

Abstract
Visualization of single mRNAs in their native cellular environment provides key information to study gene expression regulation. This fundamental biological question triggered the development of the MS2-MCP (MS2-Capsid Protein) system to tag mRNAs and image their life cycle using widefield fluorescence microscopy. The last two decades have evolved toward improving the qualitative and quantitative characteristics of the MS2-MCP system. Here, we provide a protocol to use the latest versions, MS2V6 and MS2V7, to tag and visualize mRNAs in mammalian cells in culture. The motivation behind engineering MS2V6 and MS2V7 was to overcome a degradation caveat observed in S. cerevisiae with the previous MS2-MCP systems. While for yeast we recommend the use of MS2V6, we found that for live-cell imaging experiments in mammalian cells, the MS2V7 has improved reporter properties.
Keywords: Single-molecule imaging, MS2 system, smFISH, MS2V6, MS2V7, Quantitative fluorescence microscopy, Short-lived mRNAs, Single cell
1. Introduction
The field of quantitative gene expression analysis in individual live cells was pioneered by the MS2-MCP system designed to image single mRNA molecules [1]. The MS2-MCP system has two components. The MS2 sequence is a bacteriophage-derived RNA aptamer that is integrated within the mRNA sequence as an array of 24 repeats that form 24 stem loops. Each of the stem loops is tightly bound by the second component of the system, a homodimer of the MCP (MS2 Capsid Protein) with each MCP fused to a fluorescent protein (FP). In cells expressing both components, the specific binding of the MCP to the MS2 tentatively decorates each mRNA with 48 FPs. This concentrated fluorescent signal allows for the detection of the single mRNA molecules and can inform on their fate [2, 3] (Fig. 1). Hence, it is imperative for the reporter to preserve the transcription, transport, translation, and decay properties of the tagged mRNA. Nonetheless, in yeast it was found that the tight binding of the MCP to the MS2 and the short distance between the stem loops impaired the accessibility of the mRNA decay enzymes to the MS2 array, which led to a slower degradation rate of the MS2 array. Consequently, fluorescent fragments and aggregates were formed inside cells, impairing the interpretations from studies of mRNA decay and localization [4, 5, 6, 7, 8]. Hence, this limitation prevented the use of the latest MS2 version, MS2V5, in yeast. To avoid homologous recombination, MS2V5 was made of 24 loops with synonymized sequences separated by linkers of 30 nucleotides (nts). Alike other MS2 versions, each stem loop was bound by the MCP with high affinity (Kd ≈ 0.6 nM) [8, 9].
Fig. 1.
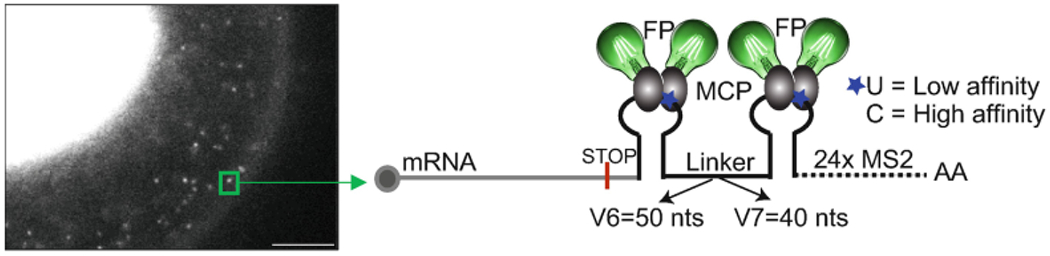
Detection of single mRNAs with the MS2-MCP system. The left panel is a snapshot of a live cell expressing mRNAs tagged with the MS2-MCP system. Expression of MCP fused to GFP is restricted to the nucleus of the cell. Single mRNA molecules are observed in the cytoplasm as discrete bright spots, as an example, one mRNA is indicated with a green square. The panel on the right is a scheme of an mRNA tagged in the 3′-UTR with the MS2-MCP system. Usually an array of 24 stem loops is inserted. The blue stars indicate the position of the nucleotide (U or C) in the loop that influences the binding affinity of the MCP and differentiates the low to the high affinity MS2 versions. The length of the linker between the loops differentiates the MS2V6 from the MS2V7 version. Scale bar is 5 μm
To solve the MS2-MCP degradation problem, we engineered the MS2V6 and MS2V7 systems. MS2V6 and MS2V7 have the same randomized sequence of the MS2 array. The only difference between these two sequences is an additional 10 nt longer linker region in MS2V6, that separates each MS2 stem-loop with a 50 nt linker. We created high and low affinity MCP-binding stem loops by replacing the cytidine (C) at position −5 of the loop, present in all high affinity MS2 versions, by uridine (U), in the low affinity stem-loops (Fig. 1). We used S. cerevisiae to compare the systems in terms of single-molecule detection and degradation kinetics [8], and showed that the low affinity version of MS2V6 (U variant) was the best system for yeast because most of the mRNAs have short half-lives. We have provided the protocol to use MS2V6 in yeast elsewhere [10]. Although MS2V6-U allows the visualization of mRNAs in mammalian cells, we learned that, in this model system, its reporting capabilities under demanding imaging conditions requiring long experimental times with high frequency acquisitions, were not optimal. Here, we show that the MS2V7 (C variant) is an accurate reporter of the life cycle of mRNAs in mammalian cells: (1) It provides brighter single molecules and for longer periods of time than the MBSV6-U version (Fig. 2), and (2) it is degraded simultaneously with the coding sequences of the tagged mRNA (Fig. 3). To assess for the simultaneous degradation of the coding sequence and the MS2V7 sequence, we have used a highly regulated mRNA, the heat shock protein 70 (HSP70) [11]. This chapter focuses on how to use MS2V7C-MCP for live-cell imaging and how to validate that the decay of the tagged mRNA is not delayed by MCP binding, by using two-color single-molecule fluorescent in situ hybridization (smFISH) [12].
Fig. 2.
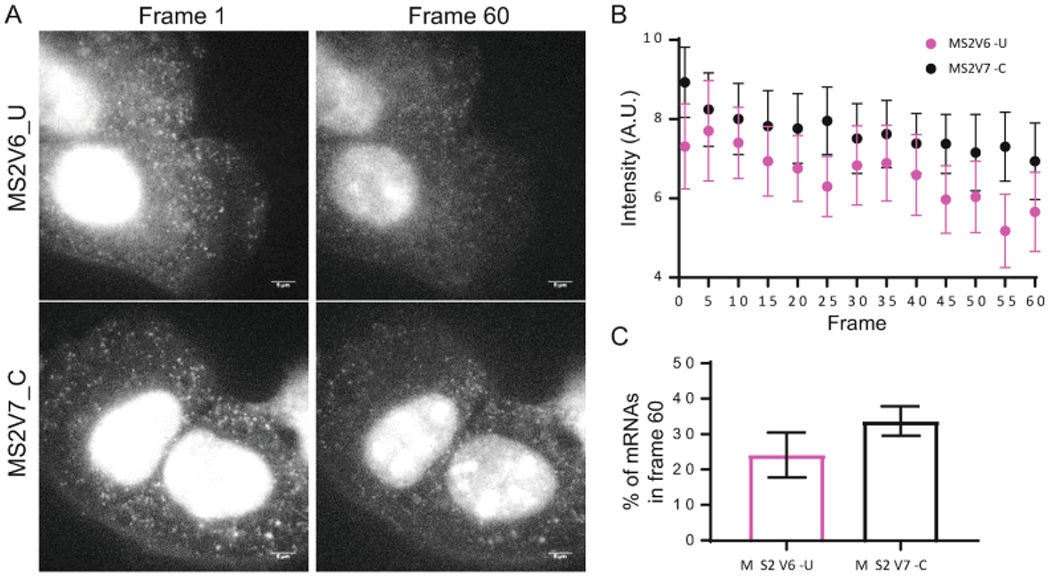
Live-cell imaging and quantification of mRNAs tagged with MSV6-U or MS2V7-C variants. (a) Maximum projection of 7 Z-stacks from the first and last frame (60) acquired during a 20-min experiment (3 f.p.s). Scale bar is 5 μm. (b) Quantification of the intensity of single mRNAs in the cytoplasm using AIRLOCALIZE. Data presented are the average and the standard deviation of n = 10 cells and ~1000 mRNAs. A.U. is arbitrary units. (c) % of mRNAs that remain visible in frame 60, relative to the first frame (after 413 acquisitions)
Fig. 3.
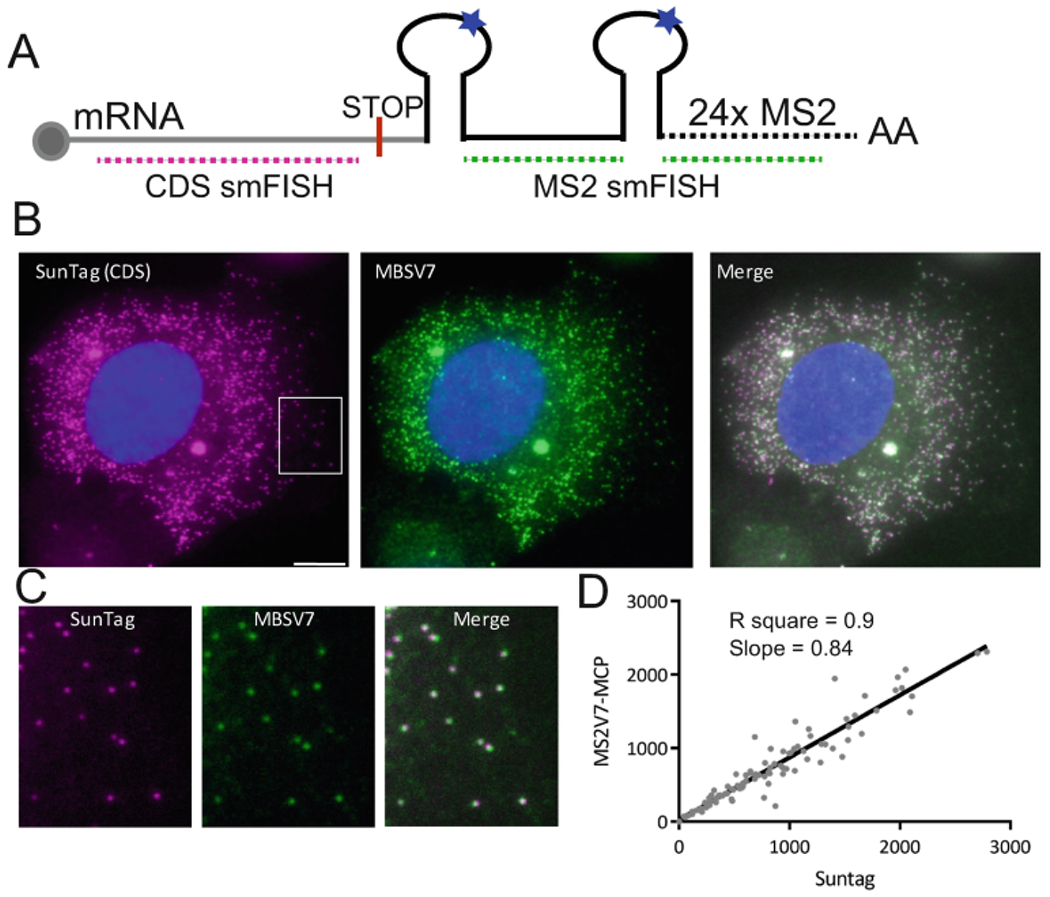
Quantification of single mRNAs by two-color smFISH. (a) Scheme of an mRNA tagged with 24×MS2 stem loops. Pink and green dotted lines indicate the localization of the smFISH probes used in the two-color smFISH experiment. (b) Image of a two-color smFISH experiment done during recovery from stress in U2OS cells expressing MCP-GFP. Name in the image indicate the sequence recognized by the probes. The SunTag sequence is in the CDS of the mRNA, and the MBSV7 sequence is in the 3′-UTR. Merge is the overlap between both signals. Nucleus is stained with DAPI. Scale bar is 10 μm. (c) Magnification of the area marked with the white rectangle. Each spot is a single mRNA that has been made visible by the two sets of probes. (d) Correlation between the number of single SunTag and MS2V7 molecules per cell. Pearson value and slope are calculated by combining two independent experiments (n = 96 cells)
2. Materials
2.1. Plasmid Cloning
Reporter plasmid containing 24×MBSV7 like MV102 or PET263 [8]. In MV102 we replaced the MS2V5 sequence in the SINAP plasmid (Addgene #84561) by the MS2V7 sequence.
Expression plasmids containing tandem MCP fused to a fluorescent protein like GFP (Addgene #98916).
Plasmids to produce second generation lentiviruses (Addgene #8454 and #12260).
High fidelity Taq polymerase like Platinum Taq DNA polymerase.
Restriction and cloning enzymes: AgeI, ClaI, StuI, CIP, and T4 DNA ligase.
Competent E-coli strain like DH5α.
Luria Broth (LB) and ampicillin (100 μg/mL) containing media and plates to grow bacteria.
DNA purification kits.
Primers to amplify the sequence of MBSV6 and MBSV7: Primer Forward (5′GATCCCAGAGCCCCCTGGCA) and Primer Reverse (5′GATCTTCCGTGTGAGGGTCTCTG).
2.2. Cell Lines and Lentivirus Production
Cell lines: HEK293T (ATCC# CRL-3216), U2OS (ATCC #HTB96), and Mouse Embryonic Fibroblast (MEFs, Immortalized using the large T antigen).
Tissue culture dishes.
Glass bottom dishes for imaging.
Cell filter for cell sorting: 30 μm filter.
Filter for lentivirus purification: PVDF 45 μm filter.
Lenti-X concentrator.
Transfection reagents: Lipofectamine 3000 and jetPRIME.
2.3. Media
Tissue culture media: DMEM containing 4.5 g/L glucose and 0.584 g/L l-glutamine, 10% fetal bovine serum (FBS), and 10 IU penicillin and 100 μg/mL streptomycin. The % of FBS is reduced to 5% for lentivirus production and infection of the cells.
Imaging media: carbon dioxide (CO2) free system, Leibovitz’s L-15 Medium without phenol red supplemented with 2% FBS and Penicillin/Streptomycin (10 IU/100 μg/mL).
Opti-MEM.
Sorting media: phosphate buffered saline (PBS) with 5% of FBS and penicillin–streptomycin (10 IU/100 μg/mL).
2.4. Microscopy
To visualize single mRNAs in live cells, we use a home-built microscope built around an IX81 stand (Olympus). The backport of the microscope was removed to allow custom laser illumination. For excitation of the GFP, a 491-nm laser (Calypso-25; Cobolt) was delivered through the back port. The laser was reflected by a four-band excitation dichroic mirror (Di01-R405/488/561/635; Semrock) to a 150× 1.45 NA oil immersion objective (Olympus). The fluorescence was collected by the same objective, passed through the dichroic mirror, a notch filter (NF01-405/488/561/635), and emission filters and was recorded on an EMCCD camera (Andor iXon3, model DU897, pixel size 16 μm). The microscope also was equipped with an automated XY stage (MS2000-XY with an extrafine lead-screw pitch of 0.635 mm and a 10-nm linear encoder resolution; Applied Scientific Instrumentation) and a piezo-Z stage (Applied Scientific Instrumentation) for fast z-stack acquisition. The AOTF, flipping mount, and piezo-Z stage were all controlled by a data acquisition board (DaqBoard/2001; IOtech, Inc.). The cells were kept at 37 °C with a stage-top incubator (INUBH-ZILCS-F1; Tokai Hit).
To visualize single mRNAs in fixed cells, we use an Olympus BX-63 epifluorescence widefield epifluorescence microscope equipped with an UPlanApo 60×, 1.35 NA, oil-immersion objective (Olympus). An X-Cite 120 PC Lamp (EXFO) and an ORCA-R2 Digital Interline CCD Camera (C10600-10B; Hamamatsu; 6.45 μm-pixel size) mounted using U-CMT and 1X-TVAD Olympus c-Mount Adapters and zero-pixel shift filter sets: DAPI-5060C-Zero, Cy3-4040C-Zero, and Cy5-4040C-Zero from Semrock. A Nomarski prism for the ×60 and the ×100 objectives is also installed. To acquire optical sections with a z-step size of 0.2 ×m, we use the ULTRASONIC STAGE for BX3/IX3, PIEZO TECH LIN ENCO.
Fluorescent microspheres: TetraSpeck™ Fluorescent Microspheres Size Kit (mounted on a slide).
2.5. Single-Molecule FISH
Stellaris probes from Biosearch Technologies. MS2V7 probes (cgcaagcgagagtgaagacg, tttgacggggaacagagtgt, gactgtacgagtagacatgc, atctgcacaccatgtatgat, gccatagcagagtgtaaact, tcgcaaggcagatgcaatac, cagaagtatccgcacgagtg, atgttctttgtagcaccgtg, ctccacatgtgagcaatacg, ggataatggtgcgatgcttc, ctcgtgaatacctgcacaac, gaagtaatgcaacggcaggg, gcgcaaatgacgacgacaga, gatagatctgtgtgagggtc, taatcctgcgtgtcgattgt, aggtagtcgagaagcgtaat, tattcctccattggcaaaaa, atgatatatgcgcggtgatc) and SunTag probes (ccacttcgttctcaagatga, ccctttttcagtctagctac, aatttttgctcagcaactcc, ttctttagtcgtgctacttc, tttcgagagtaactcctcac, ccacttcgttttcgagatga, acttcccttttttaagcgtg, tcttggatagtagctcttca, acctcgttctcaagatgata, cggaacccttcttcaaacgc, agttcttcgagagcagttcc, gatcccttttttaatcgagc, tgaaagtagttcctcaccac, cttcgttttcgaggtggtaa, ccctgaacctttctttaatc, tactcagtaattcttcaccc, tttcgatagcaactcttcgc, tttttgagcctagcaacttc, ttttcgagagcaactcctcg, acctcattttccaagtggta, tttgctcaataactcctcgc, cgcgacttcgttctctaaat, ttcgataagagttcttcgcc, ctcattttcgaggtggtagt, agtggtagttcttgctcaag, ttcaatctcgcgacctcatt, attcttgctgagcaattcct, cgacttcgttctccaaatga, cgacttcattttccaagtgg, ttgctcaataactcttcgcc, ttcgttctccaagtggtaat, agttcttcgataagagctcc, gcgacttcattctctaagtg, ttcttgctcaagagctcttc, cacctcattttccaagtggt, ttagatagtaactcttcccc, cctcgttctcgagatgataa, gatagttcttcgacaggagt, cctttttaagtcttgcaacc, ttactgagtagttcctcacc, ttcgttttccaggtggtaat, tcctgatcctttcttcaaac, cttttgagagcagttcttcg, gcaacctcattttccaaatg, tgccacttcccttttttaaa, tttcgacagaagttcctcac, gctacttcattctcgagatg, gagccagaaccctttttaag).
Fixation buffer: PBS and 5 mM MgCl2 (PBSM), and 4% PFA. Prepare fresh with ultrapure distilled water.
Quenching buffer: PBSM and 0.1 M glycine. Prepare fresh with ultrapure distilled water.
Permeabilization solution: PBS, 0.1% Triton X-100, 10 mM Ribonuclease Vanadil Complex (VRC). Prepare fresh with ultrapure distilled water.
Prehybridization buffer: 2× saline–sodium citrate (SSC) and 10% deionized formamide. Prepare fresh with ultrapure distilled water.
Hybridization solution: 2×SSC, 10% formamide, 1 mg/mL E. coli tRNA, VRC 10 mM, 10% dextran sulfate, 0.2 mg/mL ultrapure BSA. Prepare fresh with ultrapure distilled water.
ProLong Gold mounting media with DAPI.
Microscope cover glass.
2.6. Data Analysis
Fiji (Java software for image-processing analysis; freely available at https://fiji.sc/ (https://fiji.sc/)).
Image analysis software (AIRLOCALIZE [13], free software developed in the MATLAB programming language (MathWorks). Download the Airlocalize script (available upon request from Timothee Lionnet, NYU) together with the MCRInstaller, which allows one to run a MATLAB algorithm without separately installing MATLAB on the computer.
Image analysis software (FISH-quant [14], free software developed in the MATLAB programming language (MathWorks). Download the FISH-quant package (http://code.google.com/p/fish-quant/ (http://code.google.com/p/fish-quant/)).
3. Methods
3.1. Create the Plasmids to Tag mRNAs with the MS2 System
We created two different types of plasmids to (1) compare the brightness of the new MS2 systems (MBSV6-U variant and MBSV7-C variant) by live-cell imaging and (2) study the degradation of the MBSV7-C variant mRNA sequence when bound to the MCP by two-color smFISH. The first set of plasmids are based on a reporter system, SINAP system, with constitutive expression described in Wu et al. [15] (see Note 1 on where to clone the MS2 sequence in the mRNA). The second plasmid (MV71) is a highly regulated system based on the HSP70 gene tagged in the coding sequence with the SunTag system [16] and tagged in the 3′-UTR with 24×MBSV7-C variant loops (see Note 2 on how to check the integrity of the tagged mRNAs). We chose the HSP70 mRNA because it is degraded within 4 h upon recovery from stress [11].
MBSV6 and MBSV7 24× cassettes are made by duplication of the 12× cassette. 12× sequences have been synonymized and can be easily amplified by PCR. Amplify the 24× MBSV6 or MBSV7 stem-loop cassette from their plasmids using Platinum Taq DNA polymerase and specific primers. Final PCR reaction volume is 400 μL divided in eight reactions of 50 μL each, final MgCl2 concentration is 200 μM. PCR program: 1 cycle of 4 min at 94 °C, 35 cycles of 30 s at 94 °C, 30 s at 58 °C, and 2 min and 30 s at 72 °C, 1 cycle of 7 min at 72 °C.
Run the PCR product on a 1% agarose gel (wt/vol) and purify the DNA with a gel purification kit.
Digest the PCR fragments at 37 °C overnight and the cloning vector with the specific restriction enzymes. In our case we used AgeI and ClaI sites in vector (to compare the brightness of MBSV6-U variant (MV101) and MBSV7-C variant (MV102)) and StuI site in vector MV71 (with the HSP70 construct to analyze the simultaneous degradation of the coding sequence and the MBSV7-C bound to MCP).
Purify the insert and vector with a PCR purification kit. Quantify the DNA concentration using a NanoDrop. Set 20 μL of ligation reaction for 2 h at room temperature. We used a ratio 1:3 of vector: PCR insert.
Transform DH5 competent cells with 2 μL of the ligation reaction by heat shock at 42 °C for 40 s. Plate all the transformation solution on LB-Amp plates overnight at 37 °C. Grow individual colonies in 3 mL of LB-Amp for DNA extraction and screen for positive clones using restriction enzymes. Send the plasmid for sequencing with forward and reverse sequencing primers for your plasmid.
3.2. Lentivirus Production
Plate HEK 293T cells in a tissue culture treated dish of 100×20 mm at 40% confluence and grow them in complete DMEM media.
Twenty-four hours after plating, transfect with plasmids tat, rev, gag/pol, vsv-g, and MCP-GFP at a ratio of 1:1:1:2:20 using 30 μL of Lipofectmine3000 in a final transfection reaction of 37.5 μg of DNA mix and a volume of 1 mL.
Collect the supernatant at 24, 48, and 72 h posttransfection and replace with 10 mL of fresh media (see Note 3 on lentiviral manipulation).
Centrifuge the supernatants for 5 min at 1500 g and filter with a PVDF 45 μm filter. Concentrate the viruses by adding Lenti-X concentrator to a final dilution of 1:3 (v:v) in the media containing viruses. Mix gently by inversion and incubate the mixture between 2 h and overnight at 4 °C. Centrifuge for 45 min at 1500 g and 4 °C. Remove the supernatant and resuspend the pellet containing the viruses in 250 μL of DMEM. Virus aliquots of 50 μL can be stored at −80 °C or used immediately.
3.3. Create Stable Cell Lines Expressing MCP-GFP for Live Imaging
Choosing the cell line for imaging. In this paper, we used U2OS cells and immortalized MEFs because they have a flat and long cytoplasm ideal for imaging conditions. We chose GFP as the fluorescent protein, but the MCP can be fused to any other fluorescent protein (see Note 4 on other fluorescent proteins that can be fused to MCP).
Plate the cells (U2OS or MEFs) for the imaging experiments at a 30% confluence.
Twenty-four hours after plating, infect the cells by replacing the media with 6 mL of infection mix media containing 50 μL of lentivirus.
Forty-eight hours postinfection, trypsinize the cells and centrifuge for 3 min at 1000 g. Discard the supernatant and resuspend the pellet in 1 mL of sorting media. Filter the cells using a 30 μm filter before sorting. From the cell population expressing GFP, sort those cells with dim expression of GFP (see Note 5 on the expression of MCP-FP).
3.4. Expression of the MS2-Tagged mRNAs for Imaging
Experiments in this study have been performed by transient transfection of the plasmids encoding the mRNA tagged with the MS2 system. Another approach to express a reporter mRNA is by lentiviral infection. Advantages of using lentiviral infection are the long-term expression of the tagged mRNA, the selection of cells expressing the tagged mRNA, and the possibility of using primary cultures, like neurons, that are difficult to transfect. Yet the most accurate approach to investigate the life cycle of an mRNA using the MS2 system would be to tag an endogenous gene of interest using the CRISPR/Cas9 system [17] (see Note 6 on tagging of the endogenous mRNA).
3.4.1. For Live Imaging Experiments to Compare the Brightness of the Two MS2 Versions
Plate U2OS cells expressing MCP-GFP in a bottom glass dish at 30% confluence (see Note 7 on cell confluence).
Twenty-four hours after plating, transfect the plasmid to express the mRNA of interest with 24×MBS inserted in the 3′-UTR. In our case we transfect two constructs based on the SINAP system [15], SINAPV6-U or SINAPV7-C, using 2 μg of DNA and 5 μL of Lipofectamine 3000 in final transfection volume of 250 μL of Opti-MEM.
Thirty-six hours after transfection and before starting the imaging session, change the media with prewarmed (37 °C) imaging media.
3.4.2. For Experiments in Fixed Cells to Analyze the Decay of MBSV7-MCP System
Plate MEFs or U2OS cells expressing MCP-GFP in a live-cell imaging dish at 30% confluence.
Twenty-four hours after plating, transfect the plasmid to express your mRNA of interest with 24×MS2 inserted in the 3′-UTR. In our case we transfect HSP70-MBSV7 using 2 ×g of DNA and 4 ×L jetPRIME in a final transfection volume of 250 ×L (see Note 8 on transient transfection).
Thirty-six hours after transfection, heat-shock the cells at 42 °C for 1 h and let them recover for 3 h at 37 °C.
3.5. Live Imaging
3.5.1. Setting Up the Microscope and Imaging
Choosing the microscope . We use widefield fluorescence microscopy to image single mRNA molecules, but other imaging techniques can be used (see Note 9 on microscopes). The microscope is equipped with a 150× 1.45 NA oil immersion objective (Olympus) and fluorescence is recorded on an EMCCD camera. The cells are kept at 37 °C with a stage-top incubator. We use MetaMorph software to automate acquisition and for device control.
Turn on the microscope system, heater and laser (491-nm laser) at least 2 h before imaging.
Set exposure time to 50 ms. Longer times lead to blurring images due to the high motility of the mRNAs in the cell cytoplasm. Set the camera settings to the maximum gain.
Set time intervals taking into consideration the temporal resolution required for the experiment, photobleaching, and phototoxicity. Highly demanding experiments like single particle tracking and translation dynamics require high-frequency image acquisition during short periods of time. To evaluate the performance of MS2V6 and MSV7 we imaged every 20 s for 20 min (total of 427 images).
Set the Z-stack acquisition to cover the cell volume. We took seven stacks of 0.4 ×m each (see Note 10 on Z-stacks).
Set the laser power (LP) to obtain the best signal-to-noise ratio without precluding single mRNA molecule detection at the end of the time-lapse experiment or saturating the camera. We used 15% LP (1–2 mW).
Find the cells that provide optimal MCP-GFP expression and mRNA levels for imaging and analysis using a low LP (2%) to avoid photobleaching. Optimal single-molecule imaging with the MS2-MCP system is usually obtained in cells showing low GFP expression in the nucleus and few mRNAs in the cytoplasm. High mRNA concentration in the cytoplasm reduces single-molecule detection . Set the multistage position and change the LP to one optimal for imaging.
Start imaging acquisition (Fig. 2a).
3.5.2. Detection of Single mRNAs
The analysis software of choice has to enable the detection of single RNA molecules and provide information on their localization and brightness. There is commercial software, like Imaris Image Analysis Software for mRNA detection and tracking, or custom developed software, like AIRLOCALIZE which is used in this protocol (see Note 11 on AIRLOCALIZE [13]).
Install AIRLOCALIZE and open the interface. Select 3D single images from a movie.
Select detection/quantification parameters. A window to select the parameters opens and enables to modify them so they match the characteristics of the equipment and quality of the images. There are options to select an ROI for the region of interest, the units for the threshold value (intensity or absolute), the quantification method and the background correction.
Select an image from all the images of the time lapse experiment to define the characteristics of a single mRNA. Select a signal that belongs to a single mRNA with the cursor, apply a local Gaussian fit, and record the fit result. Repeat this selection with the most distinct fluorescent signals in each Z-section of the image.
Once done, a window with the detected mRNAs in yellow will appear. Each yellow signal indicates an mRNA which brightness is above the detected threshold. To determine if the detection is correct, open the Output image file in Fiji.
A text document containing five columns is automatically saved in the image folder. Each row has the information for a single mRNA. The first three columns indicate its localization in x, y, and z, the fourth one is the fluorescent intensity and the last one the identity of the cell to which it belongs (Fig. 2b). This information can be used for further analysis. We have used it to compare the intensity of single mRNAs tagged with either the MS2V6-U or the MS2V7-C systems and determine the percentage of molecules that were photobleached by the end of the experiment, (Fig. 2c) and (see Note 12 on MS2 versions).
3.6. Quality Control Analysis of the Tagged mRNAs by Two-Color smFISH
Every time that the MS2-MCP system is used to tag an mRNA (reporter or endogenous), its capacity to accurately report on the mRNA life cycle should be tested. Two possible means by which tagging an mRNA can cause perturbation are (1) altering its expression, localization, and decay, and (2) creating degradation resistant intermediates that contain only the MS2 sequence bound by the MCP. Therefore, we always compare by smFISH the behavior of the mRNA of interest before and after tagging. Once the mRNA has been tagged, a two-color smFISH experiment assesses the integrity of the mRNA and verifies that MS2 degradation intermediates do not accumulate (Fig. 1a). One set of probes should hybridize to the mRNA before the stop codon and the second set of probes should bind the MS2V7 sequence in the 3′-UTR [8] (see Note 13 for quality control experiments). To investigate the proper degradation of MS2V7 in mammalian cells, we created a reporter based on HSP70 mRNA because its half-life decreases from hours to minutes during recovery from stress (see Note 14 for half-life of HSP70 mRNA). This reporter plasmid contains the promoter and 3′-UTR of HSP70 and the SunTag sequence in the coding region and the MS2V7 in the 3′-UTR. Both probes should report on the same mRNA molecule by smFISH.
3.6.1. Single-Molecule FISH
There are different protocols to prepare probes to detect single mRNA molecules [18]. We usually use Stellaris™ FISH probes and purchase them from LGC Biosearch Technologies (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer (https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer)). Stellaris probes are usually a mix of >40 DNA oligos, each of them of 20 nts and labeled with a single fluorophore. Spectrally different labeled probes are used in two-color smFISH experiments. We recommend to order one set of probes labeled with Quasar 670 and the other one with Quasar 570.
Aspirate the culture media and wash cells once with PBSM.
Fix cells for 10 min at room temperature with 4% paraformaldehyde.
Aspirate fixation solution and incubate with quenching buffer for 10 min.
Cells can be either left over night at 4 °C in PBSM or permeabilized for 10 min at 37 °C.
Wash twice with PBSM.
Incubate with prehybridization buffer for 30–60 min at room temperature.
Aspirate well the prehybridization buffer and add 100 μL of hybridization solution to cover the well of the live-cell imaging dish where the cells were seeded. The final concentration of the SunTag and the MS2V7 probes is 150 nM (see Note 15 for choosing of smFISH probes). Incubate at 37 °C between 3 and5 h in a humidified chamber. We usually put the live-cell dishes on a petri dish with a napkin soaked in hybridization solution and we seal it with Parafilm. Incubate the remaining prehybridization solution in the same incubator.
Wash the hybridization mix with 500 μl of prehybridization solution. Incubate with 1 mL of prehybridization solution for 40 min at 37 °C.
Aspirate the prehybridization solution and do two washes of 5 min each with 2×SSC.
Let the samples air dry before mounting with an antifade agent. We use one drop of Prolong gold with DAPI.
Wait for at least 12 h before imaging.
3.6.2. Setting up the Microscope and Imaging
The optimal equipment to image smFISH experiments is a widefield microscope equipped with a digital CCD camera (e.g., we have the ORCA-R2, pixel size is 6.5 μm) and objectives with high NA. Our microscope has a ×60 NA 1.35 and a ×100 NA 1.4 Olympus objectives. Appropriate light sources are a mercury lamp or an LED. We use MetaMorph software to automate acquisition and device control.
Turn on the microscope system and let the light source warm up (see manufacturer specifications) before starting to image.
Set the order of the channels to acquire images. Always start imaging in the far red channel because far red fluorophores are more susceptible to photobleaching. Finish by imaging DAPI. For two-color smFISH experiments we image Quasar 670 (MBSV7 probes), Quasar 570 (SunTag probes) and DAPI.
Set the exposure time. Signal from the smFISH probes is weak and needs long exposure times, between 0.5 and 1 s with 100% of power of the light source. Longer exposure times could lead to increased photobleaching and background, and hence decreased the signal-to-noise ratio. They can also lead to saturation of the camera pixels by bright transcription sites or mRNA aggregates that will preclude quantification purposes. DAPI is imaged with 12% power of the light source and 50 ms exposure time. For all channels we keep the camera gain to 2 (low gain).
Set the Z-stack sections. For analysis purposes, we image 41 Z-stacks at 0.2 μm interval.
Set multiple stage positions using a lower exposure time to avoid photobleaching (i.e., 200 ms).
For two-color experiments that aim to determine colocalization, image three fields of fluorescent microspheres (~200 nm) to enable the correction of chromatic aberration.
3.6.3. Detection of Single mRNAs
Detection of the smFISH signal can be done by any software that fits the diffraction limit signal with a 3D Gaussian fitting algorithm. We choose FISH-quant [14] because of its user friendly interface, reliability and accuracy detecting fluorescent spots and their brightness. It also provides the means to analyze transcription sites, mRNA localization, intensity distribution and colocalization studies. FISH-quant is a free software developed using the MATLAB programming language (Mathworks).
Convert all your images to TIF format using Fiji.
Install FISH-quant V3.
Create folders for images, outlines and results, and choose them in the FISH-quant main window.
Set the experimental parameters, pixel size, objective amplification and NA, excitation, and emission wavelengths.
Open the image, either in Cy3 or Cy5, and create the outline. In FISH-quant_V3, outlines can be also created using CellProfiler. Open the DAPI images and determine the outline of the cell and the nucleus. Save it. Repeat this process for all the images of the experiment.
Use one image to set the filter, detection and fit spots parameters and save the detection settings.
Set batch processing mode to analyze all the images with the same settings. Once the analysis is completed, the detection of the spots can be verified for each image.
Save the FISH-quant results, detection settings, summary: mature mRNA, thresholded spots and results from each image files.
The same outlines should be used to analyze the imaging results obtained with the other wavelength. To this aim the name of each outline file and the name of the image saved within the file have to be changed accordingly (i.e., from w1Cys to w2Cy3).
The file “summary: mature mRNA” contains the information on the number of mRNAs per cell and their localization, nuclear or cytoplasmic. We have plotted the # of mRNAs detected with the SunTag vs the MS2V7 probes for each cell (Fig. 3d). This approach is the easiest to determine the correlation between the coding sequence spots and the MBSV7 spots within single cells. If there are more spots detected with the MS2V7 probes, this will indicate a delay in degradation of the MS2 array. In the case of MSV7 the number of spots detected with the MSV7 probes correlate well with those detected with the SunTag probes indicating that there is not a detectable delay in the degradation of the loops protected by the mRNA (see Note 13 for quality control experiments).
Other more accurate ways can be used to analyze spots colocalization. One of them is the spot colocalization Plugin ComDet in Fiji. For experiments that require an accurate measurement on intermolecular distances, we recommend the superregistration algorithm developed by Eliscovich et al. [19] that has been written for FISH-quant output data.
Acknowledgments
The authors are grateful to Xihua Meng and Lydia Tesfa (Grant P30CA013330) for technical help. Support was provided by National Institutes of Health Grants R01GM057071 to R.H.S. and R21AG055083 to M.V. and by the Swiss National Science Foundation for Fellowships P2GEP3_155692 and P300PA_164717 to E.T.
4 Notes
Integration of the MS2 system. MBSV6 and MBSV7 are integrated in the 3′-UTR of the gene. Their sequences lack STOP codons but have several START codons in all three reading frames. Therefore, if integrated in the 5′-UTR, the reading frame of the tagged mRNA and the sequence of the encoded protein should be taken into consideration.
Check the integrity of the mRNA. The SunTag sequence provides a unique sequence to assess for the integrity of the HSP70 reporter tagged with the MS2V7 by two-color smFISH. If there was a delay in the degradation of the MS2V7-MCP system, the number of single molecules detected with the MS2V7 probes should be higher than those detected by the SunTag probes at the time of degradation.
Lentivirus manipulation. Lentiviral vector production and manipulation requires specific biosafety considerations. The MS2V7 is shorter than the MS2V6 and therefore favors the packaging of long reporter in lentiviral particles.
Fluorescent protein fused to the MCP. The MCP has been tagged with different fluorescent proteins (i.e., GFP, mCherry, or the Halo system). Use of the Halo system requires treating cells with JF dyes [20]. JF676 and 549 have been successfully used to visualize individual mRNAs in long time lapse experiments [15]. Pick the fluorescent label for MCP depending on the experimental design of the multicolor imaging.
Expression of MCP-FP. Most MCP expression systems have an NLS to deplete the cytoplasm from unbound MCP-FP and favor the detection of single mRNA molecules by reducing the background. Likewise, sorting for cells with low levels of GFP favors the visualization of single mRNAs during live cell imaging.
System to tag mRNAs. Most experiments done with the MS2 system in mammalian cells use reporter constructs. The CRISPR/Cas9 system enables to tag endogenous mRNAs and therefore preserves the endogenous mRNA expression context [17].
Cell confluence. Single-cell experiments benefit from 60% confluence cultures because (1) it favors optimal mRNA expression levels (2) it facilitates imaging by providing a good field of view where a cell has an extended and flat cytoplasm and is isolated from other cells expressing different GFP levels and, (3) simplifies analysis by enabling efficient drawing of the cell outlines.
Transient transfection reagents. Most of transient transfection reagents lead to the formation, in some cells, of intracellular artifacts that are auto fluorescent and impair the quality of smFISH experiments.
Microscope to image single mRNAs. We use widefield fluorescence microscopy for both live and fixed experiments. Single mRNA detection is achieved through the combination of the digital camera with a proper pixel size and Quantum Efficiency (QE) and the objective with proper magnification and NA. Other modalities like TIRF [21], to image mRNAs tethered to the membranes, multipoint spinning disk microscopy [22], to image the first round of translation, and structured illumination microscopy, to define the spatial mRNA organization [23], have been successfully used.
Choose the number of Z-stacks. Acquiring high number of Z-stacks increases the photobleaching and reduces the total imaging time during live imaging experiments. Balance the number of Z-stacks, the frequency of acquisition and total experimental time.
AIRLOCALIZE. Use the ROI function to select an area in the image or in the cell with low background and optimal density of mRNAs, where single molecules can be easily distinguished from one another. Check and adjust the detection settings to be optimal for single mRNA detection and background threshold for each image in the time lapse experiment.
Comparison of the new MS2 systems, MS2V6 and MS2V7. The brightness of the mRNAs tagged with the MS2V6-U system is about 75% of that of the ones tagged with the MS2V7-C system. Therefore, MS2V6-tagged mRNAs bleach faster than MS2V7-tagged mRNA, getting undetectable sooner and limiting the capabilities of live imaging experiments. The recycling of the MCP-GFP in the U system favors the degradation of the MS2 cassette in yeast but worsens the analysis of single RNA experiments in mammalian cells.
Quality control of the MS2-MCP signal. Live imaging experiments fully rely on the fluorescent signal collected from the MCP-FP protein, which should report on the mRNA. To assure that the detected signal is not an artifact derived from the binding of the MS2 by the MCP, we perform two-color smFISH experiments. The same mRNA is detected by a set of probes complementary to the CDS and by a set of probes complementary to the MS2 sequence. Merge of both images should show a good overlapping (80–90%) of the signals [10]. Additionally, cells should not have MS2 spots brighter than a single molecule, unless the signal is also obtained with the CDS probes. Single-molecule counting should be similar with both sets of probes (usually the detection efficiency for a smFISH experiment is ~90%). The easiest is to plot these results and fit a linear regression model to obtain the coefficient of determination (R square) and the slope. An R square between 0.8 and 0.9 and a slope that does not favor the MS2 probes usually suggest that the mRNA decay occurs correctly.
HSP70 mRNA reporter. Since the caveats of the previous MS2-MCP system was its slower degradation, we use an HSP70 mRNA reporter to discard such problem with MS2V7. HSP70 mRNA is highly regulated, and it has a robust induction upon heat stress and a fast decay after 2 h of recovery [11].
smFISH probes. Stellaris lyophilized probes are resuspended in TE at a concentration of 25 μM. Prepare aliquots and store at −20 °C. The optimal concentration to use a probe usually ranges between 75 nM and 150 nM but it is determined empirically on concentrations that range between 50 and 250 nM. The higher the probe concentration, the higher the background, especially in the cell nucleus.
References
- 1.Bertrand E, Chartrand P, Schaefer M et al. (1998) Localization of ASH1 mRNA particles in living yeast. Mol Cell 2:437–445 [DOI] [PubMed] [Google Scholar]
- 2.Tutucci E, Livingston NM, Singer RH et al. (2018) Imaging mRNA in vivo, from birth to death. Annu Rev Biophys 47:85–106 [DOI] [PubMed] [Google Scholar]
- 3.Vera M, Biswas J, Senecal A et al. (2016) Single-cell and single-molecule analysis of gene expression regulation. Annu Rev Genet 50:267–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia JF, Parker R (2015) MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system. RNA 21:1393–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia JF, Parker R (2016) Ubiquitous accumulation of 3′ mRNA decay fragments in Saccharomyces cerevisiae mRNAs with chromosomally integrated MS2 arrays. RNA 22:657–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haimovich G, Zabezhinsky D, Haas B et al. (2016) Use of the MS2 aptamer and coat protein for RNA localization in yeast: a response to “MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system”. RNA 22:660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinrich S, Sidler CL, Azzalin CM et al. (2017) Stem-loop RNA labeling can affect nuclear and cytoplasmic mRNA processing. RNA 23:134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tutucci E, Vera M, Biswas J et al. (2018) An improved MS2 system for accurate reporting of the mRNA life cycle. Nat Methods 15:81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu B, Miskolci V, Sato H et al. (2015) Synonymous modification results in high-fidelity gene expression of repetitive protein and nucleotide sequences. Genes Dev 29:876–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tutucci E, Vera M, Singer RH (2018) Single-mRNA detection in living S. cerevisiae using a re-engineered MS2 system. Nat Protoc 13(10):2268–2296 [DOI] [PubMed] [Google Scholar]
- 11.Balakrishnan K, De Maio A (2006) Heat shock protein 70 binds its own messenger ribonucleic acid as part of a gene expression self-limiting mechanism. Cell Stress Chaperones 11:44–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Femino AM, Fay FS, Fogarty K et al. (1998) Visualization of single RNA transcripts in situ. Science 280:585–590 [DOI] [PubMed] [Google Scholar]
- 13.Lionnet T, Czaplinski K, Darzacq X et al. (2011) A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods 8:165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller F, Senecal A, Tantale K et al. (2013) FISH-quant: automatic counting of transcripts in 3D FISF1 images. Nat Methods 10:277–278 [DOI] [PubMed] [Google Scholar]
- 15.Wu B, Eliscovich C, Yoon YJ et al. (2016) Translation dynamics of single mRNAs in live cells and neurons. Science 352:1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanenbaum ME, Gilbert LA, Qi LS et al. (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159:635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pichon X, Bastide A, Safieddine A et al. (2016) Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J Cell Biol 214:769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichon X, Lagha M, Mueller F et al. (2018) A growing toolbox to image gene expression in single cells: sensitive approaches for demanding challenges. Mol Cell 71:468–480 [DOI] [PubMed] [Google Scholar]
- 19.Eliscovich C, Shenoy SM, Singer RH (2017) Imaging mRNA and protein interactions within neurons. Proc Natl Acad Sci U S A 114:E1875–E1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm JB, English BP, Chen J et al. (2015) A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12:244–250, 243 p following 250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katz ZB, Wells AL, Park HY et al. (2012) Beta-actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration. Genes Dev 26:1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halstead JM, Lionnet T, Wilbertz JH et al. (2015) Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347:1367–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adivarahan S, Livingston N, Nicholson B et al. (2018) Spatial organization of single mRNPs at different stages of the gene expression pathway. Mol Cell 72:727–738 e725 [DOI] [PMC free article] [PubMed] [Google Scholar]