

Abstract
After lung injury, damage-associated transient progenitors (DATPs) emerge, representing a transitional state between injured epithelial cells and newly regenerated alveoli. DATPs express profibrotic genes, suggesting that they might promote idiopathic pulmonary fibrosis (IPF). However, the molecular pathways that induce and/or maintain DATPs are incompletely understood. Here we show that the bifunctional kinase/RNase—IRE1α—a central mediator of the unfolded protein response (UPR) to endoplasmic reticulum (ER) stress is a critical promoter of DATP abundance and function. Administration of a nanomolar-potent, monoselective kinase inhibitor of IRE1α (KIRA8)—or conditional epithelial IRE1α gene knockout—both reduce DATP cell number and fibrosis in the bleomycin model, indicating that IRE1α cell-autonomously promotes transition into the DATP state. IRE1α enhances the profibrotic phenotype of DATPs since KIRA8 decreases expression of integrin αvβ6, a key activator of transforming growth factor β (TGF-β) in pulmonary fibrosis, corresponding to decreased TGF-β-induced gene expression in the epithelium and decreased collagen accumulation around DATPs. Furthermore, IRE1α regulates DNA damage response (DDR) signaling, previously shown to promote the DATP phenotype, as IRE1α loss-of-function decreases H2AX phosphorylation, Cdkn1a (p21) expression, and DDR-associated secretory gene expression. Finally, KIRA8 treatment increases the differentiation of Krt19CreERT2-lineage-traced DATPs into type 1 alveolar epithelial cells after bleomycin injury, indicating that relief from IRE1α signaling enables DATPs to exit the transitional state. Thus, IRE1α coordinates a network of stress pathways that conspire to entrap injured cells in the DATP state. Pharmacological blockade of IRE1α signaling helps resolve the DATP state, thereby ameliorating fibrosis and promoting salutary lung regeneration.
Keywords: pulmonary fibrosis, IRE1α, kinase inhibitor, lung regeneration, unfolded protein response
INTRODUCTION
The lung epithelium comprises a massive surface area facing the external environment and the attendant insults. Some injuries to the lung epithelium resolve with minimal residual deficits, whereas other injuries trigger excessive extracellular matrix deposition (fibrosis). At one extreme, most survivors of severe acute lung injury have almost no detectable long-term lung function deficits after recovery, although a minority develop fibrosis with corresponding impacts on lung function and quality of life (1). At the other extreme, patients with idiopathic pulmonary fibrosis (IPF) have progressive interstitial lung fibrosis that is believed to be caused by chronic lung epithelial injury from environmental exposures interacting with genetic risk factors, ultimately leading to respiratory failure and death (2).
Several recent studies have revealed that a discrete population of progenitor cells in a “transitional” state expand dramatically after multiple types of lung injury, including bleomycin exposure, epithelial cell ablation by diphtheria toxin, and viral infection. These cells, called damage-associated transient progenitor (DATP) cells, are derived from both airway and alveolar epithelial cells; coexpress multiple cytokeratins (Krt7, Krt8, and Krt19); and are capable of transdifferentiating into mature type 1 and type 2 alveolar epithelial cells (AT1 and AT2 cells) (3–8). After bleomycin injury, these cells exhibit a dysfunctional phenotype with reduced capacity to proliferate and differentiate and instead express genes that have been associated with fibrosis. An analogous population of cells has been found in lungs of patients with IPF (5–8), and in fatal severe lung injury caused by SARS-CoV-2 infection (9–12). DATPs seem to encapsulate the tension between effective resolution of lung injury and fibrosis, and their abnormal persistence after injury may underlie pathological lung fibrosis (13, 14). However, the molecular mechanisms that control entry into the transitional state, and those that abnormally maintain it, remain poorly understood.
An intracellular signaling pathway—the unfolded protein response (UPR)—becomes activated under endoplasmic reticulum (ER) stress, and classically induces components of the ER protein-folding machinery to adaptively restore protein-folding homeostasis. But under conditions of severe, irremediable ER stress, the UPR actively promotes a continuum of maladaptive cell fate outcomes, including transdifferentiation, senescence, and apoptosis (15). These cell fate switches are controlled in part by the ER stress sensor IRE1α, an ER transmembrane protein containing an ER luminal domain that is responsive to protein-folding status in the ER. Upon sensing ER stress, IRE1α kinase domains self-associate and transautophosphorylate, which activates the RNase domain to initiate frameshift splicing of the mRNA encoding the adaptive XBP1 transcription factor through site-specific endonucleolytic cleavage. However, continuous or high-level ER stress causes IRE1α kinase/RNAse domains to also degrade a plethora of mRNAs localizing to the ER membrane and some select micro-RNAs in a process dubbed “RIDD”—regulated IRE1α-dependent decay—leading to the aforementioned maladaptive cell fate outcomes (16–18).
We previously developed small-molecule ATP-competitive kinase inhibitors to allosterically attenuate IRE1α’s RNase catalytic activity, called kinase-inhibiting RNase attenuators—KIRAs (19, 20). We showed that KIRAs could protect mice from experimental pulmonary fibrosis induced by bleomycin exposure, even when given as late as 2 wk after bleomycin exposure (21). Given these findings, we hypothesized that IRE1α might regulate the DATP phenotype. In this study, we show that maladaptive IRE1α signaling is active in DATPs. A nanomolar-potent, monoselective kinase inhibitor of IRE1α—KIRA8—decreased DATP cell number, as did conditional gene knockout of IRE1α in the epithelium. DATPs accounted for the majority of cells expressing integrin αvβ6, a critical epithelial activator of TGF-β and thus fibroblast activation in the lung. Importantly, KIRA8 treatment decreased DATP expression of Itgb6 and TGF-β-induced gene expression, explaining how IRE1α promotes fibrosis in this model. Epithelial cell-specific IRE1α knockout or KIRA8 treatment dampened DNA damage response (DDR) signaling, a pathway known to be critical to the DATP phenotype. KIRA8 treatment accelerated the differentiation of lineage-traced DATPs into alveolar type 1 (AT1) cells. Thus, IRE1α sits in the center of a network of stress signals and gene expression programs that stalls epithelial cells in a profibrotic DATP state. Blocking IRE1α signaling helps resolve the DATP phenotype, alleviating fibrosis and accelerating normal lung repair.
METHODS
Mice
All mice used were C57BL/6 mice or congenic to the C57BL/6 background. Wild-type mice were obtained from Jax (000664). Transgenic mice were ShhCreGFP (Jax 005622), RiboTag (Jax 011029), TgCsf1r-Cre (Jax 029206), Krt19CreERT2 (Jax 026925), ROSA26Ai14 (Jax 007914), Ern1flox/flox (gift of T. Iwawaki), and TgCol1a1-EGFP (gift of D. A. Brenner). All work was approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. To reduce bias, mice were grouped into cages and ear-tagged with a serial number at weaning, before genotyping. The same tag numbers were used for sample and data analysis, thus informally blinding to genotype during handling of mice, samples, and data. Littermate controls were used where possible, particularly in the conditional knockout experiments where each litter had ∼50% flox and 50% knockout mice, and ∼50% male and female mice. To induce fibrosis, 8- to 14-wk-old mice were anesthetized with either isoflurane or ketamine and xylazine and exposed to a single dose of intranasal bleomycin (3 U/kg). Mice were euthanized by open-drop isoflurane overdose and lungs were harvested at the indicated times. KIRA8 was dissolved in a vehicle consisting of 3% ethanol, 7% Tween-80, and 90% normal saline and injected peritoneally at 50 mg/kg daily starting on the day of bleomycin exposure. The anti-integrin β6 antibody 3G9 or the inert control antibody AXUM8 was dissolved in normal saline and injected peritoneally at 10 mg/kg twice a week starting on the day of bleomycin exposure. Tamoxifen was dissolved in corn oil at 20 mg/mL and administered by gavage at 200 mg/kg at the indicated time points.
For bronchoalveolar lavage (BAL) cell count and total protein, lungs were lavaged with 1 mL saline and centrifuged; cells in the pellet were counted with a hemacytometer and the total protein in the supernatant was quantified by bicinchoninic acid assay (Pierce) against a bovine serum albumin standard. For total hydroxyproline content, lungs were processed in a rotor-stator homogenizer, protein precipitated with trichloroacetic acid, and the pellet boiled in concentrated hydrochloric acid overnight. Hydroxyproline was oxidized using chloramine T and reacted with Ehrlich’s solution and color change was measured by absorbance at 550 nm against a hydroxyproline standard.
Histology
Formalin-fixed, paraffin-embedded sections were prepared by inflating lungs with zinc formalin fixative, incubating in formalin overnight followed by 70% ethanol, then paraffin embedding and sectioning. Frozen sections were prepared by inflating lungs with 1.5% paraformaldehyde in OCT, incubating in 4% paraformaldehyde for 1 h, washing in PBS for 1 h, serial flotation in 30% sucrose/PBS and 15% sucrose/OCT, embedding in OCT, and sectioning. Picrosirius red (Abcam ab150681) or TUNEL (Invitrogen C10617) staining was performed per the respective manufacturers’ instructions. RNAscope staining (ACD Bio) was performed on formalin-fixed, paraffin-embedded sections by the manufacturer using the following probes: Mm-Krt7-C1 (511808), Mm-Krt8-C2 (424521), Mm-Itgb6-C2 (312501), and Mm-Cdkn1a-C2 (408551). Antibody staining was performed using the following primary antibodies: rabbit anti-Krt8 (Abcam EPR1628Y, 1:500), rat anti-Krt8 (DSHB TROMA-I, 1:100, RRID:AB_531826), rabbit anti-γ-H2AX (R&D 4418-APC, 1:200), rat anti-RAGE (also known as Ager, R&D MAB1179, 1:100, RRID:AB_2289349), and rat anti-CD206 (Bio-Rad MCA2235GA, 1:200, RRID:AB_322613). Heat-mediated antigen retrieval was performed at 70° for 15 min in 10 mM Tris, 1 mM EDTA, 0.05% Tween, pH 9.0 for all immunofluorescence experiments, except for sections stained for γH2AX, which were heat-retrieved in 10 mM sodium citrate, pH 6.0.
For quantification, the indicated number of representative low-power fields (1,240 µm in diameter) were extracted from one section for each mouse lung and quantified for picrosirius red staining area, cell costaining, or RNA puncta using the CellProfiler package. For bright-field in situ hybridization images, colors were deconvoluted using the UnmixColors module, followed by subtraction of nonspecific staining artifacts, resulting in separation of nuclei and stained puncta into distinct channels (Supplemental Fig. S2C; see https://doi.org/10.6084/m9.figshare.17293883). Hematoxylin-stained nuclei and stained puncta were identified using the IdentifyPrimaryObjects module. Puncta were assigned to cell/nucleus objects based on proximity. H-scores were calculated according to the RNAscope manufacturer’s instructions (ACD Bio). For single-stain immunofluorescence images, DAPI-stained nuclei overlapping antibody-stained areas were identified using the IdentifyPrimaryObjects module. For identification of double-stained cells, stained objects were separately identified and considered double-stained if and only if the centroids of both stained objects were less than 10 pixels apart.
Epithelial Cell Isolation and Reverse-Transcription PCR
Epithelial single-cell suspensions were prepared as described previously (22). Magnetic nanoparticle depletion and enrichment were performed by MACS (Miltenyi). Cells were depleted for CD16/32 (Miltenyi 130-101-895) and CD45 (130-052-301), followed by positive selection for EPCAM (Miltenyi 130-101-859). Total RNA was prepared using Tri-Reagent (Ambion). For XBP1 splice isoform detection, cDNA was synthesized using the QuantiTect Reverse Transcription Kit (Qiagen), followed by PCR using the primers ACACGCTTGGGAATGGACAC and CCATGGGAAGATGTTCTGGG and separation on 4% agarose.
RiboTag Immunoprecipitation
Lungs from ShhCre Rpl22flox/flox mice were dissected and flash-frozen in liquid nitrogen within a few minutes of respiratory arrest and stored at −80°C. All immunoprecipitation steps were performed at 4°C. Lungs were thawed into 10 volumes of supplemented homogenization buffer (100 mM KCl, 50 mM Tris-Cl pH 7.4, 12 mM MgCl2, 1% NP-40, 1 mM DTT, 0.1 mg/mL cycloheximide, 1.25 mg/mL heparin, 400 U/mL NEB murine RNase inhibitor) and dounced. The lysate was cleared by centrifugation and an aliquot taken for whole lung RNA preparation using the RNeasy Mini kit (Qiagen). The remaining lysate was incubated for 4 h with a 1:20 dilution of anti-HA magnetic beads (Pierce 88837). Beads were washed once in supplemented homogenization buffer, transferred to a fresh tube, then washed three times in high-salt wash buffer (300 mM KCl, 50 mM Tris-Cl pH 7.4, 12 mM MgCl2, 1% NP-40, 0.5 mM DTT, 0.1 mg/mL cycloheximide, 80 U/mL NEB murine RNase inhibitor). Beads were transferred to a fresh tube and immunoprecipitated RNA extracted by incubating beads in buffer RLT (Qiagen) supplemented with 2-mercaptoethanol and purified using the RNeasy Micro kit (Qiagen).
RNA Sequencing and Data Analysis
For RiboTag immunoprecipitation samples, libraries were prepared for sequencing by the UCSF Genomics CoLab using the QuantSeq Forward kit (Lexogen) and sequenced on a HiSeq 4000 (Illumina). Reads were aligned to the mouse genome (Ensembl GRCm38) using STAR 2.5.2b. Data were deposited in the Gene Expression Omnibus series GSE190821. For RNA sequencing from conditional epithelial IRE1α knockout mice, lungs of uninjured mice were flash-frozen and homogenized by bead mill in Tri-Reagent. Libraries were prepared for sequencing by Novogene using the stranded mRNA kit (Illumina) and sequenced on a HiSeq 4000 (Illumina). Reads were aligned to the mouse genome (UCSC GRCm38) using HISAT2. Data were deposited in the Gene Expression Omnibus series GSE190822.
Differential expression testing was performed by the DESeq2 package using the model ∼batch + sex + treatment group. For heatmaps, expression data were transformed using the rlog function in DESeq2 and batch-corrected using the removeBatchEffect function in the limma package. Fold-change results from DESeq2 were exported to Ingenuity Pathway Analysis (Qiagen) for upstream regulator analysis. Custom gene set statistical testing was performed using the ROAST function in the limma package.
Single-cell sequencing data were analyzed using the Seurat3 package. Gene set scores were calculated using the AUCell package and reintegrated into the corresponding Seurat object for overlay on UMAP plots, or plotted for cells over an ordered trajectory obtained from https://github.com/theislab/2019_Strunz.
Gene expression signatures were composed of the following and are available in a downloadable format in Supplemental Table S1. TGF-β (23): Ccn2, Lox, Spp1, Serpine1, Timp1, Itgb6, Actn1, Thsd1, Fbn1, Mmp14, Tgfbi, Itgav, Mmp3, Tnc, Serpine2, Epha2, Mmp3, Gja4, Rhoa, Axl, Mfap2, Fmod, Marco, Itgb5, Pck1, Ddb2, Hsp90b1, Cd44, Cdc42, Lcp1, Capn2, Ackr3, Ccr2, Cdk4, Tfap2a, Areg, Fscn1, Ctsh, S100a4, Fstl1, Mmp13, Thbs1, Igfbp4, Gadd45a, Scarb1, Tcp1, Hmgb2, Mgp, Gja1, and Rfc3. IRE1WT (16): Loc126661, Gal3st2, Serp1, Srp19, Fam18b2, Ppfibp1, Tmem82, Zbtb43, Skip, Znf189, Prrc1, Cep170, Rwdd2a, Wbscr27, Ankrd27, Bhlhb8, Sec24d, Trim5, Fbxo16, Dnajb9, Nanos3, Rarb, Dnah9, Fam57b, Kcnmb3, Dpp3, Tcf25, Tnnc1, Flj38773, Wsb1, Ush1c, Glt8d3, Tssk1b, Bcl6b, Cth, Npepl1, Tnfrsf13c, Loc120376, Tsc2, Mcemp1, Svil, Tpm3, Ccdc37, Lrch4, Prh1, Trim74, C1orf61, C10orf6, Larp2, Csf1, Lef1, Galk2, B9d1, Afm, Tncrna, Tnfrsf8, Rasal3, Prmt7, Ly6g6c, Loc283728, Hspb8, Loc401317, Cdk5rap3, Loc390564, Rad51c, Tpm2, Gtpbp2, Cdk5rap3, Ptpdc1, Cth, Usp36, Rrp7b, Larp6, Agpat7, Ddit3, Znf83, Ndrg4, Ppp1r3e, Ahsa2, Znf451, Eif4a2, Loc154860, Acyp1, Fam65a, Rln2, Ell3, Crabp2, Pramef12, Pold4, Snrpa1, Mllt11, Coro6, Cryzl1, Ttty22, Gabarapl1, Snrpa1, Irf3, Tial1, Dhtkd1, Herpud1, Btn3a1, Pcolce, Nme4, Acad11, Cryzl1, C6orf134, Cpne1, Cchcr1, Artn, Btn3a1, Irf3, Snrpa1, Trip10, Nfic, Cdkl2, Tspyl2, C1orf106, Inadl, Zfand1, Tifa, Apobec3f, Dffa, Sec61g, C5orf4, Fbxo44, Pdzd2, Or7e19p, Kiaa0895, Ccdc113, Stac3, Ttll3, C7orf46, Cep152, Gmip, Ddx11, Pter, Neil1, Golga2l1, Asgr1, Loc388160, C9orf25, Phldb3, Tial1, Serpine1, Ccdc136, Wdhd1, Sec31b, Noxa1, Nsun5c, Mark4, Aoc2, Loc92497, Jun, Hes7, Eml2, Ank3, Fkbp1a, Kif1b, Slc25a27, and Dnajc12. IRE1αI642G (16): Fam18b2, Rwdd2a, Ankrd27, Bhlhb8, Sec24d, Nanos3, Rarb, Rrp7b, Kiaa1217, Agpat7, Icam5, Ddit4, Nudt7, Mllt11, Coro6, Ttty22, Btn3a1, Btn3a1, Trip10, Nav1, C6orf57, Plekha2, Sat2, Hes7, Eml3, Eml2, Phactr1, Carm1, Il6r, Kiaa1622, Clmn, Slc25a27. Gene Ontology 0006986 “Response to unfolded protein:” Selenos, Serp2, Eif2ak4, Hsp90aa1, Hspa9, Hspa1a, Hspa2, Hspd1, Hspb1, Hsf1, Hspa1l, Atf6b, Dnajb9, Fbxo6, Tmbim6, Edem1, Hspa1b, Herpud2, Upf3b, Stt3b, Ifng, Derl1, Ubxn4, Jkamp, Creb3, Eif2a, Optn, Serp1, Ero1a, Hspa5, Hspa14, Tmed2, Vapb, Creb3l1, Syvn1, Ube2j2, Tmtc4, Eif2s1, Eif2ak3, Hspb8, Ficd, Ep300, Wfs1, Nfe2l2, Hsph1, Cdk5rap3, Edem2, Abcb10, Tmem129, Crebrf, Ccnd1, Rhbdd1, Upf3a, Edem3, Xbp1, Creb3l3, Ermp1, Thbs1, Comp, Umod, Atf3, Herpud1, Creb3l2, Upf2, Yod1, Ern1, Hspa13, Tbl 2, Casp12, Mfn2, Stub1, Eif2ak2, Faf2, Dnajc3, Ddit3, Ppp1r15a, Hspa4l, Erp44, Stc2, Derl3, Thbs4, Atf4, Parp16, Ptpn1, Pmp22, Bag3, Dab2ip, Tram1, Chac1, Tm7sf3, Atf6, Amfr, Derl2, Creb3l4, Pacrg, Qrich1, Erp27, Ern2, Hspa8, Daxx, Manf, and Bhlha15. T-UPR (15): Spa5, Ddit3, Atf4, Txnip, Bbc3, Pmaip1, Bcl2l11, and Bax. DDR-associated secretory genes (24, 25): Il7, Cxcl1, Cxcl5, Csf2, Plaur, Timp2, Icam1, Tnfrsf11b, Cxcl2, Ccl2, Igfbp2, Pigf, Tnfrsf1a, Mif, Ccl20, Tnfrsf18, Areg, Serpine1, Plat, Plau, Timp1, Mmp3, Mmp10, Mmp12, Mmp13, Mmp14, Igfbp1, Igfbp3, Igfbp4, Igfbp6, Igfbp7, Fasl, Il6st, Egfr, Il1a, Il1b, Il13, Il15, Cxcl3, Ccl8, Ccl3, Ccl11, Ccl26, Ccl25, Ccl1, Cxcl11, Csf3, Ifng, Cxcl13, Ereg, Nrg1, Egf, Fgf2, Hgf, Fgf7, Vegfa, Ang, Kitl, Cxcl12, Ngf, Ccl7, Clu, Glb1, Lama2, Mmp7, Serpine2, Cdkn2a, Cdkn1a, Tgfb1, Cdkn1c, Cdkn2b, Ccl27a, Lmnb2, Noa1, and Tnfrsf10b.
Flow Cytometry
For epithelial cells, lungs were dissociated and single-cell suspensions were prepared as described previously (22). For interstitial macrophages, lungs were incubated in RPMI supplemented with 80 U/mL DNase I, 2.5 mg/mL collagenase I, 0.4 U/mL dispase, and dissociated in C-tubes on a gentleMACS dissociator (Miltenyi). Cells were stained with CD45 (30-F11, FITC, Invitrogen 11045182, RRID:AB_465050), F4/80 (BM8, PE, Invitrogen 12480182, RRID:AB_465923), CD24 (M1/69, PE-Cy7, Invitrogen 25024282, RRID:AB_10853806), MHC-II IA/IE (M5/114, PerCP-eFluor710, Invitrogen 46532180, RRID:AB_1834440), CD11b (M1/70, APC-Cy7, Invitrogen A15390, RRID:AB_2534404), CD11c (N418, APC, Invitrogen 17011482, RRID:AB_469346), Ly6C (AL-21, V450, BD 560594, RRID:AB_1727559), Itgb6 [3G9, APC (26)], Itgb4 (346-11 A, BV421, BD 743078, RRID:AB_2741270), EPCAM (G8.8, FITC, Biolegend 118208, RRID:AB_1134107), H2-K1 (34-1-2S, APC, Biolegend 114714, RRID:AB_2734174), DRAQ7 (Biolegend 424001), and eFluor506 fixable viability dye (eBioscience 65086614). Data were acquired on a BD FACSverse and analyzed with FlowJo.
Cell Culture
A549 cells were obtained from the UCSF Cell Culture Facility (CCLZR013) from a stock that was short tandem repeat (STR) authenticated. Cells were cultured in DMEM and 10% FBS. IRE1α knockout A549 cells were generated by transient transfection of three CRISPR-Cas9 “all-in-one” plasmids bearing sgRNAs against the human ERN1 locus (ABM 194381110502). Cells were clonally selected and expanded and IRE1α loss of expression and function was verified by Western blot and XBP1 splicing assay.
Western Blotting
Lysates were prepared in RIPA buffer and blots probed for γH2AX (R&D 4418-APC), total H2AX (CST 2595, RRID:AB_10694556), and GAPDH (Santa Cruz sc-365062, RRID:AB_10847862). Blots were scanned using the LI-COR Odyssey system.
Study Approval
The animal model work described in this study was approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco (Protocol No. AN180815).
RESULTS
IRE1α Acts in the Pulmonary Epithelium to Promote Fibrosis
We previously showed that a kinase inhibitor of IRE1α, KIRA7, protected mice from bleomycin-induced lung injury when given starting on the day of bleomycin exposure (21). However, it was unclear which cell types had pathological IRE1α activation, and how IRE1α promoted lung fibrosis. We mapped a UPR gene expression signature (Gene Ontology 0006986) onto single-cell RNA sequencing data from bleomycin-exposed mice (5) and found that subpopulations of epithelial cells, monocytes/macrophages, fibroblasts, and smooth muscle cells exhibited the highest UPR gene expression (Supplemental Fig. S1A). Thus, kinase inhibitors of IRE1α could protect from fibrosis by modulating stress signaling in multiple cell types.
We chose to focus on the role of IRE1α in the lung epithelium, since the UPR has been shown to be activated in epithelial cells in human patients with IPF, adjacent to fibroblastic foci (27–29), and since enforced epithelial ER stress is sufficient to induce pulmonary fibrosis in mice without any additional injurious exposure (30, 31). We conditionally knocked out IRE1α using ShhCre, which is active starting at the foregut endoderm progenitor stage of lung development and thus efficiently induces recombination throughout all the lung epithelial lineages (32, 33) (Fig. 1A). At baseline, mice with conditional IRE1α knockout in the epithelium appeared to have histologically normal lungs (Supplemental Fig. S1B). Loss of IRE1α activity in the epithelium was confirmed by reverse-transcription PCR for XBP1 mRNA spliced isoforms in magnetically enriched CD45- EPCAM+ lung epithelial cells (Fig. 1B). UPR signaling is complex, with significant cross talk and compensation between the transcriptional responses mediated by the three canonical sensors of unfolded protein, IRE1α, PERK, and ATF6. To further define the effect of IRE1α conditional knockout on UPR signaling, we sequenced whole lung RNA from 10 uninjured mice. Using transcriptional signatures derived from combinatorial deletion of the three sensors (34), we found a decrease in IRE1α regulated genes and a small compensatory upregulation in PERK-regulated genes (Supplemental Fig. S1C).
Figure 1.
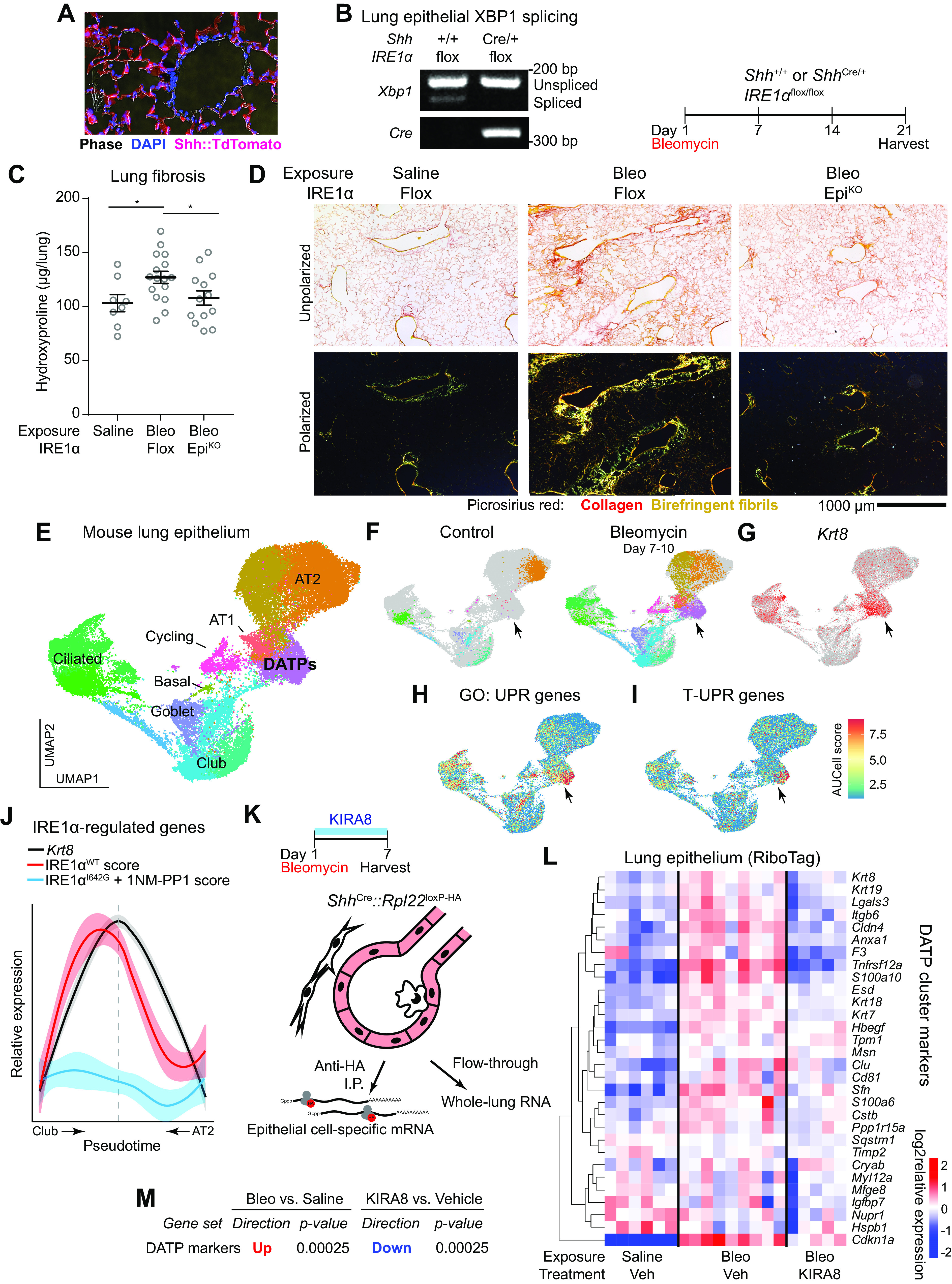
Epithelial IRE1α regulates fibrosis and DATPs exhibit maladaptive IRE1α activation. Confirmation of ShhCre epithelial expression using ROSA26Ai14 reporter mice (A) and IRE1α loss of function in the epithelium (B). Lung epithelial cells were isolated by dissociation and MACS enrichment for CD16/32− CD45− EPCAM+ cells, and IRE1α function evaluated by RT-PCR for XBP1 splice isoforms. Presence of the Cre allele was confirmed by PCR. C: bleomycin-induced fibrosis in IRE1α epithelial knockout mice. Mice were exposed to bleomycin, harvested at day 21, and hydroxyproline content measured. Each data point represents one mouse, with means ± SE indicated. Groups were compared by ANOVA with Fisher’s post hoc test. D: unpolarized and polarized images of lung sections from mice exposed as in C. To image birefringent fibrillar collagen, a circular polarizer was inserted in the illumination path and images were taken with a polarized analyzer in place. E: single-cell RNA sequencing of bleomycin-exposed lung epithelium. The UMAP projection and cluster identities were defined previously (5). F: UMAP projections of subsets of the dataset in for cells from control lungs (left) and lungs harvested at day 7 through day 10 after bleomycin exposure (right). Gray denotes cells in the data set that were collected at other time points; cluster colors and identities are as in E. G: overlay of Krt8 expression, marking some ciliated epithelial cells and the DATP population (arrowhead). Overlay of AUCell scores for Gene Ontology annotated UPR genes (H) and terminal UPR genes (I), concentrated in subset of DATPs (arrowhead). J: IRE1α-regulated gene expression over the course of distinct transdifferentiation trajectories from MHCIIhigh club cells or AT2s, converging on the DATP state marked by peak Krt8 expression (dashed line). Trajectories were defined previously (5). The relative expression level of Krt8 (black line), and AUCell scores for genes induced by wild-type IRE1α (red line) and IRE1αI642G with 1NM-PP1 administration (blue line) were mapped across the trajectory. Gene signatures were defined previously (16). K: RiboTag conditional ribosome tagging strategy. Lungs were harvested 7 days after bleomycin exposure and flash frozen. Epithelial ribosome-associated mRNA was purified by anti-HA immunoprecipitation and sequenced. L: heatmap of markers of the DATP cluster in the lung epithelium after bleomycin exposure and treatment with KIRA8. Markers were defined by the Seurat package using single-cell RNA sequencing data as described previously (5). M: unfiltered DATP marker gene expression changes and statistical testing using the ROAST function in the limma package. *P < 0.05. AUC, area under the curve; DATP, damage-associated transient progenitor; KIRA8, kinase inhibitor of IRE1α; AT2, alveolar type 2; UPR, unfolded protein response.
To determine whether IRE1α activity is required in the epithelium for pulmonary fibrosis, mice were exposed to bleomycin. Both wild-type and epithelial knockout mice exhibited the expected acute lung injury at day 8, based on bronchoalveolar lavage protein and cell count (Supplemental Fig. S1D). Compared with the wild-type, conditional knockout of IRE1α in the epithelium protected mice from lung fibrosis based on day 21 lung hydroxyproline content (Fig. 1C), a highly specific and quantitative measure of collagen. We stained histological sections with picrosirius red and captured cross-polarized images, which is highly specific for birefringent fibrillar collagen. Lungs from conditional knockout mice displayed qualitatively less prominent staining (Fig. 1D). Thus, IRE1α activity in the epithelium is required for lung fibrosis.
Terminal UPR Signaling through IRE1α Is Activated in Damage-Associated Transient Progenitors
An explanation of the aforementioned findings would be that IRE1α directs the lung epithelial response to favor fibrosis after bleomycin-induced injury. Several recent studies have revealed that damage-associated transient progenitor (DATP) cells expand dramatically in diverse models of lung injury. Paradoxically, these cells express profibrotic genes, yet possess the capacity to differentiate into mature alveolar type 1 and type 2 (AT1 and AT2) to regenerate new alveoli (3, 5–8). These cells also express stress-response programs, including those associated with the UPR (5, 6).
To further characterize the UPR and the particular role of IRE1α in DATPs, we reanalyzed a recently published single-cell RNA sequencing data set from bleomycin-exposed mice in which lung epithelial cells were sampled at daily intervals after bleomycin exposure (Fig. 1E) (5). The cluster of cells with the features of DATPs (5) (Fig. 1E) emerge by days 7–10 after bleomycin injury (Fig. 1F) and are marked by high Krt8 expression (Fig. 1G). We calculated area under the curve (AUC) scores for UPR genes annotated by Gene Ontology. Because active secretory cells physiologically express many of the genes in this signature, we calculated AUC scores for a second set of genes associated with maladaptive UPR signaling, called the “terminal” UPR (T-UPR), including the proapoptotic genes Ddit3 (CHOP) and Txnip (15). Several populations had cells with high general UPR expression, including secretory cells of the airway epithelium and DATPs (Fig. 1H). T-UPR expression scores were concentrated in a subset of DATPs (Fig. 1I), indicating that maladaptive UPR signaling is active in these cells.
In other model systems, the T-UPR can be activated by multiple upstream mediators, including IRE1α and PERK. Since kinase inhibition (21) and epithelial conditional knockout of IRE1α protected mice from fibrosis (Fig. 1C), whereas PERK inhibition did not (21), we hypothesized that IRE1α might be activated during conversion of mature epithelial cells into DATPs. Cell state trajectory analysis had previously demonstrated the transcriptional convergence of club-like airway cells and AT2 cells into a common DATP state (5), reflecting the dual airway and alveolar origins a of DATPs. We generated a gene expression signature based on genes upregulated after enforced activation of wild-type IRE1α in human cells (16) and mapped AUC expression scores for this signature onto the cell state trajectories leading to the DATP phenotype (Fig. 1J). As airway cells (left side) or AT2 cells (right side) transition into the DATP phenotype (center), expression of Krt8 increased, peaking in the DATP state. In parallel, the wild-type IRE1α expression score also increased, indicating that IRE1α is activated as cells transition into DATPs. The peak of IRE1α activation was not exactly aligned with the peak of Krt8 expression, perhaps hinting at heterogeneity within the DATP population (Fig. 1, H and I) or the differential kinetics of airway-to-DATP and AT2-to-DATP transitions (5).
Low or transient ER stress conditions trigger the “adaptive” UPR, and IRE1α’s RNase domain initiates frameshift splicing of the mRNA encoding the adaptive XBP1 transcription factor. However, persistent or high-level ER stress triggers the “terminal” UPR and causes IRE1α kinase/RNAse domains to also degrade a plethora of ER-associated mRNAs and microRNAs in a process called regulated IRE1α-dependent decay (RIDD) (16–18). These activation states are genetically and biochemically distinguishable. IRE1α with an I642G mutation in its kinase domain can be activated in the presence of the ATP analog 1NM-PP1, and is able to induce XBP1 splicing and the adaptive UPR, but is unable to catalyze RIDD or the cell phenotypes associated with the terminal UPR (16, 35, 36). To ascertain whether the adaptive arm of IRE1α signaling is activated in the DATP transition, we derived a gene expression signature from human cells with isolated enforced activation of IRE1αI642G in the presence of 1NM-PP1, which includes genes transcriptionally controlled by XBP1 (16). Remarkably, mapping the adaptive IRE1α signature onto the DATP trajectories did not reveal any significant upregulation (Fig. 1J), suggesting that the majority of the gene expression changes attributable to IRE1α in response to lung epithelial injury are part of the terminal UPR.
To further understand the role of IRE1α in the epithelial injury response, we profiled the epithelial transcriptome on day 7 after bleomycin exposure, a timepoint when markers of ER stress are first elevated in whole lung RNA (21) and when gene expression programs in injured epithelial cells, like TGF-β signaling, begin to establish the fibrotic milieu. We used RiboTag mice, in which the ubiquitous ribosomal protein Rpl22 is conditionally labeled with the HA epitope by Cre recombinase (37). This tool enables immunoprecipitation of cell-type-specific mRNAs from flash-frozen lungs and reflects both transcriptional abundance and ongoing translation while avoiding gene expression artifacts that have been described in dissociated and sorted cells, especially in stress signaling pathways (38). ShhCre was used to specifically label all epithelial subtypes in the lung (Fig. 1K and Supplemental Fig. S1E) after bleomycin exposure and treatment with KIRA8, a nanomolar potent, monoselective inhibitor of the IRE1α kinase (20, 21, 39). Upstream regulator analysis confirmed that pathways associated with IRE1α terminal UPR signaling were blocked by KIRA8 (Supplemental Fig. S1F). Furthermore, the well-characterized RIDD substrate Bloc1s1 (Blos1) (16, 17, 40) was downregulated in the lung epithelium after bleomycin exposure, and this downregulation was alleviated by KIRA8 (Supplemental Fig. S1G), confirming both that terminal UPR signaling occurs in the injured lung epithelium and can be blocked by KIRA8. Bloc1s1 expression was unchanged in the whole lung transcriptome (Supplemental Fig. S1D), suggesting that although a general UPR signature can be seen in multiple cell types (Supplemental Fig. S1A), IRE1α’s RIDD activity is more prominent in the epithelium.
To evaluate whether UPR signaling through IRE1α indeed regulates DATPs, we queried our epithelial RiboTag data for the expression of genes uniquely expressed in this cluster of cells. Inhibition of the IRE1α kinase with KIRA8 decreased expression of genes marking these cells (Fig. 1, L and M). Together, these data demonstrate that IRE1α’s terminal signaling is activated as epithelial cells transition into the DATP state, and this activation is critical to either the number or phenotype of DATPs.
IRE1α Regulates DATP Cell Number after Bleomycin Injury
To identify DATPs in lung sections, we used in situ hybridization for Krt7 mRNA. Although Krt8 has been used as a canonical marker for DATPs (5–8), it is expressed at low levels in normal ciliated bronchial epithelial cells (Fig. 2A and Supplemental Fig. S2, A and B). In situ hybridization for Krt7 mRNA was a more faithful marker for DATPs because its expression was congruent with Krt8 in DATPs (Supplemental Fig. S2A), but was not detected in the airways of uninjured lungs (Fig. 2A and Supplemental Fig. S2, B and C). Consistent with previous studies, Krt7+ DATP number increased dramatically after bleomycin exposure. This increase was blocked by KIRA8 (Fig. 2B and Supplemental Fig. S2C). To confirm these results, we stained sections for Krt8 protein and excluded recognizable airways for quantification. Krt8+ DATP numbers were decreased after KIRA8 treatment (Fig. 2, C and E).
Figure 2.
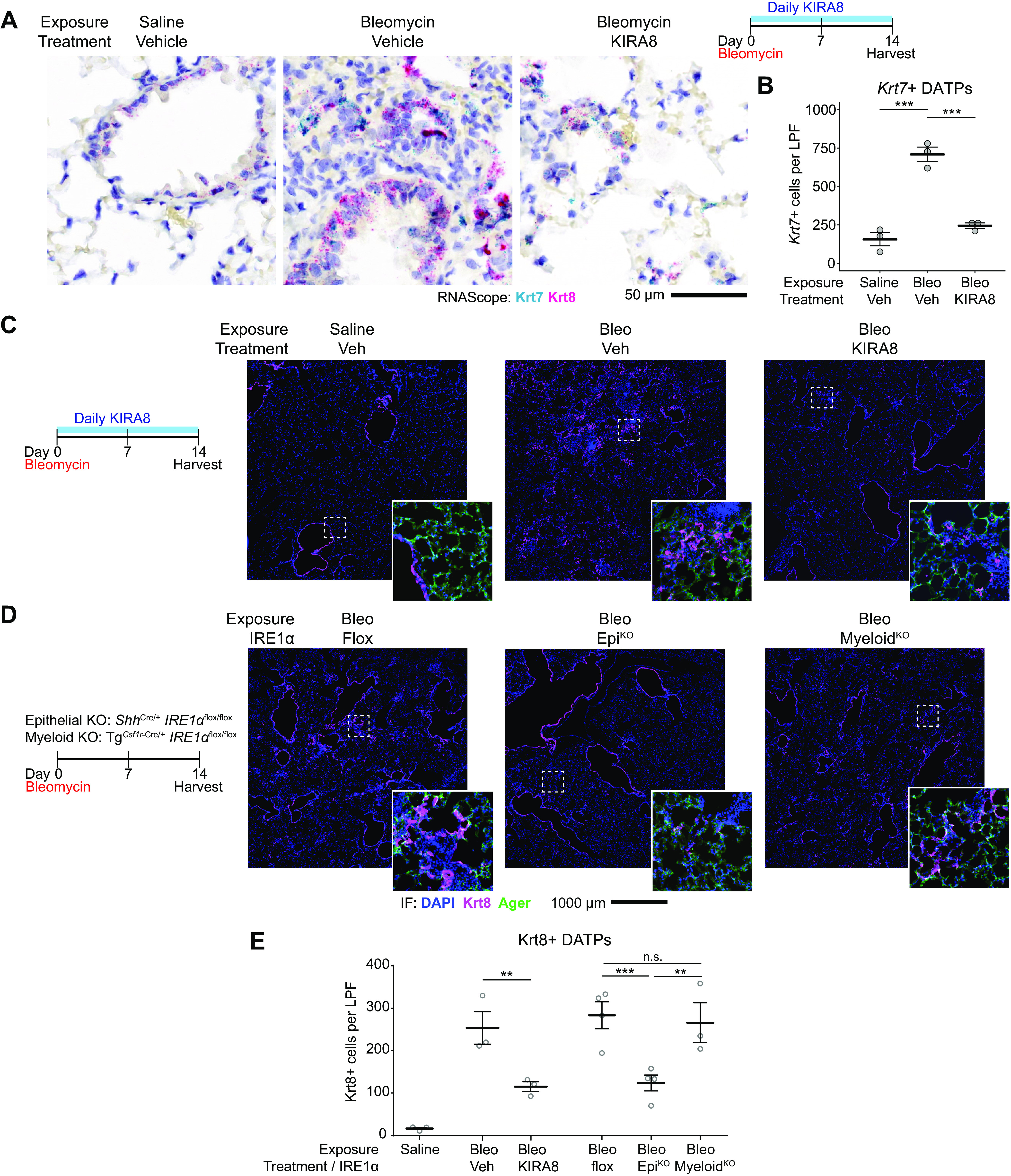
IRE1α regulates DATP number after bleomycin injury. A: RNA in situ hybridization (RNAscope) for Krt7 (teal) and Krt8 (magenta) at day 14 after bleomycin exposure and treatment with KIRA8. B: automated count of Krt7+ cells per 1,240-µm diameter low-power field (LPF). Each dot represents the average of 10 representative fields from one mouse with means ± SE indicated. Groups were compared by ANOVA with Fisher’s post hoc test. C: immunofluorescence staining for Krt8 (magenta) and Ager (green) at day 14 after bleomycin exposure and treatment with KIRA8. D: immunofluorescence staining as in (C) of mice with epithelial IRE1α conditional knockout (EpiKO) or myeloid conditional knockout (MyeloidKO). E: automated count of Krt8+ cells per low-power field. Each dot represents the average of eight representative fields from one mouse with means ± SE indicated. Groups were compared by ANOVA with Fisher’s post hoc test. **P < 0.01, ***P < 0.001. DATP, damage-associated transient progenitor; KIRA8, kinase inhibitor of IRE1α.
Surface markers and flow cytometric strategies for DATP analysis are limited. Flow sorting for EPCAM+ Itgb4+ H2K1high captures essentially all the airway-derived DATP colony-forming cells (41), but likely encompasses some cells that do not have progenitor cell capacity. Nevertheless, we found that the fraction of H2K1high cells within the Itgb4+ airway cell compartment increased dramatically after bleomycin exposure, and decreased substantially with KIRA8 treatment (Supplemental Fig. S2D).
Although we found “terminal” T-UPR signaling through IRE1α to be active in the lung epithelium, it is possible that IRE1α in other cell types might contribute to emergence of the epithelial transitional state. For example, adaptive IRE1α activation and XBP1 splicing is required for inflammatory gene expression in stimulated macrophages, including the expression of IL-1β (42); in turn, macrophage IL-1β can stimulate AT2 cells to enter the transitional state (8). We stained for Krt8 in lungs of mice with epithelial conditional knockout of IRE1α using ShhCre (epithelial KO). For comparison, we conditionally knocked out IRE1α in myeloid cells (myeloid KO) using TgCsf1r-Cre, which efficiently induces recombination in dendritic cells, monocytes, and macrophages (43). Lungs from epithelial KO mice had reduced Krt8+ DATP cell numbers compared with both control and myeloid KO mice (Fig. 2, D and E), indicating that IRE1α regulation of DATP cell number is autonomous to epithelial cells. These results do not exclude a role for IRE1α in other cell types, and loss of myeloid IRE1α may also affect the function or behavior of DATPs without altering their number.
Because adaptive IRE1α signaling promotes cell survival under stress conditions and physiological differentiation, we evaluated the possibility that loss of IRE1α signaling increased apoptosis of DATPs. This possibility was unlikely because DATPs did not exhibit significant IRE1α adaptive signaling during their emergence (Fig. 1I). On day 7, when DATPs are near their peak numbers (5), KIRA8 treatment did not cause widespread apoptosis of these cells. Indeed, although there were scattered TUNEL+ cells (Supplemental Fig. S2B, arrowheads), TUNEL+ Krt8+ double-positive cells were exceptionally rare in both vehicle and KIRA8-treated mice (Supplemental Fig. S2B).
DATPs Are the Principal Epithelial Instigators of TGF-β Signaling in the Injured Lung
To understand how IRE1α regulation of DATPs might contribute to fibrosis, we evaluated the integrin αvβ6—TGF-β axis in the DATP population. Integrin αvβ6 is expressed on injured lung epithelial cells and releases active TGF-β from the extracellular matrix, which in turn causes autocrine induction of Itgb6 transcription in a feed-forward loop that culminates in the activation of collagen-producing fibroblasts (44). Itgb6 was identified as a marker of DATPs in single-cell sequencing (5), so we evaluated the relative contribution of DATPs to integrin αvβ6 expression versus other epithelial cells. Using RNA in situ hybridization we found that approximately two-thirds of cells that expressed higher levels of Itgb6 (based on puncta counts) were marked by Krt7 mRNA (Fig. 3, A and B).
Figure 3.
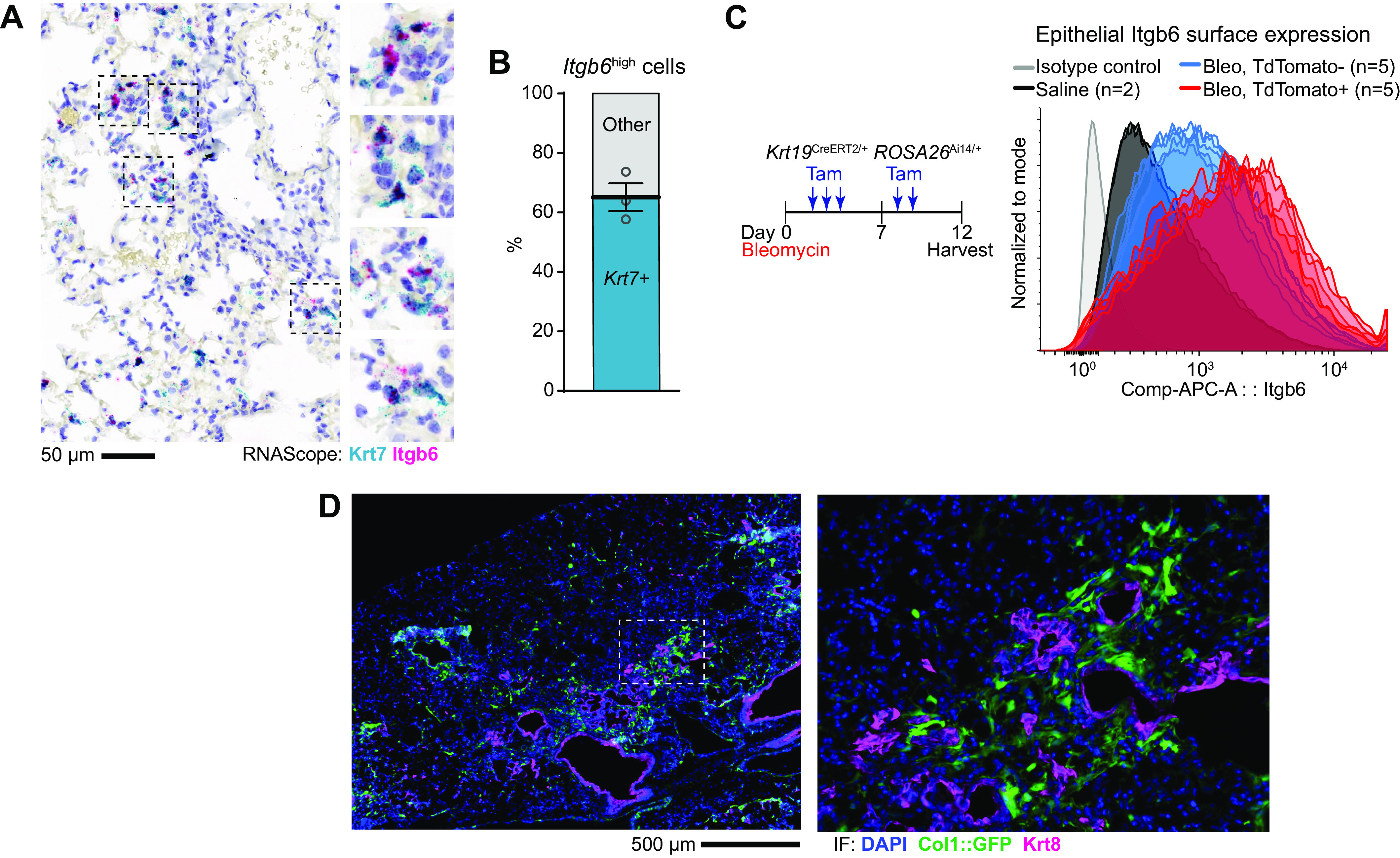
DATPs are the principal source of integrin αvβ6, a key activator of TGF-β in lung fibrosis. A: RNA in situ hybridization for Krt7 (teal) and Itgb6 (magenta) as in Fig. 2A. B: distribution of Itgb6high cells among Krt7+ or Krt7− cells. Each dot represents the average of 10 representative 1,240-µm diameter low-power fields from one mouse with means ± SE indicated. C: flow cytometric histogram of integrin αvβ6 expression on the surface of lung epithelial cells at day 12 after bleomycin. Krt19CreERT2/+ ROSA26Ai14/+ reporter mice were exposed to bleomycin and Krt19+ DATPs labeled by serial tamoxifen administration. Gated CD45− EPCAM+ epithelial cells are shown; in bleomycin-exposed mice, epithelial cells were subgated into TdTomato+ DATPs or the remaining TdTomato− epithelial cells. Each overlaid curve represents data from a single mouse. D: immunofluorescence staining for Krt8+ DATPs and collagenhigh fibroblasts (green). Col1a1::GFP reporter mice were exposed to bleomycin and harvested on day 14. DATP, damage-associated transient progenitor; GFP, green fluorescent protein; TGF-β, transforming growth factor β.
As a complementary approach, we used flow cytometry to measure surface integrin αvβ6 protein expression. DATPs express Krt19 (6), which has a nearly identical expression pattern as Krt8 (Supplemental Fig. S2A). Krt19CreERT2/+ ROSA26Ai14/+ reporter mice were exposed to bleomycin and Krt19+ DATPs labeled with TdTomato by serial administration of tamoxifen starting on day 3 after injury. After bleomycin injury, surface integrin αvβ6 expression increased in the lung epithelium, with TdTomato+ DATPs (Fig. 3C, red curves) exhibiting much higher integrin αvβ6 expression compared with TdTomato− epithelial cells (Fig. 3C, blue curves).
We reasoned that since dysfunctional progenitor cells appeared to be the principal source of integrin αvβ6-induced TGF-β activation, they would be colocalized with activated fibroblasts marked by high levels of collagen expression. Indeed, using collagen-green fluorescent protein (GFP) reporter mice exposed to bleomycin, we found that Krt8+ DATPs were surrounded by fibroblasts with high collagen 1 expression; conversely, activated fibroblasts were sparse in regions without Krt8+ cells (Fig. 3D). Together, these results indicate that DATPs are critical to establishing the fibrotic niche by signaling to fibroblasts through TGF-β.
IRE1α Regulates TGF-β Activation by DATPs
Though KIRA8 decreased DATP number, we nonetheless found clusters of Krt7+ DATPs in regions of injured lungs after KIRA8 treatment (Fig. 2A) and wondered whether IRE1α regulated the profibrotic phenotype of the DATPs that were present. In bleomycin-exposed mice we found abundant Krt7 and Itgb6 double-positive cells (Fig. 4A, closed arrowheads), surrounded by depositions of fibrillar collagen stained by picrosirius red (Fig. 4A, closed arrowheads). In KIRA8-treated mice, the number of Itgb6+ cells was significantly decreased, mostly through decreases in Krt7 and Itgb6 double-positive cells (Fig. 4B). Clusters of Krt7+ DATPs were rarely Itgb6+ (Fig. 4A), and there was a significant decrease in semiquantitative measurement of Itgb6 expression in Krt7+ DATPs by puncta counting (H-score, Fig. 4C).
Figure 4.
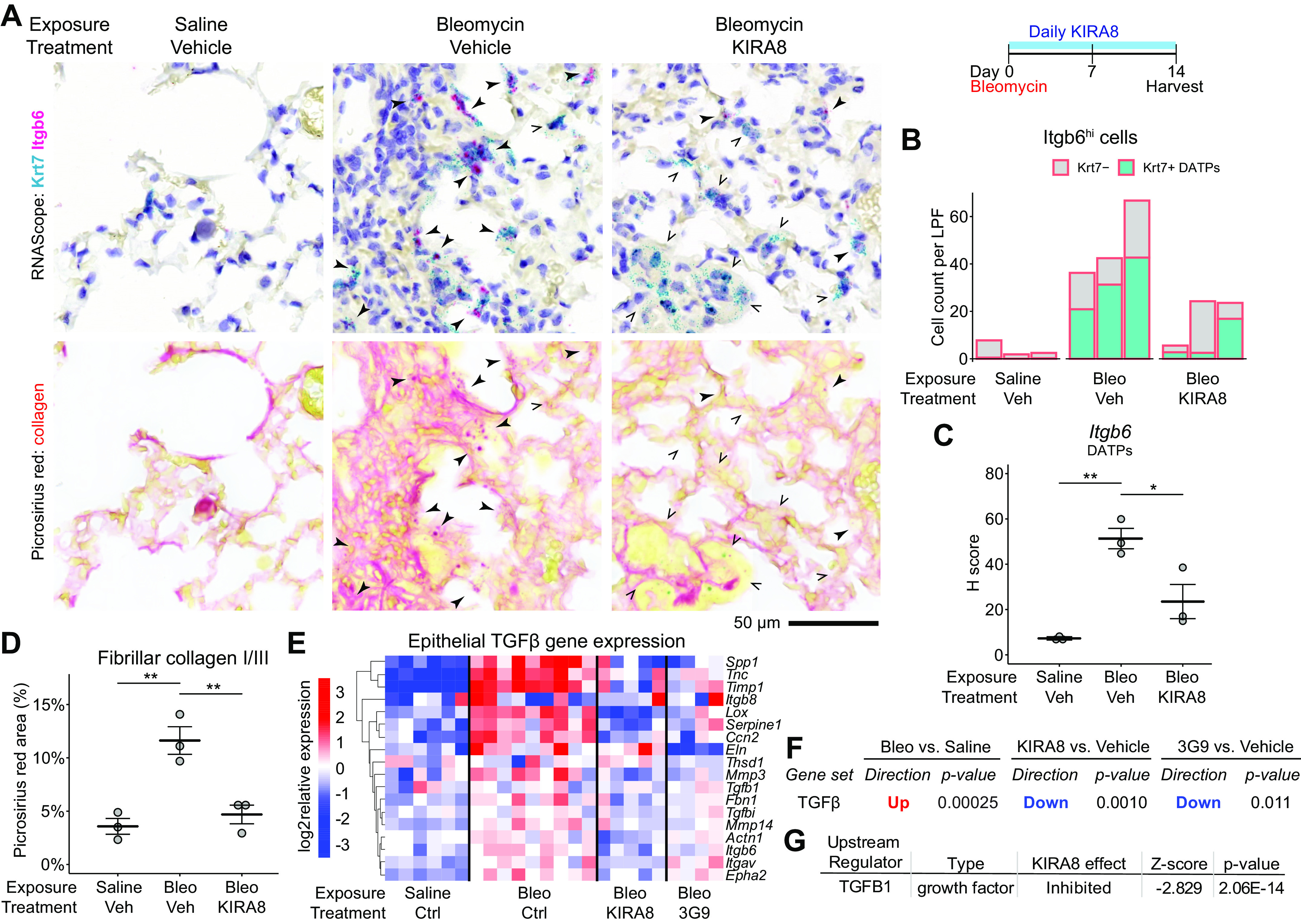
IRE1α regulates DATP expression of Itgb6 and consequent TGF-β signaling and local collagen deposition. A: RNA in situ hybridization for Krt7 (teal) and Itgb6 (magenta) as in Fig. 2A followed by washing and restaining with picrosirius red for collagen. Closed arrowheads denote Krt7 and Itgb6 double-positive cells, whereas arrow outlines indicate Krt7+ cells without significant Itgb6 expression. B: automated count of Itgb6high cells, further divided into Krt7+ (teal) or Krt7− cells (gray). Each bar represents the average of 10 representative 1,240-µm diameter low-power fields from a single mouse. C: semiquantitative H-score of Itgb6 puncta in Krt7+ DATPs, averaged over 10 representative fields per dot as in B with means ± SE indicated. D: collagen deposition measured by picrosirius red area. Each dot represents the average of 10 representative fields as in B with means ± SE indicated. E: heatmaps of batch- and sex-adjusted log2 expression values for TGF-β-induced genes in epithelial RiboTag libraries. RiboTag mice were exposed to bleomycin and treated with daily KIRA8 or twice-weekly 3G9, an inhibitory antibody against integrin αvβ6, and epithelial transcriptomes obtained as in Fig. 1I. TGF-β-induced genes were defined previously (23). F: focused statistical testing on the unfiltered TGF-β gene signature in E using the ROAST function in the limma package. G: naïve upstream regulator analysis using the Ingenuity Pathway Analysis package on epithelial RiboTag data identifies TGF-β as an inhibited pathway after KIRA8 treatment. *P < 0.05, **P < 0.01. DATP, damage-associated transient progenitor; KIRA8, kinase inhibitor of IRE1α; TGF-β, transforming growth factor β.
These decreases in Itgb6 expression corresponded to decreases in local TGF-β signaling and fibrosis. The extracellular matrix surrounding KIRA8-treated DATPs had decreased dense collagen deposition, based on picrosirius red staining (Fig. 4D). We used microarray data from bleomycin-exposed Itgb6 knockout mice to define a signature of TGF-β-induced genes (23). In our epithelial RiboTag data, these genes were upregulated after bleomycin exposure. Remarkably, KIRA8 downregulated these genes to a similar degree compared with 3G9, a direct inhibitor of αvβ6-mediated TGF-β activation (Fig. 4, E and F). Indeed, naïve upstream regulator analysis independently identified TGF-β1 as an upstream regulator downregulated by KIRA8 (Fig. 4G). Thus, IRE1α promotes fibrosis both by increasing the number of DATPs and enhancing their expression of integrin αvβ6, thereby increasing TGF-β signaling and fibroblast activation in the fibrotic niche.
IRE1α Reinforces DNA Damage Response Signaling in DATPs
It was recently proposed that DATP-like cells are persistent in the lungs of human patients with idiopathic pulmonary fibrosis, a disease of progressive interstitial fibrosis. Such persistence would require both cell-intrinsic pathways and tissue microenvironments that are self-reinforcing and thus able to durably maintain a cellular phenotype over years of disease. Instead of IRE1α regulating the DATP state as an isolated pathway, we hypothesized that IRE1α interacts with other pathways as part of a coordinated network of gene expression programs that establish and maintain the DATP phenotype.
One pathway implicated in the establishment of DATPs is DNA damage response (DDR) signaling. Lung epithelial cells undergoing the transition into DATPs exhibit histone 2AX phosphorylation (γH2AX), an upstream indicator of DNA damage, and downstream DDR-associated gene expression including the expression of Cdkn1a (p21) (6). These findings were present even if the primary lung injury would not have been expected to cause DNA damage per se. Interestingly, p53—as distinguished from other DDR downstream pathways—was required for both transition into the DATP state and differentiation into mature AT1 cells (6). Paradoxically, unremitting DDR signaling through Sin3a loss of function caused emergence and persistence of cells with a transcriptional phenotype similar to those of DATPs, and genetic ablation or pharmacological inhibition of p53 blocked fibrosis and preserved histopathologically normal appearing lungs (25), though DATP differentiation was not formally interrogated. Thus, the roles of DDR signaling in the DATP state are complex and nuances of degree and duration may be important.
IRE1α was recently shown to both prolong and enhance the magnitude of DDR signaling through its terminal signaling outputs, including RIDD-mediated downregulation of phosphatases involved in DDR signal cascades (45). Importantly, a variety of DNA damaging agents induced IRE1α’s RIDD activity without evidence of “adaptive” signaling through canonical XBP1 splicing (45), exactly the pattern we saw in cells transitioning to DATPs (Fig. 1J). We confirmed these findings by exposing A549 cells to bleomycin, which increased γH2AX levels. The induction of γH2AX was blunted in an A549 line where IRE1α was knocked out by CRISPR-Cas9 targeting of the Ern1 locus (Fig. 5A and Supplemental Fig. S3A). IRE1α knockout did not affect levels of total H2AX (Supplemental Fig. S3B). In vivo, IRE1α regulates DDR signaling in the mouse lung epithelium after bleomycin exposure; naïve upstream regulator analysis of RiboTag epithelial transcriptomes identified Tp53 (p53) and Ddx5 (p68) as pathways downregulated by KIRA8 treatment (Fig. 5B).
Figure 5.
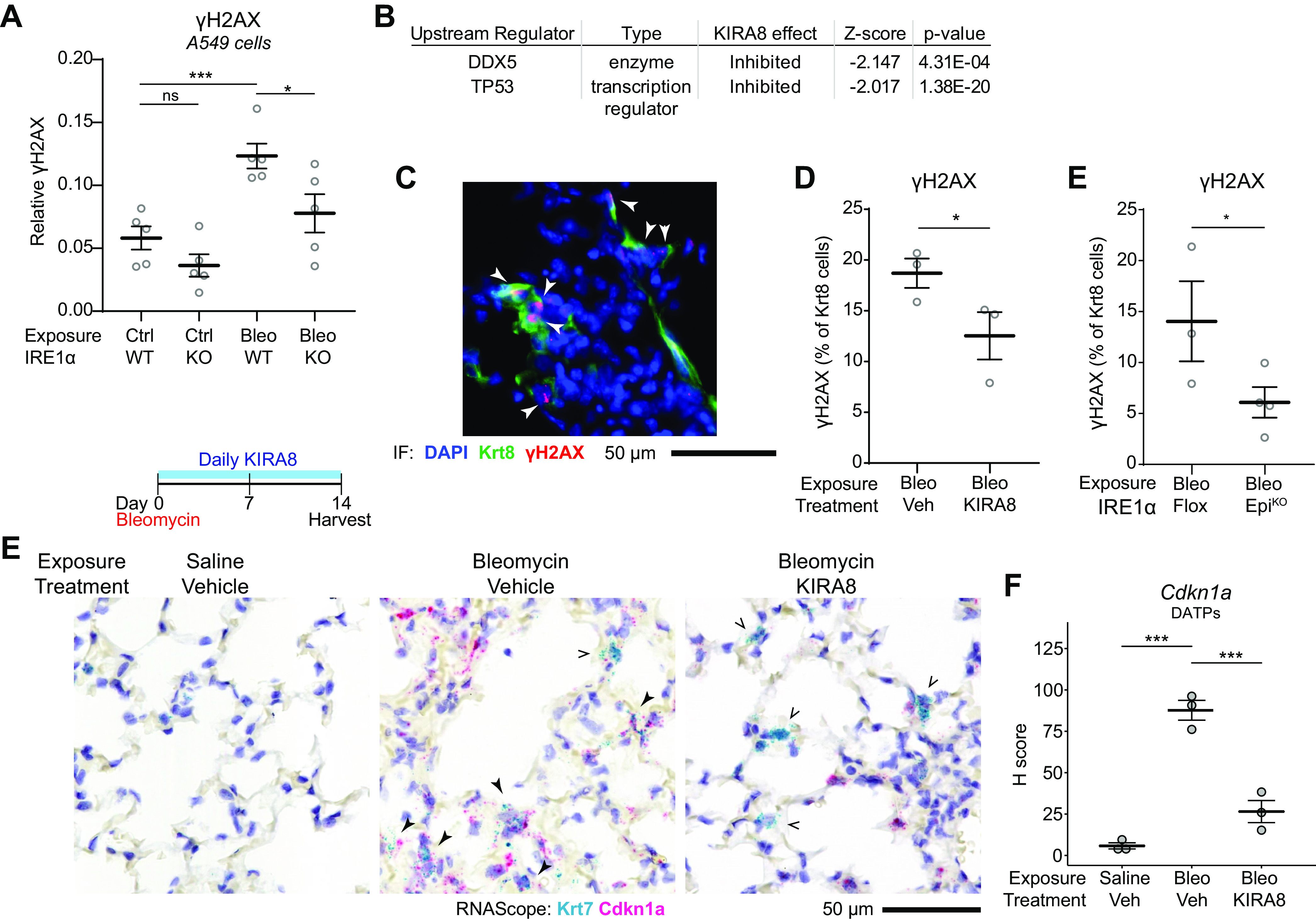
IRE1α reinforces DNA damage response signaling in DATPs. A: quantification of γH2AX expression in A549 cells or CRISPR-knockout IRE1α A549 cells. Cells were exposed to 5 µg/mL bleomycin for 4 h. Each dot represents an independent well with means ± SE indicated. Groups were compared by ANOVA with Fisher’s post hoc test. B: naïve upstream regulator analysis using the Ingenuity Pathway Analysis package on epithelial RiboTag data from bleomycin-exposed mice as in Fig. 1I, identifies p53 and DDX5 as inhibited DNA damage response pathways after KIRA8 treatment. C: representative immunofluorescence staining for Krt8 (green) and γH2AX (red) in mice exposed to bleomycin for 14 days. Arrowheads denote Krt8+ γH2AX double-positive cells. Automated counting of γH2AX and Krt8+ cells and quantification of the percentage of Krt8+ cells that are also γH2AX positive in mice treated with KIRA8 (D) or mice with conditional epithelial IRE1α knockout (E). Each dot represents the average of eight low power fields from one mouse with means ± SE indicated. Groups were compared by one-sided Student’s t test. E: RNA in situ hybridization (RNAscope) for Krt7 (teal) and Cdkn1a (red) in mice exposed to bleomycin and treated with daily KIRA8 for 14 days. Solid arrowheads denote Krt7 and Cdkn1a double-positive cells while arrow outlines indicate Krt7+ cells without significant Cdkn1a expression. F: semiquantitative H-score of Cdkn1a puncta in Krt7+ DATPs, averaged over 10 representative fields per dot with means ± SE indicated. Groups were compared by ANOVA with Fisher’s post hoc test. *P < 0.05, ***P < 0.001. DATP, damage-associated transient progenitor; KIRA8, kinase inhibitor of IRE1α.
To determine whether IRE1α enhances DDR signaling in DATPs specifically, we evaluated γH2AX staining in Krt8+ DATPs after bleomycin exposure, which showed the expected characteristic nuclear punctate pattern (Fig. 5C, arrowheads). About 20% of DATPs exhibited γH2AX foci, and this number was decreased in mice treated with KIRA8 (Fig. 5D). Consistent with our in vitro results, IRE1α regulation of γH2AX foci was cell-autonomous since conditional epithelial IRE1α knockout reduced γH2AX+ DATPs to a similar degree (Fig. 5E). We also stained for Cdkn1a (p21), a well-known cell cycle inhibitor downstream of DDR signaling that is upregulated in DATPs and may contribute to their hypoproliferative phenotype (5, 6). In bleomycin-exposed lungs, most Krt7+ DATPs were also positive for Cdkn1a (Fig. 5E, closed arrowheads), on a background of other Cdkn1a single-positive cells present in the injured lung. After KIRA8 treatment, a few Krt7+ DATPs were stained for Cdkn1a (Fig. 5E, open arrowheads). Many Cdkn1a single-positive cells remain after KIRA8 treatment, suggesting that the effect of KIRA8 on Cdkn1a expression is relatively specific to DATPs. Indeed, restricting the analysis to Krt7+ DATPs, there was a significant decrease in semiquantitative measurement of Cdkn1a expression by puncta counting (H-score, Fig. 5F).
The DDR is also known to induce the expression of secretory products that have been called “senescence-associated” although the signature is not specific to senescence and may be a general indicator of epithelial dysfunction (25, 46, 47). To the extent that these DDR-associated secretory genes were upregulated in the lung epithelium after bleomycin exposure, KIRA8 treatment blunted their expression (Supplemental Fig. S3C). Although TGF-β signaling can by itself induce senescence and some genes in this signature, the anti-integrin αvβ6 antibody 3G9 did not mitigate expression of the signature (Supplemental Fig. S3C), indicating that IRE1α regulates the DDR independently of αvβ6-mediated TGF-β activation. Several genes in the signature are well-known monocyte chemoattractants and monocyte activators, including Ccl2 (MCP-1), Ccl8 (MCP-2), Ccl7 (MCP-3), Ccl3 (MIP-1α), and Cxcl2 (MIP-2α). Recent studies have described monocytes infiltrating the lung after bleomycin injury, becoming CD11bhigh CD11c+ MHC-IIhigh “interstitial” macrophages over several days, and ultimately differentiating over weeks into monocyte-derived alveolar macrophages. These newly recruited macrophages have been proposed to help establish the fibrotic niche by expressing fibroblast growth factors such as PDGFα (48–51). In turn, IL-1β released by these macrophages can prevent DATP differentiation, instead of trapping them in the transitional state (8). On day 14 after bleomycin exposure, Krt8+ DATPs were colocalized with macrophages marked by CD206 (Supplemental Fig. S3D). Consistent with KIRA8 downregulating lung epithelial expression of monocyte chemoattractants after bleomycin exposure (Supplemental Fig. S3C), we found that KIRA8 treatment abolished CD11bhigh CD11c+ MHC-IIhigh interstitial macrophage infiltration (Supplemental Fig. S3E).
Alleviation of IRE1α Signaling Promotes DATP Differentiation and Lung Repair
Although the DATP state in mice naturally resolves in favor of differentiation and lung repair, analogous cells in human fibrotic lung disease appear to be abnormally persistent and may promote fibrosis rather than lung repair (5–7, 25). Since terminal UPR signaling through IRE1α enhances the activity of the TGF-β and DDR pathways, we hypothesized that IRE1α might promote maintenance of the DATP phenotype and delay differentiation into alveolar epithelial cells.
We used Krt19CreERT2/+ ROSA26Ai14/+ mice to label DATPs and lineage trace them into AT1 cells. As expected, in uninjured mice, this strategy largely labeled airway epithelial cells (Supplemental Figs. S2A and S4A). TdTomato+ cells were rare in alveolar regions and appeared to label nonepithelial cells, consistent with previous reports of rare recombination in Cldn4+ macrophages (6). Despite detection of Krt19 mRNA in AT1 cells in single-cell sequencing datasets and previous reports of rare recombination in uninjured AT1 cells (3, 6, 52) (Supplemental Fig. S2A), we did not find any significant labeling of AT1 cells in uninjured mice (Supplemental Fig. S4A), perhaps due to the low recombination efficiency of our tamoxifen regimen, suited for lineage tracing rather than exhaustive labeling. We then injured Krt19CreERT2/+ ROSA26Ai14/+ mice with bleomycin and pulse-labeled cells with tamoxifen on day 3 and day 4 after exposure. Mice were harvested on day 14. At this timepoint, Krt19::TdTomato-traced DATPs have begun to differentiate into Ager+ AT1 cells (Fig. 6A and Supplemental Fig. S4B), but most traced cells maintained a cuboidal/rounded morphology and stained weakly for Ager or not at all. In mice treated with KIRA8, we found clusters of lineage-traced cells, including almost complete alveoli (Supplemental Fig. S4B), with TdTomato+ cells adopting a flattened morphology and staining brightly for Ager (Fig. 6B, top row). Other alveolus-like structures were composed of lineage-traced cells with Ager staining but a more cuboidal morphology, which may represent cells that have committed to AT1 identity but have not completed differentiation (Fig. 6B, bottom row). We quantified the fraction of Krt19::TdTomato-traced cells that costained for Ager and found that DATPs in KIRA8-treated mice were approximately twice as likely to have committed to an AT1 identity based on Ager staining (Fig. 6C). Thus, DATPs with reduced IRE1α signaling more readily differentiate into alveolar cells.
Figure 6.

IRE1α impairs DATP differentiation into mature alveolar epithelial cells. Lineage tracing of DATPs after bleomycin exposure. Krt19CreERT2/+ ROSA26Ai14/+ mice were exposed to bleomycin and treated daily with vehicle (A) or KIRA8 (B). Krt19+ DATPs were pulse-labeled by tamoxifen administration on day 3 and day 4 after bleomycin injury. Lungs were harvested at day 14 and evaluated for TdTomato lineage tracing (magenta) and stained for Ager (green). C: automated quantification of Ager staining in TdTomato lineage-traced DATPs. Each dot represents the average of five representative low power fields from a single mouse. Groups were compared by two-sided Student’s t test. D: model of IRE1α at the center of a mutually reinforcing gene expression network that induces and maintains the DATP phenotype and attendant profibrotic gene expression. Solid arrows represent relationships directly interrogated in this study. Dashed arrows represent relationships described in the literature. E: model of the role of IRE1α in the induction of DATPs and their functional phenotypes. IRE1α activity contributes to the induction of the DATP state, but hyperactivation or abnormal persistence of IRE1α signaling enhances TGF-β and DDR signaling and consequent establishment of a fibrotic niche. Relief of IRE1α signaling by kinase inhibitors decreases DATP number, but also dampens TGF-β and DDR signaling in DATPs, promoting their differentiation into mature alveolar epithelial cells. **P < 0.01. DATP, damage-associated transient progenitor; DDR, DNA damage response; KIRA8, kinase inhibitor of IRE1α; TGF-β, transforming growth factor β.
DISCUSSION
An opposing dynamic has long been recognized between lung regeneration and fibrosis. Several recent papers have described a novel population of progenitor cells in the lung that emerge after injury. These cells mirror this dynamic as they progress from injured mature epithelium into damage-associated transient progenitors (DATPs) and then differentiate again into mature epithelium to repair the lung. Although in the transitional state, DATPs enact gene expression programs regarded as pathological or dysfunctional, including those associated with hypoxia, DNA damage, senescence, ER stress, and fibrosis (5–7, 41). Intriguingly, some of these programs, such as the p53 arm of the DDR, may actually be required for subsequent differentiation into mature epithelium (6), whereas others, including TGF-β signaling and perhaps also components of the DDR, restrain differentiation and instead act to maintain the DATP state (7).
These complexities suggest that the tension between regeneration and fibrosis resides in DATPs, which integrate a myriad of cell-intrinsic and extrinsic signals, and, in fibrotic lung disease, decide to abnormally remain in the transitional state, rather than proceed with normal differentiation and repair (13, 14). In the mouse bleomycin model, DATPs are the predominant cells that express integrin αvβ6, the key epithelial TGF-β activator, and are immediately adjacent to activated collagen-expressing fibroblasts (Fig. 3) (3, 5, 7, 53). In human IPF, the characteristic fibroblastic foci of the disease are well-known to be lined by “hyperplastic” epithelial cells with evidence of UPR activation (29, 54), but it has only been recently appreciated that these epithelial cells are in fact analogous to mouse DATPs (6, 25, 55). These observations suggest that, in human fibrotic lung disease, DATPs exhibiting UPR activation fail to differentiate normally and instead establish a TGF-β-rich niche and stimulate fibroblasts to deposit extracellular matrix.
We expanded on these correlative observations by modulating the IRE1α arm of the UPR using a monoselective kinase inhibitor and conditional genetic knockouts. We previously showed that interrupting the IRE1α arm of the UPR using highly potent and selective kinase inhibitors (“KIRAs”), which allosterically inhibit its RNase catalytic activity, protects mice from bleomycin-induced fibrosis (21). In this study, we found that epithelial conditional knockout of IRE1α protects mice from fibrosis. An IRE1α kinase inhibitor, KIRA8, modulates the number of DATPs, and this regulation is cell-autonomous to the epithelium. IRE1α enhances TGF-β and DDR signaling and its inhibition ameliorates the pathological consequences of those pathways (Fig. 6D, solid arrows). Other studies have shown that, in turn, both TGF-β and DNA damage can activate IRE1α (45, 56, 57). IRE1α may directly regulate these pathways through RIDD, as demonstrated in other systems (45, 57). Intriguingly, human Krt8+ cells encompass at least two subpopulations that may have distinct differentiation fates and TGF-β-activating potential (58). Our observations of IRE1α enhancement of TGF-β and DDR signaling might also reflect IRE1α-regulated conversion between occult cellular substrates within the Krt7/Krt8+ compartment. Future studies examining single-cell transcriptional states in vivo and using ex vivo organoid models of transition might help distinguish these possibilities.
We propose that IRE1α sits at the center of a network of stress signals and gene expression programs that reinforce one another and contribute to the persistence of DATPs (Fig. 6D). The feed-forward relationships between these different pathways magnify IRE1α’s influence on the overall DATP phenotype. IRE1α has also been implicated in hypoxia and IL-1β inflammatory signaling in other cell types, both shown to be regulators of the DATP phenotype (8, 18, 42, 59), suggesting that the self-reinforcing network may be even more extensive than depicted here. IRE1α activation and the associated network of stress signals are required for transdifferentiation of injured epithelial cells into DATPs. Ongoing IRE1α activation further causes cells to stall in the DATP phenotype, where they establish the fibrotic niche and concomitantly fail to execute normal lung repair programs (Fig. 6E). When lung injury is relatively mild or short-lived, we propose that most DATPs have relatively low levels of IRE1α activation, permitting rapid differentiation and resolution of lung injury by regeneration of alveoli. However, certain types or durations of lung injury cause persistent IRE1α signaling, impairing lung regeneration, and instead causing fibrotic matrix deposition. Inhibiting IRE1α would reduce fibrosis while accelerating differentiation into new AT1 cells, as we found.
The importance of IRE1α may be broadly conserved in other models of reprogramming and transdifferentiation. Our results are reminiscent of findings in induced pluripotent stem cells (iPSCs), where early and transient activation of the IRE1α pathway enhances reprogramming of fibroblasts into iPSCs, but sustained activation over days causes aberrant morphology, hypoproliferation, and eventually stem cell loss (60). We speculate that the initial transdifferentiation of lung epithelial cells into DATPs may require “adaptive” IRE1α signaling, just as fibroblast reprogramming into iPSCs requires XBP1 splicing (60). Pathological persistence of DATPs may instead depend on the “terminal” outputs of IRE1α, particularly RIDD. Consistent with this speculation, DATPs exhibited terminal IRE1α signaling without the gene expression program associated with XBP1 splicing (Fig. 1, G and H). In addition, we found RIDD-mediated downregulation of Blos1 (Supplemental Fig. S1C) but not XBP1 splicing (21), and IRE1α regulation of the DDR has been shown to require RIDD without XBP1 splicing (45).
Our model is consistent with findings from a study of enforced, persistent ER stress in AT2 cells by conditional deletion of BiP (also known as Grp78, encoded by Hspa5), which impaired AT2-derived progenitor cell function and caused fibrosis (31). Unlike studies of enforced ER stress, however, our results point to a more general model where IRE1α is activated as part of an ensemble of gene expression programs when lung epithelial cells transition into DATPs, even if the primary injury does not cause ER stress per se. Indeed, ER stress as a primary injury to lung epithelial cells is a relatively rare cause of human fibrotic lung disease; even in rare familial interstitial fibrosis, probands with surfactant mutations are a minority, and only a subset of surfactant mutations primarily cause ER stress (61). That IRE1α activation is a general feature of the transition into the DATP phenotype helps explain why epithelial cells with UPR activation have been observed in IPF without clear evidence of misfolded secretory protein (29, 54). Our model also implies that UPR activation will be important in the pathogenesis of other lung fibrotic diseases where DATP analogs have been found, including sarcoidosis, hypersensitivity pneumonitis, nonspecific interstitial pneumonia, and severe SARS-CoV-2 infection (6, 9, 12, 62). A deeper understanding of cellular stress response pathways in progenitor cells may ultimately enable therapies—such as kinase inhibition of IRE1α—that both ameliorate fibrosis and enhance regeneration in diverse human lung diseases.
DATA AVAILABILITY
The data that support this study are available upon request. Data were deposited in the Gene Expression Omnibus series GSE190821 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190821) and series GSE190822 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190822).
SUPPLEMENTAL DATA
Supplemental Figs. S1–S4 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.17293883.
GRANTS
This work was supported by the National Institutes of Health (NIH) Grants R01HL145037 (to D.S. and F.R.P.), T32HL007185 (to D.S.), F32HL145990 and K08HL157654 (to V.C.A.), and R01DK100623 (to B.J.B. and F.R.P.).
DISCLOSURES
B.J.B. and F.R.P. are founders and equity holders of OptiKira, LLC (Cleveland, OH). No funding or chemical matter from OptiKira was used for the work described in this article. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
V.C.A., M.S.D., D.S., and F.R.P. conceived and designed research; V.C.A., M.S.D., M.T., T.A.W., and B.J.B. performed experiments; V.C.A., M.S.D., M.T., T.A.W., and B.J.B. analyzed data; V.C.A., M.S.D., M.T., T.A.W., B.J.B., D.S., and F.R.P. interpreted results of experiments; V.C.A. prepared figures; V.C.A. drafted manuscript; V.C.A., M.S.D., D.S., and F.R.P. edited and revised manuscript; V.C.A., M.S.D., M.T., T.A.W., B.J.B., D.S., and F.R.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank M. Podolsky, J. Kathiriya, A. Brumwell, H. Chapman, R. Ghosh, A. Igbaria, C. Jan, and members of the Papa and Sheppard labs for helpful discussions. We thank the members of the University of California, San Francisco (UCSF) Genomics CoLab and the UCSF Histology and Biomarker Core for technical support.
A preprint is available at https://doi.org/10.1101/2021.09.16.460705.
REFERENCES
- 1.Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, Calfee CS. Acute respiratory distress syndrome. Nat Rev Dis Primers 5: 18, 2019. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolters PJ, Blackwell TS, Eickelberg O, Loyd JE, Kaminski N, Jenkins G, Maher TM, Molina-Molina M, Noble PW, Raghu G, Richeldi L, Schwarz MI, Selman M, Wuyts WA, Schwartz DA. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 6: 154–160, 2018. doi: 10.1016/S2213-2600(18)30007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, Miller AJ, Spence JR, Gerber AN, Hesselberth JR, Zemans RL. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight 5: e123637, 2019. doi: 10.1172/jci.insight.123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joshi N, Watanabe S, Verma R, Jablonski RP, Chen CI, Cheresh P, Markov NS, Reyfman PA, McQuattie-Pimentel AC, Sichizya L, Lu Z, Piseaux-Aillon R, Kirchenbuechler D, Flozak AS, Gottardi CJ, Cuda CM, Perlman H, Jain M, Kamp DW, Scott Budinger GR, Misharin AV. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J 55: 1900646, 2020. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Königshoff M, Chapman HA, Theis FJ, Schiller HB. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun 11: 3559, 2020. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, Banovich NE, Kropski JA, Tata PR. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol 22: 934–946, 2020. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, Zemans RL. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med 201: 1443–1447, 2020. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi J, Park JE, Tsagkogeorga G, Yanagita M, Koo BK, Han N, Lee JH. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell 27: 366–382.e7, 2020. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595: 107–113, 2021. doi: 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Wu H, Yu Y, Tang N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res 30: 708–710, 2020. doi: 10.1038/s41422-020-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bharat A, Querrey M, Markov NS, Kim S, Kurihara C, Garza-Castillon R, Manerikar A, Shilatifard A, Tomic R, Politanska Y, Abdala-Valencia H, Yeldandi AV, Lomasney JW, Misharin AV, Budinger GRS. Lung transplantation for patients with severe COVID-19. Sci Transl Med 12: eabe4282, 2020. doi: 10.1126/SCITRANSLMED.ABE4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ting C, Aspal M, Vaishampayan N, Huang SK, Riemondy KA, Wang F, Farver C, Zemans RL. Fatal COVID-19 and non-COVID-19 acute respiratory distress syndrome is associated with incomplete alveolar type 1 epithelial cell differentiation from the transitional state without fibrosis. Am J Pathol 192: 454–467, 2022. doi: 10.1016/j.ajpath.2021.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auyeung VC, Sheppard D. Stuck in a moment: does abnormal persistence of epithelial progenitors drive pulmonary fibrosis? Am J Respir Crit Care Med 203: 667–669, 2021. doi: 10.1164/rccm.202010-3898ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verheyden JM, Sun X. A transitional stem cell state in the lung. Nat Cell Biol 22: 1025–1026, 2020. doi: 10.1038/s41556-020-0561-5. [DOI] [PubMed] [Google Scholar]
- 15.Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell 69: 169–181, 2018. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 16.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138: 562–575, 2009. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323–331, 2009. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerner AG, Upton J-P, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16: 250–264, 2012. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh R, Wang L, Wang ES, Perera BGK, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schürer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158: 534–548, 2014. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, Mehdizadeh M, Ghosh R, Wang L, Colon-Negron K, Meza-Acevedo R, Backes BJ, Maly DJ, Bluestone JA, Papa FR. Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab 25: 883–897.e8, 2017. [Erratum in Cell Metab 25: 1207, 2017]. doi: 10.1016/j.cmet.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thamsen M, Ghosh R, Auyeung VC, Brumwell A, Chapman HA, Backes BJ, Perara G, Maly DJ, Sheppard D, Papa FR. Small molecule inhibition of IRE1α kinase/RNase has anti-fibrotic effects in the lung. PLoS One 14: e0209824, 2019. doi: 10.1371/journal.pone.0209824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaughan A, Brumwell A, Chapman H. Lung epithelial cell prep. Protoc Exch : 1–4, 2015. doi: 10.1038/protex.2015.017. [DOI] [Google Scholar]
- 23.Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci USA 97: 1778–1783, 2000. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118, 2010. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med 203: 707–717, 2021. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenkins RG, Su X, Su G, Scotton CJ, Camerer E, Laurent GJ, Davis GE, Chambers RC, Matthay MA, Sheppard D. Ligation of protease-activated receptor 1 enhances α vbeta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest 116: 1606–1614, 2006. doi: 10.1172/JCI27183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young LR, Pasula R, Gulleman PM, Deutsch GH, McCormack FX. Susceptibility of Hermansky-Pudlak mice to bleomycin-induced type II cell apoptosis and fibrosis. Am J Respir Cell Mol Biol 37: 67–74, 2007. doi: 10.1165/rcmb.2006-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng D-S, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 294: L1119–L1126, 2008. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 29.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle R-MM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178: 838–846, 2008. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo Sj, Headen AC, Basil MC, Stark JM, Mulugeta S, Deterding RR, Beers MF. . A SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 4: e126125, 2019. doi: 10.1172/jci.insight.126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borok Z, Horie M, Flodby P, Wang H, Liu Y, Ganesh S, Firth AL, Minoo P, Li C, Beers MF, Lee AS, Zhou B. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med 201: 198–211, 2020. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118: 517–528, 2004. doi: 10.1016/j.cell.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 33.Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci USA 103: 2208–2213, 2006. doi: 10.1073/pnas.0510839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167: 1867–1882.e21, 2016. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papa FR, Zhang C, Shokat K, Walter P. Bypassing a kinase activity with an ATP-competitive drug. Science 302: 1533–1537, 2003. doi: 10.1126/science.1090031. [DOI] [PubMed] [Google Scholar]
- 36.Han D, Upton J-P, Hagen A, Callahan J, Oakes SA, Papa FR. A kinase inhibitor activates the IRE1alpha RNase to confer cytoprotection against ER stress. Biochem Biophys Res Commun 365: 777–783, 2008. doi: 10.1016/j.bbrc.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 37.Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA 106: 13939–13944, 2009. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haimon Z, Volaski A, Orthgiess J, Boura-Halfon S, Varol D, Shemer A, Yona S, Zuckerman B, David E, Chappell-Maor L, Bechmann I, Gericke M, Ulitsky I, Jung S. Re-evaluating microglia expression profiles using RiboTag and cell isolation strategies. Nat Immunol 19: 636–644, 2018. doi: 10.1038/s41590-018-0110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, Dellamaggiore K, Kendall R, Beckmann H, Jaeckel P, Materna-Reichelt S, Allen JR, Lipford JR. Unfolded protein response in cancer: IRE1α inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med Chem Lett 6: 68–72, 2015. doi: 10.1021/ml500315b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore K, Hollien J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell 26: 2873–2884, 2015. doi: 10.1091/mbc.E15-02-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kathiriya JJ, Brumwell AN, Jackson JR, Tang X, Chapman HA. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell 26: 346–358.e4, 2020. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinon F, Chen X, Lee A-H, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11: 411–418, 2010. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loschko J, Rieke GJ, Schreiber HA, Meredith MM, Yao KH, Guermonprez P, Nussenzweig MC. Inducible targeting of cDCs and their subsets in vivo. J Immunol Methods 434: 32–38, 2016. doi: 10.1016/j.jim.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim KK, Sheppard D, Chapman HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol 10: a022293, 2018. doi: 10.1101/cshperspect.a022293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dufey E, Bravo-San Pedro JM, Eggers C, González-Quiroz M, Urra H, Sagredo AI, Sepulveda D, Pihán P, Carreras-Sureda A, Hazari Y, Sagredo EA, Gutierrez D, Valls C, Papaioannou A, Acosta-Alvear D, Campos G, Domingos PM, Pedeux R, Chevet E, Alvarez A, Godoy P, Walter P, Glavic A, Kroemer G, Hetz C. Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response. Nat Commun 11: 2401, 2020. doi: 10.1038/s41467-020-15694-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P-Y, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868, 2008. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J 50: 1602367, 2017. doi: 10.1183/13993003.02367-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, Van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 184: 569–581, 2011. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 49.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214: 2387–2404, 2017. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172, 2019. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Satoh T, Nakagawa K, Sugihara F, Kuwahara R, Ashihara M, Yamane F, Minowa Y, Fukushima K, Ebina I, Yoshioka Y, Kumanogoh A, Akira S. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 541: 96–101, 2017. doi: 10.1038/nature20611. [DOI] [PubMed] [Google Scholar]
- 52.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 199: 1517–1536, 2019. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, Shi M, Zhao X, Yuan J, Li J, Yang X, Bin E, Wei D, Zhang H, Zhang J, Yang C, Cai T, Dai H, Chen J, Tang N. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell 180: 107–121.e17, 2020. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 54.Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108: 10562–10567, 2011. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q, Arnett HA, Siddiqui A, Washko GR, Homer R, Yan X, Rosas IO, Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6: eaba1983, 2020. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 46: 731–739, 2012. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 57.Heindryckx F, Binet F, Ponticos M, Rombouts K, Lau J, Kreuger J, Gerwins P. Endoplasmic reticulum stress enhances fibrosis through IRE1α-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol Med 8: 729–744, 2016. doi: 10.15252/emmm.201505925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kathiriya JJ, Wang C, Zhou M, Brumwell A, Cassandras M, Le Saux CJ, Cohen M, Alysandratos K-D, Wang B, Wolters P, Matthay M, Kotton DN, Chapman HA, Peng T. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat Cell Biol 24: 10–23, 2022. doi: 10.1038/s41556-021-00809-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 508: 103–107, 2014. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simic MS, Moehle EA, Schinzel RT, Lorbeer FK, Halloran JJ, Heydari K, Sanchez M, Jullié D, Hockemeyer D, Dillin A. Transient activation of the UPRER is an essential step in the acquisition of pluripotency during reprogramming. Sci Adv 5: eaaw0025, 2019. doi: 10.1126/sciadv.aaw0025_rfseq1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J 45: 1717–1727, 2015. doi: 10.1183/09031936.00163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, Raju L, Roberson J, Ding G, Wood L, Sucre JMS, Richmond BW, Serezani AP, McDonnell WJ, Mallal SB, Bacchetta MJ, Loyd JE, Shaver CM, Ware LB, Bremner R, Walia R, Blackwell TS, Banovich NE, Kropski JA. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 6: eaba1972, 2020. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S4 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.17293883.
Data Availability Statement
The data that support this study are available upon request. Data were deposited in the Gene Expression Omnibus series GSE190821 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190821) and series GSE190822 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190822).