

Abstract
This randomized, double-blind, placebo-controlled, multiple ascending–dose study evaluated safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple doses of milvexian, an oral small-molecule FXIa inhibitor, in healthy Japanese participants. Participants received oral milvexian daily under fasted (50 mg and 200 mg) or fed conditions (500 mg) or placebo over 14 days; 24 participants (8/cohort: 6 milvexian; 2 placebo) were planned. Due to an unblinding event, participants in one cohort (200 mg daily) were discontinued, and a second cohort enrolled; 32 participants were included in safety and pharmacodynamic analyses, and 24/32 in pharmacokinetic analyses. Milvexian up to 500 mg daily for 14 days was generally well tolerated, with no deaths, serious adverse events, or discontinuations due to adverse events. Milvexian exposure increased between 50-mg and 200-mg doses. Median Tmax was similar with 50-mg and 200-mg doses (2.5–3.0 h) and delayed under fed conditions (500 mg, 7.0–8.0 h). Median T1/2 was similar across doses (8.9–11.9 h). Multiple oral milvexian administrations resulted in concentration-related prolongation of aPTT and decreased FXI clotting activity. Milvexian was generally safe and well tolerated. The pharmacokinetic and pharmacodynamic profile of milvexian demonstrates suitability for further clinical development in Japanese participants.
Subject terms: Drug discovery, Cardiology
Introduction
Anticoagulant and antiplatelet therapies are accepted in routine clinical care as prophylaxis against serious adverse thromboembolic events in patients with cardiovascular diseases, such as atrial fibrillation, or for patients with a history of ischemic stroke, arteriosclerosis, acute coronary syndrome, coronary artery disease, or peripheral artery disease1–5. However, antithrombotic therapies are also associated with an increased risk of major bleeding6. This underscores the need for novel therapies with an improved benefit/risk profile compared with currently available options.
The coagulation process encompasses a number of highly coordinated pathways that balance clot formation and dissolution7. Coagulation factors are essential for maintaining hemostasis, and their activity is mediated by thrombin, which plays an instrumental role in clot formation through the conversion of fibrinogen to fibrin and by activating platelet aggregation7,8. The zymogen factor XI (FXI) is converted by thrombin to active FXI (FXIa), a protease, through which a positive feedback loop enhances both thrombin formation and consolidation of coagulation9,10. Inhibition of key factors within the coagulation cascade is a mechanism that could be used for the prevention and treatment of thrombotic events. Spontaneous bleeding is rare in individuals with congenital FXI deficiencies, and the only clinical manifestation of FXI deficiency is a tendency for mild bleeding after a serious injury or surgery11–13. Preclinical models showed reductions in thrombus formation and clinical studies have demonstrated a reduced risk of adverse cardiovascular events and venous thromboembolism with FXI deficiency14–18. In addition, both clinical and preclinical models suggest that FXIa inhibitors can reduce thrombus formation and that normal hemostasis is not solely dependent on the FXI pathway19–22. Taken together, this evidence suggests that inhibition of FXIa can prevent thrombosis while preserving hemostasis9,10,23. Therefore, FXIa inhibitors have the potential to improve the benefit/risk profile of current anticoagulants through a safer bleeding profile in a variety of conditions where patients are predisposed to a high risk of thrombotic or bleeding events.
Milvexian is a potent orally bioavailable small molecule that binds and inhibits FXIa with high affinity and selectivity24. Milvexian is being developed to prevent and treat thrombotic events in multiple patient populations.
Milvexian has demonstrated antithrombotic activity in preclinical models of arterial and venous thrombosis while preserving hemostasis25,26. In addition, phase 1 studies showed that milvexian was generally safe and well tolerated in healthy non-Japanese participants and participants with hepatic impairment27,28. A phase 2 study is underway investigating the use of milvexian for the secondary prevention of major cardiovascular events in patients with acute ischemic stroke, and a phase 2 study on the use of milvexian for the prevention of venous thromboembolism events in patients undergoing total knee replacement surgery has been completed29,30.
Genetic variation in the metabolism of anticoagulants within ethnic groups has been well documented, underscoring the need to both demonstrate safety and to investigate the potential for dose adjustments within Japanese and other patient populations31,32. Thus, further exploration of the safety and pharmacokinetic (PK) properties of milvexian is necessary to support clinical development within the Japanese population. The primary objective of this study was to assess the safety and tolerability of multiple oral doses of milvexian in healthy Japanese participants. Secondary and exploratory objectives included assessment of multiple-dose PK and pharmacodynamics (PD) of milvexian.
Methods
Ethics
This study was conducted in accordance with Good Clinical Practice, as defined by the International Council for Harmonisation and in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study was registered with ClinicalTrials.gov (NCT03224260; registered on 07/18/2017; first posted on 07/21/2017). The protocol, amendments, and participant informed consent received appropriate approval by the Independent Ethics Committee (IEC) and the Institutional Review Board (IRB) of Alpha IRB (San Clemente, CA, USA) prior to initiation of the study at the site. The IRB/IEC ensured the protection of the rights, safety, and well-being of human participants involved in the clinical investigation. Prior to the beginning of the study, all subjects provided written informed consent. The study was conducted at 1 site in the United States, and all laws and regulatory requirements were adhered to.
Study design
This was a randomized, double-blind, placebo-controlled, multiple ascending–dose study. Participants underwent screening evaluations to determine eligibility within 21 days prior to study treatment administration. Within each panel, participants were randomized to milvexian or matched placebo according to a computer-generated randomization scheme prepared by a randomization coordinator. Randomization schedules were shipped directly to a pharmacist or other individual(s) responsible for the dispensing of blinded study treatment. Participants received once-daily study treatment from study personnel sequentially under fasted conditions at 50 mg (Cohort 1) and at 200 mg (Cohorts 2a and 2b) and under fed conditions at 500 mg (Cohort 3) on Days 1 through 14.
The dosages investigated in this study were chosen based on the results of the first-in-human study of milvexian where participants received milvexian at doses between 4 and 500 mg28. Dose-proportional plasma concentrations were observed between 20 and 200 mg, and milvexian was safe and well tolerated through doses up to 500 mg daily for 14 days. The 50-mg and 200-mg doses under fasted conditions and 500-mg dose under fed conditions were selected to capture the dose-proportional range as well as the upper end of the dosing range investigated in the first-in-human study.
An unblinding event resulted in early discontinuation of all participants in Cohort 2a, and the protocol was amended to include enrollment of a second group of participants who received milvexian 200 mg or placebo (Cohort 2b).
Participants
Eligible participants included healthy (as determined by medical history, physical examination, electrocardiogram [ECG], and clinical laboratory evaluations) men and women of non-childbearing potential who were 18 to 55 years of age and had a body mass index of 18.0 to 25.0 kg/m2. All participants were first-generation Japanese, defined as having been born in Japan and not living outside of Japan for > 10 years, and having both parents be ethnically Japanese. Women of childbearing potential or who were breastfeeding were excluded, and participants were ineligible if there was evidence of any significant acute or chronic medical illness, current or recent (within 3 months) gastrointestinal disease, or any major surgery within 4 weeks of study treatment administration.
Safety assessments
Safety assessments were based on medical review of adverse event (AE) reports and the results of vital sign measurements, ECGs, physical examinations, and clinical laboratory tests.
Pharmacokinetic and pharmacodynamic assessments
The following PK parameters were derived from plasma concentration versus time data: maximum observed plasma concentration (Cmax); time of Cmax (Tmax); area under the plasma concentration curve (AUC) from time 0 to time of last quantifiable concentration (AUC[0-T]); AUC in one dosing interval (AUC[TAU]); concentration at 24 h following dose (C24); terminal plasma half-life (T1/2); effective elimination half-life (T1/2 Eff AUC), which explains the degree of AUC accumulation observed on Day 14; effective elimination half-life (T1/2 Eff Cmax), which explains the degree of Cmax accumulation observed on Day 14; AUC accumulation index (AI AUC), which is the ratio of AUC(TAU) at steady state to AUC(TAU) after first dose; and Cmax accumulation index (AI Cmax), which is the ratio of Cmax at steady state to Cmax after first dose.
Activated partial thromboplastin time (aPTT) and FXI clotting activity were assessed as PD parameters.
All reported PK results were analyzed by validated liquid chromatography tandem mass spectrometry (LC–MS/MS) methods. Results were generated in analytical assays using appropriate calibration curves and quality control samples that met pre-established acceptance criteria and were conducted in compliance with applicable standard operating procedures in place at the time of analysis. LC–MS/MS assays had a lower limit of quantification of 1.00 ng/mL and an upper limit of quantification of 1000 ng/mL. PD parameters (aPTT and FXI clotting activity) were measured at LabCorp Colorado Coagulation (Englewood, CO, USA).
Sample collection
Blood samples were obtained at screening and on Days –1, 7 and 13 (prior to dosing), and 16 for clinical laboratory evaluations. Special laboratory tests (complete blood count with differential and coagulation factors only) were obtained prior to dosing on Days 5 and 10.
Details on the timing of sample collection for PK and PD assessments can be found in Supplementary Table S1.
Statistical analyses
Safety was assessed for all participants who received 1 dose of the study drug. The PK population was defined as all participants who received at least 1 dose of milvexian and had any available concentration–time data. The evaluable PK population was defined as all participants in the PK population with adequate PK profiles for accurate estimation of PK parameters. The PD population was defined as all participants who received any study treatment and had PD data available. Participants who received placebo in each cohort were pooled into a single placebo group.
Safety outcomes, including AEs and any AEs leading to study discontinuation or death, were summarized descriptively for all treated participants. Values for vital signs, ECGs, and clinical laboratory test results, as well as all non–serious AEs reported within 3 days of the last dose of study treatment, were recorded and summarized.
Although the number of participants was not based on statistical power considerations, administration of milvexian to 6 participants in each cohort would have provided an 80% probability of observing at least 1 occurrence of any AE that occurred with 24% incidence in the population from which the sample was drawn.
All milvexian PK data were summarized using descriptive statistics. Geometric means and coefficients of variation were presented for Cmax, C24, AUC(0-T), AUC(TAU), AI AUC, and AI Cmax. Median (minimum–maximum) values were presented for Tmax. Means and standard deviations were presented for T1/2, T1/2 Eff AUC, and T1/2 Eff Cmax. All measurements of PD markers (ie, aPTT and FXI clotting activity) were summarized descriptively.
All statistical calculations were performed by ICON (ICON Clinical Research, LLC, Gaithersburg, MD, USA) SAS programmers using SAS Version 9.3 (SAS Institute, Inc., Cary, NC, USA) software.
Results
Participants
Thirty-three participants entered the randomized portion of the study and received at least 1 dose of study treatment. Due to an unblinding event, all participants in the milvexian 200-mg group (Cohort 2a) discontinued participation early and a second group of participants who received milvexian 200 mg (Cohort 2b) was enrolled. One participant in the placebo group was discontinued after receiving 4 consecutive daily doses of placebo due to participation in multiple clinical trials; this participant was excluded from analysis and replaced. Eighteen participants were included in the PK population (excluding unblinded and early terminated participants and participants who received placebo). Thirty-two participants were analyzed in the PD population. Table 1 outlines the participant demographics and disposition.
Table 1.
Baseline characteristics for all treated participants PK, pharmacokinetics.
| Characteristica | Placebo (n = 6)b | Milvexian 50 mgc (n = 6) | Milvexian 200 mg (a)c,d (n = 8) | Milvexian 200 mgc (n = 6) | Milvexian 500 mge (n = 6) | Total (N = 32) |
|---|---|---|---|---|---|---|
| Male gender, n (%) | 6 (100.0) | 6 (100.0) | 8 (100.0) | 6 (100.0) | 5 (83.3) | 31 (96.9) |
| Age | 41.0 (23–48) | 35.5 (29–49) | 37.0 (29–54) | 29.5 (23–40) | 39.0 (30–55) | 36.5 (23–55) |
| Japanese race, n (%) | 6 (100.0) | 6 (100.0) | 8 (100.0) | 6 (100.0) | 6 (100.0) | 32 (100.0) |
| Body mass index, kg/m2 | 20.50 (19.5–23.3) | 22.90 (19.6–24.2) | – | 23.20 (19.3–24.3) | 22.90 (21.6–23.7) | 22.55 (19.3–24.3) |
aAll values presented as median (minimum–maximum) unless otherwise stated.
bOne participant in Cohort 3 (milvexian 500 mg or placebo) assigned to placebo was discontinued early due to participation in multiple clinical trials; this participant was excluded from analysis and replaced.
cMilvexian 50 mg and 200 mg were administered under fasted conditions.
dCohort 2a, milvexian 200 mg (a), includes unblinded and early terminated participants from Cohort 2 (milvexian and placebo participants). These participants were excluded from the evaluable PK population.
eMilvexian 500 mg was administered under fed conditions.
Safety and tolerability
Administration of milvexian at doses up to 500 mg once daily for 14 days was generally safe and well tolerated. No deaths or serious AEs were reported, and no AEs led to discontinuation. All reported AEs were mild to moderate in intensity and resolved with no sequelae, and none of the AEs reported were considered by the investigator to be related to study treatment. Four participants (15.4%) who received milvexian reported at least 1 AE (Table 2). There were no clinically relevant findings or trends in clinical laboratory, ECG, vital sign, or physical examination findings.
Table 2.
Adverse events for all treated participants.
| AE, n (%) | Placebo (n = 6)a | Milvexian 50 mgb (n = 6) | Milvexian 200 mg (a)b,c (n = 8) | Milvexian 200 mgb (n = 6) | Milvexian 500 mgd (n = 6) | All milvexian (n = 26) |
|---|---|---|---|---|---|---|
| Any AE | 0 | 0 | 1 (12.5) | 0 | 3 (50.0) | 4 (15.4) |
| Headache | 0 | 0 | 0 | 0 | 2 (33.3) | 2 (7.7) |
| Constipation | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.8) |
| Dermatitis contact | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.8) |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.8) |
| Presyncope | 0 | 0 | 1 (12.5) | 0 | 0 | 1 (3.8) |
| Rhinitis | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.8) |
| Urticaria | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.8) |
AE, adverse event; PK, pharmacokinetics.
aOne participant in Cohort 3 (milvexian 500 mg or placebo) assigned to placebo was discontinued early due to participation in multiple clinical trials; this participant was excluded from analysis and replaced.
bMilvexian 50 mg and 200 mg were administered under fasted conditions.
cCohort 2a, milvexian 200 mg (a), includes unblinded and early terminated participants from Cohort 2 (milvexian and placebo participants). These participants were excluded from the evaluable PK population.
dMilvexian 500 mg was administered under fed conditions.
Pharmacokinetics
Milvexian exposure increased with increasing dose between the 50-mg and 200-mg doses on Days 1 through 14 (Fig. 1 and Table 3). The median Tmax was similar for the 50-mg and 200-mg dose groups, but a delay was seen in Tmax when milvexian 500 mg was taken with food. Specifically, median Tmax for the 50-mg and 200-mg doses on Days 1 and 14 were 2.5 to 3.0 h compared to a median Tmax of 7.0 to 8.0 h when milvexian 500 mg was administered with food. The mean T1/2 following 14 days of daily dosing was similar across doses, and the effective T1/2 in terms of Cmax and AUC increased with increasing dose. Accumulation of milvexian was observed with an AI AUC of 1.14, 1.60, and 2.35, respectively, for the 50-mg, 200-mg, and 500-mg dose panels.
Figure 1.

Mean (± SD) milvexian plasma concentration profile versus time by dose group on (a) Day 1 and (b) Day 14. SD, standard deviation. Milvexian 50 mg and 200 mg were administered under fasted conditions. Milvexian 500 mg was administered under fed conditions.
Table 3.
PK parameters on Day 1 and Day 14.
| Parametera | Day 1 | Day 14 | ||||
|---|---|---|---|---|---|---|
| Milvexian 50 mgb (n = 6) | Milvexian 200 mgb (n = 6) | Milvexian 500 mgc (n = 6) | Milvexian 50 mgb (n = 6) | Milvexian 200 mgb (n = 6) | Milvexian 500 mgc (n = 6) | |
| Cmax (ng/mL) | 496 (47) | 1241 (50) | 4318 (31) | 498 (23) | 1879 (22) | 7918 (29) |
| Tmax (h) | 3.00 (2.00–4.00) | 3.00 (2.00–4.00) | 8.00 (4.00–23.9) | 2.50 (2.00–4.00) | 3.00 (3.00–4.00) | 7.00 (6.00–8.00) |
| AUC(TAU) (ng•h/mL) | 3518 (39) | 11,190 (37) | 50,619 (27) | 4007 (29) | 17,915 (22) | 119,113 (37) |
| AUC(0-T) (ng•h/mL) | – | – | – | 4562 (31) | 21,814 (23) | 181,648 (51) |
| C24 (ng/mL) | 33.9 (37) | 167 (37) | 1642 (52) | 44.2 (42) | 310 (29) | 3864 (52) |
| T1/2 (h) | – | – | – | 8.94 (1.67) | 9.46 (1.63) | 11.9 (4.60) |
| T1/2 Eff AUC (h) | – | – | – | 11.9 (4.04)d | 21.3 (11.36)e | 31.2 (11.55) |
| T1/2 Eff Cmax (h) | – | – | – | 10.8 (3.81)f | 20.6 (12.08)e | 22.6 (12.89) |
| AI AUC | – | – | – | 1.14 (27) | 1.60 (42) | 2.35 (27) |
| AI Cmax | – | – | – | 1.00 (30) | 1.51 (50) | 1.83 (35) |
PK, pharmacokinetics; Cmax, maximum observed plasma concentration; Tmax, time of maximum observed plasma concentration; AUC(TAU), area under the plasma concentration curve in one dosing interval; AUC(0-T), AUC from time 0 to time of last quantifiable concentration; C24, concentration at 24 h following dose; T1/2, terminal plasma half-life; T1/2 Eff AUC, effective elimination half-life, which explains the degree of AUC accumulation observed on Day 14; T1/2 Eff Cmax, effective elimination half-life, which explains the degree of Cmax accumulation observed on Day 14; AI AUC, AUC accumulation index, which is the ratio of AUC(TAU) at steady state to AUC(TAU) after first dose; AI Cmax, Cmax accumulation index, which is the ratio of Cmax at steady state to Cmax after first dose; %CV, coefficient of variation; SD, standard deviation.
aCmax, AUC(TAU), AUC(0-T), C24, AI AUC, and AI Cmax are presented as adjusted geometric mean (%CV); Tmax is presented as median (minimum–maximum); T1/2, T1/2 Eff AUC, and T1/2 Eff Cmax are presented as mean (SD).
bMilvexian 50 mg and 200 mg were administered under fasted conditions.
cMilvexian 500 mg was administered under fed conditions.
dn = 4.
en = 5.
fn = 3.
Pharmacodynamics
Mean baseline aPTT values were similar across groups and ranged from 27.1 to 28.6 s. Multiple doses of milvexian resulted in a concentration-related prolongation of aPTT (Fig. 2). In the fasted condition, the maximal effect was observed between 3 and 4 h postdose, which was consistent with the Tmax of milvexian for the 50-mg and 200-mg doses. When milvexian 500 mg was administered with food, the maximal effect was delayed to 12 h postdose. The observed maximal mean percent change from baseline on Day 14 was 96.0%, 185.5%, and 286.1% for the 50-mg, 200-mg, and 500-mg doses, respectively. The relationship between aPTT and serum concentration of milvexian is shown in Fig. 3.
Figure 2.

Mean (± SD) aPTT profile over time on (a) Day 1 and (b) Day 14. SD, standard deviation; aPTT, activated partial thromboplastin time. Milvexian 50 mg and 200 mg were administered under fasted conditions. Milvexian 500 mg was administered under fed conditions.
Figure 3.
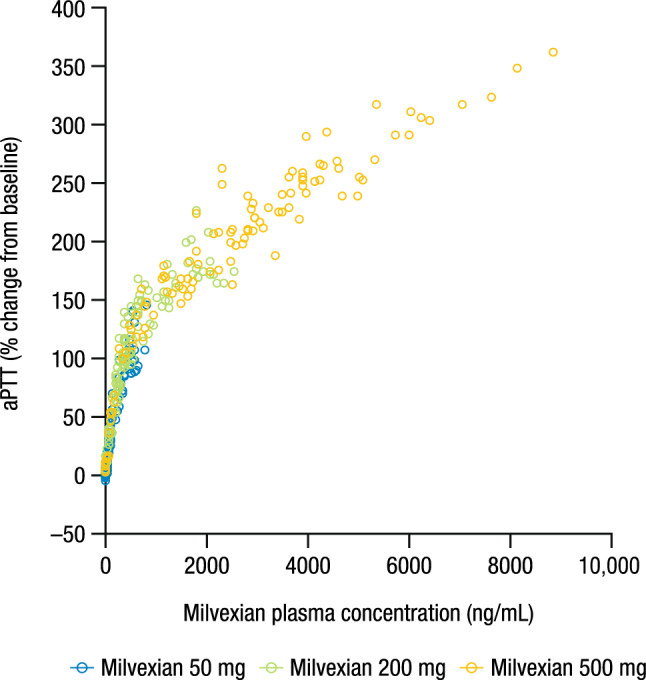
Percent change from baseline in aPTT versus milvexian plasma concentration. aPTT, activated partial thromboplastin time.
Mean baseline FXI clotting activity values were similar across groups and ranged from 86.0% to 100.3%. Milvexian reduced FXI clotting activity, and the magnitude of the reduction was related to the drug exposure (Fig. 4). The maximal mean decrease in FXI clotting activity from baseline on Day 14 was −31.5% and −74.1% for the 50-mg and 200-mg doses, respectively. The observed maximal effect occurred at 3 or 4 h postdose for the 50-mg and 200-mg doses, which was consistent with Tmax; however, when milvexian 500 mg was administered with food, the maximal effect was delayed to 12 h postdose.
Figure 4.

Mean (± SD) FXI clotting activity over time on (a) Day 1 and (b) Day 14. SD, standard deviation; FXI, factor XI. Milvexian 50 mg and 200 mg were administered under fasted conditions. Milvexian 500 mg was administered under fed conditions.
Discussion
This study investigated the safety, tolerability, PK, and PD of multiple ascending oral doses of milvexian in healthy Japanese participants. Milvexian is one of the first oral, small-molecule FXIa inhibitors being developed to prevent and treat thrombotic events in diverse patient populations24 and may offer an improvement over the benefit/risk profile of current anticoagulants. Demonstration of the safety and tolerability of milvexian in Japanese participants will allow inclusion of this population in subsequent phase 2 and 3 studies and inform whether dose adjustments will be necessary for future trials and for routine clinical care.
Milvexian was safe and well tolerated in healthy Japanese participants at doses of up to 500 mg administered with food. All observed AEs were mild in intensity, and none were considered by the investigator to be related to study treatment. There were no bleeding events reported. The safety outcomes seen here align with those from the first-in-human study of milvexian, where all treatment-emergent AEs were mild in severity and no clinically significant bleeding events were reported28.
Exposure to milvexian increased between the 50-mg and 200-mg doses, and there was a delay in Tmax when milvexian 500 mg was administered with food. The PK results from the current study in Japanese participants are similar to those observed in the single- and multiple ascending–dose study in healthy non-Japanese participants. Though not directly comparable, both studies demonstrated increasing milvexian exposure as a result of increasing dose and a delay in Tmax under fed conditions, with expected non-linearity in the PK observed at the 500-mg dose irrespective of ethnicity28. The rationale for allowing milvexian 500 mg to be taken with food was based on the observation from the first-in-human study that food increased milvexian exposure at doses beyond 200 mg and, thus, achieving high enough exposure to demonstrate safety and tolerability was important in this study28.
In Japanese participants, there was a concentration-related prolongation of aPTT and a decrease in FXI clotting activity after multiple oral administrations of milvexian, which is consistent with the mechanism of FXIa inhibition. Food intake, or potentially non-linear processes such as delayed absorption at the 500-mg dose, may have been responsible for the delayed Tmax and extended the maximal mean effect of milvexian on aPTT and FXI clotting activity. These results are similar to those observed in a study of healthy non-Japanese participants, which showed prolongation of aPTT and a decrease in FXI clotting activity as drug exposure increased28. The concentration–aPTT profile observed in this study shows a direct relationship that is similar to that seen in previous studies of primarily Caucasian participants, further confirming that there are no ethnic differences in the PD relationship of milvexian in Japanese participants28.
Based on preclinical studies and ongoing clinical pharmacology studies, the PK and absorption, distribution, metabolism, and excretion (ADME) profiles of milvexian indicate that it is a substrate of cytochrome P450 family 3, subfamily A (also known as CYP3A) and P-glycoprotein with limited renal clearance. Although there may be inherent interindividual variability associated with these pathways, there does not appear to be distinct polymorphic variability associated with these pathways, and there do not appear to be distinct polymorphic pathways that impact the disposition of milvexian (e.g., CYP2C19 or CYP2D6). Therefore, it is unlikely that there are intrinsic pharmacogenomic factors that would play a role in determining exposure in a Japanese patient population beyond body weight.
The current study was limited by the small sample size in each panel. However, a multiple ascending–dose design allowed for the stepwise evaluation of a range of doses of milvexian in Japanese participants. In addition, this study did not include an arm of non-Japanese participants for direct comparison, leading to the potential for interstudy variability.
In conclusion, milvexian was generally safe and well tolerated in healthy Japanese participants. The PK results demonstrated an increase in exposure to milvexian between the 50-mg and 200-mg doses and that administration of milvexian 500 mg with food delayed Tmax. Milvexian resulted in concentration-related prolongation of aPTT and decrease in FXI clotting activity. Taken together, the results of this study suggest that the PK and PD profiles of milvexian in Japanese participants are similar to those observed in other populations, including other phase 1 studies in volunteers and participants with hepatic impairment27,28, and demonstrate suitable dosing properties for further clinical development in Japanese participants.
Supplementary Information
Acknowledgements
Medical writing support was provided by Alana Simorellis, PhD, of Cello Health Communications/MedErgy, and was funded by Bristol Myers Squibb and Janssen Global Services, LLC.
Author contributions
V.P., Z.W., S.L., T.U., T.S., W.C., X.X., D.S., M.D., and B.M. contributed to the study design and concept, data analysis and review, data interpretation, and manuscript review and approval.
Funding
This study was sponsored by Bristol Myers Squibb.
Data availability
The data that support the findings of this study are not publicly available due to privacy or ethical restrictions.
Competing interests
V.P., Z.W., S.L., W.C., D.S., and B.M. are full-time employees of Bristol Myers Squibb. T.U. and T.S. are full-time employees of Bristol-Myers Squibb K.K. X.X. and M.D. were full-time employees of Bristol Myers Squibb when the study was conducted.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-08768-y.
References
- 1.Cavallari I, Patti G. Efficacy and safety of oral anticoagulation in elderly patients with atrial fibrillation. Anatol. J. Cardiol. 2018;19:67–71. doi: 10.14744/AnatolJCardiol.2017.8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapil N, et al. Antiplatelet and anticoagulant therapies for prevention of ischemic stroke. Clin. Appl. Thromb. Hemost. 2017;23:301–318. doi: 10.1177/1076029616660762. [DOI] [PubMed] [Google Scholar]
- 3.Gurbel PA, Fox KA, Tantry US, Ten Cate H, Weitz JI. Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease. Circulation. 2019;139:2170–2185. doi: 10.1161/CIRCULATIONAHA.118.033580. [DOI] [PubMed] [Google Scholar]
- 4.Eisen A, Giugliano RP, Braunwald E. Updates on acute coronary syndrome: A review. JAMA Cardiol. 2016;1:718–730. doi: 10.1001/jamacardio.2016.2049. [DOI] [PubMed] [Google Scholar]
- 5.Faxon DP. Use of antiplatelet agents and anticoagulants for cardiovascular disease: Current standards and best practices. Rev. Cardiovasc. Med. 2005;6(suppl 4):S3–S14. [PubMed] [Google Scholar]
- 6.Bracey A, Shatila W, Wilson J. Bleeding in patients receiving non-vitamin K oral anticoagulants: Clinical trial evidence. Ther. Adv. Cardiovasc. Dis. 2018;12:361–380. doi: 10.1177/1753944718801554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ten Cate H, Hackeng TM, de Frutos PG. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb. Haemost. 2017;117:1265–1271. doi: 10.1160/TH17-02-0079. [DOI] [PubMed] [Google Scholar]
- 8.Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18S. [DOI] [PubMed] [Google Scholar]
- 9.Lowenberg EC, Meijers JC, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J. Thromb. Haemost. 2010;8:2349–2357. doi: 10.1111/j.1538-7836.2010.04031.x. [DOI] [PubMed] [Google Scholar]
- 10.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency; clinical, coagulation, therapeutic and hereditary aspects of a new hemophilia-like disease. Blood. 1955;10:120–131. doi: 10.1182/blood.V10.2.120.120. [DOI] [PubMed] [Google Scholar]
- 12.Franchini M, Veneri D, Lippi G. Inherited factor XI deficiency: A concise review. Hematology. 2006;11:307–309. doi: 10.1080/10245330600921964. [DOI] [PubMed] [Google Scholar]
- 13.Lee SE, Choi YJ, Chi SI, Kim HJ, Seo KS. Factor XI deficiency and orthognathic surgery: A case report on anesthesia management. J. Dent. Anesth. Pain. Med. 2015;15:25–29. doi: 10.17245/jdapm.2015.15.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minnema MC, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J. Clin. Investig. 1998;101:10–14. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, et al. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. J. Thromb. Haemost. 2006;4:1982–1988. doi: 10.1111/j.1538-7836.2006.02093.x. [DOI] [PubMed] [Google Scholar]
- 16.Preis M, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210–1215. doi: 10.1182/blood-2016-09-742262. [DOI] [PubMed] [Google Scholar]
- 17.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 18.Salomon O, et al. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb. Haemost. 2011;105:269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 19.Pollack CV, Jr, Kurz MA, Hayward NJ. EP-7041, a factor XIa inhibitor as a potential antithrombotic strategy in extracorporeal membrane oxygenation: A brief report. Crit. Care Explor. 2020;2:e0196. doi: 10.1097/CCE.0000000000000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakimoto S, et al. ONO-8610539, an injectable small-molecule inhibitor of blood coagulation factor XIa, improves cerebral ischemic injuries associated with photothrombotic occlusion of rabbit middle cerebral artery. Stroke. 2017;48:WP286. doi: 10.1161/str.48.suppl_1.wp286. [DOI] [Google Scholar]
- 21.Thomas, D. et al.First Evaluation of the Safety, Pharmacokinetics and Pharmacodynamics of BAY 2433334 a Small Molecule Targeting Coagulation Factor XIa in Healthy Young Male Participants. Poster presented at: International Society on Thrombosis and Haemostasis (ISTH) Virtual Congress; July 12–14, 2020. Poster PB0243 (2020).
- 22.Weitz JI, et al. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: The FOXTROT randomized clinical trial. JAMA. 2020;323:130–139. doi: 10.1001/jama.2019.20687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb. Res. 2016;141:S8–S11. doi: 10.1016/S0049-3848(16)30354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dilger AK, et al. Discovery of milvexian, a high-affinity, orally bioavailable inhibitor of factor XIa in clinical studies for antithrombotic therapy. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00613. [DOI] [PubMed] [Google Scholar]
- 25.Wong PC, et al. Milvexian, an orally bioavailable, small-molecule, reversible, direct inhibitor of factor XIa: In vitro studies and in vivo evaluation in experimental thrombosis in rabbits. J. Thromb. Haemost. 2021 doi: 10.1111/jth.15588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang, X. et al.Antithrombotic Effects of a Novel Small Molecule FXIa Inhibitor BMS-986177/JNJ-70033093 in a Rabbit AV-Shunt Model of Thrombosis. Poster presented at: International Society on Thrombosis and Haemostasis (ISTH) Virtual Congress; July 12–14, 2020. Poster PB0179.
- 27.Perera, V. et al.Single-Dose Pharmacokinetics of BMS-986177/JNJ-70033093 in Participants with Mild or Moderate Hepatic Impairment Compared to Healthy Participants. Poster presented at: European Society of Cardiology (ESC) Congress—The Digital Experience; August 29–September 1, 2020.
- 28.Perera V, et al. First-in-human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin. Transl. Sci. 2021 doi: 10.1111/cts.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ClinicalTrials.gov. NCT03766581. A study on BMS-986177 for the prevention of stroke in patients receiving aspirin and clopidogrel (AXIOMATIC-SSP). https://clinicaltrials.gov/ct2/show/NCT03766581 (2018).
- 30.Weitz JI, et al. Milvexian for the prevention of venous thromboembolism. N. Engl. J. Med. 2021;385:2161–2172. doi: 10.1056/NEJMoa2113194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dean L, et al. Warfarin Therapy and VKORC1 and CYP Genotype. In: Pratt VM, Scott SA, Pirmohamed M, et al., editors. Medical Genetics Summaries. National Center for Biotechnology Information; 2012. [PubMed] [Google Scholar]
- 32.Obayashi K, et al. VKORC1 gene variations are the major contributors of variation in warfarin dose in Japanese patients. Clin. Pharmacol. Ther. 2006;80:169–178. doi: 10.1016/j.clpt.2006.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are not publicly available due to privacy or ethical restrictions.