

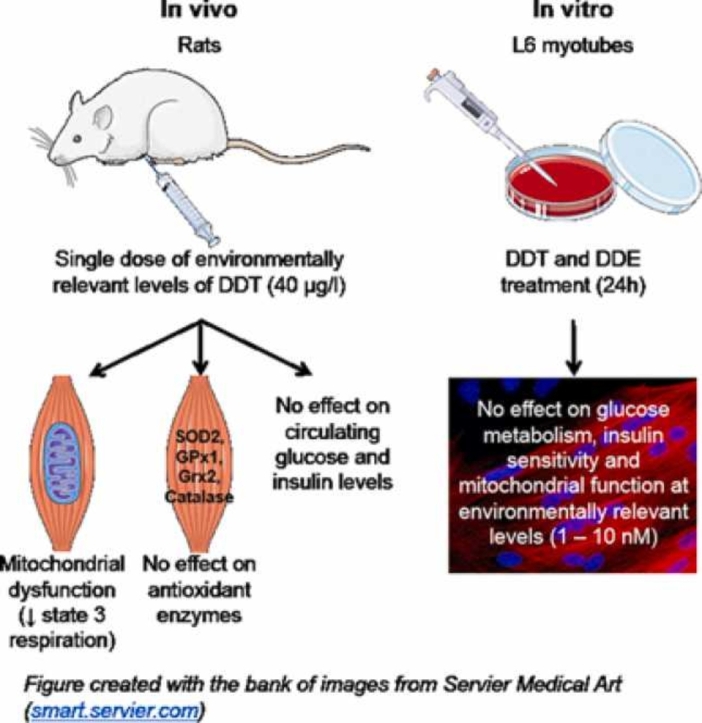
Abstract
Under insulin-stimulated conditions, skeletal muscle is the largest glucose consumer in the body. Mitochondrial dysfunction and damage to this tissue from oxidative stress are linked to the pathogenesis of type 2 diabetes. Environmental exposure to dichlorodiphenyltrichloroethane (DDT) and its metabolite, 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (DDE), has been associated with the incidence of type 2 diabetes as well as altered oxidative stress and mitochondrial dysfunction in non-muscle tissues. We hypothesized that energy metabolism and insulin sensitivity in skeletal muscle will be altered with exposure to DDT and DDE. In this pilot study, mitochondrial function was measured in permeabilized muscle fibers from Sprague-Dawley rats after one week of exposure to a single injection of DDT (40 μg/kg), a dose comparable to DDT levels in the diets of the Inuit of Northern Canada. The levels of oxidative phosphorylation chain complexes and ROS detoxification enzymes were measured in muscle tissue from these specimens. This acute in vivo exposure to DDT decreased muscle mitochondrial function by 45% without affecting the levels of mitochondrial oxidative phosphorylation chain complexes nor levels of ROS detoxification enzymes. To isolate the effects of DDT and DDE exposure on muscle, L6 myotubes were exposed to DDT or DDE (0, 10, 100, 1000, 10 000 nM) for 24 h. Only very high concentrations of DDT and DDE (1 000 – 10 000 nM) altered maximal respiration with only DDT altering basal glucose uptake in L6 myotubes. This did not alter levels of ROS detoxification enzymes or malondialdehyde (MDA) in L6 myotubes. Altogether, acute exposure to environmentally relevant doses of DDT resulted in muscle mitochondrial dysfunction in vivo in rats, but not when muscle cells were directly exposed to the pollutant or its metabolite.
Keywords: Dichlorodiphenyltrichloroethane, Glucose uptake, Glycolysis, Mitochondrial function, Oxidative stress, Persistent organic pollutants
Graphical Abstract
Highlights
-
•
A single exposure to DDT in rats alters skeletal muscle mitochondrial function.
-
•
A single exposure to DDT in rats does not alter circulating glucose and insulin.
-
•
Exposure of L6 myotubes to low levels of DTT does not alter insulin sensitivity.
-
•
Exposure of L6 myotubes to low levels of DTT does not alter mitochondrial function.
-
•
DTT exposure in rats or myotubes does not alter muscle oxidative stress markers.
1. Introduction
Discovered in the 1940s, dichlorodiphenyltrichloroethane (DDT), a highly potent and efficient pesticide, was commonly used to control the spread of vector-borne diseases [1]. On account of DDT disrupting human endocrine function and causing adverse environmental effects, it was banned thirty-years later [1], [2]. Despite this, certain countries continue to produce and use it for indoor malaria control [3]. DDT and its degradation product 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene (DDE) are persistent organic pollutants (POPs). Lipophilic in nature, they have bioaccumulated across the food chain in lipid-rich tissues posing a relevant threat to public health [4].
Resulting from a complex relationship between genetic and environmental factors such as exposure to POPs, diabetes today poses a major threat to global public health [5]. Numerous epidemiological studies have suggested an association between exposure to DDTs and type 2 diabetes (T2D) [6], [7], [8], [9], [10]. In cross-sectional and prospective studies, background exposure to DDT and DDE showed the strongest and most significant association to T2D [11], [12], [13]. Higher concentrations of DDTs, ranging between 30% and 60%, are reported in people living with T2D compared to healthy controls [11]. Also, serum levels of DDTs are positively and significantly associated with several components of the metabolic syndrome; including hyperglycemia [11], [14]. These epidemiological observations are supported with experimental evidence of metabolic syndrome and insulin resistance in animal models following exposure to a low dose of DDT [15], [16]. Though most of the studies linked DDT exposure with the development of T2D, a more recent study in India where levels of DDT are high, showed no significant association, contradicting the findings of others [17].
The mechanism of impairment caused by DDT and DDE, which potentially contributes to T2D, remains unclear. Nonetheless, these particular POPs are suggested to target the mitochondria [14], [18], [19] as noted by the alterations observed in the expression and/or functioning of mitochondrial proteins in different tissues following exposure. Early reports implicated exposure to DDT (10–200 μM) in dysfunction of mitochondrial processes, reporting decreased respiration, weak inhibition of the electron transport chain, and downstream reduction in ATP synthesis in the liver, brain, and testis [20], [21], [22]. High doses of DDT (40–50 nmol/mg of mitochondrial protein) lead to the destruction of mitochondrial structural integrity and inhibition of ATPase activity in mitochondria isolated from the liver [23]. DDE exposure at 80 nmol/mg of mitochondrial protein led to mitochondrial swelling and uncoupling oxidation from phosphorylation in liver tissue [24].
Mitochondria are a major source of reactive oxygen species (ROS) production. ROS can act as important signaling molecules; however, high levels could negatively alter cell structure and function [25]. Thus, under normal physiological conditions, ROS detoxification enzymes are the cells’ defense mechanism to scavenge ROS [26]. Hence, an imbalance between ROS production and the antioxidant capacity of a cell, known as oxidative stress, deems to be damaging. Along with the reported mitochondrial dysfunction, DDT exposure in rats (50 and 100 mg/kg) led to the elevation of ROS and renal injury [27]. Levels of scavenger enzymes, superoxide dismutase (SOD), catalase, and glutathione peroxidase 1 (GPx1) activities were all decreased, suggesting that DDT increased ROS levels, which induced apoptosis of renal tubular cells [27]. DDT also led to oxidative-stress-induced apoptosis in testes of rats and decreased levels of ROS detoxification enzymes [28] showing a consistent effect across different tissues.
Strong associations have been observed in skeletal muscle between oxidative stress, reduced mitochondrial function, and insulin resistance [29], [30]. The leading hypothesis is that muscle mitochondrial dysfunction leads to oxidative stress and downstream glucose metabolism impairment through insulin resistance [31], [32], [33], [34]. Due to its mass, skeletal muscle is a major player in the regulation of energy homeostasis. Reduced cellular response to insulin in skeletal muscle is one of the major components of the development of T2D [35]. Besides the two recent studies that have shown supraphysiological levels of DDT altering glucose metabolism in L6 myotubes [36], [37], no studies have investigated the effect of acute exposure to physiologically relevant levels of DDT and DDE on skeletal muscle glucose uptake and mitochondrial function. Therefore, this pilot study aimed to evaluate the effects of acute DDT and DDE exposure on muscle mitochondrial function, glycolysis, glucose uptake, and insulin sensitivity. This was done using levels relevant to those found in mammals that are consumed by the Inuit of Northern Canada, on the assumption that circulating serum levels of DDT and DDE in our models will be comparable once metabolised [38]. We hypothesize that DDT and its metabolite will lead to mitochondrial toxicity in muscle cells, as they have previously in other tissues, by targeting complexes and processes in the mitochondria. This will have a downstream effect on substrate utilisation by the mitochondria, overall cellular respiration and consequently insulin resistance.
2. Materials and methods
2.1. DDT exposure
Four-month-old male white Sprague-Dawley rats (Charles River, Canada) with free access to a chow diet (Harlan Teklad Global Rodent, T2018) were housed at the University of Ottawa Animal Facility and cared for according to the Canadian Council on Animal Care. Experimentations were approved by the Animal Care Committee of the University of Ottawa (ME-2230). Rats were either exposed to DDT (Ultra Scientific, USA) (40 μg/kg, n = 8) or vehicle (corn oil, n = 8) by a single intraperitoneal injection. One week after the injection, the following experiments were performed.
2.2. Tissue collection and processing
Rats were anesthetized by pentobarbital sodium intraperitoneal injection at 45 ml/kg in the fed state. Blood was drawn from the abdominal vena cava and centrifuged (3000 g for 10 min at 4 °C) in EDTA-pre-treated tubes to collect plasma. Plasma was then stored at − 80 °C until further analysis. From each rat, quadriceps, soleus, and red and white gastrocnemius muscles were then collected, flash-frozen, and stored at − 80 °C until homogenization. Part of red gastrocnemius was used fresh for the determination of oxygen consumption rate (OCR) on permeabilized muscle fibers.
2.3. Glucose and insulin measurements
According to the ELISA kit protocol, plasma insulin levels were measured (Millipore: EZRMI-13 K, Canada). Plasma glucose was measured using a glucose assay kit (GAGO-20, Sigma-Aldrich, Canada) following the manufacturer’s instructions.
2.4. Permeabilized muscle fiber oxygen consumption rate measurements
We chose red gastrocnemius muscle because of its high mitochondrial density, which, we believe, facilitates the detection of mitochondrial defects. Oxygen consumption rate (OCR) was measured at 37 °C using an Hansatech Clark-type oxygen electrode on fresh permeabilized muscle fibers, as previously described [39], [40]. Reagents used were all from Sigma-Aldrich, Canada. Muscle fibers were first isolated and permeabilized with saponin (50 μg/ml). OCR was then measured in state 2 (no ADP) and state 3 (saturating amount of ADP (2 mM)) with the following substrates: (1) complex I substrates: 5 mM pyruvate / 5 mM malate / 10 mM glutamate and (2) complex II substrate: 10 mM succinate in the presence of 0.2 μM rotenone to inhibit complex I. We then determined proton leak (state 4) by inhibiting ATP synthase with 4 μg/ml oligomycin. We measured non-mitochondrial respiration by inhibiting complex III with 5 μM antimycin A. Finally, maximal complex IV activity was measured in the presence of 0.5 mM TMPD (N,N,N′,N′-Tetramethyl-p-phenylenediamine dihydrochloride) and 2 mM ascorbate. OCR is expressed per mg of dry muscle weight.
2.5. Muscle homogenates
Flash-frozen muscle samples were homogenized using Tissue Lyser II. A homogenizing buffer containing 20 mM Trizma® base (pH 7.4), 250 mM sucrose, 50 mM NaCl, 1% Triton X-100, 2% β-mercaptoethanol, 1 mM Na-orthovanadate, 50 mM Na-fluoride (Na-F), 5 mM Na-pyrophosphate (Na-P) (anti-phosphatases) with protease inhibitors (Sigma-Aldrich, Canada) was used. The Tissue Lyser II was set at a frequency of 30 Hz for 12 min. Homogenates were then centrifuged at 12,000g for 20 min at 4 °C. The supernatant was aliquoted and frozen at − 80 °C.
2.6. Cell culture and treatments
Rat L6 myoblasts, obtained from Dr. Amira Klip (SickKids, Toronto, ON, Canada), were grown in α-minimal essential medium (α-MEM; Wisent Bioproducts, Canada) containing 10% fetal bovine serum (FBS; Wisent Bioproducts, Canada) and 1X antibiotic - antimycotic (AA). In the presence of 2% FBS α-MEM (supplemented with 1X AA: differentiation medium), myoblasts were induced to differentiate into myotubes over a 7-day period. In differentiation medium, myotubes were then exposed for 24 h to DDT or DDE (1, 10, 100, 1000, 10,000, 100,000 nM) or the vehicle (DMSO, 0.1%; Sigma-Aldrich, Canada). These concentrations allowed us to assess the effects of DDT and DDE based on comparable concentrations found in plasma or serum of populations in areas with discontinued use of these pesticides (1–20 nM) (Health Canada, 2010), as well as populations in parts of the world that actively remain exposed to it (700–1000 nM) [41], [42].
2.7. Cell viability assay
L6 myoblasts were plated at density of 20,000 cells/well of a 96-well plate. Treatment with DDT or DDE (as described above) was then performed for 24 h following the 7-day differentiation period. Myotubes were then incubated for 30 min in 1x PrestoBlue® cell viability reagent (Life Technologies, Canada) to access cell viability spectrophotometrically at 570 and 600 nm (reference wavelength).
2.8. Oxygen consumption
On XFe96 cell culture microplates (Agilent, USA), a density of 8000 cells/well of L6 myoblasts were plated and differentiated over a 7-day period. Myotubes were then incubated for 45 min at 37 °C at ambient CO2 in HCO3-free assay Dulbecco’s Modified Eagle Medium (DMEM) containing 5 mM dextrose, 1 mM sodium pyruvate, and 4 mM glutamine, at pH 7.4 (all from Sigma-Aldrich, Canada) (= Seahorse assay medium). Using the Seahorse XFe96 analyzer (Agilent, USA), OCR were measured. Rates were obtained, as previously described [43], in the basal state (no drug), after inhibition of ATP synthase with 3 μM oligomycin (= state 4 respiration), after addition of 2 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (= maximal respiration (state 3 non-coupled respiration)), and after inhibition of complex III with 4 μM antimycin A (= non-mitochondrial oxygen consumption) (all drugs from Sigma-Aldrich, Canada). Cells were then lysed using 0.05 M NaOH and the Bradford protein assay was used to assess protein content (Biorad, Canada). Mitochondrial OCR (basal, oligomycin, or FCCP OCR) are reported after the subtraction of OCR in the presence of antimycin. Mitochondrial OCR are expressed per μg of total cellular protein.
2.9. Extracellular acidification rates
On XFe96 cell culture microplates (Agilent, USA), a density of 8000 cells/well of L6 myoblasts were plated and differentiated over a 7-day period as described above. At the end of the 24-hour treatment with DDT or DDE, myotubes were then incubated for 45 min at 37 °C at ambient CO2 in HCO3-free assay DMEM containing 4 mM glutamine. Using the Seahorse XFe96 analyzer (Agilent, USA), extracellular acidification rates (ECAR, a marker of glycolysis) were measured following the Seahorse glycolysis stress test. Three compounds were added sequentially in the following order: 10 mM glucose, 1 μM oligomycin (to inhibit ATP synthase), and 50 mM 2-deoxyglucose (to inhibit glycolysis) (all from Sigma-Aldrich, Canada). Rates were obtained in the basal state (prior to the addition of glucose), and after addition of each of the three compounds. Cells were then lysed using 0.05 M NaOH and the Bradford protein assay was used to assess protein content (Biorad, Canada). ECAR values are reported in mpH/min per μg of total cellular protein. For each independent experiment, the same 0 nM control was used for DDT and DDE.
2.10. Glucose uptake
Using a 24-well plate, L6 myoblasts were plated at density of 10,000 cells/well, differentiated, and exposed to DDT, DDE, or the vehicle as described above. Then cells were starved for 3 h in serum-free α-MEM prior to glucose uptake measurement (as previously described in [44]). Insulin at 100 nM was used to treat half of the cells for 20 min at 37 °C. Cells were then washed 3 times with HEPES-Buffered Saline (HBS) and treated for 10 min with 10 μM 2-deoxy-glucose and 0.5 μCi/ml [3H] 2-deoxy-glucose in HBS, at room temperature. Non-specific glucose uptake was determined by treating few wells with Cytochalasin B (10 μM) in HBS. Cells were lysed in 0.5 ml of 0.05 M NaOH and 0.4 ml of cell lysate was counted using a Tri-Carb2910TR scintillation counter (PerkinElmer, Canada). A Bradford protein assay was used to measure protein content in the remaining 0.1 ml of the cell lysate.
2.11. Cell lysis for Western blot analysis
As described above, L6 myoblasts were plated on 6-well plates at density of 300,000 cells/well, differentiated, and exposed to DDT, DDE, or vehicle for 24 h. For Western blot analysis, L6 myotubes were lysed in lysis buffer (20 mM Tris-HCl, 250 mM sucrose, 50 nM NaCl, 1% 100X Triton, 5 mM NaP, 50 mM NaF) and kept at − 80 °C for later use.
2.12. Western blots analysis
Western blots were performed on muscle homogenates and cell lysates. The following primary antibodies were used: monoclonal anti-α-tubulin (DM1A, no. 3873, Cell Signalling, USA) diluted 1:2000, OXPHOS Rodent WB Antibody Cocktail (contains 5 mouse antibodies, CI subunit NDFB8, CII-30 kDa, CIII-Core protein 2, CIV subunit I and CV aloha subunit) (ab110413, Abcam, USA) diluted 1:1200, polyclonal anti-SOD2 (sc-30080, Santa Cruz, USA), monoclonal anti-glutathione peroxidase 1 (GPx1, ab108427), polyclonal anti-glutaredoxin 2 (Grx2, ab191292) (Abcam, USA), and monoclonal anti-catalase (CST-D5N7V, Cell Signaling Technology, Canada) diluted 1:1000. The secondary antibodies were anti-rabbit (sc-2030) and anti-mouse (sc-2005) antibodies coupled to horseradish peroxidase (Santa Cruz, USA), diluted 1:2000 and 1:3000, respectively. An enhancement luminescent reagent and Chemidoc Imager and VisionWorks LS (UVP, USA) were used to visualize the proteins. Alpha-tubulin was used as a loading control. Densitometric analysis using ImageJ program (National Institutes of Health, USA) was done to quantify the expression of proteins.
2.13. Determination of intracellular malondialdehyde (MDA) levels
Intracellular MDA levels were examined using the lipid peroxidation (MDA) assay kit (colorimetric/fluorometric) (ab118970, Abcam). L6 myoblasts were cultured in 6-well plates, then differentiated into myotubes over a 7-day period as described above. They were exposed to DDT or DDE (1, 10, 100, 1000, 10,000 nM) or the vehicle (DMSO, 0.1%) for 24 h on the sixth day of differentiation. MDA levels were then measured following the manufacturer’s instructions for cell samples and a colorimetric assay, with the following exception. To meet the manufacturer’s suggestion of 2 × 106 cells per assay, two wells in the 6-well plates were prepared for each condition. Following treatment with DDT, DDE or the vehicle, the cells were lysed using 176.75 μl of lysis buffer per well. The lysates from the 2 wells per condition were pooled, for a total lysate volume of 353.5 μl per condition. A DC protein assay (Biorad, Canada) was used to measure protein content in the lysates. For each independent experiment, the same 0 nM control was used for DDT and DDE.
2.14. Statistical analysis
Data are presented as means ± standard error of the mean (SEM). Paired t-tests, one-way analysis of variance (ANOVA), and two-way ANOVA were used to assess statistical differences with GraphPad Prism 8 Software. The post hoc test used for the ANOVA was Dunnett correction for multiple comparisons. Significance was set at p ≤ 0.05.
3. Results
3.1. Acute DDT exposure in rats did not affect body weight, plasma insulin, or glucose levels
A week following the injection of a single dose of DDT, rats did not show any significant difference in body weight compared to the vehicle-treated rats (Table S1). Furthermore, DDT exposure did not significantly alter plasma glucose or insulin levels (Table S1).
3.2. DDT exposure in rats decreased mitochondrial function in permeabilized muscle fibers without affecting the level of mitochondrial complexes
The effect of DDT on rat’s permeabilized red gastrocnemius respiration one-week post-injection is shown in Fig. 1. State 3 OCR in presence of complex I substrates (pyruvate/malate/glutamate) was significantly reduced by 45% with DDT exposure (p < 0.05, Fig. 1). A trend for a decrease in state 3 OCR in presence of complex II substrate (succinate with complex I inhibition by rotenone) was shown upon DDT exposure but it did not reach significance (Fig. 1). DDT exposure had no significant effect on state 2, state 4, non-mitochondrial OCR, and maximal complex IV activity (Fig. 1). To determine if the decrease in state 3 OCR was the result of decreased oxidative phosphorylation complex content, the levels of respiratory chain complexes were measured (Fig. 2). Levels of mitochondrial complexes in four different muscle types (white gastrocnemius, quadriceps, red gastrocnemius, or soleus muscles) were not significantly altered by DDT exposure (Fig. 2).
Fig. 1.

A single exposure to DDT in rats alters muscle mitochondrial function. Oxygen consumption rates (OCR) measured in permeabilized red gastrocnemius muscle fibers of rats exposed for a week to 40 μg/kg DDT or the vehicle (corn oil) by a single intraperitoneal injection. Oxygen consumption was measured in state 3 condition with complex I substrates (pyruvate/glutamate/malate) (state 3 complex I) and complex II substrate (succinate in the presence of rotenone to inhibit complex I) (state 3 complex II), and state 4 condition after ATP synthase inhibition by oligomycin. Non-mitochondrial oxygen consumption was assessed in the presence of antimycin A. Maximal activity of complex IV was measured in presence of TMPD/ascorbate. Mean ± SEM. n = 7 per group. *, p < 0.05.
Fig. 2.

A single exposure to DDT in rats does not affect the expression of mitochondrial respiratory chain complexes. A-D. Levels of electron transport chain complexes measured by Western blots in (A) red gastrocnemius, (B) white gastrocnemius, (C) soleus, and (D) quadriceps muscles of rats exposed to 40 μg/kg of DDT or the vehicle (corn oil) for a week by a single intraperitoneal injection. Alpha-tubulin was used as a loading control. Left panels: representative Western blots, right panels: quantification by density analysis. Mean ± SEM. n = 8 per group.
3.3. Exposure of rats to DDT did not alter levels of ROS detoxification enzymes
Protein levels of enzymes involved in ROS detoxification (GPx1, Grx2, catalase, and SOD2) were measured in both glycolytic (white gastrocnemius and quadriceps) and oxidative (red gastrocnemius and soleus) muscles of rats exposed to DDT or the vehicle (Fig. 3). GPx1, Grx2, SOD2, and catalase levels in skeletal muscle of rats were not significantly altered by DDT exposure.
Fig. 3.

A single exposure to DDT in rats does not alter the expression of ROS detoxification enzymes. A-D. Levels of reactive oxygen species detoxification enzymes (Grx2, SOD2, GPx1, and catalase) measured by Western blots in (A) red gastrocnemius, (B) white gastrocnemius, (C) soleus, and (D) quadriceps muscles of rats exposed to 40 μg/kg of DDT or the vehicle (corn oil) for a week by a single intraperitoneal injection. Alpha-tubulin was used as a loading control. Top panels: representative Western blots, bottom panels: quantification by density analysis. Mean ± SEM. n = 4–8 per group.
3.4. Effect of DDT or DDE exposure on cell viability
The effect of exposure to different concentrations of DDT and DDE for 24 h on L6 myotube viability was evaluated. As shown in Fig. 4, DDT or DDE exposure from 1 to 10,000 nM did not significantly alter the viability of L6 myotubes, while 100,000 nM significantly decreased cell viability (p < 0.0001). This concentration (100,000 nM) was therefore excluded from the rest of the experiments.
Fig. 4.

DDT and DDE exposure at 100,000 nM for 24 h results in cell death in L6 myotubes. Cell viability measured in L6 myotubes exposed for the last 24 h of differentiation to the vehicle (0.1% DMSO) or different concentrations of DDT or DDE (1 – 100,000 nM). Experiment done in triplicate for each condition. Mean ± SEM. **, p < 0.01, *** , p < 0.001 compared to the control conditions (0 nM DDT or 0 nM DDE).
3.5. DDT and DDE exposure in L6 myotubes altered maximal mitochondrial respiration
To determine if direct exposure to DDT or DDE affects muscle cell mitochondrial function, L6 myotubes were exposed for 24 h to different concentrations of DDT or DDE. DDT and DDE exposure (0, 1, 10, 100, 1000, and 10,000 nM) did not alter basal respiration or proton leak (state 4) (Fig. 5A and B). However, DDT and DDE had opposite effects on maximal respiration (OCR in presence of FCCP) at both 1000 nM and 10,000 nM (Fig. 5A and B). In the presence of DDT, maximal OCR was increased at 1000 nM, while the same concentration of DDE decreased it. An exposure to 10,000 nM of DDT led to decreased maximal OCR by 32% (p < 0.0001) while DDE had the opposite effect, 13% increased maximal OCR (p < 0.05). An increase in maximal respiration was also observed at the low concentration of 10 nM of DDE exposure (Fig. 5B).
Fig. 5.

DDT and DDE exposure in L6 myotubes alters maximal mitochondrial respiration. A-B. Oxygen consumption rate (OCR) in L6 myotubes exposed for 24 h to the vehicle (0.1% DMSO) or (A) 1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM DDT, (B) 1 nM, 10 nM, 100 nM, 1000 nM and 10,000 nM DDE. Cells were subsequently treated with oligomycin (3 μM) to assay proton leaking and FCCP (2 μM) to assay maximal OCR capacity. n = 3, each independent experiment was done in 4 replicates. Mean ± SEM. *, p < 0.05, **, p < 0.01, *** , p < 0.001 compared to 0 nM DDT or DDE.
3.6. Effect of DDT and DDE on muscle cell glycolytic function
Glycolytic function was estimated by the determination of extracellular acidification rates (ECAR) using the glycolysis stress test as described in the method section. Glycolysis was measured in absence of glucose (basal), with 10 mM glucose to stimulate glycolysis, after the inhibition of ATP synthase by oligomycin as an indirect measurement of maximal ATP production through glycolysis, and after glycolysis inhibition by 2-deoxyglucose. Direct exposure to DDT and DDE did not alter ECAR in all conditions (Fig. 6A and B).
Fig. 6.

Exposure to supraphysiological concentrations of DDT alters glucose uptake in L6 myotubes. A-B. Extracellular acidification rate (ECAR) in L6 myotubes exposed for 24 h to the vehicle (0.1% DMSO) or (A) 1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM DDT, or (B) 1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM DDE. ECAR was measured in absence of glucose (basal). Cells were subsequently treated with 10 mM glucose to activate glycolysis, 3 μM oligomycin to assay maximal glycolytic capacity, and with 50 mM 2-deoxyglucose (2-DG) to inhibit glycolysis. n = 3, each independent experiment done in 6–8 replicates. C-F. Glucose uptake in L6 myotubes exposed for 24 h to the vehicle (0.1% DMSO) and (C and E) 1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM DDT, or (D and F) 1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM DDE. Half of each condition was exposed to insulin (100 nM) for 20 min. Cells were subsequently treated with 0.5 μCi/ml [3H]2-deoxyglucose. C-D. Glucose uptake relative to the vehicle. E-F. Fold increase in glucose uptake following insulin stimulation. C-F. n = 3, each independent experiment was done in triplicate for each condition. A-F. Mean ± SEM. * , p < 0.05 compared to 0 nM DDT or DDE.
We then proceeded to measure glucose uptake in L6 myotubes exposed to DDT or DDE. Basal glucose uptake was significantly increased at the highest concentration of DDT, 10,000 nM (Fig. 6C). Insulin response, presented as the fold increase in glucose uptake in response to insulin stimulation, was mildly decreased with 10,000 nM DDT exposure but did not reach significance (Fig. 6E). Consistent with no alteration in glycolytic function post-exposure to DDE, there was no change in basal and insulin-stimulated glucose uptake (Fig. 6D). Insulin response was not affected at other tested concentrations of DDT or DDE exposure (Fig. 6E and F).
3.7. ROS detoxification enzymes and MDA levels were not altered in L6 myotubes following DDT and DDE exposure
Protein levels of different enzymes involved in ROS detoxification (GPx1, Grx2, catalase, and SOD2) and MDA levels were measured in L6 myotubes exposed to DDT, DDE, or the vehicle (Fig. 7, Fig. 8). Exposure to DDT and DDE did not affect the level of ROS detoxification enzymes (Figs. 7A-D and 8A-D). This was also validated through no significant changes in levels of MDA in L6 myotubes treated with DDT or DDE (Figs. 7E and 8E).
Fig. 7.

ROS detoxifying enzymes and MDA levels are not altered in L6 myotubes following DDT exposure. Levels of (A) Grx2, (B) SOD2, (C) GPx1, and (D) catalase measured in L6 myotubes exposed for 24 h to 0, 1, 10, 100, 1000, and 10,000 nM of DDT. Alpha tubulin was used as a loading control. Top panels: representative Western blots, bottom panels: quantification by density analysis. n = 3 independent experiments. Mean ± SEM. (E) Intracellular MDA concentration (nmol MDA/mg of protein) measured in L6 myotubes exposed for 24 h to 0, 1, 10, 100, 1000, and 10,000 nM of DDT. n = 4 independent experiments for all but 1 nM DDT (n = 3). Mean ± SEM.
Fig. 8.

ROS detoxifying enzymes and MDA levels are not altered in L6 myotubes following DDE exposure. Levels of (A) Grx2, (B) SOD2, (C) GPx1, and (D) catalase measured in L6 myotubes exposed for 24 h to 0, 1, 10, 100, 1000, and 10,000 nM of DDE. Alpha tubulin was used as a loading control. Top panels: representative Western blots, bottom panels: quantification by density analysis. n = 3 independent experiments. Mean ± SEM. (E) Intracellular MDA concentration (nmol MDA/mg of protein) measured in L6 myotubes exposed for 24 h to 0, 1, 10, 100, 1000, and 10,000 nM of DDE. n = 4 independent experiments for all but 10,000 nM DDE (n = 3). Mean ± SEM.
4. Discussion
In this pilot study, an acute in vivo exposure to 40 μg/kg of DDT in rats significantly decreased muscle mitochondrial function by 45% at the level of complex I without affecting the expression of mitochondrial complexes. Similarly, and consistent with previous studies [36], [37], our in vitro study on L6 muscle cells suggests that only supraphysiological levels of DDT and DDE (1000–10,000 nM) altered maximal respiration, glucose uptake, and response to insulin without alterations in ROS detoxification enzymes.
DDTs continue to be detected in food at concentrations reaching 1 μg/kg and plasma concentrations between 0.59 and 6.4 µg/l (1–20 nM) [45], [46]. In certain parts of the world, soil and dust levels range between 0 and 96 mg/kg for DDT [42], and serum concentrations of DDT and DDE are 55.3 and 242.2 ng/g of lipids, respectively [41]. Here, we assessed different concentrations in vivo (40 μg/kg) and in vitro (0–10,000 nM) to investigate their effects on skeletal muscle. A group investigated the effect of perinatal exposure to DDT in mice by injecting 1.7 mg/kg of DDT daily from gestational day 11.5 to postnatal day 5 [16]. The resulting internal levels of DDT were 51.1 μg/l and 2.2 μg/l of DDE in serum. Circulating DDT and DDE levels in these mice fell within the range found in the serum of contemporary people living in malaria-infested regions (~ 5 μg/l for circulating DDT) [6] and contemporary people living in northern Europe (~ 1 μg/l for circulating DDE) [47]. Based on this information, since the dose used in our study is 42.5 times lower, the estimated circulating serum concentrations would be closer to relevant maximum levels detected in the Inuit population of 0.47 and 7.78 μg/l for DDT and DDE, respectively [38], [48],59].
In conjunction with studies associating exposure to DDTs and T2D, these pollutants experimentally have been shown to alter mitochondrial function in several organs [20], [21], [22]. DDT inhibits ATPase and complex I in liver mitochondria [20], [22]. DDE interacts with complex II, decreasing mitochondrial function in the liver [24] and reduces the repolarization potential, state 3 respiration, and uncoupled respiration of testicular mitochondria [21]. The in vivo metabolism of DDT into DDE is known to occur in the liver, while DDT itself, due to its lipophilic properties mostly accumulates in adipose tissue [49]. The originality of our report lies in the fact that our study is the first, to our knowledge, to evaluate the effect of DDT on skeletal muscle mitochondrial function in rats. In accordance with studies showing altered mitochondrial function in non-muscle tissues, we demonstrate here that exposure of rats to DDT results in decreased state 3 respiration using complex I substrates in an oxidative muscle. Although data did not reach significance, oxygen consumption was also decreased using complex II substrates. This suggests that DDT may also be impacting complex V (ATP synthase) which was not evaluated in this pilot study. Thus, it is important to note that the impact of complex V on state 3 respiration cannot be excluded, a limitation to the findings of the current experiment. Besides, these observations were not linked to any alteration in the muscle expression of the electron transport chain complexes, commonly used as a mitochondrial content marker [49]. When exposed directly to L6 myotubes, maximal respiration was increased at 1000 nM DDT by 18% and decreased by 32% at 10,000 nM DDT. Interestingly so, its metabolite DDE had the opposite effect at those concentrations, with an 18% decrease in OCR at 1000 nM and a 13% increase at 10,000 nM. Taken together, these results suggest that only high concentrations of DDT or DDE alter mitochondrial function following direct exposure. To the best of our knowledge, this study is also the first one to test the effect of DDT and DDE on muscle cell mitochondrial function.
Mitochondria release ROS via complexes I and III, such that increased ROS production by the mitochondria may result in oxidative damage to mitochondrial proteins [50]. DDT exposure led to apoptosis in renal tubules and testes in relation to decreased levels of ROS detoxification enzymes [27], [28]. Vitamins C and E, antioxidants against cell oxidative stress, were shown to be protective against DDT-induced cytotoxicity via the ROS-mediated mitochondrial pathway [51]. Since muscle oxidative stress might be involved in the development of muscle mitochondrial dysfunction [34,33,52], we hypothesized that muscle mitochondrial dysfunction in response to DDT exposure in rats was the result of increased muscle oxidative stress. Acute in vivo and in vitro DDT exposure did not alter the level of ROS detoxification enzymes (GPx1, Grx2, SOD2, and catalase) nor the levels of MDA, a marker of oxidative stress. Therefore, acute exposure to doses relevant to human exposure may not directly alter ROS detoxification enzyme contents in muscle.
Xenobiotic-metabolizing enzymes (XMEs) such as arylamine N-acetyltransferases (NATs) and cytochromes P450 (CYPs) are expressed in skeletal muscle [53]. They are involved in the metabolic detoxification of chemicals. In the presence of ROS, XMEs may become impaired and thus contribute to the modification of key enzymes involved in muscle energy metabolism and lead to muscle dysfunction [54]. Besides, our lab has highlighted the induction of Cyp1a1 expression in rat muscles and in mouse C2C12 myotubes following PCB126 exposure [40], [55]. While the expression of CYPs has not been investigated in L6 myotubes or in the current study, DDT and DDE exposure could induce a similar response in muscle cells, a mechanism we can not ignore.
Due to its mass, skeletal muscle is the major site for postprandial glucose disposal, and the most important in maintaining glucose homeostasis after a meal (i.e. insulin stimulated conditions) [56], [57]. In our study, in vivo exposure to DDT in rats did not alter circulating glucose or insulin levels hinting that acute exposure may not influence muscle insulin sensitivity. However, at a supraphysiological level of 10,000 nM of DDT, L6 myotube glucose uptake was increased. Meanwhile, direct DDE exposure to L6 myotubes did not affect on glycolytic function, nor glucose uptake. Compared to another study where they had exposed L6 myotubes to supraphysiological levels of DDT (60 mg/l - 170,000 nM), 8–17x fold the highest concentration used in our study, decreased insulin-induced glucose uptake was observed [37]. Singh et al. also noted alteration in redox-sensitive kinases. Such high concentrations of DDT were shown to induce insulin resistance in L6 myotubes by impairing the phosphorylation of insulin signaling pathway molecules [37]. Furthermore, high concentrations of DDT (1 or 5 μM) on L6 myotubes for 24 and 48 h also inhibited glucose uptake, increased the production of ROS, and downregulated GLUT4 [36]. Altogether, the results indicate that very high concentrations of DDT or DDE are required to alter glucose uptake and insulin sensitivity in muscle cells upon acute exposure in vitro, but that environmentally relevant concentrations do not alter muscle glucose metabolism or insulin sensitivity.
The lack of translation from in vivo to in vitro could result from L6 myotubes not being a perfect model for mature skeletal muscle tissue. Alternatively, the effect of DDT on skeletal muscle may be the result of intermediate processes. DDT being lipophilic has limited access to lipid-poor cells such as the L6 myotubes. It can be hypothesized that in vivo, DDT could be targeting skeletal muscle through the production of cytokines or adipokines that are secreted by the adipose tissue or macrophages within the adipose tissue, which subsequently could lead to muscle mitochondrial dysfunction and insulin resistance. Disturbances in muscle fatty acid metabolism caused by pro-inflammatory cytokines are believed to contribute to the development of impaired activation of insulin receptor substrate 1 (IRS-1) and protein kinase B (known as Akt) in skeletal muscle [58]. Some studies also indicate that DDT may possess immunomodulatory properties activity by influencing cytokine production and up-regulating the gene expression levels via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transactivation [14]. Further investigations are required to test the proposed hypotheses.
There are some limitations in our study that deserve mention and consideration in the interpretation of the results. First, we were unable to assess the final concentration of circulating DDT and DDE in rat sera post-injection of 40 μg/kg of DDT. Gas chromatography using electron capture detection, a technology we unfortunately did not have access to, would have enabled us to know whether 40 μg/kg of DDT produces circulating levels in human-relevant ranges. The in vivo observations are limited in providing information relevant to human exposure. As previously mentioned, in this pilot study, we evaluated the effect of a single acute exposure, which does not truly replicate human exposure since humans are most of the time exposed chronically to this pollutant. Further experiments with chronic treatments are therefore necessary. In addition, we evaluated the effect of a single dose of DDT (40 μg/kg), which corresponds to the levels of exposure in the Inuit population in Canada [38], [48],59]. Thus, to further understand the effects of DDT on muscle metabolism, a range of doses (from low to high) must be tested that would be representative of exposures around the world. Furthermore, the highest concentrations used in the in vitro study are very high compared to what humans are exposed to. Nonetheless, the lowest concentrations (1–10 nM) are environmentally relevant, and our study demonstrates that at environmentally relevant concentrations of DDT or DDE in vitro, neither glucose uptake nor mitochondrial function is altered in L6 myotubes.
5. Conclusion
The effect of human exposure to DDTs on mitochondrial function, oxidative stress and metabolic disorders such as T2D remains unclear and widely debated. This pilot study is the first one to demonstrate that in vivo exposure of rats to DDT alters mitochondrial function in skeletal muscle specifically. However, these effects are not observed in vitro following direct acute exposure to DDT and DDE at concentrations comparable to ones found in human serum. Hence, DDT nor DDE directly alter mitochondrial function and insulin sensitivity in muscle cells at environmentally relevant concentrations.
Funding
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grants [grant numbers: 2015-06263 to CA and 418312-2012 to NAC] and operating funding from Institut du Savoir Montfort to CA. Master scholarships to LC were provided by the Institut du Savoir Montfort and Canadian Institutes of Health Research (CIHR), to HK by Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII-GSST) Program, and to AC by NSERC and Fonds de recherche Santé Québec (FRSQ).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to thank Dr. Amira Klip (SickKids, Toronto, ON, Canada) for her generous gift of L6 myoblasts and Dr. Mary-Ellen Harper (University of Ottawa, ON, Canada) for the use of the XF-96 analyzer (Seahorse Bioscience) and the provision of her lab for radiation experiments. We also thank Caroline Cachero and Cailin Outhwaite for their technical support.
Handling Editor: DR. Aristidis Tsatsakis
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.toxrep.2022.03.004.
Contributor Information
Lucia Chehade, Email: lcheh087@uottawa.ca.
Hannah Khouri, Email: hkhou006@uottawa.ca.
Julie Malatier--Ségard, Email: jmalatier@etud.univ-anger.fr.
Audrey Caron, Email: acaro076@uottawa.ca.
Jean-François Mauger, Email: jmauger@uottawa.ca.
Natalie Ann Chapados, Email: n.chapados@gmail.com.
Céline Aguer, Email: celine.aguer@mcgill.ca.
Appendix A. Supplementary material
Supplementary material.
.
References
- 1.ATSDR (Agency for Toxic Substances and Disease Registry) ATSDR; Atlanta. GA: 2002. Toxicological Profile for DDT, DDE, and DDD.〈https://www.atsdr.cdc.gov/toxprofiles/tp35.pdf〉 [PubMed] [Google Scholar]
- 2.Rogan W.J., Chen A. Health risks and benefits of bis(4-chlorophenyl)-1,1,1-trichloroethane (DDT) Lancet. 2005;366:763–773. doi: 10.1016/S0140-6736(05)67182-6. [DOI] [PubMed] [Google Scholar]
- 3.WHO (World Health Organization) World Health Organization,; Geneva: 2011. The use of DDT in Malaria Vector Control. WHO Position Statement. [Google Scholar]
- 4.Sanz-Gallardo M.I., Guallar E., Van T., Veer P., Longnecker M.P., Strain J.J., Martin B.C., Kardinaal A.F.M., Fernández-Crehuet J., Thamm M., Kohlmeier L., Kok F.J., Martín-moreno J.M. Determinants of p,p ’-Dichlorodiphenyldichloroethane (DDE) Concentration in Adipose Tissue in Women from Five European Cities. Arch. Environ. Health Int. J. 1999;54:277–283. doi: 10.1080/00039899909602486. [DOI] [PubMed] [Google Scholar]
- 5.Hu F.B. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. doi: 10.2337/dc11-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox S., Niskar A.S., Narayan K.M.V., Marcus M. Prevalence of self-reported diabetes and exposure to organochlorine pesticides among Mexican Americans: hispanic health and nutrition examination survey, 1982–1984. Environ. Health Perspect. 2007;115:1747–1752. doi: 10.1289/ehp.10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Airaksinen R., Rantakokko P., Eriksson J.G., Blomstedt P., Kajantie E., Kiviranta H. Association between type 2 diabetes and exposure to persistent organic pollutants. Dia Care. 2011;34:1972–1979. doi: 10.2337/dc10-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaacks L.M., Staimez L.R. Association of persistent organic pollutants and non-persistent pesticides with diabetes and diabetes-related health outcomes in Asia: a systematic review. Environ. Int. 2015;76:57–70. doi: 10.1016/j.envint.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Lee D.-H., Steffes M.W., Sjödin A., Jones R.S., Needham L.L., Jacobs D.R. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0015977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rignell-Hydbom A., Rylander L., Hagmar L. Exposure to persistent organochlorine pollutants and type 2 diabetes mellitus. Hum. Exp. Toxicol. 2007;26:447–452. doi: 10.1177/0960327107076886. [DOI] [PubMed] [Google Scholar]
- 11.Al-Othman A.A., Abd-Alrahman S.H., Al-Daghri N.M. DDT and its metabolites are linked to increased risk of type 2 diabetes among Saudi adults: a cross-sectional study. Environ. Sci. Pollut. Res. 2015;22:379–386. doi: 10.1007/s11356-014-3371-0. [DOI] [PubMed] [Google Scholar]
- 12.Lee D.-H., Lee I.-K., Song K., Steffes M., Toscano W., Baker B.A., Jacobs D.R. A strong dose-response relation between serum concentrations of persistent organic pollutants and diabetes: results from the national health and examination survey 1999-2002. Diabetes Care. 2006;29:1638–1644. doi: 10.2337/dc06-0543. [DOI] [PubMed] [Google Scholar]
- 13.Lee D.-H., Lee I.-K., Steffes M., Jacobs D.R. Extended analyses of the association between serum concentrations of persistent organic pollutants and diabetes. Diabetes Care. 2007;30:1596–1598. doi: 10.2337/dc07-0072. [DOI] [PubMed] [Google Scholar]
- 14.Kim K.-S., Lee Y.-M., Kim S.G., Lee I.-K., Lee H.-J., Kim J.-H., Kim J., Moon H.-B., Jacobs D.R., Lee D.-H. Associations of organochlorine pesticides and polychlorinated biphenyls in visceral vs. subcutaneous adipose tissue with type 2 diabetes and insulin resistance. Chemosphere. 2014;94:151–157. doi: 10.1016/j.chemosphere.2013.09.066. [DOI] [PubMed] [Google Scholar]
- 15.Cano-Sancho G., Salmon A.G., La Merrill M.A. Association between Exposure to p,p ′-DDT and Its Metabolite p,p ′-DDE with Obesity: Integrated Systematic Review and Meta-Analysis. Environ. Health Perspect. 2017;125 doi: 10.1289/EHP527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.La Merrill M., Karey E., Moshier E., Lindtner C., La Frano M.R., Newman J.W., Buettner C. Perinatal exposure of mice to the pesticide ddt impairs energy expenditure and metabolism in adult female offspring. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0103337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaacks L.M., Yadav S., Panuwet P., Kumar S., Rajacharya G.H., Johnson C., Rawal I., Mohan D., Mohan V., Tandon N., Barr D.B., Narayan K.M.V., Prabhakaran D. Metabolite of the pesticide DDT and incident type 2 diabetes in urban India. Environ. Int. 2019;133 doi: 10.1016/j.envint.2019.105089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee D.-H., Porta M., Jacobs D.R., Vandenberg L.N. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocrine Reviews. 2014;35:557–601. doi: 10.1210/er.2013-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim S., Cho Y.M., Park K.S., Lee H.K. Persistent organic pollutants, mitochondrial dysfunction, and metabolic syndrome: Lim et al. Annal. N. Y. Acad. Sci. 2010;1201:166–176. doi: 10.1111/j.1749-6632.2010.05622.x. [DOI] [PubMed] [Google Scholar]
- 20.Byczkowski J.Z. The mode of action of p,p′-DDT on mammalian mitochondria. Toxicology. 1976;6:309–314. doi: 10.1016/0300-483X(76)90034-2. [DOI] [PubMed] [Google Scholar]
- 21.Mota P.C., Cordeiro M., Pereira S.P., Oliveira P.J., Moreno A.J., Ramalho-Santos J. Differential effects of p,p′-DDE on testis and liver mitochondria:Implications for reproductive toxicology. Reprod. Toxicol. 2011;31:80–85. doi: 10.1016/j.reprotox.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Nishihara Y., Utsumi K. Effects of 1,1,1-trichloro-2,2-bis-(p-chlorophenyl)ethane (DDT) on ATPase-linked functions in isolated rat-liver mitochondria. Food Chem. Toxicol. 1985;23:599–602. doi: 10.1016/0278-6915(85)90185-1. [DOI] [PubMed] [Google Scholar]
- 23.Moreno A.J.M., Madeira V.M.C. Mitochondrial bioenergetics as affected by DDT. Biochim. et Biophys. Acta BBA Bioenergetics. 1991;1060:166–174. doi: 10.1016/S0005-2728(09)91004-0. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira F.M.L., Madeira V.M.C., Moreno A.J. Interactions of 2,2-bis(p-chlorophenyl)-1,1-dichloroethylene with mitochondrial oxidative phosphorylation. Biochem. Pharmacol. 1997;53:299–308. doi: 10.1016/S0006-2952(96)00689-2. [DOI] [PubMed] [Google Scholar]
- 25.Musarò A., Fulle S., Fanò G. Oxidative stress and muscle homeostasis: Current Opinion in Clinical Nutrition and Metabolic Care. 2010;13:236–242. doi: 10.1097/MCO.0b013e3283368188. [DOI] [PubMed] [Google Scholar]
- 26.Burgos-Morón Abad-Jiménez, Marañón Iannantuoni, Escribano-López L.ópez-Domènech, Salom Jover, Mora Roldan, Solá Rocha, Víctor Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: the battle continues. JCM. 2019;8:1385. doi: 10.3390/jcm8091385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marouani N., Hallegue D., Sakly M., Benkhalifa M., Ben Rhouma K., Tebourbi O. p,p′-DDT induces testicular oxidative stress-induced apoptosis in adult rats. Reprod. Biol. Endocrinol. 2017;15:40. doi: 10.1186/s12958-017-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marouani N., Hallegue D., Sakly M., Benkhalifa M., Rhouma K.B., Tebourbi O. Involvement of oxidative stress in the mechanism of p,p′-DDT-induced nephrotoxicity in adult rats. gpb. 2017;36:309–320. doi: 10.4149/gpb_2016050. [DOI] [PubMed] [Google Scholar]
- 29.Mogensen M., Sahlin K., Fernstrom M., Glintborg D., Vind B.F., Beck-Nielsen H., Hojlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 30.Asmann Y.W., Stump C.S., Short K.R., Coenen-Schimke J.M., Guo Z., Bigelow M.L., Nair K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–3319. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]
- 31.Houstis N., Rosen E.D., Lander E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 32.Montgomery M.K., Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocrine Connect. 2015;4:R1–R15. doi: 10.1530/EC-14-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zabielski P., Lanza I.R., Gopala S., Holtz Heppelmann C.J., Bergen H.R., Dasari S., Nair K.S. Altered skeletal muscle mitochondrial proteome as the basis of disruption of mitochondrial function in diabetic mice. Diabetes. 2016;65:561–573. doi: 10.2337/db15-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnard C., Durand A., Peyrol S., Chanseaume E., Chauvin M.-A., Morio B., Vidal H., Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 2008:JCI32601. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connor T., Martin S.D., Howlett K.F., McGee S.L. Metabolic remodelling in obesity and type 2 diabetes: pathological or protective mechanisms in response to nutrient excess? Clin. Exp. Pharmacol. Physiol. 2015;42:109–115. doi: 10.1111/1440-1681.12315. [DOI] [PubMed] [Google Scholar]
- 36.Park C.M., Kim K.-T., Rhyu D.Y. Low-concentration exposure to organochlorine pesticides (OCPs) in L6 myotubes and RIN-m5F pancreatic beta cells induces disorders of glucose metabolism. Toxicol. in Vitro. 2020;65 doi: 10.1016/j.tiv.2020.104767. [DOI] [PubMed] [Google Scholar]
- 37.Singh V.K., Sarkar S.K., Saxena A., Koner B.C. Effect of subtoxic DDT exposure on glucose uptake and insulin signaling in rat L6 myoblast-derived myotubes. Int. J. Toxicol. 2019;38:303–311. doi: 10.1177/1091581819850577. [DOI] [PubMed] [Google Scholar]
- 38.Muir D., Braune B., DeMarch B., Norstrom R., Wagemann R., Lockhart L., Hargrave B., Bright D., Addison R., Payne J., Reimer K. Spatial and temporal trends and effects of contaminants in the Canadian Arctic marine ecosystem: a review. Sci. Total Environ. 1999;230:83–144. doi: 10.1016/S0048-9697(99)00037-6. [DOI] [PubMed] [Google Scholar]
- 39.Kuznetsov A.V., Veksler V., Gellerich F.N., Saks V., Margreiter R., Kunz W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay-Laganière C., Garneau L., Mauger J.-F., Peshdary V., Atlas E., Nikolla A.S., Chapados N.A., Aguer C. Polychlorinated biphenyl 126 exposure in rats alters skeletal muscle mitochondrial function. Environ. Sci. Pollut. Res. 2019;26:2375–2386. doi: 10.1007/s11356-018-3738-8. [DOI] [PubMed] [Google Scholar]
- 41.Gaspar F.W., Chevrier J., Quirós-Alcalá L., Lipsitt J.M., Barr D.B., Holland N., Bornman R., Eskenazi B. Levels and determinants of DDT and DDE exposure in the VHEMBE cohort. Environ. Health Perspect. 2017;125 doi: 10.1289/EHP353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez F.D.-B., Trejo-Acevedo A., Betanzos A.F., Espinosa-Reyes G., Alegría-Torres J.A., Maldonado I.N.P. Assessment of DDT and DDE levels in soil, dust, and blood samples from Chihuahua, Mexico. Arch. Environ. Contam. Toxicol. 2012;62:351–358. doi: 10.1007/s00244-011-9700-0. [DOI] [PubMed] [Google Scholar]
- 43.Mauger J.F., Nadeau L., Caron A., et al. Polychlorinated biphenyl 126 exposure in L6 myotubes alters glucose metabolism: a pilot study. Environ. Sci. Pollut. Res. 2016;23:8133–8140. doi: 10.1007/s11356-016-6348-3. [DOI] [PubMed] [Google Scholar]
- 44.Klip Amira, Toolsie Ramlal, Denise Walker. Insulin stimulation of glucose uptake and the transmembrane potential of muscle cells in culture. FEBS Letters. 1986;205:11–14. doi: 10.1016/0014-5793(86)80855-9. [DOI] [PubMed] [Google Scholar]
- 45.Health Canada, n.d. 2009. Report on Human Biomonitoring of Environmental Chemicals in Canada.
- 46.Butlerwalker J., Seddon L., Mcmullen E., Houseman J., Tofflemire K., Corriveau A., Weber J., Mills C., Smith S., Vanoostdam J. Organochlorine levels in maternal and umbilical cord blood plasma in Arctic Canada. Sci. Total Environ. 2003;302:27–52. doi: 10.1016/S0048-9697(02)00319-4. [DOI] [PubMed] [Google Scholar]
- 47.Rylander L., Rignell-Hydbom A., Hagmar L. A cross-sectional study of the association between persistent organo-chlorine pollutants and diabetes. Environ. Health. 2005;4:28. doi: 10.1186/1476-069X-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laird D.-B., Goncharov B.-A., Chan M.-H. Body burden of metals and persistent organic pollutants among Inuit in the Canadian Arctic. Environ. Int. 2013;59:33–40. doi: 10.1016/j.envint.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Tebourbi O., Driss M.R., Sakly M., Rhouma K.B. Metabolism of DDT in Different Tissues of Young Rats. J. Environ. Sc. Health Part B. 2006;41:167–176. doi: 10.1080/03601230500364674. [DOI] [PubMed] [Google Scholar]
- 50.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin X., Song L., Liu X., Chen M., Li Z., Cheng L., Ren H. Protective efficacy of vitamins C and E on p,p′-DDT-induced cytotoxicity via the ROS-mediated mitochondrial pathway and NF-κB/FasL pathway. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0113257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguer C., McCoin C.S., Knotts T.A., Thrush A.B., Ono‐Moore K., McPherson R., Dent R., Hwang D.H., Adams S.H., Harper M. Acylcarnitines: potential implications for skeletal muscle insulin resistance. FASEB J. 2015;29:336–345. doi: 10.1096/fj.14-255901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawler J.M., Song W. Specific of antioxidant enzyme inhibition in skeletal muscle to reactive nitrogen species donors. Biochem. Biophys. Res. Commun. 2002;294:1093–1100. doi: 10.1016/S0006-291X(02)00602-2. [DOI] [PubMed] [Google Scholar]
- 54.Dairou Julien, et al. Impairment of the activity of the xenobiotic-metabolizing enzymes arylaminen-acetyltransferases 1 And 2 (NAT1/NAT2) by peroxynitrite in mouse skeletal muscle cells. FEBS Lett. 2005;579(21):4719–4723. doi: 10.1016/j.febslet.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 55.Caron Audrey, et al. Effects Of PCB126 on adipose-to-muscle communication in anin vitromodel. Environ.l Health Perspect. 2020;128(10) doi: 10.1289/ehp7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrannini E., Smith J.D., Cobelli C., Toffolo G., Pilo A., DeFronzo R.A. Effect of insulin on the distribution and disposition of glucose in man. J. Clin. Invest. 1985;76:357–364. doi: 10.1172/JCI111969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J. Progress in Molecular Biology and Translational Science. Elsevier; 2014. Enhanced skeletal muscle for effective glucose homeostasis; pp. 133–163. [DOI] [PubMed] [Google Scholar]
- 58.D.J. Dyck , Adipokines as regulators of muscle metabolism and insulin sensitivity. This paper is one of a selection of papers published in this Special Issue, entitled 14th International Biochemistry of Exercise Conference – Muscles as Molecular and Metabolic Machines, and has undergone the Journal’s usual peer review process Appl. Physiol. Nutr. Metab 34 2009 396 402 doi: 10.1139/H09-037. [DOI] [PubMed]
- 59.Walker B J., et al. Organochlorine levels in maternal and umbilical cord blood plasma in Arctic Canada. Science of The Total Environment. 2003;302(1–3):27–52. doi: 10.1016/S0048-9697(02)00319-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.