

Abstract
Aims
Epidermal growth factor receptor (EGFR) is essential to the development of multiple tissues and organs and is a target of cancer therapeutics. Due to the embryonic lethality of global EGFR deletion and conflicting reports of cardiac-overexpressed EGFR mutants, its specific impact on the adult heart, normally or in response to chronic stress, has not been established. Using complimentary genetic strategies to modulate cardiomyocyte-specific EGFR expression, we aim to define its role in the regulation of cardiac function and remodelling.
Methods and results
A floxed EGFR mouse model with α-myosin heavy chain-Cre-mediated cardiomyocyte-specific EGFR downregulation (CM-EGFR-KD mice) developed contractile dysfunction by 9 weeks of age, marked by impaired diastolic relaxation, as monitored via echocardiographic, haemodynamic, and isolated cardiomyocyte contractility analyses. This contractile defect was maintained over time without overt cardiac remodelling until 10 months of age, after which the mice ultimately developed severe heart failure and reduced lifespan. Acute downregulation of EGFR in adult floxed EGFR mice with adeno-associated virus 9 (AAV9)-encoded Cre with a cardiac troponin T promoter (AAV9-cTnT-Cre) recapitulated the CM-EGFR-KD phenotype, while AAV9-cTnT-EGFR treatment of adult CM-EGFR-KD mice rescued the phenotype. Notably, chronic administration of the β-adrenergic receptor agonist isoproterenol effectively and reversibly compensated for the contractile dysfunction in the absence of cardiomyocyte hypertrophy in CM-EGFR-KD mice. Mechanistically, EGFR downregulation reduced the expression of protein phosphatase 2A regulatory subunit Ppp2r3a/PR72, which was associated with decreased phosphorylation of phospholamban and Ca2+ clearance, and whose re-expression via AAV9-cTnT-PR72 rescued the CM-EGFR-KD phenotype.
Conclusions
Altogether, our study highlights a previously unrecognized role for EGFR in maintaining contractile homeostasis under physiologic conditions in the adult heart via regulation of PR72 expression.
Keywords: EGFR, PP2A, PR72, Cardiac contractility, Cardiac hypertrophy
Graphical Abstract

1. Introduction
The erythroblastic leukaemia viral oncogene homolog (ErbB) family of homo- and hetero-dimerizing receptor tyrosine kinases, consisting of epidermal growth factor receptor (EGFR/ErbB1) and ErbB2–4, have been studied extensively for their roles in cancer biology.1 However, ErbB receptors are also expressed in the heart where they have been recognized to regulate a number of biological processes. For instance, ErbB2 has been known for some time to relay cardiotoxicity related to cancer therapeutics,2 engagement of ErbB2/ErbB4 signalling promotes cardiomyocyte proliferation,3–5 and stimulation of EGFR enhances both survival and hypertrophic signalling in isolated cardiomyocytes.6–8 Ligands for ErbB receptors, particularly heparin-binding epidermal growth factor (HB-EGF) and neuregulins, have also been shown to be essential in the regulation of cardiac development and post-injury regeneration.9,10
Aside from direct ligand stimulation, several cardiac-expressed G protein-coupled receptors (GPCR) indirectly transactivate ErbB receptors, most commonly EGFR via autocrine release of HB-EGF, to engage pro-survival and -growth responses including activation of extracellular-regulated kinase (ERK1/2) and Akt.11 Due to enhanced levels of neurohormone released during heart failure, several studies have explored the impact of neurohormone GPCR-mediated EGFR transactivation on cardiac remodelling outcomes, as reviewed previously.11,12 In particular, our lab has investigated the molecular mechanisms and impact of the β-adrenergic receptor (βAR)-mediated EGFR transactivation in the heart,6,11,13 which have supported the notion that EGFR promotes cardiomyocyte survival signalling in the face of cardiac stress but may also promote hypertrophy.
Although EGFR has been suggested to influence several cardiac parameters, either directly or indirectly via GPCR-mediated transactivation, definitive studies into its impact on cardiac structure or function, and the mechanism(s) by which it would do so, are lacking. Genetic mouse models were previously developed to determine the role of EGFR in vivo, initially using global knockdown/knockin expression strategies that resulted in inconclusive cardiac-specific effects due to embryonic/postnatal lethality or other developmental abnormalities.14–18 Subsequent studies have employed more cardiomyocyte-selective approaches, including dominant-negative EGFR transgenic overexpression as well as EGFR deletion in combined vascular smooth muscle cell/cardiomyocyte populations, producing disparate cardiac phenotypes with mechanistically unclear causes.19–21 A more recent study using a cardiomyocyte-specific EGFR knockdown mouse model indicated that EGFR provides protection against acute ischaemic stress,22 but with no insight into cardiac function, long-term survival, or other remodelling parameters over time. Overall, there lacks a succinct in vivo analysis of the impact of cardiomyocyte-expressed EGFR on cardiac function normally or in conjunction with chronic cardiac stress in the adult heart. Here, using complimentary mouse models, we have identified a previously unrecognized role for EGFR in maintaining contractile homeostasis under physiologic conditions and shown that cardiomyocyte-expressed EGFR is a requirement for the hypertrophic response to chronic catecholamine stress.
2. Methods
2.1 Animal models and experimental procedures
All animal experiments were conducted under the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at Temple University under Animal Care and Use Protocol #4902. The mouse strain used for this research project, B6.129S6-Egfrtm1Dwt/Mmnc, identification number 031765-UNC, was obtained from the Mutant Mouse Regional Resource Center (MMRRC), a NIH-funded strain repository, and was donated to the MMRRC by David Threadgill, Ph.D., North Carolina State University. These mice contain a conditional allele of Egfr with exon 3 flanked by two loxP sites (EGFRf/f), deletion of which results in early termination of translation.23 EGFRf/f mice were crossed with αMHC-Cre transgenic mice24 to produce cardiomyocyte-specific EGFR knockdown (CM-EGFR-KD). Unless specifically stated, all experiments were initiated in adult mice between 12 and 16 weeks of age. Both male and female CM-EGFR-KD and EGFRf/f (Cre−) littermates were used throughout the study with numbers of mice used per experiment indicated in figure legends.
Recombinant AAV9 vectors encoding Cre recombinase, enhanced green fluorescent protein (eGFP), human EGFR, murine PR72, or LacZ under the control of the cardiomyocyte-specific troponin T (cTnT) promoter were generated by Vector Biolabs (Malvern, PA, USA). Anaesthetized adult EGFRf/f (Cre−) mice received either AAV9-cTnT-Cre-eGFP or AAV9-cTnT-eGFP, while adult CM-EGFR-KD mice received either AAV9-cTnT-LacZ, AAV9-cTnT-flag-EGFR, or AAV9-cTnT-PR72, via retro-orbital injection of 1 × 1012 genome copies (GC)/mouse in 150 µL.
Echocardiography, haemodynamic assessment, osmotic minipump insertion, and adult mouse cardiomyocyte isolation were performed as described previously13,25–27 and in the Supplementary material online.
Anaesthesia was induced and maintained for the duration of each procedure (echocardiography, retro-orbital injection, osmotic minipump implantation, haemodynamic assessment) via inhalation of isoflurane (3–5%, Patterson Veterinary, Greeley, CO, USA). Immediately following osmotic minipump implantation surgery, mice were given a subcutaneous injection of buprenorphine (0.1 mg/kg). At study end, mice were euthanized via inhalation of CO2 (100%) followed by cervical dislocation as a secondary confirmation of death, or removal of the heart.
2.2. Radioligand binding, skinned myocyte analysis, cardiomyocyte contractility, and Ca2+ transient analyses
Measurements of left ventricular βAR density, myocyte force generation and shortening, as well as Ca2+ sensitivity and transients were performed as described previously25,26,28 and in the Supplementary material online.
2.3 Immunohistochemistry, immunoblotting, quantitative real-time PCR (RT-qPCR), and RNA-seq analyses
Parameters of cardiac remodelling (cardiomyocyte size, cardiomyocyte death, and left ventricular fibrosis), as well as immunoblotting, RT-qPCR, and RNA-seq analyses, were carried out as described previously13,26 and in the Supplementary material online.
2.4 Statistical analyses
GraphPad Prism 9.0.2 software (GraphPad Software Inc., San Diego, CA, USA) was used for all statistical analysis. Data represent the mean ± standard error of the mean (SEM) of at least three individual samples of each group. Two-tailed unpaired t-tests were used for comparison between two groups, and ordinary one-way ANOVA with Tukey’s multiple comparisons test was used for comparison across multiple groups at a single time point. Comparison across two or more groups during time-course studies was performed using ordinary two-way ANOVA with Bonferroni’s multiple comparisons test. Intergroup differences in survival rate were tested by the log-rank (Mantel–Cox) test. Differences were considered significant at P < 0.05.
3. Results
3.1 Cardiomyocyte-specific EGFR downregulation acutely decreases cardiac function
To determine the impact of cardiomyocyte-specific EGFR expression on cardiac structure and function, we crossed mice containing a conditional allele of Egfr, with exon 3 flanked by two loxP sites (EGFRf/f), with αMHC-Cre transgenic mice to produce mice with cardiomyocyte-specific EGFR knockdown (CM-EGFR-KD). These mice displayed ∼50% knockdown of EGFR in adult (12–16-week-old) cardiac membranes (Figure 1A); however, there were no alterations in the expression of EGFR heterodimer partner ErbB2 or the phosphorylation status of known EGFR signalling kinases ERK1/2 and Akt (Supplementary material online, Figure S1A–C), indicating that EGFR knockdown does not lead to aberrant regulation of these canonical effectors. To monitor the impact of cardiomyocyte-specific EGFR knockdown on cardiac function over time, we performed serial echocardiography (Supplementary material online, Table S2). Compared to either αMHC-Cre+ or EGFRf/f (Cre−) controls, CM-EGFR-KD mice exhibited a decrease in both % ejection fraction (EF, Figure 1B) and % fractional shortening (FS, Figure 1C) by 9 weeks of age that persisted thereafter, with no differences noted between male and female CM-EGFR-KD mice (Supplementary material online, Figure S2A and B).
Figure 1.

Cardiomyocyte-specific EGFR downregulation acutely decreases cardiac function. (A) EGFR protein expression in adult EGFRf/f (Cre−) vs. CM-EGFR-KD left ventricular (LV) membrane samples normalized to α-catenin. ***P < 0.001, two-tailed unpaired t-test, n = 8 each. Ejection fraction (%EF, B) and fractional shortening (%FS, C) of EGFRf/f (Cre−) (n = 12), αMHC-Cre+ (n = 8) and CM-EGFR-KD (n = 11) mice were monitored via echocardiography from 6 to 13 weeks of age. *P < 0.05, ***P < 0.001 vs. EGFRf/f (Cre−), ordinary two-way ANOVA with Bonferroni’s multiple comparisons test. (D) EGFR protein expression in the hearts of adult EGFRf/f (Cre−) mice 4 weeks after treatment with AAV9-GFP vs. AAV9-Cre (top panels, n = 4 each) or in the hearts of adult CM-EGFR-KD mice 4 weeks after treatment with AAV9-lacZ vs. AAV9-EGFR (bottom panels, n = 6 each). *P < 0.05, two-tailed unpaired t-test. %EF (E) and %FS (F) of adult EGFRf/f (Cre−) mice treated with AAV9-GFP (n = 5) vs. AAV9-Cre (n = 6) or adult CM-EGFR-KD mice treated with AAV9-lacZ (n = 10) vs. AAV9-EGFR (n = 10) were monitored via echocardiography for 4 weeks following AAV administration (at time 0). ***P < 0.001 AAV9-EGFR vs. AAV9-LacZ in CM-EGFR-KD mice and †P < 0.05, †††P < 0.001 AAV9-Cre vs. AAV9-GFP in EGFRf/f (Cre−) mice at corresponding timepoints, ordinary two-way ANOVA with Bonferroni’s multiple comparisons test.
Although we did not observe a decrease in cardiac contractility in CM-EGFR-KD mice until ∼9 weeks of age, αMHC-Cre-mediated deletion of EGFR would occur during early developmental stages29 that could impact contractility at later timepoints. Thus, to understand whether alterations in EGFR expression in fully differentiated adult hearts alter cardiac function, we employed AAV9-mediated gene delivery in order to acutely delete or re-express cardiomyocyte-specific EGFR. To this end, we retro-orbitally injected AAV9-cTnT-Cre-eGFP to 12-week-old EGFRf/f (Cre−) mice, with AAV9-cTnT-eGFP used as a control vector in a subset of EGFRf/f (Cre−) mice (Figure 1D, Supplementary material online, Figure S1D). Indeed, AAV9-mediated induction of Cre in EGFRf/f (Cre−) mouse hearts led to cardiac dysfunction that was detectable as early as 1-week post-AAV9 injection (Supplementary material online, Table S3) and by 4 weeks post-injection achieved a similar decrease in %EF (Figure 1E) and %FS (Figure 1F) as had been observed in the CM-EGFR-KD mice. Conversely, to assess whether contractile function could be acutely restored with EGFR re-expression in adult CM-EGFR-KD mice, we delivered AAV9-cTnT-flag-EGFR to 12-week-old CM-EGFR-KD mice, after cardiac dysfunction had manifested, whereas AAV9-cTnT-lacZ was given as a control vector to a subset of CM-EGFR-KD mice (Figure 1D, Supplementary material online, Figure S1E). As early as 1-week post-delivery, AAV9-cTnT-EGFR improved cardiac function in CM-EGFR-KD mice (Figure 1E and F, Supplementary material online, Table S3), which was recovered to EGFRf/f (Cre−) levels by 4 weeks post-injection. These results demonstrate that cardiac contractility is acutely sensitive to the manipulation of cardiomyocyte-specific EGFR expression.
3.2 Cardiomyocyte-specific EGFR downregulation induces delayed cardiac remodelling
We next assessed whether CM-EGFR-KD-induced changes in contractile function were associated with structural alterations or the development of pathological cardiac remodelling. Despite the presence of cardiac dysfunction in adult CM-EGFR-KD mice (12 weeks of age), there were no changes in cardiac remodelling parameters including cardiomyocyte size, cardiomyocyte cell death, or cardiac fibrosis (Supplementary material online, Figure S3A–C). As detected via echocardiography, there were no consistent differences in diastolic wall thicknesses, though there were decreases in systolic wall thicknesses in CM-EGFR-KD mouse hearts over time (Supplementary material online, Table S2), which were also observed in the acute AAV-Cre modified mice and normalized in the acute AAV-EGFR modified mice (Supplementary material online, Table S3). However, these small changes in systolic wall thicknesses did not translate into differences in heart weights in any of the cohorts (Supplementary material online, Figure S3D) or isolated cardiomyocyte size (Supplementary material online, Figure S3E). Thus, cardiomyocyte-specific EGFR downregulation does not acutely induce cardiac remodelling despite diminished contractile function.
However, by 10 months of age CM-EGFR-KD mice displayed evidence of pathological cardiac remodelling including cardiomyocyte hypertrophy and death as well as interstitial fibrosis (Figure 2A–C). At a molecular level, there was increased expression of several genes responsive to cardiac stress and involved in remodelling and inflammatory processes, including Acta1, Myh7, Nppa, Il6, and Tnfa (Figure 2D). Additionally, compared to younger CM-EGFR-KD mice (Figure 1), contractile dysfunction was severely reduced by 10 months, as evidenced by decreases in %EF and %FS to ∼20% and 10%, respectively (Figure 2E and F). Ultimately, CM-EGFR-KD mice began to exhibit enhanced mortality vs. their littermate controls at ∼1 year of age, with median survival at 13 months of age (Figure 2G) with no significant differences between male and female CM-EGFR-KD mice amongst these parameters (Supplementary material online, Figure S2C–H). These findings demonstrate that cardiomyocyte-specific downregulation of EGFR directly mediates a rapid loss in contractile function in the absence of cardiac remodelling; however, without intervention to normalize EGFR expression, this loss in contractile function eventually promotes pathological cardiac remodelling, heart failure, and enhanced mortality.
Figure 2.

Cardiomyocyte-specific EGFR downregulation induces delayed cardiac remodelling. Cardiac structure was assessed in the LV of 10-month-old EGFRf/f (Cre−) vs. CM-EGFR-KD mice via WGA (A), TUNEL/TNNI (B), and MTC (C) staining. Panels shown are representative images from n = 6 (EGFRf/f (Cre−)) and n = 5 (CM-EGFR-KD) mouse hearts; scale bars denote 100 µm and arrowheads denote TUNEL-positive cardiomyocytes. Quantification of cardiomyocyte size, death, and cardiac fibrosis are shown in accompanying histograms. ***P < 0.001, two-tailed unpaired t-test. (D) RT-qPCR was used to measure the expression of Acta1, Myh7, Nppa, Il6, and Tnfa in the LV of 10 month-old EGFRf/f (Cre−) (n = 5) vs. CM-EGFR-KD (n = 6) mice. P < 0.05, **P < 0.01, two-tailed unpaired t-test. %EF (E) and %FS (F) of 10-month-old EGFRf/f (Cre−) (n = 10) and CM-EGFR-KD (n = 7) mice were measured via echocardiography. ***P < 0.001, two-tailed unpaired t-test. (G) EGFRf/f (Cre−, n = 19) and CM-EGFR-KD (n = 28) mice were monitored for survival over time. ***P < 0.001 vs. EGFRf/f (Cre−), Log-rank (Mantel–Cox) test.
3.3 Chronic catecholamine stimulation reversibly rescues contractile dysfunction without hypertrophy in CM-EGFR-KD mice
Adult CM-EGFR-KD mice display reduced cardiac contractility in the absence of gross morphological remodelling, suggesting that the molecular machinery for contractility remains intact, one of the major drivers of which is the βAR system. Thus, we next sought to determine whether chronic βAR stimulation could rescue the contractile dysfunction already manifested in CM-EGFR-KD mice. To test this, we infused adult CM-EGFR-KD and EGFRf/f (Cre−) littermate control mice with the βAR agonist ISO13,30 for up to 4 weeks and monitored cardiac function via weekly echocardiography. Chronic ISO infusion improved contractility in CM-EGFR-KD mice to levels not different from EGFRf/f (Cre−) littermate controls throughout the infusion period (Figure 3A and B, Supplementary material online, Table S4). Notably, the chronic catecholamine-enhanced contractility in CM-EGFR-KD mice was reversible upon cessation of ISO infusion as withdrawal of the minipumps after 2 weeks resulted in a return of contractility to baseline CM-EGFR-KD levels. Thus, the βAR system remains responsive to sympathetic stimulation in CM-EGFR-KD mice and can chronically compensate for the EGFR-dependent loss in baseline contractility.
Figure 3.

Chronic catecholamine stimulation reversibly rescues contractile dysfunction without hypertrophy in CM-EGFR-KD mice. %EF (A) and %FS (B) of adult EGFRf/f (Cre−) or CM-EGFR-KD mice infused with vehicle or ISO (3 mg/kg/day) were monitored via echocardiography for up to 4 weeks. n = 7 EGFRf/f (Cre−) + Veh, EGFRf/f (Cre−) + ISO, and CM-EGFR-KD + Veh, n = 11 CM-EGFR-KD + ISO and n = 6 CM-EGFR-KD + ISO (2 weeks on/2 weeks off). **P < 0.01 CM-EGFR-KD + Veh vs. CM-EGFR-KD + ISO, †P < 0.05, ††P < 0.01 CM-EGFR-KD + Veh vs. CM-EGFR-KD + ISO (2 weeks on/2 weeks off), #P < 0.05, ###P < 0.001 EGFRf/f (Cre−) + Veh vs. CM-EGFR-KD + ISO, ‡‡‡P < 0.001 EGFRf/f (Cre−) + Veh vs. CM-EGFR-KD + ISO (2 weeks on/2 weeks off) at corresponding timepoints, ordinary two-way ANOVA with Bonferonni’s multiple comparisons test. (C) WGA staining of the LV of adult EGFRf/f (Cre−) vs. CM-EGFR-KD mice infused with vehicle or ISO for 2 (left) or 4 weeks (right). Panels shown are representative images from n = 6 [EGFRf/f (Cre−) Veh and ISO 2 weeks], n = 8 (CM-EGFR-KD Veh and ISO 2 weeks), n = 7 [EGFRf/f (Cre−) Veh and ISO 4 weeks] and n = 7 (CM-EGFR-KD Veh and ISO 4 weeks) mouse hearts; scale bars denote 100 µm. (D) Quantification of cardiomyocyte size from (C). ***P < 0.001, ordinary one-way ANOVA with Tukey’s multiple comparisons test. (E) βAR LV membrane density was measured in adult EGFRf/f (Cre−) vs. CM-EGFR-KD mice (n = 4 each) infused with vehicle or ISO for 4 weeks. ns, not significant, ordinary one-way ANOVA with Tukey’s multiple comparisons test.
Chronic catecholamine stress-induced cardiac hypertrophy has been postulated to be EGFR-sensitive, but with variable results across studies using pharmacologic (EGFR inhibitors) or genetic (overexpression of EGFR mutants) approaches.11,13,19,21,30–33 Thus, using our CM-EGFR-KD model, we investigated whether cardiomyocyte-specific EGFR downregulation impacts chronic ISO-induced cardiac hypertrophy. Although chronic ISO infusion for either 2 or 4 weeks significantly increased cardiomyocyte size in control mice, this effect was abrogated in CM-EGFR-KD mice (Figure 3C and D). Notably, we found that βAR density was not different at baseline in Veh-infused CM-EGFR-KD and EGFRf/f (Cre−) control hearts and that receptor density became reduced to a similar extent in each mouse line by chronic ISO infusion (Figure 3E), thus EGFR downregulation does not impact βAR downregulation during chronic catecholamine stimulation. Collectively, these findings demonstrate that βAR responsiveness to catecholamine stimulation is maintained in the face of EGFR downregulation and, despite some degree of receptor desensitization, can reversibly rescue the decreased cardiac contractility in CM-EGFR-KD mice. Further, these data highlight that cardiomyocyte-specific EGFR expression is required to promote cardiac hypertrophic remodelling in response to chronic catecholamine stimulation.
3.4 Cardiomyocyte-specific EGFR downregulation leads to differential temporal effects on cardiac gene expression
To begin to attain molecular insight into the contractile dysfunction manifested in CM-EGFR-KD mice, we next investigated whether they exhibited alterations in cardiac gene expression over time. To this end, we performed RNAseq analysis of LV samples from 7-week-old (no dysfunction) and 9-week-old (earliest dysfunction manifested) CM-EGFR-KD mice vs. EGFRf/f (Cre−) littermate controls. Cardiomyocyte-specific downregulation of EGFR resulted in a distinct profile of gene expression at 7 vs. 9 weeks of age. Although 284 genes were differentially altered at 7 weeks (Supplementary material online, Table S5), with most upregulated, these changes were not sustained over time as only 67 were altered at 9 weeks (Supplementary material online, Table S6), with most downregulated (Figure 4A). Pathway analysis confirmed that a majority of the canonical signalling pathways identified increased at 7 weeks and decreased at 9 weeks in CM-EGFR-KD vs. EGFRf/f (Cre−) control hearts (Figure 4B). Several genes encoding proteins involved in regulating cardiac contractility via cAMP-dependent signalling, including Adcy5, Prkaa2, Prkab2, and Pde3a, were indicated to be altered in CM-EGFR-KD vs. EGFRf/f (Cre−) littermate control hearts at 7 weeks of age, but to be normalized by 9 weeks of age. In conjunction with our data above, these results are consistent with the contractile defect in CM-EGFR-KD mice not being a result of aberrant βAR signalling.
Figure 4.

Cardiomyocyte-specific EGFR downregulation leads to differential temporal effects on cardiac gene expression. (A) Heat maps depicting significant changes in cardiac gene expression in CM-EGFR-KD vs. EGFRf/f (Cre−) hearts (n = 3 each) at 7 (left) or 9 (right) weeks of age. Red and green colours represent upregulated and downregulated genes, respectively. (B) Top signalling pathways altered at 7 (upper panel) and 9 (lower panel) weeks of age in the LV of CM-EGFR-KD vs. EGFRf/f (Cre−) mice as determined by Ingenuity Pathway Analysis (IPA). Blue bars depict downregulation and orange bars upregulation. (C) RT-qPCR was used to measure the expression of Myl1, Myl4, Myl7, Myh7, Mybphl, and Myot in the LV of EGFRf/f (Cre−) vs. CM-EGFR-KD mice (n = 4 each). ns, not significant, *P < 0.05, **P < 0.01, two-tailed unpaired t-test. (D) Mean force as a function of calcium concentration and fitted curves for skinned myocytes isolated from adult EGFRf/f (Cre−) vs. CM-EGFR-KD mice, maximal Ca2+-activated force (Fmax, E) and sensitivity (EC50, F) shown in histograms. Measurements were attained from 3 skinned myocytes per LV isolated from three independent LV samples per genotype.
After assessing the canonical signalling pathways most significantly altered in CM-EGFR-KD hearts, we focused in particular on the Ca2+ and cytoskeleton groups as they were downregulated at 9 weeks when cardiac dysfunction had manifested. RT-qPCR analysis of genes clustered within these groups, including several myosins and myofibril-related proteins (Supplementary material online, Table S6), indeed demonstrated decreased expression at the 9-week timepoint (Figure 4C), which could suggest an abnormality in the regulation of contractile dynamics at the level of the myofilament. However, using permeabilized myocytes from adult CM-EGFR-KD or EGFRf/f (Cre−) littermate controls, we observed no difference in maximal Ca2+-activated force (Fmax) or sensitivity (EC50) (Figure 4D–F), indicating that, despite the reduction in contractility, myofilament sensitivity to Ca2+ and contractile potential remained unchanged in the absence of EGFR. Consequently, dysregulation of cardiomyocyte Ca2+ handling itself may play a role in manifesting the contractile dysfunction observed in CM-EGFR-KD mice.
3.5 Cardiomyocyte-specific EGFR downregulation alters Ca2+ handling during relaxation
Cardiomyocyte-specific EGFR downregulation induced a rapid onset of contractile dysfunction without overt cardiac remodelling or sustained changes in broad gene expression patterns, suggesting that one or more components of the cardiac excitation–contraction cycle may be altered more directly. To more precisely assess parameters of cardiac contractility, we subjected adult CM-EGFR-KD and EGFRf/f (Cre−) littermate control mice to terminal haemodynamics at baseline and upon challenge with ISO. Although there was no difference in mean systemic pressure (Figure 5A) or heart rate (Figure 5B), we observed decreased rates of contraction (Figure 5C) and relaxation (Figure 5D) at baseline. Acute ISO infusion was sufficient to enhance heart rate and rate of contraction similarly in both CM-EGFR-KD and EGFRf/f (Cre−) littermate mice, again consistent with the βAR system remaining intact when cardiomyocyte-expressed EGFR is downregulated. We next evaluated contractile parameters at a cellular level using field-stimulated isolated AMCM (Figure 5E). There were no significant differences between CM-EGFR-KD and EGFRf/f (Cre−) littermate control AMCM with respect to the magnitude of contraction produced either at baseline or following ISO stimulation (Figure 5F). However, similar to the haemodynamics data, CM-EGFR-KD AMCM displayed prolonged relaxation at baseline (Figure 5G).
Figure 5.

Cardiomyocyte-specific EGFR downregulation impairs relaxation dynamics. Haemodynamic evaluation of mean systolic pressure (SP, A), mean heart rate (HR, B), +dP/dt max (C) and -dP/dt max (D) in adult EGFRf/f (Cre−) vs. CM-EGFR-KD mice at baseline and in response to infusion with 10 ng ISO, n = 9 each. *P < 0.05, **P < 0.01, ***P < 0.001, ordinary one-way ANOVA with Tukey’s multiple comparisons test. (E) LV cardiomyocytes were freshly isolated from adult EGFRf/f (Cre−) vs. CM-EGFR-KD hearts and cardiomyocyte length measured at baseline (black line) and after stimulation with ISO (0.1 μM, red line), as shown in tracings representative of n = 9 individual cardiomyocytes isolated from n = 3 mice per genotype, with quantification of cardiomyocyte shortening (F) and time to 50% relaxation (G) summarized in histograms. ns, not significant, ***P < 0.001, ordinary one-way ANOVA with Tukey’s multiple comparisons test.
Changes in contraction/relaxation dynamics, as well as the high prediction of alterations in Ca2+ signalling from the RNASeq analysis, suggest aberrant Ca2+ handling or sensitivity. Conventional cardiomyocyte contraction–relaxation mechanisms depend on Ca2+ mobilization to and clearance from the sarcomere,12 thus we performed comparative Ca2+ transient analyses between CM-EGFR-KD and littermate EGFRf/f (Cre−) control AMCM (Figure 6A). Consistent with our findings above, the magnitude of Ca2+ transients were not different between CM-EGFR-KD and EGFRf/f (Cre−) littermate control AMCM at baseline or in response to ISO (Figure 6B), but EGFR downregulation did produce delayed Ca2+ clearance, both basally and in response to ISO (Figure 6C).
Figure 6.
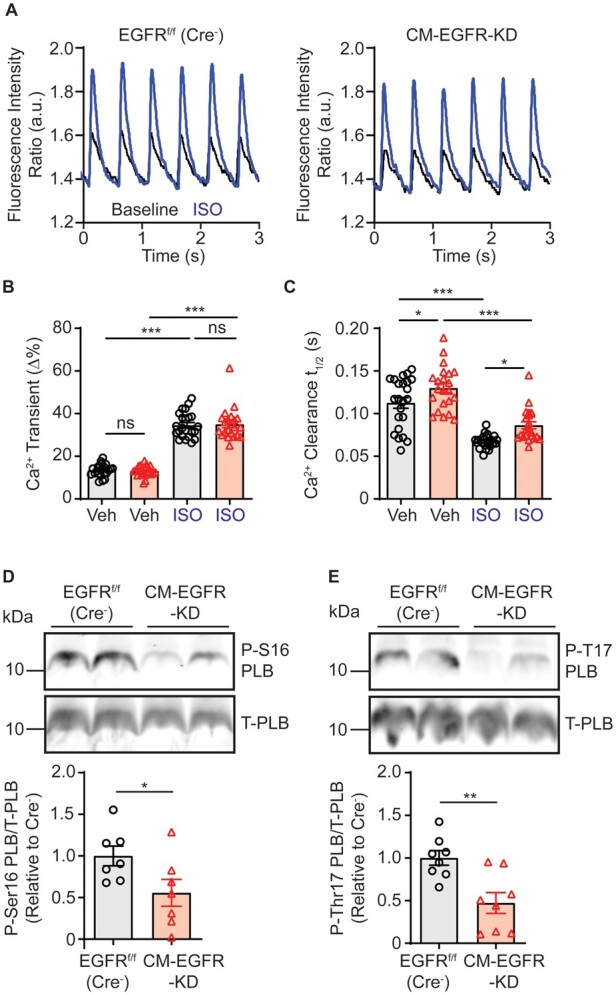
Cardiomyocyte-specific EGFR downregulation alters Ca2+ reuptake. (A) LV cardiomyocytes were freshly isolated from adult EGFRf/f (Cre−) vs. CM-EGFR-KD hearts, loaded with fura2 and stimulated at 2 Hz and Ca2+ transients measured at baseline (black line) and after stimulation with ISO (0.1 μM, red line), as shown in tracings representative of n = 23 (EGFRf/f (Cre−)) and n = 22 (CM-EGFR-KD) individual cardiomyocytes isolated from n = 3 mice each, with quantification of peak Ca2+ transients (B) and time to 50% Ca2+ clearance (C) summarized in histograms. ns, not significant, *P < 0.05, ***P < 0.001, ordinary one-way ANOVA with Tukey’s multiple comparisons test. Immunoblot analysis of adult EGFRf/f (Cre−) vs. CM-EGFR-KD cardiac LV lysates, as summarized in histograms, for Ser16 phospholamban (P-S16-PLB, D, n = 7 each) and Thr17 phospholamban (P-T17-PLB, E, n = 8 each). *P < 0.05, **P < 0.01, two-tailed unpaired t-test.
Efficient Ca2+ clearance during cardiomyocyte relaxation can be enhanced via several mechanisms, including Ca2+ reuptake into the SR by SERCA, which is enhanced by phosphorylation of the inhibitory protein phospholamban (PLB) at either Ser16 by PKA or Thr17 by CAMKII.34 Additionally, Ca2+ dissociation from TnC can be enhanced via phosphorylation of cTnI, potentially contributing to accelerated relaxation.31 Thus, we measured the phosphorylation status of PLB and cTnI in LV lysates from adult CM-EGFR-KD or EGFRf/f (Cre−) littermate control mice. Consistent with decreased cardiac contractility and Ca2+ clearance during cardiomyocyte relaxation, PLB phosphorylation at both Ser16 and Thr17 was reduced with EGFR downregulation (Figure 6D and E), as was phosphorylation of cTnI at Ser23/24 (Supplementary material online, Figure S4A). Collectively, these results demonstrate that impaired cardiac contractility caused by EGFR downregulation is associated with dysfunctional Ca2+ clearance.
3.6 Cardiomyocyte-specific EGFR downregulation decreases expression of the PP2A B″ regulatory subunit PR72
Since PLB and cTnI phosphorylation are regulated by multiple kinases downstream of βAR, including PKA and CAMKII, but βAR signalling remains intact, we hypothesized that their reduced phosphorylation levels may be due to a change in dephosphorylation dynamics. Thus, we next tested whether EGFR downregulation in cardiomyocytes impacts the expression of phosphatase subunits belonging to the protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) families, which have been shown to regulate the phosphorylation status of PLB.35,36 Our RNASeq data indicated a significant difference in transcript expression between CM-EGFR-KD and EGFRf/f (Cre−) littermate control hearts for two PP1 regulatory subunits (Ppp1r12b, Ppp1r3a) and one PP2A B” alpha regulatory subunit (Ppp2r3a) at either the 7- or 9-week timepoints. Via RT-qPCR-mediated validation experiments with an expanded sample size across early and later age groups, we found no change in expression levels of the PP1 regulatory subunits at any time point (Supplementary material online, Figure S4B and C); however, the transcript level of Ppp2r3a was consistently reduced in CM-EGFR-KD hearts at all ages tested (Figure 7A). Further, we confirmed that adult CM-EGFR-KD hearts also displayed significantly decreased Ppp2r3a protein expression, specifically the PR72 isoform (Figure 7B). While a very small decrease in the expression of the catalytic subunit PP2Ac was detected (Supplementary material online, Figure S4D), its global activity was not decreased in adult CM-EGFR-KD hearts (Supplementary material online, Figure S4E), suggesting that an alteration in its targeting to intracellular protein substrates may be a consequence of decreased PR72 expression in CM-EGFR-KD mice.
Figure 7.

Cardiomyocyte-specific EGFR downregulation decreases expression of the PP2A B” regulatory subunit PR72. (A) RT-qPCR was used to measure the expression of Ppp2r3a in the LV of EGFRf/f (Cre−) vs. CM-EGFR-KD mice at 7, 9, and 16 weeks of age (n = 6 each). ***P < 0.001, two-tailed unpaired t-test. (B) Immunoblot analysis of PR72 expression in adult EGFRf/f (Cre−) vs. CM-EGFR-KD cardiac lysates, as summarized in histogram. Blots shown are representative of n = 11 cardiac lysates per genotype. ***P < 0.001, two-tailed unpaired t-test. (C) Immunoblot analysis of PR72 expression in adult CM-EGFR-KD mouse cardiomyocytes infected for 24 h with adenoviruses encoding lacZ or PR72, as summarized in histogram. Blots shown are representative of n = 4 independent cardiomyocyte preparations. **P < 0.01, two-tailed unpaired t-test. (D) Time to 50% relaxation measured in lacZ vs. PR72-infected AMCM from CM-EGFR-KD mice. *P < 0.05, two-tailed unpaired t-test. n = 14–15 individual cardiomyocytes isolated from n = 5 mice per adenovirus infection. (E) Time to 50% Ca2+ clearance in lacZ vs. PR72-infected AMCM from CM-EGFR-KD mice. *P < 0.05, two-tailed unpaired t-test. n = 16–19 individual cardiomyocytes isolated from n = 3 mice per condition. (F) Immunoblot analysis of PR72 expression in adult CM-EGFR-KD cardiac lysates, 2 weeks after infection with AAV9-lacZ or AAV9-PR72, as summarized in histogram. Blots shown are representative of n = 6 cardiac lysates per condition. *P < 0.05, two-tailed unpaired t-test. %EF (G) and %FS (H) of adult CM-EGFR-KD mice treated with AAV9-lacZ (n = 10, reference group from Figure 1E and F, respectively) vs. AAV9-PR72 (n = 5) were monitored via echocardiography for 4 weeks following AAV administration (at time 0). *P < 0.05, **P < 0.01, ***P < 0.001 vs. reference group CM-EGFR-KD+AAV9-lacZ at corresponding timepoints, ordinary two-way ANOVA with Bonferonni’s multiple comparisons test.
Since a decrease in PR72 expression was associated with prolonged relaxation and Ca2+ clearance in CM-EGFR-KD mice, we sought to determine whether rescue of PR72 expression could restore these parameters. To test this, we infected primary AMCM isolated from CM-EGFR-KD mice with adenoviruses encoding PR72 or lacZ as a control (Figure 7C) and measured cardiomyocyte relaxation and Ca2+ clearance rates. Indeed, upregulation of PR72 expression in CM-EGFR-KD AMCM improved both parameters, with decreased time to 50% relaxation (Figure 7D) and Ca2+ clearance (Figure 7E). To next determine if the contractile dysfunction phenotype of CM-EGFR-KD mice could be rescued by re-expression of PR72, we injected adult CM-EGFR-KD mice with AAV9-cTnT-PR72 vs. AAV9-cTnT-LacZ control (Figure 7F, Supplementary material online, Figure S4F). Similar to re-expression of EGFR in these mice, treatment with AAV9-cTnT-PR72 resulted in a restoration of contractile function over time (Figure 7G and H, Supplementary material online, Table S3).
Altogether, these data demonstrate that EGFR normally exerts homeostatic control of cardiac contractility via regulation of PR72 expression, whereas EGFR downregulation in cardiomyocytes leads to the acute repression of PR72 expression and dysregulation of Ca2+ dynamics and cardiac contractility. Further, although chronic catecholamine stimulation normally acts via EGFR transactivation to induce cardiac hypertrophy, this response is absent when EGFR is downregulated and instead acts to compensate for the loss in contractility.
4. Discussion
While EGFR signalling in cardiomyocytes may provide a potentially important target to regulate cardiac remodelling during heart failure (HF), especially in relation to its role as a signalling node for neurohormone GPCRs in general, a major hurdle in defining its impact during HF is the interpretability of in vivo studies. Exploration of the impact of EGFR signalling on cardiac function and remodelling in vivo either via GPCR-mediated transactivation or otherwise have relied primarily upon pharmacologic EGFR inhibition. In vivo use of EGFR inhibitors is limited by agent selectivity profiles37 and has produced variable and conflicting reports regarding the impact of EGFR inhibition on cardiac function and remodelling either alone or in conjunction with different models of cardiac stress.13,30–33 Alternatively, genetic models were developed, initially to define the impact of EGFR on developmental processes, including global EGFR knockout (KO) mice, mice containing a point mutation in the kinase domain of EGFR rendering it almost inactive, as well as a humanized EGFR knock-in mouse line.14–18 However, while these global models demonstrated varying levels of cardiac remodelling processes over time, they either experienced embryonic/post-natal lethality or displayed a number of other developmental abnormalities throughout the body that precluded a clear understanding of the impact of EGFR specifically expressed in the heart.
Subsequently, genetic models were developed in which EGFR expression or activity was manipulated more selectively in the heart. One group crossed EGFRf/f and smooth muscle-specific protein 22-Cre mice, which resulted in downregulation of EGFR in both vascular smooth muscle cells (VSMC) and cardiomyocytes and the development of systemic hypotension with cardiac hypertrophy.20 Initially, interpretation of the cardiac phenotype distinct from the vascular phenotype in this model was unclear; however, a follow-up study using inducible VSMC-specific (smooth muscle myosin heavy chain) Cre mice showed normal cardiac EGFR expression with no hypertrophy.38 While this indirectly suggested that loss of cardiomyocyte-expressed EGFR may promote cardiac hypertrophy, our results now clearly show that cardiomyocyte-specific downregulation of EGFR does not promote hypertrophy until a later age when cardiac function has undergone severe deterioration.
Other groups have employed cardiomyocyte-specific transgenic overexpression of an intracellular domain-deficient EGFR that acted as a dominant negative (DN-EGFR) with endogenous ErbB receptors.19,21 However, these studies produced disparate effects on cardiac function and remodelling, with one group observing a lack of cardiac dysfunction at any age tested in constitutive DN-EGFR-expressing mice21 and the other group observing an immediate decline in contractility concurrent with increased cardiac dilation and hypertrophy following induction of the DN-EGFR in adult mice.19 While the differences in outcomes in these studies could be attributable to distinct methods, the extent of induction of DN-EGFR or the construct differentially altering ErbB2 activity, ultimately using two independent methods we show via echocardiography, invasive haemodynamics, and isolated cardiomyocyte contractility assays that downregulation of EGFR expression, rather than overexpression of a mutant EGFR, leads to decreased contractile function without concurrent cardiac remodelling. Instead, pathologic cardiac remodelling appears to occur in a delayed manner in these mice that only occurs following long-term contractile dysfunction. We also demonstrate that EGFR downregulation in cardiomyocytes does not alter ErbB2 expression or downstream activation status of its common effectors ERK1/2 and Akt, although these were measured in total left ventricular lysates that contain fibroblasts as well as vascular smooth muscle, immune and endothelial cells and would contribute to the signal. Alterations in cardiomyocyte-specific ErbB2 expression have been shown to directly alter cardiomyocyte growth and proliferation at various developmental stages and in response to injury,4,5,39 highlighting the essential role for ErbB2 in maintaining cardiac structure. Conversely, since αMHC turns on early,29 our data indicate that cardiomyocyte-specific EGFR is not required for cardiac post-natal development, but becomes essential in maintaining contractile function as cardiac development progresses toward adulthood. Further, acute AAV9-mediated EGFR downregulation in the adult heart demonstrated that EGFR expression is required for the continued maintenance of contractility in fully matured hearts without an overt impact on the structure. Thus, while ErbB2 is essential for cardiomyocyte proliferation and maintenance of cardiac structure, EGFR plays a more predominant role in the homeostasis of contractile function.
Cardiac contractility was decreased secondary to reduced EGFR expression, despite βAR density, responsiveness to stimulation, and downstream impact on contractility remaining intact. Further, although chronic βAR stimulation in control mice led to increased cardiomyocyte hypertrophy with a slight reduction in contractility over time, CM-EGFR-KD mice regained normal contractile function and were protected from hypertrophic remodelling. These outcomes are notable for a number of reasons. First, even though βAR and EGFR are known to associate and bi-directional regulation of their downstream signalling pathways has been reported by us and others,6,13,30,40–42 we demonstrate here that EGFR downregulation does not alter βAR density or amplitude of catecholamine-mediated contractility. Thus, β1AR and EGFR regulate cardiac contractility via distinct mechanisms and EGFR transactivation does not directly modulate β1AR-mediated cardiomyocyte contraction. Second, although EGFR transactivation by various GPCRs has been implicated in the induction of cardiac remodelling in response to chronic neurohormone stimulation, various pharmacologic and genetic models have led to conflicting interpretation of the role of EGFR in remodelling outcomes.13,30–33 However, we definitively showed that with reduced EGFR expression specifically in cardiomyocytes, chronic catecholamine stimulation is no longer able to induce a hypertrophic response in cardiomyocytes. Lastly, our study highlights that chronic βAR stimulation is able to stably and reversibly rescue the contractile dysfunction in CM-EGFR-KD mice in the absence of hypertrophy. Consistent with earlier studies showing that hypertrophy is not required for maintaining contractile function in the heart under conditions of stress,43,44 our study therefore suggests that βAR-mediated promotion of contractility in the absence of EGFR-dependent hypertrophic remodelling could provide beneficial outcomes under stress conditions in which elevated catecholamine levels would be expected to contribute to hypertrophic cardiomyopathy. However, whether the decreased hypertrophic response to chronic catecholamine stimulation in CM-EGFR-KD mice is directly due to loss of βAR-mediated transactivation-dependent hypertrophic signalling or rather to the preservation of cardiac contractility via βAR stimulation remains to be determined.
Previous studies investigating the role of cardiomyocyte-expressed EGFR on cardiac function and remodelling have been primarily phenotype-based,19–21 with little insight into mechanistic underpinnings that could explain the effects of altered cardiomyocyte-specific EGFR expression/activity on cardiac parameters. Here, we show that downregulation of EGFR in cardiomyocytes results in the altered expression of cardiac genes in a temporally dependent manner. A majority of the cardiac gene expression changes were detected at 7 weeks of age, with normalization of three quarters of these by 9 weeks of age. These data suggest that many of the gene changes observed early, before cardiac dysfunction manifested, may be elevated in the CM-EGFR-KD mice as a compensatory process that is not sustained as the mice mature, wherein relatively fewer gene changes account for the actual cardiac dysfunction. Notably, of the myofibril-related genes that were shown to be downregulated at 9 weeks of age when cardiac dysfunction was present, several encode atrial or skeletal isoforms known to be involved in development, arrhythmias or myofibrillar myopathies,45–48 suggesting that their LV downregulation could reflect an attempt to normalize cardiac function at the level of the myofilament. While we did not detect abnormalities in either cardiac structure or myofilament Ca2+ sensitivity and thus did not further investigate these changes in gene expression, future work focused on how EGFR modulates the expression and function of myofilament proteins may be warranted.
Beyond the myofilament, our data clearly pointed to dysregulated Ca2+ handling as a possible mechanism involved in the contractile dysfunction phenotype observed in CM-EGFR-KD mice. Notably, we detected a reduction in phosphorylation of PLB and cTnI, which would be expected to decrease Ca2+ reuptake into the sarcoplasmic reticulum12,34 and reduce the Ca2+ dissociation rate from the sarcomere,49 respectively, leading to slower relaxation kinetics. Indeed, we observed a decrease in cardiac relaxation at baseline and following ISO stimulation in vivo that was associated with reduced Ca2+ reuptake as measured in isolated cardiomyocytes. While reduced phosphorylation of PLB or cTnI could be secondary to decreased kinase activity, no evidence of this arose since βAR signalling pathways and functional responsiveness to βAR stimulation were not altered. Thus, we focused instead on dephosphorylation dynamics, observing that the regulatory phosphatase subunit Ppp2r3a/PR72 in particular was downregulated at all timepoints measured in CM-EGFR-KD mice.
Despite its identification as a cardiac-expressed B″ regulatory subunit of the PP2A holoenzyme over 2 decades ago,50 and its recently reported involvement in cardiac development in zebrafish,51 very little is known about the role of PR72 in the regulation of cardiac function in the adult heart, representing a limitation of our study. However, increased expression of cardiac PR72 was detected in both non-ischaemic and ischaemic human HF as well as a canine model of HF,52 which coincides with a reported increase in EGFR expression in ischaemic human heart tissue,22 though the consequences of this upregulation are unknown. Inferences can be made based on the reported localization of PR72 in adult cardiomyocytes at Z and M lines52 and PP2A at the myofilaments,53,54 which would suggest a role in the regulation of PP2A activity at the level of the sarcomere and disruption of which upon reduced PR72 expression could lead to more diffuse PP2Ac localization toward other targets such as the sarcoplasmic reticulum. However, in addition to decreased PLB phosphorylation, we also observed decreased cTnI phosphorylation which may suggest enhanced accumulation of PP2Ac at the myofilaments with reduced PR72 expression, thus the impact of PR72-dependent regulation on PP2Ac localization within cardiomyocytes requires additional study. Notably, both increased and decreased cardiomyocyte-specific PP2Ac expression has been shown to reduce contractile function and promote hypertrophy and fibrosis,53,55,56 but in our study, we observed decreased contractility in the absence of cardiac remodelling events. This would be consistent with the idea that the various B regulatory subunits of the PP2A holoenzyme specifically regulate certain pools of PP2A activity.35,57 Therefore, since the rescue of PR72 expression restored cardiac contractility both in vitro and in vivo, PR72 may normally act to restrict excessive PP2A access to targets including PLB and cTnI, which will be the focus of ongoing investigation.
In summary, using complimentary mouse models, we have for the first time identified the impact of cardiomyocyte-expressed EGFR on the homeostatic regulation of contractility via a novel regulation of the PP2A B″ regulatory subunit PR72. Further, we have conclusively demonstrated that cardiomyocyte-expressed EGFR is required for the promotion of hypertrophy under conditions of chronic catecholamine stress. Thus, strategies to uncouple β1AR-mediated transactivation of EGFR may provide a means by which to preserve both catecholamine- and EGFR-dependent regulation of cardiac contractility in the absence of hypertrophic remodelling during the development and progression of HF.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
The authors approve of this work and contributed as follows: experimental design (SG, DGT), data acquisition and analysis (SG, ADO, EM, JLS, RLC, VCP, TPT, ML, JS, AML, TGM, EG, SR), interpretation of data (SG, JAK, WJK, JYC, DGT), and drafting the work (SG, DGT).
Funding
This work was supported by National Institutes of Health grants R01 HL105414, R01 HL136219 and R01 HL139522 (D.G.T.), R01 HL136737 (J.A.K.), P01 HL147841 (W.J.K., D.G.T, E.G., S.R.), and F31 HL154814 (A.D.O).
Conflict of interest: none declared.
Data availability
The data underlying this article will be shared upon reasonable request to the corresponding authors. RNA-Seq data underlying this article are available in GEO at https://www.ncbi.nlm.nih.gov/geo/ and can be accessed with accession number GSE140407.
Supplementary Material
Contributor Information
Shuchi Guo, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Ama Dedo Okyere, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Erin McEachern, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Joshua L Strong, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Rhonda L Carter, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Viren C Patwa, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Toby P Thomas, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Melissa Landy, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Jianliang Song, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Ana Maria Lucchese, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Thomas G Martin, Department of Cell and Molecular Physiology, Loyola University Chicago, Chicago 60153, IL, USA.
Erhe Gao, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Sudarsan Rajan, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Jonathan A Kirk, Department of Cell and Molecular Physiology, Loyola University Chicago, Chicago 60153, IL, USA.
Walter J Koch, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Joseph Y Cheung, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Douglas G Tilley, Department of Pharmacology, Center for Translational Medicine, Lewis Katz School of Medicine, Temple University, Room 945A MERB, 3500 N. Broad St., Philadelphia, PA 19010, USA.
Translational perspective
Our study highlights a previously unrecognized role for epidermal growth factor receptor (EGFR) in maintaining contractile homeostasis under physiologic conditions in the adult heart via regulation of PR72, a PP2A regulatory subunit with an unknown impact on cardiac function. Further, we have shown that cardiomyocyte-expressed EGFR is required for the promotion of cardiac hypertrophy under conditions of chronic catecholamine stress. Altogether, our study provides new insight into the dynamic nature of cardiomyocyte-specific EGFR.
References
- 1. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014;25:282–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc 2014;3:e000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009;138:257–270. [DOI] [PubMed] [Google Scholar]
- 4. D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol 2015;17:627–638. [DOI] [PubMed] [Google Scholar]
- 5. Ma H, Yin C, Zhang Y, Qian L, Liu J. ErbB2 is required for cardiomyocyte proliferation in murine neonatal hearts. Gene 2016;592:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR, Tilley DG. beta-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol 2014;72:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee KS, Park JH, Lim HJ, Park HY. HB-EGF induces cardiomyocyte hypertrophy via an ERK5-MEF2A-COX2 signaling pathway. Cell Signal 2011;23:1100–1109. [DOI] [PubMed] [Google Scholar]
- 8. Rebsamen MC, Arrighi JF, Juge-Aubry CE, Vallotton MB, Lang U. Epidermal growth factor induces hypertrophic responses and Stat5 activation in rat ventricular cardiomyocytes. J Mol Cell Cardiol 2000;32:599–610. [DOI] [PubMed] [Google Scholar]
- 9. Galindo CL, Ryzhov S, Sawyer DB. Neuregulin as a heart failure therapy and mediator of reverse remodeling. Curr Heart Fail Rep 2014;11:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, Nanba D, Higashiyama S, Hori M, Klagsbrun M, Mekada E. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA 2003;100:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grisanti LA, Guo S, Tilley DG. Cardiac GPCR-mediated EGFR transactivation: impact and therapeutic implications. J Cardiovasc Pharmacol 2017;70:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res 2011;109:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grisanti LA, Repas AA, Talarico JA, Gold JI, Carter RL, Koch WJ, Tilley DG. Temporal and gefitinib-sensitive regulation of cardiac cytokine expression via chronic beta-adrenergic receptor stimulation. Am J Physiol Heart Circ Physiol 2015;308:H316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, O'Brien KD, Smyth SS, Threadgill DW. Reduced EGFR causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in C57BL/6J but not 129S1/SvImJ mice. Am J Physiol Heart Circ Physiol 2009;297:H65–H75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet 2000;24:296–299. [DOI] [PubMed] [Google Scholar]
- 16. Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkins NA, Lee DC. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev 1994;8:399–413. [DOI] [PubMed] [Google Scholar]
- 17. Sibilia M, Wagner B, Hoebertz A, Elliott C, Marino S, Jochum W, Wagner EF. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003;130:4515–4525. [DOI] [PubMed] [Google Scholar]
- 18. Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, Barnard JA, Yuspa SH, Coffey RJ, Magnuson Tet al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 1995;269:230–234. [DOI] [PubMed] [Google Scholar]
- 19. Rajagopalan V, Zucker IH, Jones JA, Carlson M, Ma YJ. Cardiac ErbB-1/ErbB-2 mutant expression in young adult mice leads to cardiac dysfunction. Am J Physiol Heart Circ Physiol 2008;295:H543–554. [DOI] [PubMed] [Google Scholar]
- 20. Schreier B, Rabe S, Schneider B, Bretschneider M, Rupp S, Ruhs S, Neumann J, Rueckschloss U, Sibilia M, Gotthardt M, Grossmann C, Gekle M. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 2013;61:333–340. [DOI] [PubMed] [Google Scholar]
- 21. Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ Res 2006;99:528–536. [DOI] [PubMed] [Google Scholar]
- 22. Lee JW, Koeppen M, Seo SW, Bowser JL, Yuan X, Li J, Sibilia M, Ambardekar AV, Zhang X, Eckle T, Yoo SH, Eltzschig HK. Transcription-independent Induction of ERBB1 through hypoxia-inducible factor 2A provides cardioprotection during ischemia and reperfusion. Anesthesiology 2020;132:763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee TC, Threadgill DW. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 2009;47:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 2015;12:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carr R 3rd, Schilling J, Song J, Carter RL, Du Y, Yoo SM, Traynham CJ, Koch WJ, Cheung JY, Tilley DG, Benovic JL. beta-arrestin-biased signaling through the beta2-adrenergic receptor promotes cardiomyocyte contraction. Proc Natl Acad Sci USA 2016;113:E4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grisanti LA, Thomas TP, Carter RL, de Lucia C, Gao E, Koch WJ, Benovic JL, Tilley DG. Pepducin-mediated cardioprotection via beta-arrestin-biased beta2-adrenergic receptor-specific signaling. Theranostics 2018;8:4664–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, Tesmer JJ, Koch WJ. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med 2015;7:277–ra231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Papadaki M, Holewinski RJ, Previs SB, Martin TG, Stachowski MJ, Li A, Blair CA, Moravec CS, Van Eyk JE, Campbell KS, Warshaw DM, Kirk JA. Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight 2018;3:e121264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res 1991;68:1742–1750. [DOI] [PubMed] [Google Scholar]
- 30. Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 2007;117:2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akhtar S, Yousif MH, Chandrasekhar B, Benter IF. Activation of EGFR/ERBB2 via pathways involving ERK1/2, P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from ischemia-reperfusion injury. PLoS One 2012;7:e39066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barrick CJ, Yu M, Chao HH, Threadgill DW. Chronic pharmacologic inhibition of EGFR leads to cardiac dysfunction in C57BL/6J mice. Toxicol Appl Pharmacol 2008;228:315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams-Pritchard G, Knight M, Hoe LS, Headrick JP, Peart JN. Essential role of EGFR in cardioprotection and signaling responses to A1 adenosine receptors and ischemic preconditioning. Am J Physiol Heart Circ Physiol 2011;300:H2161–2168. [DOI] [PubMed] [Google Scholar]
- 34. Haghighi K, Bidwell P, Kranias EG. Phospholamban interactome in cardiac contractility and survival: a new vision of an old friend. J Mol Cell Cardiol 2014;77:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol 2013;64:90–98. [DOI] [PubMed] [Google Scholar]
- 36. Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol 2009;47:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005;23:329–336. [DOI] [PubMed] [Google Scholar]
- 38. Schreier B, Hunerberg M, Rabe S, Mildenberger S, Bethmann D, Heise C, Sibilia M, Offermanns S, Gekle M. Consequences of postnatal vascular smooth muscle EGFR deletion on acute angiotensin II action. Clin Sci (Lond) 2016;130:19–33. [DOI] [PubMed] [Google Scholar]
- 39. Sysa-Shah P, Xu Y, Guo X, Belmonte F, Kang B, Bedja D, Pin S, Tsuchiya N, Gabrielson K. Cardiac-specific over-expression of epidermal growth factor receptor 2 (ErbB2) induces pro-survival pathways and hypertrophic cardiomyopathy in mice. PLoS One 2012;7:e42805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nair BG, Rashed HM, Patel TB. Epidermal growth factor stimulates rat cardiac adenylate cyclase through a GTP-binding regulatory protein. Biochem J 1989;264:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sysa-Shah P, Tocchetti CG, Gupta M, Rainer PP, Shen X, Kang BH, Belmonte F, Li J, Xu Y, Guo X, Bedja D, Gao WD, Paolocci N, Rath R, Sawyer DB, Naga Prasad SV, Gabrielson K. Bidirectional cross-regulation between ErbB2 and beta-adrenergic signalling pathways. Cardiovasc Res 2016;109:358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem 2009;284:20375–20386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 2002;105:85–92. [DOI] [PubMed] [Google Scholar]
- 44. Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation 2000;101:2863–2869. [DOI] [PubMed] [Google Scholar]
- 45. Barefield DY, Puckelwartz MJ, Kim EY, Wilsbacher LD, Vo AH, Waters EA, Earley JU, Hadhazy M, Dellefave-Castillo L, Pesce LL, McNally EM. Experimental modeling supports a role for MyBP-HL as a novel myofilament component in arrhythmia and dilated cardiomyopathy. Circulation 2017;136:1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ochala J, Carpen O, Larsson L. Maintenance of muscle mass, fiber size, and contractile function in mice lacking the Z-disc protein myotilin. Ups J Med Sci 2009;114:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Orr N, Arnaout R, Gula LJ, Spears DA, Leong-Sit P, Li Q, Tarhuni W, Reischauer S, Chauhan VS, Borkovich M, Uppal S, Adler A, Coughlin SR, Stainier DYR, Gollob MH. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun 2016;7:11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sheikh F, Lyon RC, Chen J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 2015;569:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res 2005;66:12–21. [DOI] [PubMed] [Google Scholar]
- 50. Hendrix P, Mayer-Jackel RE, Cron P, Goris J, Hofsteenge J, Merlevede W, Hemmings BA. Structure and expression of a 72-kDa regulatory subunit of protein phosphatase 2A. Evidence for different size forms produced by alternative splicing. J Biol Chem 1993;268:15267–15276. [PubMed] [Google Scholar]
- 51. Song G, Han M, Li Z, Gan X, Chen X, Yang J, Dong S, Yan M, Wan J, Wang Y, Huang Z, Yin Z, Zheng F. Deletion of Pr72 causes cardiac developmental defects in Zebrafish. PLoS One 2018;13:e0206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem 2013;288:1032–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lubbers ER, Mohler PJ. Roles and regulation of protein phosphatase 2A (PP2A) in the heart. J Mol Cell Cardiol 2016;101:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. MacDougall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem 1991;196:725–734. [DOI] [PubMed] [Google Scholar]
- 55. Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem 2004;279:40827–40834. [DOI] [PubMed] [Google Scholar]
- 56. Li L, Fang C, Xu D, Xu Y, Fu H, Li J. Cardiomyocyte specific deletion of PP2A causes cardiac hypertrophy. Am J Transl Res 2016;8:1769–1779. [PMC free article] [PubMed] [Google Scholar]
- 57. Zwaenepoel K, Louis JV, Goris J, Janssens V. Diversity in genomic organisation, developmental regulation and distribution of the murine PR72/B" subunits of protein phosphatase 2A. BMC Genomics 2008;9:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared upon reasonable request to the corresponding authors. RNA-Seq data underlying this article are available in GEO at https://www.ncbi.nlm.nih.gov/geo/ and can be accessed with accession number GSE140407.