

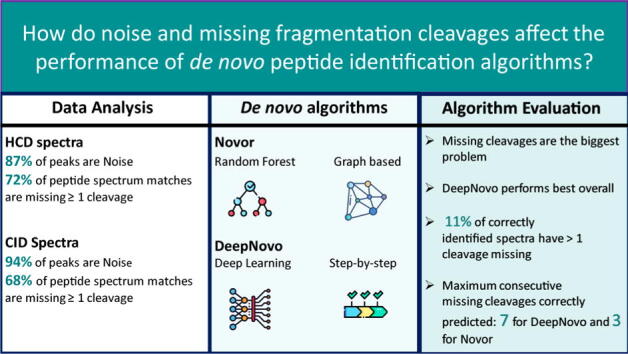
Graphical abstract
Keywords: De novo peptide sequencing, Machine learning, Peptide identification, Noise, Fragmentation cleavage sites, Peptide fragmentation
Highlights
-
•
Most correct de novo peptides have 1 missing fragmentation cleavages.
-
•
DeepNovo outperforms Novor for peptide accuracy for both data types.
-
•
Novor excels at amino acid recall when many fragmentation cleavages are missing.
-
•
Deep learning allows DeepNovo to predict amino acids without adjacent peaks.
Abstract
Proteomics aims to characterise system-wide protein expression and typically relies on mass-spectrometry and peptide fragmentation, followed by a database search for protein identification. It has wide ranging applications from clinical to environmental settings and virtually impacts on every area of biology. In that context, de novo peptide sequencing is becoming increasingly popular. Historically its performance lagged behind database search methods but with the integration of machine learning, this field of research is gaining momentum. To enable de novo peptide sequencing to realise its full potential, it is critical to explore the mass spectrometry data underpinning peptide identification. In this research we investigate the characteristics of tandem mass spectra using 8 published datasets. We then evaluate two state of the art de novo peptide sequencing algorithms, Novor and DeepNovo, with a particular focus on their performance with regard to missing fragmentation cleavage sites and noise. DeepNovo was found to perform better than Novor overall. However, Novor recalled more correct amino acids when 6 or more cleavage sites were missing. Furthermore, less than 11% of each algorithms’ correct peptide predictions emanate from data with more than one missing cleavage site, highlighting the issues missing cleavages pose. We further investigate how the algorithms manage to correctly identify peptides with many of these missing fragmentation cleavages. We show how noise negatively impacts the performance of both algorithms, when high intensity peaks are considered. Finally, we provide recommendations regarding further algorithms’ improvements and offer potential avenues to overcome current inherent data limitations.
1. Introduction
Proteomics has become an indispensable tool for biologists in the last few decades with its ability to identify system-wide protein expression. [1]. Its application is wide ranging and encompasses the identification of cancer biomarkers [2] and antigens for immunotherapy [3], as well as mechanisms underlying drought resistance in crops [4] and virulence factors in human pathogens [5], [6].
In proteomics, protein extracts are typically enzymatically digested and analysed using mass spectrometry. The corresponding mass spectra are then matched to peptides, which are short sequences of amino acids. Database search algorithms are commonly used in proteomics and aim to match theoretical peaks predicted from all possible peptides in the relevant protein databases to the peaks in actual spectra. Although database searching is the most popular technique used in protein identification, improved data quality and algorithm design mean de novo peptide sequencing is becoming increasingly popular in proteomics [7].
Recent advances in mass spectrometry (MS) have considerably raised the level of data resolution and acquisition in the field of proteomics [8], while the same database search algorithms have dominated the field for the last 20 years [9]. Typically, for shotgun proteomics, following the enzymatic digestion of proteins, the resulting complex peptide mixture is fractionated using liquid chromatography. The corresponding peptide fractions are then analysed using tandem mass spectrometry (MS/MS). Peptides are separated by mass and charge (m/z) in the first mass analyzer. Then, peaks from the resulting spectra are isolated and the associated peptides are passed through a fragmentation chamber to be charged and broken down into smaller pieces (fragment ions). These fragments pass through the second mass analyzer producing fragmentation patterns as the ions are separated. A database search or de novo peptide sequencing is then conducted to establish the most likely peptide sequence corresponding to each fragmentation pattern. Two common methods of fragmentation include collision induced dissociation (CID) and higher-energy dissociation (HCD). While similar in methodology, HCD fragmentation provides greater resolution and mass accuracy than CID [10]. Both of these methods fragment peptides by colliding them with gas molecules. This causes the cleavage of the amino acid sequence typically at a peptide (amide) bond resulting in two possible fragments; b and y ions [11]. While b and y ions themselves are the most common, peptide fragments can also suffer neutral losses of ammonia and water molecules producing different peaks with a shifted m/z value. Conventional notation enumerates the b ions according to their fragmentation site from the N-terminus to the C-terminus. Conversely, y ions are numbered from the C-terminus to the N-terminus. Although both ion types are ordered by increasing mass, it means for a peptide of length 20, the b1 ion is created from the same cleavage as the y19 ion. As the peptide mass is known, the mass of the corresponding y ion can be easily calculated given a b ion and vice versa. As these ions contain equivalent information about amino acid composition they can be grouped together. We refer to missing fragmentation cleavages from here on to indicate that neither a b or y ion, or their neutral losses, is present for a given fragmentation/cleavage site along the peptide chain. To refer to our example again, if for a peptide of 20 amino acids, neither the b1 or y19 ions were present, or peaks indicating the loss of ammonia or water from these ions, we would then consider that the first cleavage is missing.
Although popular, database searching is not straightforward due to the irregularity and incompleteness of the peptide fragmentation process which effectively means there is never a perfect match between predicted and actual peaks in the mass spectra. Even with recently developed algorithms and up-to-date, tailored databases, on average, only 25% of spectra are identified leaving the remaining 75% unclassified and thereby discarded [12], [13]. This can be partly attributed to the size of the databases, where a larger number of possible matches increases the false discovery rate [14]. This is particularly problematic for metaproteomics, where databases typically span large species diversity. Peptide identification from mass spectra can also be performed de novo, where peptides are identified based on the spectrum alone, thus removing the need for a database. Historically this approach has had a much lower sensitivity than database search methods but recent advances in machine learning and mass spectrometry have seen it become a competitive alternative [15]. Without the use of a database, de novo methods are not limited in the same way as matching algorithms are, while also being able to identify post-translational-modifications (PTMs) relatively easily [16]. PTMs expand protein function beyond the standard amino acids by both reversible and irreversible modifications. The importance of PTMs is only starting to be uncovered as evidence suggests they are involved in the regulation of almost all cellular events [17].
When database search algorithms include variable modifications, reflective of PTMs, it exponentially increases their search space as the m/z value of any peak including the modified amino acid will be shifted accordingly. This has the effect of increasing both the FDR and running time of the algorithm [18]. This is not the case for de novo peptide sequencing where the number of PTMs being searched may have little or no effect on run time [19].
Although the current state of the art de novo algorithms are still not as effective as database searching, the recent availability of big data, and the simultaneous explosion in machine learning means the field is on an upward curve. Two algorithms leading the way are Novor [20] and DeepNovo [21]. They use machine learning and dynamic programming to both learn patterns within the data and simplify the prediction process respectively. How they implement these techniques is quite different however. Novor models the spectrum as a graph, a traditional approach to de novo peptide sequencing [22]. Each node in the graph, which corresponds to a peak in the spectrum, is scored using a random forest model, trained on thousands of other spectra. Edges are created between nodes whose associated masses differ by that of an amino acid. Using dynamic programming, Novor then finds the highest scoring path through the graph, whose edges will classify the amino acids of the peptide. DeepNovo’s approach to the problem involves progressing through the spectrum step-by-step using two different deep learning architectures combined. Based on the mass of the predicted sequence so far, a convolutional neural network (CNN) is trained to encode the parts of the spectrum where the next fragment ions might appear. A long short-term memory (LSTM) recurrent neural network uses this encoding, along with all the encodings from the previous predictions, to determine the next amino acid in the sequence. DeepNovo uses dynamic programming to limit the number of possible amino acids it can predict to those that would satisfy the remaining mass of the peptide, given those already predicted.
While de novo algorithms continue to improve, their possible uses continue to increase. De novo peptide sequencing has been used successfully to both aid and confirm database search results [23], [24], [25]. To aid database methods it can be used to identify amino acid “tags” from a spectrum that can then be used to limit the size of the search space to entries that only include them, thereby decreasing the false discovery rate (FDR). More recently, advanced de novo sequencing algorithms like DeepNovo, have been used for neoantigen detection [26]. Antigens are used by the immune system to recognise pathogens and trigger a response [27]. Neoantigens are antigens previously unseen by the immune system, which may be caused by genetic mutations [28]. Identification of these neoantigens is important for the development of cancer immunotherapies as they are not expressed by healthy tissue [29].
If the continual increase in the accuracy of de novo sequencing can be sustained, it may also open up the possibility of re-mining available data. The PRIDE Repository [30] contains data from thousands of proteomics experiments and improvements in machine learning and de novo peptide sequencing could uncover new insights from previous studies. To enable de novo peptide identification to reach its full potential, it is vital to understand the underlying data [31], in order to best design de novo algorithms.
Previous studies of de novo algorithms have sought to show how these algorithms perform on different datasets while investigating what errors they are making [15], [32]. Here, we investigate the prevalence and effects of missing fragmentation cleavage sites and noise on de novo peptide sequencing using real labelled data as well as artificial data. Specifically, we address the following research questions; How prevalent are occurrences of noise and missing fragmentation cleavages in tandem MS data? What are the effects of noise and missing fragmentation cleavages on the performance of de novo peptide sequencing algorithms? How do the current state of the art approaches cope with noise and missing fragmentation cleavages? Finally, based on our findings, we propose approaches that could be implemented in the future to improve de novo peptide sequencing algorithms.
2. Methods
2.1. Data
We analysed data from eight different datasets downloaded from their respective archive on the PRIDE Public Repository [30]. A summary of each is provided in Table 1. These include the four used by Muth and Renard (2018) [15]. The eight datasets are made up of six different organisms, distributed between the two fragmentation types, CID and HCD.
Table 1.
Overview of the datasets and processing steps used in this study.
| Dataset | Pride Archive | Organism | Original Format | Mass Spectrometer | Frag Type | PrecTol | FragTol |
|---|---|---|---|---|---|---|---|
| MouseCID | PXD000790 | M. musculus | MGF | LTQ Orbitrap Elite | CID | 5 ppm | 0.50 Da |
| YeastCID | PXD002726 | S. cerevisiae | MGF | LTQ Orbitrap Velos | CID | 10 ppm | 0.80 Da |
| EcoliCID | PXD016825 | E. coli | RAW | LTQ Orbitrap Velos | CID | 20 ppm | 0.50 Da |
| StaphAurCID | PXD017932 | S. aureus | RAW | LTQ Orbitrap Velos | CID | 5 ppm | 0.60 Da |
| HeLaHCD | PXD000674 | H. sapiens | RAW | Q Exactive | HCD | 10 ppm | 0.02 Da |
| PyroHCD | PXD001077 | P. furiosus | RAW | LTQ Orbitrap Velos | HCD | 10 ppm | 0.06 Da |
| EcoliHCD | PXD008685 | E. coli | MGF | Q Exactive | HCD | 10 ppm | 0.02 Da |
| StaphAurHCD | PXD023039 | S. aureus | RAW | Q Exactive | HCD | 10 ppm | 0.06 Da |
Frag Type: Fragmentation Type.
PrecTol: Precursor Mass Tolerance.
FragTol: Fragment Mass Tolerance.
To obtain the labelled data required for this research we performed a database search using two popular search algorithms. For each organism, a protein database was downloaded from UniProt (Supp. Table 2). Just as was done by Muth and Renard (2018), all prokaryotic data were searched against the yeast proteome as well as their own. Accurate FDR estimation requires each spectrum to be compared to multiple peptides [33]. If this condition is not satisfied it can lead to an overestimation of identifications in smaller databases [34]. Therefore the small databases of prokaryotic organisms were augmented to circumvent this issue [15].
MS-GF+ [35] and X!Tandem [36] were used to search the databases through the SearchGUI platform [37]. Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine was set as a variable modification. A maximum of two missed tryptic cleavages were allowed. b and y ions were considered with precursor charge bounded between 2 and 4 inclusive. MS-GF + was set to HCD or CID mode depending on the data being used. Using an FDR of 1%, we extracted the top scoring peptide spectra matches (PSMs) from each dataset. Furthermore, we then selected from these PSMs only those for which MS-GF + and X!Tandem agreed. The results of these conditions can be found in Table 2. The data were then collated into two groups, one for each fragmentation type. This resulted in a split of 25007 HCD spectra and 23821 CID spectra. For the remainder of this research, CID data refers to the four combined CID datasets listed and HCD data refers to the four HCD datasets.
Table 2.
The number of peptides matched at the 1% FDR level for both X!Tandem and MS-GF+, as well as how many of those were in agreement (Overlap).
| Dataset | Overlap | X!Tandem | MS-GF+ |
|---|---|---|---|
| MouseCID | 12132 | 15586 | 13345 |
| YeastCID | 534 | 650 | 1519 |
| EcoliCID | 5716 | 7210 | 7752 |
| StaphAurCID | 5439 | 6020 | 6363 |
| HeLaHCD | 4061 | 4973 | 4167 |
| PyroHCD | 9719 | 12080 | 10172 |
| EcoliHCD | 5180 | 5279 | 5257 |
| StaphAurHCD | 6047 | 8279 | 6850 |
2.2. Peptide peak and noise assignment
Using the peptides assigned to the spectra following the database search, each peak was labelled as either a peptide peak or noise. To do this the assigned peptides were artificially fragmented to create b and y ions along with their neutral losses of ammonia (NH3) and water (H2O) using the Pyteomics framework [38]. These are the ion types used by both Novor and DeepNovo. If possible these were matched to peaks in the spectra and labelled as peptide peaks with a tolerance of 0.5 Da for CID data and 0.05 Da for HCD data. Thereby the ions and hence cleavage sites which were not represented in each spectrum were identified and peaks that could not be matched to a fragment ion were classified as noise. For clarity, noise was also considered in its proportion to peptide peaks [15]. When low intensity noise peaks were found not to affect performance, only those above the median of the distribution of noise peaks were included. A median normalised noise intensity value of approximately 7.2e-3 was observed for the CID data and a median of approximately 2.1e-2 for the HCD data. The number of noise peaks above this threshold was recorded for each spectrum. The noise factor was then defined as the number of high intensity noise peaks divided by the number of peptide peaks in each spectrum (#NoisePeaks/ #PeptidePeaks).
2.3. Algorithms
DeepNovo was downloaded from https://github.com/nh2tran/DeepNovo. Two models were then trained, one for CID data and one for HCD data. These two models used the parameters specified for low resolution and high resolution data in the original paper respectively [21]. The models were also trained using the same data as the original paper found at ftp://massive.ucsd.edu/MSV000081382/. The algorithm was then run through a linux terminal using Python 2.7.17. Novor was operated through the DenovoGUI interface [39] in CID or HCD mode depending on the data. Precursor precision and fragmentation tolerance were kept the same as DeepNovo for a fair comparison. Both algorithms were set to consider carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable modification.
2.4. Metrics
2.4.1. Amino acid match
For the CID data, two amino acids are considered a match if the prefix mass of the peptide before the prediction is correct to within 0.5 Da and the masses of the amino acids predicted are within 0.1 Da. For HCD data, the tolerance is lowered with an amino acid match requiring the prefix mass of the peptide before the prediction to be correct within 0.05 Da and the masses of the amino acids predicted to be within 0.01 Da.
2.4.2. AA recall
Amino acid recall is defined as the number of amino acids matched divided by the total number of amino acids in the database assigned peptide.
2.4.3. Peptide accuracy
Peptide accuracy corresponds to the number of peptide predictions that correctly match those assigned to the spectra divided by the total number of spectra.
2.4.4. Peak recall
We compare the cumulative masses generated by the amino acids in the PSM’s peptide sequence and the predicted peptide sequence which are akin to the position of cleavage sites along the peptide. For CID data a predicted fragmentation cleavage is considered correctly matched if its mass differs by less than 0.5 Da from the corresponding true peptide cleavage. We also compare if the true peptide’s cleavage sites are represented with a b or y ion in the spectrum with a tolerance of 0.5 Da. For HCD data the tolerance for both matches is reduced to 0.05 Da.
2.5. Confirmatory analysis
High scoring spectra and artificial spectra were also used in a complementary analysis to confirm the trends observed when evaluating the algorithms with respect to all of the real data.
For high scoring spectra, de novo peptides above an acceptable score were extracted for both algorithms separately. High scoring spectra are defined as those with scores above a threshold which gives 90% amino acid recall. This standard was used by Tran et al. (2019) when using DeepNovo for antigen identification. Also, similar levels of peptide accuracy or higher amino acid recall were not possible for both algorithms. 90% amino acid recall was achieved in CID data with a score threshold of 0.89 (2740 peptides) and 0.74 (10295 peptides) for Novor and DeepNovo respectively. The thresholds for Novor and DeepNovo in HCD data were 0.67 (13493 peptides) and 0.73 (16898 peptides) respectively.
Artificial data were created to match the distribution of peptides found in the real data. Prosit was downloaded fromhttps://github.com/kusterlab/prosit. A trained HCD Prosit model was then downloaded fromhttps://figshare.com/projects/Prosit/35582. The overlapping HCD peptides matched by both database algorithms were extracted and artificial spectra were created for each using this Prosit model. CID peptides were not considered as there was no available model.
The artificial data were duplicated four more times with each duplicate given a different level of noise. Therefore, for each duplicate each spectra was given additional random noise peaks corresponding to the respective noise factor of that duplicate. Noise factors of 0,4,8,12 and 16 were considered.
3. Results
3.1. Missing fragmentation cleavage sites are prevalent in mass spectra
It can be difficult to evaluate de novo algorithms as there is no such thing as real data that is 100% correctly labelled. Instead we use the results of two database search algorithms that agree at a 1% FDR. We evaluate two state of the art de novo algorithms by comparing the database PSMs to their de novo predictions. Given the assigned peptide from the database search, we establish which peaks in the spectrum are fragment ions. Those that cannot be attributed to the peptide are classified as noise. We can then quantify what fragmentation cleavage sites are present and how many are missing from the spectrum. Models are also available to create high quality artificial data [40], [41], although they only predict peaks at precise locations directly derived from the peptide sequences. They also do not include noise peaks, which affect performance when present in large volumes. We also evaluate the algorithms using these artificial data with additional random noise as a complementary analysis to provide a deeper insight into their performance.
De novo sequencing relies solely on the individual spectrum to identify the peptide that produced it. In contrast to database searching that can match peaks independently, de novo algorithms must predict and recreate each cleavage, even if no peaks from it exist in the spectrum. When available, many different fragment ions from one cleavage site serve as stronger evidence for that particular fragment as being correct. When no fragment ions from a cleavage site are present there is no direct evidence for the adjacent amino acids in the spectrum and so these are more difficult to determine.
Fig. 1 shows the distribution of fragmentation cleavage sites present for all peptide lengths in both CID and HCD data. For both data types, shorter peptides matched by the database search are more likely to have a fragment ion from each cleavage in the spectrum. As the length of the peptide increases the mean number of fragmentation cleavages in the spectra (blue line) deviates from the maximum number possible (red line). The variance, indicated by the box plots, also increases as peptide length increases. This effect is more evident in HCD data. HCD provides higher resolution peaks and the ability to use smaller fragment mass tolerance for the database search. This means random matches are less likely and so fewer matching peaks are needed by the database search algorithms for a significant match.
Fig. 1.

Number of cleavage sites present in the spectra. Box plots show the numbers of fragmentation cleavage sites present in the spectra for peptides of length 6 to 30. The combined results of all the CID spectra from this study are shown in A, with the HCD spectra from this study shown in B. The relative numbers of spectra per length are indicated by the blue dots, and the mean number of fragmentation cleavage sites present is shown by the blue line. The mode of each peptide length is highlighted by the green bar and the maximum number that could be present (peptide length – 1) is shown by the red line.
Both Novor and DeepNovo look for b and y ions, as well as peaks created from their neutral losses of both ammonia and water, to identify peptides. Using chains of fragment ions they can identify amino acids through their mass differences. For both the CID and HCD data, we consider the frequency with which spectra contain any fragment ion from the possible cleavage positions along the peptide backbone. Fig. 2 shows how likely each cleavage position in a peptide of length 20 identified through database search is to be represented by an ion in the spectra.
Fig. 2.

Fraction of spectra with one or more ions at each cleavage position. The figure shows the fraction of spectra, for length 20 peptides, that contain one or more ions at each fragmentation cleavage site. A contains all peptides of length 20 from the four CID datasets used in this study with B containing all peptides of length 20 from the four HCD datasets. Numbers on top of the bars indicate their relative frequency.
Length 20 was chosen as it revealed some interesting patterns with other peptide lengths available in Supplementary (Supp. Figs. 1–3). Just 2% of CID spectra and 6% of HCD spectra of peptides of length 20 had an ion from the first fragmentation cleavage site. The first cleavage site also had a below average rate of occurrence in other length peptides (Supp. Figs. 1–3). For peptides of length 14, the median peptide length, fragment ions from the first cleavage appeared in 37% of CID spectra and 33% of HCD spectra (Supp. Fig. 2). While 74% of HCD spectra of length 20 peptides had at least one ion from the last (19th) cleavage site, this number fell to 18% for CID spectra. Fragmentation cleavage sites closer to the centre of the peptides had a much better chance of being represented in the spectra. This trend was shared among all peptide lengths (Supp. Figs. 1–3). For both CID and HCD peptides of length 20, each cleavage site from position 3 to 18 and 2 to 19 respectively, was represented over 74% of the time.
3.2. Noise peaks outnumber peptide peaks
Further complicating the identification process is the abundance of peaks in the spectrum which do not belong to the peptide and are classified as noise [42].
The distribution of all peaks in the data is shown in Fig. 3. Each point represents the mass-to-charge ratio (m/z) and normalised intensity values of a peak in the data. A random selection of 1% of all peaks were used to make the plot readable. The peaks are categorised by those that can be explained by the assigned peptide (peptide peaks) and those that cannot (noise). Both distributions are skewed to the right with very few peaks greater than 1500 m/z. This trend is still observed even when controlling for peptide mass. Noise peaks outnumber those from the peptide approximately 15:1 in the CID data with the ratio being approximately 7:1 in the HCD data. While higher intensity ions are generally seen as more likely to come from a peptide, Fig. 3 shows how this alone is insufficient evidence. The quantity of noise peaks is equal to or above the quantity of peptide peaks at all intensity levels (Supp. Fig. 4).
Fig. 3.

Scatter plot of noise and peptide peaks. Scatter plot of the distribution of peak m/z and normalised intensities for both the four CID (A) and four HCD (B) datasets. Peaks attributable to each peptide are shown in blue with noise peaks shown in orange.
Only 6.3% of peaks in the CID data were attributable to the peptide assigned by the database search. Although this number more than doubled to 13% for HCD data as the number of noise peaks reduced, the noise peaks that were present were of a higher average intensity.
3.3. De Novo algorithm performance exponentially decreases with increasing peptide length
Fig. 4A shows the peptide length distribution of the total CID dataset, the number of peptides that had each fragmentation cleavage site represented in the spectrum and the number of peptides that each algorithm predicted correctly. In total, Novor predicted 5768 (24%) of the 23821 peptides correctly while DeepNovo managed 7870 (33%). DeepNovo performed better than Novor for all peptide lengths. Of the 2798 CID peptides of length 11, the most common length, DeepNovo correctly predicted 1243 (44%), while Novor correctly predicted 929 (33%). For length 8 peptides, the shortest peptides in the data, Novor correctly predicted 68% of the peptide sequences correctly while DeepNovo correctly predicted 76%. Novor successfully predicted just 5 peptides of length greater than 20 and none greater than 24. DeepNovo predicted 175 peptides with length greater than 20 and 37 greater than 24.
Fig. 4.

Correct peptide prediction distribution. Distribution of the correct peptide predictions of both algorithms for the four CID (A) and four HCD (B) datasets. The total number of peptides in the data of each length is shown in blue, with the number containing a fragment ion from each cleavage site shown by the hatching. Numbers of correct Novor predictions are shown in magenta with correct DeepNovo predictions shown in green.
The same distributions are shown for HCD data in Fig. 4B. DeepNovo predicted more peptides than Novor correctly for almost all peptide lengths. Novor did perform better for lengths 6 and 29, but due to the small sample size at these lengths this cannot be considered as significant. Of the 25007 HCD peptides, DeepNovo predicted 11705 (47%) correctly whereas Novor predicted 9710 (39%) correctly. The accuracy of both algorithms was greater across all peptide lengths compared to the CID data, highlighting how technological advances directly impact on algorithm performance. There were 2262 HCD peptides of length 11, again the most common length, of which DeepNovo correctly predicted 1305 (60%) and Novor correctly predicted 1202 (53%). DeepNovo and Novor correctly predicted 1564 (85%) and 1485 (81%) of the 1844 length 8 peptides respectively. The relative frequency of correct peptides across the different peptide lengths is shown in Supp. Fig. 5. Here an exponential decrease in peptide accuracy for both fragmentation types is observed as the peptide length increases.
The trends shown in Fig. 4 are not only the result of the decreased prevalence of fragmentation cleavage sites as peptide length increases. As the number of amino acids in a peptide sets the upper limit on the number of cleavages that can be missing, the two variables are correlated. However, when controlling for the number of missing cleavages, increased peptide length still negatively impacts performance. When the number of fragmentation cleavage sites that are missing is held constant, both algorithms show a linear decrease in peptide accuracy as peptide length increases for both data types (Supp. Fig. 6). For HCD data, Novor correctly predicted 86% of peptides of length 8 when no fragmentation cleavages were missing. It only predicted 36% of peptides of length 16 for the same criterion. DeepNovo’s accuracy dropped from 91% to 69% over the same interval when there were no missing fragmentation cleavages.
3.4. Increasing number of missing fragmentation cleavage sites exponentially decreases de novo peptide algorithm accuracy
Peptide ion peaks may be missing in the MS spectra for a variety of reasons. These include the random nature of the fragmentation collisions, the cut-off of the mass spectrometer or how unfavourable fragmentation at a cleavage site is given the amino acid sequence of the peptide [43], [41].
As shown in Fig. 5, the majority of the correctly identified peptides had at most one fragmentation cleavage site missing from the spectrum. Fewer than 3.6% of CID peptides correctly identified by Novor and 10% of CID peptides correctly identified by DeepNovo had more than one fragmentation cleavage missing. Novor did not predict any peptide correctly with more than 5 cleavage sites missing. CID spectra with more than one missing fragmentation cleavage account for over 36% of the total number of spectra. For HCD data, spectra with more than one missing fragmentation cleavage accounted for 11% of Novor’s correct predictions and 12% of DeepNovo’s. HCD spectra with more than one cleavage site missing account for 40% of the total.
Fig. 5.

Algorithm performance for increasing numbers of missing fragmentation cleavage sites. Bar plot showing the total number of spectra (blue), the total number of peptides correctly predicted by Novor (magenta) and the total number of peptides correctly predicted by DeepNovo (green) for each number of missing fragmentation cleavage sites. The combined CID data are shown in A with the combined HCD data shown in B.
To more easily compare the performance of the algorithms we also evaluate them using the relative frequency of the correct peptides. Fig. 6 shows the peptide accuracy and amino acid recall for the data bins shown in Fig. 5. For both CID and HCD data, there is an exponential decrease in the peptide accuracy of the algorithms as the number of missing fragmentation cleavage sites increases. DeepNovo consistently outperformed Novor in peptide accuracy for both fragmentation types and all numbers of missing cleavage sites. For CID data with 0 missing fragmentation cleavages, DeepNovo predicted 61% of the peptides correctly while Novor only predicted 51% correctly. Neither algorithm predicted any CID peptide with 9 or more missing fragmentation cleavages correctly. The accuracy of both algorithms was higher for HCD data. DeepNovo predicted 83% of peptides correctly while Novor predicted just 71% when no fragmentation cleavages were missing. For 3 missing cleavages the accuracy was 13% and 11% respectively. Once the number of fragmentation cleavage sites that are missing exceeded 3 in CID data, the probability of either algorithm correctly predicting a peptide fell below 4.3% with Novor fairing significantly worse. With HCD data, the peptide accuracy of DeepNovo fell below 7.9% and Novor below 5.0% when more than 3 fragmentation cleavage sites were missing and continued to decrease for greater numbers of missing cleavages.
Fig. 6.

Peptide accuracy and amino acid recall. Plots show both algorithms for the different fragmentation types; CID (A) and HCD (B). Peptide accuracy is shown by solid lines with amino acid (AA) recall shown by dotted lines. 95% confidence intervals surround each point with some too small to see.
To further evaluate the performance of the models, we also compare them using amino acid recall. While related to peptide accuracy, amino acid recall gives a finer resolution view of how the algorithms are dealing with missing fragmentation cleavage sites. This is particularly useful for spectra where there are many missing cleavages and peptide accuracy is extremely low.
A clear correlation can be seen in Fig. 6 between the amino acid recall of both algorithms and the number of fragmentation cleavage sites that are missing. The amino acid recall of both algorithms decreases almost continuously for both fragmentation types as the number of missing cleavages increases.
When no cleavage sites were missing, DeepNovo had an amino acid recall of 84% in CID data and 94% in HCD data. For the same data, Novor had amino acid recalls of 80% and 91% respectively. When there are 4 or fewer missing fragmentation cleavages, DeepNovo outperforms Novor with a greater amino acid recall for both fragmentation types. In contrast, when 5 or more cleavage sites were missing Novor was found to perform best. For spectra with 8 missing fragmentation cleavages, Novor correctly recalled 17% and 37% of the amino acids in CID and HCD data respectively. DeepNovo only recalled 15% and 31% of the amino acids correctly for the same respective data.
When these algorithms are used by researchers, only high-scoring peptides are included in the analysis. Therefore, we also performed a brief analysis using only these high scoring de novo peptides. We extracted all peptides above a threshold that gives 90% amino acid recall. The distribution of missing fragmentation cleavage sites in these peptides (Supp. Fig. 7) does not match that of the complete data (Fig. 5) as both algorithms favour peptides with fewer missing cleavages. Just 1.2% and 11% of Novor’s high scoring peptides in CID and HCD data respectively had more than 1 missing fragmentation cleavage site while 9.7% and 18% of DeepNovo’s high scoring peptides had more than 1 missing cleavage site for the respective fragmentation types.
To eliminate interactions between features, a further complementary analysis was carried out on artificial HCD data. The distribution of missing fragmentation cleavages in the data is shown in Supp. Fig. 8. Novor correctly predicted 8792 (88%) out of the 9839 artificial peptides with no missing cleavage site. DeepNovo correctly predicted 9342 (95%) of these peptides. Differences in the performance of the algorithms between artificial data and real data may be due to both the more accurate peak placement and lack of noise in the artificial data. It is difficult to give accurate predictions of peptide accuracy when more than 3 fragmentation cleavages are missing due to the lack of artificial spectra fitting this description.
3.5. Impact of noise changes with the number of fragmentation cleavages that are missing
The effect of noise on the accuracy of de novo peptide sequencing algorithms is sometimes difficult to elucidate. When viewed alone, the amount of noise in a spectrum did not show a clear negative correlation to performance. This is due to the much stronger influence of the number of missing fragmentation cleavages on algorithm accuracy. Also, much of the noise is at such low intensities that it does not affect the performance of the algorithms. To account for this, in the following analysis we only consider noise above a specific threshold, determined as the median of the noise distribution. We then define the noise factor as the ratio of these high intensity non-peptide noise peaks to peptide peaks. For example, a noise factor of 10 means there are 10 times as many noise peaks as peptide peaks in the spectrum.
Fig. 7 shows amino acid recall as a function of both the number of fragmentation cleavage sites that are missing and the noise factor for both algorithms and both fragmentation types. Amino acid recall was chosen over peptide accuracy as correct peptides were concentrated to where only zero or one fragmentation cleavage was missing (Fig. 5). Supp. Fig. 9 shows a similar plot for peptide accuracy. In Fig. 7 the number of missing cleavage sites increases from top to bottom while the noise factor increases from left to right. As expected, both algorithms perform best when there are very few missing fragmentation cleavages and the noise factor is low.
Fig. 7.

Amino Acid recall as a function of the number of missing fragmentation cleavage sites and the Noise Factor. Higher amino acid (AA) recall is shown in pink, with lower recall shown in cyan. Performance of Novor across the two fragmentation types are shown on the left (A and C) with the performance of DeepNovo shown on the right (B and D). CID data are shown on top (A and B) with HCD data shown on the bottom (C and D).
The distribution of the number of spectra in Fig. 7 is not uniform. Data points toward the extreme right and bottom of the graph have fewer and fewer spectra in them as these combinations of missing fragmentation cleavages and noise factor are less likely following the database search. White squares are data points where no spectra meet that particular combination. A few outliers near the white squares exhibit unusually high recall, inconsistent with the rest of the graph. These are data points where sample sizes are small and so do not reflect the trends seen in the rest of Fig. 7.
The relationship between noise and amino acid recall is linked with the absence of fragmentation cleavage sites. As the number of missing cleavage sites increases, fewer and fewer amino acids are correctly recalled from spectra with high noise factors. Performance decreases from where noise peaks and missing fragmentation cleavages are few (top left) to where both noise and missing fragmentation cleavages are more prevalent (bottom right) for each algorithm and fragmentation type. For both fragmentation types, DeepNovo is less affected by noise than Novor. Amino acid recall does not fall as sharply as with Novor as the noise factor increases. As seen in Supp Fig. 9, the peptide accuracy of Novor also decreases rapidly as the noise factor increases for both CID and HCD data. The effect is less acute for DeepNovo but still present. The trend is also much stronger for both algorithms in HCD data where the noise considered is of a higher average intensity and so has a much stronger influence on algorithm prediction.
To isolate the effect of noise from missing fragmentation cleavages we also analysed artificial data with additional noise peaks. To eliminate confounding factors, the artificial data were duplicated and each spectrum in a duplicate was given the same factor of random noise. Supp. Fig. 8B shows the linear decrease in performance as the noise factor was increased. Again, Deepnovo was less affected than Novor by the increased noise.
3.6. De novo algorithms can correctly predict amino acids missing from spectra
Earlier analyses showed the ability of both algorithms to correctly predict peptides when fragmentation cleavage sites are missing from the spectra (see Fig. 6). Although the performance of the algorithms is severely affected as the number of missing fragmentation cleavages increases, the algorithms are still able to make some accurate predictions. When one fragmentation cleavage is missing Novor had a CID peptide accuracy of 22% and a HCD peptide accuracy of 46%, while DeepNovo had a CID peptide accuracy of 35% and HCD peptide accuracy of 56%. To investigate how algorithms deal with missing fragmentation cleavages we compared how often each cleavage site was represented by a fragment ion in the spectra to how often it was correctly identified by the de novo algorithms (Fig. 8).
Fig. 8.

Algorithm cleavage site predictions compared to missing cleavage sites. The hatched blue bars represent the fraction of spectra that contain an ion from that cleavage site in the peptide. The magenta (Novor) and green (DeepNovo) bars show the fraction of peptides predicted by each algorithm that contained that same cleavage site. Numbers on top of the bars indicate their value.
As can be seen in Fig. 8A, CID peptides of length 20 are more likely to be missing a fragmentation cleavage site nearer the end of the peptide. This peptide length was selected as it highlights some interesting characteristics of the two algorithms. Other peptide lengths can be found in Supplementary (Supp. Figs. 1–3). As mentioned previously, only 2% of peptides of length 20 in CID data have an ion from the first cleavage position (b1 or y19) in the spectrum. However, both algorithms account for this fragmentation cleavage with their predicted peptides far more often than it appears in the data. Novor correctly identifies this cleavage position 29% of the time whereas DeepNovo correctly identifies it 27% of the time. Novor correctly predicted the 19th cleavage position in 63% of the peptides while DeepNovo predicted it correctly in 61%. This cleavage site was represented in only 18% of CID peptides of length 20. Even though both algorithms appear to perform similarly in Fig. 8A, DeepNovo predicted more than three times as many length 20 CID peptides correctly, when compared to Novor.
The corresponding graph for HCD data is shown in Fig. 8B. The first cleavage site is only present in 6% of spectra. Yet, Novor accounts for this site in 40% of the data and DeepNovo in 32%. Novor performs better than DeepNovo on the first and last cleavage sites in both HCD and CID data despite DeepNovo performing better overall. DeepNovo’s correct predictions are less evenly spread than Novor among all the peptides with a small subset containing most of the recalled amino acids. Other peptide lengths can be found in Supplementary for which similar trends were observed (Supp. Figs. 1–3).
4. Discussion
De novo peptide sequencing is a growing field with machine learning fuelling its development. Historically, effective design of de novo algorithms was difficult with previous methods relying on human expert knowledge. Including this knowledge in the design of machine learning algorithms is not straightforward as it is difficult to capture and may significantly increase the complexity of the corresponding algorithms [20]. In fact, most of the fragmentation rules identified by researchers are not included in proteomics identification tools [31]. Machine learning may allow algorithms to learn these features automatically as they uncover patterns in the data. However, the design of algorithm architectures that would facilitate this learning is non-trivial and requires a deep understanding of the data and fragmentation process.
As shown in our analysis and others [15], the performance of modern algorithms on artificial data far exceeds that of real data. Not only does this mean that analysis of algorithms on artificial data is not directly applicable to real data but it also highlights how the current bottlenecks lie with features of the data and the algorithms’ inability to cope with them. To elucidate some of these data features and show how they might be addressed, we evaluated two state of the art de novo sequencing algorithms on both real and artificial MS/MS data. We determined both the prevalence and effects of missing fragmentation cleavages and noise on de novo sequencing algorithms. We also investigated how the state of the art algorithms overcome these features.
We firstly analysed the performance of DeepNovo and Novor with respect to peptide length to ensure it did not confound later observations. Like in previous studies [32], [15], an increase in peptide length was found to negatively affect performance. Furthermore, we demonstrate the peptide accuracy exponentially decreases in response to an increase in length. This is likely due to the fact that de novo algorithms must predict each amino acid, meaning the likelihood of at least one incorrect prediction increases with the number of amino acids.
Missing fragmentation cleavages were found to be the main problem with the data which de novo sequencing algorithms must overcome. The vast majority of peptides correctly identified come from spectra with zero or one missing fragmentation cleavages (Fig. 5). As the number of missing cleavage sites increases, identification becomes more difficult with correct peptides from both algorithms becoming non-existent. Consequently, almost all of the peptides scored highly by the de novo algorithms have zero or one missing fragmentation cleavage sites (Supp. Fig. 7). Fragmentation cleavages were found to be more likely to be missing for longer peptides with larger mass values (Supp. Fig. 3) and toward the ends of the peptide.
Similar to other studies [32], we found amino acid recall tended to be better toward the middle of the peptide. Fig. 8 shows that the reduced cleavage prediction accuracy is a result of the reduced prevalence of fragment ions from those cleavages. This in turn leads to reduced amino acid recall. A clear relationship can be seen between the presence of a fragmentation cleavage in the spectra and the presence of that cleavage in the predicted peptide. These missing cleavages explain the equal mass multi-amino acid substitutions observed by these studies [32], [15]. A missing cleavage leaves a mass-gap in the chain of peptide fragments which can be filled by a number of equal mass amino acid sequences.
Noise has historically been seen as a major problem in de novo peptide sequencing [44]. For graph based methods in particular it also increases the complexity of the de novo sequencing problem exponentially by increasing the number of nodes and edges [45]. These additional peaks only become a major problem when present in large quantities and if of high intensity. We found that DeepNovo is better able to deal with high intensity noise compared to Novor in both real and artificial data.
Novor and DeepNovo employ a range of techniques including machine learning and dynamic programming as they attempt to overcome these challenges.
Both algorithms step through the spectrum one amino acid at a time. Using machine learning they try to learn what function of the features at that particular point distinguish peptide peaks from non-peptide peaks. The success of this approach is seen in their effectiveness against noise. Also, DeepNovo only considers nearby peaks when making each amino acid prediction, meaning many of the noise peaks are inconsequential. Similarly, Novor scores each peak independently thus limiting the effect of noise. Unlike DeepNovo however, Novor creates a graph of all peaks. As with all graph based de novo algorithms, this increases the complexity of the solution space. The difference in approaches is highlighted by DeepNovo’s greater performance with respect to noise.
A major strength of Novor’s graph based approach is its amino acid recall when many fragmentation cleavages are missing, where it outperforms DeepNovo. When many cleavage sites are missing, it is almost guaranteed that the highest scoring path in the graph does not reflect the correct peptide. This is shown by Novor’s low peptide accuracy in this range. However, the algorithm still manages to incorporate short subsequences of fragment ions that are present into the highest scoring path. While the complete path may not be present, these short subsequences will still be scored highly by the algorithm and so are likely to appear in the highest scoring path giving rise to a partially correct peptide. DeepNovo, on the other hand, has no means of rejoining the correct path once an incorrect step is made and so has more complete matches but fewer partial matches than Novor for this type of data. This is the reason DeepNovo maintains a higher peptide accuracy than Novor while having a lower amino acid recall at greater numbers of missing fragmentation cleavages.
The independent scoring of graph nodes means Novor cannot encapsulate the long range relationships of peptide fragmentation. Tiwary et al. (2019) showed that the entire peptide composition will have an impact on the peak intensity of each fragment ion [41]. Therefore accurate amino acid prediction will require the consideration of fragment ions from the entire peptide. DeepNovo uses an LSTM to keep track of fragment ions already encountered. It can then take advantage of their encodings for aiding the prediction of amino acids further along the peptide. This is particularly useful when DeepNovo is presented with a mass-gap caused by missing fragmentation cleavages. It can leverage the information encoded from the spectrum it has already encountered to replace the absence of peaks in its current position and make accurate predictions. The largest mass-gap correctly traversed by DeepNovo in this research spanned seven cleavage sites compared to a maximum of three for Novor. DeepNovo also uses dynamic programming, similar to the knapsack problem, to make up for the fact it can only see as much as one amino acid ahead at a time. As the true mass of the correct peptide is known, DeepNovo limits the number of amino acids to consider at each step by only allowing those that are possible given the remaining mass of the peptide. This is particularly useful at the end of the peptide where the number of options will be significantly reduced.
While algorithms are improving, our analysis has uncovered some limitations in their approaches. Unlike their database counterparts, the performance of the algorithms is not independent of the peptide length as both algorithms build the peptides up from individual components. Step-by-step predictions and independent peak scoring simply do not encapsulate all the necessary information from the fragment process. The whole of the peptide, and hence the whole of the spectrum is needed for exact amino acid prediction [41]. DeepNovo does incorporate some long range interactions, but only for the final amino acids predicted and only if those already predicted are correct. Graphs are the most suitable way to capture all the complex interactions but Novor’s machine learning is not applied over the graph but only on the peak scores. Thus, complete spectrum encoding would encapsulate the complex nature of peptide fragmentation leading to more accurate predictions. The combination of the strengths of these two aforementioned models can be harnessed using graph neural networks (GNNs) [46]. In GNNs, features of each node, such as its m/z value and intensity, can be encoded with a neural network and passed along the edges of the graph to other nodes. Through this mechanism, peaks from one end of the graph could influence the prediction of amino acids at the other. Similar applications have been shown such as the Graph2Seq model [47]. This model was shown to be extremely effective in tasks such as path finding, where an optimal sequence is predicted from a complex graph. This is similar to de novo sequencing where the sequence would be the peptide. Graph2Seq uses a GNN to encode the graph before an attention based LSTM is used to predict each element in a sequence. While each node will share information with its neighbours, the use of attention means the model can focus on multiple relevant parts of the graph at one time. In that way, a de novo peptide model could learn to focus on those peaks shown to be related to the sequence [41].
De novo algorithms may also benefit from a pre-processing step that removes noise peaks. Previous noise removal algorithms focus on a peak’s intensity and its rank among the other peaks [42], [48]. As shown in Fig. 3, intensity alone is an insufficient discriminator and peak interactions must be considered. Denoising spectra needs the same long range interactions, amino acid predictions does. Both tasks are essentially trying to find a function that distinguishes between peptide peaks and non-peptide peaks. Machine learning can learn such functions as shown by the performance of both algorithms. However, the noise peaks causing the problems for these algorithms are still scored highly and their removal requires more intelligent systems. The incorporation of long range interactions into a noise removal model would provide increased resolution, which in turn should improve the de novo algorithms’ performance in their current state.
5. Conclusion
The availability of large datasets and the addition of machine learning has led to notable advances in de novo peptide sequencing algorithms. Real data analyses revealed that noise peaks are far more abundant than peptide peaks while most peptides have missing fragmentation cleavages. DeepNovo was found to perform best overall with Novor surpassing it only for amino acid recall when many cleavages were missing. Missing fragmentation cleavages were found to be the biggest obstacle for both algorithms with both peptide length and noise also affecting performance. DeepNovo’s recurrent neural network helped counteract the effect of missing fragmentation cleavages. Future de novo algorithms may benefit from a complete spectrum encoding that encapsulates the long range dependencies of peptide fragmentation. While the quality of data is increasing, improvements in de novo peptide identification algorithms could allow new insights from past research. Future de novo algorithms will also benefit from the advances in the field of machine learning. Recently developed machine learning algorithms, such as graph neural networks, may help better capture the intricate relationships of peptide fragmentation thereby advancing performance in this space.
CRediT authorship contribution statement
Kevin McDonnell: Conceptualization, Methodology, Writing - original draft. Enda Howley: Writing - review & editing. Florence Abram: Conceptualization, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was supported by the Irish Research Council (grant number GOIPG/2019/1650, awarded to Kevin McDonnell).
Footnotes
Supplementary data associated with this article can be found, in the online version, athttps://doi.org/10.1016/j.csbj.2022.03.008.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Nesvizhskii A.I. Protein identification by tandem mass spectrometry and sequence database searching. Mass Spectrometry Data Anal Proteomics. 2007:87–119. doi: 10.1385/1-59745-275-0:87. [DOI] [PubMed] [Google Scholar]
- 2.Sallam R.M. Proteomics in cancer biomarkers discovery: challenges and applications. Disease Markers. 2015;2015 doi: 10.1155/2015/321370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K., et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nature Commun. 2016;7(1):1–16. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvarez S., Roy Choudhury S., Pandey S. Comparative quantitative proteomics analysis of the aba response of roots of drought-sensitive and drought-tolerant wheat varieties identifies proteomic signatures of drought adaptability. J Proteome Res. 2014;13(3):1688–1701. doi: 10.1021/pr401165b. [DOI] [PubMed] [Google Scholar]
- 5.Pocsfalvi G., Cacace G., Cuccurullo M., Serluca G., Sorrentino A., Schlosser G., Blaiotta G., Malorni A. Proteomic analysis of exoproteins expressed by enterotoxigenic staphylococcus aureus strains. Proteomics. 2008;8(12):2462–2476. doi: 10.1002/pmic.200700965. [DOI] [PubMed] [Google Scholar]
- 6.Li J., Guo M., Tian X., Liu C., Wang X., Yang X., Wu P., Xiao Z., Qu Y., Yin Y., et al. Virus-host interactome and proteomic survey of pmbcs from covid-19 patients reveal potential virulence factors influencing sars-cov-2 pathogenesis. BioRxiv. 2020 doi: 10.1016/j.medj.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muth T., Hartkopf F., Vaudel M., Renard B.Y. A potential golden age to come–current tools, recent use cases, and future avenues for de novo sequencing in proteomics. Proteomics. 2018;18(18):1700150. doi: 10.1002/pmic.201700150. [DOI] [PubMed] [Google Scholar]
- 8.White F.M. The potential cost of high-throughput proteomics. Sci Signal. 2011;4(160):pe8. doi: 10.1126/scisignal.2001813. [DOI] [PubMed] [Google Scholar]
- 9.Verheggen K., Ræder H., Berven F.S., Martens L., Barsnes H., Vaudel M. Anatomy and evolution of database search engines–a central component of mass spectrometry based proteomic workflows. Mass Spectrometry Rev. 2020;39(3):292–306. doi: 10.1002/mas.21543. [DOI] [PubMed] [Google Scholar]
- 10.Olsen J.V., Macek B., Lange O., Makarov A., Horning S., Mann M. Higher-energy c-trap dissociation for peptide modification analysis. Nature Methods. 2007;4(9):709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 11.Tabb D.L., Smith L.L., Breci L.A., Wysocki V.H., Lin D., Yates J.R. Statistical characterization of ion trap tandem mass spectra from doubly charged tryptic peptides. Anal Chem. 2003;75(5):1155–1163. doi: 10.1021/ac026122m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frank A.M., Monroe M.E., Shah A.R., Carver J.J., Bandeira N., Moore R.J., Anderson G.A., Smith R.D., Pevzner P.A. Spectral archives: extending spectral libraries to analyze both identified and unidentified spectra. Nature Methods. 2011;8(7):587–591. doi: 10.1038/nmeth.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griss J., Perez-Riverol Y., Lewis S., Tabb D.L., Dianes J.A., Del-Toro N., Rurik M., Walzer M., Kohlbacher O., Hermjakob H., et al. Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nature Methods. 2016;13(8):651–656. doi: 10.1038/nmeth.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nesvizhskii A.I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteom. 2010;73(11):2092–2123. doi: 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muth T., Renard B.Y. Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification? Briefings Bioinform. 2018;19(5):954–970. doi: 10.1093/bib/bbx033. [DOI] [PubMed] [Google Scholar]
- 16.Lu B., Chen T. Algorithms for de novo peptide sequencing using tandem mass spectrometry. Drug Discovery Today: BioSilico. 2004;2(2):85–90. [Google Scholar]
- 17.Wang Y.-C., Peterson S.E., Loring J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24(2):143–160. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahrné E., Müller M., Lisacek F. Unrestricted identification of modified proteins using ms/ms. Proteomics. 2010;10(4):671–686. doi: 10.1002/pmic.200900502. [DOI] [PubMed] [Google Scholar]
- 19.Frank A.M., Savitski M.M., Nielsen M.L., Zubarev R.A., Pevzner P.A. De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res. 2007;6(1):114–123. doi: 10.1021/pr060271u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma B. Novor: real-time peptide de novo sequencing software. J Am Soc Mass Spectrom. 2015;26(11):1885–1894. doi: 10.1007/s13361-015-1204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran N.H., Zhang X., Xin L., Shan B., Li M. De novo peptide sequencing by deep learning. Proc Nat Acad Sci. 2017;114(31):8247–8252. doi: 10.1073/pnas.1705691114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dančík V., Addona T.A., Clauser K.R., Vath J.E., Pevzner P.A. De novo peptide sequencing via tandem mass spectrometry. J Computat Biol. 1999;6(3–4):327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- 23.Kim S., Gupta N., Bandeira N., Pevzner P.A. Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra. Mol Cell Proteom. 2009;8(1):53–69. doi: 10.1074/mcp.M800103-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G.A., Ma B. Peaks db: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteom. 2012;11(4) doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank A., Tanner S., Bafna V., Pevzner P. Peptide sequence tags for fast database search in mass-spectrometry. J Proteome Res. 2005;4(4):1287–1295. doi: 10.1021/pr050011x. [DOI] [PubMed] [Google Scholar]
- 26.Tran N.H., Qiao R., Xin L., Chen X., Liu C., Zhang X., Shan B., Ghodsi A., Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 2019;16(1):63–66. doi: 10.1038/s41592-018-0260-3. [DOI] [PubMed] [Google Scholar]
- 27.Zinkernagel R.M., Hengartner H. Regulation of the immune response by antigen. Science. 2001;293(5528):251–253. doi: 10.1126/science.1063005. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Garijo A., Fajardo C.A., Gros A. Determinants for neoantigen identification. Front Immunol. 2019;10:1392. doi: 10.3389/fimmu.2019.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng M., Mo Y., Wang Y., Wu P., Zhang Y., Xiong F., Guo C., Wu X., Li Y., Li X., et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18(1):1–14. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martens L., Hermjakob H., Jones P., Adamski M., Taylor C., States D., Gevaert K., Vandekerckhove J., Apweiler R. Pride: the proteomics identifications database. Proteomics. 2005;5(13):3537–3545. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 31.Medzihradszky K.F., Chalkley R.J. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom Rev. 2015;34(1):43–63. doi: 10.1002/mas.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bringans S., Kendrick T.S., Lui J., Lipscombe R. A comparative study of the accuracy of several de novo sequencing software packages for datasets derived by matrix-assisted laser desorption/ionisation and electrospray. Rapid Communications in Mass Spectrometry: An International Journal Devoted to the Rapid Dissemination of Up-to-the-Minute Research in Mass Spectrometry. 2008;22(21):3450–3454. doi: 10.1002/rcm.3752. [DOI] [PubMed] [Google Scholar]
- 33.Cottrell J.S. Protein identification using ms/ms data. J Proteomics. 2011;74(10):1842–1851. doi: 10.1016/j.jprot.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Muth T., Kolmeder C.A., Salojärvi J., Keskitalo S., Varjosalo M., Verdam F.J., Rensen S.S., Reichl U., de Vos W.M., Rapp E., et al. Navigating through metaproteomics data: a logbook of database searching. Proteomics. 2015;15(20):3439–3453. doi: 10.1002/pmic.201400560. [DOI] [PubMed] [Google Scholar]
- 35.Kim S., Pevzner P.A. Ms-gf+ makes progress towards a universal database search tool for proteomics. Nature Commun. 2014;5(1):1–10. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig R., Beavis R.C. Tandem: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 37.Barsnes H., Vaudel M. Searchgui: a highly adaptable common interface for proteomics search and de novo engines. J Proteome Res. 2018;17(7):2552–2555. doi: 10.1021/acs.jproteome.8b00175. [DOI] [PubMed] [Google Scholar]
- 38.Levitsky L.I., Klein J.A., Ivanov M.V., Gorshkov M.V. Pyteomics 4.0: five years of development of a python proteomics framework. J Proteome Res. 2018;18(2):709–714. doi: 10.1021/acs.jproteome.8b00717. [DOI] [PubMed] [Google Scholar]
- 39.Muth T., Weilnbock L., Rapp E., Huber C.G., Martens L., Vaudel M., Barsnes H. Denovogui: an open source graphical user interface for de novo sequencing of tandem mass spectra. J Proteome Res. 2014;13(2):1143–1146. doi: 10.1021/pr4008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., Knaute T., Rechenberger J., Delanghe B., Huhmer A., et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nature Methods. 2019;16(6):509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 41.Tiwary S., Levy R., Gutenbrunner P., Soto F.S., Palaniappan K.K., Deming L., Berndl M., Brant A., Cimermancic P., Cox J. High-quality ms/ms spectrum prediction for data-dependent and data-independent acquisition data analysis. Nature Methods. 2019;16(6):519–525. doi: 10.1038/s41592-019-0427-6. [DOI] [PubMed] [Google Scholar]
- 42.Mujezinovic N., Schneider G., Wildpaner M., Mechtler K., Eisenhaber F. Reducing the haystack to find the needle: improved protein identification after fast elimination of non-interpretable peptide ms/ms spectra and noise reduction. BMC Genom. 2010;11(1):1–8. doi: 10.1186/1471-2164-11-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y., Triscari J.M., Tseng G.C., Pasa-Tolic L., Lipton M.S., Smith R.D., Wysocki V.H. Statistical characterization of the charge state and residue dependence of low-energy cid peptide dissociation patterns. Anal Chem. 2005;77(18):5800–5813. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mo L., Dutta D., Wan Y., Chen T. Msnovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry. Anal Chem. 2007;79(13):4870–4878. doi: 10.1021/ac070039n. [DOI] [PubMed] [Google Scholar]
- 45.Chen T., Kao M.-Y., Tepel M., Rush J., Church G.M. A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J Comput Biol. 2001;8(3):325–337. doi: 10.1089/10665270152530872. [DOI] [PubMed] [Google Scholar]
- 46.Scarselli F., Gori M., Tsoi A.C., Hagenbuchner M., Monfardini G. The graph neural network model. IEEE Trans Neural Networks. 2008;20(1):61–80. doi: 10.1109/TNN.2008.2005605. [DOI] [PubMed] [Google Scholar]
- 47.Xu K, Wu L, Wang Z, Feng Y, Witbrock M, Sheinin V. Graph2seq: Graph to sequence learning with attention-based neural networks, arXiv preprint arXiv:1804.00823; 2018.
- 48.Ding J., Shi J., Poirier G.G., Wu F.-X. A novel approach to denoising ion trap tandem mass spectra. Proteome Sci. 2009;7(1):1–10. doi: 10.1186/1477-5956-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.