

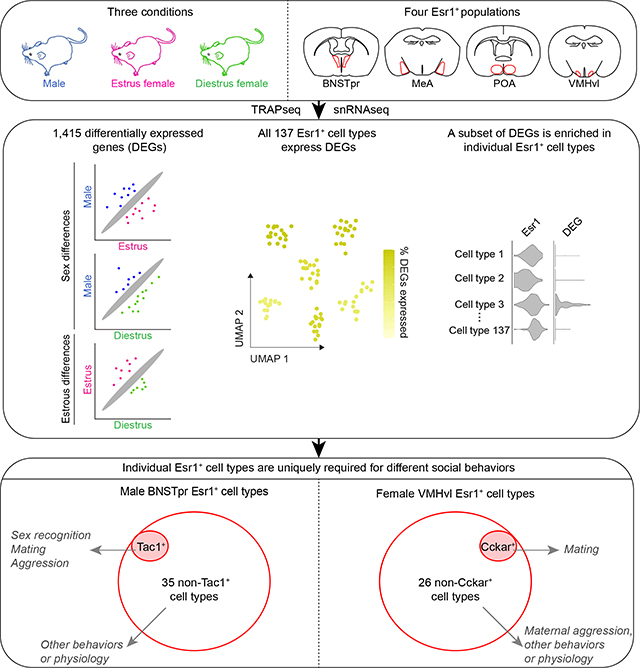
SUMMARY
Sex hormones exert a profound influence on gendered behaviors. How individual sex hormone-responsive neuronal populations regulate diverse sex-typical behaviors is unclear. We performed orthogonal, genetically-targeted sequencing of four estrogen receptor 1-expressing (Esr1+) populations and identified 1,415 genes expressed differentially between sexes or estrous states. Unique subsets of these genes were distributed across all 137 transcriptomically-defined Esr1+ cell types, including estrous stage-specific ones, that comprise the four populations. We used differentially expressed genes labeling single Esr1+ cell types as entry-points to functionally characterize two such cell types, BNSTprTac1/Esr1 and VMHvlCckar/Esr1. We observed that these two cell types, but not the other Esr1+ cell types in these populations, are essential for sex recognition in males and mating in females, respectively. Furthermore, VMHvlCckar/Esr1 cell type projections are distinct from those of other VMHvlEsr1 cell types. Together, projection and functional specialization of dimorphic cell types enables sex hormone-responsive populations to regulate diverse social behaviors.
Graphical Abstract
In brief
Differentially expressed genes in four estrogen receptor 1-expressing populations in different sexes or estrous states are identified along with a functional characterization of specific cell types that mediate social behaviors in male or female mice.
INTRODUCTION
In many vertebrates, neural circuits underlying sex or estrous (menstrual) state-typical physiology and social behaviors are regulated by sex hormones (Arnold, 2009; Beeman, 1947; Brock et al., 2011; Dey et al., 2015; Honda et al., 1998; Inoue et al., 2019; Juntti et al., 2010; Kow and Pfaff, 1998; Musatov et al., 2006; Ogawa et al., 2000; Phoenix et al., 1959; Ring, 1944; Rissman et al., 1997; Scordalakes and Rissman, 2003; Shapiro, 1937; Toda et al., 2001; Wu et al., 2009; Yang and Shah, 2014). There has been significant progress in delineating sex hormone-responsive populations that govern such context-specific physiology and behavior. In particular, neurons in the principal component of the bed nucleus of the stria terminalis (BNSTpr), medial amygdala (MeA), preoptic hypothalamus (POA), and ventromedial hypothalamus ventrolateralis (VMHvl) have been shown to regulate sex or estrous state-dependent displays of mating and aggression (Bayless et al., 2019; Hashikawa et al., 2017; Hong et al., 2014; Ishii et al., 2017; Karigo et al., 2020; Lee et al., 2014; Lin et al., 2011; Musatov et al., 2006; Osakada et al., 2018; Unger et al., 2015; Wei et al., 2018; Yang et al., 2013, 2017; Yao et al., 2017). Surprisingly, each of these regions regulates multiple such behaviors or physiological responses. How this diversity of function is implemented within individual sex hormone-responsive populations is an open question.
Sex or estrous state-typical gene expression could endow subsets of neurons with distinct functional properties within the BNSTpr, MeA, POA, and VMHvl to drive multiple outputs. However, recent studies of these regions have uncovered many transcriptomically-defined cell types (cell types, hereafter) but few or no sex or estrous state-dependent gene expression differences in these cell types (Chen et al., 2019; Kim et al., 2019; Moffitt et al., 2018; Welch et al., 2019; Xu et al., 2012). These findings immediately raise the question as to how such cell types generate sex or estrous stage dependent functional outputs. It is possible that the small number of gene expression differences between sexes or estrous states is sufficient to functionally diversify individual regions to drive multiple sex hormone-dependent outputs. Alternatively, prior efforts have underestimated the number of differentially expressed genes (DEGs) because they did not enrich for sex hormone-sensitive neurons. This is a nontrivial possibility because only a subset of neurons expresses sex hormone receptors in these regions, leading to under-sampling of the relevant neurons.
In order to distinguish between these possibilities, we wished to first identify DEGs between sexes (sDEGs) using a high throughput sequencing approach that affords deep and reliable coverage of sex hormone-sensitive neurons. We reasoned that if this yielded many sDEGs, we would use single cell RNA sequencing to map their distribution within the individual cell types that comprise sex hormone-sensitive populations. Esr1 (or ERα) is essential for sex typical behaviors in both sexes and expressed in subsets of neurons within the BNSTpr (BNSTprEsr1), POA (POAEsr1), MeA (MeAEsr1), and VMHvl (VMHvlEsr1) (Kim et al., 2019; Ogawa et al., 2000; Paxinos and Franklin, 2003; Rissman et al., 1997; Scordalakes and Rissman, 2003; Wu and Tollkuhn, 2017; Xu et al., 2012; Yang et al., 2013). To identify sDEGs in these 4 Esr1+ populations, we performed translating ribosome affinity purification and sequencing (TRAPseq) (Heiman et al., 2008; Sanz et al., 2009), which enables rapid mRNA extraction and deep, reliable coverage of translated mRNAs. Having identified numerous DEGs, we sought to define their distribution within these Esr1+ populations with single nucleus-RNA sequencing (snRNAseq), using a modified INTACT approach (Mo et al., 2015).
We profiled Esr1+ neurons from females modeled to be in two distinct stages of the estrous cycle, estrus and diestrus (Ring, 1944). Estrus and diestrus states reflect changes in circulating estrogen and progesterone with corresponding maxima and minima of sexual behavior. We reasoned that our approach would enable discovery of sDEGs that differ between females in distinct estrous states and males; in addition, it would reveal any potential DEGs that differ between these two estrous states (eDEGs).
We have identified 1,415 DEGs between sexes or estrous stages. Unique subsets of these DEGs are distributed across all 137 cell types that comprise the four Esr1+ populations we surveyed. Such diversification of gene expression between sexes and estrous states provides potential mechanisms whereby individual Esr1+ populations influence multiple gendered behaviors. We used such DEGs to genetically access individual Esr1+ cell types and developed an intersectional genetic approach to assess behavioral functions of single cell types and their complementary counterparts. For each Esr1+ cell type we manipulated, we observed a restricted contribution to sex or estrous state-dependent social interactions that was distinct from that of the other Esr1+ cell types in that region. Together, our findings reveal a large diversity of sexually differentiated and estrous state-dependent cell types in the mammalian brain. The modular contribution to gendered behaviors of the cell types that we have characterized provides a mechanism whereby individual sex hormone-responsive populations regulate diverse behaviors. Such partitioning of behavioral functions to different cell types within a population may be a general feature of the many transcriptomically-defined cell types that have been identified throughout the brain.
RESULTS
Identification of sex-differential gene expression
Adult males maintain testosterone at a steady state and reliably initiate sex hormone-dependent social behaviors whereas females exhibit cyclic changes in sex hormone titers, with peak titers at estrus opening a window of sexual receptivity. Such cyclic changes in gene expression can confound sDEG identification, and we therefore collected tissue from estrus and diestrus females. We generated sexually receptive, estrus (FR) mice with estrogen and progesterone priming and diestrus, sexually non-receptive (FNR) mice with vehicle priming (Fig. S1A). Our protocol for FR and FNR mice both mimics estrogen and progesterone titers corresponding to natural estrus and diestrus and recapitulates these cycle stages behaviorally: FR mice are sexually receptive like estrus females, and FNR mice are non-receptive like diestrus females (Inoue et al., 2019; Ring, 1944).
We performed TRAPseq on BNSTpr, MeA, POA, and VMHvl from adult FR, FNR, and gonadally intact male (M) mice bearing an Esr1Cre and a Cre-dependent allele (Rpl22HA) encoding an HA-tagged ribosomal subunit (Fig. 1A, S1B,C; Table S1) (Lee et al., 2014; Sanz et al., 2009). In order to identify sDEGs (M v FR and M v FNR comparisons; v, versus), we employed a negative binomial model for RNAseq data analysis (DESeq2) (Love et al., 2014), focusing on DEGs that exhibited ≥1.5-fold change and passed a False Discovery Rate (FDR) criterion of <0.05. This revealed 1,541 differential gene expression events between sexes (890 sDEGs, M v FR; 651 sDEGs, M v FNR) (Fig. 1B, Table S2). We identified numerous female-upregulated sDEGs even in regions such as the BNSTpr and POA that have more neurons in males, thereby validating the sensitivity of our approach (Fig. S1E) (Table S2). sDEG-encoding loci reside on all chromosomes, and many sDEGs appear insensitive to estrous cycle state (Fig. S1D,E), in that they are differentially expressed between males and females irrespective of hormone priming. We next used hybridization chain reaction based in situ hybridization (ISH, hereafter) to visualize sDEGs spanning a range of expression levels (1–220 transcripts per million or TPM) and fold changes (1.5–15) (Fig 1C, S1F) (Choi et al., 2018). Our studies confirmed differential expression for all 23 sDEGs that we had selected for in vivo analysis (Fig. 1C, S1F and data not shown). Together, we have uncovered large scale, genome-wide regulation of gene expression by sex.
Figure 1: TRAPseq identification of sex differences in gene expression.
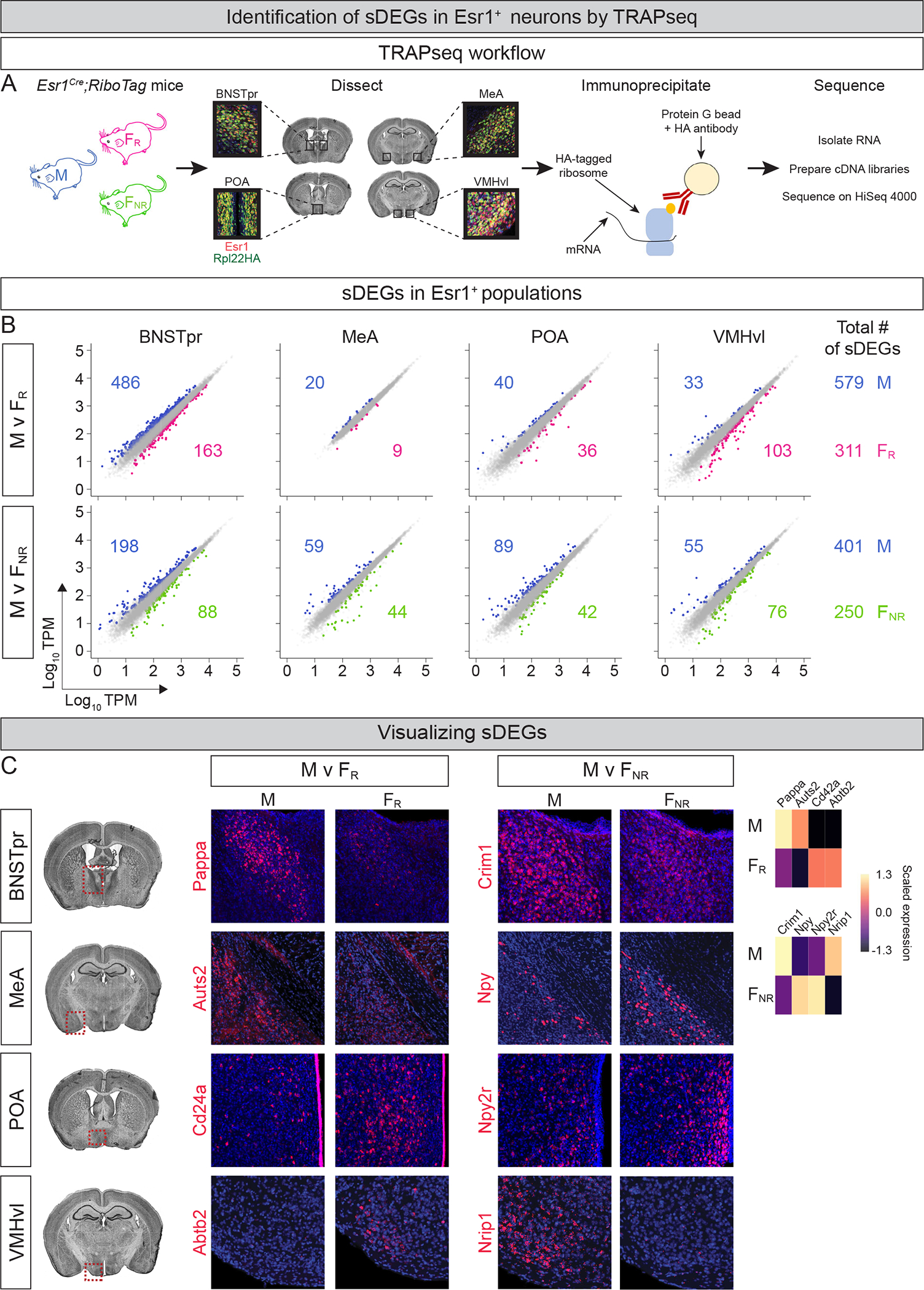
A. Schematic of TRAPseq workflow.
B. Scatter plots of sDEGs in different Esr1+ populations. Dots represent DEGs with >1.5-fold change and FDR-adjusted p value < 0.05 (colored) or genes that did not meet both criteria (gray). Colored numbers show number of sDEGs upregulated in that condition and comparison for each region and (far right) for all regions combined. In total, TRAPseq identified 890 and 651 sDEGs in M v FR and M v FNR comparisons. v, versus; TPM, transcripts/million. n = 3/condition.
C. ISH for sDEGs in coronal sections through regions enclosed within red dotted areas on Nissl-stained sections on left. ISHs confirm TRAPseq data showing higher expression of Pappa, Auts2, Criml, and Nripl in M and other genes upregulated in FR or FNR mice. Heatmap at far right shows scaled expression of DEGs that were visualized by ISH. Scale = mean z-scored expression of DEGs centered at zero for individual comparisons between conditions (sex or estrous state) and regions; analogous heatmaps provided for ISH studies in all figures. Coronal plane as well as dorsoventral and mediolateral orientations preserved for histological panels in all figures. n = 2/condition/probe. Red, mRNA; blue, DAPI. Scale bars = 100 μm.
See also Fig. S1.
Identification of estrous cycle stage-differential gene expression
Even for regions such as the BNSTpr, MeA, POA, and VMHvl that regulate female behaviors, prior gene expression profiling studies have not compared males with females in distinct estrous stages in naturally cycling or FR and FNR mice (Chen et al., 2019; Kim et al., 2019; Moffitt et al., 2018; Welch et al., 2019; Xu et al., 2012). We analyzed our dataset for eDEGs, or genes that differed significantly in expression between FR and FNR mice. We identified 770 such differential expression events using inclusion criteria we used for sDEGs (Fig. 2A). Similar to sDEGs, eDEG-encoding loci reside on all chromosomes (Fig. S2A). Our ISH studies confirmed differential expression for all eDEGs (n = 12 eDEGs) that we had selected for in vivo analysis (Fig. 2B, S2B). Esr1 and progesterone receptor (PR) typically upregulate gene expression through recruitment of coactivator complexes (Ellmann et al., 2009; Green and Carroll, 2007). Nevertheless, a large fraction of eDEGs was upregulated in FNR mice, suggesting de-repression as a mode of gene regulation in these Esr1+ neurons. Irrespective of underlying mechanisms, our data reveal large scale genome-wide regulation of gene expression by estrous state in multiple brain regions.
Figure 2: TRAPseq identification of estrous stage-dependent differences in gene expression.

A. Scatter plots of eDEGs in different Esr1+ populations. Dots represent DEGs with >1.5-fold change and FDR-adjusted p value < 0.05 (colored) or genes that did not meet both criteria (gray). Colored numbers show number of eDEGs upregulated in that condition and comparison for each region and (far right) for all regions combined. In total, TRAPseq identified 770 eDEGs. n = 3.
B. ISH for eDEGs. ISHs confirm TRAPseq data showing higher expression of Pgr15l in FNR and other genes upregulated in FR mice. n = 2/condition/probe. Scale bars = 100 μm.
C. Heatmap of p values of individual DEGs for the relevant pairwise comparison (with darker green colors indicating more significant p values) illustrating that most DEGs are restricted to one Esr1+ population.
D. Heatmap of p values of individual DEGs for the relevant pairwise comparison (with darker green colors indicating more significant p values) illustrating that most DEGs within an Esr1+ population are specific to one comparison between sexes or estrous states.
See also Fig. S2.
General properties of genes differentially expressed between sexes or estrous stages
Many DEGs are both sDEGs and eDEGs such that 1,415 unique genes are differentially expressed between sexes or estrous states (Table S2). Each Esr1+ population expresses a unique complement of DEGs, with only a subset of DEGs common to >2 regions (Fig. 2C; Tables S2, S3). Within individual Esr1+ populations, most DEGs are unique to M v FR, M v FNR, or FR v FNR comparisons (Fig. 2D, Table S3). Accordingly, principal component analysis (PCA) of DEGs within each region shows that M, FR, and FNR occupy distinct regions in PCA space, suggesting that FR and FNR mice are different from each other even in comparison to males (Fig. S2C). DEG-encoding loci are significantly enriched for sex hormone receptor binding DNA elements, suggesting a shared transcriptional regulatory framework underlying these expression patterns (Fig. S3A).
Gene Ontology analyses revealed a significant enrichment of DEGs in synaptic transmission, steroid hormone related processes, behavior, peripheral reproductive organ processes, and regulation of gene expression (Fig. S3B,Table S4) (Ashburner et al., 2000; Gene Ontology Consortium, 2021). These DEGs are not particularly enriched in developmental processes, consistent with the fact that TRAPseq was done on adult neurons. There are sex biases in many diseases, and we wondered if these DEGs were associated with such illnesses. A Disease Ontology analysis revealed enrichment for association with diverse sex-biased disorders, including autism spectrum disorder (ASD) (Fig. S3C, Table S4) (Schriml et al., 2019). Many associations were for diseases affecting non-neural tissues, suggesting that these DEGs are also differentially expressed peripherally.
ASD is extremely sex-biased (male:female :: 4:1) and many genes have been identified as risk-conferring with high confidence (Abrahams et al., 2013; Werling and Geschwind, 2013). Moreover, deficits in fundamental social interactions are a core feature of ASD. The four Esr1+ populations we surveyed are critical for social behaviors, and we therefore examined potential associations between DEGs and high confidence ASD risk-conferring genes. We found that a significant number of such genes are DEGs in the adult mouse brain (39/207; p = 3×10−5) (Fig. 3A) (Abrahams et al., 2013). We could also visualize differential expression of ASD risk-conferring DEGs in vivo with ISH (Fig. 1C, 3B). Together, our findings suggest the possibility that ASD risk-conferring DEGs may contribute to disease-relevant behavioral domains by influencing Esr1+ subcortical neural pathways.
Figure 3: ASD-association and distribution of DEGs in vivo.

A. Heatmap of expression of ASD risk-conferring DEGs. Scale = z-scored expression of DEGs centered at zero for individual comparisons between conditions (sex or estrous state) and regions. See Fig. 1C for heatmap and ISH for Auts2, an ASD-risk conferring DEG.
B. ISH for ASD-risk conferring DEGs confirm TRAPseq data showing upregulation of Tcf4, Ptchdl, Sox5, Rfx3, and Ube3a in males. n = 2/condition/probe. Scale bar = 100 μm.
C. ISHs for DEGs show that they are distributed in a few (sparse) or most (dense) Esr1+ neurons in each region. Quantification of these ISHs revealed that ‘sparse’ and ‘dense’ DEGs were expressed in ≤10% and ≥70% of Esr1+ cells. n = 2/condition/probe. Scale bar = 100 μm.
See also Fig. S3.
Identification of cell types that constitute the four Esr1+ populations
Our ISH studies showed broad or restricted expression of DEGs within individual Esr1+ populations (Fig. 1C, S1F, 2B, S2B). Dual color ISH indeed confirmed that individual DEGs are expressed in most or a few Esr1+ cells (dense and sparse expression patterns in Fig. 3C). These findings suggested considerable heterogeneity of cell types in the four Esr1+ populations we surveyed. Accordingly, we sought to characterize the distribution of DEGs at cellular resolution with snRNAseq of Esr1+ neurons from BNSTpr, MeA, POA, and VMHvl of adult FR, FNR, and male mice. To this end, we utilized mice bearing Esr1Cre and SunTag (Sunl/sfGFP, which encodes a nuclear membrane targeted fluorescent protein) alleles (Fig. S4A–C) (Mo et al., 2015). We analyzed data from the 49,776 Esr1+ neurons that passed quality control criteria (Table S5).
We pooled sequencing data from all conditions/region and employed unsupervised graph-based clustering with Seurat to classify 137 Esr1+ cell types (Hafemeister and Satija, 2019). Each cell type (36, BNSTpr; 34, MeA; 40, POA; 27, VMHvl; median cell #/cell type = 304) was uniquely labeled with one or a few genes (Fig. 4, S4D, Table S6). Importantly, Esr1+ cell type classification was essentially unchanged even if DEGs were excluded from the clustering pipeline (data not shown). We cross-validated our Esr1+ cell type classification using additional approaches. We obtained similar clustering results without pooling data from all conditions/region, indicating that clustering was not skewed by any particular condition. Indeed, analysis of all Esr1+ cell types combined revealed co-clustering by brain region rather than batch, sex, or estrous stage (Fig. S4E). Subsampling and bootstrapping approaches further confirmed that Esr1+ cell types were reproducibly identifiable (Table S5) (Tang et al., 2020). Finally, the cell types we have identified align with those previously reported for the adult BNSTpr, POA, MeA, and VMHvl using diverse sequencing platforms and analytical pipelines (Chen et al., 2019; Kim et al., 2019; Moffitt et al., 2018; Welch et al., 2019). As expected, absent from our dataset are Esr1− cell types identified by previous studies that performed single cell/nucleus RNAseq without enriching for Esr1+ populations. Sequencing purified Esr1+ cells also allowed us to identify rare, biologically significant cell types or cell type-marker genes within the population. For example, we identified the Kiss1+ cell type in POA, which is known to regulate ovulation and female mating (Hellier et al., 2018; Popa et al., 2008); we also identified aromatase+ cell types in BNSTpr that are known to regulate sex recognition in males but were not assigned to this neuronal pool in a prior RNAseq study that did not enrich for Esr1+ cells (Fig. 4A,C, S4G) (Bayless et al., 2019; Welch et al., 2019).
Figure 4: Categorizing Esr1+ populations into cell types with snRNAseq.
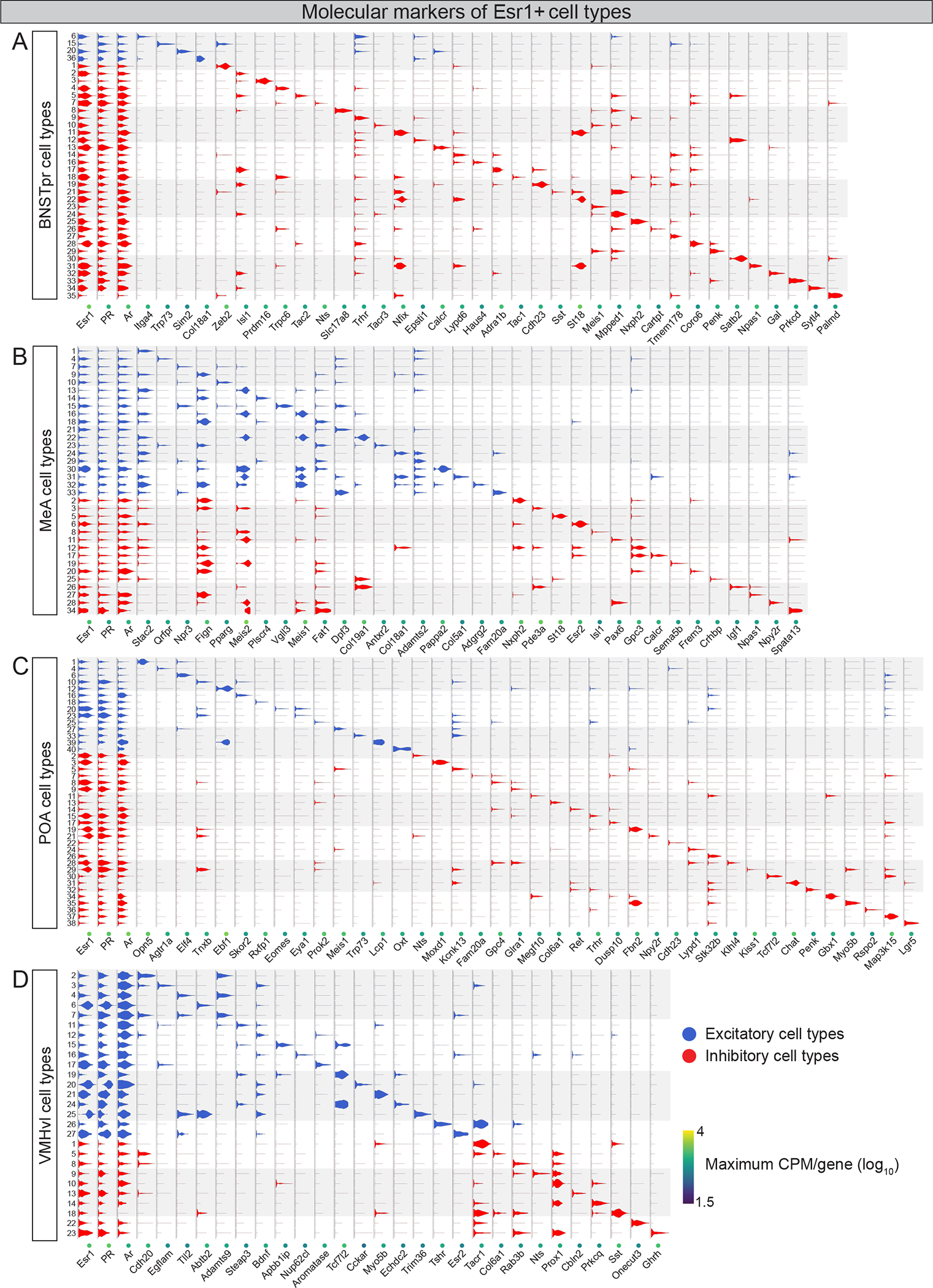
A-D. Violin plots classifying Esr1+ cell types (rows) by virtue of expression of sex hormone receptors, major neuronal neurotransmitter type (excitatory and inhibitory), and enriched genes in the BNSTpr (A), MeA (B), POA (C), and VMHvl (D). CPM, counts per million. n = 10,392 (BNSTpr), 15,929 (MeA), 17,784 (POA), and 5671 (VMHvl) Esr1+ neurons.
All cell types we identified expressed canonical neuronal markers suggesting that most Esr1+ cells in these regions are neurons (Fig. S4G). Nearly all Esr1+ cell types express PR and the androgen receptor (Fig. 4), indicative of a broad influence of sex hormones on these populations. Most cell types in the BNSTpr and POA are inhibitory whereas the MeA is composed of an equal mix of excitatory and inhibitory cell types (Fig. 4A–C, S4G). Although the VMHvl is largely glutamatergic, there is a small population of inhibitory neurons in this region (Fig. S4F) (Lein et al., 2007). Consistent with this, we detected inhibitory Esr1+ cell types in the VMHvl (Fig. 4D, S4G).
We confirmed expression of ~98% of DEGs in our snRNAseq data, in close concordance with the overall overlap (~95%) of the mRNAs between TRAPseq and snRNAseq. Although in principle, DEGs could localize to one, a few, or most cell types, we observed DEGs in all cell types (Fig. 5A–D). Each cell type expressed ≥20% of DEGs but none expressed the entire set of DEGs, and most DEGs were expressed in >1 cell type (Fig. 5D, S5A). All Esr1+ cell types harbored sDEGs and eDEGs. Although most harbored proportionally equivalent numbers of DEGs upregulated in M, FR, or FNR mice, a few were highly enriched for DEGs from one of these conditions (Fig. 5E). This distribution of sDEGs and eDEGs was observed in excitatory as well as inhibitory cell types. While most DEGs were expressed in >1 cell type, many exhibited significant enrichment or depletion in ≥1 cell types, consistent with our ISH findings (Fig. 3C, S5B).
Figure 5: Distributive representation of DEGs across Esr1+ cell types.
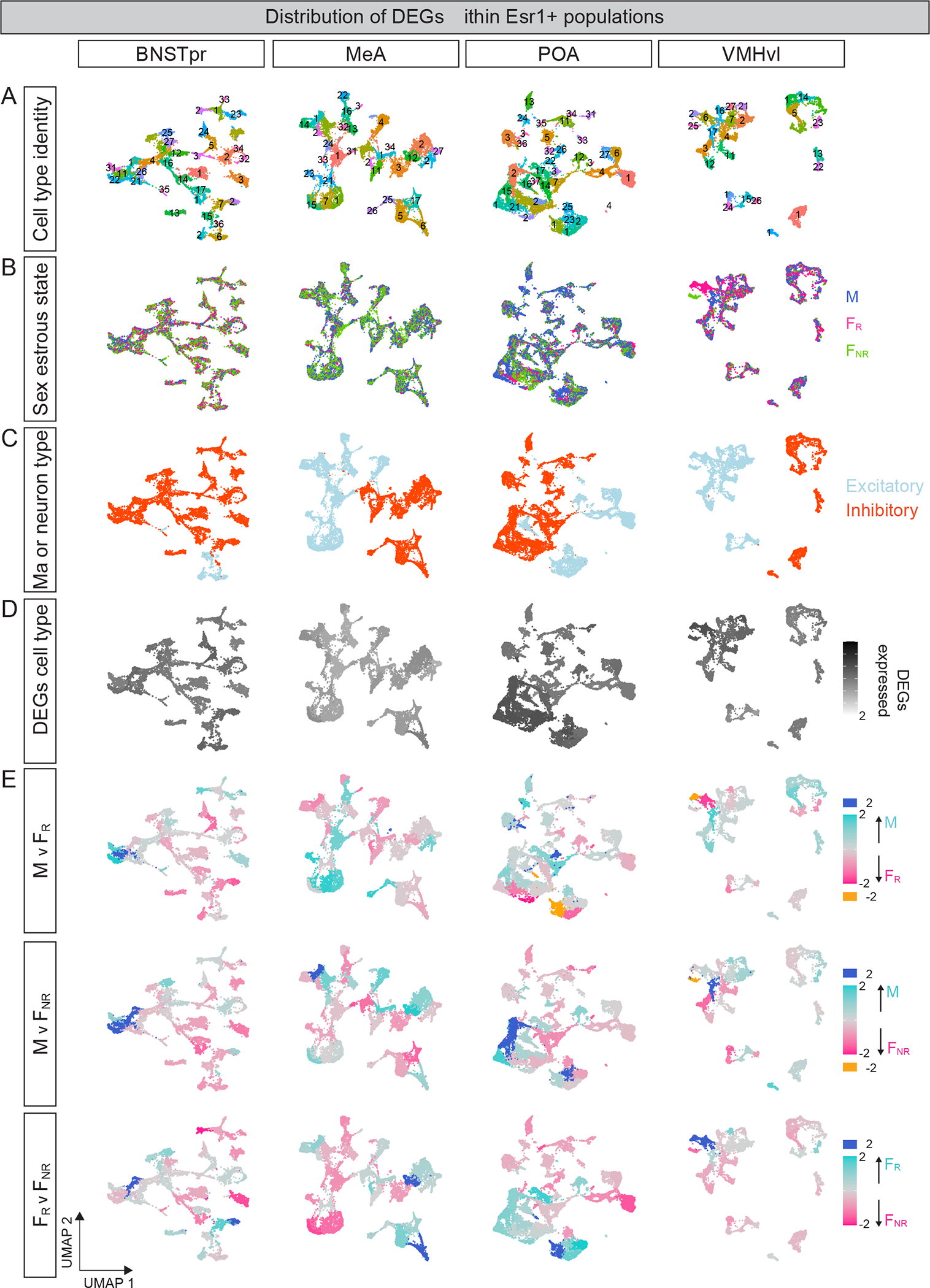
A. UMAP representation of cell types in Esr1+ populations, with numbers corresponding to those in Fig. 4.
B. UMAP representation of Esr1+ cell types by sex or estrous state shows that most of them are proportionally equivalently distributed.
C. UMAP representation of excitatory and inhibitory cell types.
D. UMAP representation shows that each cell type expresses ≥20% of DEGs in the respective Esr1+ population.
E. UMAP representation with scaled distribution of DEGs. Individual cell types express both sDEGs and eDEGs, with different cell types expressing proportionally more DEGs upregulated in a given condition (sex or estrous stage). Gray cell types (scaled value = “0”) express equivalent proportion of DEGs upregulated in both conditions being compared.
We next tested whether individual cell types were differentially represented between M, FR, and FNR mice. We imposed stringent criteria, including a ≥3-fold cell number difference between ≥2 conditions as well as significant difference in relative abundance of cells, and identified 17 such cell types (Fig. S5C). Most of these cell types occur in all conditions. However, the Cckar+ cell type in the VMHvl (VMHvlCckar/Esr1) and the VMHvlTrim36/Esr1 (expressing the eDEG Trim36) cell type appeared to be restricted to FR and FNR mice, respectively. A previous report identified the VMHvlCckar/Esr1 cell type, albeit annotated with Tsix and not assigned to the estrus phase, but not the VMHvlTrim36/Esr1 cell type because VMHvl cells from FNR mice were not profiled (Kim et al., 2019). It is possible that sequencing with higher depth of coverage will reveal additional condition-biased cell types. In any event, our current approach reveals several sex or estrous state-biased cell types and two that appear unique to FR or FNR mice.
The BNSTprTac1/Esr1 cell type mediates the exclusively male requirement for aromatase+ BNSTpr neurons in social behaviors
We sought to determine the behavioral role of single cell types in relation to the other Esr1+ cell types in the population. Within the BNSTprEsr1 population, the sex-shared BNSTprTac1/Esr1 cell type is GABAergic and displays the highest enrichment for DEGs, with most DEGs upregulated in M compared to FR and FNR (Fig. 6A, S6A,B, Table S7). This cell type is also one of seven enriched for aromatase expression (Fig. 4A, S4G), and we showed previously that aromatase+ BNSTpr neurons are critical for male but not female social behaviors (Bayless et al., 2019). In males, these neurons are essential for sex recognition and the ensuing display of mating with females and aggression toward males. By contrast, their female counterparts are not active during social interactions nor are they essential for mating or maternal displays.
Figure 6: The BNSTprTac1/Esr1 cell type, among all BNSTprEsr1 cell types, is essential for sex recognition, mating, and aggression in males.

A. Violin plots of a subset of DEGs in the BNSTprTac1/Esr1 cell type. Table S7 lists all DEGs in this cell type.
B. Salt and pepper distribution of Tac1 mRNA in Esr1+ BNSTpr neurons. Scale bar = 100 μm.
C. Schematic of intersectional chemogenetic strategy to inhibit BNSTprTac1/Esr1 (top) or BNSTprTac1−/Esr1 cell types.
D. Inhibition of the BNSTprTac1/Esr1 cell type, but not BNSTprTac1−/Esr1 cell types, abolishes male preference for female urine.
E. Inhibition of the BNSTprTac1/Esr1 cell type reduces the probability of resident males initiating mating toward receptive females as well as the number of mount or intromission events/test.
F. Inhibition of the BNSTprTac1/Esr1 cell type reduces the probability of resident males attacking intruder males as well as the number of attacks/test.
G. Inhibition of BNSTprTac1−/Esr1 cell types does not alter mating of resident males with receptive females.
H. Inhibition of BNSTprTac1−/Esr1 cell types does not alter aggression of resident males toward intruder males.
Mean ± SEM. n = 2 (B), 8 (D-H). *p < 0.05, **p < 0.01, ***p < 0.001.
See also Fig. S6.
Tac1, encoding the neuropeptide tachykinin 1 or substance P, is an sDEG that is upregulated in males and expressed in a salt-and-pepper fashion, with an expression bias for the dorsal BNSTpr (Fig. 6A,B, S1F, S6C, Table S7). Tac1 signaling has been implicated in urine preference or social behaviors in males in diverse taxa (Asahina et al., 2014; Berger et al., 2012; Halasz et al., 2009; Katsouni et al., 2009; Siegel et al., 1997). This suggested the possibility that the BNSTTac1/Esr1 cell type, uniquely among all aromatase+ cell types within the BNSTprEsr1 population, is critical for male social interactions.
We tested the functional role of the BNSTprTac1/Esr1 cell type in male social interactions. We delivered a virally encoded, Cre-dependent, inhibitory chemogenetic actuator (AAV-flex-DREADDi:mCherry) to the BNSTpr of adult males (Tac1Cre mice) bearing a Cre recombinase inserted in a gene-conserving manner into the Tac1 locus (Fig. 6C) (Harris et al., 2014). The vast majority of Tac1+ neurons co-labels for Esr1 in the BNSTpr (97% ±2; 1,066 neurons analyzed, n = 2 mice), and our strategy therefore affords specific manipulation of the BNSTprTac1/Esr1 cell type. We first tested whether, like aromatase+ BNSTpr neurons, activity of the BNSTprTac1/Esr1 cell type is required to distinguish between the sexes. Mice use pheromones to recognize the sexes, and male mice show a clear preference for urine, a rich source of pheromones, from females (Fig. 6D). Chemogenetic inhibition of BNSTprTac1/Esr1 neurons in sexually naive males abrogated this preference (Fig. 6D). As with aromatase+ BNSTpr neurons (Bayless et al., 2019), there was no diminution in total time spent sniffing urine, indicating that inhibiting BNSTprTac1/Esr1 neurons eliminates sex recognition rather than simply reducing motivation to investigate pheromones (Fig. S6E). Sex recognition is essential to direct behavioral responses in a target-typical manner, and accordingly inhibiting aromatase+ BNSTpr neurons eliminates the ability to distinguish between sexes and suppresses the ensuing attempts to mate with a female or attack a male (Bayless et al., 2019). Indeed, chemogenetic inhibition of BNSTprTac1/Esr1 cells diminished the propensity to mate or fight and also reduced mounting and intromissions (penetration) during mating and attacks during aggression (Fig. 6E,F, S6J). Together, inhibiting the BNSTprTac1/Esr1 cell type recapitulated the behavioral deficits obtained following the disabling of all aromatase+ BNSTpr neurons.
We wondered whether these functions of BNSTEsr1 neurons were restricted to the BNSTTac1/Esr1 cell type. We therefore tested whether the entire complementary set of Tac1− cell types in this Esr1+ population (BNSTprTac1−/Esr1) also modulated sex recognition, mating, and aggression. We engineered mice harboring Flpo in the 3’ UTR of the Esr1 locus and a virally encoded INTRSECT inhibitory chemogenetic actuator (AAV-Coff/Fon-DREADDi) that would only be expressed in cells that were Cre− and Flpo+ (Fig. S6F–I). We observed that inhibition of BNSTTac1−/Esr1 cell types with this chemogenetic actuator did not disrupt performance in urine preference, sexual behavior, or territorial aggression (Fig. 6D,G,H, S6D,K). These findings further demonstrated that, as observed previously, CNO does not alter social interactions unless DREADD is expressed in a behaviorally salient cell type (Bayless et al., 2019; Inoue et al., 2019; Unger et al., 2015; Yang et al., 2017). In summary, the BNSTprTac1/Esr1 cell type regulates sex recognition, mating, and aggression in males whereas other cell types in the BNSTprEsr1 population are not essential for these behaviors.
The VMHvlCckar/Esr1 cell type, among all VMHvlEsr1 cell types, is required for female mating
The VMHvlCckar/Esr1 cell type is glutamatergic, sexually dimorphic, and FR-restricted such that the genes that define it do not cluster into an identifiable cell type in males, and it is highly enriched for eDEGs that are upregulated in FR mice (Fig. S5C, 7A,B, S7A, Table S7). In addition, it expresses genes that are shared with VMHvlTrim36/Esr1, which appears restricted to FNR mice (Fig. S5C, S7B). Within the VMHvl, Cckar, a receptor for cholecystokinin, is expressed in a salt and pepper fashion in a ventrolateral and posterior subset of VMHvlEsr1 cells (Fig. 7A,C; Table S7), in agreement with our previous work (Xu et al., 2012; Yang et al., 2013). VMHvlEsr1 neurons coexpress PR (Fig. 3C, 4D) (Yang et al., 2013), and these neurons are essential for mating behavior in both sexes, maternal aggression, and male territorial aggression (Hashikawa et al., 2017; Lee et al., 2014; Yang et al., 2013, 2017).
Figure 7: The VMHvlCckar/Esr1 and VMHvlCckar−/Esr1 cell types are required for female mating and maternal aggression, respectively.
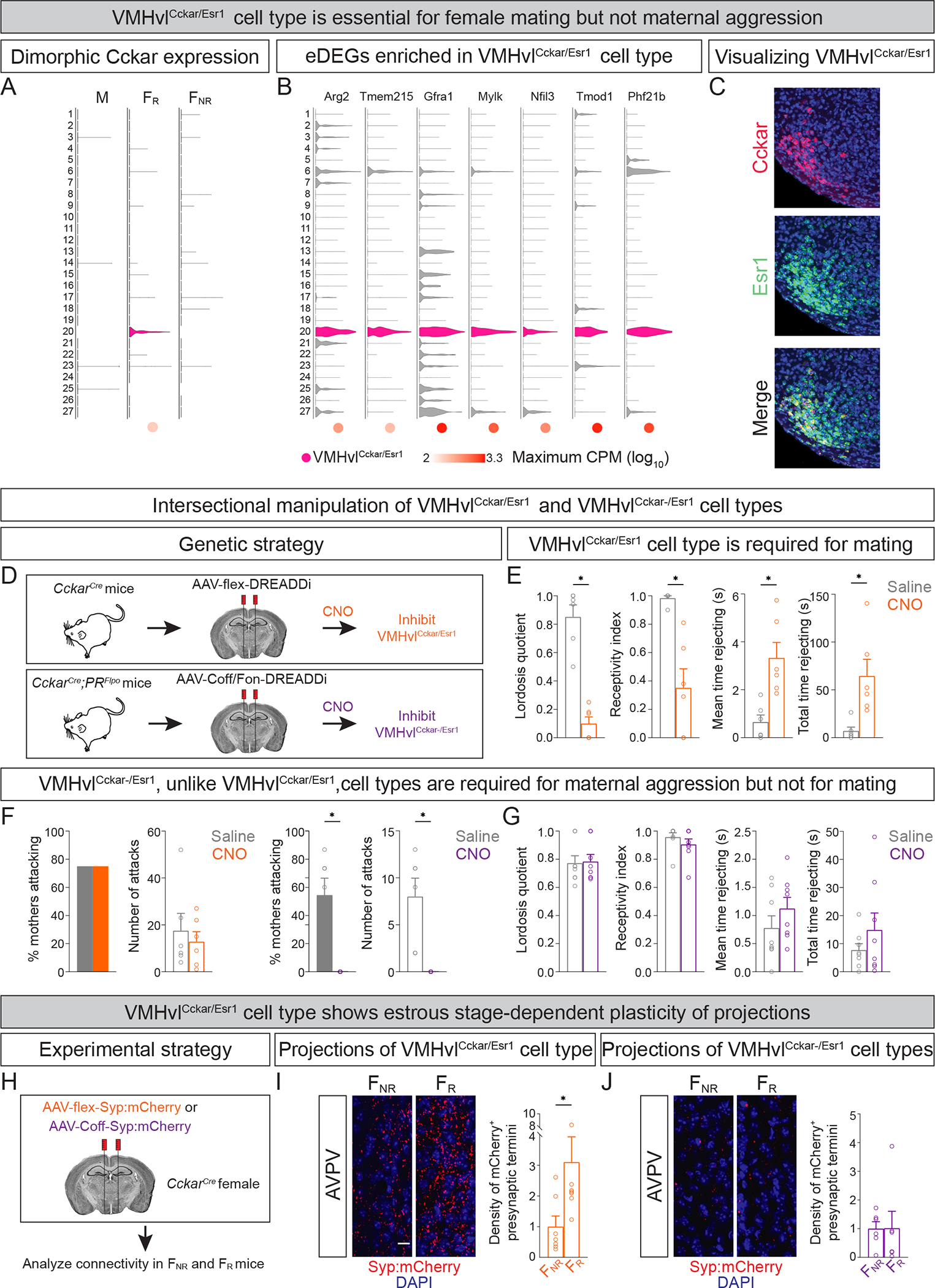
A. Violin plot showing FR-specific expression of Cckar in the VMHvlCckar/Esr1 cell type (#20).
B. Representative eDEGs significantly enriched in VMHvlCckar/Esr1 cell type compared to VMHvlCckar−/Esr1 cell types.
C. Cckar expression is restricted to the ventrolateral component of the Esr1+ VMHvl population, in agreement with previous work (Hashikawa et al., 2017; Xu et al., 2012). Scale bar = 100 μm.
D. Schematic of intersectional chemogenetic strategy to inhibit VMHvlCckar/Esr1 (top) or VMHvlCckar−/Esr1 (bottom) cell types.
E. Inhibition of VMHvlCckar/Esr1 cell type in Fr mice significantly reduces lordosis, receptivity index and increases rejection behavior. Lordosis quotient, (lordosis events/# of mounts); Receptivity index (# of intromissions/# of mounts).
F. Inhibition of the VMHvlCckar−/Esr1, but not VMHvlCckar/Esr1, cell types abrogates maternal aggression.
G. Inhibition of VMHvlCckar−/Esr1 cell types in FR mice does not disable mating behavior.
H. Schematic of strategy to label presynaptic termini of VMHvlCckar/Esr1 and Cckar− neurons.
I. Higher density of mCherry+ termini of VMHvlCckar/Esr1 neurons in AVPV of FR compared to FNR mice. For all findings related to mCherry+ termini in this and subsequent Figures, density of termini is normalized to the number of mCherry+ soma in the VMHvl as described previously (Inoue et al., 2019).
J. No change in density of mCherry+ termini of VMHvlCckar− neurons in AVPV between FR and FNR mice.
Mean ± SEM. n = 2 (B), 6 (F), ≥7 (G), 8 (H), ≥7 (J), and 6 (K). *p < 0.05.
Cckar signaling modulates female mating such that females null for Cckar or treated with Cckar antagonists mate poorly (Micevych and Sinchak, 2001; Xu et al., 2012). Here, we sought to determine the contribution of the VMHvlCckar/Esr1 cell type and its complementary set (VMHvlCckar−/Esr1) to female mating and other social interactions regulated by the VMHvlEsr1 population. To extend our characterization of VMHvlEsr1 neurons with PRCre mice (Inoue et al., 2019; Yang et al., 2013, 2017), we generated PRFlpo mice with Flpo inserted into the 3’ UTR of PR in a gene conserving manner; we also generated a CckarCre mouse strain with a similar targeting strategy (Fig. S7C,D). An overwhelming majority of Cckar+ neurons in the VMHvl co-label for Esr1 (99% ± 1.1; 787 neurons analyzed, n = 2 mice), thereby enabling use of CckarCre mice to manipulate the VMHvlCckar/Esr1 cell type.
Chemogenetic inhibition of the VMHvlCckar/Esr1 cell type suppressed sexual behavior of CckarCre Fr mice, with an ~8-fold diminution in lordosis (dorsiflexed posture adopted by females to enable mating), and increased rejection behavior (Fig. 7E, S7E). Inhibition of this cell type in lactating CckarCre mice did not alter pup retrieval or maternal aggression (Fig. 7F, S7F). Together, these findings show that the VMHvlCckar/Esr1 cell type is essential for female sexual behavior but not maternal displays.
Our findings do not preclude a requirement for VMHvlCckar−/Esr1 cell types in female mating, and they predict that these cell types are essential for maternal aggression. We employed our chemogenetic reagents to test this in CckarCre;PRFlpo mice (Fig. S6H,I, 7D). We observed complementary phenotypes upon chemogenetic inhibition of VMHvlCckar−/Esr1 cell types such that female mating was unaffected and maternal aggression was abolished (Fig. 7F,G). There was no effect on retrieval of wandering pups, consistent with work showing that this behavior is regulated by distinct neural pathways (Fig. S7F) (Kohl et al., 2018; Wu et al., 2014). Thus, VMHvlCckar/Esr1 and VMHvlCckar−/Esr1 cell types are required for female sexual behavior and maternal aggression, respectively, in a complementary manner.
Male VMHvlEsr1 neurons are a central hub for aggression, with a more subtle requirement in mating (Falkner et al., 2016; Lee et al., 2014; Yang et al., 2013, 2017). Although the VMHvlCckar/Esr1 cell type is absent in males (Fig. S5C, S7A), we observed a few neurons following expression of a Cre-dependent DREADDi in male CckarCre mice, consistent with our snRNAseq data (Fig. 7A, S7E). We therefore tested if these neurons regulated male social behaviors. Chemogenetic inhibition of VMHvlCckar/Esr1 neurons in CckarCre males did not disrupt mating or aggression, whereas inhibition of VMHvlCckar−/Esr1 cell types in CckarCre;PRFlpo males disabled both behaviors (Fig. S7G–I). Together, the VMHvlCckar/Esr1 cell type is essential for female sexual behavior whereas VMHvlCckar−/Esr1 cell types are required for maternal aggression, male sexual behavior, and male aggression.
We showed that projections of VMHvlEsr1 neurons to the hypothalamic anteroventral periventricular nucleus (AVPV) are essential for mating in Fr mice (Inoue et al., 2019). Given the requirement of the VMHvlCckar/Esr1 cell type for female mating, we wondered if this cell type preferentially projects to the AVPV. We delivered a virally-encoded, Cre-dependent synaptophysin:mCherry fusion (Syp:mCherry) that labels presynaptic termini to the VMHvl of CckarCre females (Fig. 7H) (Inoue et al., 2019). We also delivered a virally-encoded Syp:mCherry that can only be expressed in Cckar− cells (AAV-Coff-Syp:mCherry) to the VMHvl of a separate set of CckarCre females (Fig. 7H). This strategy enables testing connectivity of Cre− VMHvl neurons (Inoue et al., 2019). We observed more mCherry+ termini in the AVPV from Cckar+ compared to Cckar− neurons even though the latter far outnumber the former (Fig. 7I,J, S7A). To cross-validate this observation, we delivered cholera toxin B (CTB), a retrograde tracer (Dederen et al., 1994), to the AVPV and Cre-dependent Syp:mCherry to the VMHvl of CckarCre females and enumerated the overlap between CTB and mCherry. We found that CTB+ soma in the VMHvl were more likely to be Cckar+ than not (71.8% ± 4.6 and 32.2% ± 2.3 CTB+ soma were Cckar+ and Cckar−, respectively; n = 4, p = 0.03). The projection of VMHvlEsr1 neurons to the AVPV is strikingly dimorphic and essentially absent in males (Yang et al., 2013). Indeed, there was a >10-fold reduction in mCherry+ presynaptic termini of male Cckar+ VMHvl neurons in the AVPV compared to those of female VMHvlCckar/Esr1 neurons (density of mCherry+ presynaptic termini: males, 0.2 ± 0.04; females, 2.1 ± 0.5; n ≥ 7; p < 10−4). Together, these findings show that the female VMHvlCckar/Esr1 cell type preferentially projects to the AVPV in comparison to both male Cckar+ VMHvl neurons and female VMHvlCckar−/Esr1 cell types.
Projections of VMHvlEsr1 neurons to the AVPV expand and shrink 3-fold across the estrous cycle, with peak anatomical and functional connectivity at estrus (Inoue et al., 2019). We tested whether projections of VMHvlCckar/Esr1 neurons exhibit such structural plasticity and observed a ~3-fold increase in mCherry+ presynaptic termini in the AVPV of FR compared to FNR mice expressing Syp:mCherry in Cckar+ neurons (Fig. 7I). By contrast, there was no discernible change in the number of mCherry+ termini between FR and FNR mice when Syp:mCherry was expressed in Cckar− neurons (Fig. 7J). There was also no change in the number of presynaptic termini to the PAG or POA, the other major projection targets of VMHvlPR neurons, between FR and FNR mice in either cohort (Fig. S7J,K). Thus, we observe preferential projections and structural plasticity of the female VMHvlCckar/Esr1 cell type to the AVPV in accordance with its requirement in female sexual behavior.
DISCUSSION
We find that 1,415 genes exhibit 2,311 differential expression events between sexes or estrous states in Esr1+ neurons, with a median fold difference of 1.9. This corresponds to males or females in distinct estrous states essentially having an extra copy of 6% of their coding genes in these cells. The limited commonality in differential gene expression patterns between the 4 Esr1+ populations in the BNSTpr, MeA, POA, and VMHvl suggests that there is not a generic signature of sex or estrous cycle state across the brain. The availability of unique molecular classifiers of cell types also affords genetic access to understand how individual cell types influence gender-typical behaviors. This approach allowed us to identify modular, molecularly-specified neural pathways emanating from discrete cell types that, respectively, are critical for sex recognition and social interactions in males and sexual behavior in females.
Sex differences in gene expression
Most behaviors are shared between sexes, suggesting that neurons governing sex-specific behaviors are embedded within shared populations. Indeed, our purification schemes show that sex hormone-sensitive neurons represent 5–15% of cells within the regions that we surveyed. Circulating titers of sex hormones are sexually dimorphic, suggesting that their target neurons should exhibit vastly sex-divergent transcriptomes. In agreement with this prediction, by enriching for adult sex hormone-sensitive neurons we have identified ~10-fold more differential gene expression events than previously reported (Chen et al., 2019; Kim et al., 2019; Moffitt et al., 2018; Welch et al., 2019; Xu et al., 2012). The number of DEG-encoding loci on autosomes far exceeds that on the X chromosome, indicating that this chromosome does not play a privileged role in harboring DEGs. Our preliminary analysis suggests that many transcription factor-encoding DEGs regulate expression of other DEGs (data not shown), suggesting a hierarchical control of DEG regulation, with sex hormone receptors at the apex.
A large subset of sDEGs is restricted to a single Esr1+ population, sex, or estrous state pointing to complex and dynamic gene regulatory mechanisms. Many sDEGs are also insensitive to estrous states, suggesting that these sex differences result from the organizational effect imposed by sex hormones during critical developmental periods (Arnold, 2009; Sisk and Zehr, 2005). We have sampled only a subset of sex hormone-sensitive populations, and based on these findings, we anticipate that many more unique DEGs remain to be identified. If true, an even larger fraction of the coding genome is expressed in a sex or estrous state-specific manner. Many DEGs we have identified are implicated in hormone-regulated processes in non-neural tissues, and we speculate this to mean that sex hormones regulate expression of a discrete network of genes that is shared across diverse target organs in health and disease. Once we extend our analysis of these DEGs using spatial transcriptomics, we may find additional regions in the brain in which they are expressed in dimorphic manner. In addition, it is likely that sex differences in post-transcriptional processes will further modulate, potentially dampening or further amplifying, differential cellular function between males and females.
Constitutive deletion of genes expressed in a sex-typical manner in the brain can result in sexually dimorphic behavioral phenotypes, indicative of the importance of sex differences in expression of individual DEGs (Xu et al., 2012). How such sex differences in gene expression alter neural circuit physiology or function is an open question. Based on our gene ontology analysis, we speculate that such dimorphisms most often differentially regulate synaptic architecture, transmission, or plasticity, response to extracellular signals, neuronal excitability and homeostasis, and gene expression. The DEGs we have identified likely provide at least some of the molecular substrates that underlie the numerous observations of sex differences in neuronal projections and synaptic plasticity and density (Cooke et al., 1998). In any event, given that individual sDEGs can measurably impact behavior, the large number of sDEGs we have uncovered may enable exquisitely specific regulation of sexually dimorphic social interactions.
Many sDEGs are linked to disorders with strong sex-biases, including Alzheimer’s disease (Col25a1) (Forsell et al., 2010; Tong et al., 2010), multiple sclerosis (Cd24a) (Zhou et al., 2003), and most strikingly, ASD. Our findings support studies that have implicated testosterone and estrogen signaling in generating the sex bias in ASD and suggest potential underlying molecular mechanisms (Baron-Cohen et al., 2011; Hoffman et al., 2016; Willsey et al., 2021). Moreover, most well characterized mutations in ASD-associated DEGs are predicted to generate loss of function alleles in male-upregulated genes. Together, this suggests a model in which sex or estrous state-related expression levels of disease risk-conferring DEGs support neural circuits that, if disrupted, result in sex-biased disease outcomes.
Estrous state-dependent differences in gene expression
Estrous states exert a profound influence on diverse female behaviors, presumably by cyclic modification of neural circuit morphology and function (Cooke and Woolley, 2005; Dias et al., 2021; Inoue et al., 2019). We have identified several hundred genes that change expression between diestrus and hormonally induced estrus in Esr1+ neurons. VMHvlEsr1 neurons, whose projections to the AVPV exhibit structural plasticity across the cycle, also harbor the largest number of eDEGs. Many of these genes label the FR-restricted VMHvlCckar/Esr1 cell type that is essential for female mating and whose projections peak at estrus. Together, the VMHvlCckar/Esr1 cell type affords a facile genetic platform in which to study the relation between genes, synaptic plasticity, and behavior.
As discussed above for sDEGs, we imagine that eDEGs regulate cell biological processes that enable changes in structural plasticity, synaptic communication, responses to extracellular signals, and neuronal excitability. More generally, we have uncovered a large-scale dynamic neuronal gene expression changes in females. These eDEGs should enable characterization of neural circuit physiology and behavior across the cycle with exceptional precision. The cyclic changes in eDEG expression reflect an underlying activational role of estrogen and progesterone signaling. We imagine that, as for sDEGs, equally complex and dynamic regulatory mechanisms specify the regionally restricted patterns of eDEG expression. At least in the VMHvl, Esr1 signaling upregulates PR, and a recent study showed that a single dose of estrogen rapidly induced significant gene expression changes in this region (Krause et al., 2021; Musatov et al., 2006). Such rapid changes could set the stage for additional gene expression changes induced by PR signaling.
Although we and others have identified genes or cell types expressing such genes whose expression is sex-specific (Kim et al., 2019; Xu et al., 2012), our analysis of females in distinct estrous states has further enabled the identification of molecularly-specified cell types that are restricted to diestrus or estrus. We cannot presently distinguish whether Cckar+ and Trim36+ VMHvl cell types represent temporal snapshots of the same cell type or rather coalesce with other cell types in obverse phases of the cycle. Targeted profiling and computational analyses developed for lineage mapping should disambiguate between these possibilities (Fletcher et al., 2018). Regardless, our findings show the importance of analyzing gene expression in defined estrous states. We imagine that many additional estrous cycle state-specific cell types remain to be identified in the brain and elsewhere.
Cellular logic of sexually dimorphic social behaviors
Many transcriptomically-defined cell types have been identified in diverse brain regions (Miller et al., 2020). Whether individual cell types and their complementary counterparts within a given brain region contribute differently to specific behaviors is an open question. Our analysis of two Esr1+ cell types and their complementary counterparts provides support for a modular architecture of neural pathways underlying sexually dimorphic social behaviors. Our findings raise the possibility that cell types in other brain regions that regulate non-social, sex-shared behaviors also function in a similar, compartmentalized manner.
We find that most DEGs are expressed in multiple cell types. Such DEGs may endow different cell types in a population with shared dimorphic properties or they may perform context-specific functions in each cell type. In any event, the multi-cell type spanning expression of many DEGs likely reflects the fact that sexual differentiation of the brain, which is initiated perinatally and continues into puberty (Arnold, 2009; McCarthy and Arnold, 2011; Sisk and Zehr, 2005), is superimposed upon the earlier program of neuronal cell fate specification. Indeed, the expression of aromatase and androgen receptor only resolves into its adult dimorphic pattern following the perinatal window of sexual differentiation (Juntti et al., 2010; Wu et al., 2009).
Functional specialization of individual cell types regulating sexually dimorphic social behaviors can arise from gene expression or connectivity patterns that differ among cell types within the population and between sexes. We imagine that DEGs provide functional specialization to Esr1+ cell types to enable modular control of sexually dimorphic social behaviors. Our study also provides evidence for functional specialization arising from differential connectivity. The VMHvlCckar/Esr1 cell type preferentially projects to the AVPV and its projection termini exhibit structural plasticity across the estrous cycle. Thus, the modular function of the VMHvlCckar/Esr1 cell type in female mating may arise from its set of female and cell-type restricted DEGs and specialized connectivity. Together, the specific set of connections, DEGs, and, potentially, post-transcriptional processes may enable each cell type to contribute in a modular manner to different sexually dimorphic behavioral displays. Similar conclusions have also been made for neurons regulating locomotion (Ferreira-Pinto et al., 2021). Thus, such specificity may be a general feature of neuronal cell types participating in sexually dimorphic as well as sex-shared behaviors. It is also possible that some cell types may “reflect facets of cell identity (or cell state) distinct from projection specificity and behavioral function” (Kim et al., 2019). Regardless of such an alternative, our findings show that both excitatory and inhibitory neuronal populations contain cell types that are transcriptomically discrete and functionally specialized.
We have interrogated the behavioral role of two Esr1+ cell types and find that each is essential for specific facets of sex-typical social behaviors. We imagine that other Esr1+ cell types regulate other sex-typical behaviors and physiological processes or inhibit those of the opposite sex (Correa et al., 2015; De Vries and Boyle, 1998; Krause and Ingraham, 2017; van Veen et al., 2020; Zhang et al., 2020). Even innate sex-typical social behaviors are modifiable by context or experience, and some Esr1+ cell types may regulate such behavioral flexibility (Dey et al., 2015; Falkner et al., 2016; Li et al., 2017; McHenry et al., 2017; Remedios et al., 2017; Todd et al., 2018; Yang et al., 2017). We anticipate that detailed studies will reveal subtly different contributions of individual cell types to behavior or physiology and that some cell types may also participate in multiple functional domains. Such studies may also reveal that these Esr1+ populations participate in many processes not previously ascribed to them.
We previously showed that individual genes expressed in a sexually dimorphic manner are required for one or a few components of sex-typical social behaviors (Xu et al., 2012). Here we demonstrate that individual cell types expressing such genes are also required for specific aspects of sex-typical social behaviors. Together, our findings suggest a model in which the seemingly unitary constellation of behaviors that comprises maleness or femaleness is constructed from separable genetic and cellular networks that differ between the sexes.
Limitations of the study
Our use of Esr1Cre mice can potentially isolate neurons that are Esr1− in adult mice so that some DEGs may reside in such cells. However, we note that all cell types we report here express Esr1. The use of Esr1Cre mice eliminates concerns regarding estrous cycle-regulated changes in Esr1 expression because we used adult females who had undergone multiple estrous cycles and whose Esr1+ neurons therefore permanently expressed genetic tags required for purification. We used females primed with hormones or vehicle to model estrus and diestrus because mice have irregular cycles and the act of checking cycle stage can itself alter estrous states, making it impractical to harvest sufficient estrous stage-matched material for sequencing. By contrast, our approach, standard in the field, affords tissue collection from precisely synchronized females. Although our priming protocol generates hormone titers indistinguishable from natural estrus (Inoue et al., 2019), it is possible that some eDEGs may exhibit more subtle changes across the cycle in gonadally intact females. Future studies employing in vivo activity monitoring and optogenetic actuators in different social contexts will afford richer, temporally precise, and context-specific insights into how the Esr1+ cell types we have characterized here regulate sex-typical behaviors. Finally, our data do not shed light on spatial distribution of all Esr1+ cell types. Future studies entailing spatial transcriptomics should enable visualization of these cell types in situ.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Nirao M. Shah (nirao@stanford.edu).
Materials availability
Plasmids generated for this study (INTRSECT virus) have been deposited at Addgene (#177669). New transgenic mouse lines generated for this study will be deposited at Jackson Laboratories upon publication. Probe sets used for ISH are available from Molecular Instruments (Pasadena, CA). Oligonucleotide primers used for RT-qPCR are listed in Table S1. This study did not generate new unique reagents not listed here.
DATA AND CODE AVAILABILITY
TRAPseq and snRNAseq data have been deposited at Gene Expression Omnibus and are publicly available upon publication (GEO:GSE183092 and GEO:GSE183093 respectively).
This paper does not report original code. All MATLAB and R scripts used in this manuscript are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Animal studies were conducted following Institutional Animal Care and Use Committee guidelines and protocols. RiboTag, Esr1Cre, SunTag, and Tac1Cre mice were purchased from Jackson Labs and bred in our mouse colony. We engineered Esr1Flpo, PRFlpo, and CckarCre mice as described below and bred them in our mouse colony. Mice were housed under a reversed 12:12 hr light:dark cycle. Food and water were available ad libitum. Wild-type C57BL6/J males and females used for ISH were purchased from Jackson Laboratories. WT females were purchased from Jax (C57B16/J), and WT males (B6129SF1/J) used as stimulus animals in tests of sexual receptivity were bred in the animal facility or purchased from Jackson Laboratories. All mice were group housed by sex after weaning. Group housed mice 10–12 weeks of age were used for deep sequencing and ISH studies. For behavioral studies, mice were 12–16 weeks of age at the time of testing. Male mice used for behavioral testing were single housed for ≥1 week prior to testing. Female mice used for mating assays were group-housed.
Cell lines
HEK293FT cells (Thermo Fisher) were maintained in a 5% CO2 humid incubator with DMEM media (GIBCO) supplemented with 10% FBS (Invitrogen), 1% Glutamax (Invitrogen), and 1x Penicillin-Streptomycin (Invitrogen). They were enzymatically passaged at 90% confluence by trypsinization.
Viruses
AAV-EF 1α-flex-DREAADi:mCherry (serotype DJ) and AAV5-EF1a-fDIO-hChR2(H134R)-EYFP-WPREa (serotype 5) were purchased from the UNC Vector Core. AAV-EF1a-Coff-Syp:mCherry (serotype 8.2) was purchased from the MGH viral core. AAV-EF1a-DIO-Syp:mCherry (serotype 1) was custom packaged by Virovek (Hayward, CA). AAV-hEF-Coff/Fon-DREAADi:mCherry (serotype 8) was custom packaged by Stanford Virus Core (Stanford, CA). AAV titers were 1.5 × 1012 – 2.5 × 1013 genomic copies/mL.
METHOD DETAILS
Hormonal priming
For all sequencing and ISH studies, female mice were ovariectomized and estrus was induced as we described previously; this estrus induction protocol leads to estrogen and progesterone titers indistinguishable from naturally occurring estrus (Inoue et al., 2019; Yang et al., 2013). Briefly, we delivered subcutaneously 10 μg of estradiol benzoate dissolved in 100 μl of sesame oil on day −2, 5 μg of estradiol benzoate in 50 μl sesame oil on day −1, and 50 μg of progesterone in 50 μl of sesame oil on day 0 (FR). FNR mice received identical volumes of sesame oil on each day. Females were euthanized for dissection or histological analysis 4–6 hours after the final injection for all studies.
Dissection for TRAPseq and snRNAseq
All mice used for TRAPseq and snRNAseq were F1 Esr1Cre/+;RiboTag/+ or Esr1Cre/+;SunTag/+ and generated by mating homozygous male Esr1Cre/Cre mice with homozygous female SunTag or RiboTag mice. Adult mice were deeply anesthetized by injection with 2.5% Avertin and euthanized by decapitation. Brains were sectioned into 500 μm coronal slices using a brain matrix mold (BrainTree Scientific) chilled on ice. Sections were floated in either chilled TRAPseq homogenization buffer (100 mM KCl, 50 mM Tris-HCL pH 7.4, 12 mM MgCL2) or nuclear homogenization buffer (snRNAseq; see below). The BNSTpr, MeA, POA and VMHvl were identified using landmarks from the mouse brain atlas (Paxinos and Franklin, 2003) and dissected using a Zeiss microscope. Tissue was then either frozen on dry ice and stored at −80C until further processing (TRAPseq) or stored in cold homogenization buffer (snRNAseq) for subsequent nuclear extraction. For each condition, we pooled tissue from 3–5 mice/sex/region/replicate for TRAPseq and similarly 4–6 mice for snRNAseq.
Fixation for histology
Mice were deeply anesthetized by injection of 2.5% Avertin and then trans-cardially perfused with 20 mL ice-cold PBS followed by 20 mL ice-cold 4% paraformaldehyde. Following overnight fixation in 4% paraformaldehyde at 4°C, brains were sectioned coronally on a vibrating microtome (Leica) at 50 μm (ISH) or 65 μm (immunohistochemistry).
Immunohistochemistry
Brains from Esr1Cre/RiboTag and Esr1Cre/SunTag mice were immunolabeled using the following primary antisera: mouse anti-HA (1:1,000), sheep anti-GFP (1:2,000), rabbit anti-Esrl (1:1,000), and rat anti-mCherry (1:2,000). Primary antisera were detected with the following secondary antibodies: donkey anti-sheep Alexa Fluor 488 (1:300), donkey anti-rat Cy3 (1:800), donkey anti-mouse Alexa Fluor 488 (1:300), and donkey anti-rabbit Alexa Fluor Cy3 (1:800) (Jackson Immunoresearch). Sections were counterstained with DAPI (0.2 μg/mL) and imaged with a confocal microscope (Zeiss). Images were processed using ImageJ.
TRAPseq
We followed previously published protocols to perform TRAPseq (Sanz et al., 2009). In brief, frozen brain regions were thawed in a pestle homogenizer in 1 mL of ice-cold buffer containing 100 mM KCl, 50 mM Tris-HCL pH 7.4, 12 mM MgCL2, 1% IGEPAL, 1 mM dithiothreitol (DTT), 1 mg/mL heparin, 100 μg/mL cycloheximide (CHX), cOmplete mini protease inhibitor (Sigma-Aldrich), and 40 U/μl murine RNAse inhibitor (RNasin; NEB). Brains were homogenized using 10–20 strokes of the pestle and homogenate transferred to a 1.5 mL microcentrifuge tube. The homogenate was clarified by centrifugation at 10,000 g for 10 minutes at 4°C and the post-mitochondrial supernatant transferred to a fresh tube. 40 μl of the supernatant was added to 0.35 mL of buffer RLT (QIAgen RNeasy micro kit) supplemented with 10 μl/mL β-mercaptoethanol (Input sample) and stored at −80°C overnight. 2.5 μl of rabbit anti-HA antibody (Cell Signaling) was added to the remaining homogenate (IP sample) and the sample was incubated in an end-over-end rotator at 4°C for four hours. The sample was then added to 50 μl of pre-washed paramagnetic protein G Dynabeads (Invitrogen) and incubated overnight at 4°C on an end-over-end rotator. After incubation, beads were washed three times for ten minutes at 4°C with high-salt buffer (300 mM KCl, 50 mM Tris-HCL pH 7.4, 12 mM MgCL2, 1% IGEPAL, 1 mM DTT, 300 μg/mL cycloheximide). After the final wash, beads were transferred to a new tube, collected by magnet and the supernatant removed. RLT buffer (0.35 mL) was added directly to the beads and total RNA from Input and IP samples was extracted using the QIAgen RNeasy Micro kit according to manufacturer’s instructions, including on-column DNase digestion. RNA was eluted in 20 μl RNAse-free water and stored at −80°C. RNA yield and quality was evaluated by Bioanalyzer 2000 (Agilent) using the Total RNA Pico kit. To determine enrichment of Esr1 mRNA, 1 ng of total RNA from each Input and IP sample was used for the Protoscript cDNA synthesis kit with random hexamer priming followed by real-time quantitative PCR (RT-qPCR) on either an ABI StepOne Plus or GeneQuant qPCR machine (Biorad). We performed RT-qPCR for Esr1, Gfap and Gapdh (oligonucleotide primers listed in Table S1). For each IP sample we calculated enrichment of Esr1 and depletion of Gfap relative to Input using the Δ/ΔCt method. We then used 10 ng of total RNA as input for the Nugen Universal mRNA kit (Tecan) to generate indexed cDNA libraries according to manufacturer’s instructions. We also loaded equivalent starting material at each subsequent step so as to preclude potential biases introduced by cell number differences between sexes. Libraries were sequenced at 50 bp single-end on a HiSeq4000 (average of 22 million deduplicated reads/biological replicate; see Table S1).
snRNAseq
We modified the INTACT protocol (Mo et al., 2015) to isolate nuclei as follows. Microdissected brain regions were suspended in ice-cold nuclear homogenization buffer (HB) consisting of 0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, and 20 mM Tricine-KOH pH 7.8 supplemented with 1 mM DTT, 0.15 mM spermine, 0.5 mM spermidine, cOmplete mini protease inhibitor (EDTA-free)(Sigma-Aldrich,) and murine RNAse inhibitor. The tissue was homogenized in a Dounce homogenizer by 5–10 strokes, 60 μl of HB/5% IGEPAL was added, and the suspension was homogenized with an additional 5–10 strokes before being passed through a 40 μm filter. The resulting suspension was mixed with 1 mL (1 volume) of 50% Optiprep (Sigma-Aldrich) containing 25 mM KCl, 5 mM MgCl2, and 20 mM Tricine-KOH pH 7.8 and transferred to an ultracentrifuge tube. The suspension was underlaid with a 30% Optiprep solution and spun at 10,000g in a swinging-bucket ultracentrifuge for 20 minutes at 4°C to pellet the nuclei. The supernatant was removed and nuclei were resuspended in 1 mL HB supplemented with 1 mM DTT, 40 U/mL RNasin, and 0.1% IGEPAL. Nuclear integrity and the quality of nuclear dissociation were verified by mixing 10 μl of the above suspension with 10 μl Trypan Blue dye and viewing with a hemocytometer using a light microscope. Nuclei were sorted using an ARIA FACS sorter at the Stanford Shared FACS facility. Gates were set by first identifying a side population with 1) high 488 nm (green) signal and low 546 nm (red signal; indicative of auto-fluorescent particles), back-gating to identify events corresponding to nuclei based on side scatter, and further filtering doublets based on forward scatter versus side scatter. Nuclei were sorted into 1 mL of 0.1% IGEPAL HB supplemented with 1 mM DTT and 40 U/mL RNAsin. In a pilot experiment we dissected BNSTpr from 4 SunTag;Esr1Cre males and sorted both GFP+ and GFP− nuclei. We then used the RNeasy Micro Kit to extract total RNA from GFP+, GFP− and input (unsorted) nuclei for RT-qPCR to determine whether this GFP+ population was enriched in Esr1 and Gfp mRNA. For snRNAseq, after sorting was completed the yield of intact nuclei was verified by counting Trypan Blue-stained nuclei on a hemocytometer. Nuclei were pelleted by centrifugation at 2,000 g for 10 min at 4°C and resuspended in 60 μl of sterile PBS with 2% bovine serum albumin (BSA). They were again inspected and quantified by Trypan Blue staining prior to submission to the Stanford Functional Genomics Facility for generation of snRNA libraries using the 10X Genomics Chromium platform. Libraries were prepared using the 10X Genomics RNA 3’ v3 kit. Completed libraries were sequenced on either a HiSeq4000 or NovaSeq6000 to a target depth of 80,000 reads/cell (Table S5).
ISH
Probes and fluorescently conjugated amplifiers were purchased from Molecular Instruments (Pasadena, CA). We designed as many probe pairs as possible for each gene, up to a maximum of 40 pairs/gene. Probes were designed to be detected with hairpin amplifiers conjugated to either AlexaFluor-488, 546 or 647 fluorophores for high-, medium- and low-expressing genes respectively (estimated from TRAPseq data). ISH hybridization, wash and amplification buffers were made following manufacturer’s recommendations for mammalian cells on a slide (Choi et al., 2018). Coronal sections were collected at 50 pm on a vibrating microtome (Leica), and the sections were washed with DEPC/PBS and incubated overnight in 70% ethanol/PBS at 4°C. Sections were washed twice in PBS and cleared by incubating for 45 minutes in 5% SDS/PBS at room temperature. After clearing, sections were washed twice in 2X SSC, then incubated twice in 2X SSC for 15 min at room temperature with gentle shaking. They were then incubated in hybridization buffer at 37°C for two hours. After prehybridization probes were added to a final concentration of 10 nM and sections were hybridized at 37°C overnight. They were then washed four times in ISH wash buffer for 15 min at 37°C, followed by two washes in 2X SSCT for five minutes. The sections were incubated in ISH amplification buffer for 30 minutes at room temperature. Fluorescently conjugated hairpins were denatured for 5 min at 95°C in a thermocycler followed by snap-cooling in a dark drawer at RT for 30 min. Amplifiers were added to amplification buffer and sections were incubated overnight at room temperature in the dark. They were washed twice with 2X SSCT, incubated for 15 min in 2X SSC/DAPI, and washed twice with 2X SSC. Sections were mounted on Superfrost slides, coverslips sealed with Aquamount, and imaged with a confocal microscope (Zeiss). Images were processed using ImageJ with identical parameters used for all pairwise comparisons. To calculate the degree of overlap between select DEGs and Esr1 in vivo we used CellProfiler (McQuin et al., 2018). We segmented cells with the IdentifyPrimaryObjects function by identifying nuclei based on DAPI staining and defining the soma as occupying a 20-pixel radius beyond the border of the nucleus. Fluorescent ISH signal for Esr1 and the DEG of interest was detected with the IdentifyPrimaryObjects function using the “Global” threshold strategy, the “Robust Background” thresholding method, the “Mean” averaging method, and the “Standard Deviation” variance method. We used an intensity criterion of 2–3 standard deviations above the mean and a minimum pixel width of 3–5 to differentiate ISH signal from background, with the exact value optimized for each ISH probe.
Bioinformatics
TRAPseq data analysis
Single-end reads were first run through the bbtools clumpify.sh program (https://sourceforge.net/projects/bbmap) to remove exAmp duplicates caused by the HiSeq4000 patterned flow cell with the following options: tossbrokenreads dedupe optical spantiles adjacent dupedist=2500. Remaining reads were pseudoaligned and quantified against all mouse polyadenylated transcripts using Kallisto (mm10, gencode vM21) with the following options: -bias -b 40 --single -l 180 -s 35 (Bray et al., 2016). Transcript-length normalized counts were loaded into R using the tximport package and differential expression analyzed using DESeq2 (Love et al., 2014; Soneson et al., 2016). We eliminated from analysis Y-chromosome genes and X-linked genes with Y paralogs because these tend to be expressed broadly and are sexually dimorphic by virtue of chromosomal complement rather than differential gene regulation. We also eliminated mitochondrial genes and genes with fewer than 5 counts in at least 7 samples from analysis. A generalized linear model was constructed for each Esr1+ population using libraries from all three conditions with default DESeq2 parameters. Individual pairwise comparisons (M v FR, M v FNR, FR v FNR) were extracted using the ‘contrast’ function in DESeq2. Genes were considered differentially expressed if they passed a false discovery rate-adjusted p value < 0.05 in at least one pairwise comparison. Unless otherwise noted, for subsequent analyses we only considered genes with an absolute fold change > 1.5 in either direction. To visualize all samples in DEG space we plotted PC1 and PC2 for each Esr1+ population on all DEGs identified in that population using pcatools (https://github.com/kevinblighe/PCAtools). To visualize overlap of DEGs across regions or pairwise comparisons within regions (Fig. 2C,D), we first compiled false discovery rate-adjusted p values for each DEG for all comparisons. We then calculated −log10 of these p values and plotted them as heatmaps, sorting from most to least significant. The color transition for these heatmaps was set to 1.3 (p = 0.05) to highlight whether or not genes were differentially expressed in a given comparison. For our ISH studies, we wished to determine if the expression differences we observed in situ between M, FR, and FNR were concordant with the direction of fold change detected by TRAPseq. Accordingly, for each DEG we also visualized by ISH, we calculated the scaled (z-scored) expression for each TRAPseq run for that particular DEG and then averaged the z score across all three TRAPseq runs. We plotted these average z-scores as heatmaps and provided them alongside the respective ISH images to display visually the direction of expression change predicted by TRAPseq (Fig. 1C, S1F, 2B, S2B, 3A,B). Gene names and known or putative functions of each DEG visualized by ISH were obtained from GeneCards and are available in Table S3 (Stelzer et al., 2016).
Identification of sex hormone response elements in DEG-encoding loci
To test for enrichment of sex hormone response elements near genes encoding DEGs, we downloaded consensus motifs for androgen receptor (ARE), progesterone receptor (PRE) and estrogen receptor (ERE) from the MEME suite website (https://meme-suite.org/meme/doc/fimo.html). We scanned the mm10 build of the mouse genome for occurrences of each motif and quantified such motifs within a 250 kb genomic region flanking both the transcription start site and 3’ UTR for loci encoding DEGs and all genes expressed in each individual Esr1+ population. Enrichment of each motif at DEG loci relative to all expressed genes in each population was then calculated using Fisher’s Exact Test. We performed this test separately for sDEGs and eDEGs.
Gene Ontology analysis
Gene Ontology analysis was conducted using topGO (Alexa and Rahnenfuhrer, 2021). Non-redundant, enriched GO:Biological Process categories were identified using the following options: algorithm =”weight01”, statistic-’fisher”, nodeSize=5. The union of all DEGs (M v FR, M v FNR, FR v FNR) from each Esr1+ population was used as input, with all genes expressed in that Esr1+ population used as background. We used topGO default settings to identify significantly enriched categories. For visualization (Fig. S3) we examined the top 100 most significant Gene Ontology categories by p value in each Esr1+ population and selected those present in >1 region to highlight shared functions of sex and estrous-regulated genes. A complete list of enriched Gene Ontology categories passing p<0.05 for each Esr1+ population is available in Table S4.
Disease Ontology analysis
For Disease Ontology analysis, we converted mouse gene symbols to human Entrez IDs using the biomaRt package (Durinck et al., 2009). Disease Ontology categories were identified using enrichDO in R with the following parameters: pvalueCutoff=0.05, pAdjustMethod=“BH”, minGSSize=5, maxGSSize=500, qvalueCutoff=0.05 (Yu et al., 2015). Significantly enriched diseases were checked in the literature for evidence of sex bias; a complete list of references is available in Table S4. For visualization we omitted redundant categories (diseases with similar terminology that were identified because of the same set of DEGs being enriched, e.g. “breast cancer” and “breast carcinoma”). A complete list of enriched Disease Ontology terms is available in Table S4. To check for enrichment of high-confidence ASD risk genes in our list of DEGs we downloaded the list of ASD risk genes from SFARI Gene (https://gene.sfari.org/). We then calculated the overlap between the full list of DEGs and the list of genes assigned a score of ‘1’ (highest confidence association with ASD) using a hypergeometric test.
snRNAseq data analysis and cell type identification
10X genomics libraries were pseudoaligned to the mouse transcriptome (mm10, gencode version vm21) using kallisto-bustools (Melsted et al., 2021). Reads were aligned to a modified transcriptome index that included introns to allow for quantification of unspliced transcripts. After alignment a cell x gene matrix was loaded into R using the BusParse package (https://github.com/BUStools/BUSpaRse) and used as input to Seurat. Each library (M, FR, FNR) from each Esr1+ population was initially analyzed individually for quality control. Cells were retained if at least 500 Unique Molecular Identifiers (UMIs) were detected, while a gene was retained if ≥1 UMI was detected for that gene in ≥ 3 cells. The MeA FNR library contained a large number of barcodes with low UMI counts that likely corresponded to empty droplets. To eliminate these prior to analysis, we used the EmptyDrops function from the DropUtils package to differentiate high-quality cells from ambient RNA using 100,000 iterations and a FDR cutoff of p <0.0001 (Griffiths et al., 2018; Lun et al., 2019), after which identical criteria were used for retention of genes and cells. Count data was variance stabilized in Seurat using the sctransform function (Hafemeister and Satija, 2019). The abundant non-coding RNA Malat1 accounted for 1–4% of all UMIs/cell and showed systematic differences in relative abundance between libraries (not shown); we therefore calculated the percentage of reads mapping to this gene for each cell (%Malat1) to use as a regression variable for sctransform. The top 3000 variable features were identified and used as input for principal component analysis (PCA) followed by identification of cell types using the Jaccard-Louvain community detection algorithm. An initial round of clustering was performed on individual libraries using the top 30 PCs and a resolution of 1.5 to identify non-neuronal cell types and cell types consisting of low-quality cells or probable doublets. We identified and removed non-Esr1+, non-neuronal cell types based on enriched expression of the genes Plp1, Mbp, Inpp5d, and Cxcrl. We filtered cell types consisting of low-quality cells or probable doublets based on one or more of the following criteria: uniformly low UMI count (median >2-fold lower than that of all other cell types), high mitochondrial content (any mitochondrial transcript detected in >25% of cells), and/or lack of significantly enriched markers relative to all other cells. After removing such cell types we merged all libraries from all conditions and visualized all cells as an UMAP on the top 50 PCs to check for batch effects. Libraries were then split by Esr1+ population and M, FR and FNR merged into Seurat. The sctransform function was run on the merged libraries with %Malat1 and library of origin included as regression variables to correct for batch effects. For each Esr1+ population the top 3,000 variable genes of the merged set of cells were identified, filtered to remove Y-chromosome genes, mitochondrial genes and the X-inactivation-related transcripts Xist and Tsix, and used as input for PCA and graph-based clustering by the Jaccard-Louvain algorithm (Hafemeister and Satija, 2019). We performed an initial round of clustering on the merged libraries using the top 30 PCs and a clustering resolution of 1.5. We then removed any residual cell types consisting of low-quality cells, non-neuronal cells, and any cell types in which <25% of cells expressed Esr1. Libraries were filtered iteratively, with sctransform and PCA re-run at each filtering step, until all remaining cell types were neuronal and expressed Esr1 in >25% of cells. Following the final filtering iteration, we identified a consensus set of 137 Esr1+ cell types distributed across the four Esr1+ populations using 30 PCs and a resolution of 1.2. For the VMHvl, we removed cell types localized to the arcuate nucleus or ventral premamillary nucleus (PMV) based on expression of the following markers: Rxfp1, Avp, Pomc, Foxa1, Sall3 and Hcrt; for the BNSTpr, we removed a cell type of likely thalamic origin expressing Drd3. These markers and their expression patterns were obtained from images from the Allen mouse brain atlas (Lein et al., 2007).
To evaluate cell type stability we used the scclusteval package to calculate the Jaccard Similarity Index (Tang et al., 2020). We systematically tested a range of PCs (10, 20, 30 and 40) and resolution parameters (0.6–1.6 in increments of 0.2). For each combination of PC number and resolution we performed 20 bootstraps wherein we randomly subsampled 80% of the cells, identified cell types using the same parameters as for the full dataset, and matched the subsampled cell types to those identified in the full set of cells using the Jaccard Similarity Index. Cell types were classified as ‘stable’ if the median Jaccard Similarity Index across 20 bootstraps was >0.6.
Alignment of snRNAseq data with published datasets
To determine the degree to which our cell types aligned with published datasets profiling the same regions we obtained single nucleus RNAseq data from the BNST (Welch et al., 2019) and single cell RNAseq data from the MeA, POA, and VMH (Chen et al., 2019; Kim et al., 2019; Moffitt et al., 2018) (see Key Resources Table). For each dataset, count data was variance stabilized in Seurat using the sctransform function (Hafemeister and Satija, 2019). The top 3000 variable features were identified and used as input for PCA followed by identification of cell types using the Jaccard-Louvain community detection algorithm. For VMH and MeA datasets we filtered low-quality and non-neuronal cells exactly as described for our own datasets. The BNST dataset was available as a pre-filtered set of neurons, while the authors of the POA dataset provided cell type annotations detailing which cells were neuronal and nonneuronal, so we did not perform any preliminary filtering apart from excluding non-neuronal cells. For each region we aligned neurons from the external dataset with our corresponding snRNAseq data using Canonical Correlation Analysis (CCA) as implemented in Seurat (Butler et al., 2018). As integration features, we used the union of the top 3000 variable features identified in each dataset. We then projected the joint dataset into low-dimensional UMAP space, identified cell types, and determined the proportion of cells from each dataset present in each cell type. For the BNST, POA and VMHvl, we found that our Esr1+ cell types co-clustered with cells in the external dataset such that the cell types we report matched cell types present in the joint analyses. Notably, both the VMHvlCckar/Esr1 and BNSTprTac1/Esr1 cell types were observed with this approach. Each external dataset contained Esr1− cells that did not correspond to any cell type present in our dataset. For the MeA, 12 cell types that were present in our dataset had no clear match in the external dataset, because the external dataset (Chen et al., 2019) had extremely low sequencing depth (lower than the external and our datasets for the BNST, POA, and VMH as well as our dataset for the MeA).
Further annotation of cell types
We first classified cell types expressing Slc17a6 or Slc17a7 in >25% of cells as excitatory neurons and those expressing Gad1 in >25% as inhibitory neurons. We further classified them as aromatase+ neurons if they expressed Aromatase (Cyp19a1) in >25% of cells and as Tyrosine hydroxylase+ neurons if they expressed Th in > 25% of cells. To identify genes enriched in individual cell types, we used Model-based Analysis of Single-cell Transcriptomics (MAST), as implemented in the FindAllMarkers function in Seurat with the following parameters: min.pct=0.25, lgfc.thesh=0.223, min.diff.pct=0.25, test.use=”MAST” (Finak et al., 2015). This translates into enriched marker genes 1) being expressed in >25% of the cells in the cell type of interest, 2) exhibiting a 25-percentage point difference in number of cells expressing the gene between the cell type and all other cells, and 3) exhibiting a 1.25-fold difference in log-normalized expression per cell in that cell type versus all other cells (Table S6). To assign marker gene annotations to cell types, we sought to identify a gene that met these criteria in the cell type of interest and no others within its corresponding Esr1+ population. We annotated cell types lacking such genes based on marker genes enriched in as few other cell types as possible. All markers used to annotate cell types were identified as being expressed in both TRAPseq and snRNAseq datasets. To identify DEGs enriched in BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types, we selected the intersection of genes enriched in these cell types with DEGs identified in their respective regions (Table S7).
Mapping DEGs on to cell types
We defined a DEG as ‘expressed’ in a cell type if at least one UMI was detected in ≥10% of cells in that cell type and used this criterion to generate UMAPs showing the distribution of DEGs across all cell types in each Esr1+ population (Fig. 5D). In order to determine whether individual cell types expressed disproportionately more DEGs in one sex or estrous comparison (Fig. 5E), we performed the following analysis for each cell type within a given Esr1+ population. We first normalized the number of DEGs upregulated in both conditions for each comparison (M v FR; M v FNR; FR v FNR) and then obtained the ratio of the two conditions for that particular comparison. This ratio for each cell type was scaled across all cell types for that Esr1+ population to obtain a z-score. To assess the number of cell types a DEG was expressed in (Fig. S5A), we used the same criterion as above to define DEG expression within a cell type (≥1 UMI in ≥10% cells in the cell type). To determine whether individual DEGs were significantly enriched or depleted in a given cell type, we calculated scaled (z-scored) expression of all DEGs across all cell types in each Esr1+ population and visualized it as a heatmap (Fig. S5B). Columns (DEGs) were hierarchically clustered in pheatmap using default parameters to highlight sets of DEGs enriched in individual cell types.
Identification of condition-biased cell types
To identify condition-biased cell types in each Esr1+ population we examined the percentage of cells from each condition (M, FR, FNR) assigned to that cell type. We classified cell types as condition-biased if they met the following criteria: 1) significant difference (p<0.05) in counts of cells assigned between all libraries (Fisher’s exact Test) 2) >3-fold difference in percentage of cells contributing to the cell type between the most-represented and least-represented conditions 3) comprised at least 1% of total cells in the most-represented condition (Table S5).
Identification of sex differences within BNSTprTacl/Esr1 cell type
To identify sex differences within the BNSTprTac1/Esr1 cell type, read counts in all cells in the BNSTpr dataset were downsampled to a maximum of 40,000 UMIs and filtered to omit cells with <7,500 UMIs to minimize the influence of batch differences in sequencing depth and gene detection. The BNSTprTac1/Esr1 was split by condition, and DESeq2 was run on each pairwise comparison using default parameters, with individual cells treated as replicates. We used DESeq2 to run a Benjamini-Hochberg FDR correction only on BNSTpr DEGs, thereby effectively restricting our search to genes identified as sDEGs and eDEGs by TRAPseq. We used a cutoff of FDR-adjusted p value < 0.05 and no FC cutoff for this analysis. Genes with strong DE patterns between conditions patterns were visualized as violin plots. A complete list of DEGs differentially expressed specifically in BNSTprTac1/Esr1 neurons is available in Table S7.
To further define sex differences in the BNSTprEsr1 population, we used the AddModuleScore function in Seurat to calculate relative expression of sets of DEGs across cell type. We used the set of BNST DEGs upregulated in males compared to females (M score) or females compared to males (F score) as a feature set. Using this method we observed that BNSTTac1/Esr1 neurons exhibit a higher M score (enrichment of male-upregulated genes) compared to nearly all other BNSTprEsr1 cells (data not shown). The BNSTTac1/Esr1 cell type also contained the largest set of sDEGs when each cell type within the BNSTEsr1 population was examined for sDEGs between the two comparisons (M v FR and M v FNR).
Spatial analysis of BNSTprTac1/Esr1 and VMHvlckar/Esr1 cell types
To analyze the BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types in vivo, we used dual-color ISH to visualize Tac1/Esr1 and Cckar/Esr1 expression in the BNSTpr and VMHvl, respectively. To quantify the overlap between Esr1 and Tac1 or Cckar we manually counted cells expressing either or both genes and calculated the fraction of Tac1+ or Cckar+ cells that were also Esr1+ (n=2 animals/gene). We analyzed the spatial distribution of the BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types by comparing the rostrocaudal and dorsoventral distribution of Tac1 and Cckar expression with that of Esr1. The rostrocaudal distribution of the cell types was assessed by manually counting the number of cells expressing each gene (Esr1, Cckar or Tac1) per 50 μm section across the full extent of the region of interest (6 sections for the BNSTpr and 7 sections for the VMHvl at 50 μm thickness in the coronal plane) and calculating the percent of total cells expressing the gene present in each section. We then assessed the dorsoventral distribution of each cell type by manually counting the number of cells expressing the marker gene in the dorsal, middle and ventral thirds (BNSTpr) or dorsal and ventral halves (VMHvl) of the relevant region in each section (n=2 animals/region). The spatial distribution of BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types based on these analyses is summarized in Table S7.
Generation of PRFlpo, Esr1Flpo and CckarCre mice
PRFlpo, Esr1Flpo and CckarCre mice were generated using CRISPR/Cas9-mediated homologous recombination (Miura et al., 2018). Guide RNAs (gRNAs) were designed using the Integrated DNA Technology ALT-R CRISPR HDR design tool (idtdna.com/pages/tools/alt-r-crispr-hdr-design-tool). Targeting constructs were designed to contain 100 bp of 5’ and 3’ homology arms flanking the 2A-Flpo or 2A-Cre sequences. Homology arms were designed to target the 3’UTR of each gene (corresponding to exon 10 for Esr1, exon 5 for Cckar and exon 8 for PR). Single-stranded DNA (ssDNA) doners were co-injected with trans-activating CRISPR RNA (tracrRNA) at the Gladstone Institute (San Francisco, CA). Candidate founders were screened by PCR and backcrossed to C57BL6/J >3 generations prior to being used for behavioral experiments. To quantify overlap between PR and Flpo or Cckar and Cre, we performed dual color ISH and quantified overlap using the dotdotdot package (Maynard et al., 2020).
Generation of INTRSECT viral vectors
Subcloning
INTRSECT variants of DREAADi-mCherry were produced and validated as previously described (Fenno et al., 2020). Briefly, the reading frame was split into three pieces (referred to as ‘exon 1’, ‘exon 2’, and ‘exon 3’) after being analyzed for naturally occurring splice-site-like sequences using a public bioinformatics tool (http://www.cbs.dtu.dk/services/NetGene2/) (Brunak et al., 1991) and choosing a splice site in each of the reading frames, designed as possible to have the splice sites disrupt the reading frame to decrease the chance of partial protein synthesis from exon 2 or exon 3 in the absence of the pre-defined expression criteria. A derivative of the CMV Towne Variant intron B (Fenno et al., 2014) containing a cDIO cassette was inserted between exon 1 and exon 2, with the donor and acceptor sites fused directly to the 3’ terminus of exon 1 and 5’ terminus of exon 2, respectively. A derivative of the mouse IgE intron 3 (Fenno et al., 2014) containing a cDIO cassette was inserted between exon 2 and exon 3, with the donor and acceptor sites fused directly to the 3’ terminus of exon 2 and 5’ terminus of exon 3, respectively. Separate fDIO cassettes were added directly after the promoter (5’ to the entire coding sequence) and directly before the WPRE (3’ to the entire coding sequence). To generate Coff/Fon-hM4Di-mCherry, the exon order is exon 3, exon 2, exon 1, with all exons in the reverse complement orientation. All of these plasmids are constructed in an AAV-nEF backbone with a 3’ WPRE.
mRNA isolation and cDNA synthesis
HEK293FT cells at 50% confluence were transfected with endotoxin-free DNA using Lipofectamine 3000 (Thermo Fisher) following the manufacturer’s protocol. Five days post-transfection, RNA extraction was performed using reagents from the RNeasy Mini Kit (Qiagen). Cells were disrupted with lysis buffer and homogenized using QiaShredder homogenizer columns. Combined first-strand cDNA/PCR using the SuperScript III One-Step RT-PCR System (Thermo Fisher) was performed with the following initial reaction conditions: 45 °C .30 min, 94 °C .2 min, 40 cycles of 94 °C .15 s, 45 °C .30 s, 68 °C .1 min, ending with 68 °C .5 min using primers (Forward 5’-CATCAGCTTTGACCGCTACTTC-3’; Reverse 5’-CTTCAGCTTCAGCCTCTGCT-3’). The PCR product was electrophoresed on a 0.8% agarose gel, photographed, and DNA bands purified from the gel and sequenced to determine splice junctions.
Flow cytometry
HEK293FT cells (Thermo Fisher) were grown in 24-well tissue culture plates to 50% confluence and transfected in duplicate with 500 ng total DNA with Lipofectamine 3000 (Thermo Fisher) following the manufacturer protocol. Five days post transfection, cells were removed by enzymatic dissociation (TrypLE, Gibco), resuspended in cold PBS, pelleted at 200g for 5 min and resuspended in 500 μL PBS supplemented with 5μM 4’6-diamidino-2-phenylindole (DAPI; Thermo Fisher), and then placed on ice under aluminum foil until analysis. Flow cytometry was completed on a Novocyte Quanteon analyzer at the Stanford Shared FACS Facility using settings optimized for side scatter (SS), forward scatter (FS), vital dye (DAPI) and fluorophore (mCherry) acquisition using positive (non-recombinase-dependent DREADDi-mCherry), negative (empty transfection) and dead (heat-killed; 95 °C for 3 min) conditions as controls. Live-cell, singlet populations used in comparisons were isolated from debris and dead cells in post hoc analysis using FlowJo 10.7.2 (FlowJo) by (i) positively gating for the high-density population in plotting FS vs. SS, (ii) positively gating for singlets, and (iii) negatively gating for vital dye+ cells.
Chemogenetic studies
Chemogenetic studies were performed exactly as described previously (Bayless et al., 2019; Inoue et al., 2019; Yang et al., 2017). Clozapine N-Oxide (CNO; Enzo) was dissolved in sterile saline at 5 mg/mL and kept as single use frozen aliquots. On the day of behavioral testing, CNO was freshly diluted in sterile saline to achieve the following doses: 1 mg/kg body weight (Tac1Cre and Tac1Cre;Esr1Flpo), 15 mg/kg (female CckarCre and CckarCre;PRFlpo), 3 mg/kg (male CckarCre) and 5 mg/kg (male CckarCre;PRFlpo). These doses of CNO were optimized for each set of experiments and are within the ranges used in previous studies (Bayless et al., 2019; Inoue et al., 2019; Ray et al., 2011; Sasaki et al., 2011; Unger et al., 2015; Yang et al., 2017). Experimental mice were injected intraperitoneally (IP) with CNO or sterile saline 30 min (experimental males) or 90 min (experimental females) prior to behavioral assays. Mice were tested on each behavioral assay once each with CNO and saline, with the order of CNO and saline administration counterbalanced across animals.
Stereotaxic surgery
Viruses were delivered into brains of mice at 10–16 weeks of age exactly as described previously (Bayless et al., 2019; Inoue et al., 2019; Yang et al., 2017). The following volumes of virus were injected bilaterally: 0.5 μl (BNSTpr), 1 μl (male VMHvl), 0.3 μl (female VMHvl). For the BNSTpr we used the following previously established coordinates: ±0.85 mm mediolateral, −0.20 mm, and −4.30 mm dorsoventral relative to bregma. For the VMHvl we used the following coordinates: ±0.78 mm mediolateral, −5.75 mm dorsoventral, 1.3 mm anteroposterior. Following surgery, mice were allowed to recover individually over a heat pad and then returned to their home cages. Females used for mating behavior assays or viral tracing studies were ovariectomized at the time of stereotaxic surgery. Animals were allowed at least 2 weeks of recovery following surgery prior to being tested in behavioral assays.
Behavioral Assays
All behavioral assays were performed in the dark cycle (≥1 hr after lights out) and recorded using a webcam under infrared illumination. Videos were played at 30 frames per second and manually annotated using custom software as described previously (Bayless et al., 2019; Inoue et al., 2019; Juntti et al., 2010; Wu et al., 2009; Xu et al., 2012; Yang et al., 2017).
Urine preference assay
Prior to testing, males were single housed for ≥ 7 days. Males were tested on the pheromone preference assay once each with CNO and saline injected 30 min prior to the assay, with the order of CNO and saline administration counterbalanced across animals. During the preference assay, two 1”x1” cotton swabs (one wetted with 80 μl of undiluted, WT, group housed, hormonally primed C57BL/6J female urine and the other wetted with 80 μl of undiluted WT, group housed C57BL/6J male urine) were placed at opposite ends of their home cage. Males were given 5 min to investigate the urine swabs, and the duration, latency, and number of sniffs directed to each swab were scored. Following completion of this test, males were tested for performance in mating and aggression.
Male mating and territorial aggression assays
Experimental males were tested for mating behavior once each with CNO and saline injected 30 min prior to the assay, with the order of CNO and saline administration counterbalanced across animals. A WT group housed, hormonally primed receptive female was inserted into the home cage of a singly housed male for 30 min. For territorial aggression, we used the resident-intruder paradigm in which a WT group-housed male was inserted into the home cage of a singly housed experimental male for 15 min. Males were tested for aggression behavior once each with CNO and saline injected 30 min prior to the assay, with the order of CNO and saline administration counterbalanced across animals. Latency to initiate mating or aggression was defined as the first instance of mounting or physical attack from the start of the test, and in tests where mating or aggression did not occur, we used the maximal duration of the behavioral test (1,800 s for mating and 900 s for aggression). Aggression was scored as occurring when we observed physical attacks (episodes of biting, wrestling, tumbling, chasing).
Female sexual behavior
Female mice can exhibit low levels of sexual receptivity during the first mating experience (Thompson and Edwards, 1971; Xu et al., 2012), and to reduce variability in behavior, we subjected all experimental females to a round of mating experience with a WT sexually experienced male. In this setting, we primed the female, inserted her into home cage of a singly housed male for 30 min, and verified that mounting and intromission occurred. Following this, females were tested and analyzed for behavioral performance in mating tests with WT sexually experienced males. Lordosis was defined by the display of a dorsiflexed back while braced on all four legs during a mount or intromission. In contrast to the lordosis quotient, the receptivity index measures whether or not the female allows mounts to proceed to intromission. Rejection behavior was defined by the display of postures (running or walking away and rearing up against male) during mounting that delayed or precluded subsequent mating events.
Maternal behaviors
Following recovery from surgery, females used for tests of maternal behavior were housed with a WT male until visibly pregnant. Males were then removed and females allowed to deliver their litter in single housing. To test performance in pup retrieval, all pups were briefly removed from the cage 2 and 4 days following birth and 4 of them were subsequently placed separately in each cage corner. Pup retrieval was assayed for 5 min, allowing sufficient time for mothers to retrieve all pups. Following the assay, remaining pups were returned to the cage. To assay maternal aggression, pups at postnatal day 6 and 8 were removed, and an unfamiliar WT group housed male was inserted into the cage for 15 min. Pups were returned to the cage following completion of the assay. Maternal aggression was defined as physical attacks towards the intruder male (episodes of biting, wrestling, tumbling, chasing).
Projection mapping studies
To analyze projection termini of VMHvlCckar/Esr1 and VMHvlCckar−/Esr1 neurons, sections containing AVPV, POA, and PAG were collected, immunolabeled for mCherry, and imaged with a confocal microscope using a 63x objective, with image stacks containing 3 optical sections at 4 μm intervals. This imaging protocol was chosen to rigorously image individual presynaptic termini (Inoue et al., 2019). Sections containing VMHvl were imaged using a 20x objective, with image stacks containing 5 optical sections at 10 μm intervals. The number of presynaptic termini was enumerated using Image J software (Analyze Particle plugin) and divided by the area imaged to obtain the density of these termini in any given region. This estimate of the density of presynaptic termini was then normalized to the number of mCherry+ VMHvlCckar/Esr1 or VMHvlCckar−/Esr1 neurons to correct for subtle variations in infection in each animal.
To label AVPV-projecting VMHvlCckar/Esr1 and VMHvlCckar−/Esr1 neurons, AAV-EFla-Flex-Syp:mCherry or AAV-EF1a-Coff-Syp:mCherry were injected into the VMHvl of CckarCre female mice. One week after AAV injection, we injected 0.3 μL of the retrograde tracer CTB (conjugated to Alexa Fluor 488) into the AVPV. Mice were perfused 5 days after CTB injection. The middle three sections through the VMHvl were imaged by confocal microscopy using a 63x objective, with image stacks containing 5 optical sections at 1 pm intervals. The number of CTB/mCherry+ cells in the VMHvl was quantified using Photoshop (manual counting) and ImageJ software (Analyze Particle plugin for automated enumeration). CTB+ VMHvlCckar/Esr1 or VMHvlCckar−/Esr1 neurons are represented as a fraction of the CTB+ neuron counts in the mCherry+ neurons of WT females.
QUANTIFICATION AND STATISTICAL ANALYSIS
TRAPseq and snRNAseq data
All statistical analyses for sequencing data were performed in R v3.5.1 or R v3.6.3. Statistical tests and criteria used for TRAPseq and snRNAseq analyses are described in the relevant Method Details sections. The number of biological replicates for each experiment is stated in the main text or relevant figure legends.
Behavioral and projection mapping data
Behavioral video recordings and histological samples were analyzed blind to relevant variables, including sex, genotype, CNO administration, surgical procedure, and virus injected. Statistical analysis was performed using GraphPad PRISM (GraphPad Software). To compare categorical data, including percentage of mice displaying a behavior, Fisher’s exact test was performed from a 2×2 contingency table. To compare non-categorical data, we first determined if the data values were from a normal distribution using the D’Agostino-Pearson omnibus normality test. In experiments with paired samples, we used a paired t test or repeated measures ANOVA for parametric data and a Wilcoxon matched-pairs signed rank test or Friedman test for non-parametric data. In all other experiments, we used a t test or ANOVA for parametric data and a Mann-Whitney or Kruskal-Wallis test for non-parametric data. Differences in synaptic bouton density in the AVPV were tested using a Mann-Whitney U-test. All multiple comparisons were corrected for using Bonferroni corrections. The number of biological replicates for each experiment is stated in the text or figure legends.
Supplementary Material
A. Schematic of workflow to generate FR and FNR mice.
B. Enrichment of RNA obtained following immunoprecipitation (IP) of the L22 ribosomal subunit encoded by the RiboTag allele from the BNSTpr of Esr1Cre;RiboTag (experimental) mice compared to RiboTag (control) mice. y-axis, Bioanalyzer fluorescence signal in arbitrary units (AU); x-axis, RNA size in nucleotides (nt).
C. Enrichment of Esr1 mRNA and depletion of GFAP mRNA following IP of dissected regions from Esr1Cre;RiboTag mice. n = 3 replicates. Mean ± SEM.
D. sDEGs are distributed across all autosomes and the X chromosome. M>F, sDEGs upregulated in M compared to FR or FNR in any Esr1+ population; F>M, sDEGs upregulated in FR or FNR compared to M in any Esr1+ population; Mixed, sDEGs upregulated in M compared to FR or FNR in ≥1 Esr1+ population and upregulated in FR or FNR compared to M in ≥1 Esr1+ population.
E. Number of sDEGs upregulated in females (top) or males (bottom) in one or more comparisons between M, FR, and FNR.
F. ISH for sDEGs in coronal sections. ISHs confirm TRAPseq data showing higher expression of Tac1, Lamp5, Nripl, Gm39653, Cckar, and Criml in M and other genes upregulated in FR or FNR mice. n = 2/condition/probe. Scale bar = 100 μm.
A. eDEGs are distributed across all autosomes and the X chromosome. FR > FNR, eDEGs upregulated in FR compared to FNR in ≥1 Esr1+ populations; FNR > FR, eDEGs upregulated in FNR compared to FR in ≥1 Esr1+ populations; Mixed, eDEGs upregulated in FR and FNR each in ≥1 Esr1+ population.
B. ISH for eDEGs. ISHs confirm TRAPseq data showing higher expression of Pgr15l and Esr1 in FNR and other genes upregulated in FR mice. n = 2/condition/probe. Scale bars = 100 μm.
C. Analysis of DEGs shows that Esr1+ populations from M, FR, and FNR occupy distinct regions in PCA space.
D. Distribution of all DEGs that pass FDR<0.05 regardless of fold change in expression difference. Dots represent genes with FDR-adjusted p value < 0.05 (colored) or > 0.05 (gray) between comparisons; triangles represent genes whose expression change between conditions exceeds the scale. Dashed black lines indicate 1.5 fold differential expression within the comparison. Colored numbers show number of DEGs upregulated in that condition and comparison. n = 3.
A. Schematic of genome scan for estrogen, progesterone, and androgen response elements (ERE, PRE, ARE) using the consensus DNA sequence motifs shown (left panel). Significant fold enrichment for these sex hormone response elements in sDEGs (middle panel) and eDEGs (right panel). Black box indicates no significant enrichment for sex hormone response element for DEGs in that Esr1+ population.
B. Significant enrichment of DEGs in specific Gene Ontology categories. Gray cell, no enrichment for Gene Ontology category.
C. Significant enrichment of DEGs in specific Disease Ontology categories for disease processes with sex-bias as well as some without any apparent sex-bias.
A. Schematic of snRNAseq workflow. Dashed oval contains GFP+ nuclei that were collected and sequenced.
B. Enrichment of GFP+ nuclei with Fluorescence Activated Nuclear Sorting (left) and total yield of GFP+ nuclei (right) per condition. Horizontal gray bar = Mean.
C. Enrichment of Esr1 and GFP and depletion of GFAP mRNA in GFP+ nuclei.
D. Violin plots with distribution of cell number in cell types from each of the four Esr1+ populations.
E. Universal manifold approximation and projection (UMAP) shows that Esr1+ cells cluster by region but not by batch or condition (sex or estrous stages).
F. Schematic of coronal section through VMHvl (left) and inhibitory (Vgat mRNA expressing) cells labeled in the VMHvl (right). (Image credit: Allen Institute). Scale bar = 100 μm.
G. Violin plots classifying cell types (rows) into major neuronal types: excitatory (Slc17a6, Slc17a7), inhibitory (Gad1), aromatase-expressing, estrogen receptor 2 (Esr2 or ERβ), and tyrosine hydroxylase (TH).
*BNSTTac1/Esr1 cell type; ‡VMHvlCckar/Esr1 cell type.
A. Distribution of DEGs across Esr1+ cell types.
B. Many DEGs (columns) show enrichment or depletion in specific excitatory or inhibitory cell types (rows) in each Esr1+ population.
C. A subset of cell types in each Esr1+ population displays sex or estrous stage-bias. Bar graphs show percent cells in individual cell types for each condition (M, FR, FNR). Violin plots to the right of each bar graph show whether the cell type is excitatory or inhibitory and highlight a marker gene enriched in that cell type.
A. Violin plots showing enriched sex and estrous stage-shared genes in the BNSTprTac1/Esr1 cell type (highlighted in yellow, #18).
B. Violin plots showing expression of genes in panel A in M, FR, and FNR conditions.
C. Scatter plot of M v FR sDEGs in the BNSTprEsr1 population. This panel is identical to the one shown in Fig. 1B except that we have highlighted Tac1 (black circle) in this plot.
D. Expression of mCherry+ (DREADDi) in BNSTpr of Tac1Cre and Tac1Cre;Esr1Flpo mice after delivery of AAV-flex-DREADDi and AAV-Coff/Fon-DREADDi to this region, respectively.
E. No change in net time spent investigating urine following chemogenetic inhibition of BNSTprTac1/Esr1 or BNSTprTac1−/Esr1 cell types.
F. Schematic showing CRISPR-mediated targeting of Flpo to the 3’ UTR of Esr1.
G. Flpo-dependent expression of virally encoded EYFP (AAV-fDIO-hChR2-EYFP) in BNSTpr neurons of Esr1Flpo mice shows extensive overlap between EYFP and Esr1-expressing cells.
H. PCR of Coff/Fon INTRSECT hM4Di-mCherry plasmid DNA generates an amplicon larger than WT, while PCR of cDNA from cells co-transfected with the same plasmid and Flpo recombinase generates an amplicon equivalent to WT; a smaller PCR product is also present in the WT version (not shown). cDNA amplicon sequences are seamless across the exon junctions.
I. Flow cytometry of cells transfected with INTRSECT DREADDi-mCherry and activating Flpo recombinase shows expression comparable to WT (constitutive DREADDi-mCherry) and more than that observed in control conditions (negative, no transfection; alone, INTRSECT transfection without recombinase; +Cre +Flpo, INTRSECT transfection with Cre and Flpo). Diminished, but residual, expression is observed post-transfection when co-transfected with Cre and Flpo.
J. Chemogenetic inhibition of BNSTprTac1/Esr1 cell type increases latency of resident males in initiating mounts or intromission with receptive females and attacks toward intruder males.
K. No change in latency of mating or aggression routines in resident males upon chemogenetic inhibition of BNSTprTac1−/Esr1 cell types.
Mean ± SEM. n = 2 (D), 4 (G), and 8 (J,K). Scale bars = 100 μm (D and left panel in G) and 10 μm (inset panels in G).
A. UMAP representation of male and female VMHvlEsr1 cell types (left and middle panels), showing that VMHvlCckar/Esr1 and VMHvlTrim36/Esr1 cell types (#20 and 25, respectively) are restricted to females. Right panel shows scatter plot of M v FR sDEGs in the VMHvlEsr1 population. This panel is identical to the one shown in Fig. 1B except that we have highlighted Cckar (black circle) in this plot.
B. Shared expression of genes between VMHvlCckar/Esr1 and VMHvlTrim36/Esr1 cell types.
C. Targeting strategy (top) and validation (bottom) of PRFlpo mice. CRISPR-mediated targeting of Flpo to the 3 ‘ UTR of Pr. Extensive overlap between PR and Cre revealed by ISH in pRFlpo/+ FR mice. n =2.
D. Targeting strategy (top) and validation (bottom) of CckarCre mice. CRISPR-mediated targeting of Cre to the 3’ UTR of Cckar. Extensive overlap between Cckar and Cre revealed by ISH in CckarCre/+ FR mice. n =2.
E. Expression of mCherry+ (DREADDi) in VMHvl of CckarCre and CckarCre;PRFlpo mice after delivery of AAV-flex-DREADDi and AAV-Coff/Fon-DREADDi to this region, respectively.
F. No change in pup retrieval upon chemogenetic inhibition of VMHvlCckar/Esr1 or VMHvlCckar−/Esr1 cell types.
G. No change in mating or aggression of resident males upon inhibition of the VMHvlCckar/Esr1 cell type.
H. Reduced mating by resident males upon inhibition of VMHvlCckar−/Esr1 cell types.
I. Reduced aggression by resident males upon inhibition of VMHvlCckar−/Esr1 cell types.
J. No change in density of mCherry+ termini of VMHvlCckar/Esr1 neurons in POA and PAG between FR and FNR mice.
K. No change in density of mCherry+ termini of VMHvlCckar− neurons in POA and PAG between FR and FNR mice.
Mean ± SEM. n = ≥8 (F), 9 (G), 12 (H,I), ≥7 (J), and 6 (K). Scale bars=100 μm.
All genes differentially expressed between sexes or FR and FNR in each of the four Esr1+ populations. All genes listed passed FDR < 0.05.
All genes differentially expressed in >1 comparison by condition (M, FR, FNR) or region (BNSTpr, MeA, POA, VMHvl). All genes listed passed FDR < 0.05.
Gene names and putative functions of all DEGs visualized by ISH.
Gene Ontology and Disease Ontology categories enriched in DEGs and references for sex-bias in Disease Ontology categories.
Analysis of snRNAseq libraries, including sequencing metrics (such as UMI, read depth, # of genes/cell), cell type composition, and Jaccard Similarity Index.
Genes enriched in BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types.
Summary of spatial distribution of these two cell types.
HIGHLIGHTS.
1,415 genes are dimorphic by sex or estrous state in 4 Esr1+ neuronal populations
All 137 Esr1+ cell types within these 4 populations express subsets of these genes
Only 1 male BNSTpr Esr1+ cell type is needed to recognize sexes, mate, and fight
Only 1 female VMHvl Esr1+ cell type has dynamic projections and is needed to mate
ACKNOWLEDGMENTS
We thank Liqun Luo for comments on the manuscript, Fabian Müller for help with data analysis, Shah lab members for discussions, and Lily Duong for administrative support. This work was supported by grants to CD (NSF GRFP 2018265046; PD Soros Fellowship for New Americans), KD (NSF NeuroNex Program and AE Foundation), NL (NIH F32MH125593; Walter V. and Idun Berry Postdoctoral Fellowship, Stanford), SP (HFSP), AR (CZI, CZF2019-002460; NIH U01DA052783, R01MH123047), NMS (R01NS049488, R01HD104565), and GW (Bio-X Undergraduate Summer Research Program, VPUE Major Grant). Nuclear sorting was done in the Stanford Shared FACS Facility (NIH S10RR025518-01).
Footnotes
DECLARATION OF INTERESTS
KD is a member of Cell’s Advisory Board. DWB, SI, JRK, and NMS are named Inventors on a patent application to be filed on the subject matter.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, and Packer A (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, and Rahnenfuhrer J (2021). topGO: Enrichment Analysis for Gene Ontology.
- Arnold AP (2009). The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm Behav 55, 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K, Watanabe K, Duistermars BJ, Hoopfer E, González CR, Eyjólfsdóttir EA, Perona P, and Anderson DJ (2014). Tachykinin-Expressing Neurons Control Male-Specific Aggressive Arousal in Drosophila. Cell 156, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, and Knickmeyer R (2011). Why are autism spectrum conditions more prevalent in males? PLoS Biol 9, e1001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayless DW, Yang T, Mason MM, Susanto AAT, Lobdell A, and Shah NM (2019). Limbic Neurons Shape Sex Recognition and Social Behavior in Sexually Naive Males. Cell 176, 1190–1205.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeman EA (1947). The effect of male hormone on aggressive behavior in mice. Physiol. Zool 20, 373–405. [DOI] [PubMed] [Google Scholar]
- Berger A, Tran AH, Dida J, Minkin S, Gerard NP, Yeomans J, and Paige CJ (2012). Diminished pheromone-induced sexual behavior in neurokinin-1 receptor deficient (TACR1(−/−)) mice. Genes Brain Behav. 11, 568–576. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brock O, Baum MJ, and Bakker J (2011). The development of female sexual behavior requires prepubertal estradiol. J Neurosci 31, 5574–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, and Knudsen S (1991). Prediction of human mRNA donor and acceptor sites from the DNA sequence. Journal of Molecular Biology 220, 49–65. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Hu RK, Wu YE, Pan L, Huang S, Micevych PE, and Hong W (2019). Sexually Dimorphic Control of Parenting Behavior by the Medial Amygdala. Cell 176, 1206–1221.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke BM, and Woolley CS (2005). Gonadal hormone modulation of dendrites in the mammalian CNS. J. Neurobiol. 64, 34–46. [DOI] [PubMed] [Google Scholar]
- Cooke B, Hegstrom CD, Villeneuve LS, and Breedlove SM (1998). Sexual differentiation of the vertebrate brain: principles and mechanisms. Front Neuroendocrinol 19, 323–362. [DOI] [PubMed] [Google Scholar]
- Correa SM, Newstrom DW, Warne JP, Flandin P, Cheung CC, Lin-Moore AT, Pierce AA, Xu AW, Rubenstein JL, and Ingraham HA (2015). An Estrogen-Responsive Module in the Ventromedial Hypothalamus Selectively Drives Sex-Specific Activity in Females. Cell Reports 10, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries GJ, and Boyle PA (1998). Double duty for sex differences in the brain. Behavioural Brain Research 92, 205–213. [DOI] [PubMed] [Google Scholar]
- Dederen PJ, Gribnau AA, and Curfs MH (1994). Retrograde neuronal tracing with cholera toxin B subunit: comparison of three different visualization methods. Histochem J 26, 856–862. [DOI] [PubMed] [Google Scholar]
- Dey S, Chamero P, Pru JK, Chien M-S, Ibarra-Soria X, Spencer KR, Logan DW, Matsunami H, Peluso JJ, and Stowers L (2015). Cyclic Regulation of Sensory Perception by a Female Hormone Alters Behavior. Cell 161, 1334–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias IC, Gutierrez-Castellanos N, Ferreira L, and Lima SQ (2021). The Structural and Electrophysiological Properties of Progesterone Receptor-Expressing Neurons Vary along the Anterior-Posterior Axis of the Ventromedial Hypothalamus and Undergo Local Changes across the Reproductive Cycle. ENeuro 8, ENEURO.0049–21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmann S, Sticht H, Thiel F, Beckmann MW, Strick R, and Strissel PL (2009). Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell. Mol. Life Sci. 66, 2405–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkner AL, Grosenick L, Davidson TJ, Deisseroth K, and Lin D (2016). Hypothalamic control of male aggression-seeking behavior. Nat. Neurosci. 19, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno LE, Mattis J, Ramakrishnan C, Hyun M, Lee SY, He M, Tucciarone J, Selimbeyoglu A, Berndt A, Grosenick L, et al. (2014). Targeting cells with single vectors using multiple-feature Boolean logic. Nat Methods 11, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenno LE, Ramakrishnan C, Kim YS, Evans KE, Lo M, Vesuna S, Inoue M, Cheung KYM, Yuen E, Pichamoorthy N, et al. (2020). Comprehensive Dual- and Triple-Feature Intersectional Single-Vector Delivery of Diverse Functional Payloads to Cells of Behaving Mammals. Neuron 107, 836–853.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Pinto MJ, Kanodia H, Falasconi A, Sigrist M, Esposito MS, and Arber S (2021). Functional diversity for body actions in the mesencephalic locomotor region. Cell 184, 4564–4578.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biology 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher RB, Das D, and Ngai J (2018). Creating Lineage Trajectory Maps Via Integration of Single-Cell RNA-Sequencing and Lineage Tracing: Integrating transgenic lineage tracing and single-cell RNA-sequencing is a robust approach for mapping developmental lineage trajectories and cell fate changes. Bioessays 40, e1800056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsell C, Björk BF, Lilius L, Axelman K, Fabre SF, Fratiglioni L, Winblad B, and Graff C (2010). Genetic association to the amyloid plaque associated protein gene COL25A1 in Alzheimer’s disease. Neurobiol Aging 31, 409–415. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, and Carroll JS (2007). Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat Rev Cancer 7, 713–722. [DOI] [PubMed] [Google Scholar]
- Griffiths JA, Richard AC, Bach K, Lun ATL, and Marioni JC (2018). Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun 9, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasz J, Zelena D, Toth M, Tulogdi A, Mikics E, and Haller J (2009). Substance P neurotransmission and violent aggression: the role of tachykinin NK(1) receptors in the hypothalamic attack area. Eur J Pharmacol 611, 35–43. [DOI] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. (2014). Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front. Neural Circuits 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashikawa K, Hashikawa Y, Tremblay R, Zhang J, Feng JE, Sabol A, Piper WT, Lee H, Rudy B, and Lin D (2017). Esr1+ cells in the ventromedial hypothalamus control female aggression. Nat. Neurosci. 20, 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellier V, Brock O, Candlish M, Desroziers E, Aoki M, Mayer C, Piet R, Herbison A, Colledge WH, Prevot V, et al. (2018). Female sexual behavior in mice is controlled by kisspeptin neurons. Nat Commun 9, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EJ, Turner KJ, Fernandez JM, Cifuentes D, Ghosh M, Ijaz S, Jain RA, Kubo F, Bill BR, Baier H, et al. (2016). Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 89, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S, Harada N, Ito S, Takagi Y, and Maeda S (1998). Disruption of sexual behavior in male aromatase-deficient mice lacking exons 1 and 2 of the cyp19 gene. Biochem. Biophys. Res. Commun. 252, 445–449. [DOI] [PubMed] [Google Scholar]
- Hong W, Kim D-W, and Anderson DJ (2014). Antagonistic control of social versus repetitive self-grooming behaviors by separable amygdala neuronal subsets. Cell 158, 1348–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Yang R, Tantry A, Davis C-H, Yang T, Knoedler JR, Wei Y, Adams EL, Thombare S, Golf SR, et al. (2019). Periodic Remodeling in a Neural Circuit Governs Timing of Female Sexual Behavior. Cell 179, 1393–1408.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KK, Osakada T, Mori H, Miyasaka N, Yoshihara Y, Miyamichi K, and Touhara K (2017). A Labeled-Line Neural Circuit for Pheromone-Mediated Sexual Behaviors in Mice. Neuron 95, 123–137.e8. [DOI] [PubMed] [Google Scholar]
- Juntti SA, Tollkuhn J, Wu MV, Fraser EJ, Soderborg T, Tan S, Honda S-I, Harada N, and Shah NM (2010). The androgen receptor governs the execution, but not programming, of male sexual and territorial behaviors. Neuron 66, 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karigo T, Kennedy A, Yang B, Liu M, Tai D, Wahle IA, and Anderson DJ (2020). Distinct hypothalamic control of same- and opposite-sex mounting behaviour in mice. Nature 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouni E, Sakkas P, Zarros A, Skandali N, and Liapi C (2009). The involvement of substance P in the induction of aggressive behavior. Peptides 30, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Kim D-W, Yao Z, Graybuck LT, Kim TK, Nguyen TN, Smith KA, Fong O, Yi L, Koulena N, Pierson N, et al. (2019). Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell 179, 713–728.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl J, Babayan BM, Rubinstein ND, Autry AE, Marin-Rodriguez B, Kapoor V, Miyamishi K, Zweifel LS, Luo L, Uchida N, et al. (2018). Functional circuit architecture underlying parental behaviour. Nature 556, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kow L-M, and Pfaff DW (1998). Mapping of neural and signal transduction pathways for lordosis in the search for estrogen actions on the central nervous system. Behavioural Brain Research 92, 169–180. [DOI] [PubMed] [Google Scholar]
- Krause WC, and Ingraham HA (2017). Origins and Functions of the Ventrolateral VMH: A Complex Neuronal Cluster Orchestrating Sex Differences in Metabolism and Behavior. Adv Exp Med Biol 1043, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause WC, Rodriguez R, Gegenhuber B, Matharu N, Rodriguez AN, Padilla-Roger AM, Toma K, Herber CB, Correa SM, Duan X, et al. (2021). Oestrogen engages brain MC4R signalling to drive physical activity in female mice. Nature 599, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Kim D-W, Remedios R, Anthony TE, Chang A, Madisen L, Zeng H, and Anderson DJ (2014). Scalable control of mounting and attack by Esr1 + neurons in the ventromedial hypothalamus. Nature 509, 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, and Byrnes EJ (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445. [DOI] [PubMed] [Google Scholar]
- Li Y, Mathis A, Grewe BF, Osterhout JA, Ahanonu B, Schnitzer MJ, Murthy VN, and Dulac C (2017). Neuronal Representation of Social Information in the Medial Amygdala of Awake Behaving Mice. Cell 171, 1176–1190.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Boyle MP, Dollar P, Lee H, Lein ES, Perona P, and Anderson DJ (2011). Functional identification of an aggression locus in the mouse hypothalamus. Nature 470, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, Marioni JC, and participants in the 1st Human Cell Atlas Jamboree (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biology 20, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard KR, Tippani M, Takahashi Y, Phan BN, Hyde TM, Jaffe AE, and Martinowich K (2020). dotdotdot: an automated approach to quantify multiplex single molecule fluorescent in situ hybridization (smFISH) images in complex tissues. Nucleic Acids Research 48, e66–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, and Arnold AP (2011). Reframing sexual differentiation of the brain. Nat Neurosci 14, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHenry JA, Otis JM, Rossi MA, Robinson JE, Kosyk O, Miller NW, McElligott ZA, Budygin EA, Rubinow DR, and Stuber GD (2017). Hormonal gain control of a medial preoptic area social reward circuit. Nat Neurosci 20, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLOS Biology 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min K.H. (Joseph), da Veiga Beltrame E, Hjorleifsson KE, Gehring J, and Pachter L (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol 1–6. [DOI] [PubMed] [Google Scholar]
- Micevych P, and Sinchak K (2001). Estrogen and endogenous opioids regulate CCK in reproductive circuits. Peptides 22, 1235–1244. [DOI] [PubMed] [Google Scholar]
- Miller JA, Gouwens NW, Tasic B, Collman F, van Velthoven CT, Bakken TE, Hawrylycz MJ, Zeng H, Lein ES, and Bernard A (2020). Common cell type nomenclature for the mammalian brain. ELife 9, e59928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Quadros RM, Gurumurthy CB, and Ohtsuka M (2018). Easi -CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat Protoc 13, 195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, Rubinstein ND, Hao J, Regev A, Dulac C, et al. (2018). Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musatov S, Chen W, Pfaff DW, Kaplitt MG, and Ogawa S (2006). RNAi-mediated silencing of estrogen receptor {alpha} in the ventromedial nucleus of hypothalamus abolishes female sexual behaviors. Proc. Natl. Acad. Sci. U.S.A 103, 10456–10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Chester AE, Hewitt SC, Walker VR, Gustafsson JA, Smithies O, Korach KS, and Pfaff DW (2000). Abolition of male sexual behaviors in mice lacking estrogen receptors alpha and beta (alpha beta ERKO). Proc. Natl. Acad. Sci. U.S.A 97, 14737–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada T, Ishii KK, Mori H, Eguchi R, Ferrero DM, Yoshihara Y, Liberles SD, Miyamichi K, and Touhara K (2018). Sexual rejection via a vomeronasal receptor-triggered limbic circuit. Nat Commun 9, 4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, and Franklin KBJ (2003). The Mouse Brain in Stereotaxic Coordinates: Compact Second Edition, Second Edition (Academic Press; ). [Google Scholar]
- Phoenix CH, Goy RW, Gerall AA, and Young WC (1959). Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65, 369–382. [DOI] [PubMed] [Google Scholar]
- Popa SM, Clifton DK, and Steiner RA (2008). The role of kisspeptins and GPR54 in the neuroendocrine regulation of reproduction. Annu. Rev. Physiol. 70, 213–238. [DOI] [PubMed] [Google Scholar]
- Ray RS, Corcoran AE, Brust RD, Kim JC, Richerson GB, Nattie E, and Dymecki SM (2011). Impaired Respiratory and Body Temperature Control Upon Acute Serotonergic Neuron Inhibition. Science 333, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remedios R, Kennedy A, Zelikowsky M, Grewe BF, Schnitzer MJ, and Anderson DJ (2017). Social behaviour shapes hypothalamic neural ensemble representations of conspecific sex. Nature 550, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring JR (1944). The estrogen-progesterone induction of sexual receptivity in the spayed female mouse. Endocrinology 34, 269–275. [Google Scholar]
- Rissman EF, Early AH, Taylor JA, Korach KS, and Lubahn DB (1997). Estrogen receptors are essential for female sexual receptivity. Endocrinology 138, 507–510. [DOI] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. U.S.A. 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Suzuki M, Mieda M, Tsujino N, Roth B, and Sakurai T (2011). Pharmacogenetic Modulation of Orexin Neurons Alters Sleep/Wakefulness States in Mice. PLOS ONE 6, e20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriml LM, Mitraka E, Munro J, Tauber B, Schor M, Nickle L, Felix V, Jeng L, Bearer C, Lichenstein R, et al. (2019). Human Disease Ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Res 47, D955–D962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scordalakes EM, and Rissman EF (2003). Aggression in male mice lacking functional estrogen receptor alpha. Behav. Neurosci 117, 38–45. [PubMed] [Google Scholar]
- Shapiro H (1937). Effect of Testosterone Propionate on Mating. Nature 139, 588–589. [Google Scholar]
- Siegel A, Schubert KL, and Shaikh MB (1997). Neurotransmitters regulating defensive rage behavior in the cat. Neurosci Biobehav Rev 21, 733–742. [DOI] [PubMed] [Google Scholar]
- Sisk CL, and Zehr JL (2005). Pubertal hormones organize the adolescent brain and behavior. Front Neuroendocrinol 26, 163–174. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2016). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al. (2016). The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Current Protocols in Bioinformatics 54, 1.30.1–1.30.33. [DOI] [PubMed] [Google Scholar]
- Tang M, Kaymaz Y, Logeman BL, Eichhorn S, Liang ZS, Dulac C, and Sackton TB (2020). Evaluating Single-Cell Cluster Stability Using The Jaccard Similarity Index. Bioinformatics btaa956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson ML, and Edwards DA (1971). Experiential and strain determinants of the estrogen-progesterone induction of sexual receptivity in spayed female mice. Hormones and Behavior 2, 299–305. [Google Scholar]
- Toda K, Saibara T, Okada T, Onishi S, and Shizuta Y (2001). A loss of aggressive behaviour and its reinstatement by oestrogen in mice lacking the aromatase gene (Cyp19). J. Endocrinol. 168, 217–220. [DOI] [PubMed] [Google Scholar]
- Todd WD, Fenselau H, Wang JL, Zhang R, Machado NL, Venner A, Broadhurst RY, Kaur S, Lynagh T, Olson DP, et al. (2018). A hypothalamic circuit for the circadian control of aggression. Nat. Neurosci. 21, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Xu Y, Scearce-Levie K, Ptácek LJ, and Fu Y-H (2010). COL25A1 triggers and promotes Alzheimer’s disease-like pathology in vivo. Neurogenetics 11, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger EK, Burke KJ, Yang CF, Bender KJ, Fuller PM, and Shah NM (2015). Medial amygdalar aromatase neurons regulate aggression in both sexes. Cell Rep 10, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen JE, Kammel LG, Bunda PC, Shum M, Reid MS, Massa MG, Arneson DV, Park JW, Zhang Z, Joseph AM, et al. (2020). Hypothalamic oestrogen receptor alpha establishes a sexually dimorphic regulatory node of energy expenditure. Nat Metab 2, 351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y-C, Wang S-R, Jiao Z-L, Zhang W, Lin J-K, Li X-Y, Li S-S, Zhang X, and Xu X-H (2018). Medial preoptic area in mice is capable of mediating sexually dimorphic behaviors regardless of gender. Nat Commun 9, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, and Macosko EZ (2019). Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177, 1873–1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling DM, and Geschwind DH (2013). Sex differences in autism spectrum disorders. Curr Opin Neurol 26, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey HR, Exner CRT, Xu Y, Everitt A, Sun N, Wang B, Dea J, Schmunk G, Zaltsman Y, Teerikorpi N, et al. (2021). Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 109, 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MV, and Tollkuhn J (2017). Estrogen receptor alpha is required in GABAergic, but not glutamatergic, neurons to masculinize behavior. Hormones and Behavior 95, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MV, Manoli DS, Fraser EJ, Coats JK, Tollkuhn J, Honda S-I, Harada N, and Shah NM (2009). Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 139, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Autry AE, Bergan JF, Watabe-Uchida M, and Dulac CG (2014). Galanin neurons in the medial preoptic area govern parental behaviour. Nature 509, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Coats JK, Yang CF, Wang A, Ahmed OM, Alvarado M, Izumi T, and Shah NM (2012). Modular genetic control of sexually dimorphic behaviors. Cell 148, 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CF, and Shah NM (2014). Representing sex in the brain, one module at a time. Neuron 82, 261–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CF, Chiang MC, Gray DC, Prabhakaran M, Alvarado M, Juntti SA, Unger EK, Wells JA, and Shah NM (2013). Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Yang CF, Chizari MD, Maheswaranathan N, Burke KJ, Borius M, Inoue S, Chiang MC, Bender KJ, Ganguli S, et al. (2017). Social Control of Hypothalamus-Mediated Male Aggression. Neuron 95, 955–970.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Bergan J, Lanjuin A, and Dulac C (2017). Oxytocin signaling in the medial amygdala is required for sex discrimination of social cues. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Yan G-R, and He Q-Y (2015). DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 31, 608–609. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Reis FMCV, He Y, Park JW, DiVittorio JR, Sivakumar N, van Veen JE, Maesta-Pereira S, Shum M, Nichols I, et al. (2020). Estrogen-sensitive medial preoptic area neurons coordinate torpor in mice. Nat Commun 11, 6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Rammohan K, Lin S, Robinson N, Li O, Liu X, Bai X, Yin L, Scarberry B, Du P, et al. (2003). CD24 is a genetic modifier for risk and progression of multiple sclerosis. Proc Natl Acad Sci U S A 100, 15041–15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Schematic of workflow to generate FR and FNR mice.
B. Enrichment of RNA obtained following immunoprecipitation (IP) of the L22 ribosomal subunit encoded by the RiboTag allele from the BNSTpr of Esr1Cre;RiboTag (experimental) mice compared to RiboTag (control) mice. y-axis, Bioanalyzer fluorescence signal in arbitrary units (AU); x-axis, RNA size in nucleotides (nt).
C. Enrichment of Esr1 mRNA and depletion of GFAP mRNA following IP of dissected regions from Esr1Cre;RiboTag mice. n = 3 replicates. Mean ± SEM.
D. sDEGs are distributed across all autosomes and the X chromosome. M>F, sDEGs upregulated in M compared to FR or FNR in any Esr1+ population; F>M, sDEGs upregulated in FR or FNR compared to M in any Esr1+ population; Mixed, sDEGs upregulated in M compared to FR or FNR in ≥1 Esr1+ population and upregulated in FR or FNR compared to M in ≥1 Esr1+ population.
E. Number of sDEGs upregulated in females (top) or males (bottom) in one or more comparisons between M, FR, and FNR.
F. ISH for sDEGs in coronal sections. ISHs confirm TRAPseq data showing higher expression of Tac1, Lamp5, Nripl, Gm39653, Cckar, and Criml in M and other genes upregulated in FR or FNR mice. n = 2/condition/probe. Scale bar = 100 μm.
A. eDEGs are distributed across all autosomes and the X chromosome. FR > FNR, eDEGs upregulated in FR compared to FNR in ≥1 Esr1+ populations; FNR > FR, eDEGs upregulated in FNR compared to FR in ≥1 Esr1+ populations; Mixed, eDEGs upregulated in FR and FNR each in ≥1 Esr1+ population.
B. ISH for eDEGs. ISHs confirm TRAPseq data showing higher expression of Pgr15l and Esr1 in FNR and other genes upregulated in FR mice. n = 2/condition/probe. Scale bars = 100 μm.
C. Analysis of DEGs shows that Esr1+ populations from M, FR, and FNR occupy distinct regions in PCA space.
D. Distribution of all DEGs that pass FDR<0.05 regardless of fold change in expression difference. Dots represent genes with FDR-adjusted p value < 0.05 (colored) or > 0.05 (gray) between comparisons; triangles represent genes whose expression change between conditions exceeds the scale. Dashed black lines indicate 1.5 fold differential expression within the comparison. Colored numbers show number of DEGs upregulated in that condition and comparison. n = 3.
A. Schematic of genome scan for estrogen, progesterone, and androgen response elements (ERE, PRE, ARE) using the consensus DNA sequence motifs shown (left panel). Significant fold enrichment for these sex hormone response elements in sDEGs (middle panel) and eDEGs (right panel). Black box indicates no significant enrichment for sex hormone response element for DEGs in that Esr1+ population.
B. Significant enrichment of DEGs in specific Gene Ontology categories. Gray cell, no enrichment for Gene Ontology category.
C. Significant enrichment of DEGs in specific Disease Ontology categories for disease processes with sex-bias as well as some without any apparent sex-bias.
A. Schematic of snRNAseq workflow. Dashed oval contains GFP+ nuclei that were collected and sequenced.
B. Enrichment of GFP+ nuclei with Fluorescence Activated Nuclear Sorting (left) and total yield of GFP+ nuclei (right) per condition. Horizontal gray bar = Mean.
C. Enrichment of Esr1 and GFP and depletion of GFAP mRNA in GFP+ nuclei.
D. Violin plots with distribution of cell number in cell types from each of the four Esr1+ populations.
E. Universal manifold approximation and projection (UMAP) shows that Esr1+ cells cluster by region but not by batch or condition (sex or estrous stages).
F. Schematic of coronal section through VMHvl (left) and inhibitory (Vgat mRNA expressing) cells labeled in the VMHvl (right). (Image credit: Allen Institute). Scale bar = 100 μm.
G. Violin plots classifying cell types (rows) into major neuronal types: excitatory (Slc17a6, Slc17a7), inhibitory (Gad1), aromatase-expressing, estrogen receptor 2 (Esr2 or ERβ), and tyrosine hydroxylase (TH).
*BNSTTac1/Esr1 cell type; ‡VMHvlCckar/Esr1 cell type.
A. Distribution of DEGs across Esr1+ cell types.
B. Many DEGs (columns) show enrichment or depletion in specific excitatory or inhibitory cell types (rows) in each Esr1+ population.
C. A subset of cell types in each Esr1+ population displays sex or estrous stage-bias. Bar graphs show percent cells in individual cell types for each condition (M, FR, FNR). Violin plots to the right of each bar graph show whether the cell type is excitatory or inhibitory and highlight a marker gene enriched in that cell type.
A. Violin plots showing enriched sex and estrous stage-shared genes in the BNSTprTac1/Esr1 cell type (highlighted in yellow, #18).
B. Violin plots showing expression of genes in panel A in M, FR, and FNR conditions.
C. Scatter plot of M v FR sDEGs in the BNSTprEsr1 population. This panel is identical to the one shown in Fig. 1B except that we have highlighted Tac1 (black circle) in this plot.
D. Expression of mCherry+ (DREADDi) in BNSTpr of Tac1Cre and Tac1Cre;Esr1Flpo mice after delivery of AAV-flex-DREADDi and AAV-Coff/Fon-DREADDi to this region, respectively.
E. No change in net time spent investigating urine following chemogenetic inhibition of BNSTprTac1/Esr1 or BNSTprTac1−/Esr1 cell types.
F. Schematic showing CRISPR-mediated targeting of Flpo to the 3’ UTR of Esr1.
G. Flpo-dependent expression of virally encoded EYFP (AAV-fDIO-hChR2-EYFP) in BNSTpr neurons of Esr1Flpo mice shows extensive overlap between EYFP and Esr1-expressing cells.
H. PCR of Coff/Fon INTRSECT hM4Di-mCherry plasmid DNA generates an amplicon larger than WT, while PCR of cDNA from cells co-transfected with the same plasmid and Flpo recombinase generates an amplicon equivalent to WT; a smaller PCR product is also present in the WT version (not shown). cDNA amplicon sequences are seamless across the exon junctions.
I. Flow cytometry of cells transfected with INTRSECT DREADDi-mCherry and activating Flpo recombinase shows expression comparable to WT (constitutive DREADDi-mCherry) and more than that observed in control conditions (negative, no transfection; alone, INTRSECT transfection without recombinase; +Cre +Flpo, INTRSECT transfection with Cre and Flpo). Diminished, but residual, expression is observed post-transfection when co-transfected with Cre and Flpo.
J. Chemogenetic inhibition of BNSTprTac1/Esr1 cell type increases latency of resident males in initiating mounts or intromission with receptive females and attacks toward intruder males.
K. No change in latency of mating or aggression routines in resident males upon chemogenetic inhibition of BNSTprTac1−/Esr1 cell types.
Mean ± SEM. n = 2 (D), 4 (G), and 8 (J,K). Scale bars = 100 μm (D and left panel in G) and 10 μm (inset panels in G).
A. UMAP representation of male and female VMHvlEsr1 cell types (left and middle panels), showing that VMHvlCckar/Esr1 and VMHvlTrim36/Esr1 cell types (#20 and 25, respectively) are restricted to females. Right panel shows scatter plot of M v FR sDEGs in the VMHvlEsr1 population. This panel is identical to the one shown in Fig. 1B except that we have highlighted Cckar (black circle) in this plot.
B. Shared expression of genes between VMHvlCckar/Esr1 and VMHvlTrim36/Esr1 cell types.
C. Targeting strategy (top) and validation (bottom) of PRFlpo mice. CRISPR-mediated targeting of Flpo to the 3 ‘ UTR of Pr. Extensive overlap between PR and Cre revealed by ISH in pRFlpo/+ FR mice. n =2.
D. Targeting strategy (top) and validation (bottom) of CckarCre mice. CRISPR-mediated targeting of Cre to the 3’ UTR of Cckar. Extensive overlap between Cckar and Cre revealed by ISH in CckarCre/+ FR mice. n =2.
E. Expression of mCherry+ (DREADDi) in VMHvl of CckarCre and CckarCre;PRFlpo mice after delivery of AAV-flex-DREADDi and AAV-Coff/Fon-DREADDi to this region, respectively.
F. No change in pup retrieval upon chemogenetic inhibition of VMHvlCckar/Esr1 or VMHvlCckar−/Esr1 cell types.
G. No change in mating or aggression of resident males upon inhibition of the VMHvlCckar/Esr1 cell type.
H. Reduced mating by resident males upon inhibition of VMHvlCckar−/Esr1 cell types.
I. Reduced aggression by resident males upon inhibition of VMHvlCckar−/Esr1 cell types.
J. No change in density of mCherry+ termini of VMHvlCckar/Esr1 neurons in POA and PAG between FR and FNR mice.
K. No change in density of mCherry+ termini of VMHvlCckar− neurons in POA and PAG between FR and FNR mice.
Mean ± SEM. n = ≥8 (F), 9 (G), 12 (H,I), ≥7 (J), and 6 (K). Scale bars=100 μm.
All genes differentially expressed between sexes or FR and FNR in each of the four Esr1+ populations. All genes listed passed FDR < 0.05.
All genes differentially expressed in >1 comparison by condition (M, FR, FNR) or region (BNSTpr, MeA, POA, VMHvl). All genes listed passed FDR < 0.05.
Gene names and putative functions of all DEGs visualized by ISH.
Gene Ontology and Disease Ontology categories enriched in DEGs and references for sex-bias in Disease Ontology categories.
Analysis of snRNAseq libraries, including sequencing metrics (such as UMI, read depth, # of genes/cell), cell type composition, and Jaccard Similarity Index.
Genes enriched in BNSTprTac1/Esr1 and VMHvlCckar/Esr1 cell types.
Summary of spatial distribution of these two cell types.
Data Availability Statement
TRAPseq and snRNAseq data have been deposited at Gene Expression Omnibus and are publicly available upon publication (GEO:GSE183092 and GEO:GSE183093 respectively).
This paper does not report original code. All MATLAB and R scripts used in this manuscript are available from the lead contact upon reasonable request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.