

Abstract
Purpose:
Monoclonal antibodies (mAbs) such as anti-CD20 rituximab, are proven therapies in B-cell malignancies, yet many patients develop resistance. Novel therapies against alternative targets are needed to circumvent resistance mechanisms. We sought to generate mAbs against human B-cell activating factor receptor (BAFF-R/TNFRSF13C), which has not yet been targeted successfully for cancer therapy.
Experimental Design:
Novel mAbs were generated against BAFF-R, expressed as a natively folded cell-surface immunogen on mouse fibroblast cells. Chimeric BAFF-R mAbs were developed and assessed for in vitro and in vivo monotherapy cytotoxicity. The chimeric mAbs were tested against human B-cell tumor lines, primary patient samples, and drug-resistant tumors.
Results:
Chimeric antibodies bound with high affinity to multiple human malignant B-cell lines and induced potent antibody-dependent cellular cytotoxicity (ADCC) against multiple subtypes of human lymphoma and leukemia, including primary tumors from patients who had relapsed after anti-CD20 therapy. Chimeric antibodies also induced ADCC against ibrutinib-resistant and rituximab-insensitive CD20-deficient variant lymphomas, respectively. Importantly, they demonstrated remarkable in vivo growth inhibition of drug-resistant tumor models in immunodeficient mice.
Conclusions:
Our method generated novel anti-BAFF-R antibody therapeutics with remarkable single-agent antitumor effects. We propose that these antibodies represent an effective new strategy for targeting and treating drug resistant B-cell malignancies and warrant further development.
Keywords: Immunotherapy, Antibody, Lymphoma, Drug-resistance, BAFF-R
Introduction
Monoclonal antibody (mAb) immunotherapy is highly successful against hematological malignancies.(1–7) However, mAb treatment alone is not curative, and emerging resistance remains a problem.(8,9) A key example is resistance to anti-CD20, rituximab, thought to be caused by multiple mechanisms including down-regulation of CD20.(10) Thus, there is an urgent need to develop new therapies against B-cell malignancies.
One alternative target on B-cell tumors is B-cell activating factor receptor (BAFF-R/TNFRSF13C), a tumor necrosis factor receptor superfamily member specifically involved in B lymphocyte development and mature B cell survival.(11,12) BAFF-R is expressed almost exclusively on B cells and its surface expression has been documented on various human B-cell lymphomas.(13–17) Although much of the biology of the BAFF/BAFF-R axis is known,(18–21) its initial promise as a target for cancer therapy has not been met; however, it remains an attractive target for B-cell lymphomas, especially drug-resistant tumors.
Conventional recombinant immunogen proteins produced in bacteria for developing mAbs lack post-translational modifications and are simplistically folded because compared with eukaryotes, prokaryotes lack chaperone proteins and oxidizing environments. As a result, such proteins may differ in conformational structure from the corresponding plasma membrane-anchored native proteins. Furthermore, antibodies may be raised against off-target domains such as transmembrane or intracellular domains of the target protein. Here, we applied a strategy of generating mAbs against a natively folded, glycosylated immunogen expressed on eukaryotic cells.(22,23) Specifically, we expressed human BAFF-R as a native protein on mouse fibroblast cells, and used the engineered cell clone as an immunogen in mice. We report on the generation of novel mAbs that specifically bound and lysed human malignant B cell lines and primary lymphomas in vitro, and inhibited growth of drug-resistant lymphoma in xenogenic tumor models in vivo.
Materials and Methods
Animals, cell lines, and primary human tumor samples
Mice:
BALB/c mice for antibody development and NOD scid gamma (NSG) breeding pairs were purchased from The Jackson Laboratory (Bar Harbor, ME). The NSG breeding colony was maintained by the Animal Resource Center at City of Hope. Mice were housed in a pathogen-free animal facility according to institutional guidelines. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC: 15020).
Tumor lines:
Malignant, human hematological cell lines including JeKo-1, Mino, REC-1, JVM-13, SU-DHL-6, Raji, OCI-LY3, RL, RS4;11, MEC-1, SKNO-1, Jurkat and U266 were purchased from either ATCC (Manassas, VA) or DSMZ (Germany). Z-138 line was provided by Dr. Michael Wang (MD Anderson Cancer Center). Ibrutinib-resistant SP49-IR line was developed and provided by Dr. Jianguo Tao (University of South Florida)(24). Ibrutinib-resistant SP49 cell lines (SP49-IR) were established by treating cells with escalating doses of ibrutinib. IC50 was 5 nM for parental SP49 compared to >100 nM for SP49-IR. At 100 nM ibrutinib ~5% of SP49 cells were viable compared with >90% of SP49-IR cells. Human NK-92 176V cells were obtained from Conkwest Inc. (San Diego, CA).
Human blood and tumor samples:
Non-cultured, primary human lymphomas were obtained as cryopreserved, viable single cell suspensions in 10% DMSO from the Lymphoma Satellite Tissue Bank at MD Anderson Cancer Center under an Institutional Review Board approved protocol (IRB: 2005-0656). Primary patient samples included leukapheresis or blood from patients with mantle cell lymphoma (MCL) or chronic lymphocytic leukemia (CLL), and excised lymph nodes from patients with diffuse large B-cell lymphoma (DLBCL) or follicular lymphoma (FL). Tumor cells in each sample ranged from 80% to 98% for leukapheresis or blood, and from 50% to 60% for lymph node biopsies. Peripheral blood mononuclear cells (PBMC) were provided by the Michael Amini Transfusion Medicine Center at City of Hope (IRB: 15283).
Generation of human-BAFF-R expressing mouse fibroblast cells
Human (h)BAFF-R cDNA was from human B cells and cloned in-frame with GFP gene on pEGFP-N1 vector (Takara/Clotech, Mountain View, CA). (h)BAFF-R cDNA sequence was confirmed against the NCBI gene sequence database (Gene ID: 115650). The cDNA encoding (h)BAFF-R-GFP fusion was subsequently cloned into a lentivirus gene delivery system (pLenti6/V5-DEST Gateway Vector kit, Life Technologies, Grand Island, NY) to produce (h)BAFF-R-GFP fusion proteins when transduced into mouse fibroblast (L) cells. Single cell clones were established from sorted GFP-positive L cells, and (h)BAFF-R-GFP-expressing L cell clone D2C was used in further studies.
Antibody-producing hybridomas
Two 6-week-old BALB/c mice were immunized with live, untreated mouse fibroblast (L) cell clone (D2C) engineered to express human BAFF-R on the cell membrane by five subcutaneous injections at the foot pad once every three days. Blood samples were obtained from both mice to confirm serum antibodies against D2C by ELISA. Splenic tissue from the immunized mice was harvested on day 20. Harvested splenocytes were fused with Sp2/0 myeloma to establish hybridomas and ELISA screened for antibodies against the antigen using D2C or parental L cell-coated plates. Immunization and hybridoma procedures were conducted at the Antibody Core Facility at MD Anderson Cancer Center.
Chimeric antibody production
cDNA from selected hybridomas encoding the variable regions of antibody light and heavy chains were engineered onto expression vectors containing respective human IgG1 constant regions. Vectors were co-transfected into the FreeStyle 293 Expression System (Life Technologies) according to manufacturer’s directions. Antibodies in culture supernatant were purified by HiTrap Protein A affinity chromatography columns (GE Healthcare, Marlborough, MA) according to the manufacturer’s directions.
Cytotoxicity assays
Target cells (L cells, human tumor lines, primary patient samples) were labeled with chromium-51 (51Cr, Perkin Elmer, Waltham, MA) for a 51Cr release assay. Briefly, antibodies and effectors (NK cells or complement serum standard [Sigma Aldrich, St. Louis, MO]), were added to labeled target cells and incubated up to18 hours. NK cells were enriched from PBMC (NK cell enrichment kit, Stemcell Technologies, Vancouver, Canada). Controls included effector cells only or effectors cells + control IgG (Human IgG Isotype Control; Invitrogen, Carlsbad, CA). Counts per minute (CPM) of 51Cr released into supernatant was detected with a Wizard Automatic Gamma Counter (Perkin Elmer). Percent lysis was calculated by | SR: CPM of spontaneous release; MR: CPM of maximum release.
Generation of JeKo-1-CD20-KO
FACS-sorted, stable JeKo-1-CD20-KO were generated using CD20-CRISPR/Cas9 and HDR Plasmid Systems (Santa Cruz Biotechnology, Santa Cruz, CA) according to manufacturer’s directions. CD20 knock-out was verified by flow cytometry and Western blots.
In vivo studies
Tumor Models:
Stable, luciferase-expressing tumor lines were established for bioluminescent imaging in mouse models. Briefly, a luciferase gene was introduced into tumor lines by a lentivirus gene delivery system (pLenti7.3/V5-DEST Gateway Vector Kit, Life Technologies). The minimum lethal dose was determined for each tumor cell line by dose titration (1 × 106 JeKo-1, 5 × 105 RS4;11, 5 × 105 JeKo-1-CD20-KO, or 2.5 × 104 Z-138 cells). Tumor cells were injected intravenously (IV), and tumor development was continuously monitored by in vivo bioluminescence imaging.
Bioluminescent Imaging:
Mice were anesthetized with isoflurane and administered 150 mg/kg D-luciferin (Life Technologies) via intraperitoneal (IP) injection 10 min prior to imaging. Imaging was performed on an AmiX imaging system (Spectral Instruments Imaging, Tucson, AZ).
Antibody Studies:
Mice (n=5 per group) were IV tumor challenged three days prior to four treatments once every five days. Treatments were 300 μL IV injection: 200 μg treatment antibody, 10 × 106 effector human NK-92-176V cells, and 5 × 104 IU IL-2 (Prometheus Laboratories, San Diego, CA). Control groups received the same volume injections with saline or NK cells alone. Bioluminescent imaging was performed weekly up to 80 days. Survival was tracked up to 100 days post tumor challenge.
Results
Generation of monoclonal antibodies against human BAFF-R
In order to generate a therapeutic antibody to a biologically relevant epitope of BAFF-R we used a eukaryotic cell-surface expression system in which endogenous cell-surface proteins are presented in their native conformation with appropriate post-translational modifications. We engineered a mouse fibroblast (L) cell clone expressing cell-surface GFP-tagged, human BAFF-R. BAFF-R-expressing L cell clones were generated and characterized for GFP expression (Figure 1A). Clone D2C was expanded and successfully used to immunize BALB/c mice according to Methods and immunization schedule in Supplementary Figure 1A.
Figure 1. Generation and specificity of novel monoclonal antibodies against human BAFF-R.
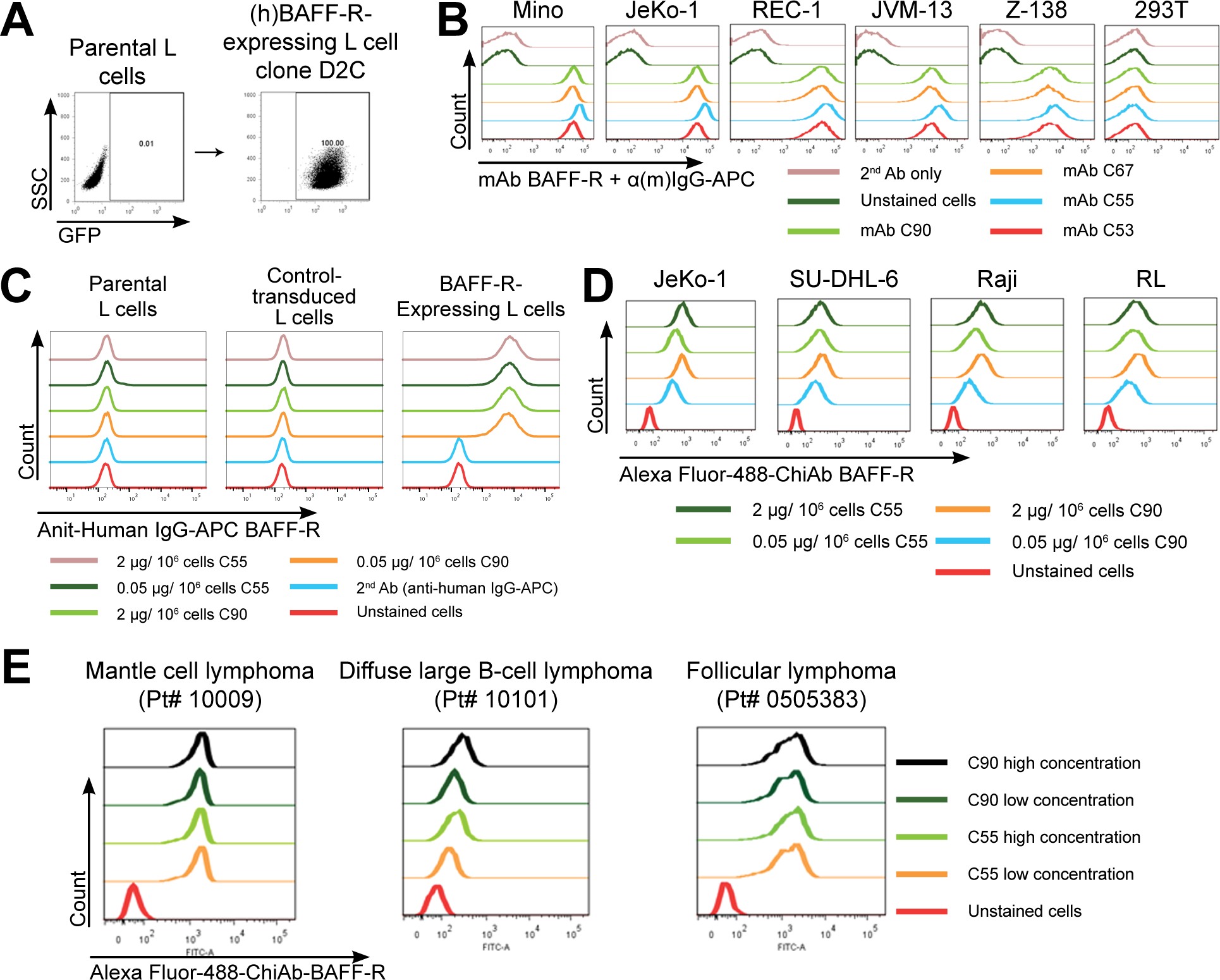
(A) FACS analysis of cell surface expression of (h)BAFF-R-GFP fusion protein in mouse fibroblast L cells. Gated on GFP-positive cells, engineered L cell clone (right plot) is compared to parental L cells (left plot). Clone D2C was selected for further studies. (B-E) FACS histograms of anti-BAFF-R monoclonal antibodies binding cell lines and patient samples: (B) Affinity purified hybridoma mAb (C90, C67, C55, and C53) binding BAFF-R-positive, human MCL lines including Mino, JeKo-1, REC-1, JVM-13, and Z-138 at a concentration of 0.05 μg mAb/106 cells. BAFF-R-negative 293T embryonic kidney cell line was used as a control. (C) Chimeric antibodies C55 and C90 at high and low concentration binding (h)BAFF-R-expressing L cells. Parental L cells, control-transduced L cells, and secondary anti-(h)IgG-APC antibodies only were used as controls. (D) Alexa fluor 488-conjugated chimeric antibodies binding a panel of NHL cell lines. (E) Chimeric antibodies binding three types of NHL primary patient samples. The data are representative of three independent experiments.
After generating and screening hybridoma clones, we identified four clones (53, 55, 67, and 90) as producing antibodies that specifically bound BAFF-R-expressing, but not parental, L cells (Supplementary Figure 1B). Supernatants of all four clones contained antibodies that bound BAFF-R-expressing Mino cell line (MCL) in a dose-dependent manner. No antibody binding was detected in BAFF-R-negative control cell line, 293T (Supplementary Figure 2).
Antibodies from the four hybridoma supernatants were purified by protein A affinity chromatography. Purified antibodies bound Mino cells in a dose-dependent manner (Supplementary Figure 3), as well as other human MCL lines, including JeKo-1, REC-1, and ibrutinib-resistant JVM-13 and Z-138 (Figure 1B).
An analysis of the complementarity determining regions (CDRs) on the four antibodies revealed that clones 53, 55, and 67 had nearly identical sequences, whereas clone 90 was unique. Therefore, we selected clones 55 and 90 for further investigation. Both clones 55 and 90 effectively bound JeKo-1 (MCL), SU-DHL-6 (DLBCL), Raji (Burkitt lymphoma), and RL (FL) at both high (2 μg/106 cells) and low (0.05 μg/106 cells) concentrations (Supplementary Figure 4).
Chimeric mAb against human BAFF-R induced anti-tumor effects both in vitro and in vivo
Clones 55 and 90 were further developed into their respective chimeric mAbs containing human IgG1 constant regions (termed C55 and C90). The chimeric antibodies retained specific dose-dependent binding to BAFF-R-expressing L cells but not parental or control-transduced L cells (Figure 1C). C55 and C90 were conjugated to Alexa Fluor 488 and exhibited direct binding to non-Hodgkin lymphoma (NHL) lines JeKo-1, SU-DHL-6, Raji, and RL (Figure 1D). Importantly, both fluorochrome-labeled chimeric mAbs (Figure 1E) and original mouse mAbs (Supplementary Figure 5) readily bound MCL, DLBCL, and FL patient primary tumor samples. Binding of our mAbs correlated with the expression of BAFF-R on human NHL cell lines (Supplementary Figure 6A) and primary tumors (Supplementary Figure 6B) further supporting antigen specificity.
C55 and C90 elicited antibody-dependent cell-mediated cytotoxicity (ADCC) specifically against BAFF-R-expressing L cells and JeKo-1, but not BAFF-R negative parental L cells nor the BAFF-R negative human multiple myeloma line (U266; Figure 2A). In contrast, antibodies did not elicit in vitro complement dependent cytotoxicity (CDC, Figure 2B). Cytotoxicity required the addition of NK cells, as shown for various types of human NHL cell lines (Figure 2C, Supplementary Figure 7), suggesting ADCC as a principal mechanism of antibody-mediated cytotoxicity. We found the antibodies inhibited BAFF/BAFF-R binding in a dose-dependent manner (Figure 2D), suggesting potential disruption of BAFF/BAFF-R interaction in tumor cells and further support for their specificity. Furthermore, C55 and C90 exhibited limited internalization upon binding BAFF-R (Supplementary Figure 8). In vitro ADCC was confirmed in our panel of malignant, human B-cell lines including: JeKo-1, SU-DHL-6, Raji, RL (Figure 2E), RS4;11 and MEC-1 (Supplementary Figure 9A). No cytotoxicity was observed in BAFF-R negative lines including U266, T-cell leukemia (Jurkat), and acute myeloid leukemia (SKNO-1 Supplementary Figure 9B). Importantly, chimeric antibodies elicited ADCC against primary patient tumor samples (Figure 3A, Supplementary Figure 10).
Figure 2. BAFF-R monoclonal antibodies exhibited specific in vitro cytotoxicity against B-cell tumor lines.
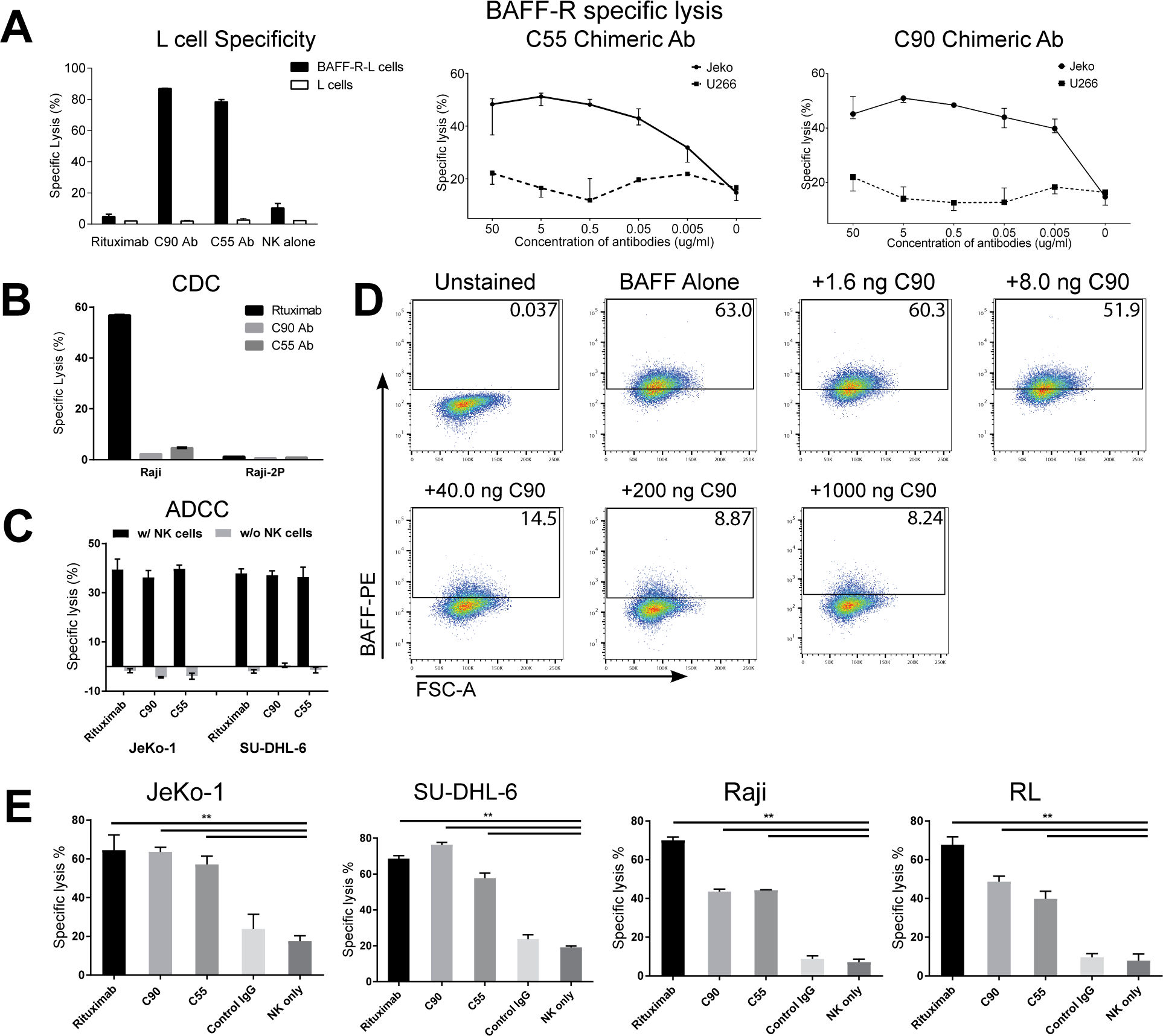
(A-D) Antibody BAFF-R specificity was determined by specific lysis and ligand blocking assays. Antibody-dependent cytotoxicity (ADCC) was measured by chromium-51 release assay and specific lysis of target cells was calculated as described in methods. Antibodies (C55, C90, or rituximab) and effectors (NK cells or complement containing serum) were incubated with chromium-51 labeled target cells. (A) Specific and dose response lysis are show for various target cells. BAFF-R expressing cells included BAFF-R L cells and JeKo-1, and BAFF-R negative controls include parental L cells and U266 human multiple myeloma cell line. NK cells were included in an effector to target ratio (E:T) of 20:1. (B) CDC specific lysis is show for CDC sensitive Raji and CDC-resistant Raji-2P. Active complement-containing human serum was incubated with target cells at 1:3 dilution. (C) ADCC specific lysis is shown for JeKo-1 and SU-DHL-6. Target cells and antibodies were incubated with or without effector NK cells (E:T = 20:1). (D) FACS plot gates show percentage of BAFF/BAFF-R binding signal in the presence of C90 mAb. BAFF-R-expressing D2C L cell clones were incubated with C90 (0–1000 ng/106 cells) at 4°C for 45 min followed by incubation with recombinant BAFF ligand (0.5 μg/106 cells) at 4 °C for 90 min. Flow cytometry was performed and gated for anti-BAFF-PE. (E) ADCC effects were measured on a panel of NHLs. Chromium-51 labeled NHL lines were incubated with antibodies rituximab, C90, C55, or Control IgG and effector NK cells (E:T = 20:1). NK cells only (without antibodies) was an additional control. Calculated specific lysis is shown as the mean ± s.d. of triplicate samples. **P < 0.05 compared with NK cell only and Control IgG by two-tailed Student’s t-test.
Figure 3. BAFF-R monoclonal antibodies induced in vitro ADCC against primary B-cell tumors.

Antibody-dependent cell-mediated cytotoxicity (ADCC) effects were measured by chromium-51 release after incubation with rituximab, C90, C55, Control IgG, and NK cells only at E:T = 20:1. Results were calculated as percentage of cell specific lysis of target cells: (A) NHL patient samples **P < 0.05 compared with NK cells only and Control IgG by two-tailed Student’s t-test; (B) primary CLL and MCL samples from rituximab-treated, refractory patients. Data are shown as the mean ± s.d. of triplicate samples. *P < 0.05 compared with controls two-tailed Student’s t-test.
In vivo, we challenged NSG mice with luciferase knock-in JeKo-1 MCL cell line followed by antibody treatments. Treatment followed the schedule in Figure 4A. Mice receiving either C55 or C90 demonstrated significant retardation of tumor growth, compared with PBS or NK cells alone control groups (Figure 4B). Similarly, C55 and C90 also markedly retarded tumor growth in RS4;11 (acute lymphoblastic leukemia, ALL) challenged NSG mice, compared with no inhibition by rituximab or controls (Figure 4C).
Figure 4. Chimeric antibodies targeting human BAFF-R elicited in vivo therapeutic effects against B-cell tumors.
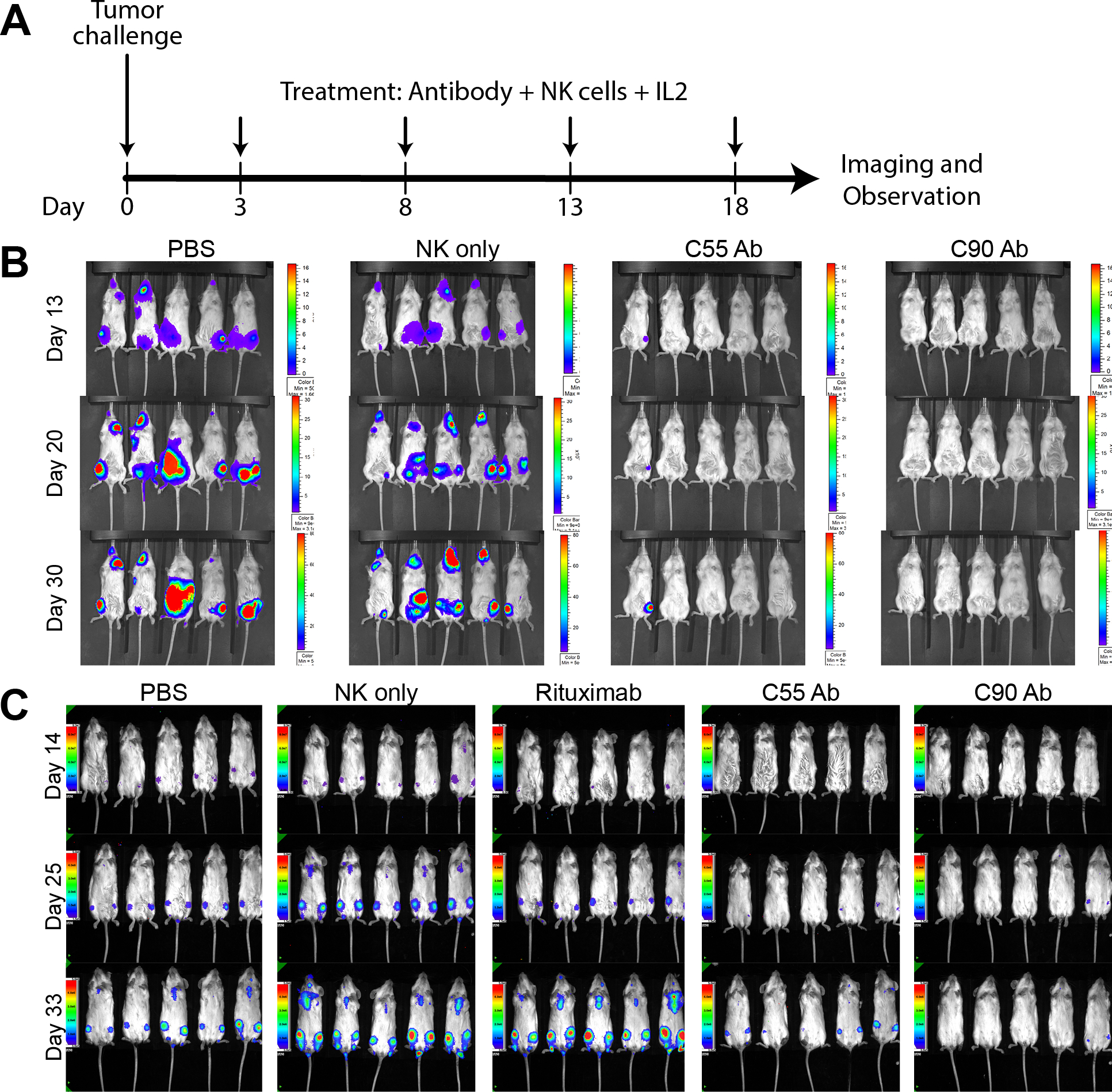
(A) Treatment schedule following Day 0 tumor challenge with minimum lethal dose of tumors. Antibody treatments were given by IV tail vein injections: 200 μg treatment antibody, 10 × 106 effector human NK-92-176V cells, and 5 × 104 IU IL-2. Bioluminescence imaging monitored mice challenged with luciferase-expressing tumors: (B) JeKo-1 (MCL) or (C) RS4;11 (ALL). Experimental groups received treatment of chimeric BAFF-R mAbs (C55 or C90, as indicated). Control group mice received PBS, NK cells alone, or rituximab on the same schedule. Data are representative of three independent experiments.
Chimeric mAb induced potent antitumor effects against drug-resistant lymphomas in vitro and in vivo.
We further tested our antibodies against primary CLL (n=3) and MCL (n=2) samples from relapsed patients who had been previously treated with rituximab. All five primary samples were sensitive to killing by ADCC with C55 and C90, suggesting their effectiveness against tumors which progressed clinically after exposure to rituximab (Figure 3B).
We sought to model drug resistance due to the loss of target and developed a model to simulate CD20 down-regulation and antigen-shaving – phenomena sometimes observed clinically following rituximab treatment.(25) Accordingly, we generated a rituximab-insensitive, stable CD20 knock-out (KO) clone of JeKo-1 using a CRISPR/HDR system. CD20-KO clones were confirmed for absence of CD20 surface expression with unaffected viability and growth rate (Figure 5A, Supplementary Figure 11A–C) and presence of BAFF-R surface expression by flow cytometry (Supplementary Figure 11D). JeKo-1-CD20-KO clone 25, selected for further studies, retained sensitivity to C55- and C90-mediated ADCC, but became insensitive to cytotoxicity mediated by anti-CD20 rituximab (Figure 5B).
Figure 5. Chimeric BAFF-R antibodies induced ADCC on drug insensitive/resistant lymphoma models in vitro.
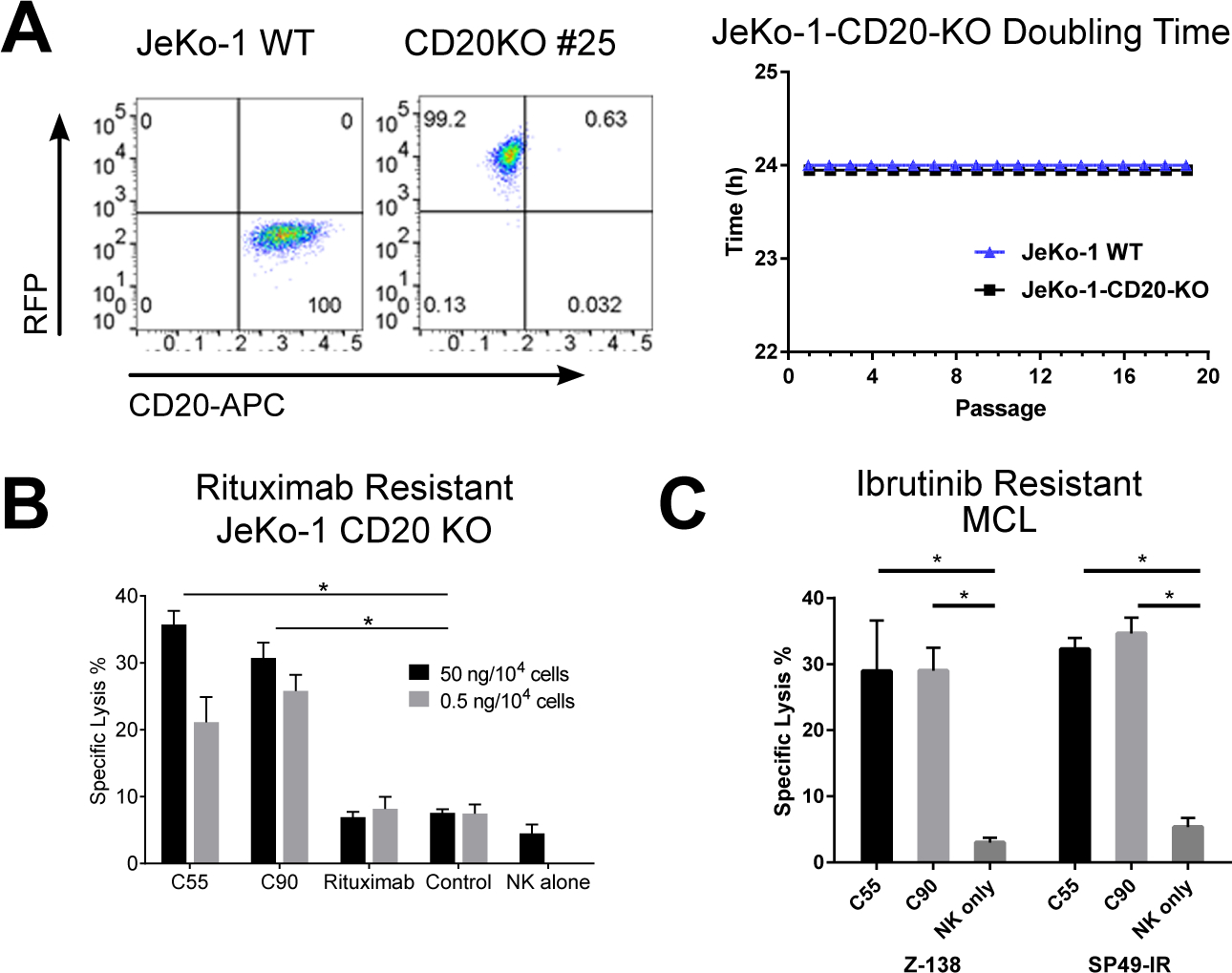
(A) Verification of JeKo-1 cells following CRISPR/HDR knock-out of CD20 gene compared with JeKo-1 wildtype (WT). FACS analysis showed CD20 binding on selected clone and WT. Viability and doubling time of JeKo-1-CD20-KO cells were monitored over 20 passages. ADCC effects measured by chromium-51 release after incubation with C55, C90, or rituximab and effectors NK cells (E:T = 20:1). Percentage of cell specific lysis of target cells: (B) rituximab-resistant JeKo-1-CD20-KO and (C) ibrutinib-resistant Z-138 and SP49-IR. All data are representative of two or more identical experiments. Data are shown as the mean ± s.d. of triplicate samples. * P < 0.05 compared with NK cells by two-tailed Student’s t-test.
As a second model of drug-resistant lymphomas, we tested our chimeric BAFF-R mAb for ADCC against the naturally ibrutinib resistant human MCL line, Z-138(26), and the induced ibrutinib resistant MCL line, SP49-IR, which had been induced in vitro for resistance to ibrutinib.(24) Significant in vitro ADCC was observed with the antibodies against both ibrutinib resistant lines (Figure 5C). Z-138 was further confirmed to be ibrutinib resistant in vivo using ibrutinib-sensitive JeKo-1 as a control (Supplementary Figure 12A–B).
Finally, three days following IV challenge with JeKo-1-CD20-KO tumor cells in vivo, NSG mice (n=5 per group) received BAFF-R antibody treatments (C55 or C90) or rituximab as described in Methods and according to the schedule in Figure 4A. Bioluminescent imaging on Day 20 revealed substantial tumor burden in controls and rituximab treated mice, but no visible tumors in BAFF-R antibody treatment groups (Figure 6A). Monitoring tumor free and long-term overall survival confirmed the significant antitumor effects of both BAFF-R antibodies, but not rituximab (Figure 6C). Similarly, significant effects were observed following treatment of ibrutinib-resistant Z-138 tumor-bearing mice with either BAFF-R antibody, compared with controls (PBS, NK only, or ibrutinib only) (Figure 6B–C, Supplementary Figure 13). Specifically, while progressive tumor growth and death were observed in all controls, tumor growth was delayed significantly in mice treated with BAFF-R antibodies and some of these developing tumors even regressed subsequently. In all in vivo studies, mice were closely monitored daily for toxicities, weight loss, and general signs of distress. No toxicities were observed in antibody-treated mice.
Figure 6. Chimeric antibodies targeting human BAFF-R elicited in vivo therapeutic effects against drug resistant B-cell tumors.

Bioluminescence images of mice challenged with luciferase-expressing tumors (A) Rituximab-insensitive JeKo-1-CD20-KO cells or (B) ibrutinib-resistant Z-138 cells followed by antibody treatments as in Figure 4. Control group mice received PBS, NK cells alone, or (A) rituximab (following antibody treatment in Figure 4A) or (B) ibrutinib (25mg/kg IP every other day on days 3–17). (C) Tumor-free and overall survival curves of the mice shown in (A) and (B), respectively. Tumor free rate and survival differences between experimental and all control groups were analyzed by log-rank test (** P<0.001). Data are representative of three independent experiments.
BAFF-R mAbs also bind normal B cells
When tested against normal PBMC, anti-BAFF-R antibody C90 exhibited specific binding (Supplementary Figure 14A–B) and lysing (Supplementary Figure 15) of B cells, as expected, without staining any T cells, NK cells, granulocytes, or monocytes. The positive staining results were verified on purified B cells (Supplementary Figure 14C). Again, purified T cells, NK cells, and gated myeloid cells showed no binding.
Discussion
BAFF-R protein is expressed almost exclusively on the surface of B cells, and is particularly up-regulated on B-cell tumors, making it an attractive target for immunotherapies. The BAFF/BAFF-R axis has been targeted successfully for autoimmune diseases, particularly with mAbs against the BAFF ligand,(27,28) however the initial promise for B-cell tumor therapy has not yet been realized. Specifically, at least one mAb against the receptor has also been developed to deplete auto-reactive B cells.(29) The antibody was examined for its ability to deplete normal B cells, but no information on activity against malignant B cells was provided. More recently, a BAFF-R antibody was developed and tested in Philadelphia-positive pre-B ALL models.(30) This antibody demonstrated in vitro cytotoxicity against drug-sensitive tumor cells, but was not tested against drug resistant ALL lines. Importantly, it yielded modest single agent anti-tumor effects in comparison to nilotinib, and the combination of antibody with this tyrosine kinase inhibitor did not confer increased efficacy. Our BAFF-R mAbs, in contrast, elicited robust in vivo antitumor effects as a single agent against multiple B-cell tumor types, including NHL, CLL, and ALL. Furthermore, our antibodies eradicated established tumors, which led to long-term, tumor-free survival in vivo.
The distinctive features of our BAFF-R mAbs may be due to the approach we used to generate them. In contrast to prior antibodies that were generated using recombinant immunogen, our approach was to express human BAFFF-R as a native surface protein on mouse fibroblast cells for immunization, increasing the likelihood of presenting a natively folded, glycosylated immunogen. Therefore, it is very likely that our antibodies are binding an accessible human BAFF-R epitope distinct from the other antibodies described. Outside of the scope of the current study, X-ray crystallography studies are planned to identify the precise binding epitopes of our antibodies on BAFF-R.
Thus, we demonstrated a technical strategy for generating monoclonal antibodies against a natively folded, eukaryotically glycosylated human BAFF-R that is able to specifically bind, lyse, and inhibit B-cell tumors in vivo. Our results suggest the main anti-tumor mechanism of our mAbs is ADCC, as NK cells were required in addition to mAbs for in vitro activity (Figure 2); we observed no evidence of CDC. The antibodies were able to competitively inhibit BAFF ligand binding to BAFF-R (Figure 2D). Although an earlier report showed blocking BAFF/BAFF-R survival signal is not sufficient to cause complete B-cell depletion,(31) potential signal blockade by C55 and C90 merits further investigation.
The current portfolio of B-cell lymphoma treatments has benefitted from the availability of several FDA approved agents, including rituximab, and the Bruton’s tyrosine kinase inhibitor, ibrutinib.(32) Although these treatments are clinically effective, many patients will develop drug resistance, especially to monotherapies.(33–35) Thus, there remains an urgent need for additional new therapeutic agents. One clinically relevant mechanism of resistance to rituximab is down-regulation of CD20.(10) We modeled this phenomenon of drug resistance with a CRISPR edited MCL line, JeKo-1, which is deficient in CD20. The significant in vivo antitumor effects of C55 or C90, but not rituximab treatment, against this line and similarly against the naturally ibrutinib resistant Z-138 MCL suggests efficacy against drug-resistant lymphomas (Figure 5). Taken together with the in vitro cytotoxicity of these antibodies against primary tumors from lymphoma patients who were previously treated with, and progressed in response to rituximab, these data suggest C55 and C90 as a potential treatment strategy to overcome drug resistance (Figure 3).
Finally, these results strongly support further development of these BAFF-R antibodies for clinical application against lymphomas, including humanization and IND-enabling studies. Future studies should also investigate the combination of our agents with other targeted therapies such as rituximab and ibrutinib.
Supplementary Material
Translational Relevance.
Developing novel strategies against drug-resistant B-cell tumors addresses the urgent need of patients experiencing relapse or resistance following first-line therapies. BAFF-R is a suitable alternative target for therapeutic antibody development because of its nearly exclusive expression on the surface of B cells and notable prominent expression on malignant B cells. Although previous attempts were made to target BAFF-R, our antibodies have greater potential to be translated into the clinic. We demonstrated the antibodies elicited potent antitumor effects against a broad range of B-cell tumors. Moreover, the pronounced therapeutic efficacy seen in drug-resistant human lymphoma xenograft models further supports translation of our antibodies to meet this urgent need.
Acknowledgements
Research reported in this publication included work performed in the Analytical Cytometry, Integrative Genomics, and Small Animal Imaging Cores supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Special thanks to the Antibody Core Facility at the University of Texas MD Anderson Cancer Center for their hybridoma production and antibody screening work.
We are grateful for the generous support from the Toni Stephenson Lymphoma Center at Beckman Research Institute of City of Hope.
Footnotes
Disclosure of Potential Conflicts of Interest:
H. Qin and L.W. Kwak hold ownership interest (including patents) in and are consultant/advisory board members for Innolifes Inc. and PeproMene Bio Inc. S.S. Neelapu is a consultant/advisory board member for Celgene, Kite Pharma, Merck and Novartis. No potential conflicts of interest were disclosed by the other authors.
References Cited
- 1.Carter P. Improving the efficacy of antibody-based cancer therapies. Nature reviews Cancer 2001;1(2):118–29 doi 10.1038/35101072. [DOI] [PubMed] [Google Scholar]
- 2.Reichert JM. Antibody-based therapeutics to watch in 2011. mAbs 2011;3(1):76–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997;90(6):2188–95. [PubMed] [Google Scholar]
- 4.Berinstein NL, Grillo-Lopez AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, et al. Association of serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 1998;9(9):995–1001. [DOI] [PubMed] [Google Scholar]
- 5.Enblad G, Hagberg H, Erlanson M, Lundin J, MacDonald AP, Repp R, et al. A pilot study of alemtuzumab (anti-CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy-refractory peripheral T-cell lymphomas. Blood 2004;103(8):2920–4 doi 10.1182/blood-2003-10-3389. [DOI] [PubMed] [Google Scholar]
- 6.Kaminski MS, Tuck M, Estes J, Kolstad A, Ross CW, Zasadny K, et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. The New England journal of medicine 2005;352(5):441–9 doi 10.1056/NEJMoa041511. [DOI] [PubMed] [Google Scholar]
- 7.Fanale MA, Horwitz SM, Forero-Torres A, Bartlett NL, Advani RH, Pro B, et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32(28):3137–43 doi 10.1200/JCO.2013.54.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turturro F. Constitutive NF- kappa B Activation Underlines Major Mechanism of Drug Resistance in Relapsed Refractory Diffuse Large B Cell Lymphoma. BioMed research international 2015;2015:484537 doi 10.1155/2015/484537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y, Zhang P, Gao Y, Fan H, Zhang M, Wu J. Evaluation of AKT phosphorylation and PTEN loss and their correlation with the resistance of rituximab in DLBCL. International journal of clinical and experimental pathology 2015;8(11):14875–84. [PMC free article] [PubMed] [Google Scholar]
- 10.Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood 2009;113(20):4885–93 doi 10.1182/blood-2008-08-175208. [DOI] [PubMed] [Google Scholar]
- 11.Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity 2001;15(2):289–302. [DOI] [PubMed] [Google Scholar]
- 12.Li YJ, Jiang WQ, Rao HL, Huang JJ, Xia Y, Huang HQ, et al. Expression of BAFF and BAFF-R in follicular lymphoma: correlation with clinicopathologic characteristics and survival outcomes. PloS one 2012;7(12):e50936 doi 10.1371/journal.pone.0050936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. The Journal of experimental medicine 1999;189(11):1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novak AJ, Grote DM, Stenson M, Ziesmer SC, Witzig TE, Habermann TM, et al. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: correlation with disease activity and patient outcome. Blood 2004;104(8):2247–53 doi 10.1182/blood-2004-02-0762. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura N, Hase H, Sakurai D, Yoshida S, Abe M, Tsukada N, et al. Expression of BAFF-R (BR 3) in normal and neoplastic lymphoid tissues characterized with a newly developed monoclonal antibody. Virchows Archiv : an international journal of pathology 2005;447(1):53–60 doi 10.1007/s00428-005-1275-6. [DOI] [PubMed] [Google Scholar]
- 16.Rodig SJ, Shahsafaei A, Li B, Mackay CR, Dorfman DM. BAFF-R, the major B cell-activating factor receptor, is expressed on most mature B cells and B-cell lymphoproliferative disorders. Human pathology 2005;36(10):1113–9 doi 10.1016/j.humpath.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Paterson JC, Tedoldi S, Craxton A, Jones M, Hansmann ML, Collins G, et al. The differential expression of LCK and BAFF-receptor and their role in apoptosis in human lymphomas. Haematologica 2006;91(6):772–80. [PubMed] [Google Scholar]
- 18.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 2001;293(5537):2108–11 doi 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 19.Mackay F, Schneider P. Cracking the BAFF code. Nature reviews Immunology 2009;9(7):491–502 doi 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 20.Hildebrand JM, Luo Z, Manske MK, Price-Troska T, Ziesmer SC, Lin W, et al. A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling. The Journal of experimental medicine 2010;207(12):2569–79 doi 10.1084/jem.20100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, et al. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 2013;38(3):475–88 doi 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spiller OB, Harris CL, Morgan BP. Efficient generation of monoclonal antibodies against surface-expressed proteins by hyperexpression in rodent cells. Journal of immunological methods 1999;224(1–2):51–60. [DOI] [PubMed] [Google Scholar]
- 23.Dreyer AM, Beauchamp J, Matile H, Pluschke G. An efficient system to generate monoclonal antibodies against membrane-associated proteins by immunisation with antigen-expressing mammalian cells. BMC biotechnology 2010;10:87 doi 10.1186/1472-6750-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nature communications 2017;8:14920 doi 10.1038/ncomms14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez-Callejo D, Gonzalez-Rincon J, Sanchez A, Provencio M, Sanchez-Beato M. Action and resistance of monoclonal CD20 antibodies therapy in B-cell Non-Hodgkin Lymphomas. Cancer treatment reviews 2015;41(8):680–9 doi 10.1016/j.ctrv.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nature medicine 2014;20(1):87–92 doi 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 27.Davidson A. Targeting BAFF in autoimmunity. Current opinion in immunology 2010;22(6):732–9 doi 10.1016/j.coi.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzova D, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis and rheumatism 2011;63(12):3918–30 doi 10.1002/art.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CV, Hymowitz SG, Wallweber HJ, Gordon NC, Billeci KL, Tsai SP, et al. Synthetic anti-BR3 antibodies that mimic BAFF binding and target both human and murine B cells. Blood 2006;108(9):3103–11 doi 10.1182/blood-2006-03-011031. [DOI] [PubMed] [Google Scholar]
- 30.Parameswaran R, Lim M, Fei F, Abdel-Azim H, Arutyunyan A, Schiffer I, et al. Effector-mediated eradication of precursor B acute lymphoblastic leukemia with a novel Fc-engineered monoclonal antibody targeting the BAFF-R. Molecular cancer therapeutics 2014;13(6):1567–77 doi 10.1158/1535-7163.MCT-13-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin WY, Gong Q, Seshasayee D, Lin Z, Ou Q, Ye S, et al. Anti-BR3 antibodies: a new class of B-cell immunotherapy combining cellular depletion and survival blockade. Blood 2007;110(12):3959–67 doi 10.1182/blood-2007-04-088088. [DOI] [PubMed] [Google Scholar]
- 32.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. The New England journal of medicine 2015;373(25):2425–37 doi 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nature communications 2016;7:11589 doi 10.1038/ncomms11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. The New England journal of medicine 2014;370(24):2286–94 doi 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib resistance in chronic lymphocytic leukemia. The New England journal of medicine 2014;370(24):2352–4 doi 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.