

Abstract
Purpose:
Pathogenic variants in Lysyl-tRNA synthetase 1 (KARS1) have increasingly been recognized as a cause of early-onset complex neurological phenotypes. To advance the timely diagnosis of KARS1-related disorders, we sought to delineate its phenotype and generate a disease model to understand its function in vivo.
Method:
Through International collaboration, we identified 22 affected individuals from 16 unrelated families harboring biallelic KARS1 likely pathogenic or pathogenic variants. Sequencing approaches ranged from disease-specific panels to genome sequencing. We generated loss of function alleles in zebrafish.
Results:
We identify ten new and four known biallelic missense variants in KARS1 presenting with a moderate-to-severe developmental delay, progressive neurological and neurosensory abnormalities, and variable white matter involvement. We describe novel KARS1-associated signs such as autism, hyperactive behavior, pontine hypoplasia, and cerebellar atrophy with prevalent vermian involvement. Loss of kars1 leads to upregulation of p53, tissue-specific apoptosis, and downregulation of neurodevelopmental related genes, recapitulating key tissue-specific disease phenotypes of patients. Inhibition of p53 rescued several defects of kars1−/− knockouts.
Conclusions:
Our work delineates the clinical spectrum associated with KARS1 defects and provides a novel animal model for KARS1-related human diseases revealing p53 signaling components as potential therapeutic targets.
INTRODUCTION
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed and essential enzymes required for the aminoacylation of specific amino acids onto their cognate tRNAs. Biallelic variants in ARS genes have been shown to cause a variety of severe and early-onset human diseases1. These diseases appear with diverse clinical manifestations such as Charcot-Marie-Tooth disease2, leukodystrophies1,3–5, cardiomyopathies6, hearing loss7 and other CNS-related pathologies8–11. ARS variants can lead to reduced aminoacylation activity, decreased translation accuracy and defects in non-canonical processes1, but the underlying mechanisms leading to pathology remain poorly understood12. As a result, there are no effective treatment strategies for these pathologies.
Of the 37 ARS genes in humans, 18 encode cytoplasmic enzymes, 17 encode mitochondrial enzymes, and two encode bifunctional enzymes; KARS1 (lysyl-tRNA synthetase) is one of the bifunctional ARS enzymes. In humans, a single KARS1 gene (MIM 601421)13 encodes both the cytoplasmic and mitochondrial lysyl-tRNA synthetases, which are generated by alternative splicing. Biallelic variants in KARS1 have been reported in patients with a broad spectrum of clinical manifestations including Charcot-Marie-Tooth2, non-syndromic hearing loss14,15, peripheral neuropathy2, congenital visual impairment16, progressive microcephaly16, hypertrophic cardiomyopathy17, leukoencephalopathies15,18,19, leukodystrophy20 and severe neurological and neurosensory disease with optic neuropathy21. Twenty-eight pathogenic variants in 30 affected individuals from 25 families have been identified to date, following an autosomal recessive inheritance pattern.
Here, we report 10 new and 4 known biallelic missense variants in KARS1 in 22 affected individuals from 16 unrelated families. Having included data from 30 previously published KARS1 cases, we provide a cumulative and comprehensive phenotypic characterization of 52 affected individuals. A mouse knockout of Kars1 is embryonic lethal and dies before organogenesis occurs22. Nevertheless, the function of Kars1 in specific tissues in vivo remained unknown. Here, we generated loss of function alleles in zebrafish using CRISPR/Cas9 to understand the function of kars1 in vivo.
Materials and Methods
Genetic and phenotypic analysis
Blood samples were collected from all participants and genomic DNA was extracted using standard methods. Sequencing methods and genetic analysis summaries per individual sequenced in each family are summarized in Table S1. Clinical information was collected using standardized templates and completed by collaborating geneticists and clinicians. Brain MRI/CT images from all previously published reports and our cohort were systematically analyzed by a pediatric neuroradiologist. Detailed clinical methods are described in supplementary information.
Zebrafish Functional Studies
Zebrafish (Danio rerio) were raised and maintained in an AALAC accredited facility at the Oklahoma Medical Research Foundation (OMRF) under standard conditions, and all experiments were performed as per protocol 17–01 approved by the Institutional Animal Care Committee of OMRF (IACUC). All zebrafish work was carried out in wild-type strain NHGRI-1. Detailed methods related to zebrafish work are described in supplementary methods.
Statistical analysis
Each experiment was repeated three-times, and sample sizes are described in the Supplementary Information. Data are presented as mean value ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism version 8.4 (GraphPad Software, San Diego, CA, USA). In all analyses, the significance level was set to 0.05. The p-value was determined as follows: The larval survival curve (Kaplan-Meier representation) was assessed using the Log Rank Mantel-Cox test. One-way ANOVA with Tukey’s multiple comparisons test for eye and head size comparisons. Two-tailed unpaired Student’s t-test with nonparametric Mann-Whitney test for hair cell and motor exon diameter calculation. Two-tailed unpaired Student’s t-test for the comparison of gene expression levels between two groups, and Holm-Šídák multiple comparisons correction is for multiple groups comparison. Two-tailed unpaired t-test with Welch’s correction was used for the VSR and auditory evoked behavior response (AEBR) analyses.
Results
Patients with KARS1 variants show multisystem abnormalities primarily involving the nervous system
We investigated 22 affected individuals from 16 unrelated families who harbored biallelic KARS1 variants (Fig. 1a). Clinical summaries and variant details are presented in the Case Reports in Supplementary information, Table 1, Figure 1C, and Tables S2 and S3. All patients underwent either exome, genome or targeted gene sequencing and analyses, which excluded other functionally relevant variants compatible with Mendelian diseases, based on mode of inheritance and clinical presentation. Variants were Sanger sequence-confirmed (Fig. S1). We identified 10 previously unpublished variants and four previously reported variants18,19,21,23,24 as listed in Table 1, and Figure S2. Each substitution affected a conserved amino acid, and all the variants are predicted to be deleterious (Supplementary information, Fig. S3).
Fig. 1: Identification of Pathogenic Variants in KARS1 in 16 families and Clinical Summary.
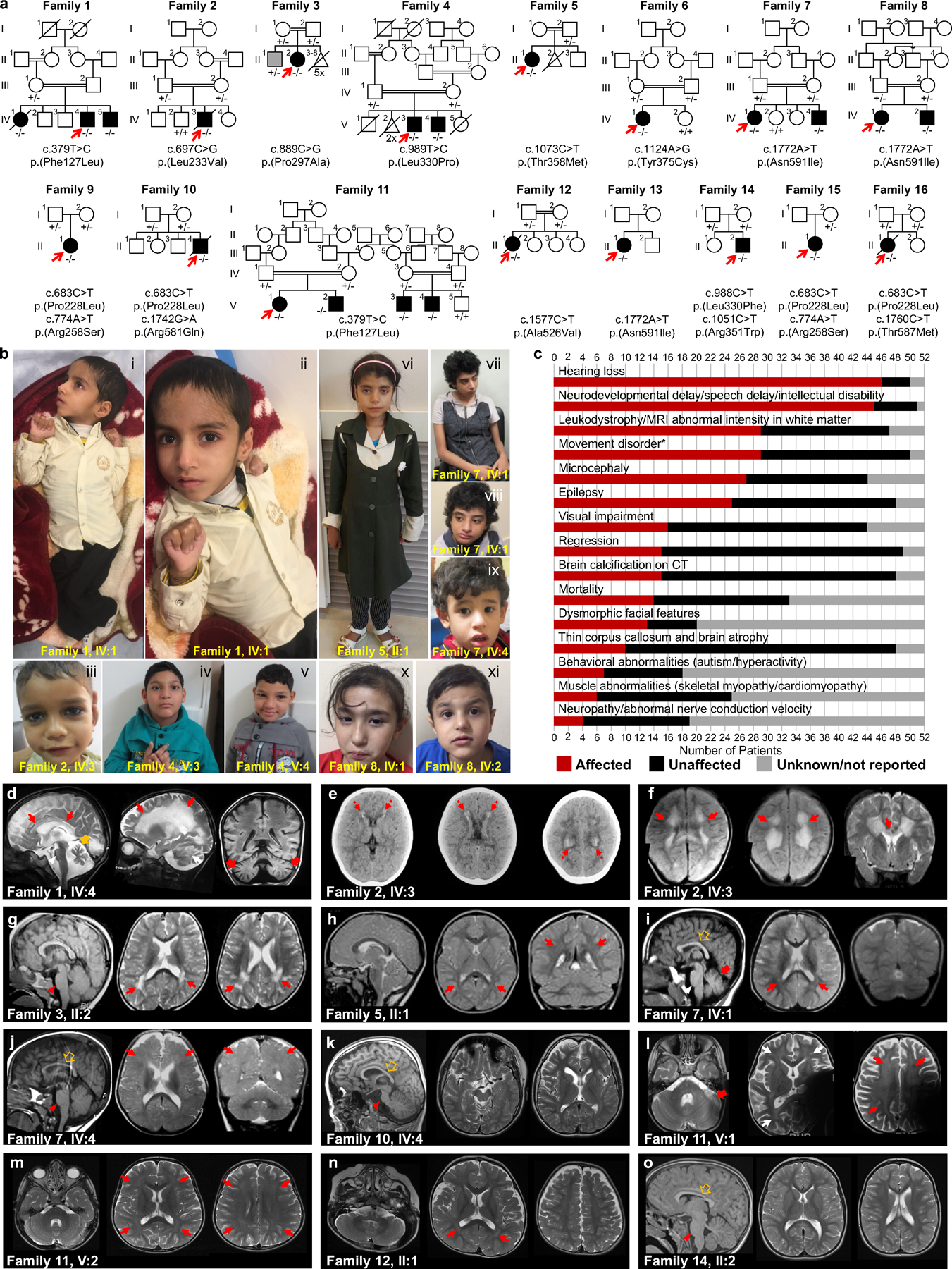
(a) Pedigrees and segregation data for the 16 families included in this study. Affected and unaffected individuals are indicated by filled and open squares(males)/circles(females), respectively. Probands are marked with red arrows. Double lines, consanguinity. Genetic diagnoses were made in 22 individuals. (b) Clinical characteristics of patients with homozygous KARS1 variants, including affected individuals from family 1 (IV:4) (i, ii), family 2 (IV:3) (iii), family 4 (V:3) (iv) and (V:4) (v), family 5 (II:1) (vi), family 7 (IV:1) (vii, viii) and IV:4 (ix), and family 8 (IV:1) (x) (IV:2) (xi). Frequent clinical features include: spasticity and contractures in the limbs with clenched hands (i, ii), high forehead (iv, v, vi, vii, x), prominent nose (i, ii, iv, v, vi, vii, viii), low-set ears (i, ii, iv, v, vii, viii, ix) and short philtrum (i, ii, iv, v, vi, vii, ix, x). (c) Phenotype summary of features associated with KARS1 pathogenic variants. Asterisk denotes movement disorders that include ataxia, spasticity, quadriplegia, dystonia, or chorea/tremor. (d-o) Neuroimaging features associated with KARS1 variants, with variable patterns of white matter (WM) involvement (red arrows), calcifications (red dotted arrows), pontine hypoplasia (red arrowheads), cerebellar atrophy (red thick arrows), enlargement of the cerebral CSF spaces, and corpus callosum hypoplasia (brown empty arrows).
Table 1.
Summary of molecular and key clinical findings
| F1 IV:1/ F11 V:1/V:2/ V:3/V:4 | F2 IV:3 | F3 II:2 | F4 V:3/V:4 | F5 II:1 | F6 IV:1 | F7 IV:1/IV:4/F8 IV:1 /IV:2/F13 II:2 | F9 II:1/ F10 II:4/ F15 II:1 | F9 II:1/F15 II:1 | F10 II:4 | F12 II:1 | F14 II:2 | F16 II:1 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular genetics summary | ||||||||||||||
| Chr16 g. position | g.75674175A>G | g.75669866G>C | g.75668181C>G | g.75668081A>G | g.75665680G>A | g.75665629T>C | g.75662474T>A | g.75669880G>A | g.75669683T>A | g.75662504C>T | g.75663371G>A | g.75668082G>A | g.75665702G>A | g.75662486G>A |
| KARS1 c. position | c.379T>C | c.697C>G | c.889C>G | c.989T>C | c.1073C>T | c.1124A>G | c.1772A>T | c.683C>T | c.774A>T | c.1742G>A | c.1577C>T | c.988C>T | c.1051C>T | c.1760C>T |
| KARS1 p. position | F127L | L233V | P297A | L330P | T358M | Y375C | N591I | P228L | R258S | R581Q | A526V | L330F | R351W | T587M |
| phyloP | 8.92 | 5.66 | 9.87 | 9.34 | 9.84 | 8.02 | 7.95 | 9.59 | 0.18 | 7.77 | 5.94 | 5.86 | 2.14 | 6.10 |
| CADD | 29.7 | 22.2 | 27.0 | 31 | 29.4 | 32 | 28.1 | 25.5 | 25.2 | 28.6 | 31 | 29.4 | 32 | 32 |
| MT | DC | DC | DC | DC | DC | DC | DC | DC | DC | DC | DC | DC | DC | DC |
| PP2 | PrD | B | PrD | PrD | PrD | PrD | PrD | PrD | PrD | PrD | PrD | PrD | PrD | PrD |
| SIFT | D | D | D | D | D | D | D | T | D | D | D | D | D | D |
| gnomAD MAF | 0 | 3.98e-6 | 0 | 0 | 4.95e-5 | 3.98e-6 | 7.97e-6 | 1.415e-4 | 1.971e-5 | 0 | 3.184e-5 | 0 | 7.070e-6 | 5.966e-5 |
| In house MAF* | 1/171678 | 0/171678 | 1/171678 | 1/171678 | 18/257204 | 3/212968 | 1/229162 | 39/262730 | 3/179738 | 1/181974 | 0/38634 | 2/222356 | 3/149466 | 17/244700 |
| Clinical summary | ||||||||||||||
| DD | +/+/+/+/+ | + | − | +/+ | − | + | +/+/+/+/− | +/+/+ | +/+ | + | + | + | + | + |
| ID | +/+/+/+/+ | + | − | +/+ | − | − | +/+/+/+/− | −/−/NA | −/NA | − | + | + | + | + |
| Hearing loss | +/+/+/+/+ | + | + | +/+ | − | + | +/+/+/+/+ | +/NA/+ | +/+ | NA | + | + | + | + |
| Regression | +/+/−/−/− | + | − | −/− | − | − | −/−/−/−/− | −/+/+ | −/+ | + | − | − | − | + |
| Seizures | +/−/+/−/− | + | − | +/− | − | − | −/−/+/+/+ | +/+/+ | +/+ | + | + | + | + | − |
| Ataxia | +/−/−/−/− | − | + | −/− | − | + | +/−/−/−/− | +/−/+ | +/+ | − | − | + | + | − |
| Hypotonia | −/−/−/−/− | − | − | +/+ | − | + | +/+/−/+/− | −/−/+ | −/+ | − | + | + | + | + |
| Spasticity | +/+/−/+/+ | + | − | −/− | − | − | −/−/−/−/− | +/+/− | +/− | + | − | − | − | + |
| Visual impairment | −/+/+/+/− | − | NA | −/− | − | − | −/−/−/−/− | +/+/− | +/− | + | NA | + | + | + |
| Absent speech | −/+/NA/+/+ | − | − | −/+ | + | + | +/+/−/−/− | +/+/− | +/− | + | NA | − | − | + |
| Mortality | −/−/−/−/− | + | − | −/− | − | − | −/−/−/−/− | −/+/− | −/− | + | + | − | − | + |
| Dysmorphic facial features | +/+/+/+/+ | + | − | +/+ | + | − | +/+/+/+/− | −/−/NA | −/NA | − | − | + | + | − |
| Behavioral abnormalities | −/+/−/−/− | − | − | −/+ | − | − | +/+/+/+/− | −/−/NA | −/NA | − | − | + | + | − |
| Leukodystrophy | +/+/+/+/− | + | + | −/+ | − | NA | +/+/−/−/− | +/+/− | +/− | + | + | − | − | − |
Genomic coordinates are listed according to GRCh37 genome build., Transcript: NM_001130089.1, MAF = minor allele frequency, phyloP scale [−14.1;6.4]
no homozygous individuals were identified; Abbreviations: B, benign; D, Deleterious; DC, Disease causing; DD, developmental delay; F, family; ID, intellectual disability; MT, MutationTaster; NA, not assessed; PP2, PolyPhen-2; PrD, Probably damaging; T, Tolerated. The in house database consists of aggregated multiethnic cohorts from study groups.
Clinical description
The cohort mostly presented with a moderate-to-severe developmental delay, progressive neurological and neurosensory impairment, and white matter involvement, variably associated with developmental regression, intellectual disability, behavioral abnormalities, and additional extra neurological signs. The disease typically manifested during early childhood with developmental delay and/or hearing impairment. Developmental regression was reported in 7/22 cases (32%). Four cases died at the mean age of 13.9 years due to respiratory infection (2/4), sepsis (1/4) or post-febrile illness associated with severe deterioration (1/4). All parents were asymptomatic and did not exhibit neurological symptoms. Developmental delay was present in 86% of cases (19/22) followed by intellectual disability (15/21, 71%), speech delay (11/19, 58%), absent speech (11/20, 55%), failure to thrive (4/22, 18%), and behavioral abnormalities such as autism (3/21, 14%), and hyperactive behavior (6/21, 29%).
All cases uniformly expressed neurological symptoms (22/22, 100%), frequently involving sensorineural hearing loss (20/21, 95%), seizures (13/22, 59%), hypotonia (9/22, 41%), cerebellar ataxia (7/22, 32%), spasticity (8/22, 36%), strabismus (6/20, 30%) and nystagmus (4/20, 20%), and acquired joint contractures (4/22,18%). Hearing loss was severe-to-profound in 16/21 cases (76%), and 5/10 (50%) of them had cochlear implants. Other variable neurological features included visual impairment/optic atrophy (7/20, 35%), quadriplegia (3/22, 14%), dystonia and tremor (2/22, 9%), neuropathy (1/10, 10%), neurophysiologically confirmed skeletal myopathy (2/22, 9%) (Video S1–2), generalized muscle atrophy (2/22, 9%), and incontinence (4/22, 18%). None of the cases expressed isolated hearing loss or sensory-motor neuropathy as the main clinical feature.
While not every case was uniformly examined, roughly half were clinically diagnosed with microcephaly (10/18, 56%). Interestingly, dysmorphic facial features were common in 67% (14/21) of the cohort and this included a high or narrow forehead, prominent nose, short philtrum, low-set ears, broad nasal bridge, thin upper lip, epicanthus, and telecanthus (Fig. 1b; Supplementary Case Report includes detailed descriptions ). Other extra-neurological signs included feeding difficulties (6/22, 27%), and neonatal vomiting with diarrhea (2/22, 11%). Single isolated cases displayed hypertrophic cardiomyopathy (1/11, 9%), and neonatal vomiting with diarrhea (2/22, 11%). The case with hypertrophic cardiomyopathy did not have available levels of lactate and mitochondrial respiratory chain enzymes measured.
Neuroimaging findings
Brain MRI studies were available for 21/22 (95%) individuals (with more than one examination in 2 cases), while head CT and spinal MRI were available for three and one subjects, respectively (Fig. 1d–o). White matter involvement was noted in 13/21 (61.9%) individuals. In 5/13 (38.4%) subjects, there were confluent T2 hyperintensities in the periventricular white matter, with prevalent involvement of parieto-occipital regions; one of these subjects studied at 53 years of age had a previous normal brain MRI (performed 8 years before). In 4/13 (23%) subjects, there was mild diffuse T2 hyperintensity of the supratentorial white matter, with sparing of the corpus callosum and U fibers; one of these individuals studied at 4 years of age had a previous normal brain MRI (performed 2 years before). Two subjects (2/13, 15.3%) presented diffuse leukodystrophy with corpus callosum involvement, sparing of the U fibers, and calcifications; in one of them the cerebellar white matter was also involved. Delayed myelination was noted in 2 other subjects (2/13, 15.3%). White matter volume loss with ventricular enlargement was present in 10/21 (47.6%) cases. Enlargement of the cerebral CSF spaces, mainly in the fronto-temporal regions was observed in 9/21 (42.8%) individuals. Hypoplasia of the pons and corpus callosum was noted in 7/21 (33.3%) and 6/21 (28.5%) of cases, respectively. Cerebellar atrophy, with prevalent or isolated vermian involvement was noted in 5/21 (23.8%) of cases. Finally, in 4/21 (19%) subjects, the brain MRI was normal.
Genotype-phenotype correlation
Inter- and intrafamilial phenotypic variability was noticed among individuals with the same variants in the present KARS1 cohort. For instance, three affected siblings in Family 11 each harbored a homozygous KARS1 c.379T>C (p.(Phe127Leu)) variant, and although they shared common symptoms such as developmental delay, infantile-onset profound hearing loss, dysmorphic facial features, spasticity, and varying degree of joint contractures, several important symptoms were expressed only by one of them. This included regression, epilepsy, optic atrophy, failure to thrive, and hyperactivity. The same KARS1 variant was homozygous in the proband from Family 1, who differed from Family 11 by a preserved vision, cerebellar ataxia, classic leukodystrophy on MRI, and the basal ganglia calcification. Similarly, 5 affected members from three independent families were homozygous for the KARS1 c.1772A>T (p.(Asn591Ile)) variant and they had variable intra- and interfamilial expression of epileptic seizures, cerebellar ataxia, intellectual disability, hypotonia, polyneuropathy, behavioral abnormalities, and impaired speech. Significant phenotypic variability was also noticed between families 9 and 15 harboring the similar compound heterozygous KARS1 variants c.683C>T (p.(Pro228Leu)) and c.774A>T (p.(Arg258Ser)).
kars1 zebrafish disease model using CRISPR/Cas9-mediated targeted mutagenesis.
Zebrafish has a single kars1 gene (NCBI Gene ID: 280647) generating two transcript variants via alternative RNA splicing, confirmed by RT-PCR from multiple developmental stages (Fig. S4, and Supplementary information). To unravel the function of KARS1, we examined kars1 mRNA expression in zebrafish embryonic development using whole-mount in situ hybridization (WISH). kars1 mRNA was initially ubiquitously expressed but gradually became more prominent in the central nervous system (CNS), eye, inner ear, muscles, and digestive system (liver, intestine and pancreas) (Fig. 2a–c, Supplementary information, and Fig. S5a–g). We generated kars1 loss-of-function zebrafish mutant lines using CRISPR/Cas9 and identified 3 independent alleles kars1om1del7, kars1om2del8 and kars1om3del7 (Fig.S6 a–d). Homozygous kars1−/− animals showed morphological abnormalities starting at approximately 3 dpf including heart edema (black arrow), as well as smaller heads (blue line), eye (red line) and otic vesicle (red arrowhead) when compared to WT animals (Fig. 2d and e, and Fig. S7a–c). Kars1 homozygous larvae also failed to inflate the swim bladder (black arrowhead) and showed abnormal trunk muscle fibers. We observed 100% mortality by 10 dpf for all three kars1 homozygous mutant larvae, possibly because of the inability to feed (Fig. 2f and Fig. S7d). We quantified the eye and head axial length which were significantly reduced in kars1−/− mutants compared to control animals (Fig. 2g and h). Additionally, the kars1−/− larvae failed to respond to touch and displayed a loss of spatial orientation (Fig. S7e, f and Video S3). The side-laying position observed in kars1−/− mutant larvae might reflect loss of vestibular function, severe muscle control failure, and/or absence of an inflated swim bladder which all affect balance. Given the kars1−/− mutants showed morphological defects in the eyes and ears, and failed to response to touch, we further quantified their visual startle response (VSR) and AEBR, kars1−/− mutants showed completed loss of locomotor activity in response to light or acoustic startle (Fig. 2i and j). Overall, the tissue/organ-specific morphological defects in kars1−/− mutants found in the eye, inner ear, and trunk muscles appeared to directly correlate with kars1 mRNA expression patterns during embryo development, strongly suggesting these phenotypes and behaviors arose from Kars1 loss-of-function.
Fig. 2: The Expression of kars1 mRNA in Embryos and kars1 Knockouts show Gross Morphological and Behavioral Defects.
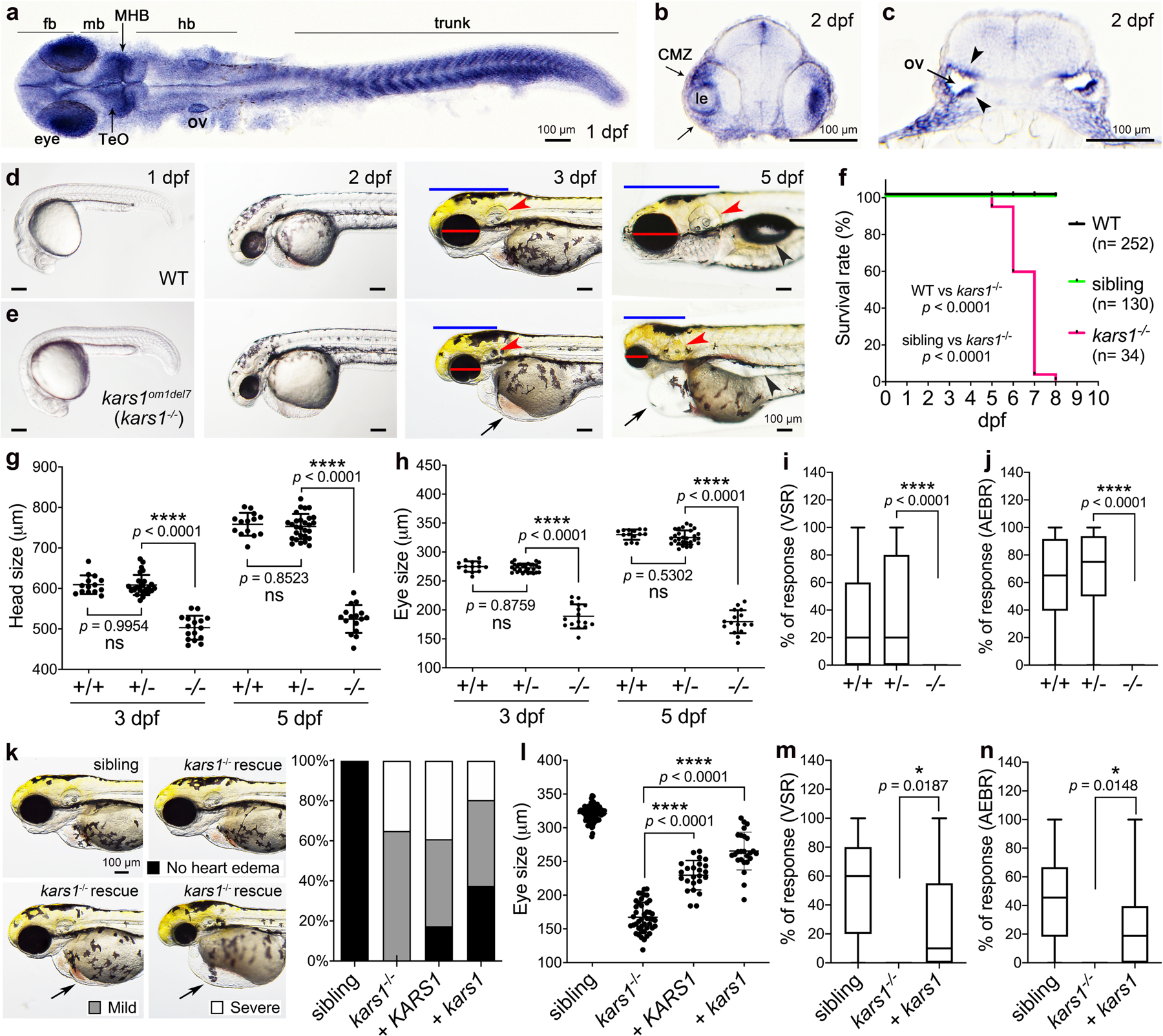
(a) kars1 expression in 1 dpf (days post-fertilization) embryo. Dorsal view. (b, c) The cross-section of 2 dpf embryo. forebrain (fb), midbrain (mb), midbrain and hindbrain boundary (MHB), hindbrain (hb), optic tectum (TeO), otic vesicle (ov), lens (le), ciliary marginal zone (CMZ). Black arrowheads indicating the otic vesicle epithelium of otic vesicles. (d, e) Representative images of WT and kars1om1del7 (kars1−/−) from 1 dpf to 5 dpf. Lateral view, anterior to the left. Red lines: eye diameter. Blue lines: brain size. Red arrowheads: inner ear. Black arrows: heart edema. Black arrowhead: swim bladder. (f) Kaplan-Meier survival curves. Time is shown in days. The Log Rank test was used for statistical analysis. (g, h) Quantification of eye and head size from kars1+/− mutant in-cross at 3 and 5 dpf. (i, j) The VSR and AEBR analyses of animals at 6 dpf from kars1+/− mutant in-cross. (k) Representative images of kars1−/− mutant rescue experiments and the quantification of heart edema phenotype. Animals were collected by defined heart edema categories at 3 dpf as shown in pictures and calculated in percentage of total animals. (l) Eye size quantification of mRNA rescue experiments at 5 dpf. (m, n) The VSR and AEBR analyses after RNA rescue at 6 dpf. n = number of animals. In (g, h and l), each dot represents one animal and error bars are presented as mean ± SD. One-way ANOVA with Tukey’s multiple comparisons test: ****p < 0.0001. In (i, j, m and n), data are plotted by box and whiskers plot and error bars indicate values from minimum to the maximum. Two-tailed unpaired t-test with Welch’s correction: ns, not significant p ≥ 0.05, *p < 0.05 and ****p < 0.0001.
To confirm the phenotypes of the kars1−/− mutant animals arise from loss of kars1 function, we performed mRNA rescue experiments by injecting either human WT KARS1 or the zebrafish WT kars1 mRNA. Co-injection of mRNAs encoding the mitochondrial and cytoplasmic isoforms into 1-cell stage showed reduced frequencies of heart edema (Fig. 2k) and significantly restored eye size in kars1−/− mutants (Fig. 2l). Furthermore, microinjection of zebrafish kars1 mRNA further rescued the startle responses compared to kars1−/− mutant animals (Fig. 2m and n) suggesting mutant phenotypes are caused by kars1 loss-of-function.
The kars1 loss-of-function zebrafish model recapitulates patient symptoms.
Histological analysis of the brain of 5 dpf larvae revealed a vacuolated spongiosus appearance with areas of reduced cell density and disorganized segment boundaries in kars1−/− mutants compared to WT siblings (Fig. 3a–c). Moreover, eye volume was reduced and the retinal layer organization was completely lost in the kars1−/− mutants compared to WT siblings (Fig. 3b) strongly suggesting impaired vision. Furthermore, the number of neuronal cells appeared strongly reduced in the brain and retina in kars1−/− mutants compared to controls (Fig. 3a and b). Immunohistochemical analysis of neuronal synapses revealed reduced staining in all brain and eye regions pointing to a significant reduction of synapses in kars1−/− mutants (Fig. 3d), suggesting impaired neuronal transmission. Additionally, staining revealed abnormal motor neuron morphology, including shrinkage of motor neuron axon projections and reduction of terminal axonal branching in the mutants (Fig. 3e), further indicating strong alteration of locomotor function in those animals.
Fig. 3: The kars1−/− Larvae Display Neurological, Muscle and Inner Ear Defects by Histological Analysis at 5 dpf.
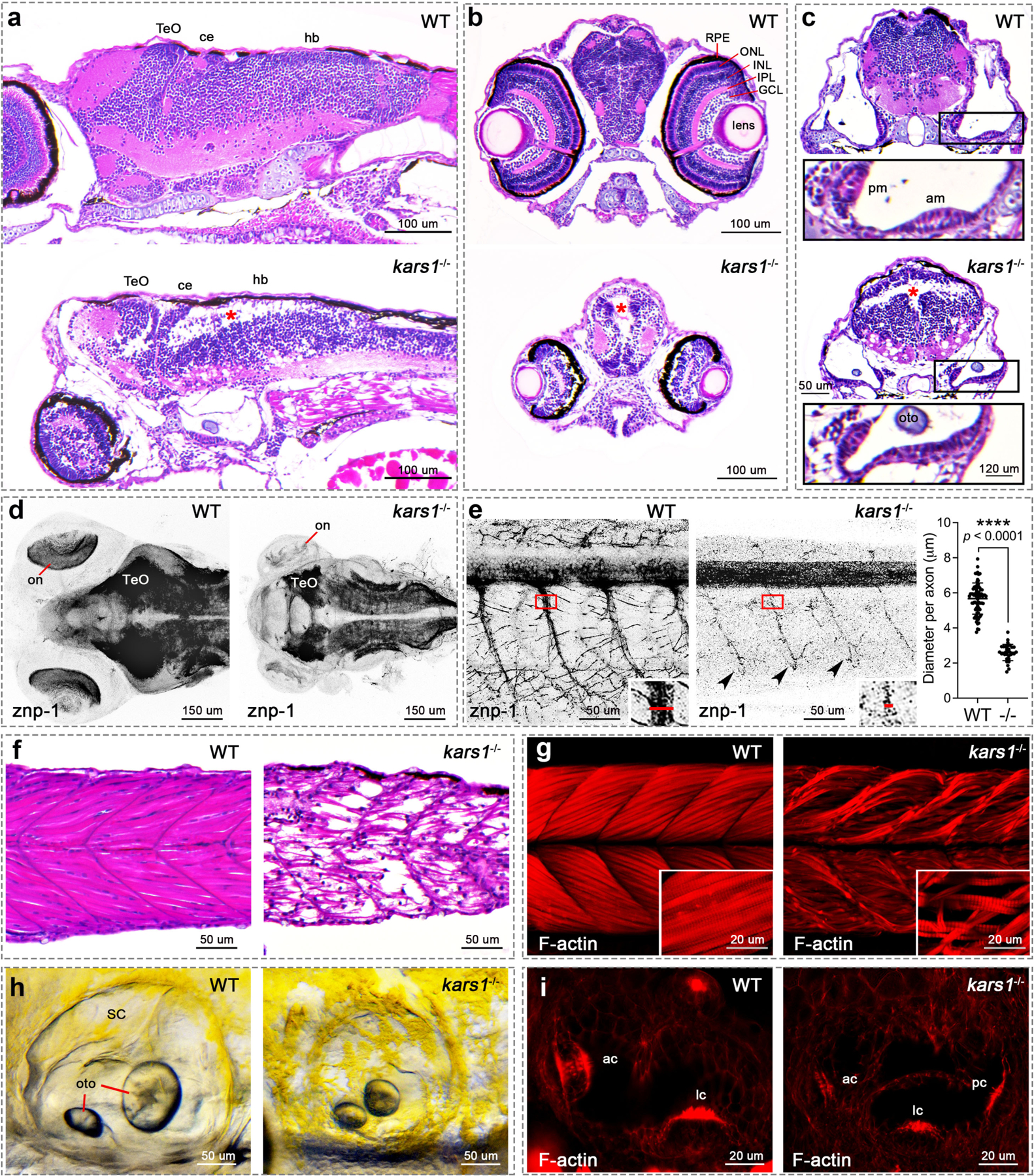
(a) Head region of WT and kars1−/− mutant by sagittal section. Red asterisks indicate massive loss of cell density, as well as in (b) and (c). Anterior to the left and dorsal to the top. (b) Eye region of WT and kars1−/− mutant by cross section. Dorsal to the top. (c) Inner ear region of WT and kars1−/− mutant by cross section. Dorsal to the top. Lower panels were enlarged picture from black box. (d) Head region of WT and kars1−/− larvae immunostained with anti-znp-1 antibody. The black-and-white fluorescent signals were inverted to negative film for a clear presentation. Dorsal view, anterior to the left. on, optic nerve. (e) Trunk region of WT and kars1−/− larvae which were immunostained with anti-znp-1 antibody. ImageJ was used to measure the diameter of primary motor axons as indicated by red line in the right-down panels (enlarged from red box). And the statistics shown on right hand side. Black dot indicates the diameter of each motor axon. Error bars = mean ± SD. Two-tailed unpaired nonparametric Mann-Whitney test: ****p < 0.0001. Black arrowheads indicate the reduced terminal axonal branching compared to WT. Anterior to the left and dorsal to the top. (f) The trunk region of WT and kars1−/− larvae revealed by sagittal section. Anterior to the left and dorsal to the top. (g) The trunk region of WT and kars1−/− larvae revealed by confocal projections of phalloidin stained muscle fiber. Anterior to the left and dorsal to the top. The right-down panels are the higher-magnification view. (h) Representative bright-field images of WT and kars1−/− inner ear. Anterior to the left and dorsal to the top. Semicircular canal (SC), otolith (oto). (i) The red fluorescent conjugated phalloidin staining was performed to visualize the bundles (stereocilia) of hair cells in inner ear. Anterior to the left and dorsal to the top. Optic tectum(TeO), cerebellum (ce), hindbrain (hb), retinal pigment epithelium (RPE), outer nuclear layer (ONL), inner nuclear layer(INL), inner plexiform layer (IPL). ganglion cell layer (GCL), posterior macula (pm), anterior macula (am), anterior crista (ac), lateral crista (lc), posterior crista (pc).
Individuals with biallelic KARS1 pathogenic variants usually present with combined neuronal and muscular dysfunction. Indeed, our WISH data revealed kars1 mRNA expression in trunk muscles (Fig. 2a) and a loss of touch-evoked responses in the kars1−/− mutants (Fig. S7e and Video S3). Phalloidin staining that labels actins showed weaker staining in the myotomes and presented misaligned and detached muscle fibers from the myotendinous junction (MTJ) (Fig. 3f and g) suggesting severe neuromuscular dysfunction leading to absence of coordinated locomotion.
Several individuals with KARS1 variant alleles also display hearing disorders reminiscent of the inner ear defects observed in kars1−/− mutant larvae, such as perturbed sensory epithelia and loss of AEBR startle response. Morphological analysis of the mutants showed smaller otic vesicles and otoliths (Fig. 3h) and flattened sensory epithelia in both anterior and posterior maculae (Fig. 3c). The phalloidin staining in the inner ear showed strong reduction in the number of F-actin-rich stereocilia, which are crucial for the formation of hair cell bundles and thus for mechanoelectrical transduction (Fig. 3i). Vital stain Yo-PRO-1 iodide, showed fewer hair cells in the zebrafish lateral line which are morphologically and functionally similar to those of the inner ear25 (Fig. S8a and b). Taken together, these findings suggest that kars1 plays a crucial role in hair cell formation in zebrafish, reminiscent of hearing disorders found in a subset of individuals affected with KARS1 variants.
Variants in KARS1 have been shown to be associated with seizures (Table S2). Seizure activity positively correlates with the expression of c-fos (fosab in zebrafish), a marker for general neuronal activity. We used classical pro-chemoconvulsant drug pentylenetetrazol (PTZ) to induce seizure activity and elevated expression of fosab mRNA (Fig. S8c). Intriguingly, untreated kars1−/− larvae, when compared with untreated WT controls, exhibited two-fold at 3 dpf and ten-fold increase in fosab mRNA levels (Fig. S8d) suggesting seizure-like activity in homozygous animals.
Together, our morphological, behavioral and histological analyses demonstrate that the kars1−/− zebrafish mutant larvae exhibit visual impairment, neuromuscular dysfunction, sensorineural hearing loss, and increased expression of seizure marker, c-fos, thus recapitulating a number of pathologies found in individuals with KARS1 variants.
Kars1 loss-of-function triggers p53-mediated apoptosis and downregulation of key neurodevelopmental related genes
To determine the consequences of Kars1 loss-of-function on gene expression, we performed RNA-sequencing (RNA-Seq) on WT and kars1−/− mutants at 3 dpf and 4 dpf and compared gene expression profiles. We found 1,616 and 1,409 differentially expressed genes (DEGs) at 3 dpf and 4 dpf respectively, 563 genes overlapped between 3 dpf and 4 dpf (Fig. S9a–c, and Table S5). KEGG pathway analysis showed kars1 loss-of-function dysregulated many pathways including the p53 and cell apoptosis pathway (Fig. S10–12). ARSs have been shown to regulate cell death pathways26,27. To test whether the morphological defects found in kars1−/− mutants were due to abnormal cell apoptosis, we performed a TUNEL assay and observed an increase in TUNEL-positive cells in the brain, eye, trunk and inner ear of kars1−/− mutants compared with sibling animals at 5 dpf (Fig. S13a and b), suggesting distinct cell types are particularly sensitive to loss of Kars1. Next, to determine whether increased cell apoptosis was mediated by p53 activation, we knocked down tp53 by microinjecting kars1−/− mutant animals with tp53 anti-sense morpholinos (MO) and subsequently performed the TUNEL assays at 3 dpf. Results showed a significant reduction in the TUNEL-positive signals in the brain, eye, ear and trunk in the MO-injected kars1−/− mutant (Fig. 4a–c). Strikingly, reduction in eye size (Fig. 4d), and up-regulation of several apoptosis markers were restored to WT levels (Fig. 4e), therefore confirming the p53 pathway was successfully knocked down, and furthermore implicating it in reduction of eye size. To further characterize the functional rescue at the molecular level after inhibiting the p53 pathway, we examined the expression of six genes we previously found to be downregulated in kars1−/− mutants (Fig. 4f). All six genes showed at least partially restored expression upon p53 knockdown in kars1−/− mutants. Taken together, the loss of Kars1 upregulated the p53 pathway thus leading to apoptosis thereby causing multiple phenotypic abnormalities.
Fig. 4: Cell Apoptosis was Activated by kars1 Loss-of-function through p53 Pathway.
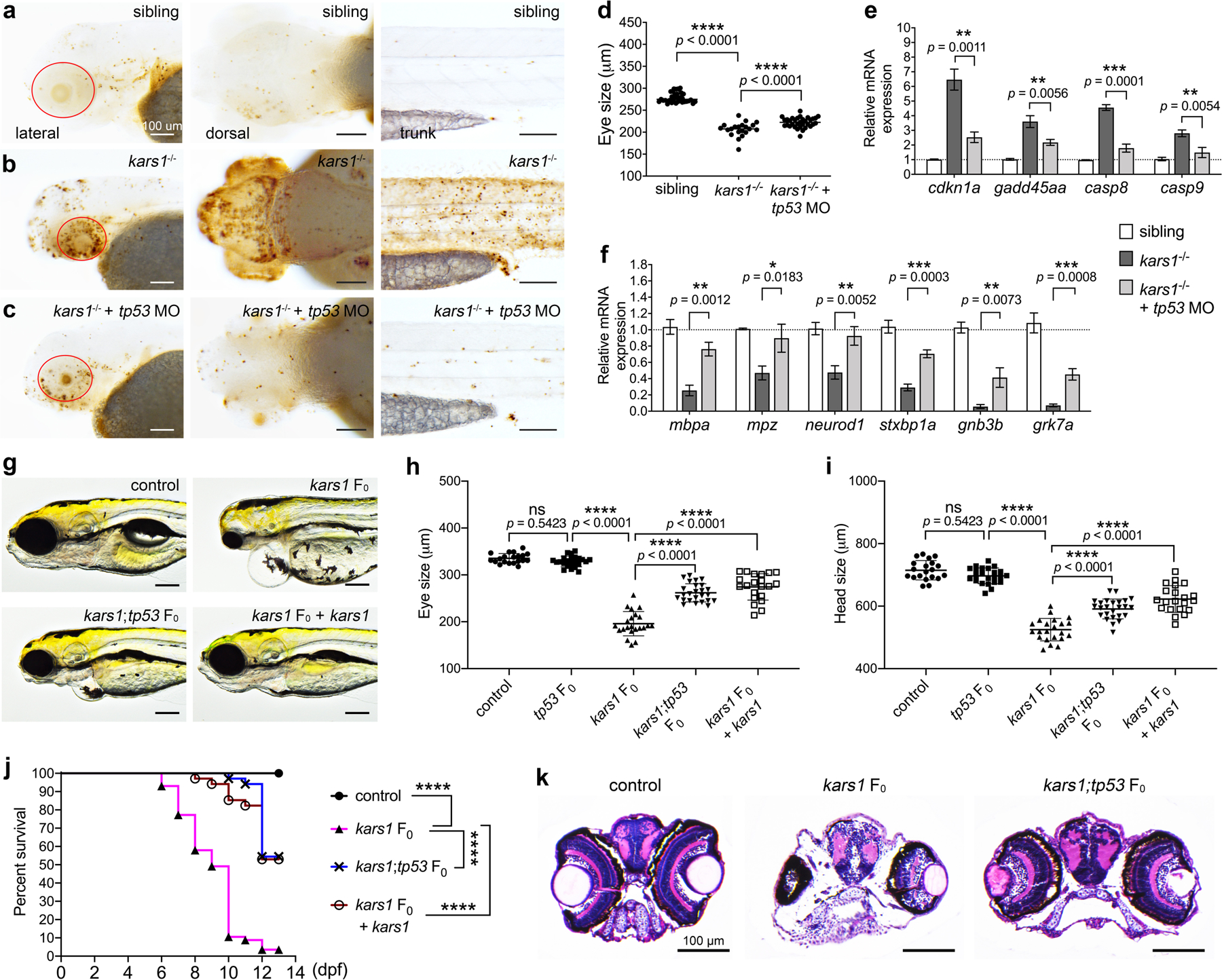
(a-c) Representative images of sibling, kars1−/− and kars1−/− + p53 MO at 3 dpf after TUNEL staining. Red circle indicates eye region. Scale bars = 100 μm. (d) The eye size measurements of sibling, kars1−/− and kars1−/− + p53 MO at 3 dpf. (e) The expression levels of p53 pathway genes were examined by RT-qPCR after p53 MO injection. (f) Those down-regulated genes in RNA-seq data were examined by RT-qPCR after p53 MO injection. For (e, f), the expression levels were normalized to 18S housekeeping gene. Error bars = mean ± SD. Two-tailed unpaired Student’s t-test with Holm-Šídák multiple comparisons correction: ns, not significant p ≥ 0.05, *p < 0.05, **p < 0.01 and ***p < 0.001. (g) Representative images of uninjected control, kars1 F0 mutant, kars1;tp53 F0 mutant, and kars1 F0 mutant co-injected with kars1 mRNA at 5 dpf. Lateral view, anterior to the left. (h, i) Measurement of eye and head size in control, tp53 F0 mutant, kars1 F0 mutant, kars1;tp53 F0 mutant, and kars1 F0 mutant co-injected with 150 picogram (pg) and 200 pg of kars1 mRNA at 5 dpf. For (d, h and i), each symbol represents one animal. Error bars = mean ± SD. One-Way ANOVA with Tukey’s multiple comparisons test: ns, not significant p ≥ 0.05 and ****p < 0.0001. (j) Kaplan-Meier survival curves. Time is shown in days. Log Rank test: ****p < 0.0001. (k) Histology analysis of uninjected control, kars1 F0 mutant and kars1;tp53 F0 mutant by cross section.
To validate our findings, we used CRISPR/Cas9 technology to generate biallelic variants. Co-injecting WT kars1 mRNAs with the kars1 sgRNAs rescued these phenotypes, confirming they arise from Kars1 loss-of-function (Fig. 4g). Importantly, co-injection of tp53 sgRNAs with kars1 sgRNAs restored the eye and head size of the kars1 F0 mutants (Fig. 4g–i). Moreover, whereas 90% of kars1 F0 mutant animals died by 10 dpf, only 20% of animals co-injected with either kars1 mRNAs or tp53 sgRNAs died by 10 dpf (Fig. 4j). Histological analysis showed vacuolated spongiosus appearance in the brain was significantly restored in kars1;tp53 F0 mutants compared to kars1 F0 mutants, as was eye volume and retinal layer organization (Fig. 4k) and vision/hearing startle responses (Fig. S13c and d). At the molecular level, kars1 F0 mutants showed significantly reduced kars1 expression and increased tp53 expression compared to the uninjected control, whereas kars1;tp53 F0 mutants showed significantly reduced expression of both kars1 and tp53 (Fig. S13e). Additionally, kars1 F0 mutants showed increased casp8 and decreased mbpa, neurod1 and stxbp1a expression compared to controls that was restored in kars1;tp53 F0 mutants (Fig. S13f). Together, our results showed kars1 F0 mutants phenocopy the kars1−/− mutant, and p53 depletion mitigates the morphological, behavior, and molecular phenotypes due to loss of Kars1.
Discussion
Deciphering how variants in different ARS genes cause diverse organ-specific phenotypes is crucial to inform therapeutic approaches for ARS-related disorders. We report 10 novel and four known variants in KARS1 from 22 patients belonging to 16 unrelated families. Having included the clinical and neuroimaging data from our cohort and the corresponding data from the previously published 30 KARS1 cases (25 families), we made a cumulative phenotypic characterization for 52 cases with biallelic KARS1 variants (Fig. 1c).
Notably, our cohort expands the phenotypic spectrum of KARS1 variants to include autism/hyperactivity. Additionally, we highlight a variety of KARS1-associated dysmorphic facial features, as facial dysmorphism has rarely been described in previous KARS1 reports. It is reported in 11/52 individuals, where 10 cases were identified in our cohort and 1 case from McLaughlin et al2. We also provide further supporting evidence for skeletal myopathy, a phenotype that has previously been reported in one case17. Our case with hypertrophic cardiomyopathy (Family 4) seems to also highlight the importance of this phenotype as in the previous few reports17,28. Finally, we highlight cerebellar ataxia might be a frequent KARS1-associated feature that was previously reported only in 3/30 (10%) cases but present in 32% (7/22) of our cohort. Our cumulative phenotypic analysis showed that similar to other ARSs, KARS1 expresses predominant neurological and neurosensory phenotypes associated with facial dysmorphism29.
The systematic analysis of all available neuroimaging data, including the present cases, suggests the spectrum of manifestations associated with KARS1 variants is wide, ranging from normal brain appearance to a severe leukodystrophy with rapid brain atrophy. Slightly more than one third of cases (18/52, 34.6%) presented with a progressive leukodystrophy, involving the corpus callosum and sparing the U fibers, variably extending to the spinal cord and cerebellar white matter, often associated with peculiar cerebral and spinal cord calcifications18–20. Less specific patterns included faint diffuse (6/52, 11.5%) or multifocal periventricular (7/52, 13.4%) white matter signal alterations. Interestingly, a third of the present patients had callosal hypoplasia, a likely underestimated feature previously described in three subjects with KARS1 variants16,30. This finding is only partly related to the loss of cerebral white matter volume, as the corpus callosum in these subjects is thin but also shorter than normal. In addition, we uncovered hypoplasia of the pons in one third of subjects, and atrophy of cerebellum with prevalent vermian involvement in 20% of cases. Taken together, these neuroimaging features expand the phenotypes related to KARS1 variants, indicating a prenatal onset of the disease often associated with a rapid neurodegenerative course31,32.
Although a clear genotype-phenotype correlation in KARS1-related disease is lacking, we noticed some interesting associations. The KARS1 c.379T>C (p.(Phe127Leu)) variant was present in the case from Family 1 in a homozygous state, and the same variant was present in compound heterozygosity in one of the cases from the Ardissone et al. report18. Besides the typical developmental delay and hearing loss, both cases shared other phenotypic similarity including seizures, psychomotor regression, hypotonia, spastic tetraparesis, and brain calcifications. The KARS1 c.683C>T (p.(Pro228Leu)) variant was present in compound heterozygosity in Family 9 and a report by Scheidecker et al.21, and both families manifested ataxia, visual impairment, and dystonia. Identifying and characterizing homozygous individuals with these variants in the future would be of interest to further evaluate this potential association.
Biallelic KARS1 variants have emerged as a cause of rare early-onset childhood neurodegenerative disorders presenting with a multiorgan dysfunction suggestive of mitochondrial disorders but usually with normal biochemical assays. Therefore, in addition to the initial association of biallelic KARS1 variants with Charcot-Marie-Tooth disease and non-syndromic hearing loss in single isolated cases (2/50), an early-onset complex neurological and syndromic phenotype should be assigned to KARS1 in an Online Catalog of Human Genes and Genetic Disorders (OMIM). This might yield more families with KARS1-related disease and improve understanding of its neurobiology and phenotypic spectrum.
The physiological relevance of several ARS genes has been assessed in vivo using zebrafish33,34,35. Here, we showed kars1 homozygous (kars1−/−) zebrafish larvae exhibit the phenotypic hallmarks associated with pathogenic variants reported in patients. In agreement with our study, Kars1 knockdown in Xenopus caused developmental defects of head and eyes; loss of myelination and reduced expression of myelin binding protein, leading to incomplete formation of white matter, and reduced brain volume suggesting an important role of KARS1 across the species20.
Loss of ARS enzymes have been shown to activate the p53 signaling pathway36. Likewise, in zebrafish, overexpression of an editing-defective valyl-tRNA synthetase (VARS1) activates the p53 signaling pathway, and increased expression of targets such as cyclin-dependent kinase inhibitor p21 (cdkn1a) and gadd45 leading to cell death37. Kars1−/− mutants show upregulation of p53 signaling and apoptosis pathway genes. Interestingly, up-regulation of p53 signaling has been connected to apoptosis of human oligodendrocytes (a type of myelin-forming cells)38, and cuprizone-39 or compressed spinal cord injury-40 induced demyelination (one of the categories in leukodystrophy classification) in mice. Genetic or pharmacological inhibition of p53 decreased the susceptibility of cuprizone-induced demyelination and increased the survival rate of oligodendrocytes39. These data and our current findings emphasize that p53 pathway inhibitors might represent potential therapeutics for KARS1 patients.
One of the limitation of this study is lack of functional variant analysis in zebrafish. However, our structural modeling data shows 27/36 variants occur in the aminoacylation domain, therefore, likely affect enzyme activity. Since KARS1 is a multifunctional protein, variants in this study could affect the canonical as well as noncanonical functions of the protein.
In summary, our findings underscore a conserved and unique requirement for ARS during nervous system and muscle development to prevent p53-mediated apoptosis and disease. Our disease model offers promising avenues to explore the biallelic contributions of KARS1, and similar approaches could be exploited to understand a number of diseases associated with variants in essential genes.
Supplementary Material
Acknowledgements
This research is supported by a grant from NIH GM103636 (Project 3), and the Presbyterian Health Foundation (PHF) (GKV), NIH grants, R01NS048453, R01NS052455 ( J.G.G.), Intramural Funding (fortüne) at the University of Tübingen (2545-1-0 to B.V.) and the Ministry of Science, Research and Art Baden-Württemberg (to B.V.). Sequencing and analysis were provided by the Broad Institute (UM1 HG008900, R01HG009141) & Yale Center for Mendelian Disorders (U54HG006504 to M. Gunel). This research was made possible in part through the 100,000 Genomes Project, UK.
Footnotes
Genomics England Research Consortium
Ambrose, J. C.1; Arumugam, P.1; Bleda, M.1; Boardman-Pretty, F.1,2; Boustred, C. R.1; Brittain, H.1; Caulfield, M. J.1,2; Chan, G. C.1; Fowler, T.1; Giess A.1; Hamblin, A.1; Henderson, S.1,2; Hubbard, T. J. P.1; Jackson, R.1; Jones, L. J.1,2; Kasperaviciute, D.1,2; Kayikci, M.1; Kousathanas, A.1; Lahnstein, L.1; Leigh, S. E. A.1; Leong, I. U. S.1; Lopez, F. J.1; Maleady-Crowe, F.1; Moutsianas, L.1,2; Mueller, M.1,2; Murugaesu, N.1; Need, A. C.1,2; O’Donovan P.1; Odhams, C. A.1; Patch, C.1,2; Perez-Gil, D.1; Pereira, M. B.1; Pullinger, J.1; Rahim, T.1; Rendon, A.1; Rogers, T.1; Savage, K.1; Sawant, K.1; Scott, R. H.1; Siddiq, A.1; Sieghart, A.1; Smith, S. C.1; Sosinsky, A.1,2; Stuckey, A.1; Tanguy M.1; Thomas, E. R. A.1,2; Thompson, S. R.1; Tucci, A.1,2; Walsh, E.1; Welland, M. J.1; Williams, E.1; Witkowska, K.1,2; Wood, S. M.1,2.
1. Genomics England, London, UK
2. William Harvey Research Institute, Queen Mary University of London, London, EC1M 6BQ, UK.
Competing Interests
HZE, TBP, ET, SM, and HY are employees of GeneDx, Inc. All other authors declare no competing interests.
Ethics Declaration
Informed written consent including use of pictures and videos was obtained prior to study inclusion. This study was performed under the tenets of the Declaration of Helsinki and approved by the Ethics Commission of the University of Wuerzburg (46/15), University of Tübingen (197/2019BO01), and University of California San Diego (140028). All experimental animal care was performed in accordance with institutional and NIH guidelines and regulations. The study protocol was approved by the Institutional Animal Care and Use Committee of Oklahoma Medical Research Foundation, protocol 17–02.
Disclaimer: The contents of this publication are the sole responsibility of the author(s) and do not necessarily reflect the views, opinions or policies of Uniformed Services University of the Health Sciences (USUHS), The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., the Department of Defense (DoD), the Departments of the Army, Navy, or Air Force. Mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Data Availability
Human variant data included in this study have been deposited in Leiden Open Variation Database (LOVD) and available through following accession numbers: 0000685774 (https://databases.lovd.nl/shared/variants/0000685774), 0000685798 (https://databases.lovd.nl/shared/variants/0000685798), 0000685800 (https://databases.lovd.nl/shared/variants/0000685800), 0000685801 (https://databases.lovd.nl/shared/variants/0000685801), 0000685802 (https://databases.lovd.nl/shared/variants/0000685802), 0000685803 (https://databases.lovd.nl/shared/variants/0000685803), 0000685804 (https://databases.lovd.nl/shared/variants/0000685804), 0000685805 (https://databases.lovd.nl/shared/variants/0000685805). Reagents related to zebrafish experiments described in this manuscript are available upon request.
References
- 1.Meyer-Schuman R, Antonellis A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. 2017;26(R2):R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLaughlin HM, Sakaguchi R, Liu C, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87(4):560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39(4):534–539. [DOI] [PubMed] [Google Scholar]
- 4.Messmer M, Florentz C, Schwenzer H, et al. A human pathology-related mutation prevents import of an aminoacyl-tRNA synthetase into mitochondria. Biochem J. 2011;433(3):441–446. [DOI] [PubMed] [Google Scholar]
- 5.Maffezzini C, Laine I, Dallabona C, et al. Mutations in the mitochondrial tryptophanyl-tRNA synthetase cause growth retardation and progressive leukoencephalopathy. Mol Genet Genomic Med. 2019;7(6):e654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gotz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88(5):635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almalki A, Alston CL, Parker A, et al. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim Biophys Acta. 2014;1842(1):56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latour P, Thauvin-Robinet C, Baudelet-Mery C, et al. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2010;86(1):77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLaughlin HM, Sakaguchi R, Giblin W, et al. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat. 2012;33(1):244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez M, McLaughlin H, Houlden H, et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry. 2013;84(11):1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safka Brozkova D, Deconinck T, Griffin LB, et al. Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain. 2015;138(Pt 8):2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113:139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolkunova E, Park H, Xia J, King MP, Davidson E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J Biol Chem. 2000;275(45):35063–35069. [DOI] [PubMed] [Google Scholar]
- 14.Santos-Cortez RL, Lee K, Azeem Z, et al. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet. 2013;93(1):132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou XL, He LX, Yu LJ, et al. Mutations in KARS cause early-onset hearing loss and leukoencephalopathy: Potential pathogenic mechanism. Hum Mutat. 2017;38(12):1740–1750. [DOI] [PubMed] [Google Scholar]
- 16.McMillan HJ, Humphreys P, Smith A, et al. Congenital Visual Impairment and Progressive Microcephaly Due to Lysyl-Transfer Ribonucleic Acid (RNA) Synthetase (KARS) Mutations: The Expanding Phenotype of Aminoacyl-Transfer RNA Synthetase Mutations in Human Disease. J Child Neurol. 2015;30(8):1037–1043. [DOI] [PubMed] [Google Scholar]
- 17.Verrigni D, Diodato D, Di Nottia M, et al. Novel mutations in KARS cause hypertrophic cardiomyopathy and combined mitochondrial respiratory chain defect. Clin Genet. 2017;91(6):918–923. [DOI] [PubMed] [Google Scholar]
- 18.Ardissone A, Tonduti D, Legati A, et al. KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature. Orphanet J Rare Dis. 2018;13(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun C, Song J, Jiang Y, et al. Loss-of-function mutations in Lysyl-tRNA synthetase cause various leukoencephalopathy phenotypes. Neurol Genet. 2019;5(2):e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh M, Dai H, Horike SI, et al. Biallelic KARS pathogenic variants cause an early-onset progressive leukodystrophy. Brain. 2019;142(3):560–573. [DOI] [PubMed] [Google Scholar]
- 21.Scheidecker S, Bär S, Stoetzel C, et al. Mutations in KARS cause a severe neurological and neurosensory disease with optic neuropathy. Hum Mutat. 2019. [DOI] [PubMed] [Google Scholar]
- 22.Dickinson ME, Flenniken AM, Ji X, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537(7621):508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray CR, Abel SN, McClure MB, et al. Novel Causative Variants in DYRK1A, KARS, and KAT6A Associated with Intellectual Disability and Additional Phenotypic Features. J Pediatr Genet. 2017;6(2):77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80(19):1762–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fay RR, Popper AN. Evolution of hearing in vertebrates: the inner ears and processing. Hear Res. 2000;149(1–2):1–10. [DOI] [PubMed] [Google Scholar]
- 26.Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci U S A. 2008;105(32):11043–11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hyeon DY, Kim JH, Ahn TJ, Cho Y, Hwang D, Kim S. Evolution of the multi-tRNA synthetase complex and its role in cancer. J Biol Chem. 2019;294(14):5340–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohda M, Tokuzawa Y, Kishita Y, et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016;12(1):e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuchs SA, Schene IF, Kok G, et al. Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet Med. 2019;21(2):319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vargas A, Rojas J, Aivasovsky I, et al. Progressive Early-Onset Leukodystrophy Related to Biallelic Variants in the KARS Gene: The First Case Described in Latin America. Genes (Basel). 2020;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81(4):857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendes MI, Green LMC, Bertini E, et al. RARS1-related hypomyelinating leukodystrophy: Expanding the spectrum. Ann Clin Transl Neurol. 2020;7(1):83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malissovas N, Griffin LB, Antonellis A, Beis D. Dimerization is required for GARS-mediated neurotoxicity in dominant CMT disease. Hum Mol Genet. 2016;25(8):1528–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siekierska A, Stamberger H, Deconinck T, et al. Biallelic VARS variants cause developmental encephalopathy with microcephaly that is recapitulated in vars knockout zebrafish. Nat Commun. 2019;10(1):708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman J, Smith DE, Issa MY, et al. Biallelic mutations in valyl-tRNA synthetase gene VARS are associated with a progressive neurodevelopmental epileptic encephalopathy. Nat Commun. 2019;10(1):707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukushima K, Motomura S, Kuraoka A, Nakano H, Nishimoto T. A single point mutation of hamster aminoacyl-tRNA synthetase causes apoptosis by deprivation of cognate amino acid residue. Genes Cells. 1996;1(12):1087–1099. [DOI] [PubMed] [Google Scholar]
- 37.Song Y, Shi Y, Carland TM, et al. p53-Dependent DNA damage response sensitive to editing-defective tRNA synthetase in zebrafish. Proc Natl Acad Sci U S A. 2016;113(30):8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ladiwala U, Li H, Antel JP, Nalbantoglu J. p53 induction by tumor necrosis factor-alpha and involvement of p53 in cell death of human oligodendrocytes. J Neurochem. 1999;73(2):605–611. [DOI] [PubMed] [Google Scholar]
- 39.Li J, Ghiani CA, Kim JY, et al. Inhibition of p53 transcriptional activity: a potential target for future development of therapeutic strategies for primary demyelination. J Neurosci. 2008;28(24):6118–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma L, Yu HJ, Gan SW, et al. p53-Mediated oligodendrocyte apoptosis initiates demyelination after compressed spinal cord injury by enhancing ER-mitochondria interaction and E2F1 expression. Neurosci Lett. 2017;644:55–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Human variant data included in this study have been deposited in Leiden Open Variation Database (LOVD) and available through following accession numbers: 0000685774 (https://databases.lovd.nl/shared/variants/0000685774), 0000685798 (https://databases.lovd.nl/shared/variants/0000685798), 0000685800 (https://databases.lovd.nl/shared/variants/0000685800), 0000685801 (https://databases.lovd.nl/shared/variants/0000685801), 0000685802 (https://databases.lovd.nl/shared/variants/0000685802), 0000685803 (https://databases.lovd.nl/shared/variants/0000685803), 0000685804 (https://databases.lovd.nl/shared/variants/0000685804), 0000685805 (https://databases.lovd.nl/shared/variants/0000685805). Reagents related to zebrafish experiments described in this manuscript are available upon request.