

Abstract
Efforts to safely and effectively treat acute myeloid leukemia (AML) by targeting a single leukemia associated antigen with chimeric antigen receptor T (CAR T) cells have had limited success, in part due to heterogeneous expression of myeloid antigens. We hypothesized T cells expressing CARs directed to two different AML-associated antigens would eradicate tumors and prevent relapse. For co-transduction with our previously optimized CLL-1 CAR currently in clinical study (NCT04219163), we generated two CARs targeting either CD123 and CD33. We then tested the antitumor activity of T cells expressing each of the three CARs alone or after co-transduction. We analyzed CAR-T cell phenotype, expansion and transduction efficacy; assessed function by in vitro and in vivo activity against AML cell lines expressing high (MOLM 13: CD123 high, CD33 high, CLL-1 intermediate), intermediate (HL60: CD123 low, CD33 intermediate, CLL-1 intermediate/high) or low (KG1a: CD123 low, CD33 low, CLL-1 low) levels of the target antigens. The in vitro benefit of dual expression was most evident when the target cell line expressed low antigen levels (KG1a). Mechanistically, dual expression was associated with higher pCD3z levels in T cells compared to single CAR T cells on exposure to KG1a (p<0.0001). In vivo, combinatorial targeting with CD123 or CD33 and CLL-1 CAR T cells improved tumor control and animal survival for all lines (KG1a, MOLM13 and HL60); no antigen escape was detected in residual tumors. Overall, these demonstrate combinatorial targeting of CD33 or CD123 and CLL-1 with CAR T cells can control growth of heterogeneous AML tumors.
Keywords: Acute Myeloid Leukemia, chimeric antigen receptor, C type lectin-like molecule 1, CD33, CD123
INTRODUCTION:
Treatment resistance and relapse remain major causes of treatment failure in acute myeloid leukemia (AML) (1). Therefore, new targeted therapies for AML are necessary. The success of chimeric antigen receptor (CAR)-modified T cells against relapsed-refractory B-cell malignancies, initially targeted toward CD19 and more recently toward CD20, BCMA, and to CD30 in Hodgkin Disease, increased interest in extending CAR T cells for the treatment of AML (2,3,4,5).
As for other malignancies, CAR T-cell mediated antitumor activity against AML requires antigen expression homogenously above the detection and activation threshold of the targeting CAR T cell. Highly and consistently expressed target antigens that are also AML-restricted and suited to CAR targeting remain elusive, as AML is a phenotypically heterogenous disease. Malignant cell phenotype and susceptibility to treatment are influenced by cell of origin, critical driver mutations, oncogenic translocations and epigenetic alterations (7,8). Thus, unlike B-cell malignancies, most AMLs are heterogeneous in surface marker expression even within a single individual, corresponding to AML’s genetic and epigenetic instability. While several potential targets have been identified, including CD33, CD123 and CLL-1 (C-type lectin like molecule 1, C-type lectin domain family 12 (CLEC 12A); CD371), the heterogeneity of AML means CAR T-cell therapies directed to single targets have had limited effectiveness (9,10,11). Combining CARs against different AML targets (12,13,14) is one potential solution to this issue, and these combinatorial approaches have shown promise in B-cell malignancies and in pre-clinical solid tumor models (15,16).
There are several ways to engineer multi-specific T-cell products, including single bicistronic vectors expressing two CARs, tandem vectors in which a single CAR contains two binder sequences, or co-transduction of CAR T cells with two separate CAR-encoding vectors (17). Co-transduction creates three subpopulations of T cells expressing one, the other, or both CARs at different molar ratios, potentially allowing in vivo selection of the “optimal fit” for each target cell (17,18,19). Results from a clinical trial evaluating the efficacy of CAR T cells co-transduced with CD19 and CD22 CARs to treat B-acute lymphoblastic leukemia support this potential therapeutic benefit. In this trial, complete remission was achieved in five of seven patients and investigators found no evidence of dual antigen escape (20). Similarly, co-transduction of two CARs targeting BCMA and GPRC5D enhanced anti-tumor activity compared to single targeting in a multiple myeloma model (21).
Here, we tested whether co-transduction of CLL-1 and CD33 or CD123 CARs with two separate vectors eradicates AML tumors with heterogeneous myeloid antigen expression. We chose CLL-1, CD33 and CD123 as potential target antigens based on their differential expression levels on most AML cells versus normal hemopoietic stem cells or non-hemopoietic tissues (22–27). As the majority of AML samples express either CD33, CD123, or both (28), we generated CARs directed to these two antigens and expressed them on T cells alone or in combination with our optimized CLL-1 CAR. We found that co-expression of CLL-1 CAR (29,30) with either CD33 or CD123 CARs enhanced anti-tumor responses compared to single antigen targeting and that this effect was most evident against targets with low antigen density in vitro. Finally, dual targeting with either CD33 or CD123 and CLL-1 CAR T-cells prolonged survival across three murine models of AML with heterogeneous levels of target antigen expression.
MATERIALS AND METHODS:
Cell lines
AML cell lines THP-1, KG-1a, HL-60, and MOLM-13 were purchased from ATCC and maintained as described in the Supplementary Methods Appendix. All cell lines were transduced with a gammaretroviral vector encoding enhanced GFP-FFluc fusion protein as previously described (29). We used flow cytometry to evaluate each cell line’s MFI of AML target expression (CLL-1, CD33 and CD123). MFI normalized to isotype control of up to 5 was scored as low (+), 5-20 as intermediate (++), >20 as high (+++) (Table 1).
Table 1.
The target expression profile of cell lines according to MFI ratios (+:Low, ++:Intermediate,+++:High)
| Cell Line | CLL-1 | CD33 | CD123 | |
|---|---|---|---|
| Molm13 | ++ | +++ | +++ |
| HL-60 | ++ | ++ | + |
| KG1a | + | + | + |
| THP-1 | +++ | ++ | ++ |
Plasmid construction and retrovirus production
We used a CLL-1 CAR previously described by our group (29). CD33 CAR was designed using humanized sequence of a CD33-specific antibody (clone my96) which was gene-synthesized. CD123 CAR was created using a CD123-specific scFv binder derived from clone 26292 (kindly provided by S. Gottschalk, MD) (31). CD33 and CD123 CARs were prepared using in-Fusion Cloning (Takara/Clontech, CA, USA). We generated a panel of CD33/CD123 CARs with either 4-1BB or CD28 costimulatory domains, CD28 transmembrane domain and CD3ζ chain which also contained a sequence from NGFR and CH3 (Figure 1A, figure 2A, supplementary figure 1A, supplementary figure 2A). Retroviral vector production and T-cell transduction have been described previously (29). CAR T-cells were expanded in complete medium (CM) consisting of 45% RPMI 1640 (Hyclone, Waltham, MA), 45% Click’s media (Irvine Scientific, Santa Ana, CA, USA), 2mM L-glutamine (Gibco-BRL, San Francisco, CA) with added penicillin and streptomycin (Gibco-BRL, San Francisco, CA, USA) and 10% FBS (Hyclone, Waltham, MA, USA) in the presence of 10 ng/mL IL-7 (Peprotech, Rocky Hill, NJ) and 10 ng/mL 1L-15 (Peprotech, Rocky Hill, NJ, USA). We co-transduced CLL-1 CAR with either CD33 CAR or CD123 CAR for combinatorial targeting.
Figure 1. Combinatorial Targeting with CLL-1 CAR and CD33 CARs in vitro.

1A. Schematic figures of CLL-1 CAR, CD33 CAR.CD28z constructs. 1B. CAR expression after single and co-transduction shown at day 3 after transduction. 1C. T cell expansion of NTR, CLL-1 CAR, CD33 CAR. CD28z, single and doubly transduced T cells in culture. 1D. The residual tumor cell numbers in THP-1 (left), HL-60 (middle), MOLM13 (middle), and KGA1 (right) after co-culture with effector cells for 72 hours at E:T (1:4). 1E. pCD3z expression in T cells with single vs combinatorial CAR targeting after exposure to KG1a (NTR: Non-Transduced). 1F. Total CD3+ T cell counts after 72 hrs co-culture with KG1a at E:T (1:4) (P>0.05). Residual CAR T cell counts with CLL-1 CAR +/− CD33 CAR.CD28 (NTR:Non Transduced).
Figure 2. Combinatorial Targeting with CLL-1 CAR and CD123 CARs in vitro.

2A. Schematic figures of CLL-1 CAR, CD123 CAR.CD28z constructs. 2B. CAR expression in single and co-transduced T cells shown on day 3 after transduction. 2C. T cell expansion of NTR, CLL-1 CAR, CD123 CAR. CD28z, single and doubly (combinatorial) transduced cells in culture. 2D. The residual counts of tumor cells in THP-1 (left), HL-60 (middle), MOLM13 (middle), and KGA1 (right) after co-culture at 72 hours E:T (1:4). (NTR: Non-Transduced). 2E. pCD3z expression in single vs combinatorial targeting when exposed to KG1a. 2F. Total CD3+ T cell counts after 72 hrs co-culture with KG1a at E:T =1:4 (P>0.05). Residual CAR T cell counts with CLL-1 CAR +/− CD123 CAR.CD28 (NTR:Non Transduced).
CAR expression and immunophenotyping of transduced cells
We detected expression of the CLL-1 CAR using AF647-conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA, USA). CD33 CAR and CD123 CAR were detected by Alexa Fluour 647 AffiniPure Goat Anti-Human IgG, Fcγ fragment specific antibody was used (Jackson ImmunoResearch, PA, USA). These construcs also contained NGFR as a surrogate marker and detected by using anti-human CD271 (NGFR) (Biolegend, San Diego, CA, USA). The MFI of expression of both CLL-1 CAR and CD33/CD123 CAR in co-transduction were detected by AF647-conjugated goat anti-mouse IgG in combination with anti-human CD271. Anti-human CD45, CD4, CD8, CD3, CD45RA, CD19, CCR7, CD271, CD371 (Clec12A, CLL-1) and Mouse IgG2alpha isotype were obtained from BD Biosciences (San Jose, CA, USA), Beckman Coulter (Indianapolis, IN, USA), Life Technologies (Carlsbad, CA, USA), or Biolegend (San Diego, CA, USA). Flow cytometric data were acquired on a Beckman Gallios (Beckman Coulter, Indianapolis, IN, USA) or BD FACS Canto II (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo 10 (BD).
Co-culture, detection of cytokine secretion, T cell exhaustion, and pCD3z activity
The details of co-culture assays are included in the Supplementary Methods Appendix. Human TNF-α, IFN-g and IL-2 were measured using a Luminex xMAP Multiplex Assay with Millipore Sigma antibodies (Burlington, MA, USA). T cell “exhaustion” markers were detected using human TIM-3 (Biolegend, San Diego CA, USA), LAG 3 (BD Biosciences, San Jose, CA, USA) and PD-1 (Biolegend, San Diego CA, USA) antibodies. We measured pCD3z activity in a 5-hour short-term cytotoxicity assay by expression of phospho-CD247 by flow cytometry and isotype control background was subtracted (Invitrogen, Waltham, MA, USA). The details of the western blot assay are included in the Supplementary Methods Appendix.
Xenogeneic AML mouse models
NOD.SCID IL-2Rg−/− (NSG-SGM3) mice were bred in the Baylor College of Medicine Transgenic Mouse Facility using breeder pairs obtained from Jackson Laboratories. Tumor burden in mice engrafted with FFluc-modified cells was measured using IVIS imaging as described before (29). For in vivo experiments, mice were bled at the specified time points. Mouse experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Patient Samples
Patient samples were obtained from Dr. Premal Lulla after informed consent on an IRB-approved protocol.
Statistical analysis
Unpaired two-tailed Student’s t test and ANOVA was used for group comparisons and values of p<0.05 were considered significant. Survival was calculated using Mantel-Cox log-rank test (Graph Pad Prism).
RESULTS:
Engineering CAR T cells directed to CD33 or CD123
For all combinatorial experiments below, we used a CLL-1 CAR with CD28 and CD3 zeta endodomains that was optimized as described in previous studies and is now in Phase I clinical trials for AML (23) (NCT04219163). We combined expression of this CAR with expression of CARs targeting the CD33 or CD123 antigen and containing a CD28 (CD33 CAR.CD28z, CD123 CAR.CD28z) co-stimulatory endodomain (Figure 1A). We compared the expansion, persistence and anti-tumor activity of each of these two CARs alone and after joint expression with our optimized CLL-1 CAR (16, 23).
We measured the MFI of each CAR expressed alone or in combination with the CLL-1 CAR as described in methods, and found no significant differences (Figure 1B). T cell expansion was higher with CLL-1-CD28 CAR alone at day 14 compared to both the CD33 CAR.CD28z alone or the combination of CLL-1 CAR with CD33 CAR.CD28z (p<0.0001) (Figure 1C). Next, we co-cultured singly transduced CAR T-cells with AML cell lines THP-1, HL-60, and MOLM-13, which express high/intermediate levels of CD33 and CLL-1. Cell line origin and phenotypic characteristics are summarized in Table 1. We found all three cell lines to be identically susceptible to singly transduced CD33 CAR.28z or CLL-1 CAR T cells. Moreover, dual targeting with T cells expressing CLL-1 CAR together with CD33 CAR.28z did not increase tumor cell killing compared to either CAR alone (Figure 1D). However, the potential benefits of dual targeting are evident following exposure to the KG-1a tumor cell line, which has low expression of both target antigens (Figure 1D) (p=0.0006). Mechanistically, following co-culture with KG-1a, we found dual CAR expression (CD33 CAR.28z and CLL-1 CAR) was associated with higher pCD3z levels (p<0.0001) compared to single CAR T cells (CD33 CAR.28z or CLL-1 CAR) (Figure 1E). Compared to single CARs, combinatorial targeting with CLL-1 CAR and CD33 CAR.CD28z enhanced T-cell expansion (Figure 1F). After 72-hour co-culture with the low-expressing KG1a cell line, levels of IFNγ, IL-2 and TNFα in supernatants of co-cultures with single or dual CAR-T cells were not significantly different (Suppl Fig 1G).
We repeated the above experiments and compared the effects of T cells expressing CD123 CAR.CD28z alone or in combination with a CLL-1 CAR (Figure 2A). As for our CD33 CAR, MFI of CAR expression was comparable (Figure 2B) and T cells expanded better with CLL-1 CAR alone at day 14 compared to both CD123 CAR.CD28z alone or in combination with CLL-1 CAR (p<0.0001) (Figure 2C). Again, the benefit of dual targeting in short term cytotoxicity assays was apparent only against KG1a (p=0.0005) but not THP-1, HL-60, and MOLM-13 (Figure 2D). As in CD33 and CLL-1 dual targeting CAR-T cells, dual expression of CD123 CAR and CLL-1 CAR was associated with higher pCD3z levels (p<0.0001) compared to single-CAR T cells following exposure to the KG-1a tumor cell line (Figure 2E). The constitutive (antigen independent) levels of pCD3z were comparable irrespective of the construct used (Suppl Fig 3). In coculture with KG1a, we found a half log more CD3+ T cells with combinatorial targeting (CLL-1 CAR and CD123 CAR.CD28z) compared to single targeting CAR T cells (Figure 2F) (p>0.05). As above, however, this increased cytotoxicity of dual compared to single targeting CAR T-cells did not significantly increase release of IFNγ or IL-2, although TNFα increased to levels approaching conventional significance (p=0.056) (Suppl Fig 2G).
We next compared the effects of combining the same CLL-1 CAR with CD33 and CD123 CARs expressing a the 41BB co-stimulatory endodomain (Supplementary Figures 1 and 2). We found no difference in transgene expression (Supplementary Fig 1B and Supplementary Fig 2B), phenotype of the cells (Supplementary Fig 1E and Supplementary Fig 2E), or CD3+ T cell expansion in co-culture with KG1a between CARs harboring either endodomain (Supplementary Fig 1F and Supplementary Fig 2F). There was, however, a consistent difference in T-cell expansion between the CD33 CARs harboring CD28 and 41BB at day 14. At this time point, expansion was 10-fold higher for T-cells with the CD33-CD28 CAR compared to the CD33-4-1BB CAR, regardless of whether the CD33 CAR was expressed alone or with CLL-1 CAR (p<0.0001) (Supplementary Fig 1C). Similarly, CD123-CD28 CAR T cells expanded 5-fold more than CD123-41BB CAR T cells at day 14 (Supplementary Fig 2C). Supplementary Fig 1D and Fig 2D show a comparison of single and dual CAR targeting in low target expressor cell line KG1a, and demonstrate enhanced antitumor activity with combinatorial targeting with CLL-1 CAR and CD33/CD123 CAR that occurred irrespective of CD28 or 4-1BB co-stimulation. PD-1, TIM-3, LAG-3 expression were similar in cells receiving CD28 or 4-1BB co-stimulation or expressing single vs dual CARs (Suppl Fig 4). Based on data showing accelerated expansion with CD28 co-stimulation and given the rapidly progressive nature of AML in the intended treatment group, we restricted further evaluation of combinatorial targeting with CLL-1 CAR T cells to constructs embedding the CD28 co-stimulatory sequence.
Effects of Combinatorial Targeting on Primary Human AML Cells
In order to test the anti-tumor effect of our CAR T cells against primary human AML cells, we analyzed CLL-1, CD33, CD123 target antigen expression on peripheral blood blast cells from 15 AML patients (characterized in Table 2). We assessed cytotoxicity at an E:T ratio of 1:1 and compared single CAR (CLL-1, CD33 or CD123) to combinatorial CAR (CLL-1 CAR T+CD33 CAR T cells or CLL-1 CAR T + CD123 CAR T cells) expression. The MFI of target expression correlated with the percentage of killing by single or combinatorial CAR T cells (Figure 3). As in our cell line studies, we found combinatorial CAR T cells had higher relative levels of cytotoxicity compared to single CAR T when the MFI of the targeted antigen(s) was low.
Table 2.
Patient Characteristics (ID: Identification number, MLLr:MLL gene rearrangement). When gated on CD45 dim/side scatter (low) populations enriched for AML the relative MFI ratios of CLL-1, CD33, CD123 were between as follows: 1-40; 1-20.6; 4-12.1.
| AML Sample ID | Blast % | Cytogenetics | CLL-1 Relative MFI | CD33 Relative MFI | CD123 Relative MFI |
|---|---|---|---|---|---|
| 1 | 93 | MLLr (6;11) | 19.8 | 20.6 | 10.5 |
| 2 | 92 | MLLr (9:11) | 1 | 1.3 | 4.3 |
| 3 | 96 | t(16:21) | 3.1 | 12 | 10 |
| 4 | 88 | MLLr (6:11) | 9.5 | 7 | 6 |
| 5 | 92 | MLLr (9:11) | 13.2 | 8.4 | 5.9 |
| 6 | 79 | Not available | 6.2 | 11.8 | 7.8 |
| 7 | 64 | inv( 16). extra copy of MLL (unbalanced t(7:11) | 2.8 | 5.7 | 5.9 |
| 8 | 74 | Not available | 3.7 | 6.7 | 8 |
| 9 | 79 | Not available | 2.5 | 6 | 4.4 |
| 10 | 50 | Not available | 40 | 20 | 12 |
| 11 | 2 | Not available | 6 | 5 | 5 |
| 12 | 84 | Not available | 3 | 1 | 4 |
| 13 | 8 | Not available | 3.4 | 4.2 | 4 |
| 14 | 86 | Not available | 8.4 | 5.9 | 12.1 |
| 15 | 71 | Not available | 2 | 14.6 | 4.3 |
Figure 3. Effects of Combinatorial Targeting on Primary Human AML Cells.
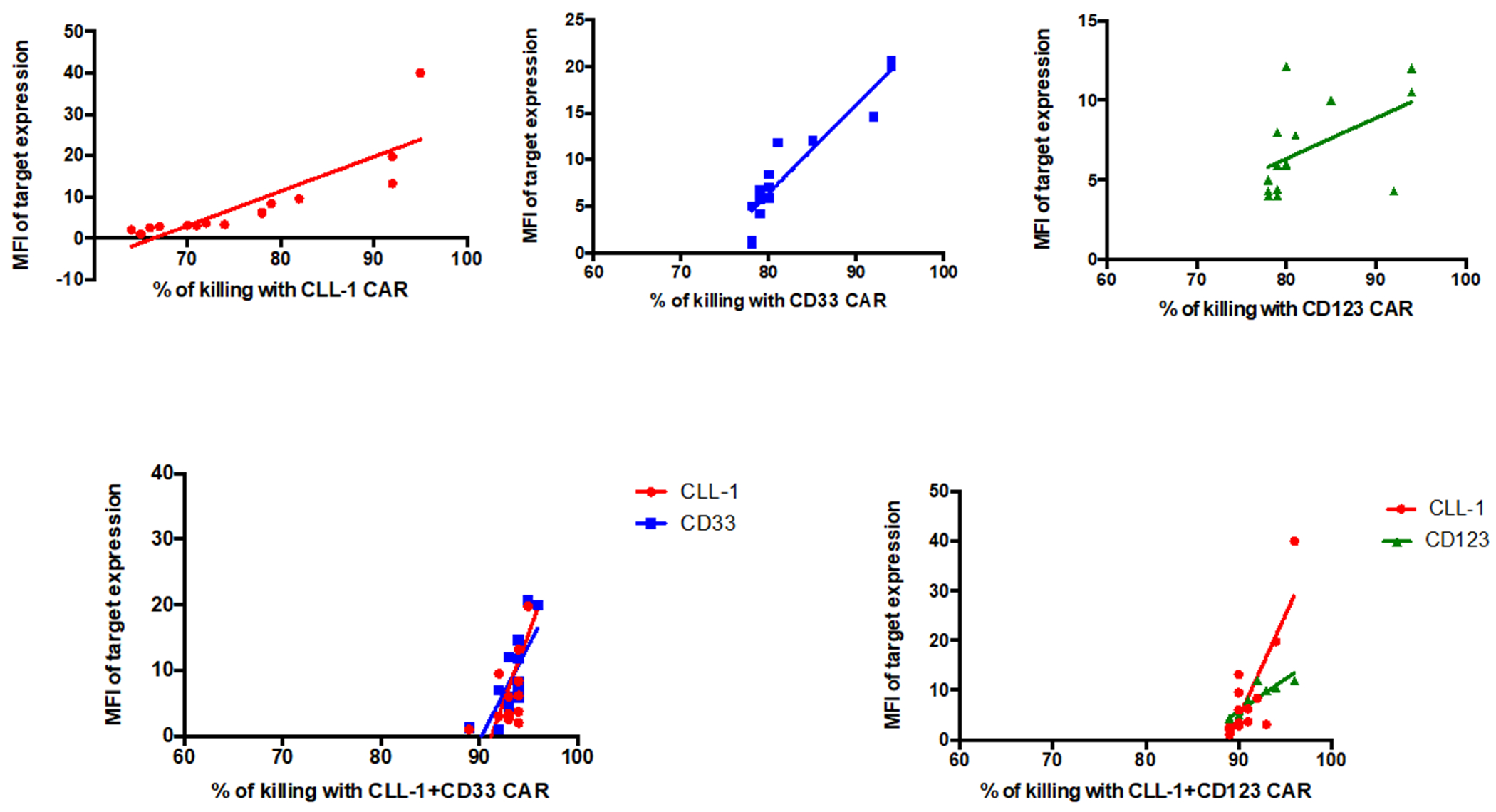
MFI of target antigen expression on patient samples vs percent killing by CAR T cells expressing each targeting moiety (E:T=1:1, 24 h assay).
Combinatorial Targeting Increases Anti-tumor Activity and Prolongs Survival in High and Intermediate AML Animal Models
We compared the ability of singly and doubly transduced CAR T cells to control established AML in animal models engrafted with human AML cell lines (MOLM13, HL60 and KG-1a), each of which has a different level of target antigen expression.
In the first model, NSG-SGM3 mice were sublethally irradiated (1.16Gy) and injected with FFLuc-modified MOLM 13 cells (1x106 cells per mouse), which expresses high levels of CD33 and CD123, and intermediate CLL-1. Six days later, we injected 1x106 CART cells targeting single and combinatorial antigens (CLL-1 CAR T, CD33 CAR T, CD123 CAR T, CLL-1 CAR T + CD33 CAR T, CLL-1 CAR T + CD123 CAR T) (Figure 4A). CLL-1 CAR T + CD33 CAR T cells and CLL-1 CAR T + CD123 CAR T cells decreased tumor growth and demonstrated sustained anti-tumor activity compared to single CAR T cells and negative controls (Figure 4B). At 15 days following infusion, CAR T cell expansion was more rapid in mice receiving doubly transduced CAR T cells (p<0.0001) compared to recipients of singly transduced cells, regardless of CD33 or CD123 targeting (Figure 4C). Survival was significantly prolonged in both groups receiving doubly transduced CAR-Ts compared to groups receiving singly transduced CAR T cells (Figure 4D). Malignant cells from animals that relapsed after administration of singly or doubly transduced CAR T cells continued to express all targeted antigens, suggesting no antigen escape (Figure 4E).
Figure 4. In vivo MOLM3 and HL60 models.
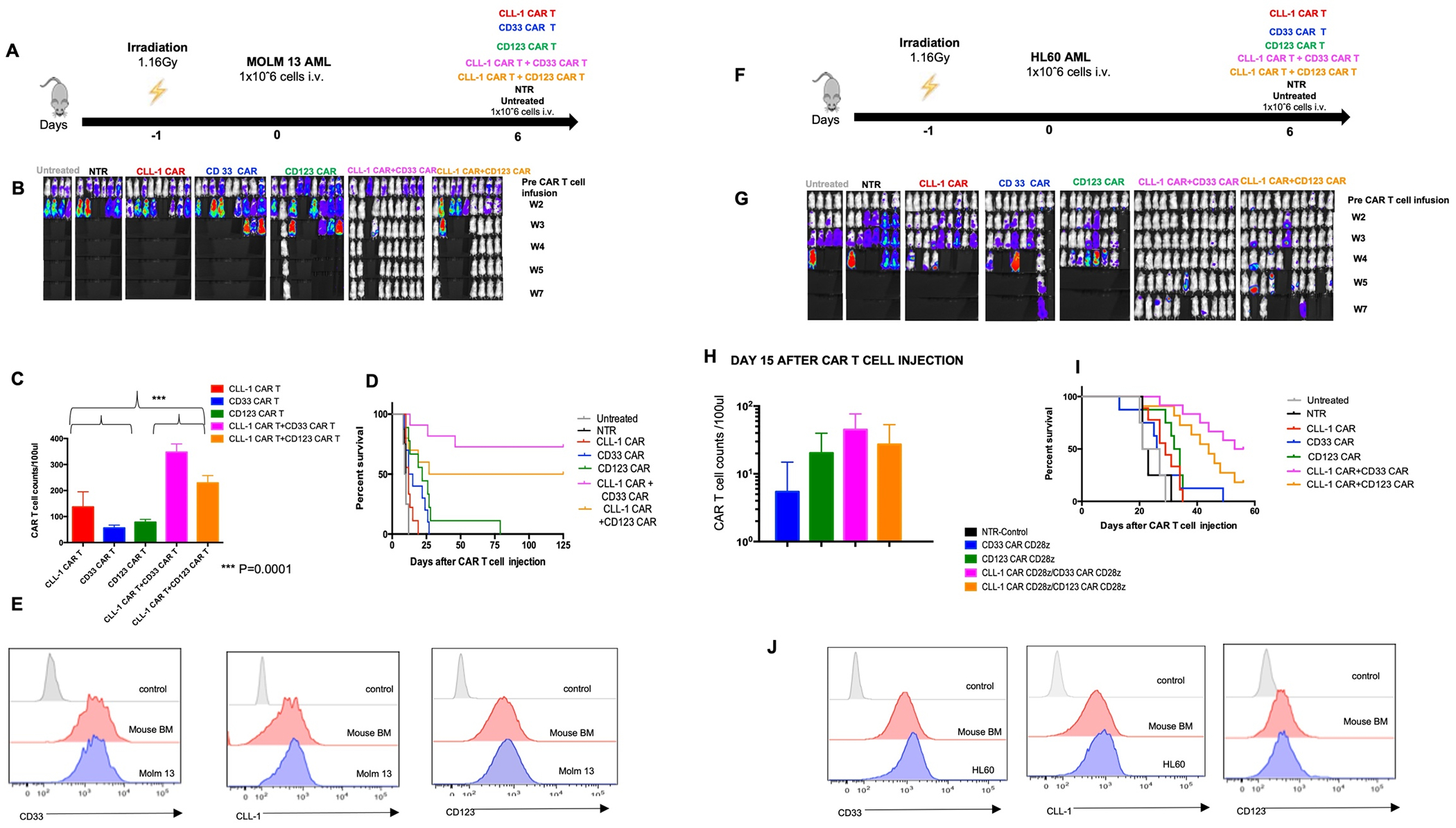
4A. Schematic figure of MOLM13 in vivo model comparing single vs combinatorial targeting. 4B. Representative images showing leukemia progression in each experimental group from week 1 to week 7. 4C. CAR T cell counts in animals receiving single vs dual (combinatorial )transduced effector cells on day 15 after CAR T cell injection. 4D. Kaplan-Meier curve showing the survival of mice in each experimental group. 4E.Target cell expression profile in animal with recurrence of MOLM13 tumor. 4F. HL60 in vivo model to compare effects of single vs combinatorial targeting on tumor growth and CAR T expansion and persistence. 3G. Representative images showing leukemia progression in groups from week 1 to week 7. 4H. CAR T cell counts in animals receiving single vs dual (combinatorial) transduced effector cells on day 15 after CAR T cell injection. 4I. Kaplan-Meier curve showing the survival of mice in each experimental group. p values were determined by log-rank Mantel-Cox test.(NTR:Non Transduced). 4J. Target cell expression profile in animal with recurrence of HL60 tumor.
In our intermediate AML model (HL60: CLL-1 intermediate, CD33 intermediate, CD123 low), NSG-SGM3 mice were sublethally irradiated (1.16Gy) and injected with FFLuc-modified HL60 cells (1x106 cells per mouse). Six days later, we injected 1x106 CART cells targeting single and combinatorial antigens (Figure 4F). Once again, we observed superior tumor control after combinatorial targeting of CAR T cells (Figure 4G), and increased CAR T cell expansion with doubly transduced CAR T compared to single targeting (Figure 4H). Increased expansion correlated with prolonged survival (Figure 4I). As above, target antigen expression on malignant cells remained similar to pre-treatment in relapsed animals (Figure 4J).
Combinatorial Targeting Significantly Increases Anti-Tumor Activity in Mouse Models of AML Expressing Low Levels of Target Antigens
We used the promyeloblastic cell line KG1a to evaluate the benefits of dual targeting when expression of each targeted antigen was low. NSG-SGM3 mice were sublethally irradiated (1.16Gy) and injected with FFLuc-modified KG1a AML cell line (1x106 cells per mouse). Six days later, we injected 1x106 CART cells targeting single and combinatorial antigens (Figure 5A). We found mice that received either CLL-1 CAR T + CD33 CAR T cells or CLL-1 + CD123 CAR T cells had significantly improved and sustained tumor control compared to recipients of T cells expressing a single CAR (Figure 5B, 5C and 5D). Although doubly transduced CAR T cells were present in greater numbers in peripheral blood on day 15 than in recipients of single CAR T cell infused, the difference did not reach statistical significance (Figure 5E).
Figure 5. In vivo study of KG1a model.
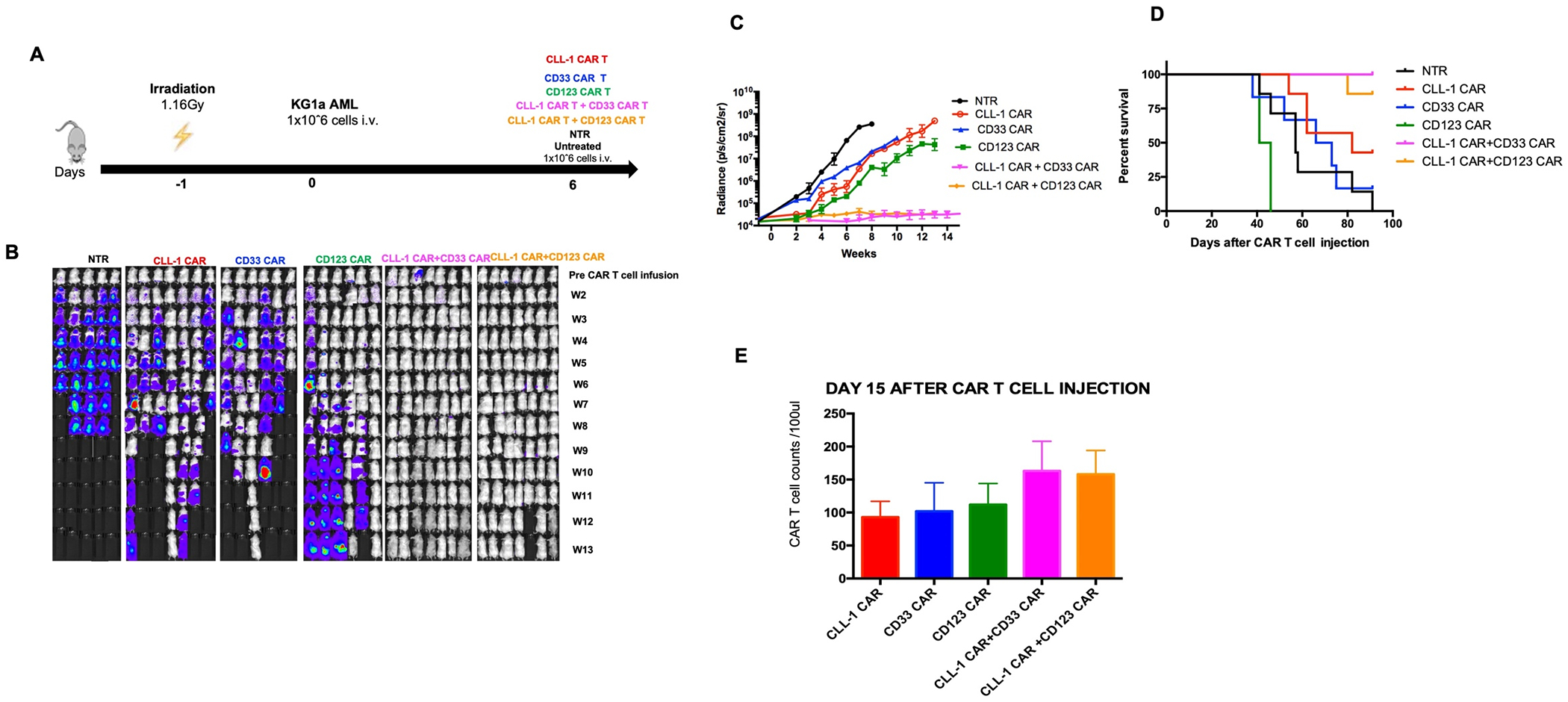
5A. Schematic figure of KG1a in vivo model comparing single vs combinatorial targeting. 5B. Representative images showing leukemia progression in groups from week 1 to week 13. 5C. Bioluminescence of mice in single vs combinatorial targeting 5D. Kaplan-Meier curve showing the survival of mice in each experimental group. 5E. CAR T cell counts in animals receiving single vs combinatorial targeting after day 15 CAR T cell injection.
DISCUSSION:
Here, we demonstrate that dual targeting with either a CD33-CAR or CD123-CAR and CLL-1 CAR increased T cell antitumor activity against AML expressing heterogenous levels of the target antigens. In vitro, the benefits of dual targeting were most profound when target antigen expression on the tumor cells was low, an effect associated with enhanced CAR T activation and antigen dependent pCD3z expression in double versus single transduced cells. In vivo, however, our dual targeting strategy significantly improved anti-tumor activity irrespective of target antigen expression.
In this study, we chose to target CLL-1, CD33 and CD123 — three antigens highly expressed with high frequency in AML. Haubner et al demonstrated that CD33 expression was positive in 96.4% of AML at initial diagnosis and 98.1% at relapse with high MFI ratios (initial diagnosis:27.1/relapse:34.1). Similarly, CD123 or CLL-1 are frequently expressed ( CD123 initial diagnosis: 97%, relapse: 98.1%; CLL-1 initial diagnosis: 80.1%, relapse: 71.4%) at moderate levels (CD123 initial diagnosis: 8.5, relapse: 9.2; CLL-1 initial diagnosis: 13.5, relapse:3.6) (32). Crucially, >70% of AML samples are dual positive for CLL-1 and CD33 (9,17,18), while >75% of patients co-express CLL-1 and CD123 (32).
One major reason for resistance to CAR T-cell therapy in both hematologic and solid tumors is loss of expression of the targeted tumor antigen (32,33,34). CAR T cells combining more than one targeting moiety have overcome this antigen escape in preclinical and some clinical studies. Though we did not observe loss of antigenic targets in the current or our previously reported pre-clinical studies (23), even in this context of treatment failure, dual targeting was associated with consistently greater anti-tumor activity than single targeting. Regardless of antigen expression levels, the superior functionality of dual targeting CAR T cells was associated with increased T cell activation (as measured by pCD3z) and proliferation. Thus, in the presence of heterogeneity of expression of one or both target antigens dual targeting may increase the avidity of CAR-T interaction with the target cell, favoring immune synapse formation and subsequent T cell recruitment (35,36).
In AML, combinatorial antigen targeting has been successfully tested with CD123 and CD33 bicistronic CAR T cells in vivo and in vitro (18). These investigators did not compare the antitumor activity of single CAR T cells vs bicistronic CAR T cells but showed significant antitumor effect of bicistronic CAR T cells in artificially created cell lines (CD33+CD123−, CD33+CD123−). In addition, Liu et al demonstrated remarkable results in a phase-1 trial of CLL-1 and CD33 bicistronic CAR T cells in patients with refractory AML (37), although this study did not include patient target antigen expression profiles. In our study, we chose to modify T cells with two separate vectors. Although manufacturing two separate vectors is initially more expensive, this approach may ultimately prove more cost effective in a heterogeneous disease like AML. Bicistronic or tandem CARs may be most successful when the molar ratio of each target is fixed. By contrast, when the expression of each target antigen is highly variable it may be preferable to manufacture a mixed product in which binder expression is equally heterogeneous in terms of their ratios and absolute numbers. Certainly, our in vitro studies show the additional anti-tumor effects elicited by superior T cell activation in dual targeting CAR T cells was especially pronounced when the tumor expressed low levels of target antigen. Consistent with this, we observed combination therapy had higher anti-tumor efficacy in all in vivo models. These results further the notion that dual targeting by separate vectors may enable expression of the range of avidities for each binder component required for optimal destruction of phenotypically heterogenous AML populations (19).
We also investigated whether the anti-tumor activity of two distinct CARs might be further improved by using potentially complementary costimulatory endodomains. For example, clinical trials of CD19 CAR T have revealed that cells with the CD28z costimulatory endodomain generally expand faster than 41BB CD19 CAR T cells. This advantage, however, may be offset by reduced persistence due to exhaustion (38,39). Although interpretation of these analyses has been confounded by different classes of gene-transfer vectors (16), our current studies using only gammaretroviral vectors confirmed more rapid CAR-T cell expansion without significant T cell exhaustion when a CD28 co-stimulatory sequence was substituted for 41BB. These results are in line with our previous report demonstrating detrimental effect of tonic 4-1BB signaling from CARs expressed by gammaretroviral vectors, which can be alleviated by either switching costimulation to CD28 or by substituting a self-inactivating lentiviral vector (40). In addition, pre-clinical studies confirmed CLL-1 CAR with CD28z endodomain had a more rapid and higher peak response compared to 4-1BB endodomain, leading to superior control of relapsed bulky disease in vivo (16). Nonetheless, there remains an argument for “mixing and matching” 41BB and CD28 when a T cell contains two distinct targeting moieties. In principle, the combination of co-stimulatory endodomains may allow superior initial expansion while sustaining long-term persistence. Indeed, CD33 and CD123 CARs with 4-1BB endodomains retained significantly higher proportions of the central memory T cells associated with longer term persistence compared to similar CARs harboring CD28z (41,42). Nonetheless, we found that CARs with 41BB expanded poorly compared to CD28z and a combination of the two endodomains produced generally inferior anti-leukemic activity in vitro compared to cells in which both the CLL-1 and complementary CARs incorporated CD28z.
Although CLL-1, CD33 and CD123 are all highly expressed on a large percentage of AML cells, CD33 and CD123 are both also expressed on normal hematopoietic tissues, risking potential on target-off tumor hemato-toxicity. In a phase I trial with CD33 CAR T cells, one patient suffered from pancytopenia due to myelosuppression (43), as well as a cytokine release syndrome due to engagement of the effector cells with their targets. CD33 is also expressed by hepatocytes, which may cause additional organ toxicity such as veno-occlusive liver disease (44). Similarly, CAR T cells targeting CD123 eliminate not only AML cells but also mature normal myeloid subsets and disrupt myelopoiesis, causing profound myelosuppression. A phase II trial of Talocotuzumab (IgG1 monoclonal antibody targeting CD123) was prematurely terminated due to high organ toxicity rates and lack of efficacy (45). Although these toxicities do not preclude the use of CAR T cells to treat high-risk or refractory AML, they require consideration in terms of protocol design and patient selection. One approach to overcome these risks is the use of safety or suicide genes to abbreviate toxicity from effector CAR T cells (46). Should unacceptably severe or prolonged toxicities occur, administration of the relevant prodrug can eliminate the CAR T cells (23,47,48). Alternatively, administration of cross-reactive CAR T cells can be a bridge to allogeneic stem cell transplant, reducing disease burden sufficiently to allow the patient to receive donor marrow. Potentially the most elegant approach, however, is to knock-out the targeted antigen in normal marrow cells, which could then repopulate a patient even in the presence of the respective CAR-T cells. The feasibility of this approach has been demonstrated for CD33 and it will be of interest to see if a similar approach will allow removal of CLL-1 and CD123, or whether removal of these structures will adversely affect the growth, trafficking or function of normal myeloid progenitor cells (49,50). Another concern for clinical application might be an excessive viral copy number in transduced cells when dual vectors are used. In previous clinical studies for lymphoid malignancies and solid tumors in which we used dual vectors, we nonetheless consistently restrict vector copy numbers to limits required by regulatory agencies. If necessary, the transduction protocol can be adjusted to meet these constraints (NCT03635632, NCT04377932).
The heterogeneity of AML and the phenotypic overlap with normal marrow precursor cells continues to make this disease a difficult target for safe and effective CAR-T cell therapy. Nonetheless, we have shown that combinatorial antigen targeting of a single T cell by two separate CARs targeting CLL-1 and either CD33 or CD123 can successfully control even bulky disease in several preclinical models. For clinical application, each patient’s tumor should be evaluated for target antigen expression and dual targeting with CD33-CAR or CD123-CAR and CLL-1 CAR considered in patients expressing low levels of the target antigens.
Supplementary Material
Acknowledgments
We thank Catherine Gillespie for critical review of the manuscript.
Funding
This work was supported by Leukemia and Lymphoma Society SCOR (#7001-19)( M.K.B.). M.K.B. is a Stand Up to Cancer grantee. P.A.A. and E.A. were supported by Scientific and Technological Research Council of Turkey 2219 grant.
Disclosure of Conflict of Interests
M.K.B. is a co-founder and equity holder, and receives fees from Allovir, Marker Therapeutics and Tessa Therapeutics PTE. M.K.B. is a scientic advisory board member reports personal fees from Allogene, Abintus, Bellicum Pharmaceuticals, Coya Therapeutics, Kuur/Athenex Therapeutics, Marker Therapeutics, Memgen LLC, Tessa Therapeutics, Turnstone Biologics, Tscan Therapeutics, Walking Fish Therapeutics. M.K.B. has Stock Options from Allogene, Abintus, Coya Therapeutics, Tscan Therapeutics, Walking Fish Therapeutics.
REFERENCES:
- 1.Lee JB, Chen B, Vasic D, Law AD, Zhang L. Cellular immunotherapy for acute myeloid leukemia: How specific should it be? Blood Rev. 2019. May;35:18–31. doi: 10.1016/j.blre.2019.02.001. Epub 2019 Feb 23. [DOI] [PubMed] [Google Scholar]
- 2.Song MK, Park BB, Uhm JE. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. Int J Mol Sci. 2019. Oct 10;20(20):5010. doi: 10.3390/ijms20205010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG,et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012. Apr 26;119(17):3940–50. doi: 10.1182/blood-2011-10-387969. Epub 2012 Feb 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017. Sep 1;127(9):3462–3471. doi: 10.1172/JCI94306. Epub 2017 Aug 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016. Sep 29;128(13):1688–700. doi: 10.1182/blood-2016-04-711903. Epub 2016 Jul 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012. Jun;122(6):2257–66. doi: 10.1172/JCI45851. Epub 2012 May 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012. Mar 10;30(8):820–9. doi: 10.1200/JCO.2011.35.6394. Epub 2012 Feb 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kavanagh S, Murphy T, Law A, Yehudai D, Ho JM, Chan S, et al. Emerging therapies for acute myeloid leukemia: translating biology into the clinic. JCI Insight. 2017. Sep 21;2(18):e95679. doi: 10.1172/jci.insight.95679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perna F, Berman SH, Soni RK, Mansilla-Soto J, Eyquem J, Hamieh M, et al. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell. 2017. Oct 9;32(4):506–519.e5. doi: 10.1016/j.ccell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan M, Li M, Gao L, Geng S, Wang J, Wang Y, et al. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J Hematol Oncol. 2017. Aug 29;10(1):151. doi: 10.1186/s13045-017-0519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansrivijit P, Gale RP, Barrett J, Ciurea SO. Cellular therapy for acute myeloid Leukemia - Current status and future prospects. Blood Rev. 2019. Sep;37:100578. doi: 10.1016/j.blre.2019.05.002. Epub 2019 May 11. [DOI] [PubMed] [Google Scholar]
- 12.Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr. 2014. Feb;26(1):43–9. doi: 10.1097/MOP.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016. Feb 11;164(4):770–9. doi: 10.1016/j.cell.2016.01.011. Epub 2016 Jan 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol. 2018. Nov 8;9:2593. doi: 10.3389/fimmu.2018.02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers (Basel). 2020. Apr 26;12(5):1075. doi: 10.3390/cancers12051075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morsink LM, Walter RB, Ossenkoppele GJ. Prognostic and therapeutic role of CLEC12A in acute myeloid leukemia. Blood Rev. 2019. Mar;34:26–33. doi: 10.1016/j.blre.2018.10.003. Epub 2018 Nov 1. [DOI] [PubMed] [Google Scholar]
- 17.Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018. Oct;8(10):1219–1226. doi: 10.1158/2159-8290.CD-18-0442. Epub 2018 Aug 22. [DOI] [PubMed] [Google Scholar]
- 18.Petrov JC, Wada M, Pinz KG, Yan LE, Chen KH, Shuai X, et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018. Jun;32(6):1317–1326. doi: 10.1038/s41375-018-0075-3. Epub 2018 Feb 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Lopez RA, Yu W, Cabral KA, Creasey OA, Lopez Pazmino MDP, Tonai Y, et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science. 2021. Mar 12;371(6534):1166–1171. doi: 10.1126/science.abc1855. Epub 2021 Feb 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner R, Annesley C, Finney O, Summers Co, Lamble AJ, Rivers J, et al. Early clinical experience of CD19 x CD22 dual specific CAR T cells for enhanced anti-leukemic targeting of acute lymphoblastic leukemia. Blood. 2018; 132 (Suppl 1):278 [Google Scholar]
- 21.de Larrea CF, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, et al. Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov. 2020. Sep;1(2):146–154. doi: 10.1158/2643-3230.bcd-20-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004. Nov 15;64(22):8443–50. doi: 10.1158/0008-5472.CAN-04-1659. [DOI] [PubMed] [Google Scholar]
- 23.Ataca Atilla P, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating TNFα activity allows transgenic IL15-Expressing CLL-1 CAR T cells to safely eliminate acute myeloid leukemia. J Immunother Cancer. 2020. Sep;8(2):e001229. doi: 10.1136/jitc-2020-001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. 2015. Mar;100(3):336–44. doi: 10.3324/haematol.2014.112748. Epub 2014 Dec 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jilani I, Estey E, Huh Y, Joe Y, Manshouri T, Yared M, et al. Differences in CD33 intensity between various myeloid neoplasms. Am J Clin Pathol. 2002. Oct;118(4):560–6. doi: 10.1309/1WMW-CMXX-4WN4-T55U. [DOI] [PubMed] [Google Scholar]
- 26.Pardanani A, Lasho T, Chen D, Kimlinger TK, Finke C, Zblewski D, et al. Aberrant expression of CD123 (interleukin-3 receptor-α) on neoplastic mast cells. Leukemia. 2015. Jul;29(7):1605–8. doi: 10.1038/leu.2015.16. Epub 2015 Feb 2. [DOI] [PubMed] [Google Scholar]
- 27.Liu K, Zhu M, Huang Y, Wei S, Xie J, Xiao Y. CD123 and its potential clinical application in leukemias. Life Sci. 2015. Feb 1;122:59–64. doi: 10.1016/j.lfs.2014.10.013. Epub 2014 Nov 21. [DOI] [PubMed] [Google Scholar]
- 28.Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014. Jun 13;4(6):e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B, et al. Treatment of Acute Myeloid Leukemia with T Cells Expressing Chimeric Antigen Receptors Directed to C-type Lectin-like Molecule 1. Mol Ther. 2017. Sep 6;25(9):2202–2213. doi: 10.1016/j.ymthe.2017.05.024. Epub 2017 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ataca Atilla P, McKenna MK, Watanabe N, Mamonkin M, Brenner MK, Atilla E. Combinatorial Antigen Targeting Strategy for Acute Myeloid Leukemia. Blood (2020) 136 (Supplement 1):22–23. [Google Scholar]
- 31.Bonifant CL, Szoor A, Torres D, Joseph N, Velasquez MP, Iwahori K, et al. CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol Ther. 2016. Sep 29;24(9):1615–26. doi: 10.1038/mt.2016.116. Epub 2016 Jun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haubner S, Perna F, Köhnke T, Schmidt C, Berman S, Augsberger C, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019. Jan;33(1):64–74. doi: 10.1038/s41375-018-0180-3. Epub 2018 Jun 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timmers M, Roex G, Wang Y, Campillo-Davo D, Van Tendeloo VFI, Chu Y, et al. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front Immunol. 2019. Jul 16;10:1613. doi: 10.3389/fimmu.2019.01613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. 2019. Jul;290(1):60–84. doi: 10.1111/imr.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu YH, et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol Ther. 2018. Apr 4;26(4):963–975. doi: 10.1016/j.ymthe.2018.01.020. Epub 2018 Mar 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015. Jan;23(1):184–91. doi: 10.1038/mt.2014.164. Epub 2014 Sep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR-T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. 2018;132(Suppl 1):901. [Google Scholar]
- 38.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015. Feb 7;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. Epub 2014 Oct 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015. Jun;21(6):581–90. doi: 10.1038/nm.3838. Epub 2015 May 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes-Silva D, Murherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017. Oct 3;21(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018. Feb 1;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017. Jun 22;129(25):3322–3331. doi: 10.1182/blood-2017-02-769208. Epub 2017 Apr 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015. Jan;23(1):184–91. doi: 10.1038/mt.2014.164. Epub 2014 Sep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giles FJ, Kantarjian HM, Kornblau SM, Thomas DA, Garcia-Manero G, Waddelow TA, et al. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer. 2001. Jul 15;92(2):406–13. doi: 10.1002/1097-0142(20010715)92:2<406::aid-cncr1336>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 45.Kubasch AS, Schulze F, Giagounidis A, Götze KS, Krönke J, Sockel K, et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia. 2020. Apr;34(4):1182–1186. doi: 10.1038/s41375-019-0645-z. Epub 2019 Dec 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. 2012. Dec;23(6):376–86. doi: 10.1089/hgtb.2012.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005. Jun 1;105(11):4247–54. doi: 10.1182/blood-2004-11-4564. Epub 2005 Feb 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foster MC, Savoldo B, Lau W, Rubinos CA, Grover N, Armistead PM, et al. Utility of Safety Switch to Abrogate CD19.CAR T Cell-Associated Neurotoxicity. Blood. 2021. Feb 23:blood.2021010784. doi: 10.1182/blood.2021010784. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell. 2018. May 31;173(6):1439-1453.e19. doi: 10.1016/j.cell.2018.05.013. Epub 2018 May 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. 2019. Mar;33(3):762–808. doi: 10.1038/s41375-018-0277-8. Epub 2018 Oct 5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.