

SUMMARY:
Quantitative subcellular metabolomic measurements can explain the roles of metabolites in cellular processes, but are subject to multiple confounding factors. We developed Stable Isotope Labeling of Essential nutrients in cell Culture - Subcellular Fractionation (SILEC-SF), which uses isotope labeled internal standard controls that are present throughout fractionation and processing to quantify acyl-Coenzyme A thioesters in subcellular compartments by liquid chromatography-mass spectrometry. We tested SILEC-SF in a range of sample types and examined the compartmentalized responses to oxygen tension, cellular differentiation, and nutrient availability. Application of SILEC-SF to the challenging analysis of the nuclear compartment revealed a nuclear acyl-CoA profile distinct from that of the cytosol, with notable nuclear enrichment of propionyl-CoA. Using isotope tracing we identified the branched chain amino acid isoleucine as a major metabolic source of nuclear propionyl-CoA and histone propionylation, thus revealing a new mechanism of crosstalk between metabolism and the epigenome.
Keywords: Subcellular, metabolomics, acyl-CoA, internal standard, matrix effects, nucleus, mitochondria, branched chain amino acids, isoleucine, propionylation, histone
eTOC Blurb
Trefely et al., developed and applied a technique termed SILEC-SF to measure acyl-coenzyme A thioesters in subcellular compartments by liquid chromatography-mass spectrometry. SILEC-SF was applied to different cell and tissue types to examine the compartmentalized responses across the cytosol, mitochondria, and nucleus to oxygen tension, cellular differentiation, and nutrient availability
Graphical Abstract
INTRODUCTION:
Quantitative measurements of metabolites can yield crucial insights into their roles in cellular processes and nutrient sensing mechanisms. However, metabolism is highly compartmentalized within cells, and the subcellular distribution of metabolites determines their use (Boon et al., 2020; Trefely et al., 2020). Whole cell analyses often provide insufficient information on compartment-specific metabolism, particularly when the compartment of interest accounts for a small fraction of the whole cell quantity of a given metabolite. Indeed, recent advances in fractionation methodologies for compartment-specific metabolite quantitation by mass spectrometry have yielded important biological insights (Chen et al., 2017; Wang et al., 2021; Wyant et al., 2017). Yet, particular challenges in measuring metabolites in subcellular compartments remain, including that subcellular fractionation is inherently disruptive and metabolic analysis after fractionation is subject to confounding factors (Dietz, 2017; Lu et al., 2017; Trefely et al., 2019).
Acyl-Coenzyme A thioesters (acyl-CoAs) are a family of metabolites that are involved in diverse metabolic pathways across subcellular compartments (Trefely et al., 2020). Acetyl-CoA, a prototypical acyl-CoA, is the product of several catabolic processes in the mitochondria, whilst in the cytosol it is the primary substrate for anabolic processes including de-novo fatty acid and cholesterol synthesis. Acetyl-CoA is the acyl-donor for acetylation in all compartments. In the nucleus, acetyl-CoA is a substrate for epigenetic regulation via histone acetylation(Sivanand et al., 2018).
Metabolite availability in the nucleus can link environmental cues with nuclear gene regulation via nutrient-sensitive chromatin modifications (Campbell and Wellen, 2018; Dai et al.; Ryan et al., 2019). The nucleus and cytosol have been generally considered a continuous metabolic compartment since the nuclear pores are permeable to small molecule metabolites. Nevertheless, whole cell acetyl-CoA abundance and histone acetylation can be uncoupled (e.g., in cells deficient in the acetyl-CoA generating enzyme ATP citrate lyase (ACLY)(Zhao et al., 2016)), and recent studies have provided evidence of distinct regulation of acetyl-CoA production within the nucleus (Li et al., 2017; Mews et al., 2017; Sivanand et al., 2017, 2018; Sun et al., 2019; Sutendra et al., 2014). Other histone modifications including succinylation, malonylation, propionylation, crotonoylation, and butyrylation are correlated with the cellular abundance of their respective acyl-CoAs (Simithy et al., 2017), but the subcellular distribution and regulation of these metabolites, as well as their metabolic sources within the nucleus, remain poorly understood. Rigorous mass spectrometry-based methodologies for determination of nuclear metabolite pools have not been reported, though such approaches are needed to advance mechanistic understanding of the links between metabolism and chromatin modification.
We developed Stable Isotope Labeling of Essential nutrients in cell Culture - Subcellular Fractionation (SILEC-SF) to measure acyl-CoAs in subcellular compartments. This approach builds on the SILEC approach using 15N113C3–vitamin B5 (VB5, pantothenate) to generate cell lines highly enriched for 15N1 13C3 –labeled acyl-CoAs (Basu et al., 2011; Snyder et al., 2014). SILEC-SF applies SILEC-labeled cells as rigorous internal standard controls, which are introduced prior to fractionation. Thus, in addition to correcting for a range of factors in analysis by liquid chromatography-mass spectrometry (LC-MS) including variability in processing, analyte loss, extraction inefficiency, and ion suppression (Ciccimaro and Blair, 2010; Mann, 2006; Ong, 2012), SILEC-SF can also account for metabolic disruptions and sample losses during the fractionation process. We applied SILEC-SF to the analysis of mitochondrial, cytosolic, and nuclear metabolites and reveal distinct metabolic regulation within each these compartments.
DESIGN:
SILEC-SF uses whole cell internal standards to control for sample processing
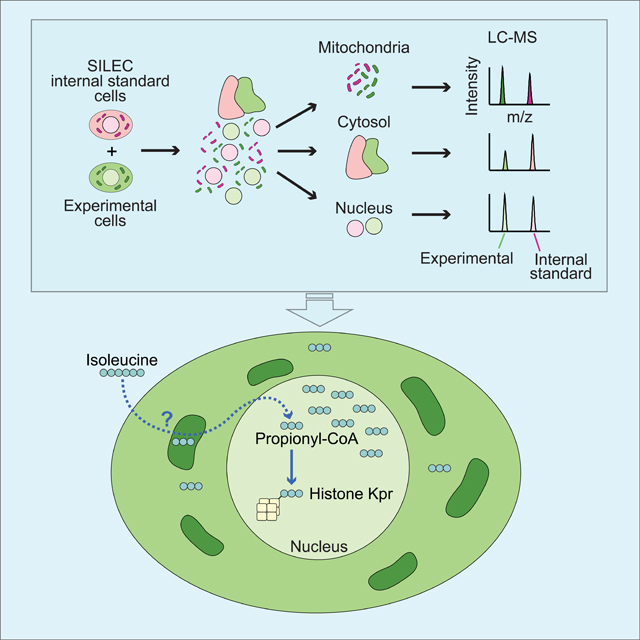
We hypothesized that inclusion of internal standards throughout fractionation and processing would enable accurate relative quantification of metabolites in subcellular compartments. SILEC labeling with 15N113C3-VB5 results in isotope label incorporation into CoA, which can be detected by LC-MS (Figure 1A)(Basu et al., 2011; Frey et al., 2016; Snyder et al., 2014). Since this allows generation of 15N113C3 labeled internal standards at >99% efficiency within living cells (Basu et al., 2011; Snyder et al., 2015), we designed SILEC-subcellular fractionation (SILEC-SF) to achieve relative acyl-CoA quantitation in subcellular compartments (Figure 1B). For this, SILEC cells are harvested in fractionation buffer and added in equal quantity to each experimental sample before fractionation to separate subcellular compartments. This strategy ensures the internal standard is present as whole cells before any processing for fractionation occurs, as well as in every relevant compartment after fractionation. Each fraction is then extracted and analyzed by LC-MS and the ratio of light (acyl-CoA molecules from experimental cells) to heavy (SILEC isotope labeled internal standard molecules) signal intensity is used to determine the relative quantities within each subcellular compartment across different experimental samples. Co-elution and simultaneous analysis of the analyte with the 15N113C3 labeled internal standard improves quantitative performance (Frey et al., 2016). This approach is complementary to, and can be adapted for use with a variety of fractionation approaches including differential centrifugation and immunoprecipitation of organelles. Thus, SILEC-SF applies internal standards as living whole cells to enable rigorous metabolite quantification in subcellular compartments.
Figure 1: SILEC-SF uses whole cell internal standards to quantify acyl-CoAs in subcellular compartments.

A) 15N113C3-isotope labeled vitamin B5 (VB5) is incorporated into the coenzyme A (CoA) moiety such that acyl-CoAs across all acyl (R group) species are isotope labeled. This can be detected after fragmentation (MS2), which results in neutral loss of an unlabeled fragment (NL 507). B) SILEC-SF workflow: internal standards were generated through isotope labeling, internal standards were added to samples as whole cells prior to cell lysis and separation of subcellular compartments by fractionation. The analyte and internal standard in each fraction were analyzed simultaneously by LC-MS and relative quantities are determined between samples.
SILEC-SF is calibrated using standard curves specific to each fraction
We generated separate standard curves for each fraction to quantify metabolites recovered within each fraction. Standard curves were generated by fractionation of additional aliquots of SILEC internal standard cells in parallel with experimental samples, followed by addition of known quantities of unlabeled standards (Figure S1A & S1B). Curves were strikingly different between subcellular fractions (Figure S1C). These distinct standard curves likely reflect several factors that vary across different fractions including the relative enrichment of specific acyl-CoA species (i.e. different quantities of SILEC internal standard), extraction efficiency, and matrix effects (Ciccimaro and Blair, 2010).
We next assessed the impact of matrix effects. Since the matrix is defined as all the components of the sample except the analyte (Guilbault and Hjelm, 1989), and those components are unique to each fraction, we reasoned that the effects of the matrix might be different across fractions. To examine this directly, we measured the impact of different subcellular matrices on raw signal intensity. SILEC subcellular matrices were generated by fractionation of fully labeled SILEC internal standard cells, similar to standard curve generation (Figure S2A, Figure S2B). Unlabeled acyl-CoA standards were added to the SILEC matrix extracts immediately before LC-MS analysis, as opposed to before extraction, to specifically assay the effects of matrix. Signal intensity for unlabeled acyl-CoA standards varied with the addition of different matrices in a manner that was specific to each metabolite (Figure S2C). This demonstrates that matrix-specific effects contribute to standard curve variation and highlight the importance of using matrix matched calibration.
RESULTS:
Distinct acyl-CoA profiles define mitochondria and cytosol in diverse cell and tissue types
SILEC-SF was applied to determine the acyl-CoA distribution in mitochondria and cytosol in diverse cells and tissues including cultured adipocytes, fibroblasts, mouse liver, and human heart (Figure 2A-E). For maximal adaptability across sample types, we employed a classical differential centrifugation protocol for isolation of mitochondria and cytosol simultaneously (Clayton and Shadel, 2014; Frezza et al., 2007), with adaptations for speed and purity (Figure 2A, S2B, S3A-C). SILEC-SF reveals distinctive acyl-CoA profiles defining cytosol and mitochondria. Succinyl-CoA, acetyl-CoA, and CoASH are the most abundant short chain acyl-CoA species on a whole cell level, consistent with data from direct extraction of matching whole cells and tissues in this study and previous studies (Figure S3D)(Bedi et al., 2016; Sadhukhan et al., 2016; Simithy et al., 2017). Notably, CoASH was enriched in the cytosol and succinyl-CoA was generally the dominant acyl-CoA species in the mitochondria, while acetyl-CoA was at similar or greater abundance than succinyl-CoA in the cytosol across these different cell/tissue types (Figure 2B-E). The cellular debris is enriched in high-density cellular material including unbroken cells and nuclei (Figure 2A, S2B, S3A-C). One of the strengths of SILEC-SF is that it can be applied in conjunction with different fractionation strategies. We also adapted SILEC-SF to a mitochondrial immunoprecipitation method (Mito-IP), which offers speed and purity advantages (Chen et al., 2017) (Figure 2F & G), and for which we generated matrix specific standard curves by standard addition (Figure S1B). SILEC-SF by Mito-IP also identified succinyl-CoA as prominent in the mitochondria and acetyl-CoA enriched outside of mitochondria (Figure 2G). Thus, the SILEC-SF method discerns distinct acyl-CoA profiles in mitochondria versus cytosol.
Figure 2: SILEC-SF reveals compartment-specific acyl-CoA profiles.

A) Differential centrifugation method for mitochondria and cytosol isolation. B-E) SILEC-SF acyl-CoA quantitation in whole cell lysate (WCL), mitochondria, cytosol and high-density debris B) Brown adipocytes in cell culture (n=4 replicate samples) C) Acly−/− mouse embryonic fibroblasts (MEFs) were incubated in DMEM supplemented with 10% dialyzed FBS and acetate (1 mM) for 4 h before cell harvest (n=4 replicate samples) D) Mouse liver tissue (n=6 mice) E) Transmural left ventricle of human heart (n=5 replicate samples from a single heart). F) Mito-IP protocol with SILEC-SF G) SILEC-SF using Mito-IP was applied to HepG2 cells incubated in serum free DMEM for 2 h before harvest (n=3 replicate samples). All panels display mean with error bars representing standard deviation. Data for all acyl-CoA species that were quantified in each fraction are displayed. Those that were not quantified showed insufficient signal intensity for the analyte, the internal standard or both. Some metabolites indicated in the legend for C-G were not quantified in the mitochondrial fraction. These were B) HMG-CoA, D) Malonyl-CoA and E) CoASH, G) all except for acetyl-CoA succinyl-CoA and CoASH. (iso)Butyryl-CoA = Butyryl-CoA + Isobutyryl-CoA, (iso)Valeryl-CoA = Valeryl-CoA + Isovaleryl-CoA (isomers are not distinguished in analysis). HMG-CoA = 3-hydroxy-3-methylglutaryl-CoA.
SILEC-SF detects mitochondria-specific response to hypoxia
We next sought to validate the SILEC-SF method using a biologically relevant perturbation that would impact acyl-CoAs in subcellular compartments in a predictable manner. Hypoxia promotes reductive carboxylation of α-ketoglutarate (αKG) in the mitochondria, directing glutamine-derived carbons to citrate production and away from succinyl-CoA production (Metallo et al., 2011) (Figure 3A). Thus, reduced mitochondrial succinyl-CoA was anticipated under hypoxia. We first assessed the effect of hypoxia exposure on acyl-CoAs in whole HepG2 cells with rapid direct extraction. Succinyl-CoA was substantially reduced (∼50%) under hypoxia, whilst acetyl-CoA was minimally impacted (Figure 3C). SILEC-SF analysis revealed that succinyl-CoA was markedly reduced specifically in the mitochondrial compartment in hypoxia (Figure 3B, D). In contrast, cytosolic succinyl-CoA was only modestly impacted by hypoxia, indicating an independently regulated metabolic pool (Figure 3D). Mitochondrial acetyl-CoA was also markedly reduced by hypoxia despite a minimal change in whole cell pools, consistent with the majority of cellular acetyl-CoA being cytosolic and demonstrating that subcellular analysis can uncover metabolic changes in compartments that contain smaller pools of a given metabolite. We also assessed compartmentalized abundance of a panel of short chain acyl-CoAs (log transformed data Figure 3E, untransformed data Figure S4A), finding that hypoxia resulted in reduced abundance of multiple acyl-CoAs in mitochondria but not in cytosol. These data demonstrate that SILEC-SF detects compartment-specific changes in acyl-CoA abundance in response to biological perturbation.
Figure 3: SILEC-SF detects distinct mitochondrial response to hypoxia.

HepG2 cells were incubated under 20% (normoxia) or 1% (hypoxia) oxygen. A) Schematic comparing glutamine metabolism in the TCA cycle under hypoxic and normoxic conditions. B) SILEC-SF involves introduction of internal standard before fractionation. C) Whole cell analysis after direct extraction of metabolites. D) Succinyl-CoA and acetyl-CoA quantitation by SILEC-SF using mitochondrial/cytosolic differential centrifugation procedure from a representative experiment (one of the replicate experiments incorporated in Figure 5C, Figure 3E, and Figure 5E). Symbols indicate individual replicate samples (n=4). E) Fold-change (hypoxia/normoxia) from mean quantitation for 3 independent SILEC-SF experiments conducted on separate days. Symbols indicate the mean from each experiment. F) Schematic for conventional addition of internal standard post-fractionation. G) Fold-change from mean quantitation for 3 independent conventional experiments conducted on separate days. Symbols indicate the mean from each experiment. Abbreviations: BH(I)B-CoA = 3-Hydroxybutyryl-CoA + 3-Hydroxyisobutyryl-CoA, (iso)Butyryl-CoA = Butyryl-CoA + Isobutyryl-CoA (isomers are not distinguished in analysis). For all graphs, error bars represent standard deviation. Statistical comparisons were made by two-tailed Student’s t-test with Welch’s correction and statistical significance was defined as p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
SILEC-SF improves on internal standard addition after fractionation
To further validate the method, we next tested the extent to which SILEC-SF improves on metabolite quantification over conventional addition of internal standards after fractionation, and accounts for sample loss and metabolic activity during processing. For these tests, we leveraged the compartmentalized response to hypoxia. First, to assess the effectiveness of SILEC-SF compared to conventional addition of internal standard after fractionation (Figure 3B versus Figure 3F), data for each method were compared across 3 separate experiments carried out on separate days. Although the conventional method reproduced the core result of mitochondrial-specific succinyl-CoA reduction under hypoxia, the variability was greater and data on other lower abundance acyl-CoAs was inconsistent between experiments (Figure 3G). Therefore, SILEC-SF improves quantification over conventional addition of internal standard after fractionation.
We next asked whether SILEC-SF accounts for metabolic activity during processing. We previously demonstrated that post-harvest metabolism occurs during fractionation by assessing the incorporation of isotope labeled substrates introduced during fractionation into metabolic products (Trefely et al 2019). We applied post-harvest labeling to assess the extent to which SILEC internal standards account for post-harvest metabolism (Figure S5). 13C5-glutamine was used since it is a substrate for succinyl-CoA generation. 13C5-glutamine was incorporated as an additional 13C4-label into succinyl-CoA M4 (unlabeled CoA derived from experimental cells) and SILEC succinyl-CoA M4 (heavy CoA derived from internal standard cells). Differences in post-harvest metabolism were apparent between fractions (5–11% in mitochondria and debris versus <1% in cytosol), but were comparable within each fraction in the experimental and internal standard molecules. This indicates that SILEC internal standards undergo compartment-specific post-harvest metabolic changes mirroring those in experimental cells, and can thus account for these changes by retaining the differences between internal standard and experimental analyte across samples. Together these data indicate that SILEC-SF improves subcellular acyl-CoA quantitation in part by accounting for post-harvest metabolism.
Cytosolic HMG-CoA is exquisitely sensitive to acetate supply
Having validated that SILEC-SF can measure acyl-CoA abundance changes within mitochondria, we examined the detection of changes in cytosolic acyl-CoA pools. We leveraged two models of ATP-citrate lyase (ACLY) deficiency, Acly-/- mouse embryonic fibroblasts (MEFs) (Zhao et al., 2016) and Acly-/- murine liver cancer cells, which both rely on acetate to supply cytosolic acetyl-CoA via Acyl-CoA Synthetase Short Chain Family Member 2 (ACSS2): (Figure S6A). Within the cytosol, acetyl-CoA is used for generation of malonyl-CoA for fatty acid synthesis and HMG-CoA in the mevalonate pathway for synthesis of sterols (Figure 4A). We first investigated the relationship between acetate dose and acyl-CoA abundance in whole cells using direct rapid extraction of metabolites. Cells were incubated in 0, 0.1, or 1 mM acetate, and the dose response was compared across 8 short chain acyl-CoA species quantified (Figure 4B, Figure S6B-D). This analysis revealed that HMG-CoA is severely depleted upon acetate withdrawal and increases 10–40 fold with increasing acetate supplementation in Acly-/- cells (Figure 4C). Acetyl-CoA was also sensitive to exogenous acetate, but the concentration changed by only 2–3 fold in the presence or absence of acetate (Figure 4C). In contrast, malonyl-CoA was not depleted upon acetate withdrawal, even though it is also produced from cytosolic acetyl-CoA (Figure 4B, Figure S6B-D). Acyl-CoAs in control (Aclyf/f) cells were largely unaffected by acetate supplementation (Figure 4B-C, Figure S6D).
Figure 4: Cytosolic HMG-CoA is a sensitive readout of cytosolic acetate supply.

A) Acetate supplies cytosolic acetyl-CoA through upregulation of ACSS2 in ACLY deficient cells. B-D) Cells were incubated in DMEM supplemented with 10% dialyzed fetal calf serum and indicated acetate concentrations for 4 h. Whole cell acyl-CoA concentrations were determined in ACLY deficient mouse embryonic fibroblasts (Acly-/- MEFs) and liver cancer cell line (Acly-/- liver cancer cells) as well as matched ACLY sufficient control (Aclyf/f liver cancer). B) Whole cell direct extraction fold-change analysis. C) Whole cell direct extraction (data from B) displayed as cellular concentration for acetyl-CoA and HMG-CoA). D) SILEC-SF using mitochondrial/cytosolic differential centrifugation procedure. For all panels, symbols represent individual replicate samples from representative experiments, error bars display standard deviation and statistical comparisons between two groups were made by two-tailed Student’s t-test with Welch’s correction and statistical significance defined as p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
We predicted that the dose-responsive change in HMG-CoA abundance occurs specifically in the cytosol, since our prior tracing analyses demonstrated a close kinetic relationship between cytosolic acetyl-CoA and HMG-CoA labeling from acetate (Trefely et al., 2019). We performed SILEC-SF to compare acyl-CoAs in the cytosolic and mitochondrial compartments in cells cultured in low versus high concentrations of acetate (Figure 4D). Importantly, the direct extraction and SILEC-SF whole cell data reported consistent trends (Figure S6B & C, Figure S7A & B). Acetyl-CoA was not significantly different between low and high acetate in either mitochondrial or cytosolic compartments (Figure 4D). HMG-CoA, however, responded to acetate dose specifically in the cytosolic and not in the mitochondrial compartment of Acly-/- cells (Figure 4D). The significant response of cytosolic HMG-CoA to acetate dose was unique amongst a panel of 6 to 7 short chain acyl-CoA species quantified in the cytosol of Acly-/- MEF and liver cancer cells (Figure S7A & B). Together, the data indicate that SILEC-SF can report on perturbations to cytosolic acyl-CoA pools and show that in the absence of ACLY, cytosolic HMG-CoA is highly sensitive to and dependent on exogenous acetate availability.
SILEC-SF reveals distinct nuclear acyl-CoA profiles, including enrichment of propionyl-CoA
We next applied SILEC-SF to the quantitation of acyl-CoAs within the nucleus, with the goals of understanding if the nucleus is metabolically distinct from the cytosol, and if this approach can be used to gain novel insights into links between metabolism and chromatin modification. Nuclear metabolite quantitation is a particular challenge due in part to large nuclear pores through which small molecules can diffuse. We reasoned that this leak could be accounted for by parallel changes in internal standard nuclei in the SILEC-SF approach.
We applied SILEC-SF using a rapid differential centrifugation protocol to separate nuclei from non-nuclear fractions (Figure 5A). We first applied this to quantitation of nuclear-specific acetyl-CoA in an adipocyte differentiation model, since histone acetylation and whole cell acetyl-CoA abundance increase markedly during this process (Fernandez et al., 2019; Wellen et al., 2009). A 5-fold increase in nuclear acetyl-CoA was measured upon differentiation induction (day 3 versus day 0), correlating with histone acetylation (Figure 5B). Perhaps unsurprisingly, acetyl-CoA increased during differentiation not only in the nucleus, but also in whole cell lysate and the non-nuclear fraction (Figure 5B). Thus, SILEC-SF applied to adipocyte differentiation demonstrated the correlation between nuclear acetyl-CoA abundance and histone acetylation, but did not allow for discernment of whether nuclear acyl-CoA profiles are distinct from that in other compartments.
Figure 5: SILEC-SF identifies enrichment of propionyl-CoA in the nucleus.

A) Differential centrifugation method for nuclear isolation. B) Preadipocytes (5A) were harvested at day 0 and day 3 following induction of differentiation. Western blots of non-nuclear fraction (left panel) and acid extracted histones (middle panel) and representative SILEC-SF analysis of acetyl-CoA (right panel). C,D) HepG2 cells were incubated under 20% (normoxia) or 1% (hypoxia) oxygen. Mean values and error for n=4 replicate samples from a representative experiment are displayed. C) Short chain acyl-CoA species quantified after SILEC-SF by mitochondrial/cytosolic differential centrifugation procedure (one of the replicate experiments incorporated in Figure 3C, Figure 3D and Figure 5E). Those that were not quantified showed insufficient signal intensity for the analyte, the internal standard or both. D) Short chain acyl-CoA species quantified after SILEC-SF using the nuclear differential centrifugation procedure (one of the replicate experiments also incorporated into Figure 5E). Some metabolites indicated in the legend for C and D were not quantified in all fractions, specifically, crotonoyl-CoA was quantified only in non-nuclear fraction, and glutaryl-CoA, (iso)butyryl-CoA, and HMG-CoA were not quantified in the nuclear fraction. E) Fold-change (propionyl-CoA/acetyl-CoA) calculated from n=3 independent experiments conducted on separate days. Symbols indicate mean from each experiment. For all graphs, error bars show standard deviation.
To further investigate the potential for nuclear-specific acyl-CoA regulation, we applied SILEC-SF to nuclear acyl-CoA quantification in HepG2 cells with hypoxia (Figure 5D, Figure S8, Figure S4B). We first confirmed that whole cell lysate data from the nuclear and mitochondrial fractionation protocols were consistent, indicating the validity of comparisons between nuclear acyl-CoA pools and those in other compartments across these experiments (Figure 5C-D). Remarkably, comparison of acyl-CoA quantification across different fractions revealed that the nuclear metabolite profile was distinct from whole cells and other compartments including the cytosol (Figure 5C-D). Notably, propionyl-CoA was substantially enriched in the nucleus compared to the cytosol, highlighted by examination of acetyl-CoA: propionyl-CoA ratio across subcellular fractions (Figure 5E). Acetyl-CoA and propionyl-CoA are approximately equimolar in the nucleus, whereas in the whole cell lysate, cytosolic and non-nuclear fractions, acetyl-CoA is more abundant. The non-nuclear acyl-CoA profile closely reflected that of the whole cell lysate, indicating that the nuclear acyl-CoA pool did not contribute substantially to the whole cell acyl-CoA abundance (Figure 5D, Figure S8). The wash fraction also reflected the non-nuclear and whole cell lysate (Figure 5D, Figure S8), suggesting that the wash was necessary to remove residual non-nuclear material from the purified nuclear pellet. We ruled out artifactual release of propionate from chromatin during processing as an explanation for high nuclear propionyl-CoA by including deacetylase inhibitors in fractionation buffer (Figure S9). Mitochondrial contamination was not indicated since the profile of acyl-CoAs in the mitochondrial fraction and the nuclear fraction were distinct (Figure 5C vs Figure 5D). Thus, SILEC-SF reveals reproducible nuclear acyl-CoA profiles with a unique enrichment for propionyl-CoA within the nucleus.
Isoleucine catabolism contributes to nuclear propionyl-CoA generation and histone lysine propionylation (Kpr) marks
We next addressed the metabolic origin of nuclear propionyl-CoA. Pathways for endogenous propionyl-CoA generation are annotated to the mitochondria, where its metabolic fate is to enter the TCA cycle via succinyl-CoA (Figure 6A), although it may also leave the mitochondria via the carnitine shuttle (Trefely et al., 2020). We first assessed the substrate contribution to total cellular propionyl-CoA by stable isotope tracing of a panel of uniformly 13C-labeled substrates followed by direct extraction of whole cells. Strikingly, isoleucine labeling into propionyl-CoA M3 contributed ∼30 – 50% of the propionyl-CoA pool across three cancer cell lines in the absence of propionate (Figure 6B). This accounts for the majority of cellular propionyl-CoA since the tracer was 50% diluted with unlabeled isoleucine. In contrast, the contribution of isoleucine to the cellular acetyl-CoA pools was <1%, and to succinyl-CoA pools varied across cell lines (<1% to 15%) (Figure S10). This reflects that other major carbon sources such as glucose and glutamine feed acetyl-CoA and succinyl-CoA pools, in contrast to propionyl-CoA (Figure 6A, Figure S10). When supplemented as a positive control, propionate also fed into propionyl-CoA pools in all 3 cell lines (Figure 6B). Since isoleucine was the dominant substrate for propionyl-CoA generation in the absence of supraphysiological propionate, we tested the potential for isoleucine to contribute to nuclear propionyl-CoA using subcellular kinetic analysis with isotope post-labeling to correct for post-harvest metabolic activity (Trefely et al., 2019). Nuclear propionyl-CoA was labeled ∼20% by 1:1 13C6-isoleucine: unlabelled isoleucine in KPC cells, indicating that the propionyl-CoA pool in the nucleus can be substantially derived from isoleucine (Figure 6C). Thus, nuclear propionyl-CoA pools are derived, at least in part, from BCAA catabolism.
Figure 6: Isoleucine is a major substrate for nuclear propionyl-CoA generation and histone lysine propionylation.

A) Propionyl-CoA is generated in the mitochondria from multiple sources including isoleucine and valine. 3 of the 6 carbons in isoleucine contribute to the acyl group of propionyl-CoA. B) Incorporation of various substrates into propionyl-CoA M3 was compared in whole cells by direct extraction of HepG2, HeLa and pancreatic adenocarcinoma (KPC) cells incubated in serum free media containing uniformly (U) 13C-labeled substrates for 18 h. C) Incorporation of 13C6-isoleucine into propionyl-CoA M3 in KPC cells incubated in serum free media containing 13C6-isoleucine diluted 1:1 with unlabeled isoleucine for the indicated times with post-labeling to account for post-harvest metabolism. D) Histone lysine propionylation reaction catalyzed by histone acyl transferase (HAT) enzymes. E) 13C6-isoleucine incorporation into 13C3-Kpr marks determined by histone acyl proteomic analysis of HepG2 cells incubated in DMEM supplemented with 10% dialyzed FBS in which unlabelled isoleucine was entirely replaced for the indicated times. For all experiments, n=3 replicate samples per condition from a single experiment. Error bars are standard deviation. p < 0.05 (*), p < 0.01 (**).
Since acyl-CoAs are acyl-donors for histone lysine acylation marks, we hypothesized that isoleucine-derived propionyl-CoA in the nucleus could be used a substrate for histone propionylation (Figure 6D). To directly interrogate this, we examined the incorporation of 13C6-isoleucine into histone propionyl-lysine (Kpr) marks. 13C3 label incorporation into Kpr was measured using the 4 best detectable peptides modified with endogenous propionylation (H2AK5pr, H4K16pr, H3K23pr and H2K14pr). Increasing label incorporation was observed in H2AK5pr, H4K16pr and H3K23pr, but not in H2K14pr, in cells incubated in 13C6-isoleucine (Figure 6E), demonstrating the direct contribution of propionate acyl-chains with all three carbons from isoleucine into histone Kpr marks.
Nuclear propionyl-CoA responds to BCAA supply
We hypothesized that BCAA availability would impact both nuclear propionyl-CoA and histone Kpr marks since isoleucine directly supplies carbons for both. To test this, cells were incubated in media lacking isoleucine and valine. Nuclear acyl-CoA analysis revealed a substantial (4–5 fold) decrease in propionyl-CoA after 24 h of deprivation of these amino acids (Figure 7A-B). Propionyl-CoA in the whole cell and non-nuclear fraction mirrored the nuclear trends (Figure 7A). Nuclear acetyl-CoA, in contrast, was only modestly impacted under these conditions (Figure 7A-B). Withdrawal of isoleucine and valine also resulted in strikingly reduced cellular propionylcarnitine (Figure 7C), as well as extrusion of propionylcarnitine into the extracellular media, correlating with nuclear propionyl-CoA (Figure 7D). These data indicate nuclear propionyl-CoA pools are sensitive to the availability of BCAAs and also suggest a plausible pathway for nuclear propionyl-CoA generation via propionylcarnitine.
Figure 7: BCAAs support nuclear propionyl-CoA and histone lysine propionylation HepG2 cells were incubated in DMEM with 10% dialyzed FBS for 24 h and switched to dropout media at the indicated times before harvest.

A) SILEC-SF analysis with nuclear/non-nuclear fractionation. B) SILEC-SF analysis of nuclear propionyl-CoA and acetyl-CoA after 24 h incubation with indicated dropout media. C) Total cellular acylcarnitine. D) Extracellular acylcarnitine in media. E) proteomic analysis of acid extracted histones. Volcano plots compare intensity of specific histone peptide modifications for each time point to t=0 control. Table summarizes histone modification classes and sites that were significantly regulated at any timepoint. Single and double modifications of a peptide that could not be assigned to a specific residue are counted as distinct sites. F) relative intensity over time for 4 Kpr sites identified as significantly regulated in E at any timepoint and for corresponding Kac marks. Statistical significance in E & F was determined by comparison to t=0 control for each mark or metabolite and is indicated above or below the specific timepoint in the color corresponding to the data set. For all experiments, n=3 replicate samples per condition from a single representative experiment, error bars are standard deviation. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
Isoleucine and valine supply controls specific histone lysine propionylation (Kpr) marks
Since nuclear propionyl-CoA was responsive to isoleucine and valine withdrawal, we used this as a model to test the relevance of nuclear propionyl-CoA in regulating histone Kpr. Western blot analysis of acid extracted histones using pan Kpr and lysine acetylation (Kac) antibodies showed no notable change in either modification (Figure S11A). This was surprising given the substantial metabolic shift detected in nuclear propionyl-CoA and raised the possibility that there might be cross reactivity in the Kpr antibodies (Simithy et al., 2017) and/or selective sensitivity in Kpr marks to isoleucine availability.
Acyl proteomic analysis (Sidoli et al., 2019a) was employed to analyze specific histone peptide modifications. A total of 171 distinctly modified histone peptides comprising 10 different chemical modifications across 15 histone peptides were quantified, including 15 distinct histone Kpr sites (Figure 7E, Figure S11B). Kpr marks were proportionally the most significantly regulated of all marks with 4 out of 15 quantified Kpr sites being significantly downregulated, H3K23, H3K18, H3K64 and the H4 peptide from amino acid 4 to 17, which contains four possible modifiable residues (K5, K8, K12 or K16). The progressive loss of Kpr at these sites over a 24 h time-course corresponds to the drop in nuclear propionyl-CoA (Figure 7F). Consistent with the relatively unchanged acetyl-CoA, the degree of change in Kac is small compared to Kpr, as seen by comparing the Kac and Kpr regulation in the 3 sites with significantly downregulated Kac (Figure S12A). Ranking of Kpr marks by average percent intensity gives a semi-quantitative indication of their abundance (Figure S12B). Interestingly, the Kpr sites top ranked by signal intensity are not regulated by isoleucine and valine withdrawal, which is consistent with the negative pan Kpr antibody data. Thus, nuclear propionyl-CoA supply is sensitive to the supply of BCAAs and regulates Kpr abundance at specific histone lysine sites.
Discussion:
We present SILEC-SF, a rigorous isotope dilution approach to subcellular quantitation of acyl-CoAs. This represents a critical advance that enables direct analysis of the mechanistic relationship between acyl-CoA supply and functional outputs of acyl-CoA metabolism including protein acylation and metabolic pathway activity, which are distinct between subcellular compartments.
Quantification of compartmentalized metabolite pools was previously approached by several methods. Genetically encoded fluorescent metabolite sensors can be targeted to specific compartments, and have been applied to a limited set of metabolites (Jaffrey, 2018; Okumoto et al., 2012; Zhang et al., 2018). However, these methods have technical limitations in that they require highly engineered experimental settings, and probes generally do not allow simultaneous monitoring of multiple metabolites. Mass spectrometry (MS) has the advantage of direct and highly multiplexed analyses. Imaging MS has shown utility in measurement of intracellular drug concentration (Lee et al., 2017) as well as endogenous metabolites in extracellular vesicles (Legin et al., 2014; Thomen et al., 2020), and membrane-specific measurements of cholesterol and lipid species (Dueñas et al., 2017; Niehaus et al., 2019) but the processing, sensitivity, and spatial resolution required to achieve robust organelle-specific quantitation for many metabolites is limiting (Bowman et al., 2020). Cellular fractionation can be scaled for appropriate quantitation of a range of biomolecules and organelles in multiple biological systems (Bayraktar et al., 2019; Chen et al., 2016, 2017; Dietz, 2017; Fly et al., 2015; Krueger et al., 2014; Ray et al., 2020; Satori et al., 2012; Wyant et al., 2017; Xiong et al., 2019). As discussed, however, fractionation is disruptive to metabolism and can introduce artifacts (Trefely et al., 2019). The innovation of SILEC-SF mitigates these problems by introducing internal standards as whole cells before fractionation.
By applying SILEC-SF to nuclear analysis, we make the intriguing observation that propionyl-CoA is enriched within the nucleus relative to other compartments. This is compelling because histone lysine propionylation, which uses propionyl-CoA as a substrate, has been identified as a dynamically regulated chromatin modification associated with active gene transcription (Kebede et al., 2017; Lagerwaard et al., 2021; Liu et al., 2009; Simithy et al., 2017), although the abundance and sources of propionyl-CoA in the nucleus are unclear (Trefely et al., 2020). We identify isoleucine as a source of nuclear propionyl-CoA that also feeds into histone lysine propionylation. These findings open up numerous questions for future investigation. Firstly, what are the mechanisms of nuclear propionyl-CoA production? Isoleucine catabolism occurs in the mitochondria and mechanisms for transport of metabolic intermediates in this pathway to the nucleus are unknown. Carnitine acetyltransferase (CrAT) have been implicated in the supply of isoleucine derived propionyl-CoA for cytosolic odd chain fatty acid synthesis (Crown et al., 2015; Green et al., 2015; Wallace et al., 2018), and similar mechanisms could plausibly feed into nuclear propionyl-CoA pools. Consistent with this possibility, we find that propionylcarnitine abundance is highly sensitive to BCAA availability in cells and media. Secondly, how is nuclear enrichment of propionyl-CoA maintained versus the cytosol? Future studies can address whether propionyl-CoA is generated within the nucleus or whether there exist mechanisms that allow for its nuclear enrichment relative to other acyl-CoAs.
Our discovery that Kpr marks at specific histone sites are correlated to nuclear propionyl-CoA, raises the question of how such specificity is achieved. One possibility is that enzymes responsible for propionyl-CoA generation are physically linked to multiprotein complexes targeted to specific histone sites as has been reported for histone succinylation (Wang et al., 2017). Another possibility is that acyl transferase enzymes sensitive to propionyl-CoA concentration (or relative concentration in competition with other acyl-CoA species) preferentially modify specific histone sites. Another question is how these chromatin modifications are linked to cell function and disease, particularly since BCAAs play an important role in a multiple disease processes including cancer and insulin resistance (Neinast et al., 2019; Sivanand and Vander Heiden, 2020; White et al., 2021). Interestingly, recent evidence indicates that isoleucine is a major driver of the adverse metabolic response to dietary BCAAs (Yu et al., 2021). Future investigations will illuminate the roles of isoleucine metabolism in regulating Kpr marks in disease processes.
In summary, SILEC-SF provides a rigorous approach for measurement of subcellular metabolite pools. This approach can be applied to probe compartmentalized metabolic responses, including that within the nucleus. This study illustrates application of this method to elucidate a connection between nutrient metabolism and chromatin modification.
Limitations of the study:
Significant limitations of the approach that should be considered include 1) number of metabolites that can be analyzed and 2) relative versus absolute quantification within compartments. SILEC-SF depends on internal standards that can be labeled within cells from essential nutrients, thus limiting analysis to specific classes of metabolites such as acyl-CoAs. In principle, SILEC-SF has the potential to be applied to other classes of metabolites that can be completely labeled such as nicotinamide adenine dinucleotide (Frederick et al., 2017). Secondly, since incomplete recovery of compartments is inherent in fractionation, SILEC-SF does not at this stage allow for absolute quantification of the concentration of metabolites within distinct compartments. Therefore, we report relative molar amounts of acyl-CoAs recovered within compartments.
It is also important with this approach to carefully consider cell lines and conditions used to generate internal standards, since SILEC-SF accuracy depends on the extent to which internal standards recapitulate artifacts generated by fractionation. Experimental conditions have the potential to change the nature of those artifacts, and in some cases, it may be useful to consider subjecting internal standard cells to the same manipulations as experimental cells. Second, there are limits to matching SILEC cells for tissues since cell lines are required for the generation of SILEC internal standards. Similar limitations have been noted for stable isotope labeling of amino acids in cell culture (SILAC) based protein internal standardization. Although SILAC mice have been generated (Krüger et al., 2008), practitioners still commonly use cell based internal standards (Monetti et al., 2011).
STAR METHODS:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact (Nathaniel W. Snyder, NateWSnyder@temple.edu).
Materials availability
Unique reagents generated in this study are murine liver cancer cell lines Aclyf/f (clone D42) and Acly−/− (clone D42C4), available upon request.
Data and code availability
All raw mass spectrometry data files from histone acyl-proteomic analysis in this study have been submitted to the Chorus repository (https://chorusproject.org/pages/index.html) under project number 1724.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Cells were maintained at 37 °C and 5% CO2 and passaged every 2–3 days at 80% confluence. Refer to Key Resources Table for cell line details. All cells were tested using MycoAlert Mycoplasma Detection Kit (Lonza Cat. #LT07–418) and mycoplasma-free. Specific culture conditions for each cell line used in this study are detailed in Table S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-Tubulin | Sigma | Cat #T16199 |
| ACSS2 | Cell Signaling Technology | Cat #3658, clone D19C66, lot 2 |
| ACLY | Proteintech | Cat #15421–1-AP, lot 00040639 |
| Histone H3 | Abcam | Cat #ab1791 |
| Histone H4 | Millipore | Cat #05–858 |
| FASN | Cell Signaling Technology | Cat #3189, lot 2 |
| Citrate Synthase | Cell Signaling Technology | Cat #14309, clone D7V8B, lot 1 |
| Lamin A/C | Cell Signaling Technology | Cat #2032 |
| OGDH | Proteintech | Cat #1512–1-AP |
| SUCLA2 | Proteintech | Cat #12627–1-AP |
| SUCLG1 | Proteintech | Cat #14923–1-AP |
| Acetylated lysine (pan Kac) | Cell Signaling Technology | Cat # 9441S, lot 14 |
| Pan anti propionyllysine (pan Kpr) | PTM Biolabs | Cat #PTM-201 |
| Anti-HA magnetic beads | Thermo-Fisher Scientific | Cat #88837 |
| Chemicals | ||
| Acetyl coenzyme A lithium salt | Sigma-Aldrich | Cat #A2181 |
| DL-β-Hydroxybutyryl coenzyme A lithium salt | Sigma-Aldrich | Cat #H0261 |
| Butyryl coenzyme A lithium salt hydrate | Sigma-Aldrich | Cat #B1508 |
| Coenzyme A trilithium salt | Sigma-Aldrich | Cat #C3019 |
| 2-Butenoyl coenzyme A lithium salt | Sigma-Aldrich | Cat #C6146 |
| Glutaryl coenzyme A lithium salt | Sigma-Aldrich | Cat #G9510 |
| DL-3-Hydroxy-3-methylglutaryl coenzyme A sodium salt hydrate | Sigma-Aldrich | Cat #H6132 |
| Isovaleryl coenzyme A lithium salt hydrate | Sigma-Aldrich | Cat #I9381 |
| Malonyl coenzyme A tetralithium salt | Sigma-Aldrich | Cat #63410 |
| n-Propionyl coenzyme A lithium salt | Sigma-Aldrich | Cat #P5397 |
| Succinyl coenzyme A sodium salt | Sigma-Aldrich | Cat #S1129 |
| [13C6,15N2]-Vitamin B5 (calcium pantothenate-13C6,15N2]) | Isosciences | Cat #5065 |
| [15N2,13C5]-Glutamine | Cambridge Isotope Laboratories | Cat #CNLM-1275-H-PK |
| [13C6]-Glucose | Cambridge Isotope Laboratories | Cat #CLM-1396–1 |
| [13C5,15N1]-Valine | Cambridge Isotope Laboratories | Cat #CNLM-422, lot PR-22329 |
| [13C6,15N1]-Leucine | Cambridge Isotope Laboratories | Cat #CNLM-281, lot PR-22831 |
| [13C6]-Isoleucine | Cambridge Isotope Laboratories | Cat #CLM-2248, lot PR-21540 |
| [13C3]-Propionate | Cambridge Isotope Laboratories | Cat #CNLM-1865, lot PR30847/09249SP-1 |
| Propionic anhydride-d10 | Sigma-Aldrich | Cat #615692 |
| Nicotinamide | Sigma-Aldrich | Cat #N0636 |
| Trichostatin A | A.G Scientific | Cat #T-1052 |
| Trichloroacetic acid (TCA) | Sigma-Aldrich | Cat #T6399 |
| Sequencing grade trypsin | Promega | Cat #V5111 |
| Experimental Models: Cell Lines | ||
| Acly−/− mouse embryonic fibroblasts (Clone: PC9) | K.E. Wellen Lab (Zhao et al., 2016) | NA |
| HepG2 | ATCC | Cat. #HB-8065 |
| Murine pancreatic adenocarcinoma (KPC, Clone: 2838c3) | B.Z. Stanger Lab (Li et al., 2018) | NA |
| Hepa1c17 | ATCC | Cat. #CRL-2026 |
| Brown preadipocytes | P. Seale Lab (Harms et al., 2014) | NA |
| Murine liver cancer Aclyf/f (Clone: D42) | K.E. Wellen Lab, this paper | NA |
| Murine liver cancer Acly−/− (Clone: D42C4) | K.E. Wellen Lab, this paper | NA |
| Murine preadipocytes (Clone: 5A) | K.E. Wellen Lab (Fernandez et al., 2019) | NA |
| Phoenix-AMPHO | ATCC | Cat. #CRL-3213 |
| Plasmids | ||
| pMXS-3XHA-EGFP-OMP25 | Addgene (Chen et al., 2016) | Cat. #83356 |
| Software and Algorithms | ||
| Tracefinder v4.1 | Thermo Scientific | |
| FluxFix v1.0 | http://www.flufix.science (Trefely et al., 2016) | NA |
| Graphpad PRISM v8 | GraphPad | |
| EpiProfile 2.0 software | https://github.com/zfyuan/EpiProfile2.0_Family (Yuan et al., 2018) | NA |
| Adobe Illustrator software v23.0.1. | Adobe | NA |
Murine liver cancer cell generation
Aclyf/f liver cancer cell line (clone D42) was generated from an Aclyf/f C57Bl/6J mouse (Zhao et al., 2016) that had liver tumors induced by a single-administration of diethylnitrosamine (25 mg/kg) via intraperitoneal injection at 2 weeks of age and high-fructose diet (Tekland TD.89247) starting at 6 weeks of age. Following euthanasia at 9 months of age, a palpable tumor was excised from the liver avoiding surrounding tissue. The tumor was digested using Liver Dissociation Kit (Miltenyi Cat. #130–105-807), and subject to manual dissociation by pipette to form a single cell suspension in DMEM F-12, 10% heat-inactivated FBS, penicillin/streptomycin, and ITS+ Premix Universal Culture Supplement mix (Corning Cat. # 354352). The single cell suspension was seeded onto a collagen-coated 6 cm tissue culture dish. Fibroblasts were depleted by differential trypsinization – cells were trypsinized and detached cells were removed by gentle rinsing with PBS and aspiration. The remaining adherent cells were trypsinized and reseeded using a limiting dilution to derive a proliferating clonal population. After clonal expansion, cells were maintained in DMEM/F-12 with standard 10% calf serum and then DMEM 10% calf serum. Acly−/− liver cancer (clone D42C4) cells were generated from Aclyf/f liver cancer cells (clone D42) infected with adenoviral Cre recombinase obtained from the University of Pennsylvania Vector Core. Single cell clonal D42 Acly−/− cell lines were then generated by limiting dilution and ACLY loss was validated by Western blot. Cell lines were generated while cultured in DMEM/F-12 media (Gibco #11320033) supplemented with 10% (v/v) fetal calf serum and penicillin/streptomycin.
Human heart tissue
Heart tissue was derived from a male organ donor with no history of heart failure, diabetes mellitus or obesity. All study procedures were approved by the University of Pennsylvania Hospital Institutional Review Board, and prospective informed consent for research use of heart tissue was obtained from organ donor next-of-kin. The heart received in-situ cold cardioplegia and was placed on wet ice in 4 °C Krebs-Henseleit Buffer. Transmural left ventricular samples, excluding epicardial fat, were cut into 20 mg pieces and each piece was immediately transferred to a pre-chilled 1 ml Potter-Elvehjem Tissue Grinder (Corning cat. #7725T-1) containing SILEC Hepa1c17 cells in buffer (2.7 E7 cells/sample) and subjected to fractionation as described below.
Mouse liver tissue
All animal studies were carried out in accordance with the IACUC guidelines of the University of Pennsylvania. Male C57Bl6 mice fed ad libitum on a chow diet (Laboratory Autoclavable Rodent Diet 5010, LabDiet cat #0001326) were sacrificed at 5 months old by cervical dislocation. The median lobe was removed to a dish on ice and a 15 mg piece cut. After weighing, the piece was immediately transferred to a pre-chilled 1 ml Potter-Elvehjem Tissue Grinder containing SILEC HepG2 cells (∼1E7 cells/sample) in fractionation buffer and subjected to mitochondria and cytosol isolation by differential centrifugation as described below.
Hypoxic treatment
HepG2 cells were incubated under 20% (normoxia) or 1% (hypoxia) oxygen with 5% CO2 and 70% humidity for 24 h before harvest. Media was equilibrated before cell treatment by preincubation in hypoxic or normoxic incubator for 24 h. Cells were serum starved in DMEM (Gibco cat #A1443001) containing 5 mM glucose and 2 mM glutamine under their respective oxygen tensions for 2 h before harvest. Serum starvation was used to avoid potential variability in metabolic response to serum batches, which could vary between experiments.
Isoleucine and valine withdrawal
Cells were washed with PBS and media replaced with withdrawal media. Withdrawal media: DMEM base lacking amino acids, glucose, pyruvic acid and phenol red (Biological Life Science D9800–26) was reconstituted with glucose (4.5 g/L), glutamine (2 mM), and other amino acids except for isoleucine and valine according to DMEM standard formulation (Dulbecco and Freeman, 1959). NaOH and HEPES (25 mM) was added to adjust to pH7.4 and dialyzed fetal bovine serum (Gemini Biosciences Cat. # 100–108) was added to 10% (v/v).
METHOD DETAILS
Whole cell direct extraction
Media was removed by decanting and cell dishes were placed on ice at an angle to drain and aspirate residual media. Cells were extracted by addition of 1 ml 10% trichloroacetic acid (w/v) in water as described previously (Trefely et al., 2019). Internal standard (100 μl internal standard generated in yeast (Snyder et al., 2015)/sample) was added to cell dishes. Cells were scraped into the trichloroacetic acid and transferred to 1.5 ml tubes on ice then either processed directly or stored at −80 °C. Standard curves were generated in parallel by addition of equal aliquots of internal standard to serial dilutions of acyl-CoA standard mixture (Table S2).
Stable isotope tracing in whole cells
For experiments involving 50% diluted isoleucine, valine, or leucine tracer, cells were preincubated in serum-free DMEM (Thermo Fisher Scientific, Gibco #11965084) for 8 hours before media was exchanged for tracing media. Cells were incubated in tracing media at 37 °C and 5% CO2 for 18 hours before whole cell direct extraction. Tracing media was prepared using DMEM base lacking glucose or glutamine (Thermo-Fisher scientific, Gibco cat. #A1443001). Glucose was replaced at (4.5 g/L), and glutamine at (584 mg/L) and additional isoleucine (105 mg/L), valine (94 mg/L) and leucine (105 mg/L) were added. Except for the targeted experimental tracer (U13C, see Key Resouces Table), all additives were unlabeled. Total substrate concentrations were equal across all samples except propionate, which was added only to the U13C-propionate tracing samples at 1 mM.
For experiments with undiluted isoleucine labeling, cells were washed with PBS and tracing media was added before incubation at 37 °C and 5% CO2. Tracing media was prepared from DMEM base lacking Amino Acids, Glucose, Pyruvic Acid and Phenol Red (Biological Life Science D9800–26) reconstituted with glucose (4.5 g/L), glutamine (2 mM), and other unlabeled amino acids according to DMEM standard formulation (Dulbecco and Freeman, 1959) except for isoleucine which was added as 13C6-isoleucine to (105 mg/L). NaOH and HEPES (25 mM) was added to adjust to pH7.4 and dialyzed fetal bovine serum (Gemini Biosciences Cat. # 100–108) was added to 10% (v/v) to avoid potential propionate contamination from undialyzed serum.
Subcellular stable isotope tracing with post-labeling
Murine pancreatic ductal adenocarcinoma (KPC) cells were incubated with U13C-isoleucine tracer before fractionation as described above. Fractionation with post-labeling was carried out as previously described (Trefely et al., 2019) by addition of U13C-isoleucine (525 mg/L) and unlabeled isoleucine (525 mg/L) to fractionation buffer.
SILEC-SF protocol
SILEC cell generation
SILEC labeling was performed in multiple cell lines to match experimental cells. This is achieved through the passaging of cells in 15N113C3-pantothenate (Vitamin B5) for at least 9 passages as previously described (Basu et al., 2011; Frey et al., 2016; Snyder et al., 2015). Labeling of SILEC cells should be >99% to avoid contamination of unlabeled signal. Each batch of charcoal:dextran stripped fetal bovine serum was tested for sufficient labeling efficiency since this can vary from batch to batch (Snyder et al., 2014). The ‘standard 0’ sample, which contained only SILEC cells, was used to validate labeling efficiency in each experiment. SILEC labeling efficiency was calculated after acyl-CoA analysis using formula 1 below.
| (1) |
SILEC cells should be matched as closely as possible to experimental cells to ensure that biochemical fractionation will perform equivalently and that postharvest metabolic capacity is appropriately matched. Cell lines used as SILEC internal standards for tissues should be chosen based on the following criteria: 1) similar properties in fractionation 2) amenable to cell culture and SILEC labeling (fast growing cell lines allow fast approach to complete SILEC labeling and sufficient material to match tissue). For SILEC-SF experiments in cultured cell lines, the same cell line was used to generate SILEC internal standards.
SILEC media was prepared by the addition 15N113C3-pantothenate (Isosciences) (1 mg/L) to custom pantothenate free DMEM (Gibco, Thermo Fisher Scientific) containing 25 mM glucose and 4 mM glutamine with all other components at concentrations according to the standard formulation (Dulbecco and Freeman, 1959). Charcoal:dextran stripped fetal bovine serum (Gemini Biosciences cat. #100–199) was added up to 10% (v/v).
Mitochondrial/cytosolic differential centrifugation protocol
Mitochondrial and cytosolic fractions were isolated by classical differential centrifugation protocol (Clayton and Shadel, 2014; Frezza et al., 2007), with adaptations for speed and purity (illustrated in Figure 2A). SILEC cells matched to the experimental cell type were harvested before experimental cells. To reduce potential for interference from the small fraction of unlabeled acyl-CoA in SILEC cells, and the expense of each experiment, SILEC internal standards were added at lower abundance than experimental cells but maintained within the same order of magnitude for analytical robustness. Thus, one third to one half the amount of SILEC cells were used for each experimental cell sample (n). Experimental cell numbers used per sample in this study: ∼4E5 brown adipocytes (confluent 10 cm dish), ∼0.6E5 MEFs/sample (80% confluent 15 cm dish), ∼1E7 HepG2 cells/sample (80% confluence 10 cm dish). See Data S1 for step-by step protocol.
Nuclear isolation by differential centrifugation protocol
Nuclear isolation was achieved by detergent assisted hypoosmotic lysis and differential centrifugation (illustrated in Figure 5A). SILEC cells matched to the experimental cell type were harvested before experimental cells. Experimental cell numbers used per sample in this study: ∼0.5E7 KPC cells/sample (80% confluent 10 cm dish), ∼1E7 HepG2 cells/sample (80% confluence 10 cm dish). See Data S1 for step-by step protocol.
Mito-IP protocol
Mitochondrial immunoprecipitation (Mito-IP) was performed as described (Chen et al., 2017) with adjustments to accommodate SILEC internal standard addition and acyl-CoA extraction in trichloroacetic acid. HepG2 cells were infected with retrovirus generated in Pheonix-AMPHO cells transfected with pMXS-3XHA-EGFP-OMP25 construct. Following selection with blasticidin (5 ug/ml) Mito tag expressing cells were sorted into 3 categories (low/no expression, medium and high) based on GFP expression level using a BD FACSJazz cell sorter. Medium expression cells were cultured for further use. SILEC cells were generated by passaging these cells in SILEC media as described above. ∼1E7 HepG2cells/sample (80% confluent 15 cm dish) were used in this study. See Data S1 for step-by step protocol.
Acyl-CoA sample processing and analysis by LC-MS
Samples were thawed and kept on ice throughout processing. Cell and fraction samples in 10% (w/v) trichloroacetic acid in water were sonicated for 12 × 0.5 s pulses, protein was pelleted by centrifugation at 17,000 ×g from 10 min at 4 °C. The supernatant was purified by solid-phase extraction using Oasis HLB 1cc (30 mg) SPE columns (Waters). Columns were washed with 1 mL methanol, equilibrated with 1 mL water, loaded with supernatant, desalted with 1 mL water, and eluted with 1 mL methanol containing 25 mM ammonium acetate. The purified extracts were evaporated to dryness under nitrogen then resuspended in 55 μl 5% (w/v) 5-sulfosalicylic acid in water.
Acyl-CoAs were measured by liquid chromatography-high resolution mass spectrometry. 5–10 μl of purified samples in 5% SSA were analyzed by injection of an Ultimate 3000 Quaternary UHPLC coupled to a Q Exactive Plus (Thermo Scientific) mass spectrometer in positive ESI mode using the settings described previously (Frey et al., 2016). Quantification of acyl-CoAs was via their MS2 fragments and the targeted masses used for isotopologue analysis are indicated in Table S3. Data were integrated using Tracefinder v4.1 (Thermo Scientific) software. Isotopic enrichment in tracing experiments was calculated by normalization to unlabeled control samples using the FluxFix calculator (Trefely et al., 2016).
Histone acid extraction
Histones acid extraction was performed as previously described (Sidoli et al., 2016). For each sample, a 10 cm dish of 80% confluent cells was washed with ice-cold PBS on ice. Cells were scraped in 0.25 ml of NIB-250 buffer (15 mM Tris-HCl (pH 7.5), 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 1 mM DTT, 10 mM sodium butyrate, 1× protease inhibitor cocktail (Sigma-Aldrich P8340)) + 0.1% NP-40. Nuclei were pelleted by centrifugation at 600 ×g for 5 min at 4°C. The pellet was washed twice by addition of 0.25 ml NIB-250 buffer without NP40, centrifugation at 600 ×g, and removal of supernatant. Nuclear pellet was resuspended in 0.6 ml of 0.4N H2SO4 and extraction with rotation at 4°C for 16 h. Centrifugation at 11,000 ×g for 10 min at 4°C sedimented precipitate. The supernatant was taken to a new 1.5 ml tube and histones were precipitated on ice for 16 h by addition of 100% (w/v) trichloroacetic acid to a final concentration of 20%. The histone pellet was sedimented by centrifugation at 11,000 ×g for 10 min at 4°C. Supernatant was removed and the histone pellet was washed with 1 ml acetone with 0.1% 12N HCl and centrifuged at 11,000 ×g for 10 min at 4°C. Pellet was washed again with 1 ml acetone and air dried.
Histone acyl proteomic analysis
Histone digestion
Acid extracted histone pellets were dissolved in 50 mM ammonium bicarbonate, pH 8.0, and histones were subjected to derivatization as previously described (Sidoli et al., 2019b). Briefly, intact histones were mixed with 5 μL of deuterium-labeled propionic anhydride (propionic anhydride-d10) rapidly followed by 14 μL of ammonium hydroxide (Sigma-Aldrich) to adjust to pH 8.0. The mixture was incubated for 15 min and the procedure was repeated. Histones were then digested with 1 μg of sequencing grade trypsin (Promega) diluted in 50 mM ammonium bicarbonate (1:20, enzyme:sample) overnight at room temperature. Derivatization reaction was repeated to derivatize peptide N-termini. The samples were dried in a vacuum centrifuge. Prior to mass spectrometry analysis, samples were desalted using a 96-well plate filter (Orochem) packed with 1 mg of Oasis HLB C-18 resin (Waters). Briefly, the samples were resuspended in 100 μl of 0.1% TFA and loaded onto the HLB resin, which was previously equilibrated using 100 μl of the same buffer. After washing with 100 μl of 0.1% TFA, the samples were eluted with a buffer containing 70 μl of 60% acetonitrile and 0.1% TFA and then dried in a vacuum centrifuge.
LC-MS/MS acquisition
Samples were resuspended in 10 μl of 0.1% TFA and loaded onto a Dionex RSLC Ultimate 3000 (Thermo Scientific), coupled online with an Orbitrap Fusion Lumos (Thermo Scientific). Chromatographic separation was performed with a two-column system, consisting of a C-18 trap cartridge (300 μm ID, 5 mm length) and a picofrit analytical column (75 μm ID, 25 cm length) packed in-house with reversed-phase Repro-Sil Pur C18-AQ 3 μm resin. Histone peptides were separated using a 30 min gradient from 1–30% buffer B (buffer A: 0.1% formic acid, buffer B: 80% acetonitrile + 0.1% formic acid) at a flow rate of 300 nl/min. The mass spectrometer was set to acquire spectra in a data-independent acquisition (DIA) mode using isolation windows as previously described (Sidoli et al., 2015). Briefly, the full MS scan was set to 300–1100 m/z in the orbitrap with a resolution of 120,000 (at 200 m/z) and an AGC target of 5×10e5. MS/MS was performed in the orbitrap with sequential isolation windows of 50 m/z with an AGC target of 2×10e5 and an HCD collision energy of 30.
Histone acyl proteomics LC-MS/MS analysis
Histone peptides raw files were imported into EpiProfile 2.0 software (Yuan et al., 2018). From the extracted ion chromatogram, the area under the curve was obtained and used to estimate the abundance of each peptide. In order to achieve the relative abundance of post-translational modifications (PTMs), the sum of all different modified forms of a histone peptide was considered as 100% and the area of the particular peptide was divided by the total area for that histone peptide in all of its modified forms. The relative ratio of two isobaric forms was estimated by averaging the ratio for each fragment ion with different mass between the two species. The resulting peptide lists generated by EpiProfile were exported to Microsoft Excel and further processed for a detailed analysis. To assess the incorporation rate of the 13C3-Kpr in Kpr marks (in Figure 6E), we performed manual signal extraction using Xcalibur QualBrowser (Thermo) and the area under the curve was used as representative of the peptide abundance.
Western blotting
Western blotting was performed using mini gel tank system (Life Biotechnologies) with 4–12% gradient Bis-Tris gels (NuPage, Invitrogen cat. #NP0335) and 0.45 μm pore size nitrocellulose membranes (BioRad cat. #1620115) or 0.22 μm pore size for histone blots. Membranes were probed with antibody (see Table 1) according to the manufacturer’s instructions. An Odyssey CLx imaging system with Image Studio v2.0.38 software (LI-COR Biosciences) was used to acquire images which were exported as TIFF files then cropped and arranged using Adobe Illustrator software v23.0.1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Standard curve generation for differential centrifugation protocols
Separate standard curves were generated for each sub-cellular fraction in mitochondria/cytosol and nuclear fractionation by differential centrifugation as illustrated in Figure S1A. Step-by step procedure in Data S1.
Standard curve generation for Mito-IP protocol
For Mito-IP, standard curves were generated separately for each experimental group in each fraction by standard addition during acyl-CoA sample processing (illustrated in Figure S1B). Step-by step procedure in Data S1.
Normalization
For cell samples, data (pmol/sample) for all fractions were normalized to cell counts from replicate cell dishes for each condition. Cells were trypsinzed and total cell number and volume was determined by Coulter counter (Coulter) or cell number was determined by haemocytometer. Tissue weight (determined before fractionation at +/− 0.01 mg accuracy) was used to normalize data from tissue samples. Values were calculated as pmol/cell or pmol/mg tissue for each fraction and reported as relative molar abundance.
Graphing and statistical analyses
Graphpad Prism software (v.8) was used for graphing and statistical analysis. Data presented are shown either of mean ± standard deviation or, for curve fits, mean ± 95% confidence intervals. For comparison between two groups, datasets were analyzed by two-tailed Student’s t-test with Welch’s correction and statistical significance defined as p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
Supplementary Material
HIGHLIGHTS.
SILEC-SF allows metabolite quantitation in subcellular compartments by LC-MS.
Acyl-Coenzyme A thioesters from mitochondria, cytosol, and nucleus were measured.
Nuclear profile was distinct from other compartments and enriched in propionyl-CoA.
Isoleucine is a major source of nuclear propionyl-CoA and histone propionylation.
Acknowledgements
NWS was supported by R01GM132261 and P30ES013508. KEW is supported by R01CA228339, R01DK116005, R01CA174761 and R01CA248315. ST was supported by the American Diabetes Association through post-doctoral fellowship 1-18-PDF-144. KH and JB-S were supported by the Austrian Science Fund grants FWF W1226 and FWF P27108. CDL is supported by NIH T32 GM07170. LI was supported by T32 GM-07229 and 2-T32-CA-115299-13. SZ was supported by F99CA222741. SS and SS are supported by the Leukemia Research Foundation (Hollis Brownstein New Investigator Research Grant), AFAR (Sagol Network GerOmic Award), Deerfield (Xseed award), the NIH P30 grant CA01333047, and the shared instrument grant NIH 1 S10 OD030286-01. We thank Dr. Kenneth B Margulies and the Gift of Life organization for allowing the use of human tissue for this project.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Basu SS, Mesaros C, Gelhaus SL, and Blair IA (2011). Stable Isotope Labeling by Essential Nutrients in Cell Culture for Preparation of Labeled Coenzyme A and Its Thioesters. Anal. Chem 83, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar EC, Baudrier L, Özerdem C, Lewis CA, Chan SH, Kunchok T, Abu-Remaileh M, Cangelosi AL, Sabatini DM, Birsoy K, et al. (2019). MITO-Tag Mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl. Acad. Sci 116, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi KC, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, et al. (2016). Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon R, Silveira GG, and Mostoslavsky R. (2020). Nuclear metabolism and the regulation of the epigenome. Nat. Metab 1–14. [DOI] [PubMed]
- Bowman AP, Bogie JFJ, Hendriks JJA, Haidar M, Belov M, Heeren RMA, and Ellis SR (2020). Evaluation of lipid coverage and high spatial resolution MALDI-imaging capabilities of oversampling combined with laser post-ionisation. Anal. Bioanal. Chem 412, 2277–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SL, and Wellen KE (2018). Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 71, 398–408. [DOI] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016). Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, and Sabatini DM (2017). Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc 12, 2215–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccimaro E, and Blair IA (2010). Stable-isotope dilution LC-MS for quantitative biomarker analysis. Bioanalysis 2, 311–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DA, and Shadel GS (2014). Purification of mitochondria by sucrose step density gradient centrifugation. Cold Spring Harb. Protoc 2014, pdb.prot080028. [DOI] [PubMed] [Google Scholar]
- Crown SB, Marze N, and Antoniewicz MR (2015). Catabolism of Branched Chain Amino Acids Contributes Significantly to Synthesis of Odd-Chain and Even-Chain Fatty Acids in 3T3-L1 Adipocytes. PLoS One 10, e0145850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Ramesh V, and Locasale JW The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet [DOI] [PMC free article] [PubMed]
- Dietz K-J (2017). Subcellular Metabolomics: The Choice of Method Depends on the Aim of the Study. J. Exp. Bot 68, 5695–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueñas ME, Essner JJ, and Lee YJ (2017). 3D MALDI Mass Spectrometry Imaging of a Single Cell: Spatial Mapping of Lipids in the Embryonic Development of Zebrafish. Sci. Rep 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulbecco R, and Freeman G. (1959). Plaque production by the polyoma virus. Virology 8, 396–397. [DOI] [PubMed] [Google Scholar]
- Fernandez S, Viola JM, Torres AM, Wallace M, Trefely S, Zhao S, Affronti HC, Gengatharan JM, Guertin DA, Snyder NW, et al. (2019). Adipocyte ACLY Facilitates Dietary Carbohydrate Handling to Maintain Metabolic Homeostasis in Females. Cell Rep. 27, 2772–2784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fly R, Lloyd J, Krueger S, Fernie A, and van der Merwe MJ (2015). Improvements to defi ne mitochondrial metabolomics using nonaqueous fractionation. Methods Mol. Biol 1305, 197–210. [DOI] [PubMed] [Google Scholar]
- Frederick DW, Trefely S, Buas A, Goodspeed J, Singh J, Mesaros C, Baur JA, and Snyder NW (2017). Stable isotope labeling by essential nutrients in cell culture (SILEC) for accurate measurement of nicotinamide adenine dinucleotide metabolism. Analyst 142, 4431–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, and Snyder NW (2016). LC- quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and (13)C-isotopic labeling of acyl-coenzyme A thioesters. Anal. Bioanal. Chem 408, 3651–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L. (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, and Metallo CM (2015). Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol 12, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbault GG, and Hjelm M. (1989). Nomenclature for automated and mechanised analysis (Recommendations 1989). Pure Appl. Chem 61, 1657–1664. [PubMed] [Google Scholar]
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, and Seale P. (2014). Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 19, 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR (2018). RNA-Based Fluorescent Biosensors for Detecting Metabolites in vitro and in Living Cells. In Advances in Pharmacology, (Academic Press Inc.), pp. 187–203. [DOI] [PubMed] [Google Scholar]
- Kebede AF, Nieborak A, Shahidian LZ, Le Gras S, Richter F, Gómez DA, Baltissen MP, Meszaros G, Magliarelli H. de F, Taudt A, et al. (2017). Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol 24, 1048–1056. [DOI] [PubMed] [Google Scholar]
- Krueger S, Steinhauser D, Lisec J, and Giavalisco P. (2014). Analysis of subcellular metabolite distributions within arabidopsis thaliana leaf tissue: A primer for subcellular metabolomics. Methods Mol. Biol 1062, 575–596. [DOI] [PubMed] [Google Scholar]
- Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fässler R, and Mann M. (2008). SILAC Mouse for Quantitative Proteomics Uncovers Kindlin-3 as an Essential Factor for Red Blood Cell Function. Cell 134, 353–364. [DOI] [PubMed] [Google Scholar]
- Lagerwaard B, van der Hoek MD, Hoeks J, Grevendonk L, Nieuwenhuizen AG, Keijer J, and de Boer VCJ (2021). Propionate hampers differentiation and modifies histone propionylation and acetylation in skeletal muscle cells. Mech. Ageing Dev 196. [DOI] [PubMed] [Google Scholar]
- Lee RFS, Riedel T, Escrig S, Maclachlan C, Knott GW, Davey CA, Johnsson K, Meibom A, and Dyson PJ (2017). Differences in cisplatin distribution in sensitive and resistant ovarian cancer cells: A TEM/NanoSIMS study. Metallomics 9, 1413–1420. [DOI] [PubMed] [Google Scholar]
- Legin AA, Schintlmeister A, Jakupec MA, Galanski M, Lichtscheidl I, Wagner M, and Keppler BK (2014). NanoSIMS combined with fluorescence microscopy as a tool for subcellular imaging of isotopically labeled platinum-based anticancer drugs. Chem. Sci 5, 3135–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee J-HH, Li W, Lyu J, Rao G, Zhang X, et al. (2017). Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 66, 684–697.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Lin Y, Darwanto A, Song X, Xu G, and Zhang K. (2009). Identification and Characterization of Propionylation at Histone H3 Lysine 23 in Mammalian Cells. J. Biol. Chem 284, 32288–32295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Su X, Klein MS, Lewis IA, Fiehn O, and Rabinowitz JD (2017). Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem 86, 277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. (2006). Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol [DOI] [PubMed]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017). Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monetti M, Nagaraj N, Sharma K, and Mann M. (2011). Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 8, 655–658. [DOI] [PubMed] [Google Scholar]
- Neinast M, Murashige D, and Arany Z. (2019). Branched Chain Amino Acids. Annu. Rev. Physiol 81, annurev-physiol-020518–114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus M, Soltwisch J, Belov ME, and Dreisewerd K. (2019). Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat. Methods 16, 925–931. [DOI] [PubMed] [Google Scholar]
- Okumoto S, Jones A, and Frommer WB (2012). Quantitative Imaging with Fluorescent Biosensors. Annu. Rev. Plant Biol 63, 663–706. [DOI] [PubMed] [Google Scholar]
- Ong SE (2012). The expanding field of SILAC. Anal. Bioanal. Chem 404, 967–976. [DOI] [PubMed] [Google Scholar]
- Ray GJ, Boydston EA, Shortt E, Wyant GA, Lourido S, Chen WW, and Sabatini DM (2020). A PEROXO-Tag enables rapid isolation of peroxisomes from human cells. BioRxiv 2020.03.10.984948 [DOI] [PMC free article] [PubMed]
- Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O’Neill LA, and Mills EL (2019). Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab 1, 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, et al. (2016). Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci 113, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satori CP, Kostal V, and Arriaga EA (2012). Review on recent advances in the analysis of isolated organelles. Anal. Chim. Acta 753, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Simithy J, Karch KR, Kulej K, and Garcia BA (2015). Low Resolution Data-Independent Acquisition in an LTQ-Orbitrap Allows for Simplified and Fully Untargeted Analysis of Histone Modifications. Anal. Chem 87, 11448–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: From histone extraction to data analysis. J. Vis. Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Lopes M, Lund PJ, Goldman N, Fasolino M, Coradin M, Kulej K, Bhanu NV, Vahedi G, and Garcia BA (2019a). A mass spectrometry-based assay using metabolic labelling to rapidly monitor chromatin accessibility of modified histone proteins. Sci. Rep 9, 13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Trefely S, Garcia BA, and Carrer A. (2019b). Integrated analysis of acetyl-CoA and histone modification via mass spectrometry to investigate metabolically driven acetylation. In Methods in Molecular Biology, (Humana Press Inc.), pp. 125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simithy J, Sidoli S, Yuan Z-F, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, et al. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun 8, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S, and Vander Heiden MG (2020). Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell 37, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, et al. (2017). Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 67, 252–265.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S, Viney I, and Wellen KE (2018). Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci 43, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder NW, Basu SS, Zhou Z, Worth AJ, and Blair IA (2014). Stable isotope dilution liquid chromatography/mass spectrometry analysis of cellular and tissue medium- and long-chain acyl-coenzyme A thioesters. Rapid Commun. Mass Spectrom 28, 1840–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder NW, Tombline G, Worth AJ, Parry RC, Silvers JA, Gillespie KP, Basu SS, Millen J, Goldfarb DS, and Blair IA (2015). Production of stable isotope-labeled acyl-coenzyme A thioesters by yeast stable isotope labeling by essential nutrients in cell culture. Anal. Biochem 474C, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun RC, Dukhande VV, Zhou Z, Young LEA, Emanuelle S, Brainson CF, and Gentry MS (2019). Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 30, 903–916.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson THH, Haromy A, Hashimoto K, Zhang N, Flaim E, and Michelakis EDD (2014). A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 158, 84–97. [DOI] [PubMed] [Google Scholar]
- Thomen A, Najafinobar N, Penen F, Kay E, Upadhyay PP, Li X, Phan NTN, Malmberg P, Klarqvist M, Andersson S, et al. (2020). Subcellular Mass Spectrometry Imaging and Absolute Quantitative Analysis across Organelles. ACS Nano. [DOI] [PMC free article] [PubMed]
- Trefely S, Ashwell P, and Snyder NW (2016). FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefely S, Liu J, Huber K, Doan MT, Jiang H, Singh J, von Krusenstiern E, Bostwick A, Xu P, Bogner-Strauss JG, et al. (2019). Subcellular metabolic pathway kinetics are revealed by correcting for artifactual post harvest metabolism. Mol. Metab 30, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefely S, Lovell CD, Snyder NW, and Wellen KE (2020). Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab 100941. [DOI] [PMC free article] [PubMed]
- Wallace M, Green CR, Roberts LS, Lee YM, McCarville JL, Sanchez-Gurmaches J, Meurs N, Gengatharan JM, Hover JD, Phillips SA, et al. (2018). Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol 14, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee J-H, Li X, et al. (2017). KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature. [DOI] [PMC free article] [PubMed]
- Wang Y, Yen FS, Zhu XG, Timson RC, Weber R, Xing C, Liu Y, Allwein B, Luo H, Yeh H-W, et al. (2021). SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. BioRxiv 2021.09.15.460381. [DOI] [PMC free article] [PubMed]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PJ, McGarrah RW, Herman MA, Bain JR, Shah SH, and Newgard CB (2021). Insulin action, type 2 diabetes, and branched-chain amino acids: a two-way street. Mol. Metab 101261. [DOI] [PMC free article] [PubMed]
- Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, and Sabatini DM (2017). mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 171, 642–654.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, He J, Xie WP, Hinojosa E, Ambati CSR, Putluri N, Kim HE, Zhu MX, and Du G. (2019). Rapid affinity purification of intracellular organelles using a twin strep tag. J. Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Richardson NE, Green CL, Spicer AB, Murphy ME, Flores V, Jang C, Kasza I, Nikodemova M, Wakai MH, et al. (2021). The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cell Metab. [DOI] [PMC free article] [PubMed]
- Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, and Garcia BA (2018). EpiProfile 2.0: A Computational Platform for Processing Epi-Proteomics Mass Spectrometry Data. J. Proteome Res 17, 2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen W, Zhao Y, and Yang Y. (2018). Spatiotemporal Imaging of Cellular Energy Metabolism with Genetically-Encoded Fluorescent Sensors in Brain. Neurosci. Bull 34, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo Y-M, et al. (2016). ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 17, 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw mass spectrometry data files from histone acyl-proteomic analysis in this study have been submitted to the Chorus repository (https://chorusproject.org/pages/index.html) under project number 1724.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.