

Abstract
Background
As human activity alters the planet, there is a pressing need to understand how organisms adapt to environmental change. Of growing interest in this area is the role of epigenetic modifications, such as DNA methylation, in tailoring gene expression to fit novel conditions. Here, we reanalyzed nine invertebrate (Anthozoa and Hexapoda) datasets to validate a key prediction of this hypothesis: changes in DNA methylation in response to some condition correlate with changes in gene expression.
Results
In accord with previous observations, baseline levels of gene body methylation (GBM) positively correlated with transcription, and negatively correlated with transcriptional variation between conditions. Correlations between changes in GBM and transcription, however, were negligible. There was also no consistent negative correlation between methylation and transcription at the level of gene body methylation class (either highly- or lowly-methylated), anticipated under the previously described “seesaw hypothesis”.
Conclusion
Our results do not support the direct involvement of GBM in regulating dynamic transcriptional responses in invertebrates. If changes in DNA methylation regulate invertebrate transcription, the mechanism must involve additional factors or regulatory influences.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-022-08474-z.
Keywords: Epigenetics, DNA methylation, Gene regulation, Plasticity, Transcriptomics, Gene body methylation, Coral
Background
The regulatory function of DNA methylation in invertebrates, if any, remains unclear. This contrasts with DNA methylation in mammals, where its role in transcriptional repression is well established. Mammals exhibit high genomic DNA methylation levels, with 70-80% of CpGs methylated [1]. This is hypothesized to have evolved for genomic defense, with methylation of transposons’ promoter elements silencing their expression [2]. Promoter methylation is also linked with mitotically heritable silencing of some mammalian genes, such as X-inactivated, imprinted-, and germline-specific genes [3]. Mitotically heritable methylation patterns are enabled by the maintenance methyltransferase DNMT1, which is homologous in vertebrates and invertebrates [4]. Methylation of gene bodies (gene body methylation; GBM), in contrast, is associated with actively transcribed genes [5]. Regions of accessible chromatin also tend to be weakly methylated or unmethylated [3], consistent with an influence of methylation on transcription factor binding [6].
In invertebrates, DNA methylation occurs predominantly in the form of GBM [7] and is positively correlated with expression [8]. Despite its association with active expression, there is little evidence that invertebrate methylation directly regulates transcription. For instance, several studies failed to detect substantial differences in GBM across invertebrate cell types or developmental stages [9–11] despite profound transcriptome differences (although see Liew et al. [12]). Conversely, removal of GBM by knockdown of DNMT1 enzyme did not significantly alter gene expression in a milkweed bug [13]. Similar results have been observed in plants [14–16]. Together, these studies indicate that changes in GBM are neither necessary nor sufficient to induce changes in transcription.
As invertebrate coding genes are separated into highly methylated and lowly methylated classes, we earlier hypothesized that methylation class serves as a regulatory signal and that the highly and lowly methylated classes of genes undergo group-level changes in methylation and transcription. In a previous study of A. millepora, we observed reciprocal changes in GBM and transcription depending on methylation class [17]. The study used MBD-seq and Tag-seq to examine changes in GBM and transcription in colonies of A. millepora reciprocally transplanted between two environments. In colony fragments transplanted to the environmentally favorable location, the highly methylated class of genes decreased in methylation, while the lowly methylated class increased. Looking at transcription in these colony fragments, the highly methylated class tended to be upregulated upon transplantation, while the lowly methylated class tended to be downregulated. The inverse pattern was observed in an independent set of coral samples transplanted from the more favorable to the less favorable location [17]. Based on this observation, we proposed a regulatory mechanism in which opposing class-level changes in GBM produce reciprocal class-level changes in transcription. As environmentally responsive genes tend to be in the lowly methylated class, and housekeeping genes tend to be in the highly methylated class, this mechanism could allow broad shifts between responsive, ‘problem solving’ transcriptional profiles and homeostatic house-keeping profiles. As the hypothesis involved reciprocal shifts between the two methylation classes, we refer to it as the ‘seesaw hypothesis’.
Here, we re-analyze publicly available methylomic and RNA-seq data from three Anthozoa and six Hexapoda studies (Fig. 1) to evaluate relationships between invertebrate DNA methylation and transcription. For each study, we contrast methylation- and transcriptional differences between two conditions. The Anthozoan studies contrasted polyp types in the coral Acropora millepora [18], pH treatments in the coral Stylophora pistillata [12], and symbiotic state in the sea anemone Exaiptasia pallida [19]. Hexapoda studies included different reproductive states in ants (Ooceraea biroi) [20], bumblebees (Bombus terrestris) [21], and termites (Zootermopsis nevadensis) [22], different subcastes in honeybee (Apis mellifera) [23], differences in maternal care in carpenter bee [24], and different diapause states in silkworm (Bombyx mori) [25]. Using these diverse datasets, we first confirm previous findings that baseline GBM levels are bimodally distributed across coding genes, are positively associated with baseline transcription level, and are negatively associated with transcriptional variation. We next examine the hypotheses that changes in GBM and/or promoter methylation between conditions correlate with changes in transcription. Lastly, we assess three components of the seesaw hypothesis of Dixon et al. (2018, 17]: (1) the highly- and lowly-methylated gene classes undergo reciprocal changes in GBM, (2) the two classes undergo reciprocal changes in transcription, (3) class-level changes in transcription will be in the opposite direction of class-level changes in GBM.
Fig. 1.
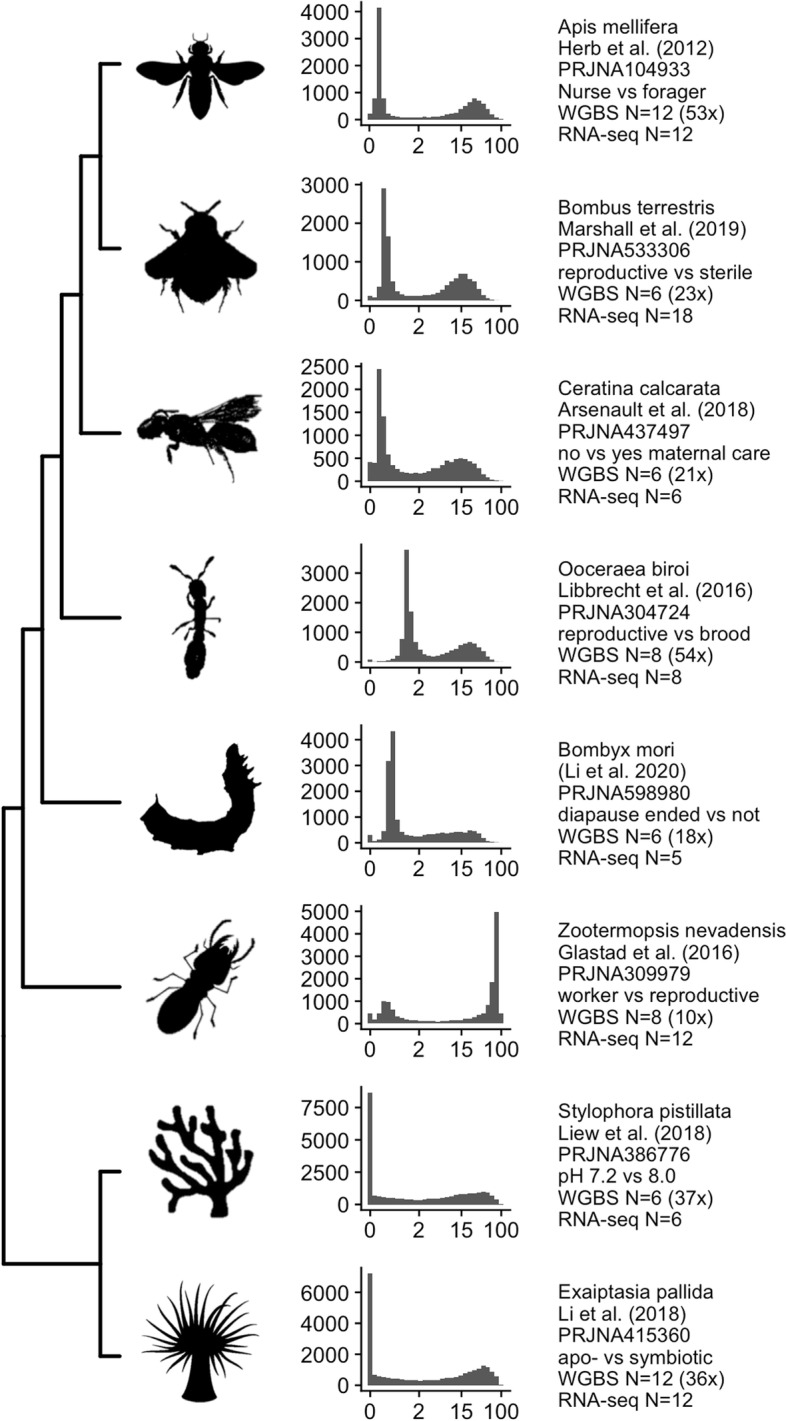
Overview of species covered by previously published datasets and the distribution of GBM levels in each. X-axes for histograms show percent methylation (summed across all CpG sites within the gene and averaged across all samples from each study) on the log2 scale. The Y-axes show the counts of genes. Text reports the species, reference, NCBI Bioproject accession, the treatment groups compared here, the Whole Genome Bisulfite sample size (with mean genomic coverage), and the RNA-seq sample size for each study
Results and discussion
Confirming previous relationships between GBM and transcription
We first sought to corroborate previous findings on the distribution of GBM, and its relationship to gene expression patterns. Using three different methylation assays, we confirmed that GBM in A. millepora shows a characteristic bimodal distribution, separating genes into highly methylated and lowly methylated classes (Fig. 2 A-C). We then confirmed that GBM level is associated with average mRNA abundance (Fig. 2 D-F), and negatively associated with differential expression between polyp types (Fig. 2 G-I). Hence, regardless of the method used to measure methylation, GBM shows the expected distribution and associations with gene expression in A. millepora.
Fig. 2.

Associations between GBM level and gene expression patterns in A. millepora based on three different methylation assays. A-C Distribution of GBM levels D-F Relationship between GBM level and mRNA abundance G-I Relationship between GBM level and the absolute value of differential mRNA expression between axial and radial polyps. X-axes for the assays are as follows. Whole Genome Bisulfite Sequencing (WGBS): percent methylation (summed across all CpG sites of each gene and averaged across all samples) on the log2 scale; Methylation Binding Domain Sequencing (MBD-seq): the log2 difference in fold coverage between the captured and unbound fractions (MBD-score, see methods); methylation dependent RAD-seq (mdRAD): Reads per Kilobase of gene sequence per Million reads on the log2 scale (log2(RPKM)). The correlation coefficient is given in the upper left hand of each plot
The other studies showed similar results. While the relative sizes and means of the peaks varied by dataset, these were also bimodally distributed (Fig. 1) and similarly associated with mRNA expression patterns (Fig. 3). The coefficient of variation (standard deviation (RPKM) / mean(RPKM) computed from control replicates) was similarly negatively related to GBM (Fig. S1). Relationships between GBM and expression level were stronger among the insects than the cnidarians. Hence, GBM was positively linked with transcription level and negatively linked with transcriptional variation in all the studies included here.
Fig. 3.

Associations between GBM level and gene expression patterns. A Relationship between GBM level and mRNA abundance (RNA-seq RPKM). B Relationship between GBM level and the absolute value of expression differences between phenotypic groups (contrasts given in Fig. 1). X-axis shows percent methylation on the log2 scale. The correlation coefficient is given in the upper left of each plot
No correlation between changes in GBM and changes in transcription across genes
As GBM is associated with elevated expression, a simple hypothesis is that increasing GBM increases transcription. Our re-analysis of GBM- and mRNA differences between phenotypic groups shows that this is not the case. Using three different methylation datasets in A. millepora, we found that measurements of GBM differences between polyp types showed no consistent association with transcriptional differences (Fig. S2). This was also the case for each of the other datasets (Fig. 4). Repeating this analysis using only differentially expressed genes (DESeq2 FDR < 0.1) or differentially methylated genes (MethylKit FDR < 0.1) produced similar results, with no clear association between differential GBM and differential transcription (Fig. S3). Hence, in Anthozoa and Hexapoda, GBM and transcription show no linear covariation between phenotypic conditions.
Fig. 4.

Changes in GBM and transcription between phenotypic conditions showed no clear relationship. X axes indicate differential GBM estimated using MethylKit. Y axes indicate differential transcription estimated using DESeq2. Both axes are on the log2 scale. Correlation coefficient is given in the upper left of each plot. Contrasts used to compute differential GBM and transcription are given in Fig. 1
No correlation between changes in promoter methylation and changes in transcription
As promoter methylation is associated with gene silencing in vertebrates, we tested whether changes in promoter methylation correlate with changes in gene expression in invertebrates. Specifically, we tested whether differences in methylation in windows 1Kb upstream of genes between phenotypic conditions predicted differences in mRNA levels. As with GBM, we found no reproducible relationship between changes in promoter methylation and changes in expression for A. millepora (Fig. S4), or any of the other studies (Fig. S5).
Class-level shifts in GBM and transcription
Regarding the seesaw hypothesis, results from the A. millepora dataset were inconclusive. The expected seesaw pattern was observed based on MBD-seq and mdRAD in axial compared to radial polyps: the highly methylated class increased in methylation level while the lowly methylated class decreased in methylation. However, this pattern was not apparent in the WGBS dataset (Fig. 5). The reason for the match between two methods but not the third one is unclear. While there might be a technical issue with this specific WGBS dataset, it is concerning that of the three GBM-detection methods the only one that failed to show the seesaw pattern is the one that is supposed to be the most reliable [26]. Looking at the transcriptional data, based on all three methylation assays, the lowly methylated class was somewhat upregulated, and the highly methylated class was somewhat downregulated (Fig. 5). While this was consistent with our previous observations [17], the overall weakness of these effects plus the disagreement with the WGBS data does not allow claiming confident support for the seesaw hypothesis.
Fig. 5.

Class-level shifts in GBM and transcription in A. millepora. X axes show baseline GBM level averaged across all samples. Y axes shows differential GBM between polyp types (top panels) and differential transcription between polyp types (bottom panels). Top panels: Differential GBM between polyp types was linked with baseline GBM level for the MBD-seq and mdRAD datasets, but not for WGBS. Bottom panels: For all three methylation datasets, the low GBM class of genes was somewhat upregulated in axial compared to radial polyps, and the high GBM class was somewhat downregulated
Among other studies, we never observed coordinated class-level changes of GBM and transcription. There were three cases (silkworm, termite, and carpenter bee) of class-level shifts in methylation, but unlike Dixon et al. (2018) [17] only the highly methylated class changed (Fig. 6). The silkworm dataset shows this most clearly, with a strong average increase in methylation level for the highly methylated class, but little or no average change in the lowly methylated class (Fig. 6). While the methylation level measurements of the lowly methylated class ranged from roughly 0 to 3% across the datasets, it seems likely that GBM in this class is essentially negligible, and does not change.
Fig. 6.

Class-level shifts in GBM. A Scatterplots of GBM change against mean GBM level. Only three studies (Silkworm, Termite, and Carpenter bee) showed group-level changes in methylation between conditions. B Density plots of GBM change for Silkworm, Termite, and Carpenter bee. Average shifts were observed in the highly methylated class, but not in the lowly methylated class
In contrast to Dixon et al. (2018) [17], class-level changes in methylation in non-Acropora studies were not associated with class-level changes in transcription (Fig. 7). Two of the eight studies did however, demonstrate class-level changes in transcription alone. In these two cases (honeybee and bumblebee), the lowly methylated class was downregulated on average, and the highly methylated class upregulated on average (Fig. 7). In summary, while some aspects of the “seesaw” hypothesis proposed by Dixon et al. (2018) [17] were detected in several cases, the hypothesis was not fully supported by any of the studies included here. We conclude that, whenever observed, GBM and gene expression seesaw patterns are unlikely to be directly functionally related. More likely, they reflect some other processes influencing the bulk of gene regulatory states, for example differences in cellular proliferation or growth.
Fig. 7.

Two cases of inverse GBM class-level shifts in transcription. A Transcriptional differences (log2 fold change) plotted against GBM level (% methylation). B Density plots of the log2 fold changes in transcription for the lowly methylated and highly methylated classes for Bumblebee (contrasting reproductive vs sterile individuals) and Honeybee (contrasting nurse vs forager subcastes). In these two cases, the lowly methylated class tended to be downregulated and the highly methylated class tended to be upregulated
A possibility that can rescue the regulatory role of invertebrate DNA methylation is that it interacts with other epigenetic modifications, which must be included to accurately model invertebrate gene expression [27]. For instance, in vertebrates, methylation of regulatory elements is known to influence the binding of transcription factors and their gene regulatory effects [28–30]. Regulatory effects of methylation could be further influenced by interactions with other chromatin features such as histone modifications [31–33]. As differential GBM has little or no power in predicting differential transcription between invertebrate phenotypic states, uncovering regulatory functions of DNA methylation in invertebrates will likely require interrogation of such additional features.
Conclusions
Here we used published methylomic and transcriptomic data from Anthozoa and Hexapoda to examine how DNA methylation relates to transcriptional variation between different phenotypic conditions. We found that, as previously reported, GBM is bimodally distributed, and that higher GBM levels are associated with elevated transcription and less transcriptional variation. However, differences in GBM between conditions showed no consistent linear association with differences in transcription. As there were often detectable differences in both GBM and transcription (Fig. S6), this indicates that changes in GBM are neither necessary nor sufficient to induce changes in transcription in invertebrates. Methylation differences 1 Kb upstream of the first exon also showed no association with differences in transcription. In conclusion, if shifting methylation patterns regulate invertebrate transcription, the mechanism is more complex than can be captured by a simple linear relationship between these two variables.
Methods
Previously published datasets
Previously published WGBS and RNA-seq datasets from invertebrate species are shown in Fig. 1. The criteria for selecting these projects were: 1) the project focused on an invertebrate species 2) the project included at least two conditions, such as environmental exposure, or caste. 3) the project characterized DNA methylation using Whole Genome Bisulfite Sequencing (WGBS) 4) the project characterized transcription using RNA-seq 5) reads were available on the NCBI SRA database. Experimental methods from some projects allowed for multiple comparisons, however for simplicity, we focused on contrasts that seemed likely to induce the greatest epigenetic change. The comparisons we made are as follows. For the anemone Exaiptasia pallida [19], we compared aposymbiotic (N = 6) to symbiotic (N = 6) individuals. For the smooth cauliflower coral Stylophora pistillata [12], we compared only the most extreme pH treatment (pH 7.2; N = 3) to controls (pH 8.0; N = 3). For silkworm Bombyx mori [25], we compared diapause terminated (N = 3) to diapause destined (N = 3) eggs. For the termite Zootermopsis nevadensis nuttingi [22], we compared winged reproductive alates of both sexes (N = 4) to larval instars (workers) of both sexes (N = 4). For the small carpenter bee Ceratina calcarata [24] we compared newly eclosed adults that developed without maternal care (N = 3) to those that received maternal care (N = 3). For bumblebee Bombus terrestris [21], we compared reproductive (N = 3) to sterile castes (N = 3). For honeybee Apis mellifera [23], we compared nurse subcastes (N = 6) to worker subcastes (N = 6). For the clonal raider ant Ooceraea biroi [20], we compared individuals in the reproductive phase (N = 4) to those in brood care phase (N = 4). For A. millepora, DNA methylation was measured using three assays, WGBS, MBD-seq, and a variation of the methylRAD assay (described by Wang et al. 2015 [34]) called mdRAD [18], and transcription was measured using Tag-based RNAseq [35]. Here we compared tissue from axial polyps (taken from the very tips of branches) to radial polyps (taken from the sides of branches). The presence of the maintenance methyltransferase DNMT1 was confirmed in each of these species by blasting the human protein sequence against each of their reference proteomes, each with an e-value of 0.
WGBS data processing
Raw reads were trimmed and quality filtered using cutadapt, simultaneously trimming low-quality bases from the 3′ end (−q 30) and removing reads below 50 bp in length (−m 50) [36]. Trimmed reads for each dataset were mapped to the appropriate reference genome (Table S1 [37–43]; using Bismark v0.17.0 [44] with adjusted mapping parameters (−-score_min L,0,-0.6). Reads from Dixon and Matz (2020) [18] were mapped using --non_directional mode as recommended by the Pico Methyl-Seq Library Prep Kit manual. PCR duplicates were removed from the Bismark alignment files using the deduplicate_bismark command. To estimate genomic coverage we computed the mean number of deduplicated reads across samples for each study, multiplied this value by the combined paired end read length, and divided by the summed length of the reference genome used. Methylation levels were extracted from the alignments using bismark_methylation_extractor with the --merge_non_CpG, −-comprehensive, and --cytosine_report arguments. Detailed steps used to process the WGBS reads are available on the git repository [45].
RNA-seq data processing
Raw reads were trimmed and quality filtered using cutadapt, simultaneously trimming low-quality bases from the 3′ end (−q 30) and removing reads below 50 bp in length (−m 50, 36]. Trimmed reads for each dataset were mapped to the appropriate reference genome (Table S1) using Bowtie2 using the --local argument [46]. PCR duplicates were removed using MarkDuplicates from Picard Toolkit [47]. Sorting and conversion from sam files were performed using Samtools [48]. The reads mapping to annotated gene boundaries were counted using FeatureCounts [49]. Detailed steps used to process the RNA reads are available in the git repository [45].
Measuring GBM level
Based on previous findings that different measures of GBM were highly similar [18], we reported GBM level as the percent methylation rate on the log2 scale. Here the percent methylation rate for a gene is the ratio of the total number of methylated read counts to read all counts summed across all CpG sites within the bounds of the gene. To allow plotting on the log scale, zero values were assigned to the lowest non-zero value for each project. Following previous studies [17, 18, 50], in the case of MBD-seq we report GBM as the log2 fold difference between the captured and unbound fractions generated during library preparation. GBM level based on mdRAD was computed as Reads per Kilobase of gene length per Million reads (RPKM) on the log2 scale. Analyses of differential methylation based on bisulfite sequencing data were done using MethylKit package [51]. Based on visual inspection of the distributions of methylation levels across genes in each species, we divided genes into highly methylated and lowly classes using a hard cutoff of 2.5% methylation.
Relationships between GBM and mRNA
For each dataset, we tested for expected relationships between GBM and mRNA expression patterns. For our dataset, generated using Tag-seq [35], we calculated mean mRNA level by averaging the regularized counts generated using the rlog function in DESeq2 across all samples [52]. For the other datasets, which used standard RNA-seq, we calculated mean mRNA level as RPKM averaged across all samples. Differences in mRNA abundance between groups were calculated using DESeq2 [52]. For our dataset, this analysis was performed including colony identity (genotype) as a factor to control for genetic effects. For simplicity, models for differential expression for the published datasets included only the treatment groups indicated in Fig. 1 (we did not include additional factors, for instance, sex or colony identity). Differences between groups are reported as log2 fold differences. General transcriptional variation was estimated based on the coefficient of variation (standard deviation / mean) in Reads Per Kilobase Million Reads (RPKM) for the control samples from each study as in Huh et al. (2013) [53].
Supplementary Information
Additional file 1: Table S1: Reference genomes used in this study.
Additional file 2: Figure S1: The coefficient of variation for transcription (standard deviation (RPKM) / mean(RPKM); computed from control replicates) was negatively related to GBM level (averaged across all samples).
Additional file 3: Figure S2: GBM and transcriptional differences between polyp types in A. millepora show no reproducible relationship. The title of each panel indicates the assay used to measure GBM differences. All axes are on the log2 scale.
Additional file 4: Figure S3: Significant differential GBM and differential transcription between phenotypic conditions show little or no relationship. The top set of panels shows the relationship between transcription and GBM differences for differentially expressed genes (DESeq2 FDR < 0.1). The bottom set of panels show the relationship for differentially methylated genes (MethylKit FDR < 0.1). All axes are on the log2 scale. The contrasts for each study are given in Fig. 1.
Additional file 5: Figure S4: Promoter methylation and transcriptional differences between polyp types in A. millepora show no relationship.
Additional file 6: Figure S5: Promoter methylation and transcriptional differences between phenotypic conditions show no relationship.
Additional file 7: Figure S6: GBM and transcriptional differences between experimental conditions show little or no relationship. Each pair of volcano plots is associated with the scatterplot below. The first volcano plot shows differences in GBM, with significant genes (q-value from MethylKit < 0.1) shown in red. The second shows differences in transcription, with significant genes in blue (FDR < 0.1). The count of significant genes is given above each volcano plot. The scatterplots are the same as those shown in Fig. 5, with expression differences on the Y axis and GBM differences on the X. The contrasts for each study are given in Fig. 1.
Acknowledgements
The authors acknowledge Evelyn Abbot and Hayley Bedwell for assistance with field work.
Abbreviations
- GBM
Gene body methylation
- RPKM
Reads Per Kilobase Million reads
- WGBS
Whole Genome Bisulfite Sequencing
- MBD-seq.
Methylation Binding Domain Sequencing
- mdRAD
methylation dependent RAD-seq
Authors’ contributions
G.D. performed field work, benchwork, analysis, and composed the manuscript. M.M. performed field work, advised the analysis and manuscript composition. All authors reviewed and approved the manuscript.
Authors’ information
Not applicable.
Funding
This research was funded by NSF grant 1755277 awarded to Mihail Matz.
Availability of data and materials
The dataset(s) supporting the conclusions of this article are available in the NCBI SRA database, PRJNA415358, PRJNA533306, PRJNA437497, PRJNA304722, PRJNA598980, PRJNA598995, PRJNA386774, PRJNA104931, PRJNA309979. https://www.ncbi.nlm.nih.gov/sra. Intermediate datasets, code, and detailed data processing steps are available on github: https://github.com/grovesdixon/invert_meth_and_transcription.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have not competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6(5):a019133. [DOI] [PMC free article] [PubMed]
- 2.Hackett JA, Azim SM. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc B Biol Sci. 2013;368:1–8. doi: 10.1098/rstb.2011.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20:590–607. doi: 10.1038/s41580-019-0159-6. [DOI] [PubMed] [Google Scholar]
- 4.Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- 5.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 6.Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, Nitta KR. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337):eaaj2239. [DOI] [PMC free article] [PubMed]
- 7.Suzuki MM, Kerr ARW, De SD, Bird A. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 2007;17:625–631. doi: 10.1101/gr.6163007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 9.Gatzmann F, Falckenhayn C, Gutekunst J, Hanna K, Raddatz G, Carneiro VC, et al. The methylome of the marbled crayfish links gene body methylation to stable expression of poorly accessible genes. Epigenetics Chromatin. 2018;11:1–12. doi: 10.1186/s13072-018-0229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris KD, Lloyd JPB, Domb K, Zilberman D, Zemach A. DNA methylation is maintained with high fidelity in the honey bee germline and exhibits global non-functional fluctuations during somatic development. Epigenetics Chromatin. 2019;12:1–18. doi: 10.1186/s13072-019-0307-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Mendoza A, Lister R, Bogdanovic O. Evolution of DNA Methylome diversity in eukaryotes. J Mol Biol. 2019;432:1687–1705. doi: 10.1016/j.jmb.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Liew YJ, Zoccola D, Li Y, Tambutte E, Venn AA, Michell CT, Cui G, Deutekom ES, Kaandorp JA, Voolstra CR, Forêt S. Epigenome-associated phenotypic acclimatization to ocean acidification in a reef-building coral. Sci Adv. 2018;(6):eaar8028. [DOI] [PMC free article] [PubMed]
- 13.Bewick AJ, Sanchez Z, McKinney EC, Moore AJ, Moore PJ, Schmitz RJ. Dnmt1 is essential for egg production and embryo viability in the large milkweed bug, Oncopeltus fasciatus. Epigenetics Chromatin. 2019;12:1–14. doi: 10.1186/s13072-018-0246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bewick AJ, Ji L, Niederhuth CE, Willing E-M, Hofmeister BT, Shi X, et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc Natl Acad Sci U S A. 2016;113:9111–9116. doi: 10.1073/pnas.1604666113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bewick AJ, Zhang Y, Wendte JM, Zhang X, Schmitz RJ. Evolutionary and experimental loss of gene body methylation and its consequence to gene expression. G3 Genes Genomes Genet. 2019;9:2441–2445. doi: 10.1534/g3.119.400365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi J, Lyons DB, Kim MY, Moore JD, Choi J, Lyons DB, et al. DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Mol Cell. 2020;1–14. 10.1016/j.molcel.2019.10.011. [DOI] [PubMed]
- 17.Dixon G, Liao Y, Bay LK, Matz MV. Role of gene body methylation in acclimatization and adaptation in a basal metazoan. Proc Natl Acad Sci U S A. 2018;115:13342–13346. doi: 10.1073/pnas.1813749115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon G, Matz MV. Benchmarking DNA methylation assays in a reef-building coral. Mol Ecol Resour. 2020;21:464–477. doi: 10.1111/1755-0998.13282. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Liew YJ, Cui G, Cziesielski MJ, Zahran N, Michell CT, et al. DNA methylation regulates transcriptional homeostasis of algal endosymbiosis in the coral model Aiptasia. Sci Adv. 2018;4:eaat2142. doi: 10.1126/sciadv.aat2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Libbrecht R, Oxley PR, Keller L, Kronauer DJC. Robust DNA methylation in the clonal raider ant brain. Curr Biol. 2016;26:391–395. doi: 10.1016/j.cub.2015.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall H, Lonsdale ZN, Mallon EB. Methylation and gene expression differences between reproductive and sterile bumblebee workers. Evol Lett. 2019;3:485–499. doi: 10.1002/evl3.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glastad KM, Gokhale K, Liebig J, Goodisman MAD. The caste- and sex-specific DNA methylome of the termite Zootermopsis nevadensis. Sci Rep. 2016;6. 10.1038/srep37110. [DOI] [PMC free article] [PubMed]
- 23.Herb BR, Wolschin F, Hansen KD, Aryee MJ, Langmead B, Irizarry R, et al. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci. 2012;15:1371–1373. doi: 10.1038/nn.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arsenault SV, Hunt BG, Rehan SM. The effect of maternal care on gene expression and DNA methylation in a subsocial bee. Nat Commun. 2018;9:3468. doi: 10.1038/s41467-018-05903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, Hu P, Zhu LB, You LL, Cao HH, Wang J, Zhang SZ, Liu MH, Toufeeq S, Huang SJ, Xu JP. DNA methylation is correlated with gene expression during diapause termination of early embryonic development in the silkworm (Bombyx mori). Int J Mol Sci. 2020;21(2):671. [DOI] [PMC free article] [PubMed]
- 26.Nair SS, Luu PL, Qu W, Maddugoda M, Huschtscha L, Reddel R, et al. Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X ten. Epigenetics Chromatin. 2018;11:1–20. doi: 10.1186/s13072-018-0194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogan SN, Strader ME, Hofmann GE. Gene regulation by DNA methylation is contingent on chromatin accessibility during transgenerational plasticity in the purple sea urchin. bioRxiv Prepr. 2021. 10.1101/2021.09.23.461091.
- 28.Wiench M, John S, Baek S, Johnson TA, Sung MH, Escobar T, et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsankov AM, Gu H, Akopian V, Ziller MJ, Donaghey J, Amit I, et al. Transcription factor binding dynamics during human ES cell differentiation. Nature. 2015;518:344–349. doi: 10.1038/nature14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Héberlé É, Bardet AF. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019;63:727–741. doi: 10.1042/EBC20190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charlet J, Duymich CE, Lay FD, Mundbjerg K, Sørensen KD, Liang G, Jones PA. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol Cell. 2016;62(3):422–31. [DOI] [PMC free article] [PubMed]
- 32.Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551–565. doi: 10.1038/nrg.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: In the right place at the right time. 2018;1340 September:1336–40. 10.1126/SCIENCE.AAT6806. [DOI] [PMC free article] [PubMed]
- 34.Wang S, Lv J, Zhang L, Dou J, Sun Y, Li X, Fu X, Dou H, Mao J, Hu X, Bao Z. MethylRAD: a simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes. Open Biol. 2015;5(11):150130. [DOI] [PMC free article] [PubMed]
- 35.Meyer E, Aglyamova GV, Matz MV. Profiling gene expression responses of coral larvae (Acropora millepora) to elevated temperature and settlement inducers using a novel RNA-Seq procedure. Mol Ecol. 2011;20:3599–3616. doi: 10.1111/j.1365-294X.2011.05205.x. [DOI] [PubMed] [Google Scholar]
- 36.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12. [Google Scholar]
- 37.Xia Q, Wang J, Zhou Z, Li R, Fan W, Cheng D, et al. The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol. 2008;38:1036–1045. doi: 10.1016/j.ibmb.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Baumgarten S, Simakov O, Esherick LY, Liew YJ, Lehnert EM, Michell CT, et al. The genome of Aiptasia , a sea anemone model for coral symbiosis. Proc Natl Acad Sci. 2015;112:201513318. doi: 10.1073/pnas.1513318112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadd BM, Barribeau SM, Bloch G, de Graaf DC, Dearden P, Elsik CG, et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015;16. 10.1186/s13059-015-0623-3. [DOI] [PMC free article] [PubMed]
- 40.Rehan SM, Glastad KM, Lawson SP, Hunt BG. The genome and methylome of a subsocial small carpenter bee, ceratina calcarata. Genome Biol Evol. 2016;8:1401–1410. doi: 10.1093/gbe/evw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voolstra CR, Li Y, Liew YJ, Baumgarten S, Zoccola D, Flot JF, et al. Comparative analysis of the genomes of Stylophora pistillata and Acropora digitifera provides evidence for extensive differences between species of corals. Sci Rep. 2017;7:1–14. doi: 10.1038/s41598-017-17484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKenzie SK, Kronauer DJC. The genomic architecture and molecular evolution of ant odorant receptors. Genome Res. 2018;28:1757–1765. doi: 10.1101/gr.237123.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuller ZL, Mocellin VJL, Morris LA, Cantin N, Shepherd J, Sarre L, Peng J, Liao Y, Pickrell J, Andolfatto P, Matz M. Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science. 2020;369(6501):eaba4674. [DOI] [PubMed]
- 44.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixon G. Invert_meth_and_transcription GitHub Page. 2020. [Google Scholar]
- 46.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broad Institute . Picard toolkit. 2019. [Google Scholar]
- 48.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 50.Dixon G, Bay LK, Matz MV. Evolutionary consequences of DNA methylation in a basal metazoan. Mol Biol Evol. 2016;33:2285–2293. doi: 10.1101/043026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE. MethylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):1–9. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15:1–21. doi: 10.1101/002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huh I, Zeng J, Park T, Yi SV. DNA methylation and transcriptional noise. Epigenetics Chromatin. 2013;6:9. doi: 10.1186/1756-8935-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Reference genomes used in this study.
Additional file 2: Figure S1: The coefficient of variation for transcription (standard deviation (RPKM) / mean(RPKM); computed from control replicates) was negatively related to GBM level (averaged across all samples).
Additional file 3: Figure S2: GBM and transcriptional differences between polyp types in A. millepora show no reproducible relationship. The title of each panel indicates the assay used to measure GBM differences. All axes are on the log2 scale.
Additional file 4: Figure S3: Significant differential GBM and differential transcription between phenotypic conditions show little or no relationship. The top set of panels shows the relationship between transcription and GBM differences for differentially expressed genes (DESeq2 FDR < 0.1). The bottom set of panels show the relationship for differentially methylated genes (MethylKit FDR < 0.1). All axes are on the log2 scale. The contrasts for each study are given in Fig. 1.
Additional file 5: Figure S4: Promoter methylation and transcriptional differences between polyp types in A. millepora show no relationship.
Additional file 6: Figure S5: Promoter methylation and transcriptional differences between phenotypic conditions show no relationship.
Additional file 7: Figure S6: GBM and transcriptional differences between experimental conditions show little or no relationship. Each pair of volcano plots is associated with the scatterplot below. The first volcano plot shows differences in GBM, with significant genes (q-value from MethylKit < 0.1) shown in red. The second shows differences in transcription, with significant genes in blue (FDR < 0.1). The count of significant genes is given above each volcano plot. The scatterplots are the same as those shown in Fig. 5, with expression differences on the Y axis and GBM differences on the X. The contrasts for each study are given in Fig. 1.
Data Availability Statement
The dataset(s) supporting the conclusions of this article are available in the NCBI SRA database, PRJNA415358, PRJNA533306, PRJNA437497, PRJNA304722, PRJNA598980, PRJNA598995, PRJNA386774, PRJNA104931, PRJNA309979. https://www.ncbi.nlm.nih.gov/sra. Intermediate datasets, code, and detailed data processing steps are available on github: https://github.com/grovesdixon/invert_meth_and_transcription.