

Abstract
Aims
Hepatic capillaries are lined with specialized liver sinusoidal endothelial cells (LSECs) which support macromolecule passage to hepatocytes and prevent fibrosis by keeping hepatic stellate cells (HSCs) quiescent. LSEC specialization is co-determined by transcription factors. The zinc-finger E-box-binding homeobox (Zeb)2 transcription factor is enriched in LSECs. Here, we aimed to elucidate the endothelium-specific role of Zeb2 during maintenance of the liver and in liver fibrosis.
Methods and results
To study the role of Zeb2 in liver endothelium we generated EC-specific Zeb2 knock-out (ECKO) mice. Sequencing of liver EC RNA revealed that deficiency of Zeb2 results in prominent expression changes in angiogenesis-related genes. Accordingly, the vascular area was expanded and the presence of pillars inside ECKO liver vessels indicated that this was likely due to increased intussusceptive angiogenesis. LSEC marker expression was not profoundly affected and fenestrations were preserved upon Zeb2 deficiency. However, an increase in continuous EC markers suggested that Zeb2-deficient LSECs are more prone to dedifferentiation, a process called ‘capillarization’. Changes in the endothelial expression of ligands that may be involved in HSC quiescence together with significant changes in the expression profile of HSCs showed that Zeb2 regulates LSEC–HSC communication and HSC activation. Accordingly, upon exposure to the hepatotoxin carbon tetrachloride (CCl4), livers of ECKO mice showed increased capillarization, HSC activation, and fibrosis compared to livers from wild-type littermates. The vascular maintenance and anti-fibrotic role of endothelial Zeb2 was confirmed in mice with EC-specific overexpression of Zeb2, as the latter resulted in reduced vascularity and attenuated CCl4-induced liver fibrosis.
Conclusion
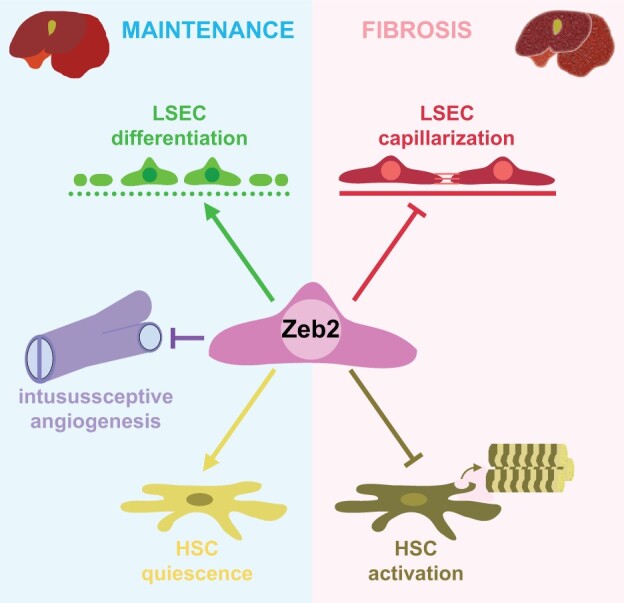
Endothelial Zeb2 preserves liver angioarchitecture and protects against liver fibrosis. Zeb2 and Zeb2-dependent genes in liver ECs may be exploited to design novel therapeutic strategies to attenuate hepatic fibrosis.
Keywords: Liver sinusoidal endothelial cells, Capillarization, Zeb2, Intussusceptive angiogenesis, Liver fibrosis
Graphical Abstract
1. Introduction
Endothelium is highly heterogeneous between different vessel types, and capillaries are lined with specialized endothelial cells (ECs) to accommodate the specific function of the organ where they reside. Transcription factors play an important role in specialization of endothelium by enhancing the expression of proteins specific for ECs in the particular organ and/or by repressing the expression of markers for EC types found in other organs or other vessel types.1–3 The liver has a unique dual blood supply: a mixture of oxygen-rich hepatic arterial blood and nutrient-rich portal venous blood enters the hepatic sinusoidal capillaries. Given the importance of the liver in metabolism, detoxification, and synthesis of macromolecules, efficient transfer between blood and hepatocytes is essential. To accommodate this transfer, liver sinusoidal ECs (LSECs) are highly specialized. They have fenestrae, they lack an organized basement membrane, and they have a high receptor-mediated endocytotic capacity.4
Maintenance of the LSEC-specific properties is important in response to liver damage. The liver is the first organ that receives blood from the intestine and is therefore frequently exposed to noxious agents that cause liver damage. Upon liver injury, hepatic stellate cells (HSCs) become activated and deposit collagen. This leads to liver fibrosis which may ultimately progress to liver dysfunction and cirrhosis. Differentiated LSECs suppress HSC activation and thus inhibit collagen deposition and attenuate fibrosis progression.5 However, shortly after liver damage, LSECs start to dedifferentiate, also known as ‘capillarization’. Characteristics of capillarization are the acquisition of an organized basement membrane, loss of fenestrae, and expression of continuous EC markers such as CD31 and CD34. Upon capillarization, LSECs lose their ability to inhibit HSC activation.3,5–7
Several studies have established unique organ-specific EC gene signatures and showed that transcription factors enriched in liver ECs co-determine the characteristic LSEC expression profiles and functions. Our previous comparative study between ECs from heart, brain, and liver confirmed the differential expression of several known LSEC-associated transcription factors (i.e. GATA4, cMAF, and TCFEC).2,3,8 Furthermore, we identified new LSEC-enriched transcription factors, most notably zinc-finger E-box-binding homeobox2 (Zeb2).8 Zeb2 is mainly known for its role in the development of the nervous system9–12 and its ability to stimulate epithelial-to-mesenchymal transition (EMT) in cancer.13 Recent studies also suggested a role for Zeb2 in liver fibrosis by showing that Zeb2 levels are increased in fibrotic livers, and the expression of Zeb2 in HSCs has been suggested to affect their activation in vitro.14,15 Furthermore, Zeb2 maintains the tissue-specific identity of Kupffer cells and its absence leads to Kupffer cell disappearance.16 Since HSCs deposit collagen and Kupffer cells are involved in inflammation, these are traditionally the main cell types studied in fibrosis. More recent studies, however, emphasized the importance of LSECs, yet, the role of endothelial factors in liver fibrosis remains largely unexplored. Therefore, we aimed to elucidate the role of endothelial Zeb2 in liver vascular maintenance and its impact on fibrosis.
2. Methods
An extended methods section and additional references are available in Supplementary material online.
2.1 Mice and human tissues/cells
Four mouse lines were used: (i) EC reporter mice specifically expressing green fluorescent protein (GFP) in blood-vascular ECs (Tie2-GFP); (ii) Zeb2 reporter mice expressing a Zeb2-enhanced (e)GFP fusion protein driven by the endogenous Zeb2 promoter (Zeb2-eGFP); (iii) tamoxifen-inducible EC-specific Zeb2 knock-out mice (ECKO) generated by intercrossing Cdh5-CreERT2 mice with mice carrying a Zeb2 exon 7 flanked by loxP sites crossed onto an R26R CAG-boosted eGFP reporter background (Supplementary material online, Figure S1A, left); and (iv) tamoxifen-inducible EC-specific Zeb2-overexpressor mice carrying the Cdh5-CreERT2 driver and two ROSA26-Zeb2tg/tg alleles (‘ECOE’; Supplementary material online, Figure S1A, right). For ECKO and ECOE mice, tamoxifen-treated Cre-negative littermates were used as wild-type (WT) controls. For recombination, mice were intraperitoneally (i.p.) injected with tamoxifen for 5 consecutive days. Recombination efficiency (expressed as the fraction of ECs gaining eGFP expression) was ∼90% and similar across organs and across the zonated liver vascular bed (Supplementary material online, Figure S1B and C). Unless indicated otherwise, 8-week-old males were used. Blood was drawn via the heart and mice were euthanized by exsanguination under ketamine (75 μg/g i.p.) and xylazine (5 μg/g i.p.) anaesthesia. Anaesthesia depth was checked by toe pinch and, if necessary, mice received another i.p. injection with 7.5 μg/g ketamine/0.5 μg/g xylazine. Mouse experiments were approved by the KU Leuven or VUB Animal Ethics Committee and performed according to the Committee’s guidelines and those from Directive 2010/63/EU. Human liver biopsies were obtained under informed consent from patients undergoing elective cholecystectomy, fixed, paraffin-embedded, and sectioned for immunofluorescence (IF) staining. Human umbilical vein ECs (HUVECs) were isolated under informed consent from the mother. The use of human material was approved by the Ethics Committee of University Hospitals Leuven and experiments were performed according to the Committee’s guidelines and the principles of the Declaration of Helsinki.
2.2 Maintenance and hepatotoxic models
To study the effect of endothelial Zeb2 on (vascular) maintenance, mice were euthanized under anaesthesia as described above 1, 2, or 4 weeks after the last tamoxifen injection (Supplementary material online, Figure S1D). Unless indicated otherwise, mRNA expression changes are shown at 2 weeks post-tamoxifen and protein expression or structural changes at 4 weeks post-tamoxifen. To study the response to acute liver injury, mice received one i.p. injection with high-dose CCl4 (0.6 μL/g in mineral oil) or with mineral oil alone (vehicle) as control and were sacrificed 24 h later (Supplementary material online, Figure S1E). To study the effect of mild fibrosis, mice were injected with low-dose CCl4 (0.2 μL/g in mineral oil) or vehicle three times with 1 day in between and were sacrificed 24 h (progression cohort) or 1 week (regression or ‘R’ cohort) after the last injection (Supplementary material online, Figure S1F). For chronic (septal) fibrosis, mice received high-dose CCl4 (0.6 μL/g in mineral oil) or vehicle three times per week for 4 weeks and were sacrificed 24 h (progression cohort) or 1 week (regression or ‘R’ cohort) after the last injection (Supplementary material online, Figure S1G). For (immuno)histological analyses, mice were euthanized under anaesthesia as described above, livers were perfusion-fixed and processed for paraffin sectioning. For other analyses, tissues were isolated, snap-frozen, and stored until further use.
2.3 Cell isolation and gene profiling
To study the organ-specific Zeb2 expression, GFP+ ECs were isolated from Tie2-GFP hearts, brains, and livers (yielding >95% pure populations consisting for >99% of microvascular ECs),2,8 and comparative gene expression was performed by quantitative (q) real-time (RT)-PCR. To simultaneously isolate four hepatic cell types by fluorescence-activated cell sorting (FACS) on an Aria-II sorter, ECKO and WT (Cre-negative littermates) monocellular suspensions were generated by enzymatic digestion as described.17 The resulting suspension was filtered and centrifuged to separate hepatocytes from the non-parenchymal cell (NPC) fraction. After lysing red blood cells, the remaining NPC fraction was stained for pan-endothelial marker Meca32 (Pan-endo) and F4/80 to enable the sorting of UV+ HSCs, UV−F4/80+ Kupffer cells, and UV−Pan-endo+ ECs (for antibodies: see Supplementary material online, Table S1). RNA was isolated using Reliaprep (Promega) and sequencing was performed by the Center for Biomics-Genomics, Erasmus MC, Rotterdam. Bioinformatic processing is described in the Supplementary material online. RNA from total liver was isolated by TRIZol. cDNA was made using the GoScript™ reverse transcription system and qRT-PCR was performed using Sybr green. Gapdh was used as housekeeping gene after validation (Supplementary material online, Note S1; for primers: see Supplementary material online, Table S2).
2.4 In vitro angiogenesis assays and lentivirus-mediated knock-down
Lentiviruses were made using plasmids containing ZEB2-shRNA or GFP in human embryonic kidney 293 cells. HUVECs were cultured in EBM2 medium supplemented with EGM2-MV in gelatin-coated flasks. Cells were transduced with viruses, and on day 6 cells were either lysed in TRIzol for RNA isolation or passaged for functional analyses (i.e. proliferation, chemotactic and scratch wound migration, tube formation, and sprouting; for details: see Supplementary material online).
2.5 Scanning electron microscopy and ultrafast ultrasound imaging
For morphometric analysis on vascular casts, mice were euthanized under anaesthesia and livers were perfused with VasQtec resin according to the manufacturer’s instructions. Livers were saponified and casts were dried and sputter-coated, and images were recorded on a JEOL scanning electron microscope. To document fenestrae in vivo, mice were perfusion-fixed with glutaraldehyde. To analyse fenestrae ex vivo, 36-h bulk cultures from Zeb2 WT and ECKO livers were fixed with glutaraldehyde. Bulk cultures were used and supplemented with 0.5 ng/mL VEGF-A in order to preserve fenestrae.18 Liver slices and cell cultures were dried, sputter-coated and pictures were recorded on a Zeiss Sigma VP scanning electron microscope. Ultrafast ultrasound imaging was customized for the liver, as described in Supplementary material online, and was performed on the left (lateral) liver lobe, regions of interest were selected manually based on anatomical landmarks and blood volume was computed. Spatiotemporal clutter filtering based on singular value decomposition of raw ultrasonic data were used to discriminate blood from tissue motion. Bandpass frequency filters were applied on the Doppler spectrum to give access to maps of blood flow at different velocity ranges. These ranges were related to vessel calibre based on a meta-analysis in different animal species and corresponding blood volumes in these different ranges were computed as described.19,20 For ultrafast ultrasound imaging, mice were anaesthetized with ketamine (75 μg/g i.p.) and medetomidine (1 μg/g once i.p.) and depth was checked by toe pinch. If necessary, mice received another i.p. injection with 7.5 μg/g ketamine and 0.1 μg/g medetomidine. After the experiment (∼45 min) anaesthesia was reversed with a subcutaneous injection of atipamezole (1 μg/g).
2.6 Morphometric analysis and assessment of liver fibrosis and function
For histology, mice were euthanized under anaesthesia as described above and perfusion-fixed with zinc-formalin via the heart. The left liver lobe, heart, and the brain were isolated, dehydrated, embedded in paraffin and 7 µm paraffin sections were prepared. To assess general morphology and liver damage, sections were stained with haematoxylin and eosin and collagen was revealed using Sirius red, Masson’s trichrome, or antibodies against fibrillar Collagen types I/III. To analyse the zonated architecture of the liver vasculature, we used antibodies against Cytokeratin 19 and Endomucin. To further characterize liver damage, sections were stained for α-smooth muscle actin, Desmin, and Cd45. To study endothelial changes, IF stainings were performed for Pan-endo, Cd31, Cd32, Cd34, von Willebrand factor (vWF), Laminin, Lyve1, Endomucin, Wheat Germ Agglutinin (WGA) lectin, and Collagen type IV. To demonstrate endothelial expression of Zeb2 in Zeb2-eGFP mice or to assess Cre-mediated recombination in ECKO mice, livers were co-stained for eGFP and ETS-related gene (Erg) or Bandeiraea simplicifolia (BS)-I lectin. To analyse recombination in periportal/pericentral LSECs, recombined ECs were counted on serial cross-sections stained for Erg/eGFP and pericentral zonation marker Cytochrome P450 Family 2 Subfamily E member 1 (Cyp2e1). Hepatocyte zonation was analysed by IF staining using antibodies against pericentral markers Cyp2e1 or glutamate ammonia ligase (Glul) and periportal/midzonal marker Arginase1 (Arg1). Where necessary, amplification was performed using Cy3- or fluorescein-tyramide kits. Human liver paraffin sections were co-stained for ZEB2 and ERG. Analyses were performed in Image J and FACS data were analysed with FACS DIVA software. Antibodies are listed in Supplementary material online, Table S1. To assess hydroxyproline content, livers were hydrolyzed and hydroxyproline was detected as detailed in Supplementary material online. To assess hepatocyte damage, plasma alanine transferase (ALT) was measured using a Spotchem EZ system analyser (Arkray) and GPT/ALT strips (Menarini diagnostics).
2.7 Statistics
Quantitative data are expressed as mean ± standard error of the mean (sem). The Student's t-test was used to compare two groups. ANOVA with Bonferroni post hoc test was used to compare more than two groups. P-value <0.05 was considered statistically significant. GraphPad Prism 8 was used for statistical analyses. Statistical analyses for the RNA sequencing data are described in Supplementary material online.
3. Results
3.1 Zeb2 is highly expressed in liver endothelium
In our previous microarray-based studies comparing gene expression of ECs from liver, heart, and brain, Zeb2 emerged as a transcription factor highly enriched in liver ECs.2,8 To confirm this finding, we sorted ECs from liver, brain, and heart from an independent group of Tie2-GFP mice. GFP+ cells mainly (>99%) represent microvascular ECs.2,8 qRT-PCR analysis revealed high enrichment of Zeb2 expression in liver microvascular ECs (i.e. LSECs) as compared to microvascular ECs from heart and brain (Figure 1A). Next, using Zeb2-eGFP reporter mice we showed high Zeb2-reporter activity in LSECs but not in heart or brain capillary ECs (Figure 1B–D). Expression of Zeb2 in LSECs was uniform across periportal and pericentral sinusoids and extended also to non-sinusoidal vessels, including the portal vein, hepatic artery, and central vein (Supplementary material online, Figure S2A). Finally, we stained human liver biopsies for ZEB2 and confirmed its presence in human LSECs and ECs from non-sinusoidal vessels (Supplementary material online, Figure S2B).
Figure 1.

Zeb2 is highly expressed in liver endothelium. (A) Zeb2 mRNA expression in ECs isolated by FACS from Tie2-GFP livers, hearts, and brains (n = 4). (B–D) Sections of Zeb2-eGFP liver (B), heart (C), or brain (D) stained for Zeb2-eGFP (red) and EC markers Erg (green) or BS-I lectin (green). Hoechst (blue) was used as nuclear counterstain. Arrowheads indicate EC nuclei. Data are expressed as mean ± sem; **P < 0.01 by one-way ANOVA with Bonferroni post hoc test. Scale bars: 50 µm. EC, endothelial cell.
3.2 EC-specific Zeb2-KO affects expression of genes related to LSEC capillarization and LSEC–HSC communication
Zeb2 mRNA expression was also detected in HSCs, Kupffer cells, and—to a lesser extent—in hepatocytes (Supplementary material online, Figure S3A and Note S2).14–16,21 To study the role of Zeb2 in the EC compartment, we generated mice, in which Zeb2 was specifically and efficiently (∼80% at mRNA level and in ∼90% of ECs) deleted upon tamoxifen treatment in endothelium (‘ECKO’ mice; Supplementary material online, Figures S1B and S3B and C). Expression of Zeb1, the family member of Zeb2, was unaltered in the ECKO ECs (expression relative to WT: 1.0 ± 0.3 for WT vs. 1.0 ± 0.1 for ECKO; n = 4–5). To study the consequences of EC-specific Zeb2 deletion on gene expression in liver ECs themselves and in the neighbouring cell types in the liver sinusoids (Supplementary material online, Figure S2A), we isolated ECs, hepatocytes, HSCs, and Kupffer cells from ECKO and WT livers (Supplementary material online, Note S3).
Purity of the isolated cell populations was verified by qRT-PCR using acknowledged lineage markers and the different cell populations segregated according to their global expression profile determined by RNA sequencing (Supplementary material online, Figure S4A). The technical quality of the sequencing was verified by qRT-PCR for a random gene set on cDNA derived from RNA from the same mice (Supplementary material online, Figure S4B). As expected, EC-specific Zeb2 deletion had most impact on the expression profile of LSECs themselves with 986 genes being differentially expressed (Figure 2A and Supplementary material online, Figure S4C). Functional annotation revealed that loss of Zeb2 affected platelet-derived growth factor (Pdgf) signalling and angiogenesis (Figure 2B). Besides Lyve1, Zeb2-deletion did not profoundly affect LSEC marker expression in vivo, which confirmed our earlier in vitro observations.8 However, the expression of a subset of continuous EC markers3 including Cd34, Endomucin (Emcn), and Apelin (Apln) was increased (Figure 2B), indicative of early capillarization. Changes in Lyve1 and Emcn were confirmed at the protein level by IF staining which revealed that their zonated expression patterns were retained upon endothelial Zeb2 deletion. Accordingly, expression of a large number of genes with a known zonated expression pattern in LSECs was unaltered by endothelial Zeb2 loss (Supplementary material online, Note S4) and preferential binding of WGA lectin in the periportal regions22 was also preserved (Supplementary material online, Figure S5A–C). Furthermore, endothelial Zeb2 loss altered gene expression in other cell types in the liver. HSCs were most profoundly affected with differential expression of 326 genes (Figure 2A and Supplementary material online, Figure S4C). Expression changes in hepatocytes or Kupffer cells were more restricted (110 and 67 genes, respectively; Figure 2A and Supplementary material online, Figure S4C). Concomitantly, genotypes segregated for LSECs and HSCs, while they clustered randomly for hepatocytes and Kupffer cells, supporting a larger impact of the EC-specific knock-out on LSEC–HSC communication than on LSEC–hepatocyte or LSEC–Kupffer cell communication (Supplementary material online, Figure S4A). To further clarify the altered LSEC–HSC communication, we performed NicheNet ligand-target prediction analysis. As input criteria for this analysis we used the downregulated ligands in ECs and detectable expression of at least one corresponding receptor in the receiver cells, here HSCs. Multiple sets of downregulated target genes in HSCs were predicted that corresponded with downregulated ligands in LSECs (Figure 2C and Supplementary material online, Figure S6), supporting that LSEC–HSC communication was strongly affected by endothelial Zeb2. Downregulated LSEC ligands included growth/differentiation factor 15 (Gdf15),23 lactoferrin (Ltf),24 and insulin-like growth factor-1 (Igf1)25 which are known to attenuate liver fibrosis. We confirmed the downregulated expression of these ligands in LSECs and a subset of their fibrosis-related target genes in HSCs on an extended set of mice (Figure 2C and Supplementary material online, Note S5).
Figure 2.

EC-specific Zeb2-KO alters expression of genes related to LSEC capillarization and HSC communication. (A) Number of differentially expressed genes in LSECs, HSCs, KCs, and HEPs (n = 2). (B) Pathways affected by Zeb2 (based on Enrichr analysis; n = 2; top). Expression of LSEC markers (determined by qRT-PCR on an extended set of mice; n = 5–7; middle) and continuous EC markers (heat map generated from the RNA sequencing dataset; n = 2; bottom). (C) NicheNet-based predictions of ligands downregulated in LSEC source cells and targets downregulated in HSC target cells (n = 2; middle). Expression of LSEC-derived ligand genes (bottom) and HSC target genes (top; highlighted in red in the middle diagram; determined by qRT-PCR on an extended set of mice; n = 5–7). Data are expressed as mean ± sem; *P < 0.05, **P < 0.01 by the Student’s t-test. EC, endothelial cell; HEPs, hepatocytes; HSC, hepatic stellate cell; KCs, Kupffer cells; LSEC, liver sinusoidal endothelial cell; WT, wild-type.
3.3 EC-specific Zeb2-KO distorts the angioarchitecture of the liver
Next, we investigated the consequences of endothelial loss of Zeb2 and impaired LSEC–HSC communication on the liver phenotype 4 weeks post-tamoxifen treatment. Body and liver weight were unaffected (Supplementary material online, Figure S7A). Despite the perturbed LSEC–HSC communication, no signs of fibrosis were detected in unchallenged ECKO livers (Supplementary material online, Figure S7B). Given the altered expression profile related to angiogenesis and LSEC capillarization of Zeb2-deficient LSECs (Figure 2B), we studied the liver vasculature in detail in ECKO mice and their WT littermates. Pan-endo staining revealed that the total vascular surface area was significantly increased in ECKO livers at 1, 2, and 4 weeks post-tamoxifen (Figure 3A and Supplementary material online, Figure S7E and Note S6). The vascular expansion occurred at the level of the small Cd32+ sinusoidal vessels but also in the medium-sized vessels, without disturbing the ratio of portal vs. central veins (Figure 3B and C and Supplementary material online, Figure S7F). Vascular expansion was not observed in brain or heart, which was likely not due to reduced recombination efficiency but rather to the low Zeb2 levels in these tissues (Supplementary material online, Figures S1B and S7G and H). Since it is unknown whether the gain of continuous EC markers is associated with loss of fenestrae,3,26 we studied fenestration by scanning EM on liver slices and cultured liver cells and we observed that ECKO mice had no obvious defects in fenestration in vivo or ex vivo, as evidenced by a preserved porosity index (Supplementary material online, Figure S7C and D). These results show that LSECs only partially dedifferentiate upon Zeb2 loss. To study how Zeb2 affects the overall ultrastructure of the vasculature, we injected mice via the portal vein with resin to create vascular corrosion casts of the liver. Analysis of these casts showed that the sinusoidal network of ECKO mice was denser and more irregular in shape with an increased vessel diameter compared to WT (Figure 3D).
Figure 3.

EC-specific Zeb2-KO distorts the angioarchitecture of the liver. (A and B) Hepatic Pan-endo+ (n = 4) and Cd32+ area (n = 8). (C) Number of medium-sized vessels counted on Pan-endo-stained sections (left; n = 4) and the ratio of portal vs. central veins (right; n = 8). (D) Representative scanning EM images of vascular corrosion casts and size distribution of sinusoidal diameter (n = 7–9; bottom). (E) Representative selection of vascular planes recorded by UFUS imaging of the left (lateral) lobe. (F) Hepatic blood volume before bandpass filtering (n = 6–8). (G and H) Representative Doppler images showing blood flow bandpass filtering in the region of interest for three different speed ranges, corresponding to three-vessel size ranges (G) and quantification of blood volume (n = 6–8) (H). Data are expressed as mean ± sem, ultrafast ultrasound data are expressed in AU as mean ± sem. *P < 0.05, **P < 0.01 by the Student’s t-test (A, B, C, and F) or two-way ANOVA with Bonferroni post hoc test (H). Scale bars: 3 mm in E and G; 100 µm in A, B, and D; and 50 µm in D, inset. AU, arbitrary units; EC, endothelial cell; UFUS, ultrafast ultrasound; WT, wild-type.
To study and confirm the vascular expansion in the liver in a more integral and functional manner in RT in living mice, we customized an ultrafast ultrasound protocol for the liver. Ultrafast ultrasound allows for highly sensitive and wide-field-of-view Doppler imaging of blood vessels far beyond conventional ultrasonography (Supplementary material online, Figure S8).19 Ultrafast ultrasound on the left lateral liver lobe revealed that, while the general anatomy of the liver vascular tree remained largely preserved upon EC-specific Zeb2-KO (Figure 3E), there was an increase in blood volume in the liver of ECKO mice as compared to their WT littermates (Figure 3F). Furthermore, bandpass frequency filtering showed that the increase in blood volume was due to a significant expansion of medium-sized vessels and small (capillary/pre-capillary) vessels (with blood flow velocity of 4–6 and 2–4 mm/s, respectively;20 Figure 3G and H) which was in accordance with our histological observations.
3.4 Endothelial Zeb2-KO promotes intussusceptive angiogenesis
To learn more about the underlying mechanism of vascular expansion, we studied the direct effect of ZEB2 knock-down (KD) in vitro on EC proliferation, migration, and tube formation. Since primary LSECs are hard to maintain in culture, we used HUVECs in which we silenced ZEB2 via a lentivirus expressing a ZEB2-shRNA. ZEB2-KD did not affect proliferation, migration, or sprouting (Supplementary material online, Figure S9A–C). Branch formation on matrigel was, however, increased in ZEB2-KD HUVECs and the network was longer compared to HUVECs with unperturbed ZEB2 expression (Supplementary material online, Figure S9D), suggesting the vascular expansion was due to altered EC organization rather than proliferation.
To evaluate whether the vascular expansion in vivo was also independent of proliferation, we quantified the total number of ECs and proliferating ECs. EC numbers (Erg+ cells) were not altered in liver sections (Figure 4A) and accordingly, the number of Ki67+ Erg+ cells was also unaffected in livers of ECKO vs. WT littermates, indicating no change in proliferation (Figure 4B). Since non-proliferative vascular expansion is indicative of intussusceptive angiogenesis,27 we used vascular corrosion casts to study the presence of signs of intussusceptive angiogenesis. Interestingly, we observed a significant increase in the number of pillars, the ultrastructural hallmark of intussusceptive angiogenesis,27 on vascular corrosion casts of ECKO as compared to WT livers (Figure 4C), suggesting that endothelial Zeb2 modulates intussusceptive angiogenesis in the liver.
Figure 4.

Endothelial Zeb2-KO promotes intussusceptive angiogenesis. (A and B) Number of Erg+ ECs (A; n = 4) and % of Ki67+ Erg+ ECs (B; n = 4). Arrowheads indicate proliferating ECs. (C) Representative scanning EM images of vascular corrosion casts (left) or a liver slice (middle) of an ECKO mouse and corresponding quantification (right) of number of intussusceptive pillars (n = 5–7). Arrowheads indicate pillars. Data are expressed as mean ± sem; *P < 0.05 by the Student’s t-test. Scale bars: 20 µm in C, left; 5 µm in C, middle. EC, endothelial cell; WT, wild-type.
3.5 Endothelial Zeb2-KO aggravates toxin-induced liver fibrosis independent of HEP zonation
In our RNA sequencing analysis we observed that endothelial Zeb2 deletion changed the LSEC–HSC communication, in part by lowering the endothelial expression of ligands previously shown to attenuate fibrosis (Figure 2B and C). Since HSC activation is a key mediator during fibrogenesis, we hypothesized that endothelial loss of Zeb2 would aggravate the response to a fibrotic challenge. Indeed, after a 1-week exposure to a low-dose of the hepatotoxin CCl4, ECKO livers had increased perivascular and parenchymal fibrosis compared to WT livers (Figure 5A). Repeated exposure to high-dose CCl4 for 4 weeks caused regional septal fibrosis in WT livers; however, septal fibrosis was more widespread in ECKO livers (Figure 5B). Increased fibrosis was confirmed by higher hydroxyproline content and more fibrillar Collagen type I and III deposition in CCl4-treated ECKO livers (Supplementary material online, Figure S10A–C). Accordingly, CCl4-treated ECKO livers had increased signs of HSC activation and inflammation in the fibrotic septa (Figure 5C–E). Increased fibrosis and HSC activation were also apparent from the significantly increased liver expression of genes related to these processes, including Tgfb1, Tgfb2, Tgfb3, Col1a1, Col1a3, and Pdgfrb after 4 weeks of CCl4 treatment (Supplementary material online, Figure S10D). In both mild CCl4 injury and chronic high-dose CCl4 injury, the difference in fibrosis was similar in the regression phase (i.e. 1 week after the last CCl4 injection; Figure 5A and B), indicating that endothelial Zeb2 mainly affects fibrosis progression rather than the regenerative response that occurs upon removal of the fibrotic challenge.
Figure 5.

Endothelial Zeb2-KO promotes toxin-induced liver fibrosis. (A) Collagen content (Sirius red+ area; n = 6–9) after treatment with oil (vehicle) or 1 week low-dose CCl4. Livers were analysed 24 h (progression cohort) or 1 week after the last oil or CCl4 injection (regression cohort ‘R’). (B) Collagen content (Sirius red+ area; n = 3–7) after treatment with oil (vehicle) or 4 weeks high-dose CCl4. Livers were analysed 24 h after last oil or CCl4 injection (progression cohort) or 1 week after last oil or CCl4 injection (regression cohort ‘R’). (C–E) α-Smooth muscle actin (SMA)+ area (C; n = 4–6), Desmin+ area (D; n = 4–6), and Cd45+ area (E; n = 4–6) after treatment with oil (vehicle) or high-dose CCl4. Data are expressed as mean ± sem; *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA with Bonferroni post hoc test. Scale bars: 100 µm. EC, endothelial cell; HSC, hepatic stellate cell; WT, wild-type.
To study if hepatocyte damage was affected by endothelial Zeb2 we measured plasma ALT levels after a single CCl4 injection. ALT levels were strongly increased but similar in both genotypes indicating that the difference in fibrotic response cannot be explained by a difference in hepatocyte injury (Supplementary material online, Figure S11A). Since the expression of Cyp2e1—the enzyme responsible for the generation of the toxic metabolite of CCl4—is zonated, we investigated whether loss of endothelial Zeb2 could affect fibrosis by skewing Cyp2e1 expression in particular and hepatocyte zonation in general. First, we found that the pattern of CCl4-induced necrosis was pericentral and similar in both genotypes (Supplementary material online, Figure S11A). Second, there was only 1 of the 67 hepatocytic DEGs (Figure 2A and Supplementary material online, Figure S4C) overlapping with a list of genes previously shown to have a zonated expression in the liver parenchyma (Supplementary material online, Figure S11B and Note S7).28 Third, Zeb2 deficiency in LSECs did not alter their expression of angiocrine factors (Wnt2, Wnt9b, Rspondin3 or Rspo3)29–31 or transcription factors (Gata4)32 known to affect hepatocyte zonation (Supplementary material online, Figure S11B). Finally, the zonated expression pattern of hepatocyte markers Glul, Cyp2e1, and Arg1 was preserved upon endothelial Zeb2 loss (Supplementary material online, Figure S11C–E).
3.6 Endothelial Zeb2-KO aggravates toxin-induced LSEC capillarization
One of the early events during toxin-induced liver fibrosis is endothelial damage and capillarization which may further stimulate HSC activation and collagen deposition.6 In our RNA sequencing analysis we observed that endothelial Zeb2 deletion in the non-challenged liver already increased the expression of a subset of continuous EC markers, indicative of early capillarization.3 Therefore, we hypothesized that capillarization of Zeb2-deficient LSECs would be intensified by a fibrotic challenge. Indeed, upon exposure to CCl4, the sinusoidal endothelium of ECKO mice showed increased vWF expression33 suggesting more EC damage (Figure 6A). Furthermore, Zeb2 ECKO livers showed more capillarization, evident from the gain of continuous EC markers Cd34 and Cd31, loss of the LSEC marker Cd32, and the acquisition of a Laminin- and Collagen Type IV-containing basement membrane (Figure 6B–F and Supplementary material online, Note S8). In agreement with the reported correlation between fibrosis progression and angiogenesis,7 CCl4-induced fibrosis was accompanied by vascular expansion (average increase in Pan-endo area of 44 ± 6%), however, this angiogenic response was not boosted by endothelial Zeb2-KO (Supplementary material online, Figure S12A). Increased capillarization was also apparent from the increased liver expression of related genes, including Cd34, Lama1, Lama4, Col4a1, and Col4a2 after 4 weeks of CCl4 treatment (Supplementary material online, Figure S12B).
Figure 6.

Endothelial Zeb2-KO promotes toxin-induced LSEC damage and capillarization. (A–F) vWF+ area (A), Cd34+ area (B), Cd31+ area (C), Cd32+ area (D), Laminin+ area (E), and Collagen type IV+ area (F) in livers after treatment with oil (vehicle) or 4 weeks high-dose CCl4. Data are expressed as mean ± sem; n = 4–7. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA with Bonferroni post hoc test. Scale bars: 100 µm. EC, endothelial cell; vWF, von Willebrand factor; WT, wild-type.
3.7 Endothelial Zeb2-overexpression reduces vascular expansion and toxin-induced fibrosis
Our data suggest that endothelial Zeb2 preserves normal liver angioarchitecture and protects against fibrosis. To directly demonstrate this via a complementary approach, we generated mice that conditionally (Cre-dependently) overexpress Zeb2 in ECs (‘ECOE’ mice; Figure 7A and Supplementary material online, Figure S1A, right). Like endothelial Zeb2-KO, gain of endothelial Zeb2 expression did not affect body or liver weight (Supplementary material online, Figure S13A). Zeb2-overexpression in ECs caused reduced vascularity in unchallenged mice (Figure 7B and Supplementary material online, Figure S13B and Note S9), and fibrosis was attenuated in ECOE mice mildly or chronically challenged with CCl4 (Figure 7C and Supplementary material online, Figure S13C) further confirming our observations in ECKO mice and directly supporting the anti-fibrotic role of Zeb2 in LSECs.
Figure 7.

Endothelial Zeb2-overexpression reduces vascular expansion and toxin-induced fibrosis. (A) Expression of Zeb2 in ECs from WT or EC-specific Zeb2 overexpressing (‘ECOE’ mice; n = 5). (B) Pan-endo+ area 4 weeks after the last tamoxifen injection (n = 3–5). (C) Collagen content (Sirius red+ area; n = 5–7) mice 24 h after the last injection of oil or CCl4 (progression cohort) or 1 week after the last CCl4 injection (regression cohort ‘R’). Data are expressed as mean ± sem; *P < 0.05, **P < 0.01, ***P < 0.001 by the Student’s t-test (A and B) or one-way ANOVA with Bonferroni post hoc test (C). Scale bars: 100 µm. EC, endothelial cell; WT, wild-type.
4. Discussion
Our organs are equipped with a microvascular network specifically adapted to support the organ's function. Identifying what determines these specific endothelial features and how they change during pathological conditions is important to design procedures to diagnose and treat organ-specific diseases. Here, we show that high levels of the transcription factor Zeb2 are expressed in the liver endothelium as compared to endothelium in heart and brain. Endothelial Zeb2 preserved the hepatic angioarchitecture and attenuated the fibrogenic response upon (chronic) exposure to a hepatotoxin.
A key observation of our study was that Zeb2 deletion in ECs of adult mice caused the liver vasculature to expand. Even though we used a generalized approach to delete Zeb2 in ECs, vascular expansion did not occur in heart or brain, which is in agreement with the very low levels of Zeb2 in endothelium in these organs and an organ-specific role for Zeb2 in the liver. In a previous study using a Tie2-Cre driver line to conditionally delete Zeb2 in ECs and haematopoietic cells, no major defects of the developing liver vasculature were found but mice died in utero because of brain haemorrhage due to the Zeb2 deletion in the haematopoietic lineage.34 Here, we used a more EC-specific and tamoxifen-inducible Cre driver line (Cdh5-CreERT2) to be able to study the role in liver ECs in adult mice. Our detailed microscopic and ultrastructural analyses of ECKO mice did reveal expansion of the adult liver's vasculature and significant irregularities in the angioarchitecture.
Previously, it was suggested that ZEB2 has a pro-angiogenic effect; however, the effects of ZEB were indirectly evaluated35 or only observed upon high-glucose exposure in vitro.36 We found that KD of ZEB2 in HUVECs did not affect their proliferative, migratory, or sprouting behaviour but did increase their ability to branch. Complementary to our in vitro findings, endothelial Zeb2-KO did not affect EC proliferation in vivo in the liver, but the presence of pillars suggested the induction of a non-sprouting/non-proliferative type of angiogenesis, known as intussusceptive angiogenesis.27 Intussusceptive angiogenesis can cause rapid vascular expansion,27 which is compatible with our observation that EC-Zeb2-deficient mice showed vascular expansion within 1 week after deletion. The molecular mechanisms underlying intussusceptive angiogenesis remain largely unknown, although a number of candidate regulators have been identified including Pdgf-b37 and Notch1.38 Pdgfb was one of the genes upregulated by Zeb2-KO in ECs while in Schwann cells Zeb2 is needed to generate anti-Notch activity.11,12 While the aberrant liver angioarchitecture of ECKO mice shared some features (e.g. large variation in sinusoidal diameter) with that in Notch1-deficient livers, expression of Notch1 or Notch target genes Hey1 and Hes1 were not significantly altered in Zeb2-deficient ECs (Supplementary material online, Figure S14A), suggesting that they work independently in liver ECs. Therefore, Zeb2-ECKO mice could be an interesting mouse model to further elucidate mechanisms of intussusceptive angiogenesis.
To obtain a more global and functional view on the liver vasculature, we optimized an ultrafast ultrasound protocol previously applied in brain19 for the use in liver, which is a soft tissue subject to significant movement, both intrinsic and extrinsic, the latter due to breathing. For this purpose, we applied a complimentary two-dimensional motion correction algorithm. Then, by applying bandpass filtering on the Doppler spectrum in each individual pixel, we showed an increase in blood volume mainly in the smaller/medium-sized vessels of ECKO livers, in accordance with our microscopic/ultrastructural findings.
Another key observation of our study was that endothelial Zeb2 protects against fibrosis in the liver. As such, our study further underscores that Zeb2 has a tissue, cell type- and context-dependent role in fibrosis. Zeb2 contributes to fibrosis in the heart by stimulating cardiac fibroblast-myofibroblast phenoconversion.39 Zeb2 expression has been shown to increase upon CCl4-exposure which may be related to non-EC cell types in the liver, including HSCs. In HSCs, Zeb2 has been suggested to have a pro-fibrotic role since its downregulation through mir145 overexpression caused apoptosis and inhibited activation of the HSC LX-2 cell line.14,15 Zeb2 also has a complex role in hepatocytes where Zeb2 deficiency supports fat accumulation but also enhances liver regeneration via increased hepatocyte proliferation.14,15,21 In Kupffer cells, Zeb2 has been shown to be an important determinant of their specific identity and differentiation state.16 Zeb2 did not affect the zonated expression pattern of LSECs but rather caused a partial LSEC dedifferentiation under baseline conditions. This dedifferentiation (known as ‘capillarization’) was more pronounced upon CCl4 challenge. This aggravated capillarization likely contributed to the increased fibrosis upon endothelial Zeb2 loss, as capillarized LSECs lose their ability to inhibit HSC activation.6 The different and sometimes opposite roles for Zeb2 in different liver cell types definitely implies that using Zeb2 as a target for treating liver disease will require a cell type-specific approach. Here, we studied the role of Zeb2 upon toxin-induced liver fibrosis. It remains to be determined whether endothelial Zeb2 would have a similar role in other liver injury models.
Zeb2 has extensively been studied in cancer because of its role in primary tumours and metastasis by promoting EMT.13,16 ECs can also change to a mesenchymal cell phenotype through a similar process, known as endothelial-to-mesenchymal transition (EndoMT).40 EndoMT has been suggested to play a role in liver fibrosis since Erg-deletion in mice was recently shown to lead to EndoMT and liver fibrosis; the extent to which EndoMT occurs during fibrosis is, however, organ-dependent and is limited in the liver.40–42 If Zeb2 would induce EndoMT, its loss in ECs would reduce fibrosis. Based on mRNA expression changes in Zeb2-deficient ECs, we found no evidence for a role of Zeb2 in EndoMT (Supplementary material online, Figure S14B).
Depending on which co-factors Zeb2 interacts with, it can either act as transcriptional repressor or activator.9 Zeb2 acts mainly as a transcriptional repressor in embryonic stem cells, haematopoietic cells, forebrain neurons, and myelinating cells,11,12,43–46 but loss of Zeb2 in ECs revealed a majority of downregulated genes (Supplementary material online, Figure S4C). This could be because of its direct aforementioned action as a transcriptional activator through interaction with activating co-factors and/or indirectly occur via Zeb2-dependent repression of EC-specific other repressors, a mechanism that has also been suggested in Schwann cells.11,12 While the pro-angiogenic and dedifferentiation effects resulting from endothelial Zeb2-KO were likely mostly the result of a cell-autonomous effect, we propose that the pro-fibrotic effect was related to the disturbed paracrine communication with HSCs. A non-autonomous effect has been observed previously in brain cortex development, where Zeb2 present in the upper layers of neurons is needed for the control of appropriate levels of factors that control neurogenesis and gliogenesis in subventricular progenitor cells45 and deletion of Zeb2 from the ventricular-subventricular zone in the developing brain affects non-targeted cells.10 In contrast to HSCs, endothelial Zeb2-KO had the least effect on the expression profiles of hepatocytes and did not change the expression of angiocrine factors known to be involved in hepatocyte zonation,29–31 suggesting the communication with hepatocytes was not much affected. Also, endothelial Zeb2 loss did not influence the hepatocytes’ regenerative response during the regression phase nor did it alter the intrinsic susceptibility of hepatocytes to CCl4-induced acute damage. Accordingly, endothelial Zeb2 deficiency did not disturb the zonated expression pattern of hepatocyte markers, including that of Cyp2e1, the enzyme responsible for the metabolization of CCl4 that is required for its hepatotoxicity. In contrast, a recent study in which Gata4, another LSEC-enriched transcription factor, was deleted in LSECs revealed altered paracrine communication with both hepatocytes and HSCs, the former involving decreased angiocrine signals supporting hepatocyte regeneration and zonation and the latter involving increased expression of Pdgfb, which is known to activate HSCs.32 We also found increased Pdgfb expression upon endothelial Zeb2 deletion, which may co-determine the increased fibrosis seen in Zeb2-ECKO mice. In addition, our NicheNet analysis predicted numerous downregulated ligands in LSECs that have previously been shown to attenuate liver fibrosis (including Gdf15, Igf1, and Ltf), hence their concerted downregulation could stimulate HSC activation upon Zeb2-KO in ECs.23–25 Although the functional importance of these ligands is supported by our observation that a large panel of their predicted targets was also downregulated in HSCs, additional experiments are required to prove their causal involvement in altered HSC behaviour upon endothelial Zeb2 deletion. While endothelial Gata4 deficiency caused spontaneous fibrosis, we did not observe this upon endothelial Zeb2 loss, even after long-term follow-up (Supplementary material online, Note S10), suggesting that compared to Gata4 deficiency, Zeb2 loss had a less severe phenotype. This aligns with our previous report8 that Gata4 has more impact on the LSEC signature than Zeb2; however, the difference could also partly be due to a different timing of the deletion which was during the embryonic stage in the Gata4 study32 vs. the adult stage in ours.
Endothelial Zeb2-KO did not provoke spontaneous fibrosis, but aggravated fibrosis upon repeated toxin exposure. We therefore propose that while endothelial Zeb2-KO can sensitize HSCs by releasing the brake on their activation, an additional challenge is necessary to induce full HSC activation and fibrosis. While endothelial Zeb2-deficiency only induced partial signs of spontaneous LSEC capillarization, upon exposure to CCl4, the loss of Zeb2 strongly aggravated this process. Finally, in line with the previous observation that fibrosis is associated with vascular expansion,7 exposure to CCl4 caused an expansion of the vasculature; however, this expansion was not further increased by endothelial Zeb2-KO. Therefore, the pro-fibrotic effect of endothelial Zeb2-KO is not likely indirectly caused by vascular expansion and the effect of Zeb2-KO on vascular expansion was only apparent in the non-challenged liver. Altogether, we therefore conclude that the increased fibrosis in ECKO mice was most likely due to increased capillarization and altered EC-HSC communication resulting in increased HSC activation.
In conclusion, we demonstrate that Zeb2 has a cell type-specific and context-dependent role in liver vascular maintenance and fibrosis. Our study also emphasizes the importance of the liver endothelium in the pathogenesis of liver fibrosis. Currently, there are no effective treatments for patients suffering from liver fibrosis, hence the identification of novel candidate targets is urgent. To exploit the protective effect of Zeb2 in ECs against liver fibrosis and bypass its pro-fibrogenic effect in other liver cells, an EC-specific approach should be designed. Alternatively, the HSC quiescence factors we identified that depend on Zeb2 in ECs could be used as new means to tackle fibrosis.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
W.d.H. designed the study, performed experiments, analysed data, and wrote the manuscript; W.D. performed experiments, analysed data, and edited the manuscript; K.A. performed ultrafast ultrasound experiments and analysed data; A.C., M.W.S., S.Vi., P.V., E.C., M.L., and I.M. performed experiments. W.F.J.v.IJ. coordinated RNA sequencing experiments. S.D.-D., T.D., and M.T. optimized ultrafast ultrasound data processing and analysis; S.Ve. performed NicheNet analysis; E.M. performed RNA sequencing data analysis; T.T. and Y.H. provided a transgenic mouse model and performed experiments; J.J. and B.T. provided human biopsies; G.B. and J.H. provided transgenic mouse models; A.Z., L.A.v.G., and F.P.G.L. provided critical intellectual input; D.H. provided transgenic mouse models and critical intellectual input; A.L. designed the study and edited the manuscript. All authors reviewed and approved the manuscript.
Conflict of interest: none declared.
Funding
This work was supported by internal funding from KU Leuven [C12/16/023 to W.d.H. and A.Z., C14/19/095 to A.L., A.Z., and D.H., and ID-N19/031 to A.L.]; a Program Financing grant [PF/10/014 to A.L.]; a European Research Council grant [FP7-StG-IMAGINED203291 to A.L.]; a Cosmetics Europe/European Commission FP7 Grant [FP7-Health-HemiBio266777 to A.L. and L.A.v.G.]; an Interuniversity Attraction Poles grant [IUAP/P7/07 to A.L., A.Z., and D.H.]; Fonds voor Wetenschappelijk Onderzoek [FWO] grants [G.0A3116 to D.H. and W001420N to A.L., F.P.G.L., and A.Z.]; a ZONMW OffRoad program [2018/23115|ZONMW to E.M.]; a grant of KAKENHI [17K09896 to Y.H.]; a Marie Sklodowska-Curie Actions post-doctoral fellowship [H2020-MSCA-IF-REZONABLE658666 to W.d.H.], a Horizon 2020 Marie Skłodowska-Curie Actions-Innovative Training Network grant [H2020-MSCA-ITN-RenalToolBox813839 to F.P.G.L. and K.A.]; a pre-doctoral FWO fellowship [1157318N to W.D.]; a post-doctoral FWO fellowship [12N5419N LV 2479 to I.M.]; the Inserm Technology Research Accelerator grant in Biomedical Ultrasound [to M.T.]; and a grant from the Dutch Cancer Society [KWF 10339 to F.P.G.L.].
Data availability
The RNA sequencing datasets underlying this article are available in the NCBI GEO repository at https://www.ncbi.nlm.nih.gov/geo/, and can be accessed with GSE150699.
Supplementary Material
Contributor Information
Willeke de Haan, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Wouter Dheedene, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Katerina Apelt, Department of Internal Medicine (Nephrology), Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, Leiden, The Netherlands.
Sofiane Décombas-Deschamps, Physics for Medicine Paris, Inserm, CNRS, ESPCI Paris, Paris Sciences et Lettres University, Paris, France.
Stefan Vinckier, Department of Oncology, Laboratory of Angiogenesis and Vascular Metabolism, KU Leuven, Leuven, Belgium; Laboratory of Angiogenesis and Vascular Metabolism, Center for Cancer Biology, Vlaams Instituut voor Biotechnologie (VIB), Leuven, Belgium.
Stefaan Verhulst, Liver Cell Biology Research Group, Vrije Universiteit Brussel, Brussels, Belgium.
Andrea Conidi, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Thomas Deffieux, Physics for Medicine Paris, Inserm, CNRS, ESPCI Paris, Paris Sciences et Lettres University, Paris, France.
Michael W Staring, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Petra Vandervoort, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Ellen Caluwé, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Marleen Lox, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Inge Mannaerts, Liver Cell Biology Research Group, Vrije Universiteit Brussel, Brussels, Belgium.
Tsuyoshi Takagi, Department of Disease Model, Institute of Developmental Research, Aichi Developmental Disability Center, Aichi, Japan.
Joris Jaekers, Abdominal Surgery, UZ Leuven, Leuven, Belgium.
Geert Berx, Molecular and Cellular Oncology Laboratory, Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium; Cancer Research Institute Ghent (CRIG), Ghent, Belgium.
Jody Haigh, Department of Pharmacology and Therapeutics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, Manitoba, Canada; Research Institute in Oncology and Hematology, Cancer Care Manitoba, Winnipeg, Manitoba, Canada.
Baki Topal, Abdominal Surgery, UZ Leuven, Leuven, Belgium.
An Zwijsen, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Yujiro Higashi, Department of Disease Model, Institute of Developmental Research, Aichi Developmental Disability Center, Aichi, Japan.
Leo A van Grunsven, Liver Cell Biology Research Group, Vrije Universiteit Brussel, Brussels, Belgium.
Wilfred F J van IJcken, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands; Center for Biomics-Genomics, Erasmus University Medical Center, Rotterdam, The Netherlands.
Eskeatnaf Mulugeta, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands.
Mickael Tanter, Physics for Medicine Paris, Inserm, CNRS, ESPCI Paris, Paris Sciences et Lettres University, Paris, France.
Franck P G Lebrin, Department of Internal Medicine (Nephrology), Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, Leiden, The Netherlands; Physics for Medicine Paris, Inserm, CNRS, ESPCI Paris, Paris Sciences et Lettres University, Paris, France.
Danny Huylebroeck, Department of Cell Biology, Erasmus University Medical Center, Rotterdam, The Netherlands; Department of Development and Regeneration, KU Leuven, Leuven, Belgium.
Aernout Luttun, Department of Cardiovascular Sciences, Center for Molecular and Vascular Biology, KU Leuven, Campus Gasthuisberg, Onderwijs & Navorsing 1, Herestraat 49, Box 911, 3000 Leuven, Belgium.
Translational perspective
Liver sinusoidal endothelial cells (LSECs) are specialized to support transport between blood and hepatocytes and to protect against fibrosis by inhibiting hepatic stellate cell (HSC) activation. Here, we show that transcription factor Zeb2 in LSECs controls their specialization and communication with HSCs and protects against fibrosis. Therefore, Zeb2 and Zeb2-dependent genes represent appealing targets to tackle liver fibrosis.
References
- 1. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res 2007;100:174–190. [DOI] [PubMed] [Google Scholar]
- 2. Coppiello G, Collantes M, Sirerol-Piquer MS, Vandenwijngaert S, Schoors S, Swinnen M, Vandersmissen I, Herijgers P, Topal B, van Loon J, Goffin J, Prósper F, Carmeliet P, García-Verdugo JM, Janssens S, Peñuelas I, Aranguren XL, Luttun A. Meox2/Tcf15 heterodimers program the heart capillary endothelium for cardiac fatty acid uptake. Circulation 2015;131:815–826. [DOI] [PubMed] [Google Scholar]
- 3. Geraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, Leibing T, Demory A, Ulbrich F, Diett M, Singh S, Sticht C, Breitkopf-Heinlein K, Richter K, Karppinen SM, Pihlajaniemi T, Arnold B, Rodewald HR, Augustin HG, Schledzewski K, Goerdt S. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest 2017;127:1099–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou PE. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol 2017;66:212–227. [DOI] [PubMed] [Google Scholar]
- 5. Marrone G, Shah VH, Gracia-Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 2016;65:608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeLeve LD, Maretti-Mira AC. Liver sinusoidal endothelial cell: an update. Semin Liver Dis 2017;37:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Natarajan V, Harris EN, Kidambi S. SECs (Sinusoidal Endothelial Cells), liver microenvironment, and fibrosis. Biomed Res Int 2017;2017:4097205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Haan W, Oie C, Benkheil M, Dheedene W, Vinckier S, Coppiello G, Aranguren XL, Beerens M, Jaekers J, Topal B, Verfaillie C, Smedsrod B, Luttun A. Unraveling the transcriptional determinants of liver sinusoidal endothelial cell specialization. Am J Physiol Gastrointest Liver Physiol 2020;318:G803–G815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conidi A, Cazzola S, Beets K, Coddens K, Collart C, Cornelis F, Cox L, Joke D, Dobreva MP, Dries R, Esguerra C, Francis A, Ibrahimi A, Kroes R, Lesage F, Maas E, Moya I, Pereira PN, Stappers E, Stryjewska A, van den Berghe V, Vermeire L, Verstappen G, Seuntjens E, Umans L, Zwijsen A, Huylebroeck D. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFbeta/BMP signaling in vivo. Cytokine Growth Factor Rev 2011;22:287–300. [DOI] [PubMed] [Google Scholar]
- 10. Deryckere A, Stappers E, Dries R, Peyre E, van den BV, Conidi A, Zampeta FI, Francis A, Bresseleers M, Stryjewska A, Vanlaer R, Maas E, Smal IV, van IJcken WFJ, Grosveld FG, Nguyen L, Huylebroeck D, Seuntjens E. Multifaceted actions of Zeb2 in postnatal neurogenesis from the ventricular-subventricular zone to the olfactory bulb. Development 2020;147:dev184861. [DOI] [PubMed] [Google Scholar]
- 11. Quintes S, Brinkmann BG, Ebert M, Frob F, Kungl T, Arlt FA, Tarabykin V, Huylebroeck D, Meijer D, Suter U, Wegner M, Sereda MW, Nave KA. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat Neurosci 2016;19:1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu LM, Wang J, Conidi A, Zhao C, Wang H, Ford Z, Zhang L, Zweier C, Ayee BG, Maurel P, Zwijsen A, Chan JR, Jankowski MP, Huylebroeck D, Lu QR. Zeb2 recruits HDAC-NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat Neurosci 2016;19:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 2001;7:1267–1278. [DOI] [PubMed] [Google Scholar]
- 14. Zhou DD, Wang X, Wang Y, Xiang XJ, Liang ZC, Zhou Y, Xu A, Bi CH, Zhang L. MicroRNA-145 inhibits hepatic stellate cell activation and proliferation by targeting ZEB2 through Wnt/beta-catenin pathway. Mol Immunol 2016;75:151–160. [DOI] [PubMed] [Google Scholar]
- 15. Yang J, Liu Q, Cao S, Xu T, Li X, Zhou D, Pan L, Li C, Huang C, Meng X, Zhang L, Wang X. MicroRNA-145 increases the apoptosis of activated hepatic stellate cells induced by TRAIL through NF-kappaB signaling pathway. Front Pharmacol 2017;8:980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, Bonnardel J, De Prijck S, Vandamme N, Cannoodt R, Saelens W, Vanneste B, Toussaint W, De Bleser P, Takahashi N, Vandenabeele P, Henri S, Pridans C, Hume DA, Lambrecht BN, De Baetselier P, Milling SWF, Van Ginderachter JA, Malissen B, Berx G, Beschin A, Saeys Y, Guilliams M. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity 2018;49:312–325.e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stradiot L, Verhulst S, Roosens T, Oie CI, Moya IM, Halder G, Mannaerts I, van Grunsven LA. Functionality based method for simultaneous isolation of rodent hepatic sinusoidal cells. Biomaterials 2017;139:91–101. [DOI] [PubMed] [Google Scholar]
- 18. Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, Gaarde WA, Deleve LD. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012;142:918–927.e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demene C, Tiran E, Sieu LA, Bergel A, Gennisson JL, Pernot M, Deffieux T, Cohen I, Tanter M. 4D microvascular imaging based on ultrafast Doppler tomography. Neuroimage 2016;127:472–483. [DOI] [PubMed] [Google Scholar]
- 20. Piechnik SK, Chiarelli PA, Jezzard P. Modelling vascular reactivity to investigate the basis of the relationship between cerebral blood volume and flow under CO2 manipulation. Neuroimage 2008;39:107–118. [DOI] [PubMed] [Google Scholar]
- 21. Yang L, Wang Y, Shi Y, Bu H, Ye F. Deletion of SIP1 promotes liver regeneration and lipid accumulation. Pathol Res Pract 2016;212:421–425. [DOI] [PubMed] [Google Scholar]
- 22. Barbera-Guillem E, Rocha M, Alvarez A, Vidal-Vanaclocha F. Differences in the lectin-binding patterns of the periportal and perivenous endothelial domains in the liver sinusoids. Hepatology 1991;14:131–139. [DOI] [PubMed] [Google Scholar]
- 23. Chung HK, Kim JT, Kim HW, Kwon M, Kim SY, Shong M, Kim KS, Yi HS. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep 2017;7:17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tung YT, Tang TY, Chen HL, Yang SH, Chong KY, Cheng WT, Chen CM. Lactoferrin protects against chemical-induced rat liver fibrosis by inhibiting stellate cell activation. J Dairy Sci 2014;97:3281–3291. [DOI] [PubMed] [Google Scholar]
- 25. Nishizawa H, Iguchi G, Fukuoka H, Takahashi M, Suda K, Bando H, Matsumoto R, Yoshida K, Odake Y, Ogawa W, Takahashi Y. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci Rep 2016;6:34605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffin CT, Gao S. Building discontinuous liver sinusoidal vessels. J Clin Invest 2017;127:790–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Styp-Rekowska B, Hlushchuk R, Pries AR, Djonov V. Intussusceptive angiogenesis: pillars against the blood flow. Acta Physiol (Oxf) 2011;202:213–223. [DOI] [PubMed] [Google Scholar]
- 28. Cheng X, Kim SY, Okamoto H, Xin Y, Yancopoulos GD, Murphy AJ, Gromada J. Glucagon contributes to liver zonation. Proc Natl Acad Sci USA 2018;115:E4111–E4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rocha AS, Vidal V, Mertz M, Kendall TJ, Charlet A, Okamoto H, Schedl A. The angiocrine factor Rspondin3 is a key determinant of liver zonation. Cell Rep 2015;13:1757–1764. [DOI] [PubMed] [Google Scholar]
- 30. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 2015;524:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wohlfeil SA, Hafele V, Dietsch B, Schledzewski K, Winkler M, Zierow J, Leibing T, Mohammadi MM, Heineke J, Sticht C, Olsavszky V, Koch PS, Geraud C, Goerdt S. Hepatic endothelial notch activation protects against liver metastasis by regulating endothelial-tumor cell adhesion independent of angiocrine signaling. Cancer Res 2019;79:598–610. [DOI] [PubMed] [Google Scholar]
- 32. Winkler M, Staniczek T, Kürschner SW, Schmid CD, Schönhaber H, Cordero J, Kessler L, Mathes A, Sticht C, Neßling M, Uvarovskii A, Anders S, Zhang X-J, von Figura G, Hartmann D, Mogler C, Dobreva G, Schledzewski K, Géraud C, Koch P-S, Goerdt S. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J Hepatol 2021;74:380–393. [DOI] [PubMed] [Google Scholar]
- 33. Sun HJ, Chen J, Zhang H, Ni B, van Velkinburgh JC, Liu Y, Wu YZ, Yang X. Von Willebrand factor protects against acute CCl4-induced hepatotoxicity through phospho-p38 MAPK signaling pathway inhibition. Immunol Res 2017;65:1046–1058. [DOI] [PubMed] [Google Scholar]
- 34. Goossens S, Janzen V, Bartunkova S, Yokomizo T, Drogat B, Crisan M, Haigh K, Seuntjens E, Umans L, Riedt T, Bogaert P, Haenebalcke L, Berx G, Dzierzak E, Huylebroeck D, Haigh JJ. The EMT regulator Zeb2/Sip1 is essential for murine embryonic hematopoietic stem/progenitor cell differentiation and mobilization. Blood 2011;117:5620–5630. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y, Banda M, Speyer CL, Smith JS, Rabson AB, Gorski DH. Regulation of the expression and activity of the antiangiogenic homeobox gene GAX/MEOX2 by ZEB2 and microRNA-221. Mol Cell Biol 2010;30:3902–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang LJ, Wu ZH, Zheng XT, Long JY, Dong YM, Fang X. Zinc finger E-box binding protein 2 (ZEB2) suppress apoptosis of vascular endothelial cells induced by high glucose through mitogen-activated protein kinases (MAPK) pathway activation. Med Sci Monit 2017;23:2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gianni-Barrera R, Butschkau A, Uccelli A, Certelli A, Valente P, Bartolomeo M, Groppa E, Burger MG, Hlushchuk R, Heberer M, Schaefer DJ, Gurke L, Djonov V, Vollmar B, Banfi A. PDGF-BB regulates splitting angiogenesis in skeletal muscle by limiting VEGF-induced endothelial proliferation. Angiogenesis 2018;21:883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dill MT, Rothweiler S, Djonov V, Hlushchuk R, Tornillo L, Terracciano L, Meili-Butz S, Radtke F, Heim MH, Semela D. Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 2012;142:967–977.e962. [DOI] [PubMed] [Google Scholar]
- 39. Jahan F, Landry NM, Rattan SG, Dixon IMC, Wigle JT. The functional role of zinc finger E box-binding homeobox 2 (Zeb2) in promoting cardiac fibroblast activation. Int J Mol Sci 2018;19:3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med 2016;5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dufton NP, Peghaire CR, Osuna-Almagro L, Raimondi C, Kalna V, Chauhan A, Webb G, Yang Y, Birdsey GM, Lalor P, Mason JC, Adams DH, Randi AM. Dynamic regulation of canonical TGFbeta signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat Commun 2017;8:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ribera J, Pauta M, Melgar-Lesmes P, Córdoba B, Bosch A, Calvo M, Rodrigo-Torres D, Sancho-Bru P, Mira A, Jiménez W, Morales-Ruiz M. small population of liver endothelial cells undergoes endothelial-to-mesenchymal transition in response to chronic liver injury. Am J Physiol Gastrointest Liver Physiol 2017;313:G492–G504. [DOI] [PubMed] [Google Scholar]
- 43. Stryjewska A, Dries R, Pieters T, Verstappen G, Conidi A, Coddens K, Francis A, Umans L, van IWF, Berx G, van Grunsven LA, Grosveld FG, Goossens S, Haigh JJ, Huylebroeck D. Zeb2 regulates cell fate at the exit from epiblast state in mouse embryonic stem cells. Stem Cells 2017;35:611–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li J, Riedt T, Goossens S, Carrillo Garcia C, Szczepanski S, Brandes M, Pieters T, Dobrosch L, Gutgemann I, Farla N, Radaelli E, Hulpiau P, Mallela N, Frohlich H, La Starza R, Matteucci C, Chen T, Brossart P, Mecucci C, Huylebroeck D, Haigh JJ, Janzen V. The EMT transcription factor Zeb2 controls adult murine hematopoietic differentiation by regulating cytokine signaling. Blood 2017;129:460–472. [DOI] [PubMed] [Google Scholar]
- 45. Seuntjens E, Nityanandam A, Miquelajauregui A, Debruyn J, Stryjewska A, Goebbels S, Nave KA, Huylebroeck D, Tarabykin V. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat Neurosci 2009;12:1373–1380. [DOI] [PubMed] [Google Scholar]
- 46. van den Berghe V, Stappers E, Vandesande B, Dimidschstein J, Kroes R, Francis A, Conidi A, Lesage F, Dries R, Cazzola S, Berx G, Kessaris N, Vanderhaeghen P, van Ijcken W, Grosveld FG, Goossens S, Haigh JJ, Fishell G, Goffinet A, Aerts S, Huylebroeck D, Seuntjens E. Directed migration of cortical interneurons depends on the cell-autonomous action of Sip1. Neuron 2013;77:70–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing datasets underlying this article are available in the NCBI GEO repository at https://www.ncbi.nlm.nih.gov/geo/, and can be accessed with GSE150699.