

SUMMARY
The response to oxygen availability is a fundamental process concerning metabolism and survival/death in all mitochondria-containing eukaryotes. However, the known oxygen-sensing mechanism in mammalian cells depends on pVHL, which is only found among metazoans but not in other species. Here, we present an alternative oxygen-sensing pathway regulated by ATE1, an enzyme ubiquitously conserved in eukaryotes that influences protein degradation by posttranslational arginylation. We report that ATE1 centrally controls the hypoxic response and glycolysis in mammalian cells by preferentially arginylating HIF1α that is hydroxylated by PHD in the presence of oxygen. Furthermore, the degradation of arginylated HIF1α is independent of pVHL E3 ubiquitin ligase but dependent on the UBR family proteins. Bioinformatic analysis of human tumor data reveals that the ATE1/UBR and pVHL pathways jointly regulate oxygen sensing in a transcription-independent manner with different tissue specificities. Phylogenetic analysis suggests that eukaryotic ATE1 likely evolved during mitochondrial domestication, much earlier than pVHL.
Keywords: oxygen sensing, hypoxic signaling, posttranslational protein modification, Arginylation, arginyltransferase, ATE1, HIF1α, degradation, glycolysis, Warburg effect
eTOC blurb
Moorthy et al. find an alternative pathway for oxygen-dependent degradation of HIF1α, mediated by ATE1 rather than pVHL. Unlike pVHL, which is not found outside metazoans, ATE1 is conserved throughout eukaryotes and its evolution likely accompanied the domestication of mitochondria.
Graphical Abstract
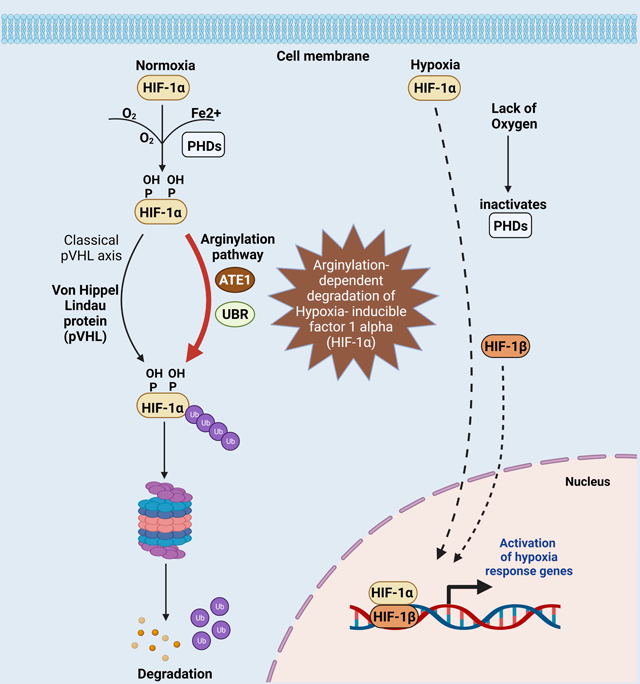
INTRODUCTION
The ability to sense oxygen availability and adapt accordingly involves switching between mitochondrial respiration and glycolysis, and influences cell survival and programmed cell death in eukaryotic systems(Al Tameemi et al., 2019). In most metazoans, a critical player in oxygen sensing is the Von Hippel–Lindau tumor suppressor protein (pVHL)-cullin E3 ubiquitin ligase(Jaakkola et al., 2001a, Iwai et al., 1999). Specifically, pVHL recognizes hypoxia-inducible factor 1α (HIF1α)(Wang et al., 1995, Semenza and Wang, 1992) with specific proline residues hydroxylated by the family of prolyl hydroxylases (PHD), which use oxygen as a substrate(Fong and Takeda, 2008). This mechanism drives a broad range of transcriptional responses to changes in oxygen availability(Ivan et al., 2001, Jaakkola et al., 2001a, Majmundar et al., 2010). Whereas several other oxygen-independent mechanisms also influence HIF1α stability(Liu et al., 2007, Koh et al., 2008, Luo et al., 2010, Jeong et al., 2002), the pVHL pathway is the only one known in animals that couples the half-life of HIF1α to the oxygen level(Ratcliffe et al., 1998, Majmundar et al., 2010, Huang et al., 1998, Jaakkola et al., 2001a, Ohh et al., 2000). However, the gene encoding pVHL (VHL) is strictly present in metazoans and no other eukaryotes. Since the need for oxygen sensing likely co-evolved with the domestication of mitochondria and thus should exist in nearly all eukaryotes(Taylor and McElwain, 2010), there should be additional oxygen-sensing pathways conserved between metazoan and non-metazoan eukaryotes.
In green plants, the oxygen-sensing mechanism appears to be regulated by N-terminal arginylation: a posttranslational addition of one extra arginine to the N-terminus. Arginylation is a conserved process among eukaryotes and is mediated by the family of arginyltransferases(Kaji et al., 1963, Kaji, 1968, Soffer, 1971). In many instances, N-terminal arginylation constitutes a signal for acute degradation according to the N-end rule theory, which associates the identity of the N-terminal residue to a protein’s half-life(Varshavsky, 2011, Bachmair et al., 1986). In plants, the arginylation of N-terminal Cys of a subset of proteins is promoted by the oxidation of this residue: a reaction catalyzed by the oxygen-sensing plant cysteine oxidases (PCOs)(Licausi et al., 2011, White et al., 2017). Through this mechanism, the half-lives of ethylene response factor (ERF)-transcription factors, which regulate metabolism, proliferation, and apoptosis in plant cells, are coupled to the oxygen concentration in an arginylation-dependent manner(Licausi et al., 2011). In animals, arginylation is solely mediated by arginyltransferase 1 (ATE1), which is ubiquitously conserved between plants, fungi, and animals(Balzi et al., 1990, Kwon et al., 1999, Rai and Kashina, 2005). Growing evidence suggests the involvements of ATE1 in the response to reactive oxygen species (ROS) and/or nitric oxide, which often result from cellular redox imbalance(Hu et al., 2005, Ingoglia et al., 2000, Kumar et al., 2016, Masdehors et al., 2000). Furthermore, in parallel to the PCOs in plants, the oxygen-dependent cysteamine (2-aminoethanethiol) dioxygenase (ADO) in animals promotes the oxidation of the N-terminal cysteine and the subsequential ATE1-dependent arginylation of several members of the Regulator of G protein signaling (RGS) family, which are important regulators of tissue development and homeostasis(Masson et al., 2019). However, currently no direct evidence shows the involvement of ATE1/arginylation in the PHD-HIF1α-pVHL axis, which is considered the main regulator of cellular metabolism, growth, proliferation, and apoptosis in response to oxygen concentrations.
In this study, we identify ATE1 as a key regulator of glycolysis and hypoxic responses. We demonstrate HIF1α as a previously unknown substrate of arginylation and found that this posttranslational modification (PTM) is promoted by PHD-mediated hydroxylation, in an oxygen-dependent manner. Furthermore, we show that the arginylated HIF1α is degraded independently of the conventional pVHL pathway but is instead dependent on the Ubiquitin Protein Ligase E3 Component N-Recognin (UBR) family enzymes. Finally, phylogenetic analysis suggests the ATE1-dependent mechanism is a conserved pathway for oxygen-sensing in many, diverse forms of life.
RESULTS
Depletion or reduction of ATE1 leads to aerobic glycolysis
Under standard culture conditions (non-confluent and non-stressed), ATE1-KO does not appear to directly affect cellular proliferation (Kumar et al., 2016, Rai et al., 2015). However, we noticed that ATE1-KO mouse embryonic fibroblasts (MEF) acidified the culture media faster than their wild-type (WT) counterparts as a sign of enhanced glycolysis (Suppl Fig. S1A). Consistently, ATE1-KO MEF showed higher rates in glucose intake and lactate production (Fig. 1A, B). Similarly, a knockdown of ATE1 expression (Fig. 1C) increased glycolytic rate and capacity (Fig.1D-F). These phenotypes are reminiscent of the classic Warburg effect, an increase of glycolysis for ATP production in the presence of oxygen(Liberti and Locasale, 2016). Indeed, the application of glycolysis inhibitor 2-deoxy-D-glucose (2-DG)(Maschek et al., 2004) led to a larger reduction of ATP in ATE1-KO MEF than in WT cells (Fig. 1G; See also Suppl Fig. S1B for the direct comparison of ATP levels in these cells). Similar results were obtained when a different glycolysis inhibitor, 2-Fluoro-2-deoxy-D-glucose (2FDG)(Som et al., 1980), was used (Fig. 1H). To ensure that these effects are ATE1-specific, we stably expressed recombinant ATE1 in ATE1-KO MEF at an amount comparable to the endogenous level(Birnbaum et al., 2019, Jiang et al., 2020), and found this restored the glycolysis-dependent ATP content comparable to WT cells (Fig. 1J and K). Consistently, ATE1-knockdown also led to a larger 2-DG-induced depletion of ATP (Fig. 1L). Therefore, our data suggests that ATE1 depletion can cause the Warburg effect, which is often associated with the pseudohypoxic state(Al Tameemi et al., 2019).
Figure 1. Depletion of ATE1 increases glycolysis.
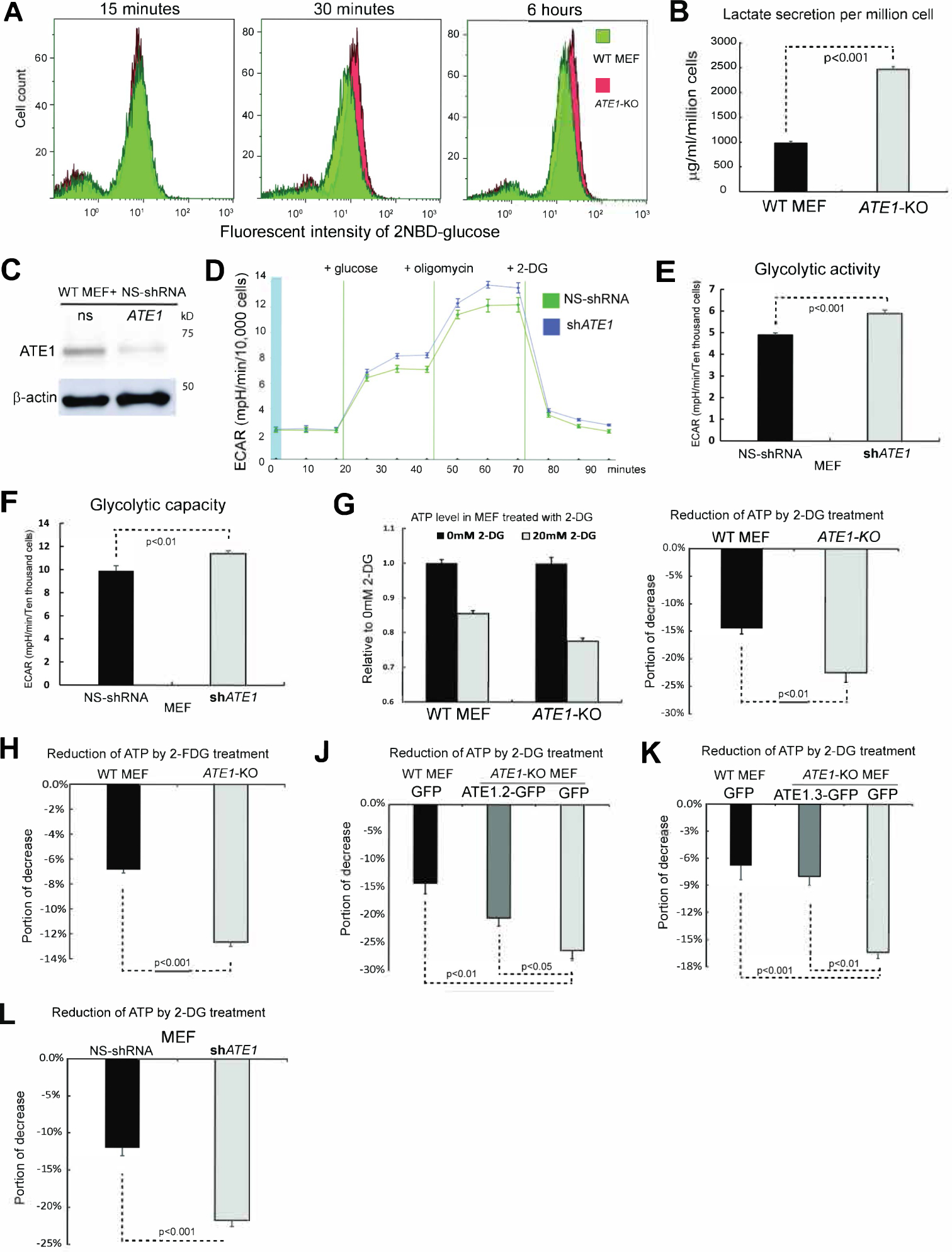
A) Glucose intake in WT and ATE1-KO MEF at increasing time points measured by flow cytometry using the green-fluorescent glucose analog, 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2 (2-NBDG).
B) Concentration of lactate secreted in the medium in 24 hrs by WT and ATE1-KO MEF (n=7). See also Suppl Figure S1A for the color of culture media. Throughout this study, error bars represent standard error of mean (SEM) and p-values were calculated by Student’s t-test. A p-value >0.05 was considered non-significant (n.s).
C) Knockdown of ATE1 by specific shRNA compared to non-silencing (NS) control.
D) The curve of extracellular acidification rate (ECAR) measured with a Seahorse flux analyzer in MEF with ATE1-knockdown or NS control (n=10).
E) Glycolytic activity and (F) maximum glycolytic capacity of MEF with ATE1-knockdown or NS control measured in Seahorse flux analyzer (n= 10).
G) Left panel: ATP concentrations in WT and ATE1-KO MEF with or without 20mM 2-DG (n=5), individually and internally normalized to a random group without 2-DG. Right panel: 2-DG-induced ATP reduction (n=5). See also Suppl Fig. S1B for the direct comparison of ATP levels in WT and ATE1-KO cells.
H) Similar as (G), except with 15 mM 2-FDG (n=5).
J) 2-DG induced ATP reduction in WT or ATE1-KO MEF expressing GFP alone or GFP conjugated ATE1 isoform-2 (an enzymatic potent and ubiquitously expressed splice variant(Rai and Kashina, 2005, Wang et al., 2011)) (n=5).
K) similar to (J), except that Ate1 isoform-3 (which is also expressed in MEF(Rai and Kashina, 2005)) was used (n=5).
L) 2-DG induced ATP reduction in MEF treated with NS- or ATE1-specific shRNA (n=5).
ATE1 controls glycolysis and hypoxia response by suppressing the level and function of HIF1α independently of its transcription regulation
To identify potential arginylation targets involved in metabolic regulation, we designed a screening assay by assuming arginylation promotes degradation. We found multiple proteins whose levels are inversely correlated with the ATE1 level in human prostate cancer cells(Birnbaum et al., 2019) (Suppl Fig. S1C). These included HIF1α: the primary regulator of hypoxia response in mammalian cells(Majmundar et al., 2010)(Suppl Fig. 1D). As a validation, the steady-state levels of HIF1α in ATE1-KO MEF appear much higher than in WT (Fig.2A and 2B). Furthermore, the expression of a recombinant GFP-fused ATE1 significantly lowered the HIF1α level in ATE1-KO MEF, while a mutant ATE1 with reduced enzymatic activity(Li and Pickart, 1995b, Li and Pickart, 1995a) resulted in a weaker effect (Fig.2C). These data indicate that a fully active ATE1 is needed to suppress HIF1α level. When comparing ATE1-KO MEF to WT cells incubated under different oxygen concentrations, we found that the HIF1α level in ATE1-KO is comparable to WT cells treated with 10% oxygen: a moderately hypoxia for cultured cells (Fig.2D). The sustained elevation of HIF1α in ATE1-KO cells is anticipated to generate pseudohypoxic effects(Al Tameemi et al., 2019, Majmundar et al., 2010), which affect glycolysis and stress response. As previously reported(Rai et al., 2015, Kumar et al., 2016), the growth rate of WT and ATE1-KO MEF are very similar when cultured in non-contact inhibited seeding density under normoxic conditions (Fig. 2F). However, under severely hypoxic culturing conditions (0.5% oxygen), while WT MEF cells underwent significant cell death, ATE1-KO cells were more resistant and continued to grow (Fig.2G). The knockdown of the HIF1α gene (HIF1A) expression in ATE1-KO MEF (Fig. 2E) was able to restore cellular sensitivity to hypoxia, while having little effect on cell growth under normoxia (Fig.2G). In addition, the HIF1A knockdown in ATE1-KO MEF reduced lactate production rate and glycolysis-derived ATP content to comparable WT levels (Fig. 3A, 3B). Therefore, the observed Warburg effect and hypoxia-resistance in ATE1-deficient cells are largely caused by the elevated HIF1α levels.
Figure 2. ATE1 regulates hypoxia response.
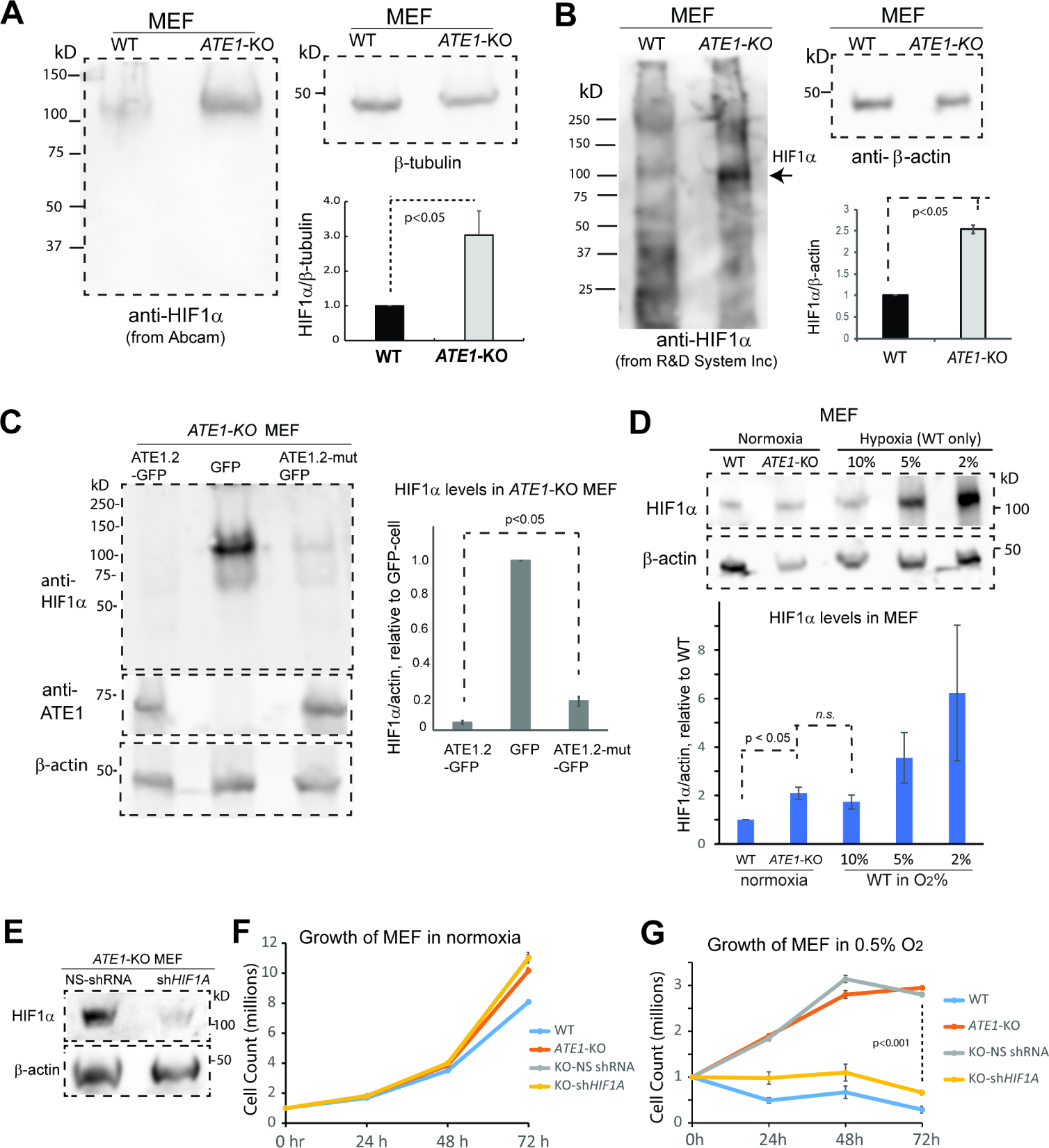
See also Suppl Fig. S1C and 1D for the identification of potential ATE1 substrates with antibody arrays.
A) HIF1α in WT and ATE1-KO MEF probed with monoclonal rabbit anti HIF1α (Abcam, Cambridge, MA, Cat# ab179483, used as elsewhere in this study unless otherwise indicated). The β-actin was used as a loading control. The graph at the bottom right shows the quantification (n=6).
(B) Similar as (A), except with mouse anti-Hif1α (R&D systems, Cat# MAB1536) (n=3).
C) Left panel: the level of HIF1α in ATE1-KO MEF stably expressing either ATE1.2-GFP, mutant ATE1.2-C23–25S (labeled as ATE1.2-mut) with a reduced enzymatic activity(Kumar et al., 2016, Berleth et al., 1992, Li and Pickart, 1995a, Li and Pickart, 1995b), or the GFP alone. The level of recombinant ATE1 was probed with anti-ATE1. β-actin is used as loading control. Graph on the right shows quantification (n=3) of HIF1α in relative to GFP-expressing cells. The levels of ATE1-GFP (probed by anti-ATE1 on a different gel with the same cell samples) were also shown.
D) The levels of HIF1α in WT and ATE1-KO MEF maintained under normoxia (18% O2 for typical culture conditions), in comparison to WT MEF exposed to different O2 concentrations for 6 hours. β-actin was used as loading control. Graph on the bottom shows quantification (n=6).
E) Representative immunoblot showing the efficiency of shRNA knockdown of HIF1A in ATE1-KO MEF.
F) Time-dependent viable cell counts (with trypan blue) under normoxia (18% O2) for WT MEF and ATE1-KO MEF (non-treated, treated with NS- or HIF1A- shRNA) (n=4). Please note that the curves for non-treated and NS-shRNA ATE1-KO nearly overlap.
(G) Similar to (F) except under hypoxia (0.5% O2) condition.
Figure 3: Ate1 regulates glycolysis and HIF1α signaling.
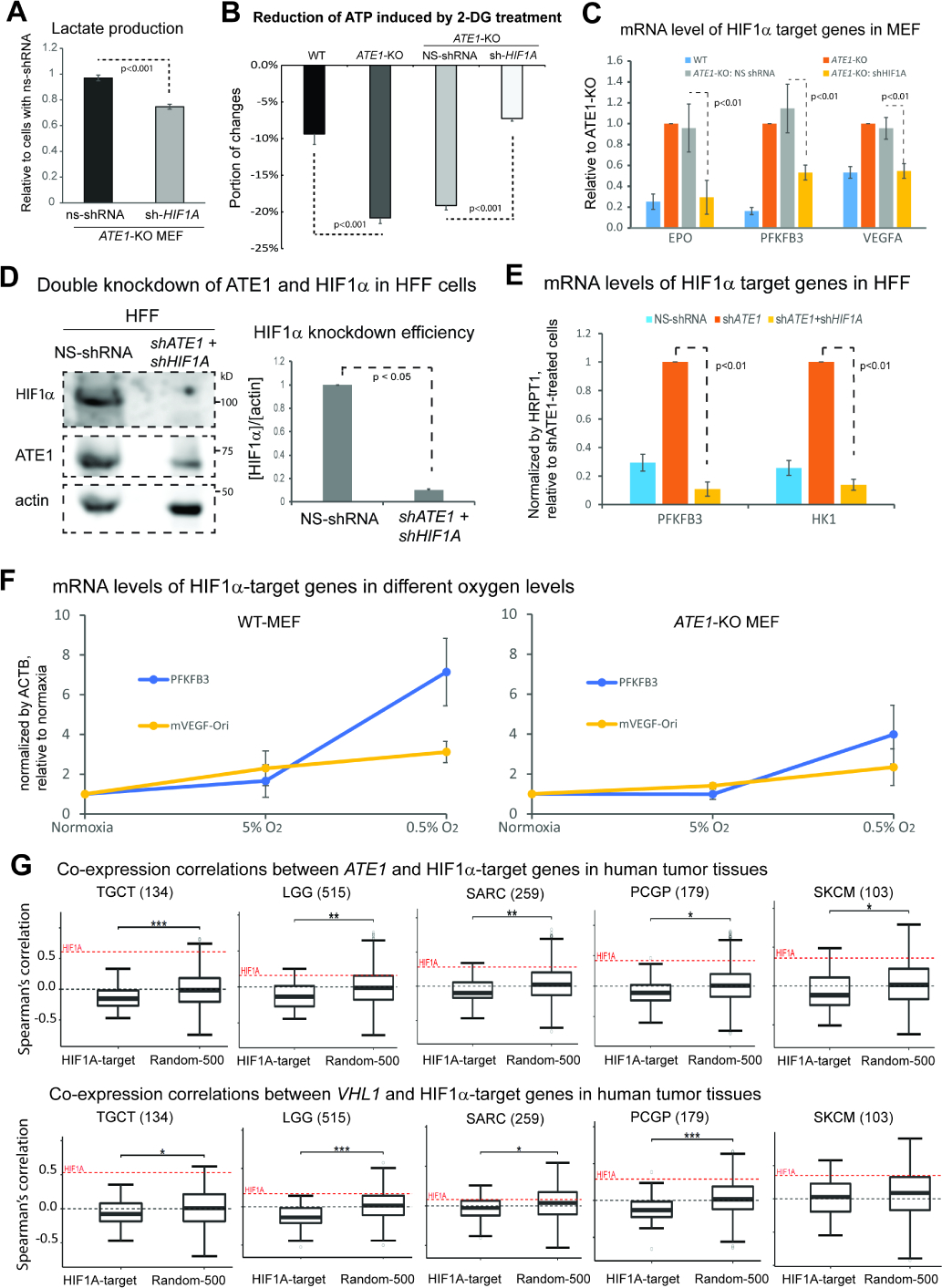
A) Lactate secretion in the media over 24 hours for ATE1-KO MEF with HIF1α-specific or NS shRNA (n=4).
B) The 2-DG-induced ATP reduction in MEF WT, ATE1-KO, ATE1-KO with NS- or HIF1α-specific shRNA (n=5).
C) The mRNA levels of HIF1α-target genes EPO, PFKFB3 and VEGFA in MEFs of WT, ATE1-KO, ATE1-KO with NS- or HIF1A-shRNA. Quantified by quantitative RT-PCR using 18S rRNA as a loading control (n=3).
D) Representative immunoblot and quantification (n=3) showing the efficiency of a double knockdown of HIF1A and ATE1, in HFF.
E) The mRNA levels of HIF1α-target genes, PFKFB3 and hexokinase-1 (HK1), in HFF treated with either: NS-shRNA, ATE1-shRNA, or ATE1- + HIF1A-shRNAs. Human hypoxanthine phosphoribosyltransferase 1 (HPRT1) was used as a loading control in quantitative PCR (n=3).
F) Graphs showing for mRNA levels measured by qPCR (n=3) of HIF1α-target genes PFKFB3 and VEGFA in WT and ATE1-KO MEF: challenged with different O2 concentrations for 6 hours. ACTB was used as a loading control. The levels of these genes in normoxia (18% in cultured cells) were used as normalization points for individual series of data.
G) Box whisker plots showing the Spearman’s correlations of mRNA expressions of ATE1 (or VHL) with 50 validated HIF1α-activating target genes (see Suppl Table S1 for the list) in tumor tissues in The Cancer Genome Atlas (TCGA). A random set of 500 genes (Random-500)(Li et al., 2012, Soler-Oliva et al., 2017, Zhao and Liu, 2019) was used as control for HIF1α-targets. The examined tissues include testicular germ cell tumors (TGCT), low grade glioma (LGG), sarcoma (SARC), Pheochromocytoma and Paraganglioma (PCPG), and skin cutaneous melanoma (SKCM); their sample sizes are indicated in parentheses. The co-expression correlation between ATE1 (or VHL) and HIF1A in each sample set are also illustrated as a red dotted line on the plot. The significance (p-value) was calculated by Mann−Whitney U test. The symbols *, **, and *** indicate p-values of <0.05, <0.005, and <0.0005, respectively.
Since HIF1a mainly regulates transcription, we measured the expression of known HIF1α-target genes in MEF, which include erythropoietin (EPO), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), and vascular endothelial growth factor A (VEGFA). We found that their mRNA levels are increased in ATE1-KO cells and this effect can be specifically reversed by HIF1A-knockdown (Fig. 3C). Similar observations were made in human foreskin fibroblasts (HFF) by the knockdown of ATE1 and HIF1A (Fig. 3D, 3E). Notably, as found in reported cases with an adaptation to pseudohypoxia(Koido et al., 2017, van Gisbergen et al., 2020), while the ATE1-KO MEF have higher basal expression levels of many HIF1α-target genes, they also appear to be less transcriptionally responsive to changes in oxygen levels (Fig. 3F). Next, we examined the co-expression relationship between ATE1 and a panel of HIF1α-activating genes (Supp Table S1) in human tumor tissues (Fig. 3G). We found that the majority of these genes have a negative correlation (or lower correlation compared to randomly-selected genes) with ATE1 in mRNA levels (Fig.3G; top panel). However, HIF1A itself shows a positive relationship with ATE1 (Fig.3G and Suppl Fig.S2A). These data indicate that ATE1 suppresses the expression of HIF1α-target genes independently of the transcription regulation of HIF1A per se. A similar behavior was found with the pVHL gene (VHL) (Fig.3G; bottom panel). Thus, ATE1 likely suppresses HIF1α-signaling in vivo independently of HIF1α transcription, in a manner similar to pVHL.
ATE1 controls the protein level of HIF1α by promoting its degradation with N-terminal arginylation
Consistent with the positive correlation between ATE1 and HIF1A expression in tissues (Fig.3G and Suppl Fig.S2A), HIF1A mRNA is lower in ATE1-KO MEF than WT (Fig.4A). This contrasts with the elevated HIF1α protein level seen in ATE1-KO cells (Fig.2A). By using cycloheximide (a translation inhibitor) to estimate protein turnover rates, we observed a slower degradation of HIF1α in ATE1-KO MEF (Fig.4B; see also Suppl Fig. S2B for DMSO control). To validate this finding, the cells were treated with MG132, a proteasome inhibitor also useful in estimating protein degradation dynamics. In agreement with the cycloheximide treatment, MG132 led to a smaller fold of accumulation of HIF1α in ATE1-KO cells than WT (Suppl Fig. S2C). Thus, ATE1 appears to influence HIF1α stability.
Figure 4. ATE1 promotes HIF1α degradation by direct N-terminal arginylation.
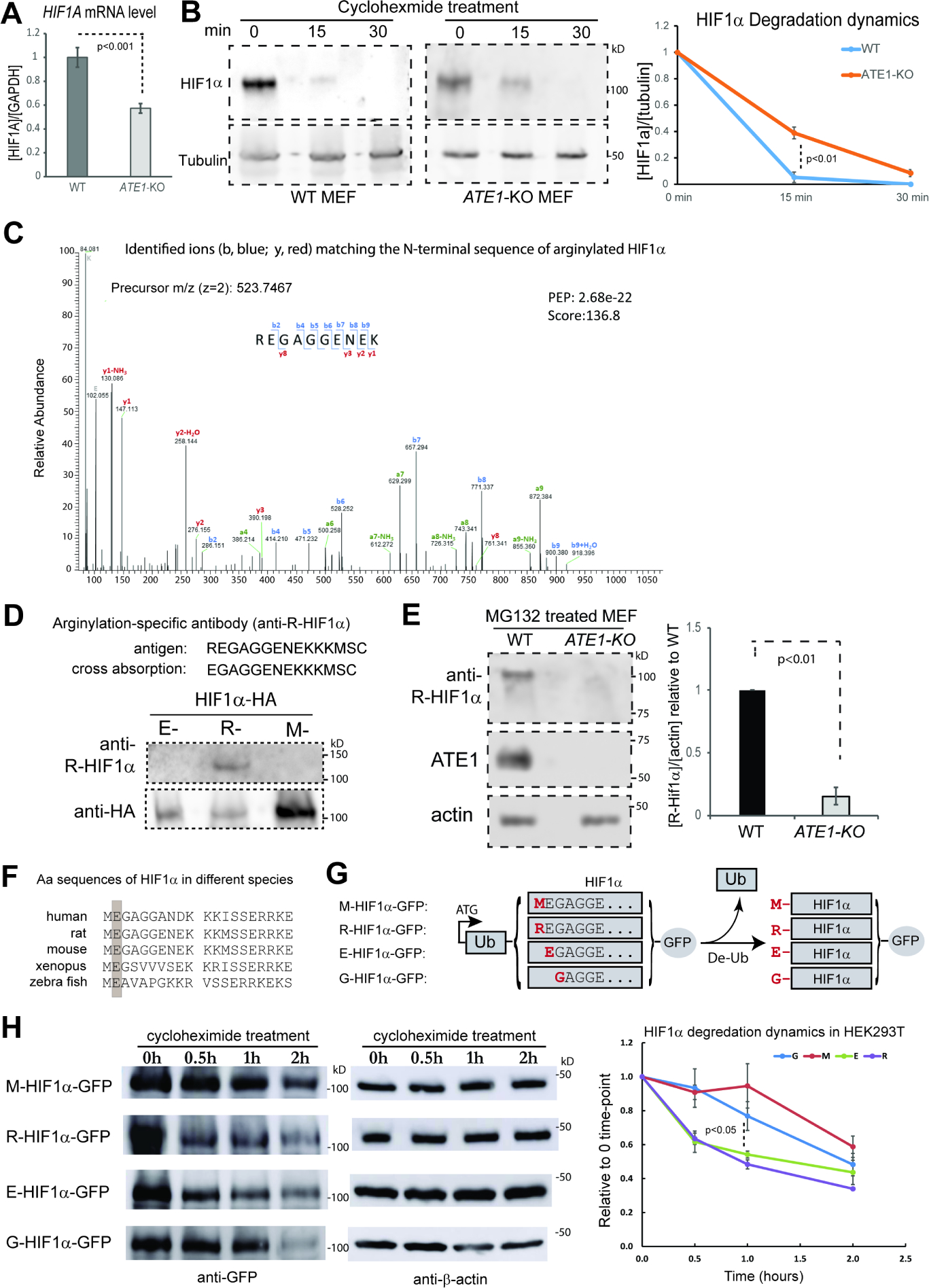
A) The levels of HIF1α mRNA in WT and ATE1-KO MEF, with GAPDH as loading controls (n=4).
B) The degradation dynamics of endogenous HIF1α in WT and ATE1-KO MEF measured in the presence of translation inhibitor cycloheximide. Tubulin was used as loading control. The graph on the right shows the quantification (n=4). See Suppl Fig S2B for the effects of DMSO as a reagent control, and Suppl Fig. S2C for the differential effects of a brief treatment of MG132 on HIF1α accumulation.
C) Mass Spec analysis of recombinant mouse HIF1α expressed in (and purified from) WT MEF harvested from SDS-PAGE (See Suppl Fig. S2D for an example of the protein bands). The shown spectrum, obtained from the proteomic core of the University of South Florida (USF), is consistent with the sequence of REGAGGENEK anticipated from an N-terminally arginylated mouse HIF1α. The detected b (blue color) and y (red color) ions were indicated on the spectrum. The posterior error probability (PEP) values and the Andromeda score, which reflect the quality of the peptide assignment in the spectrum(Cox et al., 2011, Cox and Mann, 2008), were also shown. See also Suppl Fig. S2E and S2F for additional examples of spectra from different facilities showing arginylated HIF1α from WT MEF. None of these peptide signals were detected when the protein was expressed and purified from the arginylation-deficient ATE1-KO MEF (Suppl Fig. S3).
D) Production and validation of an arginylation-specific antibody for HIF1α. The peptides representing the N-terminal sequence of arginylated HIF1α were used as antigen, while the pre-arginylated peptide was used for cross-absorption. The immunoblot on the bottom shows the reactivity of the resulting antibody on the constitutively arginylated (-R), non-arginylated (M-), and pre-arginylated (E-) forms of recombinant HIF1α expressed in ATE1-KO MEF cells. C-terminal HA fusion tag was used as a loading control (see Fig. 4G and 5A for further details of these constructs).
E) The levels of arginylated, endogenous HIF1α in WT and ATE1-KO cells treated with MG132 for 9 hrs. The graph on the right side shows the quantification (n=3).
F) N-terminal amino acid (Aa) sequences of HIF1α in different species. The position for the 2nd residue is highlighted in a grey box to show the identity.
G) Construction of recombinant mouse HIF1α with N-terminal residue M, R, E or G. Protein translation is initiated from the start codon of the Ub-coding region. After expression, the Ub will be cleaved by endogenous de-Ub machineries, leaving the penultimate amino acid as the new N-terminal end. The protein starting with an N-terminal methionine (M) is to represent the constitutively, non-arginylated form or an arginine (R), for the arginylated form. Also, the HIF1α starting with glutamic acid (E) mimics the naïve form immediately after the first M is removed, which is potentially eligible for arginylation. The one starting with glycine (G) is expected to reduce the frequency of arginylation(Varshavsky, 2011, Wong et al., 2007, Wang et al., 2018). The C-terminal tag (in this case, GFP) is used to facilitate detections.
H) Degradation dynamics of the M-, R-, E-, and G- forms of HIF1α-GFP in the cells (**unsure of which cells) in the presence of translation inhibitor cycloheximide, as shown in representative Western blot images, and quantified in the chart on the right (n=3 for M, R, E; n=6 for G).
Based on identified substrates and in vitro tests, arginylation appears to preferentially affect proteins starting with certain amino acids (D, E, N, Q, or C) after the first methionine (M) is removed(Varshavsky, 2019). While the sequence of HIF1α fits such a scenario (Fig. 4F), its arginylation was never confirmed, probably due to technical difficulties in detecting arginylation on low-abundancy proteins(Decca et al., 2006, Wong et al., 2007, Piatkov et al., 2012, Brower et al., 2013). To circumvent this issue, we fused a C-terminal 6xHis tag to mouse HIF1α and expressed it in WT MEF (Suppl Fig. S2D), which displays a normal level of arginylation(Kumar et al., 2016). By using mass spectrometry for peptide sequencing on the purified protein, we detected peptide peaks consistent with an N-terminally arginylation (Fig.4C and Suppl Fig.S2D-F). This modification was not found on the protein isolated from the arginylation-deficient ATE1-KO MEF (Suppl Fig. S3A, S3B). To further validate N-terminal arginylation on endogenous HIF1α, we custom ordered an antibody specific for the peptide sequence representing the arginylated N-terminus of HIF1α (Fig. 4D). By using this antibody, a much stronger signal was detected in WT MEF compared to the ATE1-KO cells (Fig.4E).
Arginylation does not always lead to rapid degradation(Karakozova et al., 2006, Zhang et al., 2010, Zhang et al., 2015, Carpio et al., 2010). As another validation for the arginylation-eligibility and subsequential effects on degradation of HIF1α, we produced a series of recombinant proteins starting with different residues by using N-terminal ubiquitin fusion(Bachmair et al., 1986, Zhang et al., 2010) (Fig.4G). By using cycloheximide, we found that the arginylated form (R-) of HIF1α degrades fastest while the non-arginylated (M-) form, the slowest. Furthermore, the potentially arginylation-eligible E-HIF1α degrades faster than the anticipated arginylation-retarding G-form (Fig. 4H). All together, these data suggest that HIF1α is eligible for arginylation and subsequent degradation.
ATE1-mediated arginylation of HIF1α constitutes a pVHL-independent cellular oxygen sensing pathway that involves UBR proteins
The effects of arginylation on HIF1α instability raised the question of how this mechanism is related to the classic PHD-pVHL axis, which couples HIF1α degradation to oxygen levels(Majmundar et al., 2010).
We first tested whether the arginylation-dependent degradation of HIF1α requires the action of pVHL: an E3 ubiquitin ligase that recognizes proline-hydroxylated HIF1α(Jaakkola et al., 2001b). To address this question, we generated recombinant proteins representing different eligibilities for arginylation and proline-hydroxylation (Fig. 5A). Consistently with observed differences in degradation dynamics (Fig.4H), M-HIF1α showed the highest steady-state protein level, followed by the G-form, and the R-form being the lowest (Fig.5B). When proline 402 and 577 on mouse HIF1α were mutated to alanine and glycine (termed as “PAPG-HIF1α”) to block the recognition of pVHL(Jaakkola et al., 2001b), it did not abolish the metabolic difference between the M- and R- forms (Fig.5C). As a validation, the recombinant HIF1α representing different arginylation eligibilities were expressed in UOK111 cells: a pVHL-deficient human renal carcinoma cell line (Anglard et al., 1992). In these cells, R- or E-HIF1α are still less stable than the M- or G-form (Fig.5D). Furthermore, these observed differences can be minimized by MG132 treatment (Suppl Fig. S4A). In addition, a knockdown of ATE1 in UOK111 cells is sufficient to increase the endogenous HIF1α protein level (Fig.5E). Therefore, our data suggest that the arginylation pathway is intrinsically independent of the conventional pVHL pathway. To directly compare the impacts of the ATE1- and pVHL-dependent pathways, the steady-state levels of recombinant HIF1α with different arginylation-eligibilities and PAPG mutations were compared side by side. We found much larger impacts when the arginylation eligibility is changed, regardless of the status of PAPG mutation (Fig.5F). This may explain why the drastic upregulation of pVHL in ATE1-KO cells (Fig.5G) cannot completely suppress HIF1α elevation (Fig.2A).
Figure 5. Arginylation-mediated degradation of HIF1α is independent of the pVHL pathway.
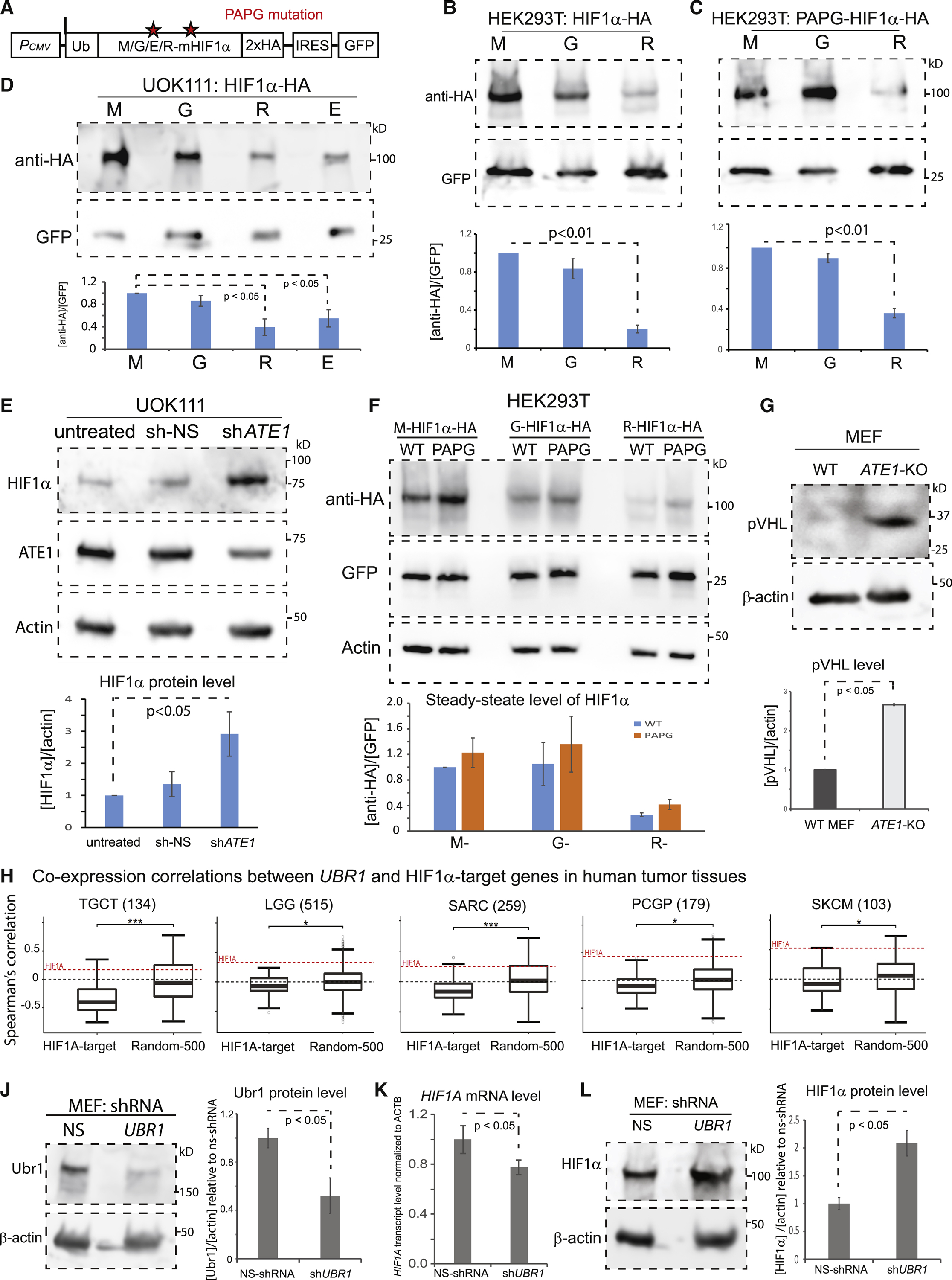
A) A diagram showing recombinant HIF1α with different eligibilities for arginylation or proline-hydroxylation. Asterisks indicate the positions of the two critical proline (P) residues, 402 and 577, on mouse HIF1α (corresponding to 402 and 564 in human HIF1α). Mutations of P402 to Alanine (A) and P577 to Glycine (G) (referred to as the PAPG mutation) are expected to block both the PHD-mediated hydroxylation and the downstream recognition by pVHL necessary for ubiquitination(Jaakkola et al., 2001b). The C-terminal 2xHA tag is to facilitate the detection. An internal ribosome-entry site (IRES) and the coding sequence of GFP were placed behind HIF1α for the normalization of transfection and translation efficiencies.
B) Steady-state levels of HIF1α (with M, G, or R on the N-terminus), transiently expressed in HEK293T cells, detected by antibody against HA tag. The level of GFP is used as a loading control to normalize differences in the expression efficiencies of the vector. On the bottom is the quantification (n=8).
C) Similar to (B), except with the PAPG mutation on HIF1α (n=8).
D) Steady-state levels of the M-, G-, R-, or E- HIF1α transiently expressed in the pVHL-deficient human renal carcinoma cell line UOK111 and quantified similar as in (B) (n=3). See also Suppl Fig. S4A for the effects of the treatment of MG132, a proteasome inhibitor, on these proteins in UOK111 cells.
E) Steady-state levels of HIF1α and ATE1 in UOK111 cells (untreated, treated with NS- or ATE1- specific shRNA) with β-actin as loading controls. The graph on the bottom shows the quantification of HIF1α levels (n=3).
F) Similar to (B), except that all different forms of HIF1α were loaded in the same gel and grouped by their states/eligibilities of arginylation. The graph on bottom shows the quantification (n=3).
G) The pVHL levels in WT and ATE1-KO MEF with β-actin as loading controls. The graph on the bottom shows the quantification (n=3).
H) Similar to Fig.3G, except showing UBR1 with HIF1α-activating target genes. See also Suppl Fig. S4B for the data of other UBR family members (UBR2–5).
J) The Ubr1 levels in MEF cells treated with NS- or UBR1-specific shRNA, with β-actin as loading controls. The quantification is shown on the right side (n=4).
K) The mRNA levels of HIF1A in MEF cells stably expressing NS- or UBR1- specific shRNA measured by quantitative PCR. The β-actin gene (ACTB) was used as a loading control (n=5).
L) The steady-state HIF1α levels in MEF cells with NS or UBR1- specific shRNA, with β-actin as loading control. The graph on the right side shows quantification (n=4). As another control, see Suppl Fig. S4C for the effects of UBR1-knockdown in ATE1-KO MEF. See also Suppl Fig. S4D-4F for UBR1-knockdown in HFF and the consequential effects on the mRNA/protein levels of HIF1α.
Many arginylated proteins were found to be degraded by the action of the UBR family E3 Ub-ligases(Tasaki et al., 2005). The mammalian genome contains at least 5 UBR members, Ubr1–5(Varshavsky, 2019), although their exact roles and/or tissue specificities remain unclear. When examining the expression correlation of these UBR genes with a group of known HIF1α-target genes (Supp Table S1) in various cancer tissues, we observed mostly negative relationships with HIF1α-target genes and a positive relationship with HIF1A itself (Fig.5H and Suppl Fig.S4B), in a manner similar to ATE1 or VHL. As a validation, we knocked down UBR1, which has an established role in ATE1-dependent degradation in different types of cells including fibroblasts(Xia et al., 2008, Hwang et al., 2010, Sultana et al., 2012) (Fig.5J). While this treatment leads to a moderate decrease in HIF1A mRNA levels in MEF (Fig. 5K), it significantly increased the steady-state HIF1α protein level (Fig.5L). As a control, UBR1-knockdown in ATE1-KO cells generated little effect on the HIF1α level (Suppl Fig. S4C). Similar observations were found in human cells HFF (Suppl Fig. S4D-F). Therefore, it is likely that UBR1 (and potentially other UBRs) mediates HIF1α degradation.
To understand how ATE1-KO leads to defects in oxygen-sensing, we next looked at the PHD enzyme. We found that the protein level of PHD2 – one of the most potent members in the PHD family – was not lowered, but rather increased in ATE1-KO cells (Fig. 6A). Also, the treatment of ascorbate – a co-substrate of PHD enzymes – generated a much smaller impact on HIF1α levels in ATE1-KO MEF compared to WT (Fig. 6B). Furthermore, under normoxic conditions, ATE1-KO MEF and WT cells appear to contain comparable amounts of hydroxylated HIF1α (Fig. 6C and Suppl Fig. S5A and S5B). Thus, the compromise of oxygen-sensing in ATE1-KO cells likely takes place in a downstream step.
Figure 6. Arginylation of HIF1α is sensitive to the oxygen-dependent hydroxylation on this protein.
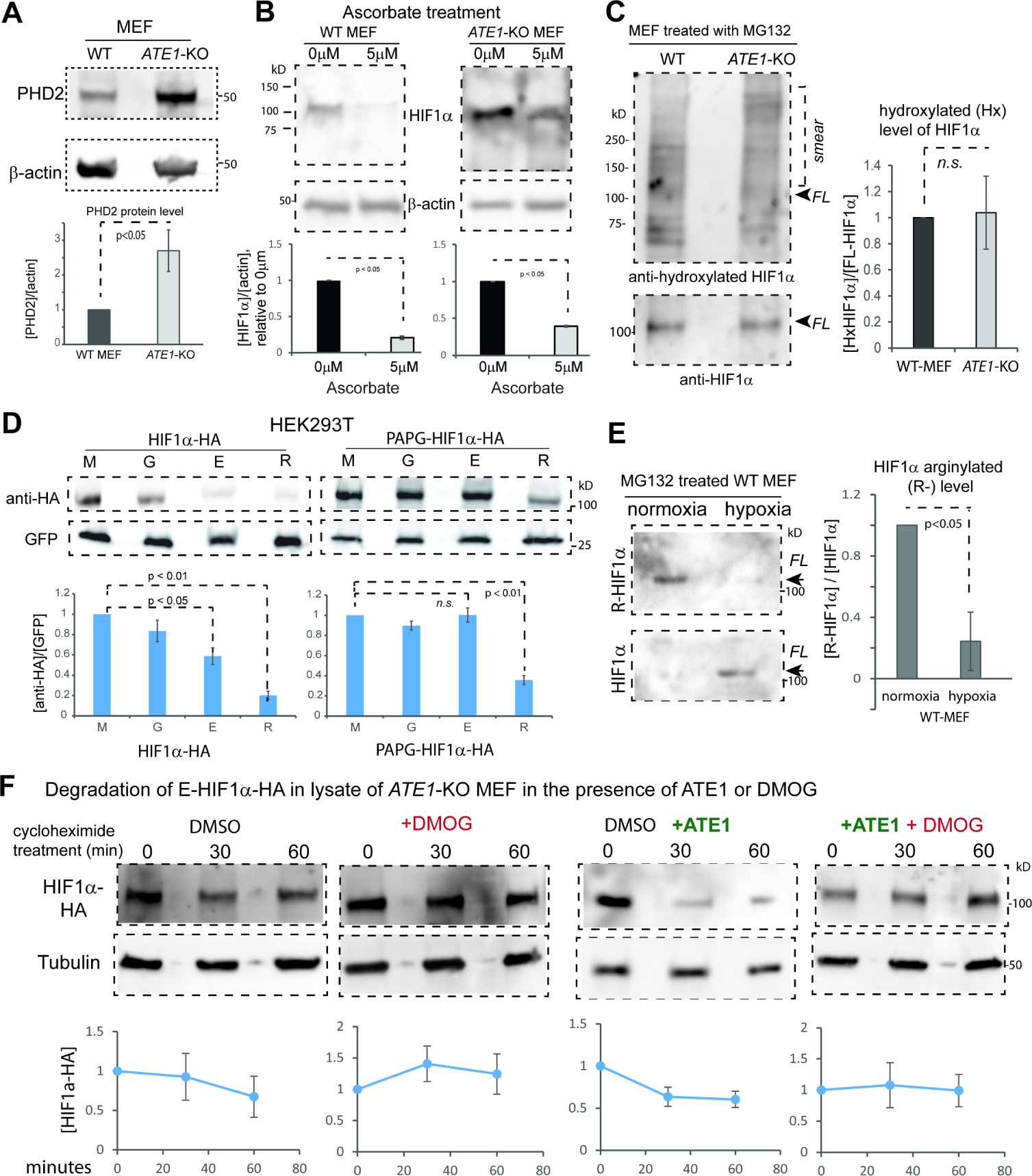
A) Immunoblots of PHD2 protein and quantification (n=3) in WT and ATE1-KO MEF with β-actin as loading controls.
B) The level of HIF1α (and β-actin) in WT and ATE1-KO MEF treated with either 5μM L-Ascorbic acid 2-phosphate (a co-factor and co-activator for PHD) or DMSO (as reagent control) for 24 hours. Under each set of immunoblots is the graph showing the fold changes of HIF1a in ascorbate-treated group (5μM), relative to the control group (0μM) (n=3).
C) Immunoblots on the left shows the P564-hydroxylated (Hx-) HIF1α, probed with rabbit anti hydroxyl-HIF1α (Cell Signaling, Danvers, MA, Cat# 3434), in WT and ATE1-KO MEF treated with MG132 for 6 hrs. The positions of full-length (FL) HIF1α and the ubiquitinylated-smear are indicated. The loading amounts were pre-adjusted to reach equivalent levels of FL HIF1α in each lane; these were determined by loading on a different gel to avoid signal bleed-through, because both the anti-HIF1α and the anti hydroxyl-HIF1α were rabbit antibodies. The graph on the right shows the level of P564-hydroxylated HIF1α in proportion to FL-HIF1α (n=4). See Suppl Fig. 5A for the validation of the specificity of the antibody for hydroxyl-HIF1α. See also Suppl Fig. S5B for the detection of hydroxyl-HIF1α with a different antibody.
D) Steady-state levels of M-, G-, E-, or R- HIF1α with or without PAPG mutations transiently expressed in HEK293T cells. Graph shows quantification with GFP as controls for loading and transfection efficiency (n=8).
E) Endogenous arginylated (R-) HIF1α in WT MEF, under different O2 levels, probed by the custom-made arginylation-specific antibody as described in Fig.3D. The cells were pre-cultured in normoxia (18% O2) or hypoxia (0.5% O2) for at least 2 hrs followed by MG132 addition and allowed to culture for 9 hrs. Arrows point to the expected size of the full-length (FL) HIF1α. The level of total HIF1α, probed by anti-HIF1α, was used to calculate the ratio of arginylation signal. The graph on the right side shows quantifications (n=3).
F) Cycloheximide time-chase for E-HIF1α–HA in the lysate of ATE1-KO MEF, with the addition of recombinant mouse ATE1 isoform 1 (or buffer) and 1mM DMOG (an inhibitor of PHD) or DMSO, as a vehicle control. Tubulin serves as a loading control. Graphs on bottom show quantification (n>=3). See also Supp Fig. S5C for the validation of the effectiveness of DMOG on endogenous HIF1α. See also Suppl Fig. S5D for the lack of effect of DMOG on the recombinant HIF1α bearing the PAPG mutation that blocks proline hydroxylation.
See also Suppl Fig. S6 for the mutation burdens in ATE1, UBRs and VHL genes in tumor samples.
Since past reports suggest that arginylation is promoted by oxidation(Zhang et al., 1998, Hu et al., 2005, Lee et al., 2005, Tasaki et al., 2012), we next tested if arginylation can be influenced by proline-hydroxylation (a form of oxidation). We examined the effects of the PAPG mutation on the arginylation-eligible E-HIF1α and found it indeed led to stabilization (Fig.6D). To validate this on endogenous HIF1α, we again used the custom-produced arginylation-specific antibody for HIF1α (Fig.4D) and found its signal in WT MEF is significantly reduced by a treatment of hypoxia (0.5% oxygen) compared to normoxia (Fig.6E). To further validate the effect of arginylation on HIF1α is dependent on the action of PHD, we performed an in-lysate degradation assay of the arginylation-eligible E-HIF1α (with a C-terminal HA tag) expressed in ATE1-KO MEF. In agreement with previous results, we found that the addition of purified recombinant ATE1 protein to the cell lysate accelerated the degradation of the HA-tagged HIF1α (Fig. 6F). However, the application of DMOG, an inhibitor of PHD family protein, significantly stabilized HIF1α both in the absence or presence of ATE1 (Fig.6F). Thus, ATE1-mediated arginylation of HIF1α is promoted by PHD-mediated hydroxylation with a sensitivity to the oxygen level.
To further understand the relationship between the ATE1-Ubr and the pVHL pathways in oxygen-sensing in vivo, we analyzed databases for mutation burdens of these genes in human cancers that commonly involve dysregulations of hypoxic signaling and metabolism(Majmundar et al., 2010). Notably, we found strong tissue specificities and a mutual complementation for these two pathways. In most examined solid tumor tissues, the UBR family and ATE1 appear to be more frequently dysregulated than VHL, while an opposite was observed in kidney cancers (Suppl Fig. S6).
The ATE1-dependent pathway emerged earlier than pVHL in molecular evolution
The need for oxygen sensing in eukaryotes likely arose during the acquisition of mitochondria, when the earth’s atmosphere transitioned from reductive to oxidative(Williams et al., 2013). Using phylogenetic analyses of the genes coding for PHD, ATE1, HIF1α, Ubr, and pVHL, we found that key functional domains of ATE1 and PHD exist in both eukaryotes and prokaryotes, while those of Ubr, HIF1α, and pVHL are predominantly present in eukaryotes (Fig.7A). Notably, the orthologs/homologs of ATE1 and PHD can be found in the class of alpha-proteobacteria including Rickettsiales: the extant relatives of the mitochondrion ancestor (Fig.7B). These data suggest that eukaryotic ATE1, together with PHD, were likely transferred from mitochondria to the nuclear genome similar to many mitochondrion-associated genes(Gray, 2012). The Ubr proteins appear to exist in all eukaryotes, suggesting the conservation of the arginylation-dependent degradation pathway. In contrast, the essential domain of pVHL only exists within metazoans. Furthermore, while the distribution of HIF1α ortholog/homologs largely overlap with pVHL, several prominent exceptions can be found. Particularly, in the phylum of Porifera – likely relative of the ancestor of most modern metazoans –, orthologs/homologs can be found for ATE1, PHD, Ubr, and HIF1α, but not for pVHL (Fig.7B). Similarly, certain crustaceans such as the Copepods appear to have a lateral loss of pVHL(Graham and Barreto, 2019), while retaining the other 4 players (Fig. 7B). Although the lack of pVHL in these organisms may be due to other reasons, including potential lower needs for oxygen sensing(Mills et al., 2018), these data nevertheless suggest that the regulation of HIF1α may not be solely dependent on pVHL. Additionally, the HIF1α proteins identified in Porifera and Copepods both contain potential arginylation-eligible residues at their N-termini (Suppl Fig. S7A and S7B).
Figure 7. Phylogenetic analysis of the amino acid sequences of ATE1, PHD, HIF1α, pVHL, and Ubr among different species.
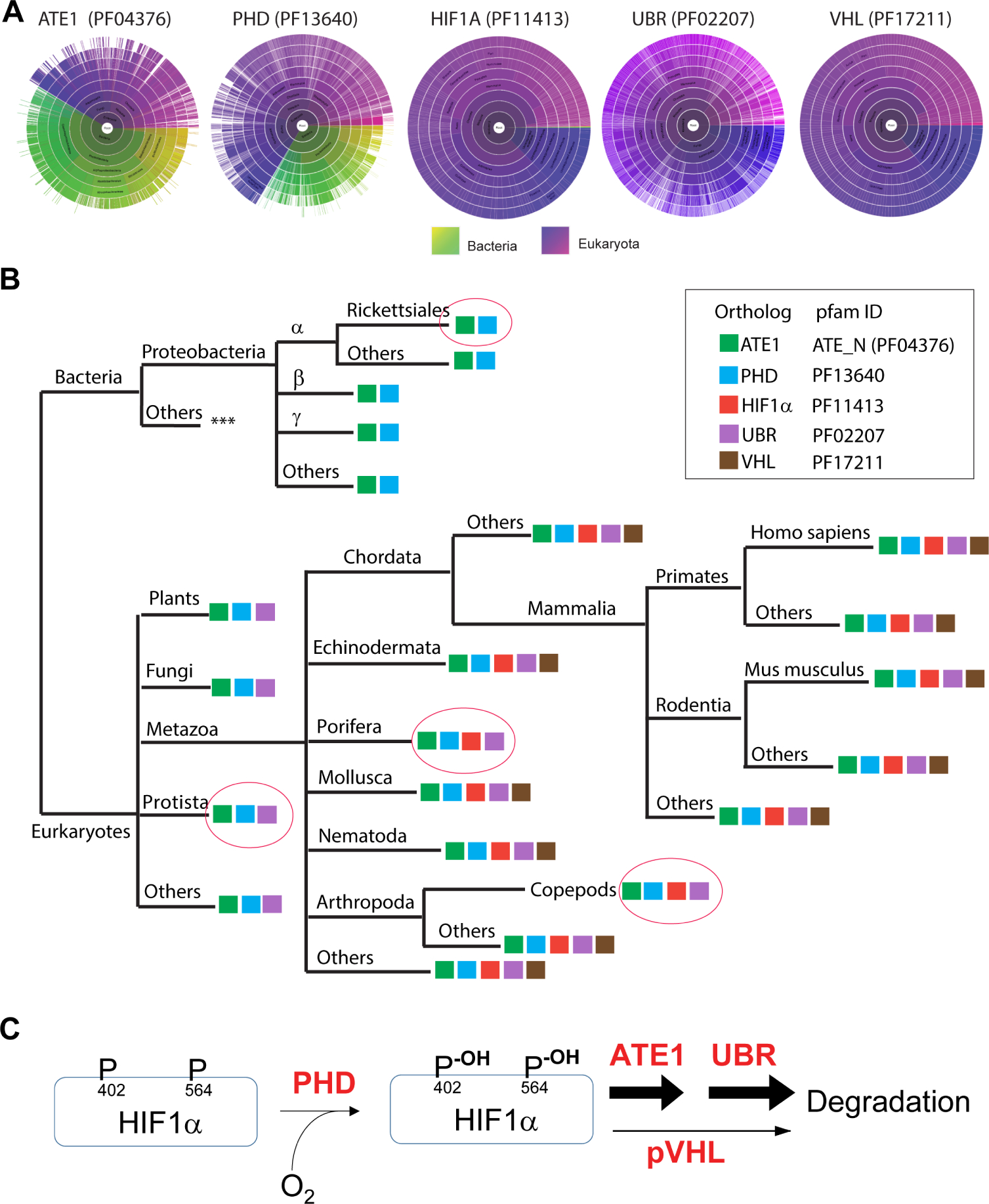
A) Sunburst graph showing the distribution of essential domains of ATE1, PHD, HIF1α, Ubr, and pVHL among eukaryotes and/or bacteria (indicated by color keys). For ATE1, the ATE-N domain (Pfam ID: 04376), which was known to be essential for its arginylation function(Kumar et al., 2016), was used as the representative. For PHD, the 2OG-Fe(II) oxygenase superfamily domain (Pfam ID: 13640), essential for catalyzing oxidation, was used. For HIF1α, Pfam ID 11413 was used. For VHL, the VHL box domain (VHL-C, Pfam ID: 17211) – required for the binding of cullin 2 protein, and thus for the ubiquitination of HIF1α – was used. For UBR, the zf-UBR domain (Pfam ID: PF02207), which is conserved among Ubr family, was used.
B) The presence of key Pfam domains of ATE1, PHD, HIF1α, pVHL, and Ubr (as indicated in the box) among different species are shown in a simplified evolution tree. Pink circles highlight several important spots. Among these, the Rickettsiales order of the alphaproteobacterial class – considered to be an extant relative to the ancestor of mitochondria – contains orthologs of ATE1 and PHD (e.g., GenBank: MBO88368.1 and GenBank: MBB20576.1, respectively, in https://www.ncbi.nlm.nih.gov/protein). Protista – considered a close relative to the ancestor of eukaryotes – contains ATE1, PHD and UBR. Further evidence can also be seen in Naegleria gruberi (Amoeba/Protista), where ATE1 (UniprotKB D2V1S2), PHD (UniprotKB D2V646), and Ubr (UniProtKB - D2V5N7) are found. Porifera is considered a close to the ancestor of metazoans, but it appears to lack pVHL. As an example, in Amphimedon queenslandica (sponge/Porifera), ATE1 (UniprotKB- A0A1X7UG82), PHD (UniprotKB- A0A1X7V6W1), HIF1α-like [NCBI accession no. XP_011403284.1], and Ubr (UniProtKB - A0A1X7UYL9) are found, but not for pVHL. A lateral loss of pVHL was also observed in Copepods (a branch of Arthropoda). As an example, in Eurytemora affinis (Copepods), we found ATE1 (NCBI accession no. XP_023347556.1), PHD (NCBI accession no. XP_023332905.1), HIF1A-like (NCBI accession no. XP_023346906.1), and Ubr (NCBI accession no: XP_023321273.1) but not pVHL. See Suppl Fig. S7A and S7B for sequences of the HIF1α-like proteins in Porifera and Copepods with putative arginylation-eligible residues. See also the figure legend of Suppl Fig. S7A and S7B for further details of the lack of VHL proteins in Protista, Porifera and Copepods.
C) The data in this study suggest that ATE1 acts downstream of PHD and parallel to pVHL for the degradation of HIF1α during oxygen sensing in mammalian cells.
DISCUSSION
Our study uncovers the ATE1/arginylation as a parallel mechanism to the canonical pVHL pathway in regulating HIF1α for oxygen sensing in mammalian cells (Fig. 7C).
Our data suggest that ATE1 likely co-evolved with mitochondria to meet the emerging needs for oxygen sensing. The broad variety of arginylation substrates(Wang et al., 2018) may allow different kingdoms to build their own strategies during separate evolutions. For example, while HIF1α is the key player in oxygen sensing in animals, green plants use a different set of proteins (eg., ERF-transcription factors) to perform similar tasks; albeit, in both cases, they are affected by arginylation(Sasidharan and Mustroph, 2011, Gibbs et al., 2011, Licausi et al., 2011).
The parallel relationship between ATE1-UBR and VHL in regulating HIF1α may explain their similar phenotypes in embryogenesis and vascularization(Kwon et al., 2002, Gnarra et al., 1997). Considering their difference in molecular evolution, it is possible that the ATE1-UBR pathway serves as a more ancient or baseline regulatory mechanism, while pVHL may have evolved laterally to accommodate unique oxygen-sensing demands in metazoans (such as the development of the circulation system, muscle, or kidney). Also of note, certain isoforms of pVHL carry N-terminal residues eligible for arginylation, although this possibility awaits closer investigation. The role of the ATE1-dependent pathway in oxygen sensing also provide a different perspective for interpreting its role in physiological or diseased conditions, which may include the cancer Warburg effect.
As an estimate of the impact that ATE1 has on HIF1α turnover, we found that ATE1-KO cells prolonged the half-life of HIF1α by at least 3–4 folds (given an unchanged hydroxylation level). A related (but separate) question is the ratio of the arginylated fraction among endogenous HIF1α: a question hard to answer due to the strong degradation-promoting effects of arginylation. In past studies, such a measurement was only performed on β-actin, an abundant protein that is not rapidly degraded by arginylation(Karakozova et al., 2006). Further investigation, hopefully powered by new and better techniques, would be needed to further address related questions.
Past studies concerning arginylation and oxidation were focused on the N-terminal cysteine(Hu et al., 2005, Lee et al., 2005, Hu et al., 2008, Masson et al., 2019, Licausi et al., 2011); our data suggests that the hydroxylation of mid-chain residues (proline, in this case) also influences arginylation. Consistent with our data, the arginylation of BSA or insulin A-chain were found enhanced by oxidation(Zhang et al., 1998, Ingoglia et al., 2000). Since these proteins contain N-terminal D or G and cannot be further oxidized (in aqueous solutions), mid-chain oxidation is likely involved. Potential explanations may include a direct sensing of the oxidized residues by ATE1 or its accessory proteins(Wang et al., 2011, Brower et al., 2014).
ATE1 may affect oxygen sensing in animals by other mechanisms. For instance, many ATE1 proteins contain Heme Regulatory Motifs (HRMs)(Yi and Ragsdale, 2007, Soto et al., 2012), which may sense the balance between hemin (Fe3+-heme) and heme (Fe2+)(Hu et al., 2008): a process affected by oxygen levels in the chronic adaptation phase. Also, the oxidation and subsequential arginylation of Cys in proteins such as RGS can be viewed as a branch of the oxygen-sensing pathway(Davydov and Varshavsky, 2000, Lee et al., 2005, Masson et al., 2019). Furthermore, ATE1 regulates mitochondrial chain complexes II (a.k.a. succinate dehydrogenase) and affects the level of succinate(Jiang et al., 2020), which can inhibit PHD enzymes(Majmundar et al., 2010). While this mechanism is not likely the cause of defects in oxygen-sensing in ATE1-KO cells (because of the unaltered HIF1α hydroxylation level), succinate may still impact metabolism through other mechanisms such as protein succinylation(Yang et al., 2021). In addition, the “moonlighting” function of ATE1 in mitochondria(Jiang et al., 2020) is likely separate from its role in the ubiquitin-proteasome system. For the upstream regulators of arginylation (besides PHD), other oxygen-sensing enzymes such as thiol oxidases can also facilitate arginylation of substrate proteins in animals(Masson et al., 2019). Likewise, in addition to HIF1α, ATE1 may regulate other proteins relevant to metabolism and/or hypoxic response(Decca et al., 2006, Wong et al., 2007). For example, ATE1 affects phosphoribosyl pyrophosphate synthetase: a regulator of purine synthesis(Zhang et al., 2015) that connects to glycolytic activity by the pentose shunt pathway(Ge et al., 2020). Finally, the HIF family of proteins includes additional members such as HIF2α and HIF3α. While they contain interacting domains with pVHL, they appear unfavorable as arginylation substrates based on their sequences. Consistent with this possibility (and in contrast to HIF1α), we found that HIF2α is significantly downregulated in ATE1-KO cells (Suppl Fig. S7C).
Limitations of Study:
This study was conducted in cultured cell lines and cancer samples with limited numbers. No validation has been performed in animal models or non-diseased human tissues.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Fangliang Zhang (fzhang2@miami.edu).
Materials Availability
All materials generated in this study are available from the lead contact without restriction.
Data and Code Availability
Data: Original data reported in this paper is available from the lead contact upon request
Code: N/A
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cells culture
Immortalized mouse embryonic fibroblasts (MEF) that are either wild-type or genomic knockout for ATE1 (ATE1-KO) were gifts from Dr. Anna Kashina (University of Pennsylvania), prepared as described elsewhere (Zhang et al., 2012). The human embryonic kidney cell line (HEK 293T; clone T7) was obtained from ATCC. Human foreskin fibroblasts (HFF) were gifts from Dr. John M. Murray (Indiana University at Bloomington). The human prostate cancer cell lines PC-3 and PC3-ML were gifts from Dr. Kerry Burnstein (University of Miami). Renal carcinoma cell line (UOK 111) deficient in pVHL pathway was a gift from Dr. W. Marston Linehan (National Cancer Institute, Urologic Oncology Branch).
For routine growth and maintenance, unless otherwise indicated, mammalian cells were cultured in media containing high-glucose DMEM supplemented with 1mM pyruvate and glutamine (Gibco, Cat# 10569) with 10% FBS (HyClone, Cat# SH30910.03). To minimize interference with cellular metabolism, no antibiotics were used for cell culture, and the cultured cells were periodically checked for bacterial or mycoplasma contamination.
For cultures under normoxia, the cells were cultured in a 5% CO2 incubator at 37 °C, unless otherwise indicated. At this condition, the concentration of oxygen in the culture apparatus is considered as 18%. To minimize any effects of contact inhibition or changes in cell cycle phase distributions in MEF or HFF, only actively growing cells in culture density of less than 50% confluency were used for any test in this study, unless otherwise indicated. As reported previously, the WT and ATE1-KO MEF have similar cell cycle distributions under such a culture condition(Rai et al., 2015).
For the treatment of hypoxia, the cells were placed in the O2 Control InVitro Glove Box (COY Lab Products, Inc) where the oxygen of desired concentrations was maintained with 5% CO2 at 37°C.
METHOD DETAILS
Cell counting and cell size measurement
The numbers of resuspended cells in solution were counted with an automated Bio-Rad TC20 cell counter (Cat#1450102) and dead cells were excluded with trypan blue, unless otherwise indicated. The diameters of the resuspended cells were also measured by the built-in feature of the cell counter. All peaks of size distributions and the associated cell numbers displayed by the counters were used to calculate the average diameter of the cell.
Cell viability - growth curve analysis
Cells of interest were seeded with the starting density of 1×105, were cultured in media containing high-glucose DMEM supplemented with 1mM pyruvate and glutamine (Gibco, Cat# 10569) and 10% FBS (HyClone, Cat# SH30910.03), and allowed to adhere for 6 hrs at 37 °C. For the normoxia condition, the cells were cultured in a 5% CO2 incubator at 37 °C, whereas for the hypoxia condition, the cells were placed in the O2 Control InVitro Glove Box (COY Lab products) where 0.5% oxygen was maintained with 5% CO2 at 37°C. The number of live cells was counted every 24 hrs by using trypan blue (Bio-Rad, Cat#1450021) and TC20 automated cell counter (Bio-Rad, Cat# 1450102)
Antibodies
Primary antibodies used in this study are (unless otherwise indicated): rat anti-ATE1 (EMD-Millipore, Billerica, MA, Cat# MABS436, clone 6F11), anti-beta-actin (Sigma-Aldrich, St. Louis, MO, Cat# A1978), mouse anti-beta-tubulin (Sigma-Aldrich, St. Louis, MO, Cat# T5201), mouse anti GFP (Roche Diagnostics, Indianapolis, IN, Cat# 11814460001), mouse anti-HA (ThermoFisher, Rockford, IL, Cat#26183), rabbit anti HIF-1α (Abcam, Cambridge, MA, Cat# ab179483; used in most shown images), mouse anti-HIF1α (R&D systems, Cat# MAB1536; only shown in Fig. 2B), rabbit anti-HIF2a (Cell Signaling, Danvers, MA, Cat # 59973), rabbit anti hydroxyl-HIF1α (recognizing hydroxylated Pro564 in human or Pro577 in mouse HIF1α, respectively) (Cell Signaling, Danvers, MA, Cat# 3434; used in Fig.6C and Suppl Fig. S5A), rabbit HIF1-alpha hydroxy 564/577 antibody (Rockland, Limerick, PA, Cat# 100–401-A25; used in Suppl Fig.S5B); mouse anti-PHD2 (Santa Cruz Biotechnology, Dallas, TX, Cat# SC271835), rabbit anti-VHL (ThermoFisher Scientific, Rockford, IL, Cat#PA5–27322), mouse anti-Ubr1 (Santa cruz, Dallas, TX, Cat#sc515753).
Secondary antibodies used in this study are (unless otherwise indicated): Anti-mouse-HRP (Pierce (now Thermos Scientific) Cat # 31430), Anti-rabbit-HRP (Thermo Fisher, Cat # 65–6120), Anti-Rat-HRP (BioLegand, Cat # 405405).
The arginylation-specific antibody for HIF1α was custom ordered from GenScript USA Inc (Piscataway, NJ) using the PTM-specific protocol in the vendor for polyclonal antibody production. In brief, a synthetic peptide representing the N-terminal sequence of arginylated mouse-HIF1α was produced in the sequence of REGAGGENEKKKMSC (an extra C-terminal cysteine was added for conjugation to KLH protein). The peptide was then conjugated to KLH protein for immunization of rabbits for three rounds. The anti-serum was then cross-absorbed with the non-arginylated synthetic peptide EGAGGENEKKKMSC. ELISA and dot-blot assays were used to validate the specificity of the resulting serum to the arginylated-peptide, before antibody purification was performed.
SDS-PAGE and immunoblot/Western Blot
Cell lysate or protein samples were generally prepared in SDS-loading buffer and boiled for 10 minutes for denaturing. For analysis of membrane-associated proteins, samples were first dissolved in 8M urea/PBS, after which SDS-loading buffer was added to the sample and subsequently denatured at 55°C for 10 minutes. The protein lysates were either used fresh or snap-frozen in liquid nitrogen and stored in −80°C freezer. The proteins were separated by electrophoresis in 4–20% or 10% SDS-PAGE (unless otherwise indicated) as needed. The proteins were then transferred to nitrocellulose membranes (LI-COR, Cat# 926–31092) for Western Blot analysis. The protein bands were examined with Chemifluorescence visualization utilizing the HRP conjugated on secondary antibodies and reagents provided in the BM Chemifluorescence Western Blotting Kit Mouse/rabbit (Roche) or the SuperSignal West Femto Chemiluminescence Kit (Thermo Fisher, Cat# 34096). Throughout this study (except for Suppl Fig. S1C), the chemiluminescent signals in immunoblots were documented by GE Amersham Imager model 600, which automatically masks saturated signal with bright pink color. For images acquired by the GE imager, an ImageQuant TL software pack (v8.1) and its 1D gel analysis module were used to quantify the intensity of signals. On the single occasion as shown in Suppl Fig.S1C, the signal of immunoblots was documented on film (from Denville). The films were scanned by an Epson Perfection 2400 photo/film scanner with at least 1200dpi resolution to convert into digital forms.
Throughout this study, unless otherwise indicated, when quantifying the level of total HIF1α (with or without PTM), all signals and bands detected by the corresponding antibodies at or exceeding the molecular weight of 100kD (the expected size of full-length HIF1α, slice variant 1) were included in measurements. In the case of UOK111 cells, we noticed that the HIF1α antibody from Abcam (Cat# ab179483) detected the main signal around 75 kDa – 85 kDa. This molecular weight is consistent to the splice variant 2 of HIF1a, which skips residues 736–826 without affecting the N-terminal region (Uniprot Identifier Q16665–2). As such in this case the measurement include all bands at or exceeding 75 kDa.
Throughout this study unless otherwise indicated, to use the same membrane for different antibodies such as loading controls, the signal of the previous antibody on the processed membrane was removed by Restore™ Western Blot Stripping Buffer (Thermo-Scientific, Cat# 21059), followed by cleaning with dH2O and PBST for three times. The cleaned membrane is then processed with the antibody incubation and detection procedures as described above.
Antibody array
Proteins from mammalian cells were extracted and then analyzed with the Proteome Profiler Human XL Oncology antibody array kit (R&D Systems), which targets 86 candidate proteins that are commonly associated with cancer metabolism and/or progression. The steady-state levels of each protein were normalized to loading controls on the same slide of the array.
Molecular cloning and preparation of plasmids
The plasmid carrying the coding sequence for mouse HIF1α (mHIF1α) [NM_010431.2] was obtained from Vectorbuilder.
All PCRs were performed with high-fidelity polymerases including Herculase and Pfu Turbo (from Agilent).
For cloning of the C-terminal 6XHis tagged mHIF1α, the following primers were used for PCR
Hif1-NotI-F
ATTGATCCGCGGCCGCATGGAGGGCGCCGGCGGCGAGAACGAGAAG
Hif1-BamH-6HIS-EcoR-R
GGCGGAATTCTCAGTGATGGTGATGGTGATGGGATCCGTTAACTTGATCCAAAGCTCTG
The PCR product was cloned into pQC-XIG vector (Addgene, Plasmid #26286) with restriction sites NotI and BamHI.
For cloning of mHIF1α starting with different versions of N-terminal amino acids (M, E, R, G) and a C-terminal GFP, the following primers were used:
Forward primers:
SacII-M-mHIF1av1
TTAACCGCGGTGGTATGGAGGGCGCCGGCGGCGAGAACGAGAAg
SacII-E-mHIF1av1
TTAACCGCGGTGGTGAGGGCGCCGGCGGCGAGAACGAGAAG
SacII-R-mHIF1av1
TTAACCGCGGTGGTAGAGAGGGCGCCGGCGGCGAGAACGAGAAG
SacII-G-mHIF1av1
TTAACCGCGGTGGTGGCGCCGGCGGCGAGAACGAGAAG
Reverse primer:
mHIF1av1-BamH1-R
TTAAGGATCCCGTTAACTTGATCCAAAGCTCTGAGTAATTC
The PCR product was then digested by SacII and BamHI and then inserted to a template vector pUB-GFP containing an N-terminal ubiquitin, a linker (nucleotide sequence – gggatccaccggcctgcttctggctct, coding for G-I-H-R-P-A-S-G-S), and a C-terminal GFP as described previously(Zhang et al., 2010).
For generating pQC-link-HA-XIG vector, the original pQC-XIG vector was modified by removing SacII restriction site for compatibility in cloning. The nucleotides were ordered from GenScript Inc. and cloned into pQC-XIG using Xba1 and Not1 restriction sites. In the final product, the HIF1α proteins were followed by a linker region (amino acid sequence G-I-L-S-G) and 2xHA tag on the C-terminus.
The mutations of P402 and P577 on mouse HIF1α (corresponding to P402 and P564 in human HIF1α) to A and G, respectively, were generated by two-step PCR method using the following primers:
Hif1A P402-F cactctgctggctGcagctgccggcga
Hif1A P402-R tcgccggcagctgCagccagcagagtg
Hif1A P577-F ggagatgctggctGGctatatcccaatg
Hif1A P577-R cattgggatatagCCagccagcatctcc
Ub Xho1-F TATATCTCGAGatgcaaattttcgtcaagactttg
Hif1 BamH1-R TTAAGGATCCcgttaacttgatccaaagctctgagtaat
The PCR products were then cloned into pQC-link-HA-XIG using Xho1 and BamH1 restriction sites.
The mutation of C2325S in mouse ATE1.2-GFP were generated by two-step PCR method using pBabe-ATE1.2 as the template and with the following primers:
NgoMIV-Kozak-F taatgccggccggatcagccgccaccatg
SalI-GFP-R atatgtcgacggcgcgccttacttgtacagctcgtccatg
C23–25-F cctccttccagtctggctactccaagaacaagttgggcagtcgc
C23–25-R gttcttggagtagccagactggaaggaggtctggccctc
The PCR product was then cloned into pBABE-puro vector using NgoMIV and SalI restriction sites.
The identities of all DNA constructs were verified by Sanger sequencing services from Eurofins Genomics USA (Louisville, KY, USA) or Genewiz (South Plainfield, NJ, USA).
RNA isolation and quantification
RNA was isolated from cells using Quick-RNA MiniPrep Kit (Genesee Scientific, Cat #: 11–328). cDNA was prepared by using Superscript First-strand synthesis system for Reverse-transcription (RT)-PCR (Invitrogen, Cat#: 11904–018) according to the instructions manual. Real-time quantitative-PCR (q-PCR) was carried out using SsoAdvanced™ Universal SYBR® Green Supermix (Biorad, Cat#: 1725271) and CFX Connect Real-Time PCR machine (Biorad). Following the MIQE (minimum information for publication of quantitative real-time PCR experiments) guidelines, we report that the samples were run as three or more replicates on Hard- Shell PCR 96 well plates from Biorad (Cat#: HSP9601). PCR conditions: initial denaturation for 30 sec at 95°C, followed by 40 cycles of 95°C for 15 seconds and 58°C for 1 minute. After each run, a melting curve was added to confirm the specificity of the amplification. The relative expression of the genes was calculated by the delta Ct method.
The primer pairs used for mouse cDNA were:
HIF1A qPCR-F - CCACAGGACAGTACAGGATG
HIF1A qPCR-R - TCAAGTCGTGCTGAATAATACC
Gapdh qPCR-F – CTGAGGACCAGGTTGTCTCC
Gapdh qPCR-R – GCCTCTCTTGCTCAGTGTCC
mEPO qPCR_F – CATCTGCGACAGTCGAGTTCTG
mEPO qPCR_R – CACAACCCATCGTGACATTTTC
PFKFB3 qPCR_F – TTGTGGCCTCCAACATCAT
PFKFB3 qPCR_R – TCCATGGCTTCTGCTGAGTT
18s rRNA qPCR_F – CGGCTACCACATCCAAGGAA
18s rRNA qPCR_R – GCTGGAATTACCGCGGCT
β-actin (ACTB)_qPCR_F – CAGCTGAGAGGGAAATCGTG
β-actin (ACTB)_qPCR_R - CGTTGCCAATAGTGATGACC
HK1-qPCR_F - GCGTGGACGGGACGCTCTAC
HK1_qPCR_R - CCTTCACTGTTTGGTGCATGATTC
Serpine_qPCR_F - AGGATCGAGGTAAACGAGAGC
Serpine_qPCR_R- GCGGGCTGAGATGACAAA
mVEGFA_Ori_qPCR_F - CTGCTGTAACGATGAAGCCCTG
mVEGFA_Ori_qPCR_R - GCTGTAGGAAGCTCATCTCTCC
The primer pairs used for human cDNA were:
h_Hif1_qPCR_F – CGCATCTTGATAAGGCCTC
h_Hif1_qPCR_R - AATCACCAGCATCCAGAAG
hHK1_qPCR_F – CTGCTGGTGAAAATCCGTAGTGG
hHK1_qPCR_R – GTCCAAGAAGTCAGAGATGCAGG
hPFKFB3_F – GGCAGGAGAATGTGCTGGTCAT
hPFKFB3_R – CATAAGCGACAGGCGTCAGTTTC
hACTB_qPCR_F – CACCATTGGCAATGAGCGGTTC
hACTB_qPCR_R – AGGTCTTTGCGGATGTCCACGT
HPRT1_qPCR_F- CATTATGCTGAGGATTTGGAAAGG
HPRT1_qPCR_R - CTTGAGCACACAGAGGGCTACA
Gene knockdown with shRNA
Lentiviral vectors carrying specific shRNA sequences or the non-targeting sequence were constructed by VectorBuilder with the pLV backbone, U6 promoter, and mCherryFP or GFP as selection markers. The specific sequence for mouse ATE1, human ATE1, mouse HIF1α and mouse UBR1 are: GCCATGCCTTACGGTGTTTAT, GATGACATCAAAGAGAGTTTA, GCCGCTCAATTTATGAATATT, and TTACCAGAGAGGAGGTTATAA, respectively. The sequence for non-silencing/non-targeting shRNA was CCTAAGGTTAAGTCGCCCTCG.
These plasmids and the VSV.g and Δ8.2 lentivirus packaging vectors were used to co-transfect HEK293T cells for virus production as described in published protocols(Kumar et al., 2016, Birnbaum et al., 2019). After 8 hours, the plasmid-containing media is replaced with fresh culture media. The supernatant containing viruses was collected at 24, 48, and 72-hour time-points, and filtered through a 0.45μM Syringe filter (Olympus, #25–246). The supernatant was then used to transduce MEF or UOK111 cells, aided by 10μg/mL polybrene. After transduction, cells expressing the red fluorescence were enriched by fluorescence-activated cell sorting (FACS). Once the selected cell line was stabilized, a western blot was run on a cell lysate to examine specific changes in protein levels.
Preparation of cells stably expressing recombinant ATE1 proteins
ATE1-KO MEF stably transfected with ATE1–2-GFP, ATE1–2GFP carrying C23S-C25S mutation, or GFP alone were generated as described previously by retroviral delivery(Zhang et al., 2015), except that cells were enriched with FACS without puromycin selection. Briefly, HEK293T cells were used to produce retrovirus with the low-expression pBabe-Puro vectors carrying coding sequence for ATE1 or GFP, with the assistance of GAG-Pol and VSV-G vectors(Stewart et al., 2003). The viruses were then allowed to infect ATE1-KO MEF in the presence of 10 mg/mL polybrene. Successfully transfected MEF were enriched by fluorescence sorting.
Analysis of cellular metabolism profile by the Seahorse analyzer
Metabolic analyses of MEF (ATE1-Knockdown or mock knockdown control with non-silencing shRNA) were performed in an Agilent Seahorse XFe24 Analyzer with corresponding assay kits and reagents from the same manufacturer, which include the XF Glycolysis Stress Test Kit (Agilent, Cat#103020–100) and XF Base Medium (Agilent, Cat# 102353–100). The measurement for extracellular acidification rate (ECAR) of the cells was performed using the protocols attached with the kit, except for these optimized conditions: 35,000 cells were seeded in each well for the 24well-plate on the first day. On the second day, the loading order and concentrations for key test reagents are as below: port A: glucose, 15.5 mM; port B: oligomycin, 2.0μM; port C: 2-DG, 50mM. At the end of the experiments, cell numbers in each well are counted for normalization. The glycolysis activity and glycolytic capacity are calculated using the built-in function of the analysis software of the XF Glycolysis Stress Test Report Generator V2 that were provided with the instrument.
Measurement of ATP level in the cells
The concentration of ATP inside the cells was measured with a thermostable luciferase-based CellTiter-Glo Luminescent assay (from Promega, Cat # G7575) according to the manufacturer’s protocol. In brief, cells were harvested by trypsin digestion, washed, and resuspended in DMEM medium (Gibco, Cat# 10569). The number of cells was adjusted to 8×105live cell/mL by using trypan blue to exclude dead cells. The cell suspension for different types of cells is then loaded into 96-well plates with an opaque wall. DMEM medium is also loaded as blank control. Solutions with known ATP concentrations dissolved in DMEM medium were used to prepare the standard curve. Each well is then added with an equal volume of premixed CellTiter-Glo Reagent, and the luminescence of the plate is promptly examined by the luminescence module in a plate reader (FLUOstar Omega from BMG Labtech, Ortenberg, Germany).
Glucose starvation assay
MEF cells (WT or ATE1-KO) were cultured in high-glucose DMEM containing 25mM glucose and 1mM pyruvate (Gibco Cat#10569) supplemented with 5% FBS (Hyclone, Cat# SH30910.03) for several generations. Right before the experiment, the cells were split and cultured for one generation (24 hours) in the same high-glucose DMEM except with 5% dialyzed FBS (life technologies 26400–044). At the time of the experiment, the cells were trypsinized, washed with dPBS, and then resuspended in either starving media with glucose-free, pyruvate-free DMEM (Gibco, Cat#11966) and 5% dialyzed FBS, or non-starving media with high-glucose, 1mM pyruvate DMEM (Gibco Cat#10569) and 5% dialyzed FBS. The cells were then inoculated into 6-cm culture dishes with 50,000 cells per dish so that the cells stay in non-confluent culture density throughout the duration of the tests. Live cells that remained attached to the plate were then counted at given time points, using trypan blue to exclude dead cells.
Glucose intake assay
Actively growing MEF cells (WT or ATE1-KO) cultured in high glucose DMEM (Life Technologies, Cat# 11995–065) with 10% FBS (HyClone, Cat# SH30910.03) were split into 6-well plates with 300,000 cells per well and allowed to grow overnight. In the next morning, the old media was removed, and the glucose-free, pyruvate-free DMEM (Gibco, Cat#11966) was added to the cells. The glucose intake test was performed with the Glucose Uptake Cell-Based Assay Kit according to the manufacturer’s protocol (Cayman, Cat#600470). In brief, 100μg/mL 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG) was added to the media. Cells at various time points were washed with assay buffer and stained with Propidium iodide. The green fluorescence of 2-NBDG in cells was then immediately analyzed on a flow cytometer by the GFP channel, and the red fluorescence of PI was used to exclude dead cells from the analysis.
Purification of recombinant ATE1 from bacterial expression systems
Escherichia coli BL21-CodonPlus harboring plasmid pET9d-ATE1 cultured in LB supplemented with 100mg/ml kanamycin and 50mg/ml chloramphenicol at 37 °C. When the optical density (OD 600nm) reached 0.4–0.5, the culture was cold-shocked on ice for 30 minutes, and then induced with 0.8 mM IPTG (isopropyl-1-thio-galactopyranoside) for 18 h at 16°C. Bacteria were collected by centrifuging at 6000rpm at 4°C for 30 min in a JA-10 rotor (or similar size) with a centrifuge bottle and the pellets were resuspended in lysis buffer (0.5 M NaCl, 1mM MgCl2, 50mm Tris pH7.5, 5 mM imidazole, with freshly added 10 mM β-mercaptoethanol, fresh 1 mM PMSF, and fresh bacterial protease inhibitor cocktails (1×100 dilution)). Lysis was carried out by sonication at level 5, 10×10s with 30s intervals on ice until the color of the bacterial mixture turn from light yellow into pale grey. The lysate was centrifuged at 20,000 ×g (16,000 rpm in the pre-chilled JA-20 rotor), for 30 minutes at 4°C. Meanwhile, Ni-NTA resin was equilibrated by passing a 5 bed-volume of lysis buffer through the column. The supernatant from the centrifugation was collected and passed through 0.45 μm filter (low protein-binding: cellular acetate or PES membrane). The equilibrated Ni-NTA agarose from the column was resuspended and added to the filtered lysate. The binding was carried out at 4°C with rotation or shaking for 0.5~1hr. The column was washed with 5 bed volume (2.5mL) of washing buffer (50mm Tris pH7.5, 1M NaCl, 1mM MgCl2, 25 mM imidazole, freshly added 10 mM β-mercaptoethanol) for 5 times and eluted with elution buffer (50mM Tris pH7.5, 0.5 M NaCl, 1 mM MgCl2, fresh added 0.5M imidazole (pH 7.5), freshly added 10 mM β-mercaptoethanol), 1 bed volume (0.5mL) each time for 5 times.
The protein concentration in the elution fractions was quantified using Bradford’s reagent (from Bio-Rad). The fractions with high protein content were collected and concentrated by using a spin column concentrator with MWCO < 20kD (such as Pierce 9K MWCO concentrator) in a swing-bucket rotor centrifuge at 4°C, ~3000×g (or according to the manufacturer’s instructions). The solution was sealed in dialysis cassettes with MWCO <20kD and dialyzed against 500ml of dialysis buffer (50 mM HEPES, pH 7.5, 1M NaCl, 0.2 mM EDTA, 2 mM MgCl2, fresh 2mM DTT,) overnight at 4°C with gentle stirring. Dialysis was repeated twice (3 hours each) with fresh dialysis buffer. The dialyzed protein was retrieved from the cassette, spun down at the maximum speed in a desktop centrifuge for 30 min at 4°C. The supernatant was collected and the protein concentration was determined by Bradford assay by using BSA standards.
In vitro arginylation and degradation assay
In vitro arginylation assay was carried out as previously described(Kumar et al., 2016). MEF ATE1-KO cells stably expressing the “E-” form of HIF1α (pQC-E-HIF1α-link-2XHA) were used. In brief, cells were pre-treated with 1mM DMOG (or DMSO as the vehicle control) for 12 hours, collected in 1.5ml Eppendorf tube, spun down in a desktop centrifuge at 5000 rpm for two minutes at room temperature, washed with 1x PBS and weighed on a scale. According to the weight of the cell pellet, it was suspended in 2x volume of ice-cold arginylation buffer (50mM Tris-cl pH 7.4, 32mM Na3PO4, 5mM MgCl2, 1m EDTA, 2.5mM ATP, 0.2mM Arginine with 0.2% NP-40) supplemented with fresh cycloheximide at a final concentration of 20μg/mL and DMOG (or DMSO as vehicle control) at a final concentration of 1mM. Recombinant ATE1 protein (1μg) or buffer control (PBS) was then added to the mixture. After that, the cells were lysed by vortexing briefly (<15 sec). Samples were spun down and placed in a water bath at 37°C to start the arginylation reaction. The reaction was terminated at different time points (0, 30, and 60-minute) by adding an equal volume of 4X SDS sample buffer and boiling for 10 minutes.
To facilitate the comparison of the degradation speeds between different conditions, the loading amounts for each condition were individually adjusted so that the level of HA-tagged HIF1α in the beginning appear to be similar across different conditions. Please note that the levels of HIF1α or tubulin cannot be cross-compared if not acquired in the same membrane due to variations in antibody treatments and image acquisitions.
Ascorbate treatment
MEF WT and ATE1-KO cells were cultured in DMEM media as described above. Once they reach ~40% confluency, both WT and ATE1-KO cells were treated with 5μM L-Ascorbic acid 2-phosphate (Sigma, Cat# A8960) for 24 hr. Cells were then harvested for protein extraction followed by Western blotting.
MG132 treatment
MG132 (Sigma, Cat# C2211) was dissolved in DMSO for a stock concentration of 20mM and stored in −80°C. Before usage, the stock solution was taken out from the freezer and thaw in room temperature. The reagent was added to the conical tube, and then dispersed by flushing it with appropriate amount of conditioned cell culture media (recovered from the cells) to a final concentration of 20μM (unless otherwise indicated). DMSO is used as a vehicle control, unless otherwise indicated. The mixture was then added to the cultured cells for the desired durations.
Post-translational degradation / cycloheximide assay
To measure the post-translational degradation dynamics of HIF1α, the cytoplasmic ribosomes were inhibited with cycloheximide to prevent the synthesis of new proteins as described before (Zhang et al., 2010, Zhang et al., 2012). Briefly, MEF cells (WT and ATE1-KO) and HEK293T cells expressing different forms of HIF1α were treated with conditioned media containing 100μg/ml cycloheximide and were collected in a series of different time points as indicated for analysis of protein levels.
HIF1α−6xHis protein purification and mass spectrometry
To purify HIF1α for protein sequence analysis, mHIF1-α was conjugated with a C-terminal 6xHis tag and cloned into a mammalian lentiviral expression vector pQC-XIP. WT and ATE1-KO MEF cell lines were stably induced as mentioned before(Zhang et al., 2015). The 6xHis-tagged HIF1α was purified as previously described(Zhang et al., 2015). In brief, stably transduced MEF cells were cultured up to 70–80% confluency, 20 μM of MG132 was added to culture media, followed by 6 hours incubation to allow accumulation on Hif1α−6xHIS. Cells were collected and lysed with 8 M urea/PBS, and the tagged HIF1α protein was purified using Ni-NTA resin and washed at least six times with alternating buffers containing 8 M urea/PBS at pH 8.0 and pH 5.6. Bound proteins were eluted with 200 mM Imidazole/PBS with 6.4M urea. The eluted proteins were then separated on SDS-PAGE and stained with colloidal Coomassie blue protocol as described on https://www.embl.de/proteomics/proteomics_services/pdf/coomassie_staining.pdf. The protein band matching the anticipated molecular weight of HIF1α was then cut from the gel and submitted for analysis of mass spectrometry.
Mass spectrometry analysis was performed at three different locations. The data presented in Fig.4C, Suppl Fig.S2E, and Suppl Fig. S3 were obtained from the Proteomics Core Facility at the University of South Florida, Tampa, FL, USA. The submitted samples were digested with trypsin in the gel and then extracted. Peptides were resuspended in H2O/0.1% formic acid for the subsequent LC-MS/MS analysis: Peptides were separated using a 75 μm × 50 cm C18 reversed-phase-HPLC column (Thermo Scientific) on an Ultimate 3000 UHPLC (Thermo Scientific) with a 60-minute gradient (2–32% ACN with 0.1% formic acid) and analyzed on a hybrid quadrupole-Orbitrap instrument (Q Exactive Plus, Thermo Fisher Scientific). Full MS survey scans were acquired at 70,000 resolution. The top 10 most abundant ions were selected for MS/MS analysis. Raw data files are processed in MaxQuant (www.maxquant.org) and searched against the current Uniprot mouse protein sequences database. Search parameters include constant modification of cysteine by carbamidomethylation and the variable modification, methionine oxidation, protein N-term acetylation, and N-term arginylation. Proteins are identified using the filtering criteria of 1% protein and peptide false discovery rate.
Data in Suppl Fig. S2F was obtained in the Proteomic Laboratory supervised by Dr. Aldrin V. Gomes at the University of California, Davis, CA, USA. The submitted samples were incubated with trypsin to digest the proteins in the gel and then the peptides were extracted from the gel. Peptides were resuspended in 2% acetonitrile/0.1% TFA for LC-MS/MS analysis. Peptides were separated on a PepMap EasySpray reverse-phase column using a Dionex Nano Ultimate 3000 UHPLC (Thermo Scientific) and analyzed on an Orbitrap Exploris 480 (Thermo Fisher Scientific). Mass spectrometry data files were analyzed with PEAKS Studio 10.6 build 20201221 using the following parameters: Fixed modifications (daltons): carbamidomethylation, 57.02, variable modifications (daltons): oxidation (M), 15.99; arginylation, 156.10; Fragment ion tolerance was set at 0.2 (daltons). All samples were searched against the current Uniprot mouse protein sequences database.
Bioinformatic analysis
The computations for co-expression correlations were performed in R (ver3.6.0, ref: R Core Team (2013). (http://www.R-project.org/). The control gene set Random-500 was selected as described in published protocols(Li et al., 2012, Soler-Oliva et al., 2017, Zhao and Liu, 2019). The mRNA data (Pan-Cancer normalized RSEM values) were obtained from https://pancanatlas.xenahubs.net.
The mutation burden in tumor tissue was analyzed by the built-in function in cBioPortal for Cancer Genomics (https://www.cbioportal.org/), which contains genomic sequencing data from multiple published sources. The results were separated by cancer study in the display.
The sunburst graphs showing the distribution of different protein domains were generated with corresponding tools from pfam.xfam.org hosted by EMBL-EBI.
The presence of the four proteins (ATE1, PHD, pVHL, and HIF1α) on the evolution tree was first established by examining the tree structure generated with corresponding tools from pfam.xfam.org hosted by EMBL-EBI. The presence of these proteins in several key species was then cross-checked by searching the presence of the corresponding key functional domains with the Blastp in NCBI website (https://blast.ncbi.nlm.nih.gov/). The database of non-redundant protein sequence (nr) was used and the name of organism was specified. For each protein, the query sequence of the key functional domain was derived from either human or mouse orthologs.
The sequence alignment was performed with T-COFFEE/M-Coffee, Version_11.00 (Version_11.00), as hosted in http://tcoffee.crg.cat and described in published reports(Di Tommaso et al., 2011). Minor adjustments on the alignment results were made manually.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of signals on immunoblots
For images acquired by the GE imager, an ImageQuant TL software pack (v8.1) and its 1D gel analysis module were used to quantify the intensity of signals.
Image processing
The raw images were processed in Adobe Photoshop (Version: 22.4.3) to choose the area of interest by “crop” and to change the dynamic range of display by the adjustment of “levels”. For the adjustment of levels, the “auto” option is used in most cases. When a manual adjustment was made, only the maximum and minimum border values were changed, while the gamma value remained “1.00”. The images and charts were assembled into figures by Adobe Illustrator (Version: 25.3.1).
Statistical analysis
Throughout this study, unless otherwise indicated, error bars represent standard error of mean (SEM) and p-values were calculated by Student’s t-test. A p-value >0.05 was considered non-significant (n.s).
Supplementary Material
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat Anti-ATE1 | EMD-Millipore | Cat# MABS436, clone 6F11 |
| Mouse anti-β-actin | Sigma Aldrich | Cat# A1978 |
| Mouse anti-GFP | Roche | Cat# 11814460001 |
| Mouse anti-HA | Thermo Fisher | Cat#26183 |
| Rabbit anti-HIF1α | Abcam | Cat# ab179483 |
| Mouse anti-HIF1α | R&D systems | Cat# MAB1536 |
| Rabbit anti-hydroxyl HIF1α | Cell Signaling | Cat# 3434 |
| Rabbit anti-hydroxyl HIF1α | Rockland | Cat# 100-401-A25 |
| Rabbit anti-VHL | Thermo Fisher | Cat#PA5-27322 |
| Rat anti-tubulin | Novus Biologicals | NB600-506SS |
| Mouse anti-tubulin | Sigma-Aldrich | Cat# T5201 |
| Anti-PHD2 | Santa Cruz | Cat# SC271835 |
| Mouse anti-UBR1 | Santa Cruz | Cat#sc515753 |
| Rabbit anti-R-HIF1 α | GenScript | Custom ordered |
| Bacterial and virus strains | ||
| TOP10 Chemically Competent E.Coli | Thermo Fisher | Cat#C404010 |
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Polyethyleneimine | Sigma-Aldrich | Cat#408727; CAS: 9002-98-6 |
| Cycloheximide | Sigma-Aldrich | Cat#01810-1G |
| L-Ascorbic acid 2-phosphate | Sigma-Aldrich | Cat# A8960 |
| Critical commercial assays | ||
| SuperSignal Femto Chemiluminescence Kit | ThermoFisher | Cat# 34096 |
| Quick-RNA Miniprep Kit | Genesse Scientific | Cat #: 11-328 |
| Superscript first-strand synthesis system for RT-PCR | Invitrogen | Cat#: 11904-018 |
| SSOAdvanced Universal SYBR green Supermix | Biorad | Cat#: 1725271 |
| XF Glycolysis Stress Test Kit | Agilent | Cat# 103020-100 |
| CellTiter-Glo Luminescent assay | Promega | Cat # G7575 |
| Glucose Uptake Cell-Based Assay Kit | Cayman | Cat#600470 |
| Restore™ Western Blot Stripping Buffer | ThermoScientific | Cat# 21059 |
| Western blot Blocking buffer | Roche | Cat#1096176001 |
| Deposited data | ||
| N/A | ||
| Experimental models: Cell lines | ||
| MEF | Gift from Dr. Anna Kashina (Kwon et al., 2002) | https://www.ncbi.nlm.nih.gov/pubmed/12098698 |
| MEF ATE1-KO | Gift from Dr. Anna Kashina (Kwon et al., 2002) | https://www.ncbi.nlm.nih.gov/pubmed/12098698 |
| HEK293T | ATCC | ATCC CRL-11268 |
| HFF | Gift from Dr. John Murray (Indiana University) | N/A |
| UOK111 | Gift from Dr. W. Marston Linehan (National Cancer Institute) | N/A |
| PC-3 and PC3-ML | Gift from Dr. Kerry Burnstein (University of Miami) | N/A |
| Experimental models: Organisms/strains | ||
| N/A | ||
| Oligonucleotides | ||
| See Supplemental Table S2 for a list of oligonucleotides. | ||
| Recombinant DNA | ||
| Delta R8.2 | N/A | Addgene 12263 |
| VSV-G | Stewart et al., 2003 | Addgene 8454 |
| pQC-XIG | N/A | Addgene 26826 |
| Mouse HIF1A coding sequence ([NM_010431.2]) | Vectorbuilder | |
| pQC-link-M-HIF1α-HA-XIG | This study | N/A |
| pQC-link-G-HIF1α-HA-XIG | This study | N/A |
| pQC-link-R-HIF1α-HA-XIG | This study | N/A |
| pQC-link-E-HIF1α-HA-XIG | This study | N/A |
| pQC-link-M-HIF1αPAPG-HA-XIG | This study | N/A |
| pQC-link-G-HIF1αPAPG-HA-XIG | This study | N/A |
| pQC-link-R-HIF1αPAPG-HA-XIG | This study | N/A |
| pQC-link-E-HIF1αPAPG-HA-XIG | This study | N/A |
| Software and algorithms | ||
| R | http://www.R-project.org/ | |
| cBioPortal for Cancer Genomics | https://www.cbioportal.org/ | |
| T-COFFEE/M-Coffee | http://tcoffee.crg.cat | |
| CFX 3.1 manager | Bio-Rad | http://www.bio-rad.com/en-us/sku/1845000-cfx-manager-software?ID=1845000 |
| FCS Express 7 | De Novo Software | https://denovosoftware.com/academic-research/ |
| ImageQuant TL software pack | Cytiva | https://www.cytivalifesciences.com/en/us/shop/molecular-biology/nucleic-acid-electrophoresis--blotting--and-detection/molecular-imaging-for-nucleic-acids/imagequant-tl-8-2-image-analysis-software-p-09518 |
| Other | ||
| N/A | ||
Highlights.
ATE1 controls glycolysis and hypoxic response in mammalian cells by affecting HIF1α
HIF1α is arginylated by ATE1 and degraded by UBRs independently of pVHL
ATE1-dependent oxygen sensing pathway evolved much earlier than the pVHL pathway
ACKNOWLEDGEMENT
The access to a Seahorse analyzer and technical supports were provided by Drs. Howard Guy, Delcroix Gaëtan, and Medhi Wangpaichitr at the University of Miami (UM). Access to hypoxia chambers was provided by Drs. Claudia Rodrigues and Roberta Soares at UM. The flow cytometer was performed in the core facility of the Sylvester Comprehensive Cancer Center. A portion of Mass Spectrometry analysis was performed by Dr. Dale Chaput in the Proteomics Core Facility at the University of South Florida.
This study is supported in part by NIGMS/NIH R01GM138557 and a University of Miami startup funding awarded to FZ. AB is supported by an NIGMS-MIRA [R35GM118141 to A.B.] and a Muscular Dystrophy Association Research Grant [MDA-381828 to A.B.].
The graphic abstract was adapted from “NFAT Signaling pathway”, by BioRender.com (2022).
Abbreviations:
- ATE1
arginyltransferase 1
- GFP
green-fluorescence protein
- HIF1α
Hypoxia-inducible factor 1α
- hrs
hours
- KO
knock-out
- LGG
low grade glioma
- MEF
mouse embryonic fibroblasts
- min
minutes
- NS
non-silencing
- n.s.
non-significant
- O.D.
Optical density
- PCPG
Pheochromocytoma and Paraganglioma
- PHD
prolyl hydroxylase
- SARC
sarcoma
- shRNA
Short Hairpin RNA
- SKCM
skin cutaneous melanoma
- TGCT
testicular germ cell tumors
- UBR
Ubiquitin Protein Ligase E3 Component N-Recognin
- VHL
von Hippel–Lindau tumor suppressor
- WT
wild-type
Footnotes
DECLARATION OF INTEREST
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AL TAMEEMI W, DALE TP, AL-JUMAILY RMK & FORSYTH NR 2019. Hypoxia-Modified Cancer Cell Metabolism. Front Cell Dev Biol, 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANGLARD P, TRAHAN E, LIU S, LATIF F, MERINO MJ, LERMAN MI, ZBAR B & LINEHAN WM 1992. Molecular and Cellular Characterization of Human Renal-Cell Carcinoma Cell-Lines. Cancer Res, 52, 348–356. [PubMed] [Google Scholar]
- BACHMAIR A, FINLEY D & VARSHAVSKY A 1986. In vivo half-life of a protein is a function of its amino-terminal residue. Science, 234, 179–86. [DOI] [PubMed] [Google Scholar]
- BALZI E, CHODER M, CHEN WN, VARSHAVSKY A & GOFFEAU A 1990. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J Biol Chem, 265, 7464–71. [PubMed] [Google Scholar]
- BENITA Y, KIKUCHI H, SMITH AD, ZHANG MQ, CHUNG DC & XAVIER RJ 2009. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res, 37, 4587–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERLETH ES, LI J, BRAUNSCHEIDEL JA & PICKART CM 1992. A reactive nucleophile proximal to vicinal thiols is an evolutionarily conserved feature in the mechanism of Arg aminoacyl-tRNA protein transferase. Arch Biochem Biophys, 298, 498–504. [DOI] [PubMed] [Google Scholar]
- BIRNBAUM MD, ZHAO N, MOORTHY BT, PATEL DM, KRYVENKO ON, HEIDMAN L, KUMAR A, MORGAN WM, BAN Y, REIS IM, CHEN X, GONZALGO ML, JORDA M, BURNSTEIN KL & ZHANG F 2019. Reduced Arginyltransferase 1 is a driver and a potential prognostic indicator of prostate cancer metastasis. Oncogene, 38, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWER CS, PIATKOV KI & VARSHAVSKY A 2013. Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol Cell, 50, 161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWER CS, ROSEN CE, JONES RH, WADAS BC, PIATKOV KI & VARSHAVSKY A 2014. Liat1, an arginyltransferase-binding protein whose evolution among primates involved changes in the numbers of its 10-residue repeats. Proc Natl Acad Sci U S A, 111, E4936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARPIO MA, LOPEZ SAMBROOKS C, DURAND ES & HALLAK ME 2010. The arginylation-dependent association of calreticulin with stress granules is regulated by calcium. Biochem J, 429, 63–72. [DOI] [PubMed] [Google Scholar]
- COX J & MANN M 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol, 26, 1367–72. [DOI] [PubMed] [Google Scholar]
- COX J, NEUHAUSER N, MICHALSKI A, SCHELTEMA RA, OLSEN JV & MANN M 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res, 10, 1794–805. [DOI] [PubMed] [Google Scholar]
- DAVYDOV IV & VARSHAVSKY A 2000. RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J Biol Chem, 275, 22931–41. [DOI] [PubMed] [Google Scholar]
- DECCA MB, BOSC C, LUCHE S, BRUGIERE S, JOB D, RABILLOUD T, GARIN J & HALLAK ME 2006. Protein arginylation in rat brain cytosol: a proteomic analysis. Neurochem Res, 31, 401–9. [DOI] [PubMed] [Google Scholar]
- DI TOMMASO P, MORETTI S, XENARIOS I, OROBITG M, MONTANYOLA A, CHANG JM, TALY JF & NOTREDAME C 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res, 39, W13–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FONG GH & TAKEDA K 2008. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ, 15, 635–41. [DOI] [PubMed] [Google Scholar]
- GE T, YANG J, ZHOU S, WANG Y, LI Y & TONG X 2020. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front Endocrinol (Lausanne), 11, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIBBS DJ, LEE SC, ISA NM, GRAMUGLIA S, FUKAO T, BASSEL GW, CORREIA CS, CORBINEAU F, THEODOULOU FL, BAILEY-SERRES J & HOLDSWORTH MJ 2011. Homeostatic response to hypoxia is regulated by the N-end rule pathway in plants. Nature, 479, 415–U172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GNARRA JR, WARD JM, PORTER FD, WAGNER JR, DEVOR DE, GRINBERG A, EMMERT-BUCK MR, WESTPHAL H, KLAUSNER RD & LINEHAN WM 1997. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A, 94, 9102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAHAM AM & BARRETO FS 2019. Loss of the HIF pathway in a widely distributed intertidal crustacean, the copepod Tigriopus californicus. Proc Natl Acad Sci U S A, 116, 12913–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY MW 2012. Mitochondrial evolution. Cold Spring Harb Perspect Biol, 4, a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU RG, SHENG J, QI X, XU Z, TAKAHASHI TT & VARSHAVSKY A 2005. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature, 437, 981–6. [DOI] [PubMed] [Google Scholar]
- HU RG, WANG H, XIA Z & VARSHAVSKY A 2008. The N-end rule pathway is a sensor of heme. Proc Natl Acad Sci U S A, 105, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG LE, GU J, SCHAU M & BUNN HF 1998. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A, 95, 7987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWANG CS, SHEMORRY A, AUERBACH D & VARSHAVSKY A 2010. The N-end rule pathway is mediated by a complex of the RING-type Ubr1 and HECT-type Ufd4 ubiquitin ligases. Nat Cell Biol, 12, 1177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INGOGLIA NA, RAMANATHAN M, ZHANG N, TZENG B, MATHUR G, OPUNI K & DONNELLY R 2000. What is the signal for the posttranslational arginylation of proteins? Neurochem Res, 25, 51–8. [DOI] [PubMed] [Google Scholar]
- IVAN M, KONDO K, YANG H, KIM W, VALIANDO J, OHH M, SALIC A, ASARA JM, LANE WS & KAELIN WG JR. 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 292, 464–8. [DOI] [PubMed] [Google Scholar]
- IWAI K, YAMANAKA K, KAMURA T, MINATO N, CONAWAY RC, CONAWAY JW, KLAUSNER RD & PAUSE A 1999. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci U S A, 96, 12436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAAKKOLA P, MOLE DR, TIAN YM, WILSON MI, GIELBERT J, GASKELL SJ, VON KRIEGSHEIM A, HEBESTREIT HF, MUKHERJI M, SCHOFIELD CJ, MAXWELL PH, PUGH CW & RATCLIFFE PJ 2001a. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science, 292, 468–72. [DOI] [PubMed] [Google Scholar]
- JAAKKOLA P, MOLE DR, TIAN YM, WILSON MI, GIELBERT J, GASKELL SJ, VON KRIEGSHEIM A, HEBESTREIT HF, MUKHERJI M, SCHOFIELD CJ, MAXWELL PH, PUGH CW & RATCLIFFE PJ 2001b. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O-2-regulated prolyl hydroxylation. Science, 292, 468–472. [DOI] [PubMed] [Google Scholar]
- JEONG JW, BAE MK, AHN MY, KIM SH, SOHN TK, BAE MH, YOO MA, SONG EJ, LEE KJ & KIM KW 2002. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell, 111, 709–20. [DOI] [PubMed] [Google Scholar]
- JIANG C, MOORTHY BT, PATEL DM, KUMAR A, MORGAN WM, ALFONSO B, HUANG J, LAMPIDIS TJ, ISOM DG, BARRIENTOS A, FONTANESI F & ZHANG F 2020. Regulation of Mitochondrial Respiratory Chain Complex Levels, Organization, and Function by Arginyltransferase 1. Front Cell Dev Biol, 8, 603688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAJI H 1968. Further studies on the soluble amino acid incorporating system from rat liver. Biochemistry, 7, 3844–50. [DOI] [PubMed] [Google Scholar]
- KAJI H, NOVELLI GD & KAJI A 1963. A Soluble Amino Acid-Incorporating System from Rat Liver. Biochim Biophys Acta, 76, 474–7. [PubMed] [Google Scholar]
- KARAKOZOVA M, KOZAK M, WONG CC, BAILEY AO, YATES JR 3RD, MOGILNER A, ZEBROSKI H & KASHINA A 2006. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science, 313, 192–6. [DOI] [PubMed] [Google Scholar]
- KOH MY, DARNAY BG & POWIS G 2008. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol, 28, 7081–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOIDO M, HAGA N, FURUNO A, TSUKAHARA S, SAKURAI J, TANI Y, SATO S & TOMIDA A 2017. Mitochondrial deficiency impairs hypoxic induction of HIF-1 transcriptional activity and retards tumor growth. Oncotarget, 8, 11841–11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMAR A, BIRNBAUM MD, PATEL DM, MORGAN WM, SINGH J, BARRIENTOS A & ZHANG F 2016. Posttranslational arginylation enzyme Ate1 affects DNA mutagenesis by regulating stress response. Cell Death Dis, 7, e2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWON YT, KASHINA AS, DAVYDOV IV, HU RG, AN JY, SEO JW, DU F & VARSHAVSKY A 2002. An essential role of N-terminal arginylation in cardiovascular development. Science, 297, 96–9. [DOI] [PubMed] [Google Scholar]
- KWON YT, KASHINA AS & VARSHAVSKY A 1999. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol, 19, 182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE MJ, TASAKI T, MOROI K, AN JY, KIMURA S, DAVYDOV IV & KWON YT 2005. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci U S A, 102, 15030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J & PICKART CM 1995a. Binding of phenylarsenoxide to Arg-tRNA protein transferase is independent of vicinal thiols. Biochemistry, 34, 15829–37. [DOI] [PubMed] [Google Scholar]
- LI J & PICKART CM 1995b. Inactivation of arginyl-tRNA protein transferase by a bifunctional arsenoxide: identification of residues proximal to the arsenoxide site. Biochemistry, 34, 139–47. [DOI] [PubMed] [Google Scholar]
- LI JY, PATTERSON M, MIKKOLA HKA, LOWRY WE & KURDISTANI SK 2012. Dynamic Distribution of Linker Histone H1.5 in Cellular Differentiation. Plos Genetics, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIBERTI MV & LOCASALE JW 2016. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci, 41, 211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LICAUSI F, KOSMACZ M, WEITS DA, GIUNTOLI B, GIORGI FM, VOESENEK LA, PERATA P & VAN DONGEN JT 2011. Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature, 479, 419–22. [DOI] [PubMed] [Google Scholar]
- LIU YV, BAEK JH, ZHANG H, DIEZ R, COLE RN & SEMENZA GL 2007. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell, 25, 207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO W, ZHONG J, CHANG R, HU H, PANDEY A & SEMENZA GL 2010. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem, 285, 3651–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAJMUNDAR AJ, WONG WJ & SIMON MC 2010. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell, 40, 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASCHEK G, SAVARAJ N, PRIEBE W, BRAUNSCHWEIGER P, HAMILTON K, TIDMARSH GF, DE YOUNG LR & LAMPIDIS TJ 2004. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res, 64, 31–4. [DOI] [PubMed] [Google Scholar]
- MASDEHORS P, GLAISNER S, MACIOROWSKI Z, MAGDELENAT H & DELIC J 2000. Ubiquitin-dependent protein processing controls radiation-induced apoptosis through the N-end rule pathway. Exp Cell Res, 257, 48–57. [DOI] [PubMed] [Google Scholar]
- MASSON N, KEELEY TP, GIUNTOLI B, WHITE MD, PUERTA ML, PERATA P, HOPKINSON RJ, FLASHMAN E, LICAUSI F & RATCLIFFE PJ 2019. Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science, 365, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLS DB, FRANCIS WR, VARGAS S, LARSEN M, ELEMANS CP, CANFIELD DE & WORHEIDE G 2018. The last common ancestor of animals lacked the HIF pathway and respired in low-oxygen environments. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHH M, PARK CW, IVAN M, HOFFMAN MA, KIM TY, HUANG LE, PAVLETICH N, CHAU V & KAELIN WG 2000. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol, 2, 423–7. [DOI] [PubMed] [Google Scholar]
- PIATKOV KI, BROWER CS & VARSHAVSKY A 2012. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci U S A, 109, E1839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAI R & KASHINA A 2005. Identification of mammalian arginyltransferases that modify a specific subset of protein substrates. Proc Natl Acad Sci U S A, 102, 10123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAI R, ZHANG F, COLAVITA K, LEU NA, KUROSAKA S, KUMAR A, BIRNBAUM MD, GYORFFY B, DONG DW, SHTUTMAN M & KASHINA A 2015. Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RATCLIFFE PJ, O’ROURKE JF, MAXWELL PH & PUGH CW 1998. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. Journal of Experimental Biology, 201, 1153–1162. [DOI] [PubMed] [Google Scholar]
- SASIDHARAN R & MUSTROPH A 2011. Plant Oxygen Sensing Is Mediated by the N-End Rule Pathway: A Milestone in Plant Anaerobiosis. Plant Cell, 23, 4173–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEMENZA GL & WANG GL 1992. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol, 12, 5447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEMENZA GL 2003. Targeting HIF-1 for cancer therapy. Nature Reviews Cancer, 3, 721–732. [DOI] [PubMed] [Google Scholar]
- SOFFER RL 1971. Enzymatic modification of proteins. 4. Arginylation of bovine thyroglobulin. J Biol Chem, 246, 1481–4. [PubMed] [Google Scholar]
- SOLER-OLIVA ME, GUERRERO-MARTINEZ JA, BACHETTI V & REYES JC 2017. Analysis of the relationship between coexpression domains and chromatin 3D organization. Plos Computational Biology, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOM P, ATKINS HL, BANDOYPADHYAY D, FOWLER JS, MACGREGOR RR, MATSUI K, OSTER ZH, SACKER DF, SHIUE CY, TURNER H, WAN CN, WOLF AP & ZABINSKI SV 1980. A fluorinated glucose analog, 2-fluoro-2-deoxy-D-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med, 21, 670–5. [PubMed] [Google Scholar]
- SOTO IC, FONTANESI F, MYERS RS, HAMEL P & BARRIENTOS A 2012. A heme-sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab, 16, 801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART SA, DYKXHOORN DM, PALLISER D, MIZUNO H, YU EY, AN DS, SABATINI DM, CHEN IS, HAHN WC, SHARP PA, WEINBERG RA & NOVINA CD 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA, 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SULTANA R, THEODORAKI MA & CAPLAN AJ 2012. UBR1 promotes protein kinase quality control and sensitizes cells to Hsp90 inhibition. Exp Cell Res, 318, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASAKI T, MULDER LC, IWAMATSU A, LEE MJ, DAVYDOV IV, VARSHAVSKY A, MUESING M & KWON YT 2005. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol, 25, 7120–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASAKI T, SRIRAM SM, PARK KS & KWON YT 2012. The N-end rule pathway. Annu Rev Biochem, 81, 261–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR CT & MCELWAIN JC 2010. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiology (Bethesda), 25, 272–9. [DOI] [PubMed] [Google Scholar]
- VAN GISBERGEN MW, OFFERMANS K, VOETS AM, LIEUWES NG, BIEMANS R, HOFFMANN RF, DUBOIS LJ & LAMBIN P 2020. Mitochondrial Dysfunction Inhibits Hypoxia-Induced HIF-1alpha Stabilization and Expression of Its Downstream Targets. Front Oncol, 10, 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARSHAVSKY A 2011. The N-end rule pathway and regulation by proteolysis. Protein Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARSHAVSKY A 2019. N-degron and C-degron pathways of protein degradation. Proc Natl Acad Sci U S A, 116, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG GL, JIANG BH, RUE EA & SEMENZA GL 1995. Hypoxia-Inducible Factor-1 Is a Basic-Helix-Loop-Helix-Pas Heterodimer Regulated by Cellular O-2 Tension. Proc Natl Acad Sci U S A, 92, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J, HAN X, SAHA S, XU T, RAI R, ZHANG F, WOLF YI, WOLFSON A, YATES JR 3RD & KASHINA A 2011. Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chem Biol, 18, 121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J, PEJAVER VR, DANN GP, WOLF MY, KELLIS M, HUANG Y, GARCIA BA, RADIVOJAC P & KASHINA A 2018. Target site specificity and in vivo complexity of the mammalian arginylome. Sci Rep, 8, 16177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE MD, KLECKER M, HOPKINSON RJ, WEITS DA, MUELLER C, NAUMANN C, O’NEILL R, WICKENS J, YANG J, BROOKS-BARTLETT JC, GARMAN EF, GROSSMANN TN, DISSMEYER N & FLASHMAN E 2017. Plant cysteine oxidases are dioxygenases that directly enable arginyl transferase-catalysed arginylation of N-end rule targets. Nat Commun, 8, 14690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS TA, FOSTER PG, COX CJ & EMBLEY TM 2013. An archaeal origin of eukaryotes supports only two primary domains of life. Nature, 504, 231–6. [DOI] [PubMed] [Google Scholar]
- WONG CC, XU T, RAI R, BAILEY AO, YATES JR 3RD, WOLF YI, ZEBROSKI H & KASHINA A 2007. Global analysis of posttranslational protein arginylation. PLoS Biol, 5, e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA Z, WEBSTER A, DU F, PIATKOV K, GHISLAIN M & VARSHAVSKY A 2008. Substrate-binding sites of UBR1, the ubiquitin ligase of the N-end rule pathway. J Biol Chem, 283, 24011–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG L, MIAO S, ZHANG J, WANG P, LIU G & WANG J 2021. The growing landscape of succinylation links metabolism and heart disease. Epigenomics, 13, 319–333. [DOI] [PubMed] [Google Scholar]
- YI L & RAGSDALE SW 2007. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J Biol Chem, 282, 21056–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG F, PATEL DM, COLAVITA K, RODIONOVA I, BUCKLEY B, SCOTT DA, KUMAR A, SHABALINA SA, SAHA S, CHERNOV M, OSTERMAN AL & KASHINA A 2015. Arginylation regulates purine nucleotide biosynthesis by enhancing the activity of phosphoribosyl pyrophosphate synthase. Nat Commun, 6, 7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG F, SAHA S & KASHINA A 2012. Arginylation-dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. J Cell Biol, 197, 819–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG F, SAHA S, SHABALINA SA & KASHINA A 2010. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science, 329, 1534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG N, DONNELLY R & INGOGLIA NA 1998. Evidence that oxidized proteins are substrates for N-terminal arginylation. Neurochem Res, 23, 1411–20. [DOI] [PubMed] [Google Scholar]
- ZHAO XH & LIU ZP 2019. Analysis of Topological Parameters of Complex Disease Genes Reveals the Importance of Location in a Biomolecular Network. Genes, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: Original data reported in this paper is available from the lead contact upon request
Code: N/A
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.