

Abstract
Background/objectives
REALITY is an international observational retrospective registry of LHON patients evaluating the visual course and outcome in Leber hereditary optic neuropathy (LHON).
Subjects/methods
Demographics and visual function data were collected from medical charts of LHON patients with visual loss. The study was conducted in 11 study centres in the United States of America and Europe. The collection period extended from the presymptomatic stage to at least more than one year after onset of vision loss (chronic stage). A Locally Weighted Scatterplot Smoothing (LOWESS) local regression model was used to analyse the evolution of best-corrected visual acuity (BCVA) over time.
Results
44 LHON patients were included; 27 (61%) carried the m.11778G>A ND4 mutation, 8 (18%) carried the m.3460G>A ND1 mutation, and 9 (20%) carried the m.14484T>C ND6 mutation. Fourteen (32%) patients were under 18 years old at onset of vision loss and 5 (11%) were below the age of 12. The average duration of follow-up was 32.5 months after onset of symptoms. At the last observed measure, mean BCVA was 1.46 LogMAR in ND4 patients, 1.52 LogMAR in ND1 patients, and 0.97 LogMAR in ND6 patients. The worst visual outcomes were reported in ND4 patients aged at least 15 years old at onset, with a mean BCVA of 1.55 LogMAR and no tendency for spontaneous recovery. The LOESS modelling curve depicted a severe and permanent deterioration of BCVA.
Conclusions
Amongst LHON patients with the three primary mtDNA mutations, adult patients with the m.11778G>A ND4 mutation had the worst visual outcomes, consistent with prior reports.
Subject terms: Optic nerve diseases, Epidemiology
Introduction
Leber hereditary optic neuropathy (LHON) is an inherited optic neuropathy caused by mitochondrial DNA (mtDNA) mutations, which affect complex I subunits of the mitochondrial respiratory chain, impairing mitochondrial respiration and increasing production of reactive oxygen species [1–4]. Retinal ganglion cells (RGCs) are particularly vulnerable to mitochondrial dysfunction, which may lead to apoptotic cell death and axonal degeneration, ultimately resulting in optic atrophy [5, 6]. LHON affects ~1 in 30,000 to 1 in 50,000 people [4, 7, 8], with a peak age of onset between 15 and 35 years, and a large male predominance [3, 9–12]. LHON is characterised by bilateral painless central vision loss, typically sequential, with the fellow eye undergoing disease conversion weeks to a few months after onset in the first eye [13–15]. There is a well-documented incomplete penetrance, implying that the mtDNA mutations are necessary, but not sufficient to precipitate visual loss. Although there is considerable interfamilial variability, approximately 50% of men and 10% of women who carry one of the primary LHON mutations will experience visual loss during their lifetimes, highlighting also a higher male prevalence [8, 14, 16].
Three mtDNA point mutations account for about 90% of LHON cases: m.11778G>A in ND4, m.3460G>A in ND1, and m.14484T>C in ND6 [3, 4]. The m.11778G>A mutation is the most common cause of LHON worldwide and it is known to be a severe mutation with less than 15% of patients experiencing some degree of visual recovery [17, 18]. Children have a better visual prognosis, especially when age of onset is before 12 years [10, 19]. Treatment options for LHON remain limited, with some improvement demonstrated in subgroups of LHON patients treated with idebenone [20–22], and some early promising results with intravitreal gene therapy [23–26].
The REALITY study is an international multicenter observational retrospective registry of LHON patients designed to evaluate the natural history of the three most common disease-causing mtDNA mutations and the factors influencing the visual outcome.
Methods
Study design
Eleven study centres in the US and Europe (France, Italy, Spain, and the United Kingdom) participated in the REALITY registry. To qualify for study inclusion, patients had to have a diagnosis of LHON confirmed by genotyping for one of the three primary mutations (m.11778G>A in ND4, m.3460G>A in ND1 and m.14484T>C in ND6). Furthermore, patients were included only if they had undergone at least two visual function assessments, performed at any time between Year 1 and Year 3 after onset of vision loss. There was no restriction on age, and patients could have received idebenone or any other treatment. The primary source of demographic and clinical data was the enroled subjects’ medical records.
The relevant local Independent Ethics Committees approved the study protocol before subjects were identified and data collected. For patients under 18 years old, permission from a legal guardian was required to participate in the study. This study was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines.
Best-corrected visual acuity (BCVA) analysis
On-chart BCVA measures, expressed in decimal fraction or Snellen notation, were converted into LogMAR values. Off-chart BCVA was assigned the following LogMAR values: 2.0 for count fingers; 2.3 for hand motion based on the Lange scale equivalence [27]; 4.0 for light perception; and 4.5 for no light perception.
Data analysis
The Full Analysis Set included all enroled subjects whose eligibility was confirmed. Subgroup analyses were performed by LHON genotype (ND4, ND1, ND6), age at onset of vision loss (two cut-offs were applied: 12 and 15 years old), and idebenone treatment status. Change in BCVA was evaluated from the presymptomatic phase to the last available clinic observation. Missing data for presymptomatic BCVA were imputed a LogMAR value of 0 (Snellen equivalent 20/20; decimal fraction equivalent 1) [28, 29]. No other data imputation were performed for missing data.
A Locally Weighted Scatterplot Smoothing (LOWESS), non-parametric, local regression model was used on the individual BCVA data points of the LHON ND4 patients who were at least 15 years old at onset. The resulting curve depicting the evolution of BCVA over time was based on a series of polynomial regressions around each data point. The regressions used a limited look back and look forward, giving distant points less weight. The starting point of the curve was set at the onset of vision loss and included presymptomatic values (missing presymptomatic data were imputed 0 LogMAR). All computations and generation of analysis datasets and tables were performed using SAS® software version 9.4 or higher (SAS Institute, Cary, NC, USA).
Results
Study population characteristics and follow-up period
A total of 44 affected LHON patients were included in the REALITY study: 34/44 (77%) from European countries (France, Italy, Spain, and the United Kingdom) and 10/44 (23%) from the United States of America (Table 1). The majority of enroled patients were male (33/44, 75%) and Caucasian (33/44, 75%) (Table 1). The mean age at onset of vision loss was 27.9 years (range: 4–71 years), with 30/44 (68%) patients ≥18 years old at the onset of vision loss. The proportion of patients who were at least 15 years old at onset was 82% (36/44). Five (11%) patients were 12 years old or younger at onset.
Table 1.
Demographics of LHON cohort.
| All patients (N = 44) |
ND4 patients (N = 27) |
ND1 patients (N = 8) |
ND6 patients (N = 9) |
ND4 patients aged ≥ 15 at onset (N = 23) |
|
|---|---|---|---|---|---|
| Age at onset of vision lossa (years) | |||||
| Mean (SD) | 27.9 (18.7) | 30.2 (20.1) | 24.9 (15.8) | 23.6 (17.7) | 34.2 (19.0) |
| Median | 21.5 | 23.0 | 17.5 | 21.0 | 25.0 |
| IQR | 16.0, 32.0 | 16.0, 39.0 | 15.5, 29.0 | 14.0, 24.0 | 19.0, 57.0 |
| Min, Max | 4, 71 | 4, 71 | 14, 61 | 8, 68 | 16, 71 |
| Categories of age at onseta | |||||
| ≤12 years old | 5/44 (11.4%) | 3/27 (11.1%) | 0/8 (0.0%) | 2/9 (22.2%) | NA |
| 13 to 14 years old | 3/44 (6.8%) | 1/27 (3.7%) | 1/8 (12.5%) | 1/9 (11.1%) | NA |
| 15 to 17 years old | 6/44 (13.6%) | 3/27 (11.1%) | 3/8 (37.5%) | 0/9 (0.0%) | 3/23 (13.0%) |
| 18 and older | 30/44 (68.2%) | 20/27 (74.1%) | 4/8 (50.0%) | 6/9 (66.7%) | 20/23 (87.0%) |
| Sex | |||||
| Male | 33/44 (75.0%) | 22/27 (81.5%) | 5/8 (62.5%) | 6/9 (66.7%) | 18/23 (78.3%) |
| Female | 11/44 (25.0%) | 5/27 (18.5%) | 3/8 (37.5%) | 3/9 (33.3%) | 5/23 (21.7%) |
| Ethnicity | |||||
| Caucasian/White | 33/44 (75.0%) | 20/27 (74.1%) | 5/8 (62.5%) | 8/9 (88.9%) | 18/23 (78.3%) |
| Black | 1/44 (2.3%) | 1/27 (3.7%) | 0/8 (0.0%) | 0/9 (0.0%) | 1/23 (4.3%) |
| Asian | 1/44 (2.3%) | 1/27 (3.7%) | 0/8 (0.0%) | 0/9 (0.0%) | 0/23 (0.0%) |
| Unknown | 9/44 (20.4%) | 5/27 (18.5%) | 3/8 (37.5%) | 1/9 (11.1%) | 4/23 (17.4%) |
| Country of origin | |||||
| France | 9/44 (20.5%) | 6/27 (22.2%) | 3/8 (37.5%) | 0/9 (0.0%) | 5/23 (21.7%) |
| Italy | 12/44 (27.3%) | 6/27 (22.2%) | 4/8 (50.0%) | 2/9 (22.2%) | 5/23 (21.7%) |
| Spain | 5/44 (11.4%) | 2/27 (7.4%) | 1/8 (12.5%) | 2/9 (22.2%) | 2/23 (8.7%) |
| United Kingdom | 8/44 (18.2%) | 5/27 (18.5%) | 0/8 (0.0%) | 3/9 (33.3%) | 4/23 (17.4%) |
| United States | 10/44 (22.7%) | 8/27 (29.6%) | 0/8 (0.0%) | 2/9 (22.2%) | 7/23 (30.4%) |
IQR inter-quartile range, NA not applicable, SD standard deviation.
aOnset in first-affected eye.
Three LHON patients had been diagnosed with an additional genetic syndrome: (i) the blepharophimosis, ptosis, and epicanthus inversus syndrome; (ii) the Ehlers–Danlos syndrome; (iii) and the Marcus–Gunn syndrome. In addition, one woman was diagnosed with multiple sclerosis following the onset of visual loss from LHON.
Most patients (27/44, 61%) were seen by an ophthalmologist or a neuro-ophthalmologist within the first 3 months of disease onset. The mean time from onset to the first visit was 5.7 months (range: 0–25.7 months), and the mean duration of follow-up was 32.5 months (range: 14.1–178.3 months) (Table 2). Seven patients (7/44, 16%) had a follow-up of more than 3 years, 6 of whom carried the m.11778G>A mutation and one the m.14484T>C mutation. The mean number of BCVA visits per patient was 7.9, with a range of 4–28 visits reported per patient.
Table 2.
BCVA data collection in REALITY.
| All patients (N = 44) |
ND4 patients (N = 27) |
ND1 patients (N = 8) |
ND6 patients (N = 9) |
ND4 patients aged ≥ 15 at onset (N = 23) |
|
|---|---|---|---|---|---|
| Time from onset to first visit (months) | |||||
| Mean (SD) | 5.7 (7.3) | 6.9 (7.8) | 3.0 (6.2) | 4.5 (6.2) | 6.8 (7.2) |
| Median | 1.9 | 2.9 | 0.6 | 1.3 | 3.0 |
| IQR | 0.5, 11.2 | 0.8, 14.5 | 0.2, 2.0 | 1.1, 6.0 | 0.8, 14.5 |
| Min, Max | 0.0, 25.7 | 0.0, 25.7 | 0.0, 18.2 | 0.0, 17.8 | 0.0, 20.5 |
| Time period of first visit post-onset | |||||
| ≤1 month | 14/44 (31.8%) | 7/27 (25.9%) | 5/8 (62.5%) | 2/9 (22.2%) | 6/23 (26.1%) |
| >1 to 3 months | 13/44 (29.5%) | 7/27 (25.9%) | 2/8 (25.0%) | 4/9 (44.4%) | 5/23 (21.7%) |
| >3 to 6 months | 4/44 (9.1%) | 3/27 (11.1%) | 0/8 (0.0%) | 1/9 (11.1%) | 3/23 (13.0%) |
| >6 to 12 months | 4/44 (9.1%) | 3/27 (11.1%) | 0/8 (0.0%) | 1/9 (11.1%) | 3/23 (13.0%) |
| >2 months | 9/44 (20.5%) | 7/27 (25.9%) | 1/8 (12.5%) | 1/9 (11.1%) | 6/23 (26.1%) |
| Time from onset to last visit (months) | |||||
| Mean (SD) | 32.5 (24.1) | 36.3 (29.8) | 26.0 (6.0) | 26.7 (8.6) | 36.8 (32.2) |
| Median | 30.3 | 30.4 | 27.3 | 30.3 | 29.3 |
| IQR | 23.3, 34.5 | 25.0, 35.4 | 22.2, 31.1 | 18.7, 35.1 | 24.4, 35.3 |
| Min, Max | 14.1, 178.2 | 15.9, 178.2 | 15.3, 31.4 | 14.1, 36.0 | 15.9, 178.2 |
| Time period of last visit post-onset | |||||
| ≤18 months | 4/44 (9.1%) | 1/27 (3.7%) | 1/8 (12.5%) | 2/9 (22.2%) | 1/23 (4.3%) |
| >18 to 24 months | 8/44 (18.2%) | 4/27 (14.8%) | 2/8 (25.0%) | 2/9 (22.2%) | 4/23 (17.4%) |
| >24 to 30 months | 9/44 (20.5%) | 8/27 (29.6%0 | 1/8 (12.5%) | 0/9 (0.0%) | 7/23 (30.4%) |
| >30 to 36 months | 16/44 (36.4%) | 8/27 (29.6%) | 4/8 (50.0%) | 4/9 (44.4%) | 6/23 (26.1%) |
| >36 months | 7/44 (15.9%) | 6/27 (22.2%) | 0/8 (0.0%) | 1/9 (11.1%) | 5/23 (21.7%) |
| Number of eyes with BCVA data available in time period | |||||
| Up to Year 1 after onset | 59/88 (67.0%) | 35/54 (64.8%) | 10/16 (62.5%) | 14/18 (77.8%) | 31/46 (67.4%) |
| Year 1 to Year 2 after onset | 76/88 (86.4%) | 44/54 (81.5%) | 16/16 (100.0%) | 16/18 (88.9%) | 39/46 (84.8%) |
| Year 2 to Year 3 after onset | 59/88 (67.0%) | 42/54 (77.8%) | 10/16 (62.5%) | 7/18 (38.9%) | 34/46 (73.9%) |
| >Year 3 after onset | 14/88 (15.9%) | 12/54 (22.2%) | 0/16 (0.0%) | 2/18 (11.1%) | 10/46 (21.7%) |
| Number of BCVA visits per patient | |||||
| Mean (SD) | 7.9 (4.9) | 7.2 (3.9) | 8.0 (3.7) | 9.9 (8.0) | 7.3 (4.2) |
| Median | 6.0 | 6.0 | 7.0 | 6.0 | 6.0 |
| IQR | 5.5, 8.0 | 5.0, 8.0 | 6.0, 9.0 | 6.0, 10.0 | 4.0, 8.0 |
| Min, Max | 4, 28 | 4, 22 | 4, 16 | 4, 28 | 4, 22 |
BCVA best-corrected visual acuity, IQR interquartile range, SD standard deviation.
Date of onset of vision loss in the first eye to be affected was used to calculate times from onset for each patient.
LHON genotypes
Twenty-seven patients (27/44, 61%) carried the m.11778G>A mutation in ND4, 8 patients (8/44, 18%) carried the m.3460G>A mutation in ND1, and 9 patients (9/44, 21%) carried the m.14484T>C mutation in ND6, with a mean age at onset of 30.2, 24.9, and 23.6 years, respectively (Table 1). Among the 36 patients who were at least 15 years old at onset, there were 23 ND4 patients (64%), 7 ND1 patients (19%) and 6 ND6 patients (17%).
Idebenone status
Twenty-five patients (25/44, 57%) had taken idebenone with a mean cumulative duration of treatment of 55.7 months (Supplementary Table S1). The treated group included 16/27 (59%) ND4 patients, 5/8 (62%) ND1 patients and 4/9 (44%) ND6 patients. Treatment was started during the first year after disease onset in 21/25 (84%) patients.
BCVA evolution based on LHON genotypes and age of onset
For the entire study cohort, the mean (standard deviation (SD)) BCVA was 1.37 (0.78) LogMAR at last observed measure (mean of 32.2 months from onset of vision loss) (Table 3). Based on the LHON genotype, the mean (SD) BCVA was 1.46 (0.63) LogMAR in ND4 patients, 1.52 (1.06) LogMAR in ND1 patients, and 0.97 (0.83) LogMAR in ND6 patients (Table 3).
Table 3.
BCVA outcomes according to LHON genotypes.
| All patients (N = 88 eyes) | ND4 patients (N = 54 eyes) | ND1 patients (N = 16 eyes) | ND6 patients (N = 18 eyes) | ND4 patients aged ≥ 15 at onset (N = 46 eyes) | ND4 patients aged ≥ 15 at onset and treated with idebenone (N = 30 eyes) | |
|---|---|---|---|---|---|---|
| Time from onset to last visit (months) | ||||||
| Mean (SD) | 32.2 (24.0) | 36.3 (29.5) | 26.0 (5.8) | 25.6 (8.6) | 36.8 (31.8) | 40.5 (38.0) |
| Presymptomatic BCVA (LogMAR)a | 0 | 0 | 0 | 0 | 0 | 0 |
| Last-observed BCVA (LogMAR) | ||||||
| Mean (SD) | 1.37 (0.78) | 1.46 (0.63) | 1.52 (1.06) | 0.97 (0.83) | 1.55 (0.62) | 1.57 (0.62) |
| 95% CI | 1.15, 1.59 | 1.17, 1.74 | 1.00, 2.04 | 0.48, 1.46 | 0.96, 1.54 | 0.71, 1.52 |
| Median | 1.30 | 1.47 | 1.60 | 0.75 | 1.60 | 1.60 |
| Minimum, maximum | 0.00, 4.00 | 0.00, 2.30 | 0.10, 4.00 | 0.00, 2.30 | 0.00, 2.30 | 0.10, 2.30 |
| Q1, Q3 | 0.80, 2.00 | 1.10, 2.00 | 0.00, 2.00 | 0.22, 2.00 | 1.20, 2.00 | 1.10, 2.00 |
BCVA best-corrected visual acuity; SD standard deviation.
aMissing presymptomatic BCVA were assigned a value of 0 LogMAR (normal visual acuity).
At last observed measure, LHON patients ≤ 12 years old at onset had a mean (SD) BCVA of 0.65 (0.52) LogMAR, compared with 1.46 (0.77) LogMAR in patients over 12 years old at onset (p = 0.0193) (Supplementary Table S2).
When considering LHON patients aged at least 15 years at onset, 55/72 (76%) of eyes had a BCVA of 1.0 LogMAR or worse at last observed measure: 39/46 (85%) for ND4 patients, 8/14 (57%) for ND1 patients and 8/12 (67%) for ND6 patients (Fig. 1). The proportion of eyes with off-chart BCVA was comparable for the three LHON genotypes: 22/46 (48%) for ND4 patients, 6/14 (43%) for ND1 patients and 5/12 (42%) for ND6 patients. The proportion of patients with a BCVA better than 1.0 LogMAR in at least one eye at the last observed measure was 5/23 (22%) for ND4 patients; 3/7 (43%) for ND1 patients and 2/6 (33%) for ND6 patients.
Fig. 1. BCVA data collection in REALITY.
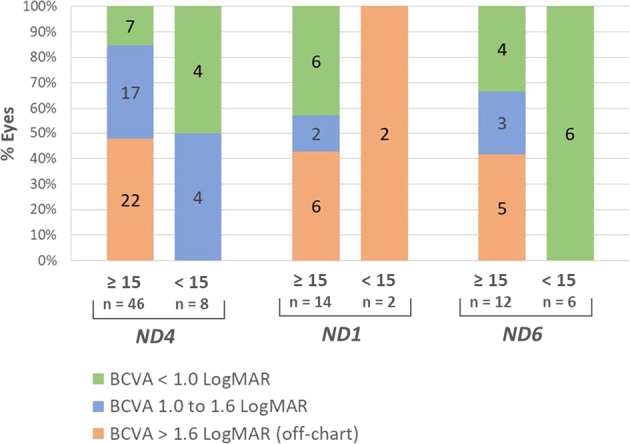
The distribution of BCVA data collected in the REALITY registry is presented according to LHON genotype and age at onset (below 15 or at least 15 years old). The number of eyes is displayed for each category.
For LHON patients who were below 15 years of age at onset, 6/16 (37%) eyes had a BCVA of 1.0 LogMAR or worse at last observed measure: 4/8 (50%) of ND4 patients, 2/2 (100%) of ND1 patients and 0/6 (0%) of ND6 patients (Fig. 1). Of note, no eyes were off-chart at the last observed measure in ND4 and ND6 patients who were less than 15 years old at onset (Fig. 1).
Subgroup analysis of LHON ND4 patients aged ≥15 years at onset
Twenty-three LHON patients carrying the m.11778G>A ND4 mutation were at least 15 years old at onset, with the majority being male (18/23, 78.3%). The mean (SD) age at onset was 34.2 (19.0) years and 3/23 (13%) patients were between 15 and 18 years old at onset of vision loss (Table 1). The mean (SD) time from onset to the first visit was 6.8 (7.2) months and the mean duration of follow-up was 36.8 (32.2) months for this subgroup (Table 2).
The mean (SD) BCVA was 1.55 (0.62) LogMAR at the last observed measure (Table 3). Fifteen (15/23, 65%) ND4 patients aged at least 15 years at onset were treated with idebenone during the observational period (Supplementary Table S1). For nine of these patients, the mean (SD) cumulative duration of treatment with idebenone was 70.0 (24.9) months. At the last observed measure, the mean (SD) BCVA for the idebenone-treated group was 1.57 (0.62) LogMAR on average 40.5 (38.0) months after onset (Table 3).
The LOWESS curve depicting the evolution of BCVA over time showed a marked loss of visual acuity in the first 18 months after disease onset (Fig. 2). Following this initial drop of vision, the deterioration of BCVA continued at a slower rate and no trend of visual recovery was observed.
Fig. 2. BCVA evolution in ND4patients aged ≥15 at onset of vision loss.
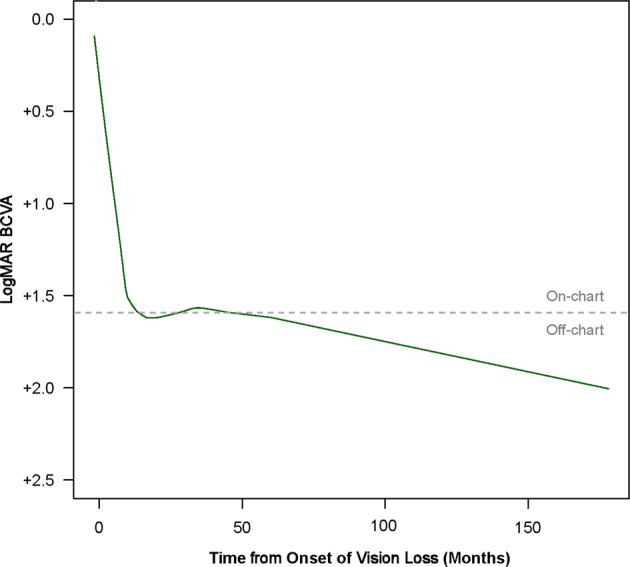
The LOWESS regression curve for ND4 patients aged at least 15 at onset of vision loss (n = 23 patients, 213 individual BCVA measures) was based on fitting a series of linear regressions around each data point collected. The regressions used a limited look back and look forward, giving distant points less weight. The starting point of the curves was set at the onset of vision loss and included presymptomatic values (missing presymptomatic data were imputed a value of 0 LogMAR).
Discussion
The REALITY study cohort is representative of the general LHON patient population with regards to the age of onset (mean of 27.9 years) and gender distribution (75% males) [3, 12]. Prior studies have estimated the peak age of onset to be between 15 and 35 years old, although the reported range for age at onset in molecularly-confirmed LHON patients varies from 2 to 87 years of age [3, 10, 11]. The proportion of patients in REALITY who were less than 15 years old at disease onset was 18% (8/44) and the predominant genotype among all included patients was the m.11778G>A ND4 mutation (61%), also in keeping with the literature. [5, 6, 15, 17, 30–32] Half of the patients enroled in REALITY had received treatment with idebenone for a cumulative period of more than 4 years.
The genetic syndromes reported in three patients included in REALITY are unlikely to be related to the underlying LHON-causative mtDNA mutation. One additional patient was diagnosed with multiple sclerosis. The co-occurrence of visual loss due to LHON and central nervous system demyelination is often referred to as Harding’s disease [33], and this overlap syndrome can occur with all three primary LHON mutations, predominantly in women [15, 33, 34].
The two most important predictors of visual outcome in LHON are the underlying causative mtDNA mutation and the age at onset of vision loss. In the REALITY study, LHON patients carrying the m.14484T>C mutation achieved better final vision compared with those carrying the m.11778G>A and m.3460G>A mutations. The literature reports that children who become affected at the age of 12 years or younger achieve significantly better vision compared with adult-onset LHON patients [13, 17, 19, 35]. Accordingly, in the REALITY registry we observed that patients ≤12 years old at onset had a mean final BCVA of 0.65 LogMAR, compared with 1.46 LogMAR for those over 12 years old at onset. Furthermore, the majority of eyes of LHON patients who were below the age of 15 at onset had a BCVA better than 1.0 LogMAR at the last observed measure, and all the ND4 and ND6 patients younger than 15 at onset had on-chart visual acuity in both eyes, confirming the better visual prognosis in these younger groups.
In the LHON ND4 subgroup of patients who were at least 15 years old at onset, the majority of eyes had a BCVA of 1.0 LogMAR or worse at the last observed measure. The m.11778G>A mutation is known to carry a poor prognosis, with <15% of patients experiencing spontaneous partial visual recovery, and few achieving visual acuities better than 20/200. [17] In REALITY, 84.8% (39/46) of eyes from LHON ND4 patients who were at least 15 at onset had BCVA of 20/200 or worse, and 47.8% (22/46) were off-chart at the last observed measure. In this subgroup, a LOWESS model curve showed a decline in two phases: a rapid decline in the first 18 months, followed by a progressive slow decline. A prospective long-term follow-up study of untreated LHON ND4 patients is needed to confirm the projection of this model, which has important implications for the treatment of chronic cases.
The REALITY registry bears a number of limitations due to the retrospective nature of its study design and the relatively small sample size of 44 LHON patients. Nevertheless, the findings of this study confirm the better visual outcome with childhood-onset LHON and the poor visual prognosis for patients with the m.11778G>A mutation who were at least 15 years old at disease onset.
Summary
What was known before
LHON is a rare mitochondrial blinding disease affecting mostly males between 15 and 35 years old.
Three primary mtDNA mutations are responsible for 90% of the cases.
Spontaneous partial visual recovery is scarcely reported in the literature.
What this study adds
This international retrospective registry study provided a better understanding of the evolution of visual acuity in LHON patients, including clinical data from 44 subjects aged 4 to 71 years at onset of vision loss.
LHON subjects carrying the ND4 mutation did not show a trend for spontaneous improvement over time.
LHON subjects aged 12 or less at onset showed the best visual prognosis.
Supplementary information
Acknowledgements
We are grateful to the study teams that contributed to the conduct of the REALITY registry in the various recruitment centres. We would also like to thank the patients who took part in this study. PYWM is supported by a Clinician Scientist Fellowship Award (G1002570) from the Medical Research Council (UK), and also receives funding from Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity, the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the International Foundation for Optic Nerve Disease (IFOND), the UK National Institute of Health Research (NIHR) as part of the Rare Diseases Translational Research Collaboration, the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014), and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. NJN and VB are supported in part by an ophthalmology department core grant from the NIH/NEI (P30EY006360). VC is supported by grants from the Italian Ministry of Health (RF-2018-12366703), the Italian Ministry of Research (20172T2MHH), and Telethon Italy (GUP15016). VC is also supported by patients’ organisations MITOCON and IFOND, and patients’ donations. JAS is supported by the Agence Nationale de la Recherche within the Programme Investissements d’Avenir, Institut Hospitalo Universitaire FOReSIGHT [ANR-18-IAHU-0001], LabEx LIFESENSES (ANR-10-LABX-65), and an unrestricted centre grant by Research to Prevent Blindness.
for the LHON REALITY Study Group
Rima Hussain3,4, Rasha Jorany3,4, Priyansha Sheel3,4, Lindreth DuBois24, Michele Carbonelli6,7, Lidia Di Vito6,7, Martina Romagnoli6,7, Adam A. DeBusk25, Maria Massini26, Rabih Hage10,11, Gad Heilweil26, Irena Tsui26, Virginia Garcia27, Antonio Morilla27, Piero Barboni28, Maria Lucia Cascavilla28, Marco Battista28, Francesca Calcagno28, Adelaide Pina28
Author contributions
All authors participated in the conceptualisation, data collection, analysis, interpretation, writing, and critical review of the manuscript.
Funding
GenSight Biologics (Paris, France) fully funded and sponsored the study.
Data and materials availability
All data associated with this study are available in the main text or the supplementary materials
Compliance with ethical standards
Conflict of interest
PYWM is a consultant for GenSight Biologics and Stealth BioTherapeutics and has received research support from GenSight Biologies and Santhera Pharmaceuticals. NJN is a consultant for GenSight Biologics, Santhera Pharmaceuticals and Stealth BioTherapeutics, has received research support from GenSight Biologics and Santhera Pharmaceuticals, served on the Data Safety Monitoring Board for the Quark NAION study and is a medical-legal consultant. VC is a consultant for Santhera Pharmaceuticals, GenSight Biologics, and Stealth BioTherapeutics and has received research support from Santhera Pharmaceuticals and Stealth BioTherapeutics. CV-C is a consultant for Santhera Pharmaceuticals and GenSight Biologics. VB is a consultant for GenSight Biologics. MM is a consultant for GenSight Biologics and has received research support from GenSight. AAS is a consultant for Stealth BioTherapeutics. LB and MT are GenSight Biologics employees. JAS is a co-founder and shareholder of GenSight Biologics, and a patent co-author on allotopic transport. Authors declare competing financial interests in relation with the work submitted (see “Acknowledgements” for details).
Footnotes
Clinical Trial Number: NCT03295071 (ClinicalTrials.gov).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the LHON REALITY Study Group are listed below acknowledgements.
Contributor Information
Patrick Yu-Wai-Man, Email: py237@cam.ac.uk.
for the LHON REALITY Study Group:
Rima Hussain, Rasha Jorany, Priyansha Sheel, Lindreth DuBois, Michele Carbonelli, Lidia Di Vito, Martina Romagnoli, Adam A. DeBusk, Maria Massini, Rabih Hage, Gad Heilweil, Irena Tsui, Virginia Garcia, Antonio Morilla, Piero Barboni, Maria Lucia Cascavilla, Marco Battista, Francesca Calcagno, and Adelaide Pina
Supplementary information
The online version contains supplementary material available at 10.1038/s41433-021-01535-9.
References
- 1.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–30. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 2.Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, et al. Idebenone Treatment in Leber’s Hereditary Optic Neuropathy. Brain. 2011;134:1–5. doi: 10.1093/brain/awr180. [DOI] [PubMed] [Google Scholar]
- 3.Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517–23. doi: 10.1016/j.ajo.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 4.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–14. doi: 10.1016/j.preteyeres.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu-Wai-Man P, Votruba M, Burté F, La Morgia C, Barboni P, Carelli V. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016;132:789–806. doi: 10.1007/s00401-016-1625-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carelli V, Carbonelli M, De Coo IF, Kawasaki A, Klopstock T, Lagrèze WA, et al. International consensus statement on the clinical and therapeutic management of leber hereditary optic neuropathy. J Neuro-Ophthalmol. 2017;37:371–81. doi: 10.1097/WNO.0000000000000570. [DOI] [PubMed] [Google Scholar]
- 7.Puomila A, Hämäläinen P, Kivioja S, Savontaus ML, Koivumäki S, Huoponen K, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15:1079–89. doi: 10.1038/sj.ejhg.5201828. [DOI] [PubMed] [Google Scholar]
- 8.Spruijt L, Kolbach DN, De Coo RF, Plomp AS, Bauer NJ, Smeets HJ, et al. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol. 2006;141:676–82. doi: 10.1016/j.ajo.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Yu-Wai-Man P, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet. 2003;72:333–9. doi: 10.1086/346066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barboni P, Savini G, Valentino ML, La Morgia C, Bellusci C, De Negri AM, et al. Leber’s hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. 2006;47:5303–9. doi: 10.1167/iovs.06-0520. [DOI] [PubMed] [Google Scholar]
- 11.Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. 2009;46:145–58. doi: 10.1136/jmg.2007.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman NJ, Biousse V. Hereditary optic neuropathies. Eye. 2004;18:114460. doi: 10.1038/sj.eye.6701591. [DOI] [PubMed] [Google Scholar]
- 13.Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–37. doi: 10.1093/brain/118.2.319. [DOI] [PubMed] [Google Scholar]
- 14.Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. Am J Med Genet. 2002;39:162–9. doi: 10.1136/jmg.39.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750–62. doi: 10.1016/S0002-9394(14)76784-4. [DOI] [PubMed] [Google Scholar]
- 16.Giordano C, Iommarini L, Giordano L, Maresca I, Pisano A, Valentino ML, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain. 2014;137:335–53. doi: 10.1093/brain/awt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman NJ, Carelli V, Taiel M, Yu-Wai-Man P. Visual outcomes in Leber hereditary optic neuropathy patients with the m.11778G>A (MTND4) mitochondrial DNA mutation. J Neuroophthalmol. 2020;40:547–57. [DOI] [PubMed]
- 18.Guo DY, Wang XW, Hong N, Gu YS. A Meta-analysis of the association between different genotypes (G11778A, T14484C and G3460A) of Leber hereditary optic neuropathy and visual prognosis. Int J Ophthalmol. 2016;9:1493–8. doi: 10.18240/ijo.2016.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majander A, Bowman R, Poulton J, Antcliff RJ, Reddy MA, Michaelides M, et al. Childhood-onset Leber hereditary optic neuropathy. Br J Ophthalmol. 2017;101:1505–9. doi: 10.1136/bjophthalmol-2016-310072. [DOI] [PubMed] [Google Scholar]
- 20.Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–86. doi: 10.1093/brain/awr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Catarino CB, Von Livonius B, Priglinger C, B0anik R, Matloob S, Tamhankar MA, et al. Real-world clinical experience with Idebenone in the treatment of Leber Hereditary Optic Neuropathy. J Neuroophthalmol. 2020;40:558–65. [DOI] [PMC free article] [PubMed]
- 23.Yu-Wai-Man P, Newman NJ, Carelli V, Moster ML, Biousse V, Sadun AA, et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med. 2020;12:eaaz7423. doi: 10.1126/scitranslmed.aaz7423. [DOI] [PubMed] [Google Scholar]
- 24.Newman NJ, Yu-Wai-Man P, Carelli V, Moster ML, Biousse V, Vignal-Clermont C, et al. Efficacy and safety of intravitreal gene therapy for Leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. 2021 In press. 10.1016/j.ophtha.2020.12.012. [DOI] [PubMed]
- 25.Guy J, Feuer WJ, Davis JL, Porciatti V, Gonzalez PJ, Koilkonda RD, et al. Gene therapy for Leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology. 2017;124:1621–34. doi: 10.1016/j.ophtha.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J, Zhang Y, Liu H, Wang D, Du Y, Tian Z, et al. Seven-year follow-up of gene therapy for Leber’s hereditary optic neuropathy. Ophthalmology. 2020;127:1125–7. doi: 10.1016/j.ophtha.2020.02.023. [DOI] [PubMed] [Google Scholar]
- 27.Lange C, Feltgen N, Junker B, Schulze-Bonsel K, Bach M. Resolving the clinical acuity categories “hand motion” and “counting fingers” using the Freiburg Visual Acuity Test (FrACT) Graefes Arch Clin Exp Ophthalmol. 2009;247:137–42. doi: 10.1007/s00417-008-0926-0. [DOI] [PubMed] [Google Scholar]
- 28.Guy J, Feuer WJ, Porciatti V, Schiffman J, Abukhalil F, Vandenbroucke R, et al. Retinal ganglion cell dysfunction in asymptomatic G11778A: Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2014;55:841–8. doi: 10.1167/iovs.13-13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang TJ, Karanjia R, Moraes-Filho MN, Gale J, Show Tran J, Chu ER, et al. Natural History of Conversion of Leber’s Hereditary Optic Neuropathy: A Prospective Case Series. Ophthalmology. 2017;124:843–50. doi: 10.1016/j.ophtha.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Hotta Y, Fujiki K, Hayakawa M, Nakajima A, Kanai A, Mashima Y, et al. Clinical features of Japanese Leber’s hereditary optic neuropathy with 11778 mutation of mitochondrial DNA. Jpn J Ophthalmol. 1995;39:96–108. [PubMed] [Google Scholar]
- 31.Van Senus AH. Leber’s disease in the Netherlands. Doc Ophthalmol. 1963;17:1162. doi: 10.1007/BF00573524. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg T, Nørby S, Schwartz M, Saillard J, Magalhães PJ, Leory D, et al. Prevalence and genetics of Leber hereditary optic neuropathy in the Danish population. Invest Ophthalmol Vis Sci. 2016;57:1370–5. doi: 10.1167/iovs.15-18306. [DOI] [PubMed] [Google Scholar]
- 33.Harding AE, Riordan-Eva P, Govan GG, Mitochondrial DNA. diseases: genotype and phenotype in Leber’s hereditary optic neuropathy. Muscle Nerve Suppl. 1995;3:S82–84. doi: 10.1002/mus.880181417. [DOI] [PubMed] [Google Scholar]
- 34.Bargiela D, Chinnery PF. Mitochondria in neuroinflammation—multiple sclerosis (MS), Leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci Lett. 2019;710:132932. doi: 10.1016/j.neulet.2017.06.051. [DOI] [PubMed] [Google Scholar]
- 35.Nikoskelainen EK, Huoponen K, Juvonen V, Lamminen T, Nummelin K, Savontaus ML. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations [published correction appears in. Ophthalmology 1996 Jul;103(7):998] Ophthalmology. 1996;103:504–14. doi: 10.1016/S0161-6420(96)30665-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are available in the main text or the supplementary materials