

Abstract
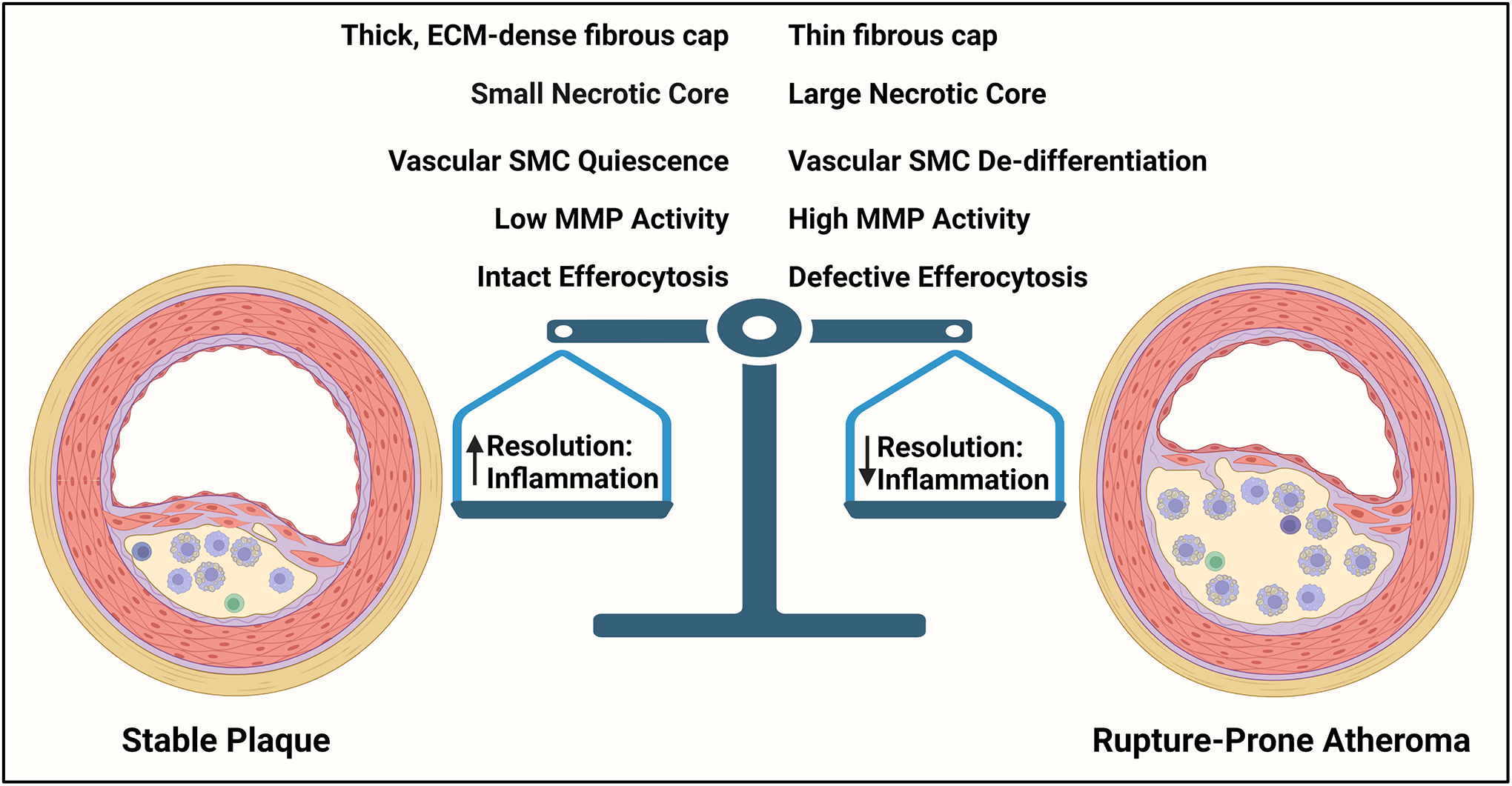
Most acute cardiovascular events are due to plaque rupture, with atheromas containing large necrotic cores and thin fibrous caps being more susceptible to rupture and lesions with small necrotic cores and thick fibrous caps being more protected from rupture. Atherosclerotic plaques are comprised of various extracellular matrix proteins, modified lipoprotein particles, and cells of different origins, i.e. vascular cells and leukocytes. Although much has been revealed about the mechanisms that lead to plaque instability, several key areas remain incompletely understood. This In-Focus Review highlights processes related to cellular crosstalk and the role of the tissue microenvironment in determining cell function and plaque stability. Recent advances highlight critical underpinnings of atherosclerotic plaque vulnerability, particularly impairments in the ability of macrophages to clear dead cells and phenotypic switching of vascular smooth muscle cells. However, these processes do not occur in isolation, as crosstalk between macrophages and vascular smooth muscle cells and interactions with their surrounding microenvironment play a significant role in determining plaque stability. Understanding these aspects of cellular crosstalk within an atherosclerotic plaque may shed light on how to modify cell behavior and identify novel approaches to transform rupture-prone atheromas into stable lesions.
Keywords: macrophages, vascular smooth muscle cells, atherosclerosis, ECM remodeling, efferocytosis
Graphical Abstract.
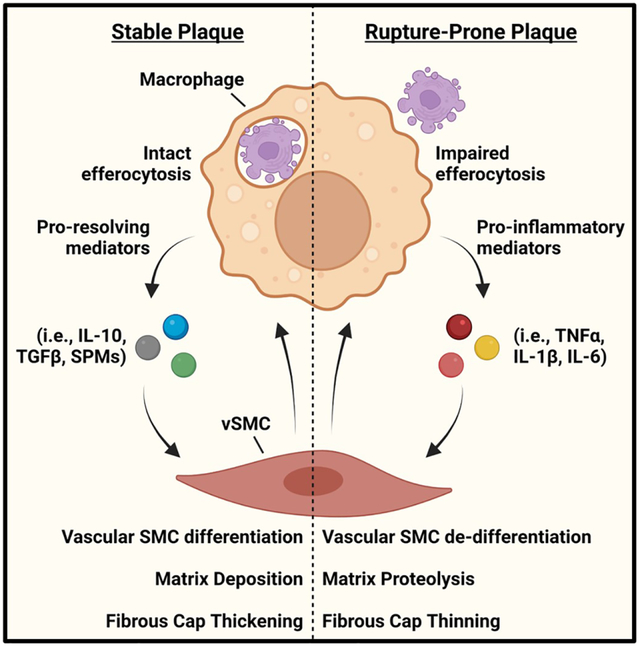
Features of Stable and Unstable Atherosclerosis. Stable plaques are characterized by a thick fibrous cap and small necrotic core. These plaques also show features of vSMC quiescence and have low MMP activity. Macrophages in stable plaques also show intact efferocytosis. Rupture-prone plaques contain large necrotic cores and thin fibrous caps. These atheromas also show features associated with vSMC de-differentiation and contain high MMP activity and ECM proteolysis. Macrophages from rupture-prone atheromas also display impaired efferocytosis. The stability of an atherosclerotic plaque is owed to a balance between resolving mediators and inflammatory factors such that an increase in resolution mediators drive a more stable plaque, whereas an increase in inflammatory factors promote a more vulnerable plaque.
Background
Exponential advancements in molecular and cellular technologies have significantly expanded our understandings of atherosclerotic cardiovascular disease (ASCVD)1. It is now evident that the interplay between soluble factors, cell-cell interactions, and the tissue microenvironment governs atherosclerosis progression and plaque stability2. Subendothelial retention of plasma-derived apolipoprotein B-containing lipoproteins in the intima of medium-to-large-sized arteries drives a low-grade inflammatory response that, if sustained, initiates atherosclerosis3. These retained lipoproteins undergo extracellular modification that activate endothelial and vascular smooth muscle cells and promote the secretion of pro-inflammatory chemokines and cytokines that drive the recruitment of leukocytes4. Monocyte-derived macrophages also secrete factors that modify lipoproteins further, promoting a feed-forward loop of lipid modification and chronic inflammation5. Furthermore, impairments in the phagocytic clearance of apoptotic cells (ACs) by macrophages also sustain inflammation, and together with the continued accumulation of modified lipoproteins, rampant cell death, and pooling of extracellular lipids, a necrotic core is formed. These necrotic cores are clinically dangerous as they often are the site of intraplaque hemorrhaging, contain highly thrombogenic material, and act as the nucleating site for calcium deposition6. These intimal inflammatory responses also cause vascular smooth muscle cells (vSMCs) in the tunica media to de-differentiate towards a synthetic phenotype, characterized by high rates of proliferation and migration, and drive plaque expansion that occludes the lumen through fibroproliferative remodeling7.
Powerful therapies exist to lower plasma LDL cholesterol and impede atherosclerosis progression. However, despite aggressive lipid-lowering strategies, ASCVD remains the leading cause of morbidity and mortality worldwide. The recent success of the CANTOS and COLCOT trials demonstrate that targeting aspects of inflammation will complement our current approaches that lower circulating cholesterol in treating ASCVD8, 9. However, individuals that are undergoing treatments to lower inflammation experience a heightened rate of infection-related adverse events. Therefore, strategies aimed specifically at transforming rupture-prone atheromas into stable plaques may be a more attractive approach (Fig. 1). In this review, we summarize recent work that is beginning to elucidate the molecular and cellular mechanisms that determine plaque stability and highlight the major questions facing the field.
Figure 1. Reciprocal Crosstalk between Macrophages and vSMCs.
Macrophages clearing dead cells produce pro-resolving mediators, such as IL-10, TGFβ, and SPMs. These in turn cause vSMCs to differentiate and deposit ECM, leading to thicker fibrous caps. However, when efferocytosis is impaired, macrophages release pro-inflammatory mediators, such as TNFα, IL-1β, and IL-6 that cause vSMC de-differentiation, ECM degradation, and fibrous cap thinning. These functions are reciprocal and can lead to either plaque stability or rupture-prone atheromas.
The Role of Vascular Smooth Muscle Cells in Fibroproliferative Remodeling as a Determinant in Fibrous Cap Formation
During homeostasis, vascular smooth muscle cells (vSMCs) regulate blood pressure and distribution and provide structural integrity for blood vessels. However, vSMCs are remarkably plastic and can significantly contribute to vascular remodeling under physiological and pathological settings. This is owed to their ability to undergo dynamic switching between a differentiated, termed “quiescent”, and de-differentiated, termed “synthetic”, phenotype10. Normally, vSMCs proliferate and migrate at an extremely low rate and express an exclusive repertoire of vSMC-specific genes, which include αSMA, γSMA, Calponin, SM22α, smoothelin, and SM-MHC11. This is dominantly controlled by myocardin-serum response factor (SRF) and kruppel-like factor 4 (KLF4)12, 13. Vascular SMC-specific genes contain the conserved DNA recognition element CC(A/T-rich)6GG (CArG), which requires the transcription factor SRF bound directly to the coactivator Myocardin to drive gene transcription. However, this mechanism can be antagonized by KLF4, which suppresses myocardin expression and prevents SRF from associating with CArG boxes by binding to G/C repressor elements14. Importantly, deletion of KLF4 in vSMCs mitigates the loss in vSMC-specific genes during injury, limits proliferation, and significantly lowers vascular remodeling during atherosclerosis15, 16. KLF4-dependent de-differentiation of vSMCs also promotes their conversion towards a macrophage-like state17.
While positive remodeling during atherosclerosis progression preserves vessel patency by expanding outwards, negative remodeling, which occurs once the vessel has expanded by more than 40%, occludes lumen diameter as atherosclerosis advances18. However, myocardial infarctions and strokes are caused by the sudden rupture of atherosclerotic plaques, which may not be overly stenotic, and subsequent luminal thrombosis19. Central to plaque stability is the composition and size of the fibrous cap overlying a necrotic core (Fig. 1). Because vSMCs are the major producer of ECM proteins in an atheroma, their population within the fibrous cap directly correlates with plaque stability20 21. Interestingly, these vSMCs that dominate the protective fibrous cap re-express genes associated with vSMC differentiation, such as αSMA and SM22. Therefore, the prevailing theory has been that while vSMC de-differentiation directly contributes to plaque size, phenotypic switching also plays a beneficial role in plaque stabilization by forming the protective ECM-dense fibrous cap. However, these beneficial functions may be negatively regulated by the inflammatory components within the fibrous cap.
Extracellular matrix (ECM) composition and organization govern cell function, and reciprocally, cells actively remodel their surrounding ECM22. During development, the ECM undergoes controlled matrix remodeling whereby organ morphogenesis occurs uninterrupted. However, uncontrolled remodeling disrupts tissue function, causes tissue fibrosis, and propagates non-resolving inflammatory diseases23. Laminins, polymerized collagen I, collagen III, collagen IV, small amounts of fibronectin, and elastin fibers comprise the ECM during the early stages of atherosclerosis. In contrast, the ECM at later stages of atherosclerosis is comprised of monomeric collagen I, fibronectin, fibrinogen, vitronectin, and osteopontin2, 24, 25. Interactions with the ECM are largely mediated by the integrin family of cell-matrix adhesion receptors. Encompassing 18 α subunits and 8 β subunits, integrins heterodimerize into 24 distinct integrin pairs and regulate a panoply of signaling pathways26. Vascular SMCs interact with the surrounding ECM through the laminin-binding integrins α3β1 and α7β1 and the collagen-binding integrins α1β1 and α2β127. However, the ECM proteins assembled during atherosclerosis bind to the integrins α5β1, αvβ3, and αvβ528. Vascular SMC function is influenced by their interactions with the ECM. For instance, the integrin α1β1, which binds to collagen I, is highly expressed in contractile SMCs, and phenotypic switching of vSMCs towards a de-differentiated state decreases integrin α1 gene expression29. Interestingly, collagen I can exist either in a polymeric form, rich in healthy tissue niches, and in a monomeric form, thought to be abundant in advanced stages of atherosclerosis due to high MMP activity30. In its polymeric form, collagen I promotes vSMC quiescence, whereas in its monomeric form, collagen I drives vSMC de-differentiation30. As another example, ligation of integrin α7β1 to laminin in vSMCs lowers ERK activation, reduces proliferation, and prevents vascular remodeling31. Consistently, inhibiting vSMC interactions with matrix proteins that are abundant in advanced stages of atherosclerosis using αvβ3-blocking peptides or function-blocking antibodies lowers vSMC proliferation and reduces atherosclerosis formation32, 33.
In addition to directly governing signaling cascades, the microenvironment also plays a role in retaining modified LDLs, pro-inflammatory cytokines, and many growth factors that perpetuate fibroproliferative remodeling and inflammation within the atheroma. Glycosaminoglycans (GAGs), polysaccharides containing repeating disaccharide units on proteoglycans, directly interact with oxidized LDL, leading to its uptake by macrophages34. Consistently, enhancing LDL interactions with proteoglycans by deleting APRIL, a cytokine known to bind proteoglycans, drives LDL accumulation, enhances macrophage recruitment to atherosclerotic lesions, and worsens necrotic core formation35. As another example, TNFα interacts with laminin and fibronectin, which sustains inflammation36, 37. Interactions between most chemokines with heparan sulfate proteoglycans drive oligomerization and favor their presentation to receptors on leukocytes and promote directed migration towards an ECM-rich chemokine gradient38. GAGs also interact and retain a wide range of growth factors, including PDGF-BB, TGFβ, HGF, FGF, and IGF39. Similar to other matrix-bound soluble factors, growth factors show disparate signaling pathways compared to their soluble-only versions. For instance, soluble growth factors are rapidly internalized and degraded upon binding to their cognate receptor. However, growth factors become immobilized once bound to the ECM and are then resistant to endocytosis and degradation, which sustains growth factor signaling. Also, growth factor receptors can associate with cell-matrix adhesion proteins that potentiate growth factor-dependent proliferation, migration, and angiogenesis40.
PDGF-BB is a dominant growth factor that drives vSMC de-differentiation, proliferation, and migration. Inhibiting PDGF-BB signaling by blocking PDGF receptors lowers fibrous cap formation in ApoE knockout mice fed a high-fat diet41. Interestingly, PDGF-BB-dependent vascular SMC migration and expansion of the neointima can be antagonized by IL-1β42. This is consistent with mouse models demonstrating that administration of an IL-1β neutralizing antibody decreased overall plaque burden and is supported by the success of the CANTOS trial, where canakinumab was shown to lower mortality from cardiovascular disease8, 43. In contrast to these findings, an elegant study demonstrated that when neutralizing antibodies against IL-1β were delivered to mice with pre-established atherosclerosis vSMC incorporation into the fibrous cap was significantly reduced, macrophage migration was enhanced, and beneficial outward remodeling was impaired44. The features associated with rupture-prone atheromas can be enhanced by IFNγ, as elevated expression of this cytokine blunts collagen synthesis by vSMC and drives apoptosis45, 46. Furthermore, destruction of the ECM by matrix metalloproteinases (MMPs) weaken the protective fibrous cap. For instance, rupture-prone plaques in humans show enhanced MMP activity compared to stable plaques, and MMPs are considered a risk factor for future cardiovascular events47. Furthermore, MMP2 and MMP9 knockout mice show reduced plaque size and exhibit features of plaque stability48, 49. Consistently, promoting vSMC differentiation by increasing TGFβ signaling or over-expressing insulin growth factor-1 specifically in vSMCs enhances fibrous cap thickening50, 51. As another example, deletion of KLF4 specifically in vSMCs enhances fibrous cap thickening and is associated with an increase in αSMA-positive cells in the fibrous cap13.
The Role of Efferocytosis by Macrophages in Inflammation Resolution as a Determinant in Necrotic Core formation
Phagocytosis of apoptotic cells, termed “efferocytosis”, prevents secondary necrosis, terminates inflammatory responses, and activates pro-resolving pathways52. These protective functions are compromised when efferocytosis is impaired, leading to non-resolving inflammatory diseases. Cells that die early during atherosclerosis formation become rapidly cleared, but through a variety of mechanisms, efferocytosis becomes defective as atherosclerosis progresses53, 54. Macrophages interact with apoptotic cells through a unique repertoire of direct and indirect receptors, yet many of these are either downregulated or cleaved as atherosclerosis advances. As one example, MerTK expression decreases and what remains on the cell surface of macrophages is cleaved by the disintegrin and metalloproteinase domain-containing protein 17 (ADAM17)55. Mice expressing a cleavage-resistant form of MerTK have plaques with smaller necrotic cores that is associated with increased lesional efferocytosis56. Importantly, MerTK also stimulates signaling pathways that dampen inflammation and drive the synthesis of specialized pro-resolving lipid mediators (SPMs), which are lipid species that significantly blunt atherosclerosis progression57–59. Another mechanism of impaired efferocytosis during atherosclerosis is oxidized LDL-dependent downregulation of LRP1. Treating macrophages in vitro with oxidized LDL drives epsin-mediated, ubiquitin-dependent internalization and degradation of LRP1. Consistently, loss of epsin 1 and 2 in myeloid cells lowers plaque size and necrotic core area by enhancing LRP1-mediated efferocytosis60. Furthermore, LRP1 in macrophages is necessary for anti-CD47 blocking antibodies to increase efferocytosis, which have been shown to reduce atherosclerosis progression61. The bridging molecule MFGE8 ligates ACs to integrin αVβ3 and transglutaminase 2 on macrophages to drive AC internalization, yet MFGE8 expression declines as atherosclerosis advances. Mice with a genetic deletion in MFGE8 in hematopoietic cells show an increase in plaque size and necrotic core areas62. Altogether, impairments in efferocytosis unequivocally lead to post-apoptotic necrosis and cause necrotic core expansion (Fig. 1).
The breakdown, metabolism, and response to AC-derived molecules by macrophages, termed “efferotabolism”, is an exciting new area of study as it continues the successive clearance of dead cells and transitions the termination of inflammation into resolution53. After an AC has been broken down in the phagolysosome, the digested components burden a macrophage with substances that must either be rapidly metabolized or exported63–67. For instance, intracellular cholesterol rises when a macrophage degrades a foamy AC. Phagolysosomal cholesterol is then trafficked to the ER and is acted on by acyl-CoA:cholesterol acyltransferase to esterify free cholesterol to cholesterol fatty acid esters, which restrict the membrane-damaging effects of elevated levels of free cholesterol68. Also, AC-derived sterols stimulate PPAR and LXR to drive the expression of ABCA1 and ABCG1 to promote cholesterol efflux69. When macrophages are polarized towards an Arg1-positive pro-resolving phenotype, arginine from an AC is converted into ornithine after efferocytosis. This then becomes decarboxylated by the enzyme ODC1 into putrescine, which drives cytoskeletal remodeling to mediate the successive clearance of multiple ACs66. Furthermore, putrescine also maintains MerTK expression on the surface of macrophages, which mediates IL-10 production upon engagement with an AC70. An inability to convert AC-derived arginine into putrescine reduces continual efferocytosis, lowers collagen deposition in the fibrous cap, and prevents a reduction in lesion size and necrotic core area during atherosclerosis regression66. Interestingly, a non-canonical route of glutaminolysis that is dependent on glutaminase 1 supports efferocytosis by promoting oxidative phosphorylation65. Disrupting this unique route of glutaminolysis in myeloid cells using Gls1fl/fl mice showed impairment in AC clearance in vivo and worsened atherosclerosis65.
The Role of vSMC and Macrophage Crosstalk in Plaque Stability
Pro-resolving macrophages, particularly ones actively clearing apoptotic cells, secrete a panoply of molecules that drive features of plaque stability. Factors released by macrophages after efferocytosis that stimulate the waning of inflammation and initiate the active process of resolution include IL-10, TGFβ, and SPMs3. In turn, these secreted factors act on surrounding vSMCs to promote specific functions relating to plaque stability (Fig. 2).
Figure 2. Crosstalk Between Macrophages and vSMCs in Atherosclerosis.
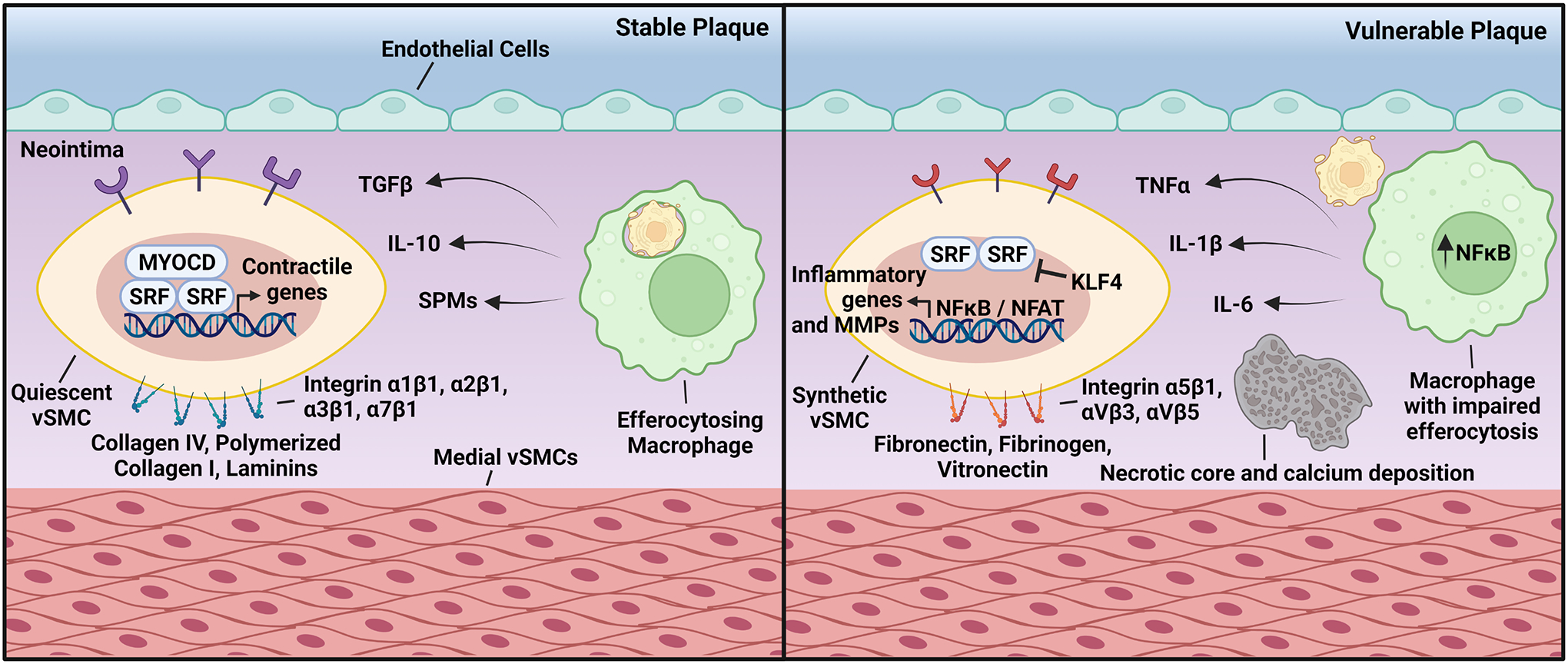
(Left) In stable atherosclerosis, lesional macrophages clearing apoptotic cells produce TGFβ, IL-10, and SPMs. These pro-resolving molecules bind to receptors present on the surface of vSMCs that drive Myocardin/SRF binding to CArG boxes, which promote the expression of genes associated with vSMC quiescence. This can be further enhanced by the interaction between vSMCs and the surrounding ECM through integrins α1β1, α2β1, α3β1, and α7β1. (Right) Plaques vulnerable to rupture contain macrophages with impaired efferocytosis, which leads to the secretion of the proinflammatory cytokines TNFα, IL-1β, and IL-6. These bind to their cognate receptors in vSMCs and downregulate Myocardin expression. Inflammatory cytokines also stimulate the association of KLF4 to G/C elements that prevent SRF binding to CArG boxes. Interactions between vSMCs and the ECM proteins fibronectin, fibrinogen, and vitronectin cause NFκB and NFAT activation and promotes the expression of MMPs and inflammatory genes.
TGFβ is one of the more well-known pro-resolving factors secreted by macrophages after efferocytosis. Human clinical studies demonstrate that higher levels of circulating TGFβ are associated with plaque stability71. Similarly, immunohistochemistry revealed that TGFβ levels were lower in rupture-prone atheromas72. TGFβ enhances vSMC contractile gene expression and also stimulates the expression of ECM genes73, 74. Furthermore, TGFβ suppresses inducible nitric oxide synthase and interleukin 6 (IL-6) in vSMCs by lowering SMAD3 signaling75. These reports suggest that TGFβ is protective during atherosclerosis by reducing inflammation and enhancing ECM production. Another pro-resolving factor released by efferocytosing macrophages is IL-10. This pro-resolving cytokine has potent protective effects in vSMCs. For instance, IL-10 inhibits LPS-induced IL-6 secretion by inactivating NFκB. Furthermore, IL-10 reduces balloon injury-induced vascular remodeling by inhibiting vSMC proliferation and migration76. Through similar mechanisms, IL-10 also inhibits monocrotaline-induced pulmonary arterial hypertension and angiotensin II-induced hypertension77, 78.
In advanced atherosclerosis, there is an imbalance between specialized pro-resolving lipid mediators (SPMs), such as resolvins and lipoxins, and pro-inflammatory factors. SPMs actively drive inflammation resolution, increase the clearance of dead cells by macrophages, and enhance features of plaque stability during atherosclerosis79. Lipoxins, resolvins, protectins, and maresins comprise the family of SPMs and are derived from n-3 fatty acid-derived eicosapentaenoic acid (EPA), docosapentaenoic acid (DPA), or docosahexaenoic acid (DHA)79. EPA, DPA, and DHA are released from phospholipids by phospholipases and then acted on by either 5-lipoxygenase, 12-lipoxygenase, or 15-lipoxygenase to yield SPMs80. In addition to the protective effects of SPMs in macrophages, SPMs also positively influence vSMC function. Resolvin D1 and D2 (RvD1 and RvD2) dose-dependently inhibit vSMC proliferation, migration, superoxide production, and proinflammatory gene expression. Furthermore, these D-series resolvins inhibit neointimal hyperplasia following arterial angioplasty81. RvE1 and lipoxins inhibit PDGF-induced vSMC migration by inhibiting PDGFR activation82. As another example, maresin 1 (MaR1) blunts CCL2 production in vSMCs by inhibiting TNFα-dependent NFκB activation83. Furthermore, MaR1 treatment also blocks vSMC migration towards PDGF and attenuates neointimal hyperplasia84.
Alterations in cell metabolism following efferocytosis also contribute to macrophage-vSMC crosstalk. Internalization of an AC stimulates aerobic glycolysis and expression of the lactate transporter Slc16a185. Upon its release by efferocytosing macrophages, lactate then acts in a paracrine manner in nearby macrophages to upregulate production of the pro-resolving mediators TGFβ and IL-10. Interestingly, lactate is also taken up by vSMCs, and vSMCs take on a more proliferative and matrix-producing phenotype in an environment high in lactate86. However, there appears to be a balance in lactate in the microenvironment as uncontrolled uptake of lactate by vSMCs accelerates calcification87. Polyamines synthesized from macrophages after efferocytosis may be secreted and act on vSMCs in atheromas to promote plaque stability. Polyamines can promote vSMC migration and proliferation, features that lead to fibrous cap thickening. Interestingly, polyamine uptake by vSMCs requires caveolin-1 (Cav-1), and vSMCs isolated from Cav-1 KO mice are highly migratory and proliferative in a manner that is polyamine-dependent88. Altogether, these studies point to a model whereby macrophages produce pro-resolving mediators to restrict necrotic core expansion, and surrounding vSMCs respond by establishing a matrix-dense fibrous cap (Fig. 2).
Substantial advances in the field of bioinformatics, particularly in single-cell RNA sequencing (scRNA-seq), has provided detailed analysis of the transcriptome in individual cells and is beginning to validate the existence of cellular crosstalk within atherosclerotic plaques89. For instance, scRNA-seq has been used to profile the cellular landscape of atherosclerotic plaques from both humans and mice, revealing a larger diversity of immune cells in lesions than previously appreciated. In individuals with stroke, macrophages show high expression of CCL5, and interestingly, its interaction with its cognate receptor CCR5 on vSMCs drives proliferation, de-differentiation, and vascular remodeling, demonstrating the presence of cell-cell communication within atherosclerotic plaques90, 91. As another example, using “scTalk”, an advanced network-based modeling method using confirmed interactions from StringDB, suggest that vSMCs signal to fibroblasts via C3 complement and MMP292. New insights provided by scRNA-seq have also confirmed that vSMCs execute de-differentiation programs and give rise to a population of SEM cells (stem cell, endothelial cell, monocyte) that may further transform into fibrochondrocyte-like cells, taking on features associated with inflammation and ECM degradation92, 93.
Concluding Remarks and Future Directions
Cholesterol-lowering therapies are an effective strategy to lower the risk of ASCVD. However, despite the availability of tools that aggressively lower LDL-cholesterol, the residual risk that remains causes ASCVD to continue being the leading cause of morbidity and mortality world-wide. The success of the CANTOS and COLCOT trials demonstrate that lowering inflammation is a key requisite for attenuating this residual risk8, 9. However, individuals on anti-inflammatory therapies remain susceptible to infection-related adverse outcomes. Therefore, therapeutic strategies that focus on transitioning a rupture-prone plaque into a stable plaque may be more desirable. The concept that macrophages and vSMCs crosstalk within atheromas and that their communication can either promote plaque stability or contribute to plaque vulnerability remains understudied. Achieving a deeper mechanistic insight into understanding the crosstalk between vSMCs and macrophages will lead to strategies that limit matrix proteolysis, enhance ECM deposition in the fibrous cap, tune vSMC phenotype, and drive the pro-resolving function of macrophages. Furthermore, defining how the tissue microenvironment contributes to the non-resolving aspect of cardiovascular disease may lead to identifying how soluble and insoluble factors guide macrophage and vSMC communication within atheromas.
HIGHLIGHTS.
Atherosclerotic plaque stability is controlled by the balance between resolution mediators and pro-inflammatory factors.
The tissue microenvironment influences macrophage and vSMC function by tuning responses to soluble and insoluble factors.
Cellular crosstalk between macrophages and vSMCs control plaque stability.
ACKNOWLEDGMENTS
Figures were created with www.BioRender.com.
SOURCES OF FUNDING
This work was supported by the NIH grant R00 HL145131.
Nonstandard Abbreviations and Acronyms:
- AC
apoptotic cell
- Arg1
arginase 1
- ASCVD
atherosclerotic cardiovascular disease
- ECM
extracellular matrix
- GAGs
glycosaminoglycans
- IL
interleukin
- KLF4
kruppel-like factor 4
- LRP1
LDL receptor related protein 1
- MaR1
maresin 1
- MerTK
MER tyrosine-protein kinase
- ODC1
ornithine decarboxylase 1
- Rv
resolvin
- SPMs
specialized pro-resolving mediator
- SRF
serum response factor
- vSMC
vascular smooth muscle cell
Footnotes
DISCLOSURES
The author declares no competing financial interests.
REFERENCES
- 1.Libby P The changing landscape of atherosclerosis. Nature. 2021;592:524–533. [DOI] [PubMed] [Google Scholar]
- 2.Yurdagul A Jr., Finney AC, Woolard MD, et al. The arterial microenvironment: the where and why of atherosclerosis. Biochem J. 2016;473:1281–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Back M, Yurdagul A Jr., Tabas I, et al. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr. and Orr AW. Blood Brothers: Hemodynamics and Cell-Matrix Interactions in Endothelial Function. Antioxid Redox Signal. 2016;25:415–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg D and Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–6. [DOI] [PubMed] [Google Scholar]
- 6.Bentzon JF, Otsuka F, Virmani R, et al. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–66. [DOI] [PubMed] [Google Scholar]
- 7.Basatemur GL, Jorgensen HF, Clarke MCH, et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16:727–744. [DOI] [PubMed] [Google Scholar]
- 8.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 9.Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 10.Gomez D and Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee MY, Park C, Berent RM, et al. Smooth Muscle Cell Genome Browser: Enabling the Identification of Novel Serum Response Factor Target Genes. PLoS One. 2015;10:e0133751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pipes GC, Creemers EE and Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–56. [DOI] [PubMed] [Google Scholar]
- 13.Shankman LS, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Sinha S, McDonald OG, et al. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem. 2005;280:9719–27. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida T, Sinha S, Dandre F, et al. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003;92:856–64. [DOI] [PubMed] [Google Scholar]
- 16.Manabe I and Owens GK. Recruitment of serum response factor and hyperacetylation of histones at smooth muscle-specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ Res. 2001;88:1127–34. [DOI] [PubMed] [Google Scholar]
- 17.Yap C, Mieremet A, de Vries CJM, et al. Six Shades of Vascular Smooth Muscle Cells Illuminated by KLF4 (Kruppel-Like Factor 4). Arterioscler Thromb Vasc Biol. 2021;41:2693–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glagov S, Weisenberg E, Zarins CK, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–5. [DOI] [PubMed] [Google Scholar]
- 19.Sakakura K, Nakano M, Otsuka F, et al. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013;22:399–411. [DOI] [PubMed] [Google Scholar]
- 20.Allahverdian S, Chaabane C, Boukais K, et al. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. 2018;114:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies MJ, Richardson PD, Woolf N, et al. Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J. 1993;69:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu P, Takai K, Weaver VM, et al. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egeblad M, Nakasone ES and Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doran AC, Meller N and McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strom A, Ahlqvist E, Franzen A, et al. Extracellular matrix components in atherosclerotic arteries of Apo E/LDL receptor deficient mice: an immunohistochemical study. Histol Histopathol. 2004;19:337–47. [DOI] [PubMed] [Google Scholar]
- 26.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. [DOI] [PubMed] [Google Scholar]
- 27.Hultgardh-Nilsson A and Durbeej M. Role of the extracellular matrix and its receptors in smooth muscle cell function: implications in vascular development and disease. Curr Opin Lipidol. 2007;18:540–5. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz MA. Integrin signaling revisited. Trends Cell Biol. 2001;11:466–70. [DOI] [PubMed] [Google Scholar]
- 29.Obata H, Hayashi K, Nishida W, et al. Smooth muscle cell phenotype-dependent transcriptional regulation of the alpha1 integrin gene. J Biol Chem. 1997;272:26643–51. [DOI] [PubMed] [Google Scholar]
- 30.Orr AW, Lee MY, Lemmon JA, et al. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welser JV, Lange N, Singer CA, et al. Loss of the alpha7 integrin promotes extracellular signal-regulated kinase activation and altered vascular remodeling. Circ Res. 2007;101:672–81. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Green J, Yurdagul A Jr., et al. alphavbeta3 Integrins Mediate Flow-Induced NF-kappaB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am J Pathol. 2015;185:2575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maile LA, Busby WH, Nichols TC, et al. A monoclonal antibody against alphaVbeta3 integrin inhibits development of atherosclerotic lesions in diabetic pigs. Sci Transl Med. 2010;2:18ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan M and Aviram M. Retention of oxidized LDL by extracellular matrix proteoglycans leads to its uptake by macrophages: an alternative approach to study lipoproteins cellular uptake. Arterioscler Thromb Vasc Biol. 2001;21:386–93. [DOI] [PubMed] [Google Scholar]
- 35.Tsiantoulas D, Eslami M, Obermayer G, et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature. 2021;597:92–96. [DOI] [PubMed] [Google Scholar]
- 36.Alon R, Cahalon L, Hershkoviz R, et al. TNF-alpha binds to the N-terminal domain of fibronectin and augments the beta 1-integrin-mediated adhesion of CD4+ T lymphocytes to the glycoprotein. J Immunol. 1994;152:1304–13. [PubMed] [Google Scholar]
- 37.Hershkoviz R, Goldkorn I and Lider O. Tumour necrosis factor-alpha interacts with laminin and functions as a pro-adhesive cytokine. Immunology. 1995;85:125–30. [PMC free article] [PubMed] [Google Scholar]
- 38.Xu D and Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 2014;83:129–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarrazin S, Lamanna WC and Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneller M, Vuori K and Ruoslahti E. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 1997;16:5600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sano H, Sudo T, Yokode M, et al. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103:2955–60. [DOI] [PubMed] [Google Scholar]
- 42.Englesbe MJ, Deou J, Bourns BD, et al. Interleukin-1beta inhibits PDGF-BB-induced migration by cooperating with PDGF-BB to induce cyclooxygenase-2 expression in baboon aortic smooth muscle cells. J Vasc Surg. 2004;39:1091–6. [DOI] [PubMed] [Google Scholar]
- 43.Bhaskar V, Yin J, Mirza AM, et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011;216:313–20. [DOI] [PubMed] [Google Scholar]
- 44.Gomez D, Baylis RA, Durgin BG, et al. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med. 2018;24:1418–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLaren JE and Ramji DP. Interferon gamma: a master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009;20:125–35. [DOI] [PubMed] [Google Scholar]
- 46.Buttice G, Miller J, Wang L, et al. Interferon-gamma induces major histocompatibility class II transactivator (CIITA), which mediates collagen repression and major histocompatibility class II activation by human aortic smooth muscle cells. Circ Res. 2006;98:472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li T, Li X, Feng Y, et al. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediators Inflamm. 2020;2020:3872367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi ET, Collins ET, Marine LA, et al. Matrix metalloproteinase-9 modulation by resident arterial cells is responsible for injury-induced accelerated atherosclerotic plaque development in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1020–5. [DOI] [PubMed] [Google Scholar]
- 49.Kuzuya M, Nakamura K, Sasaki T, et al. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1120–5. [DOI] [PubMed] [Google Scholar]
- 50.Chen PY, Qin L, Li G, et al. Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO Mol Med. 2016;8:712–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shai SY, Sukhanov S, Higashi Y, et al. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe−/− mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol. 2010;30:1916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doran AC, Yurdagul A Jr. and Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yurdagul A Jr. Metabolic Consequences of Efferocytosis and its Impact on Atherosclerosis. Immunometabolism. 2021;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kojima Y, Volkmer JP, McKenna K, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorp E, Vaisar T, Subramanian M, et al. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem. 2011;286:33335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai B, Thorp EB, Doran AC, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127:564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tibrewal N, Wu Y, D’Mello V, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283:3618–3627. [DOI] [PubMed] [Google Scholar]
- 58.Cai B, Thorp EB, Doran AC, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. 2016;113:6526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Viola JR, Lemnitzer P, Jansen Y, et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ Res. 2016;119:1030–1038. [DOI] [PubMed] [Google Scholar]
- 60.Brophy ML, Dong Y, Tao H, et al. Myeloid-Specific Deletion of Epsins 1 and 2 Reduces Atherosclerosis by Preventing LRP-1 Downregulation. Circ Res. 2019;124:e6–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mueller PA, Kojima Y, Huynh KT, et al. Macrophage LRP1 (Low-Density Lipoprotein Receptor-Related Protein 1) Is Required for the Effect of CD47 Blockade on Efferocytosis and Atherogenesis-Brief Report. Arterioscler Thromb Vasc Biol. 2022;42:e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ait-Oufella H, Kinugawa K, Zoll J, et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–77. [DOI] [PubMed] [Google Scholar]
- 63.Tabas I and Bornfeldt KE. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ Res. 2020;126:1209–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang S, Weinberg S, DeBerge M, et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019;29:443–456 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merlin J, Ivanov S, Dumont A, et al. Non-canonical glutamine transamination sustains efferocytosis by coupling redox buffering to oxidative phosphorylation. Nat Metab. 2021;3:1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yurdagul A Jr., Subramanian M, Wang X, et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020;31:518–533 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerlach BD, Ampomah PB, Yurdagul A Jr., et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. 2021;33:2445–2463 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui D, Thorp E, Li Y, et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J Leukoc Biol. 2007;82:1040–50. [DOI] [PubMed] [Google Scholar]
- 69.Viaud M, Ivanov S, Vujic N, et al. Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circ Res. 2018;122:1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yurdagul A Jr., Kong N, Gerlach BD, et al. ODC (Ornithine Decarboxylase)-Dependent Putrescine Synthesis Maintains MerTK (MER Tyrosine-Protein Kinase) Expression to Drive Resolution. Arterioscler Thromb Vasc Biol. 2021;41:e144–e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borrelli V, di Marzo L, Sapienza P, et al. Role of platelet-derived growth factor and transforming growth factor beta1 the in the regulation of metalloproteinase expressions. Surgery. 2006;140:454–63. [DOI] [PubMed] [Google Scholar]
- 72.Cipollone F, Fazia M, Mincione G, et al. Increased expression of transforming growth factor-beta1 as a stabilizing factor in human atherosclerotic plaques. Stroke. 2004;35:2253–7. [DOI] [PubMed] [Google Scholar]
- 73.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. [DOI] [PubMed] [Google Scholar]
- 74.Grainger DJ. Transforming growth factor beta and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler Thromb Vasc Biol. 2004;24:399–404. [DOI] [PubMed] [Google Scholar]
- 75.Feinberg MW, Watanabe M, Lebedeva MA, et al. Transforming growth factor-beta1 inhibition of vascular smooth muscle cell activation is mediated via Smad3. J Biol Chem. 2004;279:16388–93. [DOI] [PubMed] [Google Scholar]
- 76.Mazighi M, Pelle A, Gonzalez W, et al. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H866–71. [DOI] [PubMed] [Google Scholar]
- 77.Kim HY, Cha HJ and Kim HS. CCL5 upregulates IL-10 expression and partially mediates the antihypertensive effects of IL-10 in the vascular smooth muscle cells of spontaneously hypertensive rats. Hypertens Res. 2015;38:666–74. [DOI] [PubMed] [Google Scholar]
- 78.Ito T, Okada T, Miyashita H, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101:734–41. [DOI] [PubMed] [Google Scholar]
- 79.Kasikara C, Doran AC, Cai B, et al. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. 2018;128:2713–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Serhan CN, Chiang N and Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin Immunol. 2015;27:200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyahara T, Runge S, Chatterjee A, et al. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013;27:2220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ho KJ, Spite M, Owens CD, et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol. 2010;177:2116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chatterjee A, Sharma A, Chen M, et al. The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS One. 2014;9:e113480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akagi D, Chen M, Toy R, et al. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. 2015;29:2504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morioka S, Perry JSA, Raymond MH, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature. 2018;563:714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang L, Gao L, Nickel T, et al. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ Res. 2017;121:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu Y, Ji JJ, Yang R, et al. Lactate accelerates calcification in VSMCs through suppression of BNIP3-mediated mitophagy. Cell Signal. 2019;58:53–64. [DOI] [PubMed] [Google Scholar]
- 88.Grossi M, Rippe C, Sathanoori R, et al. Vascular smooth muscle cell proliferation depends on caveolin-1-regulated polyamine uptake. Biosci Rep. 2014;34:e00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fernandez DM and Giannarelli C. Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat Rev Cardiol. 2022;19:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin CS, Hsieh PS, Hwang LL, et al. The CCL5/CCR5 Axis Promotes Vascular Smooth Muscle Cell Proliferation and Atherogenic Phenotype Switching. Cell Physiol Biochem. 2018;47:707–720. [DOI] [PubMed] [Google Scholar]
- 92.Ma WF, Hodonsky CJ, Turner AW, et al. Enhanced single-cell RNA-seq workflow reveals coronary artery disease cellular cross-talk and candidate drug targets. Atherosclerosis. 2022;340:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pan H, Xue C, Auerbach BJ, et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]