

Abstract
Thalamocortical network dysfunction contributes to seizures and sleep deficits in Dravet syndrome (DS), an infantile epileptic encephalopathy, but the underlying molecular and cellular mechanisms remain elusive. DS is primarily caused by mutations in the SCN1A gene encoding the voltage-gated sodium channel NaV1.1, which is highly expressed in GABAergic reticular thalamus (nRT) neurons as well as glutamatergic thalamocortical neurons. We hypothesized that NaV1.1 haploinsufficiency alters somatosensory corticothalamic circuit function through both intrinsic and synaptic mechanisms in nRT and thalamocortical neurons. Using Scn1a heterozygous mice of both sexes aged P25-P30, we discovered reduced excitability of nRT neurons and thalamocortical neurons in the ventral posterolateral (VPL) thalamus, while thalamocortical ventral posteromedial (VPM) neurons exhibited enhanced excitability. NaV1.1 haploinsufficiency enhanced GABAergic synaptic input and reduced glutamatergic input to VPL neurons, but not VPM neurons. In addition, glutamatergic input to nRT neurons was reduced in Scn1a heterozygous mice. These findings introduce alterations in glutamatergic synapse function and aberrant glutamatergic neuron excitability in the thalamus as disease mechanisms in DS, which has been widely considered a disease of GABAergic neurons. This work reveals additional complexity that expands current models of thalamic dysfunction in DS and identifies new components of corticothalamic circuitry as potential therapeutic targets.
Keywords: Dravet syndrome, NaV1.1, SCN1A, somatosensory thalamus, reticular thalamus, thalamocortical neuron, glutamatergic, GABAergic, synaptic transmission, excitability
INTRODUCTION
Dravet syndrome (DS) is an infantile epileptic encephalopathy most often caused by mutations in the SCN1A gene, which encodes the alpha subunit of the NaV1.1 voltage-gated sodium channel (Claes et al., 2001; Dravet, 2011). DS is characterized by intractable convulsive and non-convulsive (absence) seizures in infancy as well as progressive symptomology including ataxia, intellectual disability, attention deficits, autistic features, sleep disruptions, and a high risk of sudden death (Berkvens et al., 2015; Darra et al., 2019; Dravet, 2011; Licheni et al., 2018; Ragona, 2011; Rodda et al., 2012; Takayama et al., 2014). NaV1.1 is expressed in the axon initial segment as well as distal axons of primarily, but not exclusively, GABAergic neuron populations (Favero et al., 2018; Hedrich et al., 2014; Ogiwara et al., 2007). More than half of DS mutations lead to haploinsufficiency of NaV1.1, which disrupts the excitatory/inhibitory balance across a variety of brain circuits in DS models (Bender et al., 2016; Kalume et al., 2007; Ogiwara et al., 2007; Ritter-Makinson et al., 2019; Tai et al., 2014). The multifaceted DS phenotype likely arises from dysfunction of specific cell-types within these brain circuits (Bender et al., 2013; Han et al., 2012; Hedrich et al., 2014; Kalume et al., 2015; Kalume et al., 2007; Ogiwara et al., 2007; Ritter-Makinson et al., 2019; Rubinstein et al., 2015a; Tai et al., 2014), which may contribute to the limited efficacy of pharmacological tools targeting brain-wide excitation or inhibition in DS (Chiron, 2011; Cross et al., 2019; Gataullina and Dulac, 2017; Takayama et al., 2014). Therefore, elucidating the precise cellular and molecular mechanisms underlying dysfunction in particular circuits may provide more specific therapeutic targets to ameliorate DS symptoms.
The somatosensory corticothalamic (CT) circuit controls somatic information flow between the periphery and the cerebral cortex, and it is involved in attention, pain processing, and sleep (Beenhakker and Huguenard, 2009; Wolff and Vann, 2019; Zikopoulos and Barbas, 2006). This circuit generates oscillatory sleep spindles that are critical for NREM sleep (Fernandez and Luthi, 2020), and under pathological conditions these oscillations can become hypersynchronous, leading to absence seizures (Cheong and Shin, 2013; Lee et al., 2004; McCafferty et al., 2018). Altered thalamic activity is evident in some DS patients (Moehring et al., 2013), and DS mouse models exhibit somatosensory CT circuit dysfunction including reduced sleep spindles, hypersynchronous oscillations, and absence seizures (Hedrich et al., 2014; Kalume et al., 2015; Ritter-Makinson et al., 2019). However, the molecular and cellular mechanisms underlying thalamic dysfunction in DS are not well understood.
The core somatosensory CT circuit comprises layer VI somatosensory cortical neurons, the ventrobasal (VB) thalamus, and the reticular nucleus of the thalamus (nRT). The VB thalamus includes two glutamatergic thalamocortical neuron populations - the ventral posterolateral (VPL) and ventral posteromedial (VPM) thalamus. Both VPL and VPM neurons receive somatic and nociceptive information from the periphery; however, VPL neurons receive input from the body via the medial lemniscus and spinothalamic tract, whereas VPM neurons receive input from the head via the trigeminothalamic tract. The VPL and VPM transmit this distinct information to the somatosensory cortex, which provides reciprocal glutamatergic feedback to the thalamus (Ab Aziz and Ahmad, 2006; Brecht and Sakmann, 2002; Lenz, 1992). These corticothalamic and thalamocortical projections send axon collaterals to the GABAergic nRT, which provides the primary inhibitory input to the VPL and VPM (Figure 1A). Neurons in the nRT, VPL, and VPM have two firing modes critical for somatosensory CT circuit function: a tonic firing mode involved in somatosensory processing, and a burst firing mode underlying intra-thalamic oscillations (Sherman, 2001; Steriade and Llinas, 1988). GABAergic nRT neurons as well as glutamatergic VPL and VPM neurons express NaV1.1 at high levels (Ogiwara et al., 2013; Papale et al., 2013). Interestingly, many glutamatergic neuron populations do not highly express NaV1.1, so its expression in VPL and VPM neurons may uniquely contribute to disease pathophysiology and provide distinctive therapeutic targets (Ogiwara et al., 2013).
Figure 1. Scn1a mRNA and NaV1.1 protein expression in the somatosensory thalamus.
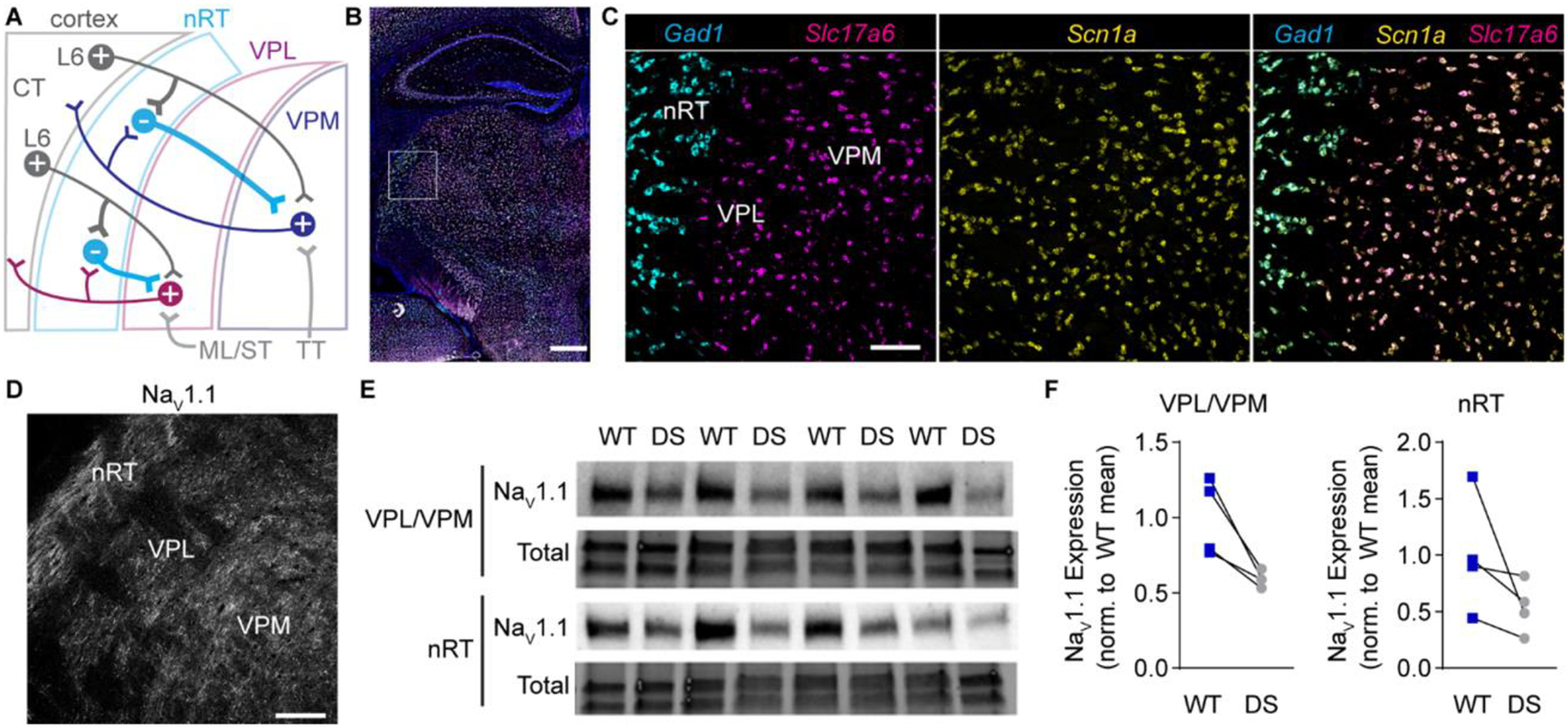
A. A circuit diagram illustrates somatosensory corticothalamic (CT) circuit connectivity. Layer 6 (L6) glutamatergic CT neurons innervate nRT, VPL, and VPM neurons. GABAergic nRT neurons innervate VPL and VPM neurons, which send glutamatergic projections to the cortex and collaterals to the nRT. Ascending glutamatergic sensory afferents from the medial lemniscus and spinothalamic tract (ML/ST) innervate VPL neurons and the trigeminothalamic tract (TT) innervates VPM neurons. B. A representative 20X tiled image of a coronal mouse brain section shows Scn1a (yellow), Gad1 (cyan), and Slc17a6 (magenta) mRNA labeled by FISH with DAPI counterstain (blue). Scale bar: 500 μm. C. 20X images of the boxed region in panel B show that Gad1+ and Slc17a6+ neurons express Scn1a mRNA. D. A representative 20X image shows NaV1.1 immunolabeling in the nRT, VPL, and VPM. Scale bar: 100 μm (C, D). E. A western blot shows NaV1.1 and total protein expression in nRT and VPL/VPM tissue punches from WT and DS mice (n = 4 littermate pairs). F. NaV1.1 protein levels were quantified by densitometry and normalized
Given the delicate balance of excitation and inhibition in the somatosensory CT circuit, changes in either intrinsic excitability or synaptic connectivity could significantly alter thalamocortical network function in DS (Shane R. Crandall, 2015; Temereanca S, 2004). To effectively target thalamic dysfunction, it is critical to understand precisely how the circuitry is dysregulated. We hypothesized that NaV1.1 haploinsufficiency alters somatosensory CT circuit function through both intrinsic and synaptic mechanisms in GABAergic nRT and glutamatergic thalamocortical neurons. Herein, we report cell-type-specific alterations to neuronal excitability and synapse function in nRT and thalamocortical neurons. These findings expand current models of somatosensory thalamic dysfunction in DS by introducing impaired thalamocortical neuron excitability and thalamic synapse dysfunction as novel disease mechanisms.
MATERIALS AND METHODS
Mouse model
Mouse studies were performed according to protocols approved by the Institutional Animal Care and Use Committee at Virginia Polytechnic Institute and State University and in accordance with the National Institutes of Health guidelines. Scn1atm1Kea mice (037107-JAX), which are on a 129S6/SvEvTac background, were purchased from the Mutant Mouse Resource and Research Center (Miller et al., 2014). The Scn1atm1Kea colony was maintained by crossing heterozygous 129S6/SvEvTac-Scn1atm1Kea/WT mice with wild type 129S6/SvEvTac mice. Experimental mice were F1 hybrids generated by crossing heterozygous 129S6/SvEvTac-Scn1atm1Kea/WT mice with C57BL/6J mice (000664, JAX). Genotyping was performed by Transnetyx using real-time PCR. Mice were housed in a 12 hour light/dark cycle with ad libitum access to food and water. Age-matched WT and Scn1a+/− littermates of both sexes aged P25-P30 were used for all experiments.
Fluorescence in situ hybridization
For mRNA detection, mice were euthanized by isoflurane overdose and brains were rapidly dissected and flash frozen in OCT. Brains were cryosectioned (20 μm), fixed with 4% paraformaldehyde (PFA), and RNAscope (ACDBio) multiplex fluorescent in situ hybridization (FISH) was performed according to the manufacturer’s instructions with probes against Scn1a, Gad1, and Slc17a6 (VGLUT2). Tiled 20X images of coronal brain sections were acquired with CellSens software and an Olympus IX83 widefield fluorescence microscope equipped with a Hamamatsu Orca Flash IV camera, X-Cite Xylis LED, and DAPI, FITC, TRITC, and Cy5 filter sets. 20X images of the thalamus were also acquired with a Zeiss 700 laser-scanning confocal and Zen Black software using 405, 488, 561, and 633 lasers. The images were processed for presentation using ImageJ software.
Western blotting
Mice were euthanized by isoflurane overdose and transcardially perfused with ice-cold 1X PBS. Brains were dissected and coronal slices (1 mm) were made using a brain matrix. Tissue punches from the VB thalamus and nRT were snap frozen in liquid nitrogen. Tissue was sonicated in buffer containing (in mM) 65 Tris base, 150 NaCl, 1 EDTA, 50 NaH2PO4, 10 Na4P2O7, 1 Na3VO4, 50 NaF, 1% Triton X-100, and 0.5% deoxycholic salt. Samples were centrifuged at 13,500 × g for 15 minutes at 4°C, denatured using Laemmli sample buffer with 200 mM dithiothreitol, and heated at 95°C. Equal amounts of protein were run on Mini-PROTEAN TGX stain-free gels, which were activated using UV light exposure to covalently bind fluorophores to protein. Protein was transferred to polyvinylidene difluoride (PVDF) membranes, and fluorescence imaging on a ChemiDoc MP System was used to image total protein content. Membranes were probed with anti-NaV1.1 antibodies (1:1000; NeuroMab; Antibodies Inc. 75–023) and goat anti-mouse HRP-conjugated secondary antibodies (1:10,000; Jackson Immunoresearch 115-035-146), and then visualized using Pico Plus substrate (ThermoFisher) and a ChemiDoc MP System. Bands were quantified using Image Lab, and band intensity was normalized to total protein levels.
Immunohistochemistry and microscopy
NaV1.1 immunohistochemistry was performed as previously described (Alshammari et al., 2016). Briefly, mice were euthanized by isoflurane overdose and transcardially perfused with 1X PBS followed by ice-cold 1% PFA and 0.5% methanol in 1X PBS. Brains were post-fixed in 1% PFA and 0.5% methanol in 1X PBS for 24 hr, transferred to 30% sucrose in 1X PBS for 48 hr, and then flash frozen in OCT. Cryosections (20 μm) were slide-mounted, treated with 100% acetone at −20°C for 7 min, blocked with FAB fragments and 10% normal donkey serum (NDS), and then immunostained with mouse anti-NaV1.1 (1:200; NeuroMab; Antibodies Inc., 75–023) and Alexa 488-conjugated anti-mouse IgG1 secondary antibodies (ThermoFisher). Images were acquired with a Zeiss 700 laser-scanning confocal, 20X objective, 488 argon laser, and Zen Black software.
For synaptic marker immunostainings, mice were transcardially perfused with 1X PBS followed by 4% PFA in 1X PBS, and the brains were post-fixed in 4% PFA for 24 hr, cryoprotected in 30% sucrose in 1X PBS, and flash frozen in OCT. Cryosections (20 μm) were treated with 0.8% sodium borohydride in 1X Tris-buffered saline (TBS) at room temperature, and then 0.01M sodium citrate buffer, pH 6.0, at 100°C for 10 min. Sections were permeabilized with 0.5% Triton X-100 in 1X TBS, pH 7.4, and immunostained with antibodies against: VGLUT1 (1:100; Synaptic Systems; 135–511), VGLUT2 (1:200; Synaptic Systems; 135–402), VGAT (1:200; Synaptic Systems; 131–103), and gephyrin (1:200; Synaptic Systems; 147–011). Sections were incubated with Alexa 555-conjugated anti-mouse IgG1 and Alexa 488-conjugated anti-rabbit secondary antibodies (ThermoFisher), as appropriate. Images were acquired with a Zeiss 700 laser-scanning confocal, a 100X objective, and 488 or 533 lasers. Three random fields were imaged from the left and right nRT, VPL, and VPM in two sections for a total of 9 – 12 images per mouse.
Image Analysis
Images were assigned numerical identifiers and analysis was performed blind to genotype. The Synapse Counter plugin from ImageJ was used to analyze the number and size of VGAT/gephyrin, VGLUT1, and VGLUT2 puncta. Synapse size restrictions in Synapse Counter were set as follows: VGAT in all regions (20–1000 px2), gephyrin in all regions (10–400 px2), VGLUT1 in all regions (10–400 px2), VGLUT2 in nRT (70–1200 px2), and VGLUT2 in VPL and VPM (60–3000 px2). Data from each synapse marker were averaged for all images from one mouse. The area occupied by cell bodies and white matter tracks were automatically detected in VGLUT1 images using ImageJ, and the number of synapses was corrected for this area. Images were processed for presentation in ImageJ, and WT and DS images within each figure are displayed with equivalent intensity settings.
Electrophysiology
Mice were deeply anesthetized with an overdose of inhaled isoflurane and transcardially perfused with ice-cold sucrose-based artificial cerebrospinal fluid (aCSF) containing (in mM) 230 sucrose, 24 NaHCO3, 10 glucose, 3 KCl, 10 MgSO4, 1.25 NaH2PO4, and 0.5 CaCl2 saturated with 95% O2 / 5% CO2. The brain was removed and glued to a vibratome stage (Leica VT1200S), and horizontal 300 μm slices were cut in an ice-cold sucrose-aCSF bath. Slices were incubated in a NaCl-based aCSF containing (in mM) 130 NaCl, 24 NaHCO3, 10 glucose, 3 KCl, 4 MgSO4, 1.25 NaH2PO4, and 1 CaCl2 saturated with 95% O2 / 5% CO2 at 32°C for 30 min. The slices were then equilibrated to room temperature (RT) for 30 minutes and maintained at room temperature until used for recordings up to 8 hr later. One cell was recorded per slice, and no more than two cells in any dataset are from the same mouse.
Recordings were made using a Multiclamp 700B amplifier (Molecular Devices), sampled at 20 kHz (Digidata 1550B, Molecular Devices), and low-pass filtered at 10 kHz using Axon pClamp 11 software (Molecular Devices). The extracellular recording solution contained (in mM) 130 NaCl, 24 NaHCO3, 10 glucose, 3 KCl, 1 MgSO4, 1.25 NaH2PO4, and 2 CaCl2 saturated with 95% O2 / 5% CO2, and was maintained at 32°C for all recordings. For whole-cell current-clamp recordings, borosilicate glass recording electrodes (4–5 MΩ) were filled with (in mM) 130 K-gluconate, 4 KCl, 2 NaCl, 10 HEPES, 0.2 EGTA, 4 ATP-Mg, 0.3 GTP-Tris, 14 phosphocreatine-K, and 0.1% biocytin, pH 7.3. Pipette capacitance neutralization and bridge balance were enabled during current-clamp recordings for capacitance and series resistance compensation. Membrane potential values were corrected for the liquid junction potential after the recording (15 mV). To analyze intrinsic membrane properties, voltage responses were elicited by 200 ms hyperpolarizing current injections between 20 – 100 pA (20 pA steps). Depolarization-induced spike firing was elicited by 500 ms depolarizing current injections between 10 – 400 pA (10 pA steps). Hyperpolarization-induced rebound bursting was elicited by 500 ms hyperpolarizing current injections between 50 – 200 pA (50 pA steps). All current-clamp experiments were conducted from the natural resting membrane potential (RMP) of the cell, and three trials were completed for each current injection experiment.
For whole-cell voltage-clamp recordings, borosilicate glass recording electrodes (4–5 MΩ) were filled with (in mM) 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 tetraethylammonium chloride, 10 HEPES, 1 EGTA, 3 Mg-ATP, 0.3 Na-GTP, 1.5 MgCl2, 1 QX-314, and 0.1% biocytin, pH 7.3. After a ten minute equilibration period, mEPSCs and mIPSCs were recorded in the presence of 1 μM TTX in two minute epochs at a holding potential of −70 mV and 0 mV, respectively. Holding commands were adjusted for the 10 mV liquid junction potential during the recordings. Series resistance and cell capacitance were monitored throughout the experiment, but were not compensated, and cells were excluded if either parameter changed >20%.
Biocytin labeling in acute brain slices
The cell location was confirmed after electrophysiology recordings by biocytin labeling (Figure S1). Briefly, brain slices were fixed with 4% PFA in 1X PBS, pH 7.4, overnight at 4°C, washed in 1X PBS, and then stored at −20°C in cryoprotectant solution containing 0.87 M sucrose, 5.37 M ethylene glycol, and 10 g/L polyvinyl-pyrrolidone-40 in 0.1 M phosphate buffer, pH 7.4. For biocytin labeling, slices were blocked with 10% NDS in 1X PBS with 0.25% Triton X-100, and then incubated with 1.0 μg/ml DyLight 594-conjugated streptavidin (Jackson Immunoresearch) in blocking solution overnight at RT. Slices were stained with DAPI for 1 hr at RT and placed on a glass slide with a coverslip and DABCO mounting media (Sigma Aldrich). 10X images were acquired on an Olympus IX83 microscope with a Hamamatsu Orca Flash 4.0 camera, X-Cite Xylis LED, and DAPI and TRITC filter sets using CellSens software.
Electrophysiology data analysis
Recordings were assigned numerical identifiers and analysis was performed blind to genotype. All current-clamp recordings were analyzed in Clampfit 11 (Molecular Devices). RMP was measured from current-clamp recordings two minutes following breakthrough of the cell membrane. Input resistance (Rin) was determined from the amplitude of voltage responses to 200 ms hyperpolarizing current injections, the time constant (τm) was determined by a mono-exponential fit of the voltage response, and cell capacitance was calculated by Cm = τm/Rin. The Clampfit 11 threshold detection module was used to quantify the number of spikes, spike frequency, and latency to the first spike in response to 500 ms depolarizing current injections or upon removal of 500 ms hyperpolarizing current injections. Rheobase was defined as the smallest depolarizing current injection that elicited an action potential. Values were averaged across three runs for all current injection experiments.
The shape of single action potentials was analyzed using the Action Potential Search module in ClampFit 11. The baseline was manually set, and single action potentials at or near rheobase were analyzed for amplitude, half width, rise and decay time, after-hyperpolarization amplitude, and threshold. Spike frequency adaptation was evaluated by measuring spike frequency across depolarizing current injections for VPL and VPM neurons. Some VPL and VPM neurons had a prolonged inter-event interval between the first two spikes following depolarization; therefore, the frequency of the first three spikes following current injection were averaged in VPL and VPM neurons and were compared to the frequency of the last two spikes. The ratio of these frequencies (last two spikes/first three spikes) is reported as the adaptation ratio. Because many DS nRT neurons fired very few spikes in response to depolarization, only the frequency of the first three spikes following depolarization was assessed. The frequencies from multiple runs in the same cell were averaged.
For mIPSC and mEPSC analysis, recordings of 4 – 6 minutes were analyzed to determine inter-event interval, amplitude, and decay kinetics using MiniAnalysis software (Synaptosoft). Data were digitally filtered at 1 kHz. Automated detection identified events with amplitude ≥ 7 pA (~5 x RMS noise), and then events ≥ 5 pA were manually detected and automated detection accuracy was assessed. The reported numbers of mEPSCs per cell were normalized to the total recording time for each cell. The decay time at 37% of the peak was determined for each mEPSC in MiniAnalysis. To group mEPSCs as Type 1 (fast) or Type 2 (slow), the decay times for all mEPSCs were plotted as a frequency distribution for each cell independently, and then distributions were tested for bimodality by Hardigan’s dip test in Matlab. All distributions were multimodal (p < 0.05). The bimodal distribution was separated at the decay time (x-value) corresponding to the minimum frequency (y-value) between the two modes. To determine this minimum y-value, each distribution was interpolated using the Akima spline method (GraphPad Prism 9), and the first derivative of the spline was plotted to determine the x-intercept between the two modes, which is the decay time corresponding to the minimum frequency. This value was determined for each cell independently. All events with decay times faster than this cutoff value were Type 1 and those with slower decay times were Type 2.
The reported amplitude and inter-event interval values for each cell are averages of the amplitude and inter-event interval measured for each Type 1 mEPSC, Type 2 mEPSC, or mIPSC recorded from the cell. The average mEPSC and mIPSC decay times for each cell were determined from ensemble averages of all Type 1 mEPSCs, Type 2 mEPSCs, or mIPSCs recorded from each cell. The 10–90% peak to baseline decay times of the ensemble averages were fitted using the following double-exponential function:
| Equation 1 |
Where τFAST is the fast deactivation time constant, τSLOW is the slow deactivation time constant, AmpFAST is the current amplitude of the fast deactivation component, and AmpSLOW is the current amplitude of the slow deactivation component. The weighted decay time constant (τW) was calculated by:
| Equation 2 |
Statistical analysis
A priori power analyses were performed in GPower 3.1 to estimate required samples sizes given appropriate statistical tests with α = 0.05, power (1 – β) = 0.8, and a moderate effect size or effect sizes based on pilot data. Statistical analyses were performed in GraphPad (Prism). All datasets were tested for normality with the Shapiro-Wilk test and equal variances using an F test (two independent groups) or the Brown-Forsythe test (three or more groups). Normal datasets with equal variances were analyzed by unpaired t-test or ANOVA with corrections for multiple comparisons. Non-normal datasets were analyzed with the Mann-Whitney U test (two independent groups) or Kruskal-Wallis test (three or more groups). Repeated measures datasets were analyzed by two-way repeated measures ANOVA, and a mixed-effects analysis was used when any data points were missing. The specific tests used and associated test statistics are reported in the respective figure legend or table. All group data are plotted as mean ± s.e.m. in the figures, and numerical data reported in the text are mean ± s.e.m., unless otherwise stated.
RESULTS
Scn1a mRNA and NaV1.1 protein expression in GABAergic and glutamatergic neurons in the thalamus
To evaluate how thalamic neuron function is impaired in DS, we utilized the F1 hybrid 129S-Scn1a+/− x C57Bl/6J mouse model at age P25-P30, hereafter referred to as DS mice. Given extensive strain differences in DS mouse model phenotypes, we first examined Scn1a mRNA and NaV1.1 protein levels to confirm high expression in GABAergic nRT and glutamatergic VPL and VPM neurons in wild type F1:129S-Scn1a+/+ x C57Bl/6J (WT) mice at P25-P30. Indeed, multiplex FISH revealed abundant Scn1a mRNA expression in Gad1-positive GABAergic nRT neurons as well as glutamatergic VPL and VPM neurons detected by Slc17a6 mRNA, which encodes VGLUT2 (Figure 1B,C). NaV1.1 protein was detected in the nRT, VPL, and VPM by immunohistochemistry (Figure 1D). In addition, western blotting of VPL/VPM and nRT tissue punches showed reduced NaV1.1 protein levels in DS mice relative to WT littermates, confirming haploinsufficiency in the thalamus of this DS model (VPL/VPM: 59.4 ± 2.6% of WT; nRT: 53.8 ± 1.1% of WT; Figure 1E,F).
GABAergic nRT neuron excitability is impaired in DS mice
Previous studies have reported opposing effects on nRT neuron excitability in different DS mouse models as well as age-dependent changes in GABAergic neuron excitability in the cortex (Favero et al., 2018; Hedrich et al., 2014; Kalume et al., 2015; Ritter-Makinson et al., 2019; Tai et al., 2014). To determine how nRT neuron excitability was affected in this DS mouse model, we examined intrinsic membrane properties, depolarization-induced spike firing, and hyperpolarization-induced rebound bursting of neurons at their RMP in acute brain slices.
In response to depolarizing current injections, WT nRT neurons fired action potentials tonically, as expected. A majority of nRT neurons from DS mice fired a low number of action potentials immediately following current injection, but a subpopulation fired long-latency low-threshold spikes with bursts of action potentials (6 of 16 neurons); we will hereafter refer to these two groups as “DS” neurons and “DS-burst” neurons, respectively (Figure 2A). We compared the intrinsic membrane properties of DS and DS-burst neurons to determine whether these might be two distinct nRT neuron populations. The DS-burst group had a significantly hyperpolarized RMP and significantly reduced input resistance (Rin) compared to both WT and DS neurons, while cell capacitance (Cm) and the membrane time constant (τ) were not significantly different (Table 1, Supplementary Figure S2A). The input resistance, cell capacitance, and the membrane time constant were not significantly different between WT and DS neurons. These data suggest that a subpopulation of nRT neurons have a hyperpolarized RMP in DS mice, which may increase the likelihood of nRT burst firing in response to depolarizing input at rest.
Figure 2. nRT neuron excitability is altered in DS mice.
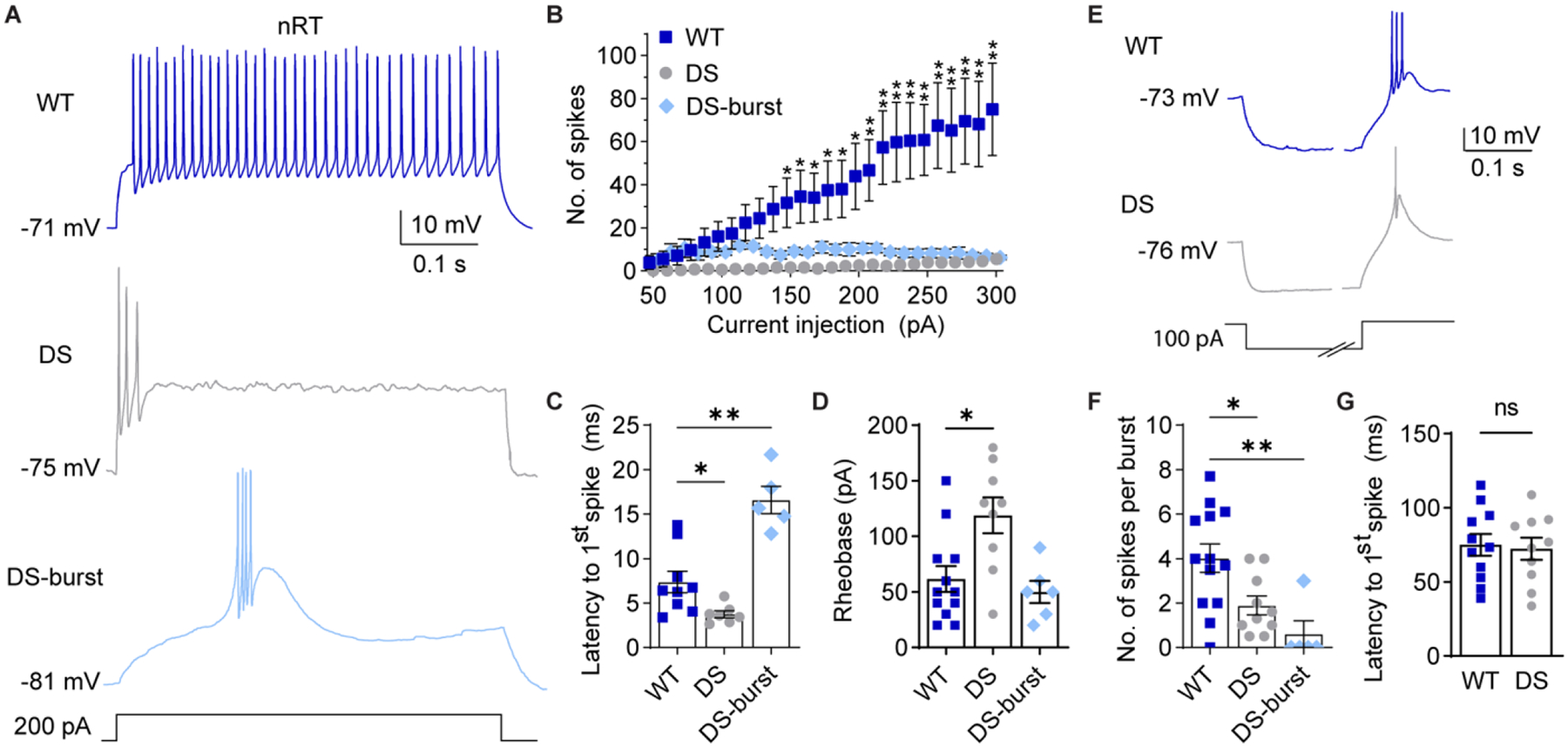
A. Representative traces show nRT neuron spike firing in response to depolarizing current injections from RMP. B. The number of spikes at each current injection for WT (n = 9 cells from 6 mice), DS non-burst firing cells (labeled DS, n = 8 cells from 6 mice), and DS burst firing neurons (DS-burst, n = 5 cells from 4 mice) were analyzed by mixed-effects analysis for repeated measures (Genotype: F(2,18) = 6.431, p = 0.008, Current x genotype: F(50,440) = 6.051, p < 0.001) with posthoc Dunnett’s tests at each current injection. *p<0.05 for WT vs. DS, **p< 0.05 for both WT vs. DS and WT vs. DS-burst. C. Latency to the first spike was quantified and analyzed by one-way ANOVA: F(2,17) = 20.38, p = 0.001; posthoc Dunnett’s tests, *p = 0.034, **p = 0.015. D. Rheobase was quantified for WT (n = 12 cells from 8 mice), DS (n = 9 cells from 7 mice), and DS-burst (n = 6 cells from 5 mice), and analyzed by one-way ANOVA: F(2,24) = 7.016, p = 0.004; posthoc Dunnett’s tests, *p = 0.007, WT vs. DS-burst: p = 0.800. E. Representative traces show rebound burst firing upon recovery from 500 ms hyperpolarizing current injections. The time axis was broken to facilitate displaying both the hyperpolarization and spike periods. F. The number of spikes per burst were compared by the Kruskal-Wallis test (H = 13.94, p = 0.001), due to non-normal distribution of the data (Shapiro-Wilk test, p < 0.001), and posthoc Dunn’s tests: *p = 0.041, **p = 0.001 (WT: n = 13 cells from 8 mice, DS: n = 10 from 8 mice, DS-burst: n = 5 from 5 mice). G. Latency to the first spike for WT and DS groups were compared by an unpaired t-test (p = 0.803). The symbols in all bar graphs represent individual neurons.
Table 1.
Membrane properties of nRT, VPL, and VPM neurons.
| nRT | VPL | VPM | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WT | DS | DS-burst | ANOVA | WT | DS | p value | WT | DS | p value | |
| RMP (mV) | −68.9 ± 1.7 | −71.2 ± 1.3 | −81.2 ± 2.0* | F (2,18) = 13.24 p < 0.001 |
−74.7 ± 1.0 | −78.1 ± 0.7 | *0.03 | −74.9 ± 0.9 | −72.4 ± 1.7 | 0.20 |
| Cm (pF) | 99.9 ± 0.9 | 107 ± 4 | 119 ± 14 | F(2,18) = 1.672 p = 0.216 |
223 ± 30 | 207 ± 28 | 0.69 | 186 ± 14 | 198 ± 31 | 0.70 |
| Rin (MΩ) | 274 ± 35 | 273 ± 21 | 158± 25** | F(2,18) = 4.781 p =0.022 |
123 ± 14 | 125 ± 15 | 0.91 | 117 ± 10 | 154 ± 12 | *0.03 |
| Tau (ms) | 27.4 ± 3.6 | 29.2 ± 2.5 | 17.9 ± 2.4 | F(2,18) = 3.517 p = 0.051 |
29.8 ± 6.9 | 25.9 ± 4.5 | 0.66 | 22.0 ± 3.1 | 29.8 ± 5.1 | 0.19 |
| N | 7 (6) | 9 (7) | 5 (5) | 10 (7) | 8 (6) | 10 (8) | 6 (5) | |||
Both DS and DS-burst neurons fired significantly fewer action potentials in response to depolarizing current injections compared to WT (Figure 2B). DS neurons had a shorter latency between current injection and the first spike, while DS-burst neurons had a longer latency due to the low-threshold spike (WT: 7.4 ± 1.2 ms, DS: 3.8 ± 0.4 ms; DS-burst: 16.6 ± 1.5 ms; Figure 2C). DS neurons required significantly more current to elicit an action potential compared to WT neurons (rheobase: WT: 62 ± 12 pA, DS: 119 ± 16 pA; DS-burst: 50 ± 10 pA; Figure 2D). However, the frequency of the first three action potentials fired by WT (164 ± 25 Hz) and DS neurons (135 ± 30 Hz) were not significantly different (unpaired t-test, p = 0.48). Furthermore, we found no significant difference in action potential amplitude, half width, rise or decay time, after-hyperpolarization amplitude, or threshold measured from single action potentials in WT and DS neurons (Table S1). These data suggest NaV1.1 haploinsufficiency impairs sustained firing in the majority of nRT neurons without altering RMP, the shape of single action potentials, or the frequency at which the initial spikes are fired.
Burst firing in nRT neurons contributes to oscillatory activity between nRT and VPL/VPM neurons, and aberrant thalamic oscillations underlie sleep impairments and absence seizures in DS. Therefore, we investigated how NaV1.1 haploinsufficiency impacted rebound burst firing after removal of hyperpolarizing current injections in nRT neurons at RMP (Figure 2E). DS and DS-burst nRT neurons fired significantly fewer action potentials per burst than WT neurons, which is reported as the median with 95% confidence intervals [WT: 4 (2.0, 6.0), DS: 1.3 (0.5, 4.0), DS-burst: 0 (0, 3.0); Figure 2F], while burst latency was not altered (WT: 75 ± 7.4 ms; DS: 72 ± 7.5 ms; Figure 2G). Burst latency was not reported for the DS-burst group as only one of five neurons exhibited a rebound burst. These data suggest that NaV1.1 haploinsufficiency impairs nRT neuron resting excitability, including alterations in both depolarization-induced spike firing and rebound burst firing following hyperpolarization, which may be due to impaired action potential generation as well as the observed hyperpolarized RMP in a subpopulation of neurons.
Aberrant excitability of glutamatergic VPL neurons in DS mice
Previous studies of the somatosensory thalamus in DS mouse models examined the VB complex, which includes both VPL and VPM neurons; however, these two cell populations have different synaptic connectivity and may have distinct intrinsic membrane properties. Therefore, we evaluated these two cell populations independently.
We determined how NaV1.1 haploinsufficiency impacted the intrinsic membrane properties, depolarization-induced spike firing, and hyperpolarization-induced rebound burst firing in VPL and VPM neurons at their RMP.
Pairwise comparisons were performed using post hoc Sidak’s tests; *nRT RMP: WT vs. DS-burst, p < 0.001, DS vs. DS-burst, p = 0.002, WT vs. DS, p = 0.547; **nRT Rin: WT vs. DS-burst, p = 0.035, DS vs. DS-burst, p = 0.029, WT vs. DS, p =0.998. VPL and VPM parameters were compared across genotype by unpaired t-tests. N values are the number of cells followed by the number of mice in parentheses.
VPL neurons in DS mice had a significantly hyperpolarized RMP relative to WT, while input resistance, cell capacitance, and the membrane time constant were not significantly different (Table 1, Supplementary Figure S2B). VPL neurons in DS mice fired significantly fewer spikes than WT mice over a range of depolarizing current injections as detected by a significant interaction effect between genotype and current injection in the ANOVA; however, no significant differences were detected by pairwise comparisons at individual current injections (Figure 3A,B). Neither the rheobase (WT: 143 ± 26 pA; DS: 172 ± 50 pA) nor spike latency (WT: 9.2 ± 1.8 ms; DS: 15.8 ± 3.5 ms) were significantly affected in VPL neurons (Figure 3C,D). No significant differences in action potential shape, including amplitude, half width, rise or decay time, after-hyperpolarization amplitude, and threshold, were detected for DS VPL neurons compared to WT (Table S1).
Figure 3. NaV1.1 haploinsufficiency alters VPL neuron excitability.
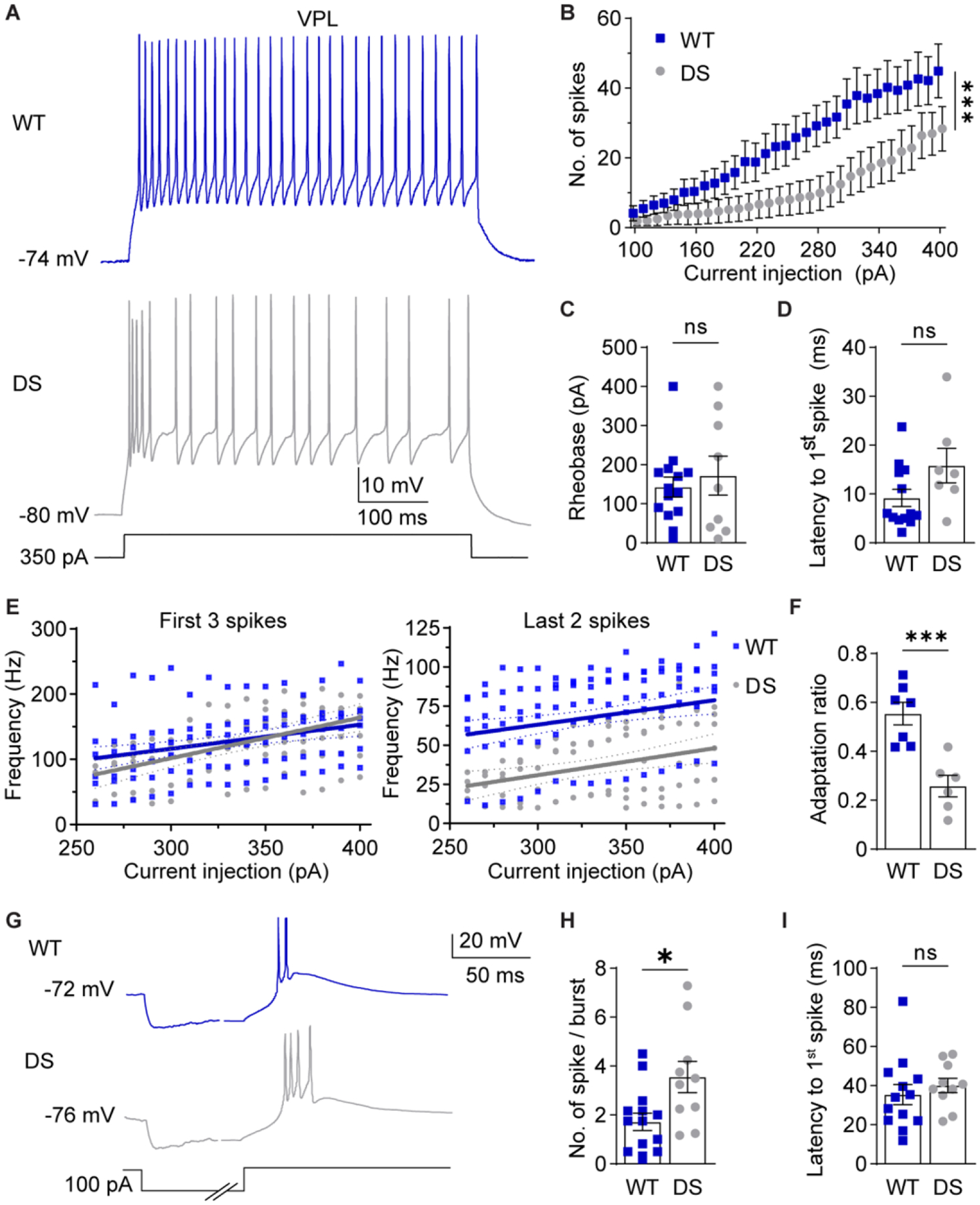
A. Representative traces show WT and DS VPL neuron spike firing in response to depolarizing current injections at RMP. B. The number of spikes fired by VPL neurons across current injections were analyzed by two-way repeated measures ANOVA (WT: n = 13 cells from 7 mice; DS: n = 8 cells from 6 mice; Genotype: F(1,19) = 3.605; p = 0.07, Interaction: F(30,570) = 2.200; ***p < 0.001) and posthoc Sidak’s tests at each current injection (p > 0.05 at each current amplitude). C. Rheobase was analyzed by unpaired t-test for WT (n = 14 cells from 7 mice) and DS (n = 9 cells from 6 mice) neurons (p = 0.571). D. Latency was analyzed by Mann-Whitney test (p = 0.183) due to failed normality (Shapiro-Wilk test, p = 0.026). E. The frequency of the first 3 spikes and last 2 spikes were plotted for each cell across current injections. Linear regression of WT and DS data yielded the plotted lines with 95% confidence interval (CI) bands, and fits were compared by sum of squares F tests. First 3 spikes: F (2,176) = 1.600, p = 0.205. Last 2 spikes: F (2,161) = 42.89, p < 0.001. F. Spike frequency adaptation ratios (last 2 spikes/first 3 spikes) were averaged across all current injections for each cell and compared by an unpaired t-test (***p < 0.001). G. Representative traces show rebound burst firing at RMP upon recovery from hyperpolarization. The time axis was broken to facilitate displaying the hyperpolarization and spike periods. H. Spikes per burst (*p = 0.01) and (I) burst latency (p = 0.49) were compared by unpaired t-tests (WT: n = 13 cells from 7 mice; DS: n = 10 cells from 6 mice). The symbols in all bar graphs
To further examine how VPL neuron spike firing was disrupted in DS mice, we measured spike frequency at the beginning and the end of depolarizing current injections, and then calculated a spike frequency adaptation ratio. Interestingly, the frequency of the first three spikes was unchanged in DS VPL neurons (120 ± 8 Hz) compared to WT (127 ± 4 Hz), while the frequency of the last two spikes was significantly reduced (WT: 68 ± 2 Hz; DS: 36 ± 2 Hz; Figure 3E). As a result, the spike frequency adaptation ratio was significantly reduced in DS VPL neurons (0.28 ± 0.01) compared to WT (0.55 ± 0.01; Figure 3F), which indicates that spiking during sustained depolarization is impaired in DS VPL neurons. In addition, VPL neurons from DS mice fired more spikes per rebound burst upon recovery from hyperpolarization compared to WT (WT: 1.7 ± 0.4 spikes; DS: 3.6 ± 0.6 spikes; Figure 3G,H), while burst latency was not significantly affected (WT: 35.4 ± 5.1 ms; DS: 40.1 ± 3.7 ms; Figure 3I). Taken together, these data demonstrate that DS mice have reduced depolarization-induced spike firing and enhanced spike adaptation as well as enhanced hyperpolarization-induced rebound burst firing in VPL neurons, which may be due, in part, to the observed hyperpolarized RMP.
Glutamatergic VPM neurons are hyperexcitable in DS mice
VPM neurons had significantly greater input resistance in DS mice compared to WT mice, while RMP, cell capacitance, and the membrane time constant were not significantly different (Table 1, Supplementary Figure S2C). Given the increased input resistance, VPM neurons would be predicted to have enhanced responses to current injections in DS mice. Indeed, VPM neurons fired significantly more action potentials in DS mice than WT at corresponding depolarizing current injections (Figure 4 A,B). In addition, DS neurons required significantly less current to elicit an action potential (WT: 185 ± 11 pA; DS: 114 ± 21 pA; Figure 4C) and had a shorter spike latency (WT: 42.0 ± 8.0 ms; DS: 16.8 ± 6.5 ms; Figure 4D). Spike firing frequency in DS VPM neurons was significantly increased for the first three spikes (WT: 84 ± 7 Hz; DS: 137 ± 4 Hz) and the last two spikes (WT: 44 ± 3 Hz; DS: 73 ± 3 Hz) compared to WT (Figure 4E), but the spike frequency adaptation ratio was similar in WT (0.47 ± 0.06) and DS neurons (0.48 ± 0.07; Figure 4F). No significant differences were found in the shape of single action potentials including amplitude, half width, rise or decay time, after-hyperpolarization amplitude, or threshold for WT and DS mice (Table S1). In addition, the number of spikes per hyperpolarization-induced rebound burst (WT: 2.5 ± 0.3; DS: 3.0 ± 0.5; Figure 4G,H) and burst latency (WT: 20.5 ± 2.2 ms; DS: 18.1 ± 1.7 ms) were not significantly different between WT and DS mice (Figure 4I). Together, these data suggest that VPM neurons have enhanced tonic firing in response to depolarization in DS mice, which may be due to altered intrinsic membrane properties as indicated by the increased input resistance.
Figure 4. NaV1.1 haploinsufficiency alters VPM neuron excitability.
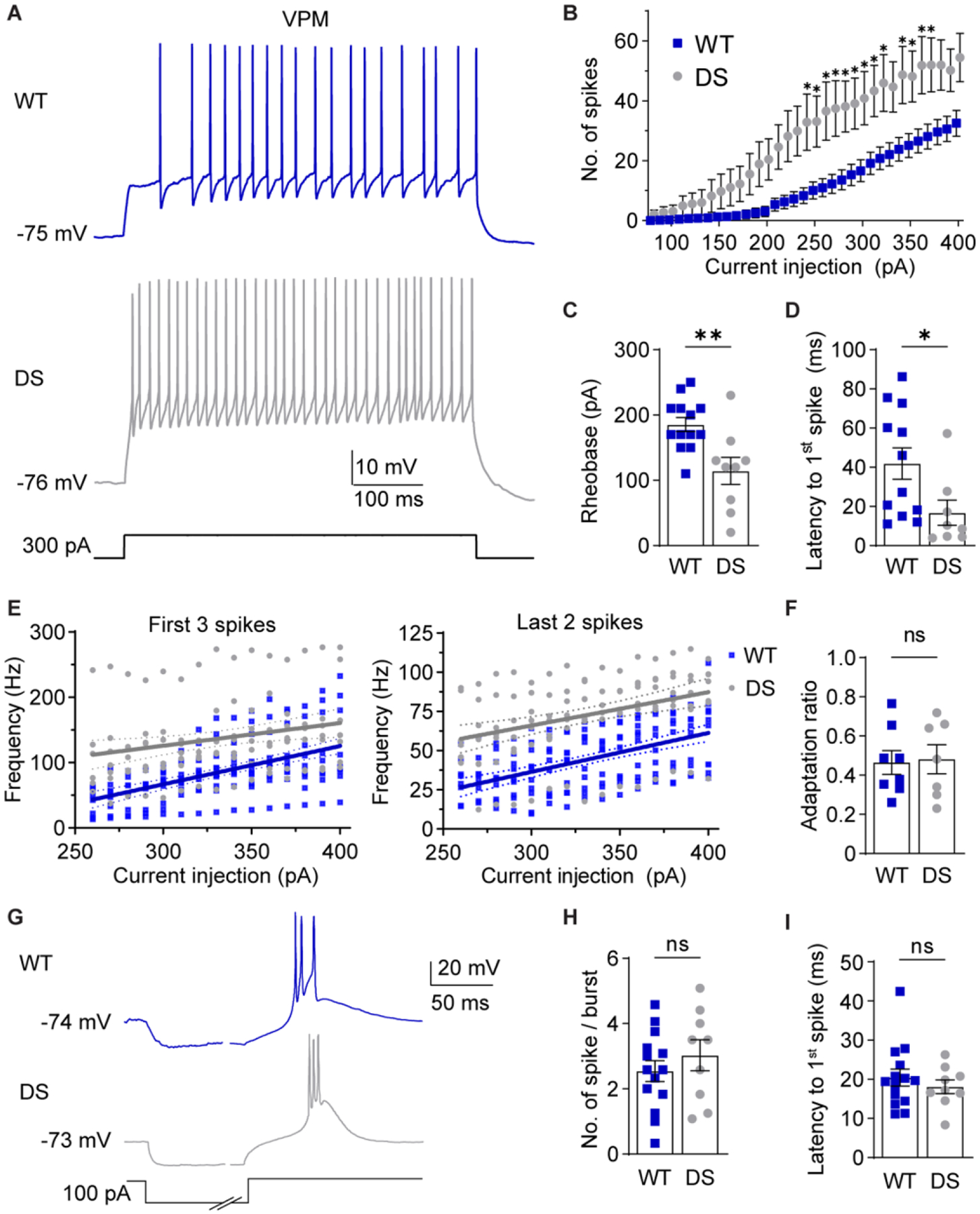
A. Representative traces show VPM spike firing in response to depolarizing current injections from RMP. B. The number of spikes at each current injection was analyzed by a two-way repeated measures ANOVA (WT: n = 12 cells from 8 mice; DS: n = 9 from 5 mice; Genotype: F(1,19) = 7.992, p = 0.011, Interaction: F(32,608) = 4.677, p < 0.001) with posthoc Sidak’s tests at each current injection (*p < 0.05). C. Rheobase for WT (n = 13 cells from 8 mice) and DS (n = 9 cells from 5 mice) neurons were analyzed by unpaired t-test (**p = 0.004). D. Spike latency for WT (n = 12 cells from 8 mice) and DS (n = 8 cells from 5 mice) neurons were compared by an unpaired t-test (*p = 0.037). E. The frequency of the first 3 spikes and last 2 spikes were plotted for each cell across current injections. Linear regression of WT and DS data yielded the plotted lines with 95% CI bands, and fits were compared by sum of squares F tests. First 3 spikes: F (2,244) = 36.50, p < 0.001. Last 2 spikes: F (2,244) = 59.13, p < 0.001. F. Spike frequency adaptation ratios (last 2 spikes/first 3 spikes) were averaged across all current injections for each cell and compared by an unpaired t-test (p =0.860). G. Representative traces show rebound burst firing upon recovery from hyperpolarization. H. Spikes per burst (p = 0.39) and (I) burst latency (p = 0.46, WT: n = 14 cells from 8 mice, DS: n = 9 cells from 5 mice) were analyzed by unpaired t-tests. The symbols in all bar graphs represent individual neurons.
Reduced glutamatergic input to nRT neurons in DS mice
We hypothesized that altered intrinsic excitability in thalamic neurons would disrupt activity-dependent synapse development and thus exacerbate thalamocortical network dysfunction in DS. The nRT receives glutamatergic inputs from corticothalamic (CT) and thalamocortical (TC) neurons. Deficits in CT-nRT and TC-nRT connectivity would disrupt feed-forward and feedback inhibition of VPL/VPM neurons, respectively (see Figure 1A). Previous work revealed that nRT EPSCs can be divided into two populations based on decay kinetics, and it was postulated that CT-nRT synapses mediate the slow decaying EPSCs and TC-nRT synapses mediate the fast decaying EPSCs (Deleuze and Huguenard, 2016). Therefore, to investigate how nRT glutamatergic inputs differed between WT and DS mice, we recorded nRT mEPSCs, grouped the events based on decay times, and analyzed the two kinetically distinct mEPSC populations (Figure 5A). Faster decaying events are referred to here as Type 1, and slower decaying events are referred to as Type 2 (Figure 5B,C).
Figure 5. Selective reduction in Type 2 nRT mEPSC frequency in DS mice.
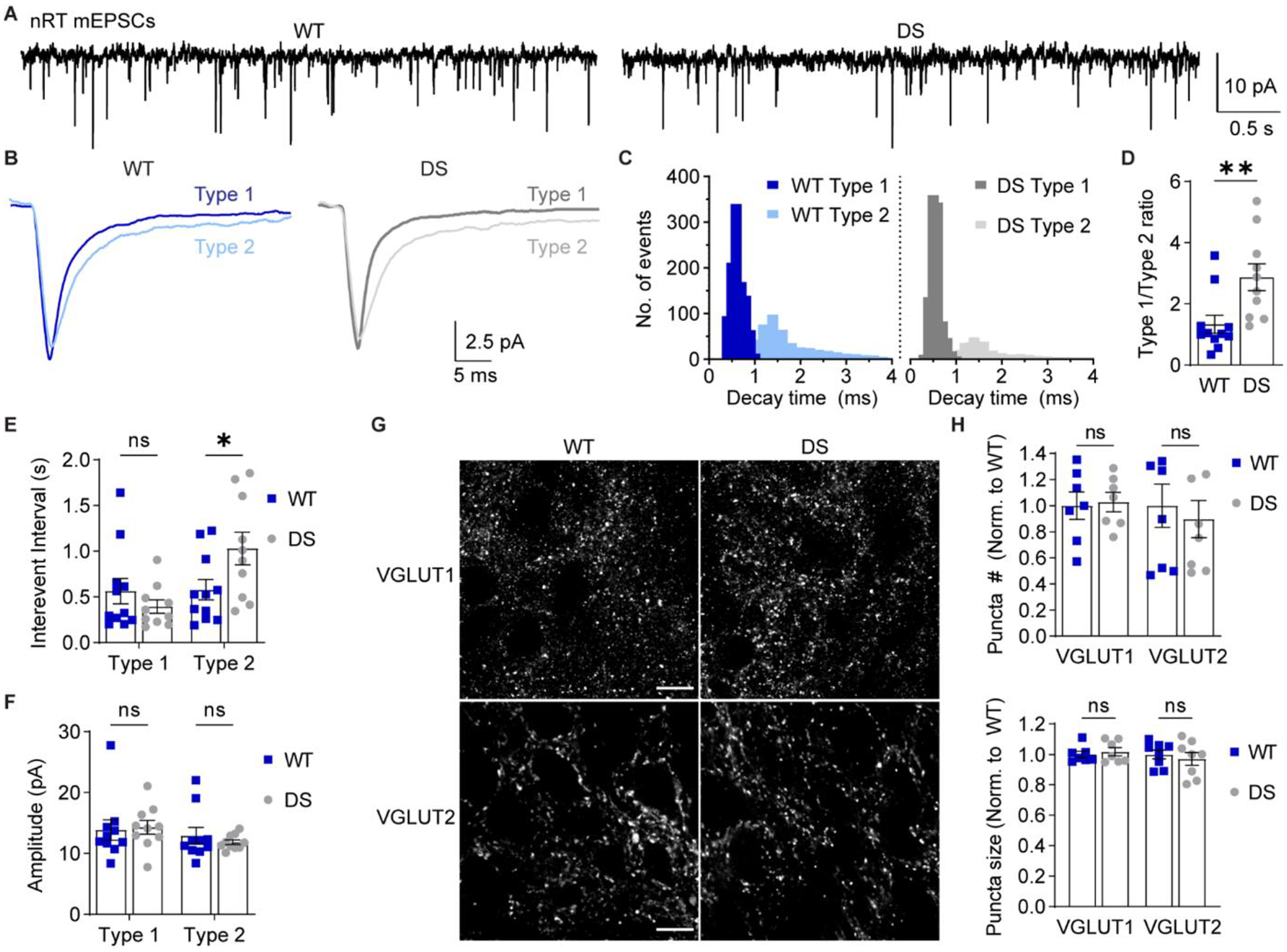
A. mEPSCs were recorded from nRT neurons in acute brain slices in the presence of TTX. B. Representative traces show ensemble averages of WT and DS Type 1 and Type 2 mEPSCs as determined by decay time. C. The decay time distributions from each cell were averaged for WT and DS groups to generate the depicted histogram of the mean number of events per cell (bin size = 0.1 ms). D. The ratio of Type 1/Type 2 events for WT (n = 11 cells from 6 mice) and DS (n = 10 cells from 6 mice) neurons were compared by unpaired t-test (*p = 0.008). E. Mean inter-event interval and (F) amplitude values for Type 1 and Type 2 mEPSCs were analyzed by two-way ANOVA with posthoc Sidak’s tests. Inter-event interval: Genotype F(1,38) = 1.169, p = 0.286; Interaction F(1,38) = 5.619, p = 0.023; Type 1 WT vs. DS: p = 0.600; Type 2 WT vs. DS: *p = 0.039. Amplitude: Genotype F(1,38) = 0.0699, p = 0.795; Interaction F(1,38) = 0.388, p = 0.538. See Supplementary Figure S3 for cumulative distributions of inter-event interval and amplitude data. G. Representative 100X images depict VGLUT1 and VGLUT2 staining in WT and DS nRT. Scale bar: 10 μm. H. The number and size of VGLUT1 and VGLUT2 puncta (n = 7 mice) were analyzed by two-way ANOVA. Puncta number: Genotype: F(1,26) = 0.081, p = 0.78; Interaction: F(1,26) = 0.250, p = 0.620. Puncta size: Genotype: F(1,26) = 0.031, p = 0.86; Interaction: F(1,26) = 0.545, p = 0.47. Data points in the bar graphs represent individual neurons (D-F) or mice (H).
The total number of mEPSCs recorded per cell was not significantly altered in DS mice (1266 ± 180) relative to WT (1533 ± 254; unpaired t-test, p = 0.38), but the ratio of Type 1 to Type 2 mEPSCs was increased in DS mice (WT: 1.3 ± 0.3; DS: 2.9 ± 0.4; Figure 5D). The inter-event interval was significantly increased for Type 2 events in DS mice (1.03 ± 0.18 s) relative to WT (0.58 ± 0.11 s), while the Type 1 inter-event interval was not significantly altered (WT: 0.56 ± 0.14 s; DS: 0.40 ± 0.07 s; Figure 5E). There was no main effect of genotype on inter-event interval, further suggesting that the overall frequency of nRT glutamatergic input was unchanged. Furthermore, mEPSC amplitude was unaltered in both Type 1 (WT: 13.9 ± 1.7 pA, DS: 14.9 ± 1.2 pA) and Type 2 (WT: 12.9 ± 1.3 pA, DS: 11.8 ± 0.4 pA) mEPSC populations in DS mice compared to WT (Figure 5F), and there was no significant change in the decay time for Type 1 events (WT: 2.6 ± 0.3 ms, DS: 3.6 ± 0.4) or Type 2 events (WT: 5.4 ± 0.7 ms; DS: 6.9 ± 0.9 ms; Supplementary Figure S3). These data suggest that DS nRT neurons have a selective decrease in the frequency of the slow decaying mEPSC population (Type 2) with no change in glutamatergic synapse strength.
To evaluate changes in nRT glutamatergic synaptic input in an input-specific manner, we immunostained brain sections from WT and DS mice for CT-specific presynaptic marker VGLUT1 and TC-specific presynaptic marker VGLUT2 (Figure 5G) (Graziano et al., 2008). DS mice exhibited no significant differences in either VGLUT1 puncta number (102.8 ± 7.4 % of WT) and size (101.8 ± 2.7 % of WT) or VGLUT2 puncta number (89.7 ± 14.3 % of WT) and size (97.1 ± 4.2 % of WT; Figure 5H). Taken together, the imaging and physiology data are consistent with reduced glutamate release at a subset of glutamatergic nRT synapses with no change in the number of CT or TC synapses onto nRT neurons.
In mice aged P25-P30 used herein, there are no GABAergic mIPSCs in nRT neurons and, therefore, we could not evaluate the frequency or strength of spontaneous GABAergic synaptic transmission. However, we did evaluate GABAergic synapse number and size by immunostaining for presynaptic marker VGAT and postsynaptic marker gephyrin in WT and DS tissue (Supplementary Figure S4A). We did not detect any significant differences in VGAT puncta number (102 ± 12% of WT) and size (102 ± 2% of WT), gephyrin puncta number (101 ± 18% of WT) and size (104 ± 2% of WT), or the number (100 ± 10% of WT) and size (103 ± 2% of WT) of VGAT/gephyrin colocalized regions (Supplementary Figure S4B). If GABAergic synapse density is indeed unchanged in the nRT, then this finding together with the observed reduction in mEPSC frequency could indicate imbalanced excitatory and inhibitory input to the nRT in DS mice.
Synapse-specific alterations in glutamatergic input to VPL neurons
VPL and VPM neurons receive descending glutamatergic CT inputs as well as ascending glutamatergic sensory inputs. Previous studies have shown that evoked EPSCs at these two inputs have distinct kinetics, with ascending inputs having faster decay times than CT input (Castro-Alamancos, 2002; McCormick and von Krosigk, 1992; Miyata and Imoto, 2006). Therefore, we hypothesized that VPL and VPM mEPSCs could be distinguished based on decay time similarly to nRT neurons. Indeed, a frequency histogram of mEPSC decay times resulted in two distinct populations in VPL (Figure 6A–C) and VPM (Figure 6G–I) neurons. We cannot unequivocally identify these kinetically distinct populations as ascending and descending inputs to VPL/VPM neurons; therefore, we refer to faster decaying mEPSCs as Type 1 and slower decaying mEPSCs as Type 2.
Figure 6. VPL neurons in DS mice exhibit reduced glutamatergic synaptic transmission.
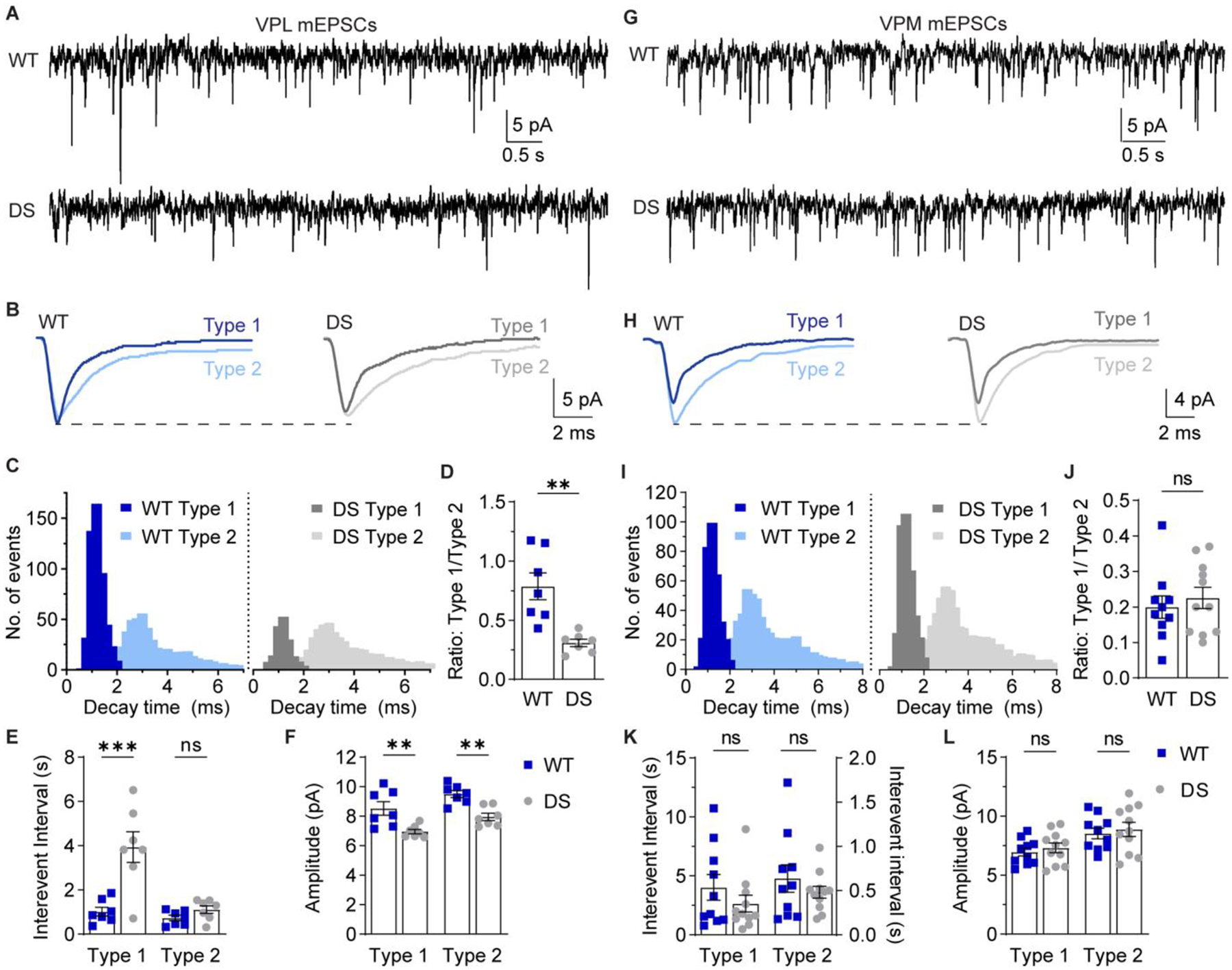
A. mEPSCs were recorded from VPL neurons in acute brain slices in the presence of 1 μM TTX. B. Representative traces show ensemble averages of WT and DS Type 1 and Type 2 VPL mEPSCs as determined by decay times. C. The decay time distributions from each cell were averaged for WT and DS groups to generate histograms of the mean number of events per cell (bin size = 0.2 ms). D. The ratio of Type 1 to Type 2 events was quantified for each WT (n = 7 cells from 6 mice) and DS neuron (n = 7 cells from 7 mice) and analyzed by unpaired t-test (**p = 0.002). E. Inter-event interval and (F) amplitude values for Type 1 and Type 2 mEPSCs in VPL neurons were analyzed by two-way ANOVA with posthoc Sidak’s tests. Inter-event interval: Genotype F(1,24) = 19.28, p < 0.001; Interaction F(1,24) = 11.42, p = 0.002; Type 1 WT vs. DS: ***p < 0.001; Type 2 WT vs. DS: p = 0.73. Amplitude: Genotype F(1, 24) = 27.15, p < 0.001; Interaction F(1,24) = 0.002, p = 0.969. Type 1 WT vs. DS: **p = 0.002; Type 2 WT vs. DS: **p = 0.003. G. mEPSCs were recorded from VPM neurons and (H) representative traces show ensemble averages of WT and DS Type 1 and Type 2 mEPSCs. I. The decay time distributions from each cell were averaged for WT and DS groups to generate histograms of the mean number of events per cell (bin size = 0.2 ms). J. Type 1/Type 2 event ratios for WT (n = 10 cells from 8 mice) and DS (n = 11 cells from 9 mice) VPM neurons were analyzed by unpaired t-test (p = 0.57). K. Inter-event interval and (L) amplitude values for Type 1 and Type 2 mEPSCs in VPM neurons were analyzed by two-way ANOVA with posthoc Sidak’s tests. Inter-event interval: Genotype F(1,38) = 1.407, p = 0.243; Interaction F(1,38) = 0.893, p = 0.351; Type 1 WT vs. DS: p = 0.261; Type 2 WT vs. DS: p = 0.982. Amplitude: Genotype F(1, 38) = 0.573, p = 0.454; Interaction F(1,38) < 0.001, p = 0.993. Type 1 WT vs. DS: p = 0.840; Type 2 WT vs. DS: p = 0.833. See Supplementary Figure S5 for cumulative distribution inter-event interval and amplitude
In VPL neurons, the total number of mEPSCs was significantly reduced in DS mice (712 ± 86) relative to WT (1102 ± 111; p = 0.01). Furthermore, the ratio of Type 1 to Type 2 mEPSCs was decreased in DS mice (WT: 0.79 ± 0.11; DS: 0.31 ± 0.03; Figure 6D), with no change in the decay times for Type 1 mEPSCs (WT: 5.9 ± 0.8 ms; DS: 6.2 ± 0.8 ms) or Type 2 mEPSCs (WT: 9.2 ± 0.8 ms; DS: 8.8 ± 1.0 ms; Supplementary Figure S5A,B). The change in relative proportions of Type 1 and Type 2 mEPSCs was driven by a significant increase in the inter-event interval for Type 1 mEPSCs (WT: 1.02 ± 0.20 s; DS: 3.95 ± 0.69 s), whereas the Type 2 mEPSC inter-event interval was not significantly affected (WT: 0.73 ± 0.12 s; DS: 1.11 ± 0.17 s; Figure 6E). Furthermore, mEPSC amplitude was significantly reduced for both Type 1 (WT: 8.5 ± 0.5 pA; DS: 7.0 ± 0.2 pA) and Type 2 (WT: 9.5 ± 0.2 pA; DS: 8.0 ± 0.2 pA) events in DS VPL neurons compared to WT (Figure 6F). These data suggest that VPL neurons in DS mice have a global decrease in glutamatergic synapse strength, but a specific reduction in the frequency of the faster decaying mEPSC population.
Interestingly, VPM neurons in WT and DS mice had similar total numbers of mEPSCs (WT: 915 ± 138; DS: 1190 ± 128; p = 0.70), and the ratio of Type 1 and Type 2 events was not significantly different (WT: 0.2 ± 0.03; DS: 0.23 ± 0.03; Figure 6J). Furthermore, mEPSC decay times were similar for Type 1 (WT: 3.5 ± 0.3 ms; DS: 4.0 ± 0.5 ms) and Type 2 mEPSCs (WT: 7.0 ± 0.7 ms; DS: 8.0 ± 0.6 ms; Supplementary Figure S5C,D). VPM neurons in WT and DS mice also exhibited no significant differences in the inter-event interval of Type 1 (WT: 4.0 ± 1.1 s; DS: 2.6 ± 0.7 s) and Type 2 mEPSCs (WT: 0.64 ± 0.16 s; DS: 0.48 ± 0.07 s) or the amplitude of Type 1 (WT: 7.0 ± 0.3 pA; DS: 7.3 ± 0.4 pA) and Type 2 mEPSCs (WT: 8.5 ± 0.5 pA; DS: 8.9 ± 0.6 pA; Figure 6K,L). Taken together, the mESPC data suggest that NaV1.1 haploinsufficiency may affect synapse development in VPL and VPM neurons differently leading to cell-type-specific roles for these glutamatergic neuron populations in thalamic dysfunction in DS.
To evaluate glutamatergic synapses in the VPL and VPM in an input-specific manner, we immunostained for VGLUT1 to label CT synapses and VGLUT2 to label ascending sensory synapses, and then quantified the number and size of synaptic puncta (Figure 7A,B). The number of VGLUT2 puncta in the VPL was decreased in DS mice relative to WT (83.7 ± 3.2% of WT), whereas the number of VGLUT1 puncta was unchanged (99.9 ± 6.7% of WT, Figure 7C). The size of VGLUT1 and VGLUT2 puncta were not significantly different between WT and DS mice (VGLUT1: 96.7 ± 3.2 % of WT; VGLUT2: 107 ± 6.1 % of WT). Furthermore, the VPM in WT and DS mice had similar VGLUT1 and VGLUT2 puncta number (VGLUT1: 112.3 ± 5.9 % of WT; VGLUT2: 87.0 ± 12.7 % of WT) and size (VGLUT1: 96.2 ± 3.2 % of WT; VGLUT2: 101.5 ± 7.2 % of WT; Figure 7D). These imaging data are consistent with a specific reduction in the number of ascending sensory synapses in the VPL.
Figure 7. DS mice exhibit reduced ascending sensory input to the VPL.
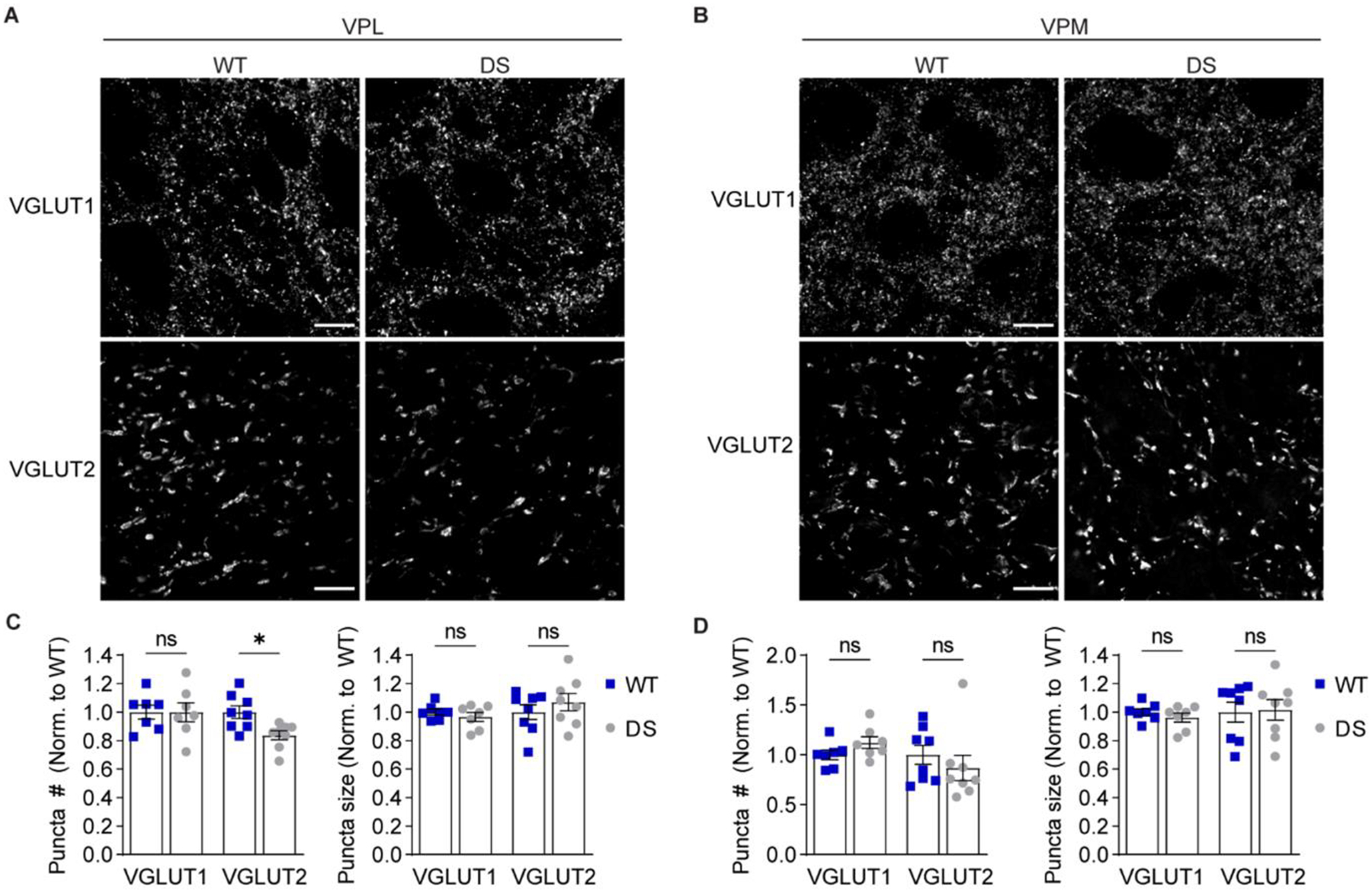
Representative 100X images show VGLUT1 (scale bar: 10 μm) and VGLUT2 (scale bar: 20 μm) immunostaining in the (A) VPL and (B) VPM. The number and size of VGLUT1 and VGLUT2 puncta were quantified for the (C) VPL and (D) VPM (n = 7) and analyzed by two-way ANOVA. VPL puncta number: Genotype F(1,26) = 2.914, p = 0.10; Interaction F(1,26) = 2.829, p = 0.10; posthoc Sidak’s tests: VGLUT1, p = 0.99; VGLUT2, *p = 0.04. VPL puncta size: Genotype: F(1,26) = 0.161, p = 0.69; Interaction: F(1,26) = 1.262, p = 0.27; posthoc Sidak’s tests: VGLUT1, p = 0.86; VGLUT2, p = 0.47. VPM puncta number: Genotype: F(1,26) = 0.002, p = 0.96; Interaction: F(1,26) = 1.9, p = 0.18; posthoc Sidak’s tests: VGLUT1, p = 0.60; VGLUT2, p = 0.52. VPM puncta size: Genotype: F(1,26) = 0.040, p = 0.84; Interaction: F(1,26) = 0.221, p = 0.64; posthoc Sidak’s tests: VGLUT1, p = 0.88; VGLUT2, p = 0.98. Data points in all bar graphs represent individual mice.
Inhibitory synaptic input is enhanced in VPL neurons
VPL and VPM neurons receive inhibitory input from nRT neurons, with VPL neurons receiving input from both somatostatin- and parvalbumin-positive neurons and VPM neurons receiving input from parvalbumin-positive nRT neurons (Clemente-Perez et al., 2017). We hypothesized that aberrant excitability within the somatosensory thalamic circuitry would alter inhibitory synaptic input from the nRT to VPL and VPM neurons. Therefore, we analyzed the decay time, frequency, and amplitude of mIPSCs in VPL and VPM neurons (Figure 8A,G). VPL neuron mIPSC decay time was significantly faster in DS mice (16.8 ± 2.4 ms) compared to WT (25.4 ± 2.1 ms; Figure 8B), which suggests that either postsynaptic receptor composition or the proximal-distal distribution of GABAergic inputs may be altered in DS mice. Moreover, VPL neurons exhibited reduced mIPSC inter-event interval in DS mice (0.133 ± 0.023 ms) compared to WT (0.266 ± 0.033 ms; Figure 8C), whereas VPL mIPSC amplitude was not significantly altered in DS mice (23.0 ± 1.1 pA) relative to WT (20.3 ± 0.6 pA; Figure 8D). VPM neurons in DS mice had no significant differences relative to WT in mIPSC decay time (WT: 29.8 ± 5.5 ms; DS: 35.8 ± 7.9 ms), inter-event interval (WT: 0.294 ± 0.06 s; DS: 0.269 ± 0.06 s), or amplitude (WT: 19.2 ± 0.7 pA; DS: 19.7 ± 0.6 pA; Figure 8H–J). These data suggest that NaV1.1 haploinsufficiency enhances the frequency of GABAergic input to VPL neurons without affecting GABAergic input to VPM neurons.
Figure 8. VPL neurons in DS mice exhibit reduced GABAergic synaptic transmission.
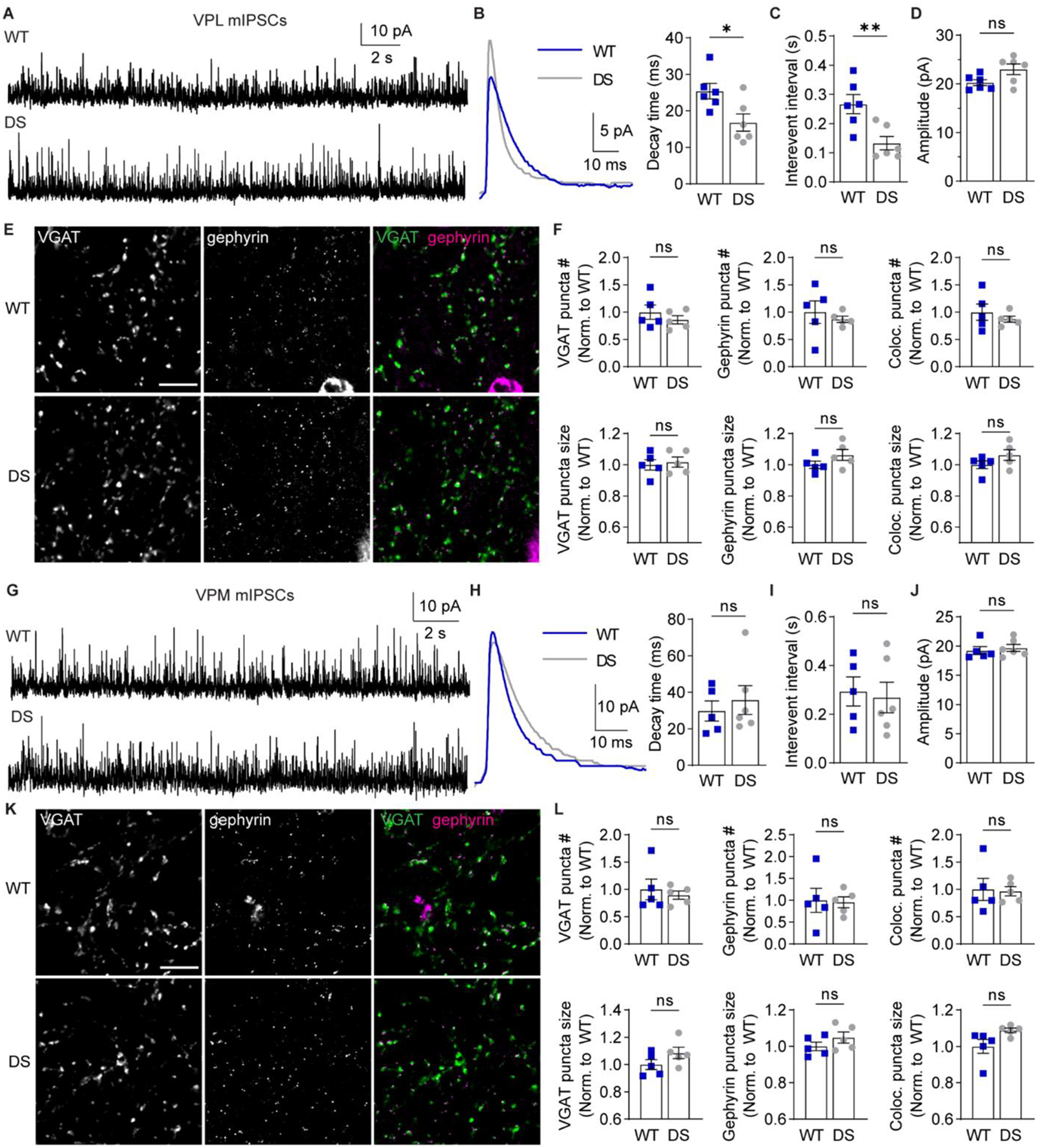
A. mIPSCs were recorded from VPL neurons in acute brain slices in the presence of 1 μM TTX. B. Representative traces show ensemble averages of WT and DS mIPSCs. mIPSC ensemble averages were fitted to determine decay time for WT and DS neurons (n = 6 cells from 5 mice) and compared by unpaired t-test (*p = 0.02). WT and DS mIPSC (C) inter-event interval (**p = 0.008) and (D) amplitude (p = 0.054) were quantified for each neuron and compared by unpaired t-tests. E. Representative 100X images show VGAT and gephyrin immunolabeling in the VPL (scale bar: 10 μm). F. The number and size of VGAT, gephyrin, and VGAT-gephryin colocalized puncta in the VPL (n = 5 mice) were compared by unpaired t-tests. VGAT puncta number: p = 0.39, size: p = 0.72; gephyrin puncta number: p = 0.56, size p = 0.17; colocalized puncta number: p = 0.44, size: p = 0.18. G. mIPSCs were recorded from VPM neurons, and (H) representative traces show ensemble averages of WT and DS mIPSCs. mIPSC ensemble averages were fitted to determine decay time for WT (n = 5 cells from 5 mice) and DS (n = 6 cells from 6 mice) neurons and compared by an unpaired t-test (p = 0.57). I. WT and DS inter-event interval (p = 0.785) and (J) amplitude (p = 0.635) were compared by unpaired t-tests. K. Representative 100X images show VGAT (scale bar: 10 μm) and gephyrin immunolabeling in the VPM. L. The number and size of VGAT, gephyrin, and VGAT-gephryin colocalized puncta were quantified in the VPM (n = 5 mice) and compared by unpaired t-tests. VGAT puncta number: p = 0.62, size: p = 0.16; gephyrin puncta number: p = 0.89, size p = 0.24; colocalized puncta number: p = 0.88, size: p = 0.06. The data points in the bar graphs represent individual neurons (B-D, H-J) or mice (F, L).
To further evaluate how GABAergic inputs were altered in DS mice, we immunolabeled VGAT and gephyrin in WT and DS thalamus and quantified the number and size of inhibitory synapses (Figure 8E,K). We detected no significant differences in WT and DS VPL when comparing VGAT puncta number (86 ± 7.5 % of WT), VGAT puncta size (102 ± 3.2 % of WT), gephyrin puncta number (87 ± 5.5 % of WT), or gephyrin puncta size (106 ± 3.5 % of WT; Figure 8F). We also quantified colocalized regions of VGAT and gephyrin puncta in the VPL and found no significant differences in the number (87 ± 5.6 % of WT) or size (106 ± 3.5 % of WT; Figure 8F). Similarly, we did not find any significant differences in GABAergic synapse labeling in VPM when we compared VGAT puncta number (90 ± 7.5 % of WT), VGAT puncta size (109 ± 4.2 % of WT), gephyrin puncta number (96 ± 12.4 % of WT), or gephyrin puncta size (105 ± 3.0 % of WT; Figure 8L). We also found no significant differences in colocalized VGAT and gephyrin puncta number (97 ± 8.6 % of WT) or size (109 ± 1.3 % of WT) in the VPM (Figure 8L). Taken together, the mIPSC and VGAT/gephyrin data are consistent with enhanced frequency and accelerated kinetics of GABAergic synaptic transmission in VPL neurons of DS mice, which may occur through changes in presynaptic release and/or postsynaptic receptor expression as opposed to changes in GABAergic synapse number.
DISCUSSION
Collectively, these data provide novel insight into the mechanisms underlying somatosensory CT circuit dysfunction in a NaV1.1 haploinsufficiency DS mouse model. The excitability of GABAergic nRT neurons and glutamatergic VPL and VPM neurons was disrupted in a cell-type-specific manner, including alterations in both depolarization-induced spike firing and hyperpolarization-induced rebound burst firing in nRT and VPL neurons. Unexpectedly, VPL and VPM neurons exhibited opposing changes to depolarization-induced firing, suggesting they may differentially contribute to circuit-wide dysfunction in DS. Glutamatergic synaptic input to both the nRT and VPL was reduced, and GABAergic synaptic input to the VPL was enhanced, further contributing to an imbalance of synaptic input to the region. Together, these results indicate that synaptic- and cellular-level changes in GABAergic and glutamatergic neuron populations contribute to somatosensory thalamic dysfunction in DS.
As expected, NaV1.1 haploinsufficiency resulted in altered excitability in GABAergic nRT neurons, including altered depolarization- and hyperpolarization-induced firing. Previous studies revealed reduced tonic and burst firing in Scn1a-haploinsufficient and NaV1.1-R1648H mouse models of DS (Hedrich et al., 2014; Kalume et al., 2015). Consistent with these studies, we observed reductions in the number of spikes fired in response to depolarization and the number of spikes per rebound burst in nRT DS neurons. Our spike firing experiments were conducted from RMP enabling us to determine how neurons responded to depolarizing or hyperpolarizing currents from their natural resting state; whereas, some previous work studied tonic and burst firing properties while holding the cells at a constant membrane potential to control for differences in RMP (Hedrich et al., 2014). Unlike these previous studies, we identified a subset of DS nRT neurons with a hyperpolarized RMP. These neurons fired low-threshold spikes with bursts of action potentials in response to depolarization, which is a unique finding compared to previous studies reporting hypoexcitability of DS nRT neurons (Hedrich et al., 2014). However, the majority of DS nRT neurons responded to depolarization with a low number of spikes immediately upon current injection, and their RMP was slightly hyperpolarized compared to WT neurons, but the difference was not statistically significant. It is thus unlikely that this small hyperpolarization in RMP caused the dramatic reduction in spike firing in these DS nRT neurons, suggesting that there are also likely impairments in their tonic firing properties. From a circuit-level perspective, DS nRT neurons responding to depolarizing input with a reduced number of spikes or in a burst firing mode could result in profound somatosensory informational processing deficits, as both the number of spikes and mode of spiking communicate critical somatosensory information to the cerebral cortex.
Both populations of DS nRT neurons exhibited a significant reduction in the number of spikes per hyperpolarization-induced rebound burst compared to WT neurons. It is possible that the observed reduction in RMP in a subset of nRT neurons may cause their T-type calcium channels to be de-inactivated at rest and thus require depolarization, not hyperpolarization, to be activated. Therefore, the hyperpolarized RMP could contribute to reduced rebound burst spiking as well as rebound bursting in response to depolarization in this subset of DS nRT neurons. However, it is unlikely that changes in RMP underlie the reduced rebound burst firing in all DS nRT neurons, as the larger population of neurons did not display a significant change in RMP. This indicates that there are also impairments in either the T-type calcium channel low threshold-spike or sodium/potassium-dependent action potential firing; however, additional studies that hold WT and DS neurons at a constant membrane potential will be required to elucidate the precise mechanism underlying reduced rebound burst firing in DS nRT neurons. Regardless of the underlying mechanisms, impaired nRT neuron burst firing could change nRT-VPL/VPM bursting patterns leading to altered intra-thalamic oscillations, which are critical for sleep spindles.
The identity of the two subsets of DS nRT neurons identified here remains unclear. There are several recent studies characterizing functionally distinct subpopulations of nRT neurons including parvalbumin-, somatostatin-, and calbindin-expressing neurons (Clemente-Perez et al., 2017; Li et al., 2020; Martinez-Garcia et al., 2020). These neuron populations are reported to have distinct distributions within the nRT and different functional properties such as rebound burst strength, intrinsic membrane properties, and tonic spike firing properties (Clemente-Perez et al., 2017; Martinez-Garcia et al., 2020). While the RMP and input resistance of the two DS nRT neuron populations identified here were significantly different, no other membrane properties differed between the populations. Further, because tonic and rebound burst states were not isolated by controlling the membrane potential in this study, it remains unknown whether these two populations have distinct intrinsic firing properties. Thus, future studies will be needed to elucidate whether these two DS nRT neuron groups represent functionally or molecularly distinct populations.
In contrast to our findings, the NaV1.1-R1407X mouse model exhibited hyperexcitability of parvalbumin-positive nRT neurons including prolonged hyperpolarization-induced rebound bursts and enhanced depolarization-induced spike firing (Ritter-Makinson et al., 2019). Discrepancies between models could be attributed to the specific etiology of the disease model or the background strain as DS models are known to have a high degree of strain-dependent phenotype variability (Miller et al., 2014; Rubinstein et al., 2015b). The age of the mice could also cause discrepancies between studies as some cortical neuron populations exhibit a transient window of altered excitability (Favero et al., 2018); however, developmental studies of thalamic dysregulation have not been reported in DS mouse models. From a therapeutic perspective, these model-specific effects indicate that successful patient treatment may largely depend on the patient’s specific genetic alteration. As truncation mutations resulting in haploinsufficiency are responsible for about half of all DS cases, the mechanisms uncovered in this study may be relevant to a large population of patients with DS.
The hyperpolarization of the RMP in both nRT and VPL neurons was an unexpected finding of this study, and how NaV1.1 haploinsufficiency leads to this change remains unknown. nRT neurons exhibit a persistent sodium current, which is a slow-inactivating current that amplifies even small depolarizations in membrane potential (Landisman, 2012). Thus, it is likely that this persistent sodium current contributes to the RMP of nRT neurons, and loss of NaV1.1 or compensatory changes in NaV channel expression or function could contribute to the observed hyperpolarized RMP. Indeed, altered persistent sodium current has been proposed as a mechanism contributing to disrupted cell excitability in epilepsies caused by sodium channel mutations including DS (Stafstrom, 2007). Whether VPL and VPM neurons exhibit a persistent sodium current has not been established, though other thalamic nuclei such as the dLGN do exhibit this current in rats (Parri and Crunelli, 1998). These findings provide impetus for investigating the persistent sodium current in the VPL and VPM and whether it contributes to the observed hyperpolarized RMP in DS. In addition, spontaneous glutamatergic input likely depolarizes the RMP of thalamic neurons, and the observed reduction in glutamatergic input could hyperpolarize nRT and VPL neuron RMP leading to altered excitability. This would provide an interesting therapeutic opportunity, as correcting the synaptic-level deficits could potentially ameliorate the hyperpolarized RMP and altered firing. Finally, altered potassium channel expression has been implicated in nRT dysfunction in other DS models (Layer et al., 2021; Ritter-Makinson et al., 2019), and it is possible that NaV1.1 haploinsufficiency alters potassium channel expression or function leading to the hyperpolarized RMP and altered excitability evident in nRT and VPL neurons.
Interestingly, this study also discovered opposing changes in glutamatergic VPL and VPM neuron excitability as well as distinct effects on intrinsic membrane properties. Many glutamatergic neuron populations express NaV1.1 at low levels and reportedly exhibit no altered excitability in DS, but recent evidence suggests that CA1 pyramidal neuron excitability is altered and changes over the time course of the disease (Almog et al., 2021). Previous investigations have reported no alterations in the excitability of VB neurons in DS models (Hedrich et al., 2014; Ritter-Makinson et al., 2019). This could be due to differences in the specific DS model, the developmental time period studied, or a lack of differentiating between the VPL and VPM, the two regions comprising the VB thalamus, which could have resulted in an averaging of cell-type-specific alterations and no overall change.
The cellular properties of VPL and VPM neurons have not yet been directly compared, and thus our understanding of how NaV1.1 haploinsufficiency leads to opposing changes in their excitability is limited. We speculate that different NaV1.1 expression levels or subcellular localization may underlie distinct roles for NaV1.1 in normal VPL and VPM neuron physiology. Alternatively, the expression of different complements of NaV, CaV, or KV channels in VPL and VPM neurons could lead to distinct compensatory mechanisms that underlie cell-type-specific alterations in the DS model. This is supported by evidence from a spinal cord injury model wherein VPL neurons exhibit increased NaV1.3 expression post-injury, while VPM neurons do not (Zhao et al., 2006). Additionally, as previously stated, reduced glutamatergic input to nRT and VPL neurons, specifically, could hyperpolarize their RMP and lead to hypoexcitability. While the specific mechanisms underlying the distinct changes in intrinsic properties are unknown, these findings suggest that the VPL and VPM may contribute differentially to circuit dysfunction and successful correction of circuit function may require cell-type-specific therapeutic targets.
An additional discovery of this study is altered synaptic connectivity in the thalamus. nRT neurons receive glutamatergic input from CT and TC neurons, and substantial evidence suggests that CT-nRT EPSCs have slower decay kinetics than TC-nRT EPSCs (Deleuze and Huguenard, 2016). VPL and VPM neurons receive glutamatergic input from CT neurons and ascending sensory afferents, and it is well-established that CT EPSCs have slower decay kinetics than sensory EPSCs (Castro-Alamancos, 2002; McCormick and von Krosigk, 1992; Miyata and Imoto, 2006). Indeed, mEPSCs recorded from nRT, VPL, and VPM neurons can be separated into two kinetically distinct populations, which we analyzed independently to assess potential synapse-specific alterations in DS mice. DS nRT neurons exhibited reduced frequency of the slow-decaying mEPSC population, which are putative CT-nRT synaptic events. Altered CT-nRT synaptic connectivity may disrupt feed-forward CT-nRT-TC inhibition, which is critical for proper CT circuit function. VPL neurons in this DS mouse model exhibited a global reduction in the amplitude of mEPSCs as well as reduced frequency of the fast-decaying population of mEPSCs, which are putative ascending sensory synaptic events. Consistent with this result, we observed a reduction in VGLUT2 synaptic puncta specifically in the VPL thalamus. VPL neurons also exhibited increased mIPSC frequency. Thus, VPL neurons receive an imbalance of synaptic input including excessive inhibition and insufficient excitation, which could contribute to dysfunctional somatosensory processing as well as altered reciprocal VPL-nRT connectivity.
The cell-type- and input-specific nature of these synaptic alterations suggests that tuning circuit excitability may require pathway-specific therapeutic targets. The distinct changes in VPL and VPM synaptic connectivity could be due to their respective changes in excitability or their distinct sources of ascending input as the VPL and VPM receive somatosensory information from the body and head, respectively. A most intriguing finding is the putative reduction in CT-nRT input without a corresponding reduction in CT input to either VPL or VPM neurons. The CT axons that innervate nRT neurons are collaterals of those innervating VPL and VPM neurons, suggesting that a postsynaptic mechanism may disrupt synapse development in the nRT. We did not detect a corresponding change in VGLUT1 puncta in the nRT, and thus our observations are consistent with alterations in glutamate release, glutamate receptor expression, or dendritic filtering. Determining the underlying mechanism will require investigating specific glutamate receptor populations as well as studying synaptic potentials under more physiological conditions in addition to the voltage-clamp studies conducted herein.
The NaV1.1-R1648H DS model previously showed reduced spontaneous IPSCs in glutamatergic thalamocortical neurons, but no changes in mIPSCs (Hedrich et al., 2014). Another DS model exhibited no change in spontaneous EPSCs in nRT or VB neurons (Ritter-Makinson et al., 2019). These discrepancies could be due to the specific cell populations that were studied as the previous studies examined VB neurons, which presumably included both VPL and VPM neurons. Furthermore, we discovered differential effects in two kinetically distinct mEPSC populations in nRT and VPL neurons, whereas previous studies evaluated all spontaneous EPSCs as one group, which may have obscured synapse-specific effects. Age-dependent changes may also contribute to different findings as studies at earlier developmental time points may not reveal such synaptic-level changes (Hedrich et al., 2014). Developmental studies will be important for elucidating the time course over which both intrinsic and extrinsic alterations occur.
From a clinical perspective, the dysfunction in glutamatergic synapses as well as glutamatergic VPL and VPM neurons observed here may present unique opportunities for therapeutic intervention. Specifically, the diversity of glutamate receptor expression in the thalamus provides a wide array of therapeutic targets. Modulating glutamate receptors is becoming increasingly feasible as a variety of subtype-selective modulators for metabotropic glutamate receptors, AMPA receptors, and NMDA receptors have been developed (Azumaya et al., 2017; Hansen et al., 2018; Hovelso et al., 2012; Kadriu et al., 2021; Mazzitelli et al., 2018). For example, a recent study indicated that the GluN2A positive allosteric modulator GNE-0723 reduces low-frequency oscillations and epileptiform activity in a DS mouse model, providing evidence that NMDA receptor modulation could be a viable therapeutic option (Hanson et al., 2020). The GluN2C and GluN2D subunits of NMDA receptors have more restricted expression patterns, including relatively high expression in the thalamus, and recently developed GluN2C/2D-specific modulators could offer a means to tune thalamic function with more limited adverse effects (Acker et al., 2011; Khatri et al., 2014; Liu et al., 2019; Mullasseril et al., 2010; Swanger et al., 2018; Yi et al., 2020). Thus, the data presented here lay the foundation to explore glutamatergic synapse modulation as a possible therapeutic strategy to correct thalamic dysfunction in DS.
Altogether, this evidence indicating cell-type-specific dysfunction advances our understanding of how GABAergic and glutamatergic neuron populations may together contribute to pathological thalamocortical network function. We posit that altered depolarization-induced spike firing may impair VPL cell output and enhance VPM cell output in response to ascending sensory information. The reduced ascending input to the VPL may further impair somatic information processing, perhaps contributing to reduced sensitivity to pain and attention deficits evident in the disease (Catarino et al., 2011; Villas et al., 2017). Burst firing in the somatosensory thalamus underlies oscillatory activity critical for thalamocortical network function, and disrupted burst firing is associated with absence seizures, sleep disorders, and chronic pain (Fogerson and Huguenard, 2016; Hains et al., 2006; Henderson et al., 2013). Intra-thalamic oscillations occur when nRT neurons at a hyperpolarized RMP receive depolarizing input from the cortex, which leads to feed-forward nRT-VPL and nRT-VPM inhibition. VPL and VPM neurons are then hyperpolarized and will burst upon recovery from hyperpolarization leading to feedback inhibition from the nRT. In this DS model, VPL neurons had a hyperpolarized RMP and fired significantly more spikes during burst firing. Some nRT neurons exhibited a hyperpolarized RMP and an increased propensity to burst in response to depolarization, which could result in augmented feed-forward inhibition of VPL and VPM neurons. Thus, together these mechanisms may result in enhanced bursting in all three cell populations that contributes to aberrant thalamic oscillations in DS (Kalume et al., 2015; Ritter-Makinson et al., 2019). A complete understanding of how these cellular and synaptic mechanisms contribute to circuit dysfunction will require elucidating how cells respond to excitatory drive from ascending and descending synaptic inputs.
Conclusions
In summary, this work discovered cell-type-specific dysregulation of synapses and excitability within the somatosensory thalamus in a DS mouse model. Specifically, the findings introduce altered glutamatergic neuron excitability and synapse function as disease mechanisms that may contribute to thalamocortical network dysfunction underlying aberrant sensory processing, disrupted sleep, and absence seizures. Cell-type-specific intrinsic and synaptic disease mechanisms could affect thalamic function distinctly and thus contribute to particular symptoms or developmental stages of DS. Further investigation is required to elucidate how these previously unidentified mechanisms contribute to DS symptomology across the course of the disease and whether modulating specific therapeutic targets in the glutamate system can restore thalamic function and ameliorate corresponding phenotypes.
Supplementary Material
Highlights.
NaV1.1 haploinsufficiency leads to cell-type-specific thalamic dysfunction
NaV1.1 haploinsufficiency impairs glutamatergic neuron excitability in the thalamus
NaV1.1 haploinsufficiency alters excitatory and inhibitory synapses in the thalamus
Thalamic excitation and inhibition are imbalanced in a Dravet syndrome mouse model
Funding
This work was supported by the National Institutes of Health [NS105804], CURE Epilepsy, the Dravet Syndrome Foundation, and Brain Research Foundation.
Abbreviations
- DS
Dravet syndrome
- nRT
reticular nucleus of the thalamus
- VPM
ventral posteromedial nucleus
- VPL
ventral posterolateral nucleus
- VB
ventrobasal complex
- CT
corticothalamic
- TC
thalamocortical
- VGLUT1
vesicular glutamate transporter 1
- VGLUT2
vesicular glutamate transporter 2
- VGAT
vesicular GABA transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
None
REFERENCES
- Ab Aziz CB, and Ahmad AH (2006). The role of the thalamus in modulating pain. Malays J Med Sci 13, 11–18. [PMC free article] [PubMed] [Google Scholar]
- Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, and Traynelis SF (2011). Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Mol Pharmacol 80, 782–795. 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almog Y, Fadila S, Brusel M, Mavashov A, Anderson K, and Rubinstein M (2021). Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol Dis 148, 105209. 10.1016/j.nbd.2020.105209. [DOI] [PubMed] [Google Scholar]
- Alshammari MA, Alshammari TK, and Laezza F (2016). Improved Methods for Fluorescence Microscopy Detection of Macromolecules at the Axon Initial Segment. Front Cell Neurosci 10, 5. 10.3389/fncel.2016.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azumaya CM, Days EL, Vinson PN, Stauffer S, Sulikowski G, Weaver CD, and Nakagawa T (2017). Screening for AMPA receptor auxiliary subunit specific modulators. PLoS One 12, e0174742. 10.1371/journal.pone.0174742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, and Huguenard JR (2009). Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron 62, 612–632. 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AC, Luikart BW, and Lenck-Santini PP (2016). Cognitive Deficits Associated with Nav1.1 Alterations: Involvement of Neuronal Firing Dynamics and Oscillations. PLoS One 11, e0151538. 10.1371/journal.pone.0151538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AC, Natola H, Ndong C, Holmes GL, Scott RC, and Lenck-Santini PP (2013). Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis 54, 297–307. 10.1016/j.nbd.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkvens JJ, Veugen I, Veendrick-Meekes MJ, Snoeijen-Schouwenaars FM, Schelhaas HJ, Willemsen MH, Tan IY, and Aldenkamp AP (2015). Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav 47, 11–16. 10.1016/j.yebeh.2015.04.057. [DOI] [PubMed] [Google Scholar]
- Brecht M, and Sakmann B (2002). Whisker maps of neuronal subclasses of the rat ventral posterior medial thalamus, identified by whole-cell voltage recording and morphological reconstruction. J Physiol 538, 495–515. 10.1113/jphysiol.2001.012334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA (2002). Properties of primary sensory (lemniscal) synapses in the ventrobasal thalamus and the relay of high-frequency sensory inputs. J Neurophysiol 87, 946–953. 10.1152/jn.00426.2001. [DOI] [PubMed] [Google Scholar]
- Catarino CB, Liu JY, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, Woodward C, Davis MB, Smith SJ, Cross JH, et al. (2011). Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain 134, 2982–3010. 10.1093/brain/awr129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong E, and Shin HS (2013). T-type Ca2+ channels in normal and abnormal brain functions. Physiol Rev 93, 961–992. 10.1152/physrev.00010.2012. [DOI] [PubMed] [Google Scholar]
- Chiron C (2011). Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 53 Suppl 2, 16–18. 10.1111/j.1469-8749.2011.03967.x. [DOI] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, and De Jonghe P (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68, 1327–1332. 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente-Perez A, Makinson SR, Higashikubo B, Brovarney S, Cho FS, Urry A, Holden SS, Wimer M, David C, Fenno LE, et al. (2017). Distinct Thalamic Reticular Cell Types Differentially Modulate Normal and Pathological Cortical Rhythms. Cell Rep 19, 2130–2142. 10.1016/j.celrep.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, and Lagae L (2019). Dravet syndrome: Treatment options and management of prolonged seizures. Epilepsia 60 Suppl 3, S39–S48. 10.1111/epi.16334. [DOI] [PubMed] [Google Scholar]
- Darra F, Battaglia D, Dravet C, Patrini M, Offredi F, Chieffo D, Piazza E, Fontana E, Olivieri G, Turrini I, et al. (2019). Dravet syndrome: Early electroclinical findings and long-term outcome in adolescents and adults. Epilepsia 60 Suppl 3, S49–S58. 10.1111/epi.16297. [DOI] [PubMed] [Google Scholar]
- Deleuze C, and Huguenard J (2016). Two classes of excitatory synaptic responses in rat thalamic reticular neurons. J Neurophysiol 116, 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravet C (2011). The core Dravet syndrome phenotype. Epilepsia 52 Suppl 2, 3–9. 10.1111/j.1528-1167.2011.02994.x. [DOI] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, Kearney JA, and Goldberg EM (2018). A Transient Developmental Window of Fast-Spiking Interneuron Dysfunction in a Mouse Model of Dravet Syndrome. J Neurosci 38, 7912–7927. 10.1523/JNEUROSCI.0193-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez LMJ, and Luthi A (2020). Sleep Spindles: Mechanisms and Functions. Physiol Rev 100, 805–868. 10.1152/physrev.00042.2018. [DOI] [PubMed] [Google Scholar]
- Fogerson PM, and Huguenard JR (2016). Tapping the Brakes: Cellular and Synaptic Mechanisms that Regulate Thalamic Oscillations. Neuron 92, 687–704. 10.1016/j.neuron.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gataullina S, and Dulac O (2017). From genotype to phenotype in Dravet disease. Seizure 44, 58–64. 10.1016/j.seizure.2016.10.014. [DOI] [PubMed] [Google Scholar]
- Graziano A, Liu X-B, Murray KD, and Jones EG (2008). Vesicular Glutamate Transporters Define Two Sets of Glutamatergic Afferents to the Somatosensory Thalamus and Two Thalamocortical Projections in the Mouse. The Journal of Comparative Neurology 507, 1258–1276. [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, and Waxman SG (2006). Alterations in burst firing of thalamic VPL neurons and reversal by Na(v)1.3 antisense after spinal cord injury. J Neurophysiol 95, 3343–3352. 10.1152/jn.01009.2005. [DOI] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, and Catterall WA (2012). Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, and Traynelis SF (2018). Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol 150, 1081–1105. 10.1085/jgp.201812032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Ma K, Elstrott J, Weber M, Saillet S, Khan AS, Simms J, Liu B, Kim TA, Yu GQ, et al. (2020). GluN2A NMDA Receptor Enhancement Improves Brain Oscillations, Synchrony, and Cognitive Functions in Dravet Syndrome and Alzheimer’s Disease Models. Cell Rep 30, 381–396 e384. 10.1016/j.celrep.2019.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich UB, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihne M, et al. (2014). Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J Neurosci 34, 14874–14889. 10.1523/JNEUROSCI.0721-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson LA, Peck CC, Petersen ET, Rae CD, Youssef AM, Reeves JM, Wilcox SL, Akhter R, Murray GM, and Gustin SM (2013). Chronic pain: lost inhibition? J Neurosci 33, 7574–7582. 10.1523/JNEUROSCI.0174-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovelso N, Sotty F, Montezinho LP, Pinheiro PS, Herrik KF, and Mork A (2012). Therapeutic potential of metabotropic glutamate receptor modulators. Curr Neuropharmacol 10, 12–48. 10.2174/157015912799362805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadriu B, Musazzi L, Johnston JN, Kalynchuk LE, Caruncho HJ, Popoli M, and Zarate CA Jr. (2021). Positive AMPA receptor modulation in the treatment of neuropsychiatric disorders: A long and winding road. Drug Discov Today. 10.1016/j.drudis.2021.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, and Catterall WA (2015). Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. Neurobiol Dis 77, 141–154. 10.1016/j.nbd.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Yu FH, Westenbroek RE, Scheuer T, and Catterall WA (2007). Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci 27, 11065–11074. 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri A, Burger PB, Swanger SA, Hansen KB, Zimmerman S, Karakas E, Liotta DC, Furukawa H, Snyder JP, and Traynelis SF (2014). Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Mol Pharmacol 86, 548–560. 10.1124/mol.114.094516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landisman J.S.H.a.C.E. (2012). State-dependent modulation of gap junction signaling by the persistent sodium current. Frontiers in Cellular Neuroscience 5 (31). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer N, Sonnenberg L, Gonzalez EP, Benda J, Hedrich UBS, Lerche H, Koch H, and Wuttke TV (2021). Dravet Variant SCN1AA1783V Impairs Interneuron Firing Predominantly by Altered Channel Activation. Frontiers in Cellular Neuroscience 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim D, and Shin HS (2004). Lack of delta waves and sleep disturbances during non-rapid eye movement sleep in mice lacking alpha1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A 101, 18195–18199. 10.1073/pnas.0408089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz FA (1992). The ventral posterior nucleus of thalamus is involved in the generation of central pain syndromes. APS Journal 1, 42–51. 10.1016/S1058-9139(06)80011-2. [DOI] [Google Scholar]
- Li Y, Lopez-Huerta VG, Adiconis X, Levandowski K, Choi S, Simmons SK, Arias-Garcia MA, Guo B, Yao AY, Blosser TR, et al. (2020). Distinct subnetworks of the thalamic reticular nuclues. Nature 583, 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licheni SH, McMahon JM, Schneider AL, Davey MJ, and Scheffer IE (2018). Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol 60, 192–198. 10.1111/dmcn.13601. [DOI] [PubMed] [Google Scholar]
- Liu J, Shelkar GP, Zhao F, Clausen RP, and Dravid SM (2019). Modulation of burst firing of neurons in nucleus reticularis of the thalamus by GluN2C-containing NMDA receptors. Mol Pharmacol. 10.1124/mol.119.116780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garcia RI, Voelcker B, Zaltsman J, Patrick S, Stevens T, Conners B, and Cruikshank S (2020). Two dynamically distinct circuits driving inhibition in sensory thalamus. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzitelli M, Palazzo E, Maione S, and Neugebauer V (2018). Group II Metabotropic Glutamate Receptors: Role in Pain Mechanisms and Pain Modulation. Front Mol Neurosci 11, 383. 10.3389/fnmol.2018.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty C, David F, Venzi M, Lorincz ML, Delicata F, Atherton Z, Recchia G, Orban G, Lambert RC, Di Giovanni G, et al. (2018). Cortical drive and thalamic feed-forward inhibition control thalamic output synchrony during absence seizures. Nat Neurosci 21, 744–756. 10.1038/s41593-018-0130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, and von Krosigk M (1992). Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A 89, 2774–2778. 10.1073/pnas.89.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AR, Hawkins NA, McCollom CE, and Kearney JA (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 13, 163–172. 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, and Imoto K (2006). Different composition of glutamate receptors in corticothalamic and lemniscal synaptic responses and their roles in the firing responses of ventrobasal thalamic neurons in juvenile mice. J Physiol 575, 161–174. 10.1113/jphysiol.2006.114413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring J, von Spiczak S, Moeller F, Helbig I, Wolff S, Jansen O, Muhle H, Boor R, Stephani U, and Siniatchkin M (2013). Variability of EEG-fMRI findings in patients with SCN1A-positive Dravet syndrome. Epilepsia 54, 918–926. 10.1111/epi.12119. [DOI] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, et al. (2010). A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat Commun 1, 90. 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, and Yamakawa K (2013). Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 22, 4784–4804. 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27, 5903–5914. 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Makinson CD, Christopher Ehlen J, Tufik S, Decker MJ, Paul KN, and Escayg A (2013). Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia 54, 625–634. 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri RH, and Crunelli V (1998). Sodium current in rat and cat thalamocortical neurons: role of a non-inactivating component in tonic and burst firing. J Neuroscience 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragona F (2011). Cognitive development in children with Dravet syndrome. Epilepsia 52 Suppl 2, 39–43. 10.1111/j.1528-1167.2011.03000.x. [DOI] [PubMed] [Google Scholar]
- Ritter-Makinson S, Clemente-Perez A, Higashikubo B, Cho FS, Holden SS, Bennett E, Chkhaidze A, Eelkman Rooda OHJ, Cornet MC, Hoebeek FE, et al. (2019). Augmented Reticular Thalamic Bursting and Seizures in Scn1a-Dravet Syndrome. Cell Rep 26, 54–64 e56. 10.1016/j.celrep.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda JM, Scheffer IE, McMahon JM, Berkovic SF, and Graham HK (2012). Progressive gait deterioration in adolescents with Dravet syndrome. Arch Neurol 69, 873–878. 10.1001/archneurol.2011.3275. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, and Catterall WA (2015a). Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233. 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, and Catterall WA (2015b). Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol Dis 73, 106–117. 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall Shane R., C. S, Connors Barry (2015). A Corticothalamic Switch: Controlling the Thalamus with Dynamic Switches. Neuron 86, 768–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM (2001). Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci 24, 122–126. 10.1016/s0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE (2007). Persistent Sodium Current and Its Role in Epilepsy. Current Review in Basic Science 7, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, and Llinas RR (1988). The functional states of the thalamus and the associated neuronal interplay. Physiol Rev 68, 649–742. 10.1152/physrev.1988.68.3.649. [DOI] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, Acker TM, Zimmerman SS, DiRaddo JO, Myers SJ, Bundgaard C, Mosley CA, Summer SL, Menaldino DS, et al. (2018). A Novel Negative Allosteric Modulator Selective for GluN2C/2D-Containing NMDA Receptors Inhibits Synaptic Transmission in Hippocampal Interneurons. ACS Chem Neurosci 9, 306–319. 10.1021/acschemneuro.7b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, and Catterall WA (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 111, E3139–3148. 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama R, Fujiwara T, Shigematsu H, Imai K, Takahashi Y, Yamakawa K, and Inoue Y (2014). Long-term course of Dravet syndrome: a study from an epilepsy center in Japan. Epilepsia 55, 528–538. 10.1111/epi.12532. [DOI] [PubMed] [Google Scholar]
- Temereanca S, S. D (2004). Functional topography of corticothalamic feedback enhances thalamic spatial response tuning in the somatosensory whisker/barrel system. Neuron 41, 639–651. [DOI] [PubMed] [Google Scholar]
- Villas N, Meskis MA, and Goodliffe S (2017). Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav 74, 81–86. 10.1016/j.yebeh.2017.06.031. [DOI] [PubMed] [Google Scholar]
- Wolff M, and Vann SD (2019). The Cognitive Thalamus as a Gateway to Mental Representations. J Neurosci 39, 3–14. 10.1523/JNEUROSCI.0479-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Rouzbeh N, Hansen KB, Xu Y, Fanger CM, Gordon E, Paschetto K, Menniti FS, and Volkmann RA (2020). PTC-174, a positive allosteric modulator of NMDA receptors containing GluN2C or GluN2D subunits. Neuropharmacology 173, 107971. 10.1016/j.neuropharm.2020.107971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, and Hains BC (2006). Sodium channel expression in the ventral posterolateral nucleus of the thalamus after peripheral nerve injury. Mol Pain 2, 27. 10.1186/1744-8069-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, and Barbas H (2006). Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci 26, 7348–7361. 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.