

Abstract
Ribavirin has recently been demonstrated to have efficacy in combination with alpha interferon for treatment of relapsed hepatitis C. The marked improvement in the response rate after treatment with the combination regimen (10-fold higher versus that from monotherapy with alpha interferon) highlights the importance of determining the absolute bioavailability of ribavirin as a first step in beginning to investigate the pharmacodynamics of the combination. The objective of this study was to determine the absolute bioavailability of ribavirin with an intravenous formulation containing ribavirin labeled with the stable isotope 13C3 (13C3-ribavirin) and unlabeled oral ribavirin. Six healthy volunteers received 150 mg of intravenous 13C3-ribavirin followed 1 h later by a 400-mg oral dose of ribavirin. Samples of blood and urine were collected up to 169 h postdosing. Concentrations of 13C3-ribavirin and unlabeled ribavirin were determined by a high-performance liquid chromatography tandem mass spectrometric method. All plasma and urine data were comodeled for labeled and unlabeled ribavirin by using both the two- and three-compartment models in the program ADAPT II. A three-compartment model was chosen for the pharmacokinetic analysis with the Akaike Information Criterion. The mean maximum concentrations of drug in plasma for intravenous and oral ribavirin were 4,187 and 638 ng/ml, respectively. The mean bioavailability was 51.8% ± 21.8%, and the mean γ-phase half-life was 37.0 ± 14.2 h. The mean renal clearance, metabolic clearance, and volume of distribution of the central compartment were 6.94 liters/h, 18.1 liters/h, and 17.8 liters, respectively. The use of the stable-isotope methodology has provided the best estimate of the absolute bioavailability of ribavirin that is currently available, as there was neither a period bias nor a washout effect to confound the data. The study demonstrated that the mean bioavailability for a 400-mg dose of ribavirin was 52%, which is higher than that previously reported in other investigations.
Hepatitis C virus produces a chronic viral infection which is widespread throughout the world and which is a cause of serious morbidity and some mortality if it is left untreated. The cornerstone of therapy for this condition, alpha interferon, has traditionally yielded response rates of 15 to 25% (10). More recently, clinical studies of the combination of ribavirin and alpha interferon suggested that the combination has a clinical benefit, with response rates reported to be 36 to 77% (1, 4, 12, 19). Most recently, a randomized, double-blind, placebo-controlled trial of the combination of oral ribavirin and alpha 2b interferon versus alpha 2b interferon alone demonstrated a response rate 10-fold higher with the combination than with alpha 2b interferon alone (50 versus 5%, respectively) in patients who had relapses after being given alpha interferon alone (7). On the basis of these and other data, ribavirin in combination with interferon is currently approved as treatment for hepatitis C.
The marked improvement in response rate with the addition of ribavirin highlights the importance of understanding the absolute bioavailability of ribavirin. While this has been estimated in the past (13, 14, 17), these findings may be questioned because of the properties of ribavirin. Ribavirin partitions into all cells rapidly and is phosphorylated to monophosphate, diphosphate, and triphosphate nucleotides (16). In nucleate cells, there is a slow dephosphorylation process (9) which contributes to an extremely long terminal phase (11). Anucleate erythrocytes lack the ability to dephosphorylate ribavirin nucleotides (16), which are sequestered intracellularly until the erythrocytes are destroyed in the reticuloendothelial system (20). In vitro studies have determined that the intracellular concentrations of ribavirin and its nucleotides are approximately nine times those seen in plasma by 6 h postdosing (13). This depot of ribavirin makes it difficult to interpret the results of studies with a traditional crossover design for the determination of absolute bioavailability. The life span of an erythrocyte is approximately 120 days, although this may be reduced by ribavirin (3, 20). Studies with a washout period that is too short can be difficult to interpret because the effects of preexisting ribavirin stores in the erythrocytes and the slow release of ribavirin from other nucleate cells may affect the pharmacokinetics of ribavirin given during the second period of the crossover study. This may have occurred in two previous studies that have estimated the bioavailability of ribavirin (14, 17), in which the washout periods were 10 and 14 days, respectively. In addition, Lertora et al. (14) estimated bioavailability by calculating the ratio of orally administered ribavirin to intravenous drug using concentrations in urine rather than concentrations in serum. In that study, the urine collection was carried out for 48 h, which is considerably shorter than ribavirin’s half-life (11). In a crossover study, administration of the second dose of ribavirin after a sufficient wait can lead to significant period bias, casting doubt on whether the patient is physiologically unchanged from the first administration. A way to avoid this problem is not to use a crossover design but, instead, to use different patients in the intravenous and oral dosage form groups (parallel group design), as was done in the study by Laskin et al. (13). The limitation of this design, however, is that interindividual pharmacokinetic differences may bias absolute bioavailability estimates.
Our objective was to determine the absolute bioavailability of ribavirin using stable-isotope methods (22). Stable-isotope-labeled ribavirin in an intravenous formulation and unlabeled ribavirin in an oral formulation were coadministered to subjects, 1 hour apart, obviating the need to use different patients for each dosage form or to have a prolonged washout period in a crossover study.
MATERIALS AND METHODS
Subject selection.
Six healthy male volunteers with a mean age of 36.8 years (age range, 31 to 44 years) and a mean weight of 78.3 kg (weight range, 58.7 to 89.8 kg) were entered into the study at the Albany Medical Center. All volunteers had normal hematologic, renal, and hepatic laboratory parameters at screening and on the day of dosing. All volunteers were negative for human immunodeficiency virus antibody, hepatitis B virus surface antigen, and hepatitis C virus antibody and had negative urine drug screens for illicit drug use. Electrocardiograms were performed at screening and were also normal. No subjects were receiving any concurrent medications, and all subjects were nonsmokers. The demographic data for these subjects are provided in Table 1. Informed consent was obtained from each subject prior to entry into the study according to Albany Medical Center Institutional Review Board guidelines.
TABLE 1.
Demographic data for individual volunteersa
| Subject no. | Age (yr) | Wt (kg) | Ht (cm) | SCr concn (mg/dl) | CLCR (ml/min) | Hb concen (g/dl) |
|---|---|---|---|---|---|---|
| 1 | 33 | 84.5 | 171.4 | 1.1 | 114.2 | 16.5 |
| 2 | 33 | 82.0 | 174.0 | 0.9 | 135.4 | 15.9 |
| 3 | 31 | 83.0 | 184.2 | 1.1 | 114.2 | 15.3 |
| 4 | 42 | 89.9 | 175.0 | 0.9 | 136.0 | 14.4 |
| 5 | 44 | 71.8 | 176.5 | 0.8 | 119.7 | 16.4 |
| 6 | 38 | 58.7 | 158.0 | 0.9 | 92.4 | 14.3 |
| Mean ± SD | 36.8 ± 5.3 | 78.3 ± 11.3 | 173.2 ± 8.6 | 0.95 ± 0.12 | 118.6 ± 16.2 | 15.5 ± 0.96 |
SCr, creatinine in serum; CLCR, estimated creatinine clearance by the Cockcroft-Gault (5) equation; Hb, hemoglobin in serum.
Drug administration and dosage.
The 13C3-labeled ribavirin (13C3-ribavirin) and unlabeled ribavirin were supplied by Schering-Plough Research Institute, Kenilworth, N.J. After a 10-h overnight fast which continued until 5 h after administration of the intravenous dose, subjects received 150 mg of 13C3-ribavirin intravenously over 1 min (5 ml of a 30-mg/ml solution in phosphate buffer), and the drug administration cannula was flushed with 10 ml of normal saline after dosing. One hour later, the subjects received orally two 200-mg capsules of unlabeled ribavirin with 200 ml of tap water.
Collection of blood and urine specimens.
Venous blood samples were collected through an indwelling catheter in the arm opposite that into which drug was infused. Catheters were flushed with normal saline after each blood draw. After drawing a separate 3-ml sample to clear the catheter of any remaining saline flush, blood samples for ribavirin concentration determinations were collected in a syringe and were immediately transferred to a prechilled heparinized tube. The tubes were retained on ice until they were centrifuged. Sampling times were based on the time of intravenous drug administration and were time zero (predosing) and then 5, 10, 20, 30, and 45 min and 1, 1.5, 2, 2.5, 3, 4, 5, 7, 9, 11, 13, 17, 25, 37, 49, 61, 73, 97, 121, 145, and 169 h postdosing. Within 15 min of collection, all venous blood samples (3 ml each) were centrifuged at 1,000 × g at 4°C for 5 min. Plasma was pipetted into a freezer tube, and the tube was frozen at −80°C until analysis.
Urine samples were collected just prior to intravenous drug administration at time zero and then in block samples at 0 to 1, >1 to 3, >3 to 7, >7 to 11, >11 to 17, >17 to 25, >25 to 49, >49 to 73, >73 to 97, >97 to 121, >121 to 145, and >145 to 169 h postdosing. In order to ensure adequate urine output, subjects drank 500 ml of tap water immediately after the intravenous infusion at time zero, 200 ml of tap water at hour 1, and 100 ml of tap water hourly from hours 2 to 13. Samples collected during each collection period were refrigerated. After thorough mixing of each block sample, the volume of the total block sample was recorded, and then two 15-ml aliquots were transferred to freezer containers and were frozen at −80°C until analysis.
Ribavirin concentrations.
The concentrations of 13C3-ribavirin and unlabeled ribavirin in plasma and urine were determined by a validated high-performance liquid chromatographic tandem mass spectrometric method. Five curves were generated for each quality control analysis, and the interassay precisions of the assay for determination of the 13C3-ribavirin concentration in plasma at 0.129, 2.057, and 4.002 μg/ml were ±5.9, ±7.2, and ±8.3%, respectively. The interassay precisions of the assay for determination of the 13C3-ribavirin concentration in urine at 0.617, 10.3, and 21.8 μg/ml were ±4.1, ±3.6, and ±1.5%, respectively. The lowest quantifiable limits for the concentrations of 13C3-ribavirin in plasma and urine were 0.0499 and 0.242 μg/ml, respectively. The interassay precisions of the assay for determination of the unlabeled ribavirin concentration in plasma at 0.130, 2.088, and 4.218 μg/ml were ±5.7, ±2.0, and ±7.6%, respectively, and those for determination of the unlabeled ribavirin concentration in urine at 0.861, 10.2, and 20.5 μg/ml were ±1.8, ±2.6, and ±1.3%, respectively. The lowest limits of detectability of unlabeled ribavirin in plasma and urine were 0.0496 and 0.248 μg/ml, respectively.
Adverse event monitoring.
All subjects had a complete history and a physical at the time of screening and at the end of the study, on day 8 postdosing. Laboratory test results were monitored and included a complete blood count plus differential and reticulocyte count, chemistry panel, and urinalysis on the evening prior to dosing, day 4, and day 8. An electrocardiogram was done at screening and on study day 8. Vital signs, including resting blood pressure, pulse, respirations, and oral body temperature, were obtained just prior to intravenous drug administration at time zero and at 1, 2, 3, 4, 5, 7, 9, 11, 13, 17, 25, 37, 49, 61, 73, 97, 121, 145, and 169 h postdosing.
Pharmacokinetic analysis.
Two- and three-compartment models were fit to the data. A three-compartment model best fit the data on the basis of Akaike’s Information Criterion (23). The parameters identified included volume of distribution of the central compartment (Vc), the absorption rate constant (ka), the intercompartmental rate constants from the central to second (k12), second to central (k21), central to third (k13), and third to central (k31) compartments, metabolic clearance (CLmet), renal clearance (CLR), and bioavailability (F). α, β, and γ half-lives (t1/2) were calculated from the estimated parameters. All data for labeled and unlabeled ribavirin concentrations in plasma and urine data were comodeled by using the following differential equations:
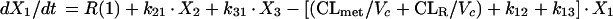 |
1 |
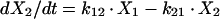 |
2 |
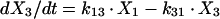 |
3 |
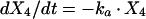 |
4 |
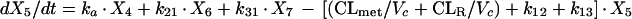 |
5 |
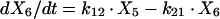 |
6 |
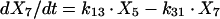 |
7 |
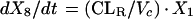 |
8 |
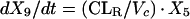 |
9 |
where R(1) is the dose rate of labeled drug, X1 is the amount of labeled drug in the central compartment, X2 is the amount of labeled drug in the second compartment, X3 is the amount of labeled drug in the third compartment, X4 is the amount of unlabeled drug in the absorption compartment, X5 is the amount of unlabeled drug in the central compartment, X6 is the amount of unlabeled drug in the second compartment, X7 is the amount of unlabeled drug in the third compartment, X8 is the cumulative amount of labeled drug excreted renally, and X9 is the cumulative amount of unlabeled drug excreted renally. Bioavailability entered into the model through the system outputs for unlabeled drug for determination of the concentration in plasma and cumulative urinary recovery in urine. Data were analyzed by use of a maximum-likelihood estimator with linear variance models in the identification subroutine of ADAPT II (6). A separate and independent variance model was used for each output.
RESULTS
Concentrations in plasma.
Six healthy volunteers were studied, and demographic data are shown in Table 1. The mean maximum concentrations in plasma (Cmaxs) for the intravenous labeled compound and oral unlabeled compound were 4,187 ± 199.4 ng/ml (mean ± standard deviation [SD]) and 638 ± 15.95 ng/ml. The dose-normalized (to 150 mg) Cmax for the oral dosage form was 239 ± 5.98 ng/ml, with a time to Cmax of 1.33 ± 0.034 h after oral dose administration. The mean area under the concentration-versus-time curve from time zero until time of final quantifiable sample for the intravenous form was 4,263 ± 99.14 ng · h/ml and that for the dose-normalized oral form was 2,758 ± 48.38 ng · h/ml.
Excretion in urine.
The mean amount of ribavirin excreted in the urine from time zero to 169 h postdosing was 40,052.5 ± 3,743.2 μg for the intravenous form (26.7% of the dose) and 62,484.2 ± 1,882.0 μg (15.6% of dose) for the oral form (Table 2).
TABLE 2.
Cumulative recovery of 13C3-ribavirin and unlabeled ribavirin in urine from time zero to 169 h postdosing
| Subject no. | Amt (μg)
|
||
|---|---|---|---|
| 13C3-ribavirin | Ribavirin | Ribavirin adjusted to 150-mg dose | |
| 1 | 37,296 | 39,255 | 14,720.6 |
| 2 | 42,790 | 44,030 | 16,511.2 |
| 3 | 34,729 | 58,535 | 21,950.6 |
| 4 | 36,978 | 88,487 | 33,182.6 |
| 5 | 45,729 | 86,211 | 32,329.1 |
| 6 | 42,784 | 58,387 | 21,895.1 |
| Mean | 40,052.5 | 62,484.2 | 23,431.5 |
| CVa (%) | 10.7 | 33.2 | 33.2 |
| % of dose | 26.7 | 15.6 | 15.6 |
CV, coefficient of variation.
Pharmacokinetic parameters.
The values of the ribavirin pharmacokinetic parameters for each individual are summarized in Table 3. The mean ± SD α-, β-, and γ-phase t1/2s in plasma were 0.040 ± 0.032, 0.480 ± 0.172, and 37.00 ± 14.20 h, respectively. The mean ± SD Vc was 17.80 ± 11.86 liters. The mean ± SD F was 51.80% ± 21.8%. The concentration-versus-time profiles of the oral and intravenous forms of ribavirin in serum for all subjects are shown in Fig. 1. Figure 2 demonstrates the cumulative recovery of both dosage forms of ribavirin in urine for the subjects.
TABLE 3.
Values of pharmacokinetic parameters for ribavirin obtained with comodeled data for plasma and urinea
|
t1/2 (h)
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject no. | CLmet (liters/h) | CLR (liters/h) | Vc (liters) | Vss (liters) | V2 (liters) | V3 (liters) | ka (h−1) | k12 (h−1) | k21 (h−1) | k13 (h−1) | k31 (h−1) | F (%) | α | β | γ |
| 1 | 8.894 | 5.359 | 6.080 | 873.4 | 844.5 | 22.86 | 0.383 | 13.22 | 0.095 | 17.64 | 4.691 | 28.2 | 0.019 | 0.332 | 49.64 |
| 2 | 23.29 | 8.323 | 39.46 | 669.3 | 619.4 | 10.40 | 0.312 | 1.981 | 0.126 | 7.095 | 26.93 | 33.1 | 0.020 | 0.308 | 19.87 |
| 3 | 30.38 | 8.797 | 12.80 | 1,113.6 | 1,047.0 | 53.82 | 0.208 | 3.415 | 0.042 | 15.62 | 3.715 | 58.4 | 0.028 | 0.703 | 35.76 |
| 4 | 19.20 | 6.815 | 14.52 | 1,120.1 | 1,077.0 | 28.60 | 0.325 | 5.279 | 0.071 | 14.14 | 7.179 | 84.6 | 0.026 | 0.354 | 39.33 |
| 5 | 20.05 | 8.139 | 11.49 | 607.0 | 566.3 | 29.21 | 0.340 | 4.479 | 0.091 | 7.512 | 2.955 | 66.5 | 0.043 | 0.530 | 22.24 |
| 6 | 6.454 | 4.239 | 22.42 | 711.5 | 667.3 | 21.27 | 0.298 | 2.165 | 0.073 | 2.447 | 2.579 | 40.0 | 0.104 | 0.649 | 55.17 |
| Mean ± SD | 18.06 ± 8.991 | 6.94 ± 1.823 | 17.80 ± 11.86 | 849.20 ± 2253 | 803.67 ± 221.1 | 27.69 ± 14.49 | 0.31 ± 0.058 | 5.09 ± 4.184 | 0.083 ± 0.028 | 10.74 ± 5.924 | 8.01 ± 9.415 | 51.80 ± 21.8 | 0.040 ± 0.032 | 0.480 ± 0.172 | 37.00 ± 14.20 |
VSS, volume of distribution at steady state; V2, volume of distribution of the second compartment; V3, volume of distribution of the third compartment. The other abbreviations were defined in the text.
FIG. 1.

Concentration-versus-time profile of intravenous and oral ribavirin. The log-transformed average concentrations of intravenous (IV) (▴) and oral (■) ribavirin versus time were obtained for subject 1 (A), subject 2 (B), subject 3 (C), subject 4 (D), subject 5 (E), and subject 6 (F).
FIG. 2.

Cumulative recovery of intravenous and oral ribavirin in urine versus time. The cumulative recoveries of intravenous (IV) (▴) and oral (■) ribavirin in urine versus time were obtained for subject 1 (A), subject 2 (B), subject 3 (C), subject 4 (D), subject 5 (E), and subject 6 (F).
Adverse drug reactions and toxicity.
No significant adverse drug reactions occurred during the study.
DISCUSSION
Use of stable-isotope methodology in a bioavailability study offers several advantages over the traditional crossover methodology (2). Intrasubject variabilities in drug dissolution, absorption rate, and metabolism are eliminated as potential confounding variables. Also, the duration of the study is decreased by half.
Bioavailability studies with stable isotopes assume that the metabolism and pharmacokinetics of the labeled and unlabeled drugs are the same, i.e., that there are no isotope effects. Isotopic substitution can cause changes in physiochemical properties, such as lipophilicity (8) and changes in the rates of enzymatic transformations (21). Changes in physiochemical properties are generally seen only with heavily deuterated compounds and are usually small. Changes in the rates of metabolic transformations, kinetic isotope effects, are more common and critical. Reaction rates of deuterated compounds can be substantially lower compared with those of the unlabeled compounds, with rate constant ratios kheavy/klight up to 8 being common (15). The much smaller difference in mass between 12C and 13C leads to k12C/k13C rate constant ratios less than 1.1. The stable-isotope-labeled compound used in our study, 13C3-ribavirin, is unlikely to exhibit any detectable isotope effects.
The use of the comodeling technique allowed data for both plasma and urine to be used in the estimation of F and other pharmacokinetic parameter values. Also, as the variance structures were different for the assays with plasma and urine, we were able to obtain proper relative weighting by using maximum-likelihood estimation with different variance models for each of the matrices (plasma and urine) as well as for the cold and labeled forms of the drug. We had used the assay performance data prior to the analysis to indicate that a linear variance model was an acceptable form (higher-order polynomials were checked), under the assumption that assay variance is an important component of total observation variance.
We observed that the total clearance of ribavirin averaged 25 liters/h (coefficient of variation, 43%), with approximately 30% (coefficient of variation, 26%) of the total clearance being accounted for as CLR. The actual value of CLR (circa 7 liters/h) is quite close to the estimated creatinine clearance in this patient population (7.12 liters/h). Whether there is any net tubular handling of ribavirin cannot be determined directly from our data, but it is doubtful that major tubular handling takes place. It remains a possibility that counterbalancing tubular reabsorption and excretion occur, but it should be noted that Laskin et al. (13) identified a mean ratio of CLR/creatinine clearance of 0.97, which is essentially identical to the ratio of 0.98 that we identified. In both studies, creatinine clearance was estimated by the method of Cockcroft and Gault (5). These findings are different from those of Lertora et al. (14), who identified a ratio greatly in excess of 1.0.
The rest of the total clearance is labeled as CLmet, but it should be realized that a fraction of CLmet involves partitioning of the drug into cells, with further anabolism to various nucleotides of ribavirin.
The finding of an overall clearance of approximately 25 liters/h with 30% being accounted for as CLR is quite in line with previous determinations of ribavirin’s pharmacokinetics (13, 17).
Of interest, ka was very reproducible, with a mean of 0.311 (coefficient of variation, 18.8%), giving an average absorption t1/2 of 2.3 h. This indicates that some absorption is ongoing throughout most of a 12-h dosing interval and suggests that absorption occurs at multiple sites throughout the length of the gastrointestinal tract. One mechanism for gastrointestinal absorption has already been identified and involves N1-sodium-dependent nucleoside transporters (18).
Our data suggest that ribavirin may exhibit a first-pass metabolism, as the amount of the oral form of the drug excreted in urine collected for 0 to 144 h was 15.6%, whereas with the intravenous form of the drug collected for 0 to 144 h, the amount excreted in urine was 26.7%. Other data were also suggestive of this, demonstrating urinary excretions of 4.4 and 16.7% for urine collected from 0 to 48 h after dosing of the oral and intravenous forms of ribavirin, respectively (9). While the ratios in the two studies differ (58 versus 26%), this may have been due to the different collection intervals.
The F of 52% is higher than that identified previously, but it is still in the range of that described previously by Lertora et al. (14) as well as by Laskin et al. (13) (45%). However, as noted previously, Lertora et al. (14) determined ribavirin’s F by examining the urinary excretion and did so with a short period between drug administrations. Laskin et al. (13) recognized the difficulties in performing a study with this drug with a two-period design and used a parallel-group design. Our study, then, is the only one which was able to circumvent the problems associated with ribavirin’s complex pharmacology through the use of a stable-isotope methodology and the application of sophisticated modeling techniques.
In summary, this study has defined a number of single-dose pharmacokinetic characteristics of ribavirin. The clearance is approximately 25 liters/h and has a between-patient coefficient of variation of 43%. CLR accounts for 30% of total clearance. The F of ribavirin is 52%, and F is modestly higher than those previously reported in other studies. This estimate of F is likely to be the most accurate on the basis of the methodology used.
ACKNOWLEDGMENTS
This work was supported by a grant from Schering-Plough Research Institute.
We thank Robert Clement and colleagues in the Drug Metabolism Group, Schering Plough Research Institute, for assay development.
REFERENCES
- 1.Brillanti S, Garson J, Foli M, Whitby K, Deaville R, Masci C, Migliotti M, Barbara L. A pilot study of combination therapy with ribavirin plus interferon alfa for interferon alfa-resistant chronic hepatitis C. Gastroenterology. 1994;107:812–817. doi: 10.1016/0016-5085(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 2.Browne T R. Stable isotopes in clinical pharmacokinetic investigations: advantages and disadvantages. Clin Pharmacokinet. 1990;18:423–433. doi: 10.2165/00003088-199018060-00001. [DOI] [PubMed] [Google Scholar]
- 3.Catlin D H, Smith R A, Samuels A I. 14C-ribavirin: distribution and pharmacokinetic studies in rats, baboons, and man. In: Smith R A, Kirkpatrick W, editors. Ribavirin: a broad spectrum antiviral agent. New York, N.Y: Academic Press, Inc.; 1990. pp. 83–98. [Google Scholar]
- 4.Chemello L, Cavalletto L, Bernardinello E, Guido M, Pontisso P, Alberti A. The effect of interferon alfa and ribavirin combination therapy in naive patients with chronic hepatitis C. J Hepatol. 1995;23(Suppl 2.):8–12. [PubMed] [Google Scholar]
- 5.Cockcroft D W, Gault M H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 6.D’Argenio D Z, Schumitzky A. ADAPT II user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Los Angeles, Calif: Biomedical Simulations Resource; 1997. [Google Scholar]
- 7.Davis G L, Esteban-Mur R, Rustgi V, Hoefs J, Gordon S, Trepo C, Shiffman M, Zeuzem S, Craxi A, Raffanel C, Reindollar R, Rizzetto M the International Hepatitis Interventional Therapy Group. Retreatment of relapse after interferon therapy for chronic hepatitis C: an international randomized controlled trial of interferon plus ribavirin vs interferon alone. Hepatology. 1997;26:247A. [Google Scholar]
- 8.Falconnet J B, El Tayar N, Bechalany A, et al. Isotope effects on lipophilicity, synthesis, and applications of isotopically labelled compounds. In: Bailie T A, Jones J R, editors. Synthesis and applications of isotopically labelled compounds 1988. New York, N.Y: Elsevier; 1989. pp. 355–360. [Google Scholar]
- 9.Glue P. Ribavirin: clinical pharmacology and mechanism of action. Semin Liver Dis. 1999;19(Suppl. 1):17–24. [PubMed] [Google Scholar]
- 10.Hoofnagle J H, DiBisceglie A M. The treatment of chronic viral hepatitis. N Engl J Med. 1997;336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- 11.Khakoo S, Glue P, Grellier L, Wells B, Bell A, Dash C, Murray-Lyon I, Lypnyj D, Flannery B, Walters K, Dusheiko G M. Ribavirin and interferon alpha-2b in chronic hepatitis C: assessment of possible pharmacokinetic and pharmacodynamic interactions. Br J Pharmacol. 1998;46:563–570. doi: 10.1046/j.1365-2125.1998.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai M Y, Yang P M, Kao J H, Wang J T, Lee H S, Chen D S. Combination therapy of α-interferon and ribavirin in patients with chronic hepatitis C: an interim report. Hepatology. 1993;18(Suppl.):93A. (abstr.). [Google Scholar]
- 13.Laskin O L, Longstreth J A, Hart C C, Scavuzzo D, Kalman C M, Connor J D, Roberts R B. Ribavirin disposition in high-risk patients for acquired immunodeficiency syndrome. Clin Pharmacol Ther. 1987;41:546–555. doi: 10.1038/clpt.1987.70. [DOI] [PubMed] [Google Scholar]
- 14.Lertora J J L, Rege A B, Lacour J T, Ferencz N, George W J, VanDyke R B, Agrawal K C, Hyslop N E., Jr Pharmacokinetics and long-term tolerance to ribavirin in asymptomatic patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 1991;50:442–449. doi: 10.1038/clpt.1991.162. [DOI] [PubMed] [Google Scholar]
- 15.March J. Advanced organic chemistry. New York, N.Y: John Wiley & Sons, Inc.; 1992. p. 226. [Google Scholar]
- 16.Page T, Connor J D. The metabolism of ribavirin in erythrocytes and nucleated cells. Int J Biochem. 1990;22:379–383. doi: 10.1016/0020-711x(90)90140-x. [DOI] [PubMed] [Google Scholar]
- 17.Paroni R, Del Puppo M, Borghi C, Sirtori C R, Kienle M G. Pharmacokinetics of ribavirin and urinary excretion of the major metabolite 1,2,4-tirazole-3-carboxamide in normal volunteers. Int J Clin Pharmacol Ther Toxicol. 1989;27:302–307. [PubMed] [Google Scholar]
- 18.Patil S D, Glue P, Unadkat J D. Intestinal absorption of ribavirin is mediated by the Na+-nucleoside purine (Ni) transporter. Pharm Res. 1998;15:950–952. doi: 10.1023/a:1011945103455. [DOI] [PubMed] [Google Scholar]
- 19.Reichard O, Norkrans G, Fryden A, Braconier J H, Sonnerborg A, Weiland O. Randomised, double-blind, placebo-controlled trail of interferon alpha-2b with and without ribavirin for chronic hepatitis C. The Swedish Study Group. Lancet. 1998;351:83–87. doi: 10.1016/s0140-6736(97)06088-1. [DOI] [PubMed] [Google Scholar]
- 20.Schulman N R. Clinical applications of ribavirin. New York, N.Y: Academic Press, Inc.; 1984. Assessment of hematologic effects of ribavirin in humans; pp. 79–92. [Google Scholar]
- 21.Simon H, Palm D. Isotope effects in organic chemistry and biochemistry. Angew Chem Int Ed. 1966;5:921–933. [Google Scholar]
- 22.Wolen R L. The application of stable isotopes to studies of drug bioavailability and bioequivalence. J Clin Pharmacol. 1986;26:419–424. doi: 10.1002/j.1552-4604.1986.tb03551.x. [DOI] [PubMed] [Google Scholar]
- 23.Yamaoka K, Nakagawa T, Uno T. Application of Akaike’s Information Criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6:165–175. doi: 10.1007/BF01117450. [DOI] [PubMed] [Google Scholar]