using High-Entropy Alloy Catalysts. Nat. Commun. 2019, 10, 4011. 10.1038/s41467-019-11848-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.; Huang Z.; Xie P.; Lacey S. D.; Jacob R. J.; Xie H.; Chen F.; Nie A.; Pu T.; Rehwoldt M.; Yu D.; Zachariah M. R.; Wang C.; Shahbazian-Yassar R.; Li J.; Hu L. Carbothermal Shock Synthesis of High-Entropy-Alloy Nanoparticles. Science 2018, 359, 1489–1494. 10.1126/science.aan5412. [DOI] [PubMed] [Google Scholar]
- Varalakshmi S.; Kamaraj M.; Murty B. S. Synthesis and Characterization of Nanocrystalline AlFeTiCrZnCu High Entropy Solid Solution by Mechanical Alloying. J. Alloys Compd. 2008, 460, 253–257. 10.1016/j.jallcom.2007.05.104. [DOI] [Google Scholar]
- Yao Y.; Liu Z.; Xie P.; Huang Z.; Li T.; Morris D.; Finfrock Z.; Zhou J.; Jiao M.; Gao J.; Mao Y.; Miao J.; Zhang P.; Shahbazian-Yassar R.; Wang C.; Wang G.; Hu L. Computationally Aided, Entropy-Driven Synthesis of Highly Efficient and Durable Multi-Elemental Alloy Catalysts. Sci. Adv. 2020, 6, eaaz0510 10.1126/sciadv.aaz0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F.; Yang Y.; Bei H.; George E. P. Relative Effects of Enthalpy and Entropy on the Phase Stability of Equiatomic High-Entropy Alloys. Acta Mater. 2013, 61, 2628–2638. 10.1016/j.actamat.2013.01.042. [DOI] [Google Scholar]