

Abstract
Small vessel disease (SVD) is a disorder that causes vascular lesions in the entire parenchyma of the human brain. At present, it is not well known how primary and secondary damage interact to give rise to the complex scenario of white matter (WM) and grey matter (GM) lesions. Using novel cross-sectional and longitudinal connectomic approaches, we unveil the bidirectional nature of GM and WM changes, that is, primary cortical neurodegeneration that leads to secondary alterations in vascular border zones, and WM lesions that lead to secondary neurodegeneration in cortical projecting areas. We found this GM-WM interaction to be essential for executive cognitive performance. Moreover, we also observed that the interlocked degeneration of GM and WM over time associates with prototypical expression levels of genes potentially linked to SVD. Among these connectomic-genetic intersections, we found that the Androgen Receptor (AR) protein coding gene, is a particularly central candidate gene that might confer key vulnerability for brain lesion development in SVD. In conclusion, this study advances in the understanding of the bidirectional relationships between GM and WM lesions, primary and secondary vascular neurodegeneration, and sheds light on the biological mechanisms which underlie the genetic signatures of SVD.
Keywords: Small Vessel Disease, Grey Matter, White Matter, Neurodegeneration, Connectomics, Genetics
Introduction
Small vessel disease (SVD) is a disorder of the small brain arterioles and capillaries that causes lesions in the brain parenchyma, including white matter (WM) and grey matter (GM) (Ter Telgte et al., 2020; Viswanathan et al., 2006; Wardlaw et al., 2019). WM hyperintensities (WMH) of presumed vascular origin, lacunar stroke, microbleeds, microinfarcts, dilated periventricular spaces, or superficial siderosis form the SVD lesions’ spectrum. These lesions are related to a high risk of stroke (both ischemic and hemorrhagic), and the development of a wide variety of motor, sensory, and cognitive impairments (Wardlaw et al., 2019). WMH is one of the most well-characterized features of the SVD neuroimaging manifestations; it is a validated biomarker and it is correlated with SVD clinical syndromes in the aging population (Li et al., 2018). It is hypothesized that leukoaraiosis could also lead to neurodegeneration in distributed areas (e.g., spatially distant from the original WM lesion), impacting both GM and WM remotely and impairing the whole-brain network functionality (Wardlaw et al., 2019). The leading hypothesis is that the cortical damage is caused by secondary degeneration after the loss of long WM tracts, and some studies have successfully correlated the degree of cortical atrophy with the amount of WMH (Habes et al., 2018; Lambert et al., 2016; Rizvi et al., 2018). Interestingly, primary vascular damage within the GM has also been suggested in recent studies of cortical microinfarcts (Ter Telgte et al., 2020), particularly relevant in genetic forms of SVD, such as CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; that is caused by mutations in the NOTCH3 gene(Viswanathan et al., 2006). This proposed bidirectional relationship between GM and WM pathology, despite introducing complexity to the study of SVD, also presents exciting new challenges to develop frameworks for elucidating its primary and secondary damage mechanisms implicated in this pathology.
At present, the etiology of the SVD is not well understood. The most reported abnormality in the SVD is a diffuse arteriopathy affecting the smaller perforating vessels that display histopathological changes in the vascular-cell integration without consensus about where the primary damage occur (arteriolar smooth muscle cell, endothelium, pericytes and/or blood-brain barrier (BBB); Figure 1-I)(Craggs et al., 2014). It has been hypothesized that the perforating vessels arteriopathy causes chronic ischemia in WM due to a reduction in cerebral perfusion and cerebral autoregulation with primary affectation of the mural vessel cells (mostly, smooth muscle cells)(Craggs et al., 2014; Markus, 2008). Other recent hypothesis suggests that diffuse intrinsic microvascular arteriopathy manifests throughout the brain as endothelial dysfunction (Wardlaw et al., 2019). The endothelial dysfunction increases BBB permeability and leads to leakage of plasma material into the vessel wall, causing inflammation and loss of structure and integrity of the WM, both at the microscopic level (only seen with advanced MRI) and macroscopic level producing leukoaraiosis (Li et al., 2018; Wardlaw et al., 2019). The lack of identifiable causal triggers in sporadic SVD (compared to genetically associated forms) – along with multiple susceptibility genes identified in GWAS studies and complex relationships between vascular risk factors in aging populations- make it challenging to disentangle specific vascular vulnerability within the GM or/and the WM of the human brain (e.g. vascular regulation in pial, perforant, terminal arteries/capillaries, or other postulated BBB factors)(Paternoster et al., 2009; Tan et al., 2017; Tran et al., 2016; Xu et al., 2010).
Figure 1.
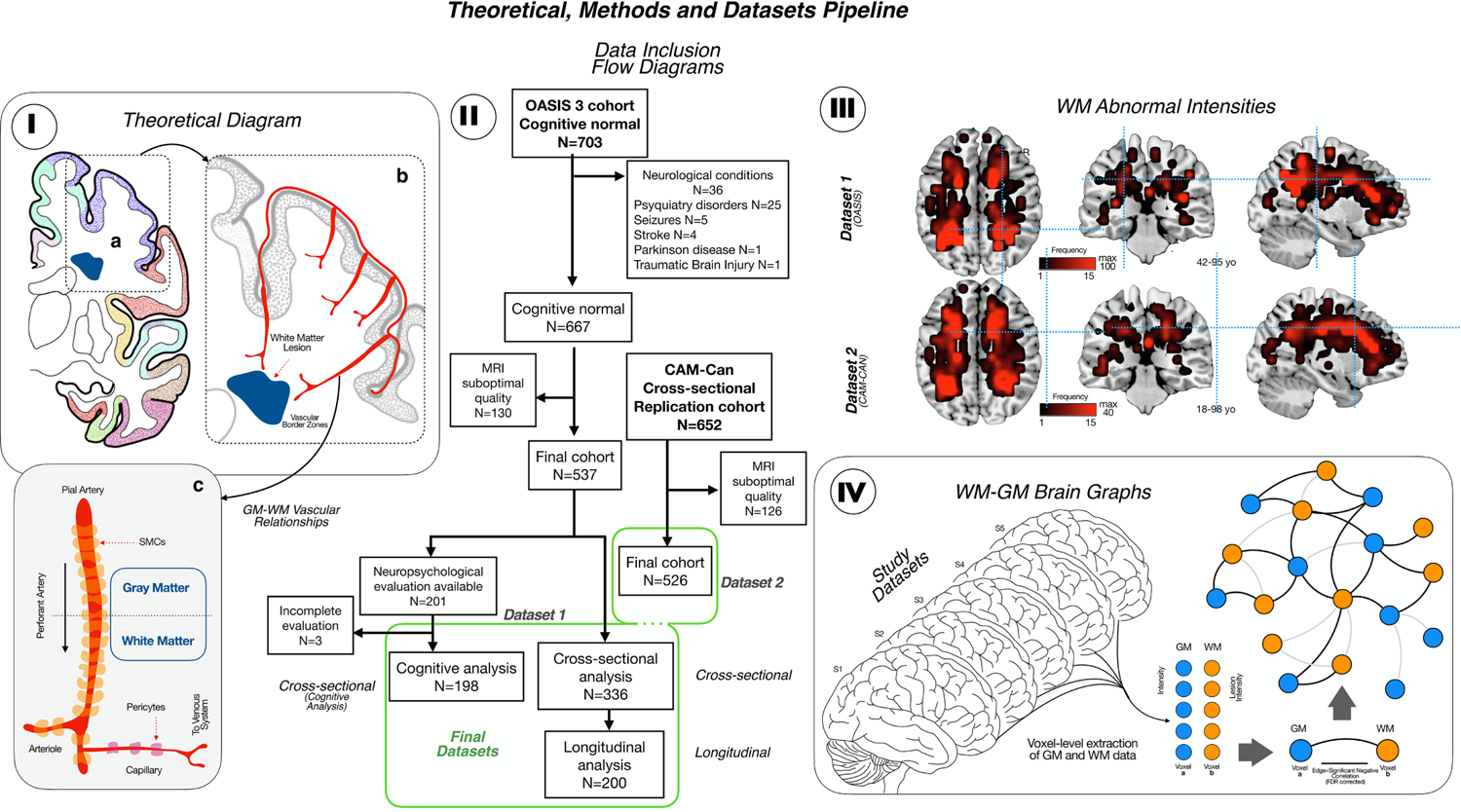
I. The theoretical diagram shows the hypothesis for the SVD damage, with primary affectation of the cerebral blood flow regulation in perforant arteries from cortex to white matter in the vascular border zones. II. Flow chart of the participants included in both datasets. III. Detection of WM lesions in the T1 MRI sequences of both cohorts following by the creation of a WM lesion frequency map (the brightest the color, the higher the frequency of the lesions with a maximum of 100 for Dataset 1 and 40 for Dataset 2). For each Dataset, the age range is described. IV. WM-GM brain graph. First, GM and WM data were voxel-level extracted, being each voxel a node of the graph. Then, we create a Pearson correlation matrix between each GM and WM node and each significant negative correlation (after FDR correction) between GM and WM in understand as an edge, so we can construct a GM-WM connectivity graph.
In this study, we aimed to analyze the interacting and degenerating patterns of WM and GM in SVD in vivo, using novel MRI graph-theory analytical approaches in cognitively normal participants. Graph-based analysis in neuroimaging is generally used to evaluate the complex network relationships between brain areas (Diez et al., 2020; Hu et al., 2015; Sepulcre et al., 2018) Here, we integrate weighted-degree and link-level analyses to quantify the centrality influence of specific cerebral areas (voxels), as well as to characterize connectivity strength relationships in the brain’s network architecture of GM and WM intersecting neurodegeneration. We also implement a graph theory strategy to study the longitudinal neurodegenerative changes between WM and GM in SVD. Finally, to improve the biological knowledge of connectomic findings, we used GM-WM connectomic phenotypes and genomic brain-wide gene expression atlas, the Allen Human Brain Atlas (AHBA; (Shen et al., 2012)), in searching of molecular correlates of vascular degeneration vulnerability. This strategy has improved our understanding of individual brain differences (Xin et al., 2018), brain plasticity (Ortiz-Terán et al., 2017), cortical organization (Bueichekú et al., 2020; Diez et al., 2020) and neurogenetic contributions to neurological diseases, including dementia (Sepulcre et al., 2018), among others (Diez and Sepulcre, 2021; Fornito et al., 2019). The overall approach of this study helped us: 1) to bridge the gap between the brain phenotypes and the specific genetic factors involved in SVD and 2) to validate the biological plausibility of the cortical maps for GM and WM primary or secondary vascular neurodegeneration.
Methods
Participants
We used two independent MRI datasets to investigate the network relationships between GM neurodegeneration and clinically silent WM lesions (flow chart in Figure 1-II): the Open Access Series of Image Study (OASIS 3; (LaMontage et al., 2019)(Dataset 1) and the stage 2 data of the Cambridge Center for Ageing and Neuroscience (Cam-CAN project; (Shafto et al., 2014)(Dataset 2). For our study, we considered the whole spectrum of WMH - as a proxy to cerebral SVD. Briefly, the OASIS 3 dataset includes 703 participants between 42 and 95-year-old with normal cognition (controls), that we used in this analysis. Medical history and cognitive assessment were revised for each participant included. Patients with a previous history of stroke or neurological conditions different from SVD were excluded. Within the control subjects, we only selected the stable cognitive participant, with a mean centiloid values of amyloid deposition at baseline less than 10 and less than 2 rate of growth in the follow up (LaMontage et al., 2019), to avoid the inclusion of preclinical Alzheimer Disease. We use 2 subsets of participants: one subset for the cross-sectional study and for the longitudinal study (that includes participants within the cross-sectional subset with at least one follow-up MRI session with a mean of 4.5 (+/− 2.38) years between baseline and follow-up); and another one for the cognitive analysis, which was non overlapped with the cross-sectional subset. The cognitive evaluation included the following tests: MiniMental State Examination (MMSE), logical memory from Weschler Memory test (WMT) immediate and delayed (units recall and time elapse), digit span forward and backward (units and longest length), trail making A and B, one-minute verbal fluency test (animals and vegetables), Boston naming test, and Weschler Adult Intelligence Scale (WAIS)-R Digit Symbol test. Then, Dataset 2 was used for validation/replication purposes. Briefly, the Cam-CAN project is a community-based prospective study to analyze the influence of age in the structure and function of the brain across the lifespan. It includes 700 participants older than 18-year-old without cognitive decline (MMSE > 24) and without history of neurological conditions such stroke, neurodegenerative diseases, brain tumor, multiple sclerosis, or epilepsy. We use this cohort for the cross-sectional validation analysis, as follow-up MRIs were not available.
Image Acquisition and Preprocessing
For the Dataset 1 and Dataset 2, we used the images obtained in 3T MRIs. High resolution structural sequences, including T1-weighted images, were acquired in both protocols. For CAM-Can subjects, a TIM Trio 3T (Siemens Medical Solutions USA, Inc) with 32 coils was used to acquire a Magnetization Prepared Rapid Gradient Echo (MP-RAGE) sequence (TR=2250ms; TE=2.99; FA=9u and 1mm isotropic voxel) (Shafto et al., 2014). For OASIS-3, a TIM Trio 3T (Siemens Medical Solutions USA, Inc) with 20 coils was used to acquire a MP-RAGE sequence TR=2400ms; TE=3.1ms; FA=8u and 1mm isotropic voxel) (LaMontage et al., 2019).
We used the T1 sequence in both cohorts to perform voxel-based morphometry (VBM) analysis and spatial detection of WM lesions; reflecting irreversible axonal damage in the white matter and avoiding the inclusion of potentially transient lesions in T2 and T2-FLAIR sequences (Al-Janabi et al., 2019) and in agreement with the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) consensus (Wardlaw et al, 2013). An optimized VBM protocol (Good et al., 2001) carried out with FSL tools (Douaud et al., 2007; Smith et al., 2004) was used to study GM and WM maps. First, structural images were brain-extracted and GM-segmented before being registered to the MNI 152 standard space using non-linear registration (Andersson et al., 2007). The resulting images were averaged and flipped along the x-axis to create a left-right symmetric, study-specific GM template. Second, all native GM images were non-linearly registered to this study-specific template and “modulated” to correct for local expansion (or contraction) due to the non-linear component of the spatial transformation. The modulated GM images were then smoothed with an isotropic Gaussian kernel with a sigma of 3 mm. Finally, we use the modulated GM partial volume estimation maps derived from VBM to identify WM lesions. Further details about the process could be found in Supplementary Figure 1. Briefly, while WM shows partial volume estimates values close to 0, WM lesions that are visually hypointense in the T1 show higher partial volume estimates than normal WM. An optimal threshold for WM lesion and normal WM separation in our data was observed at 0.2 partial volume estimate. Thus, voxel in the WM that reached a higher PVE value than 0.2 were considered as WM lesion. Each resulting WM lesions map was registered into the MNI152 standard space and down-sampled to 6mm. Individual lesion maps were automatically extracted by using a PVE algorithm for WM lesion segmentation in T1-MRIs (see example in Supplementary Figure 1). Lesion frequency map for all the subjects was computed as the sum of lesions at the voxel-level (Figure 1-III).
Cross-sectional Analysis
1. Structural connectivity analysis
Whole brain GM and WM lesions maps underwent specific analysis to investigate voxel-level network relationships (Figure 1-IV). First, we compute a partial correlation coefficient to create association matrices using all between (not within) GM and WM voxels’ pairs across the sample of individuals. The association matrices were controlled by age, brain size, whole brain GM mean density and WM lesions mean density using partial correlation. We focused on negative coefficients, as our investigation targeted the inverse association between GM signal intensity and WM lesions (the more WM lesions, the less GM volume). Association matrices were corrected using a false discovery rate (FDR) at q-level < 0.05 (Benjamini and Hochberg, 1995). An additional analysis corrected by sex and the diagnosis of hypertension were performed in the Dataset 1 and shown in supplementary material. Using these association matrices, we computed the weighted degree of GM-WM lesions relationships by summing all coefficients (Figure 2-I and 2-II) obtaining two matrices: one for the degree from each GM voxel to WM lesions connections and another one from each WM lesions voxel to GM connections. Additionally, we visualize the connectivity profiles and topology distributions of given GM of WM voxels (those with the highest weighted degree) (Figure 2-III) using a seed-based approach.
Figure 2.
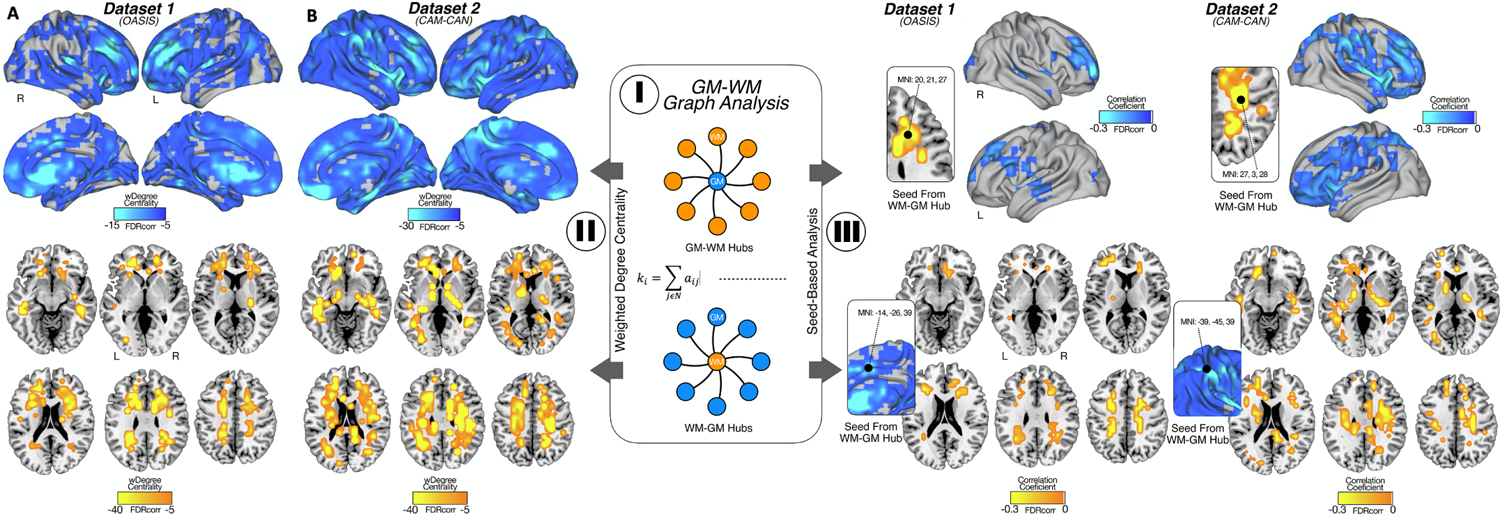
I. GM and WM graph analysis. Here, we calculate the weighted degree as the sum of the correlations for each node of GM (top) and each node of WM (bottom). II. Weighted degree centrality maps for GM and WM of Dataset 1 (A) and Dataset 2 (B): GM regions most influenced by WM lesions are located in orbitofrontal cortex, dorsolateral frontal cortex, insula and anterior cingulate cortex (the highest weighted degree centrality). For WM lesions maps, those regions most connected with GM are located in supratentorial white matter in middle cerebral artery (MCA) and anterior cerebral artery (ACA) watershed territories. III. Seed based analysis: for both cohorts, the WM lesions voxel with highest weighted degree was located in border zone areas, projecting with the highest correlation coefficient in dorsolateral frontal cortex and insula. The voxel of highest weighted degree in GM were located in cingulate cortex for OASIS and supra marginal gyrus for CAM-CAN, both projecting with the highest correlation coefficient widespread in supratentorial WM.
2. GM-WM lesions Bipartite Interactions and Modularity
An interesting aspect of GM and WM connections, in the context of the appearance of network neurodegeneration and WM lesions, is whether they form close interacting neighborhoods (also known as communities or modules). We know that, for instance, within WM, voxels tend to cluster together to form specific areas or conglomerates of WM lesions. However, it is not well-known whether GM and WM form tight interacting modules interacting between them. In this analysis, we investigate GM-WM modules by searching first for robust bipartite network motifs among the GM-WM association matrices (Figure 3-I) using a MATLAB toolbox for bipartite network analysis (Flores et al., 2016). First, we identified quadrangle motifs by selecting nodes that connects to other nodes directly (1-Step connectivity), and indirectly in 3-steps. This strategy allowed us to obtain GM-WM modules that showed close neighborhood relationships, disregarding GM-WM connections without graph neighborhood properties. Second, we applied LP-BRIM bipartite modularity algorithm to visualize all different communities within that GM-WM network. Third, a WM degree map was computed counting the number of GM voxels related to that white matter voxel that were assigned to different communities normalized by the total amount of links of that WM voxel. Finally, we perform a Pearson correlation analysis between each motifs’ cluster (GM and WM together, GM and WM alone) and the punctuation in the cognitive test described before. The analysis was adjusted by age and the years of education.
Figure 3.
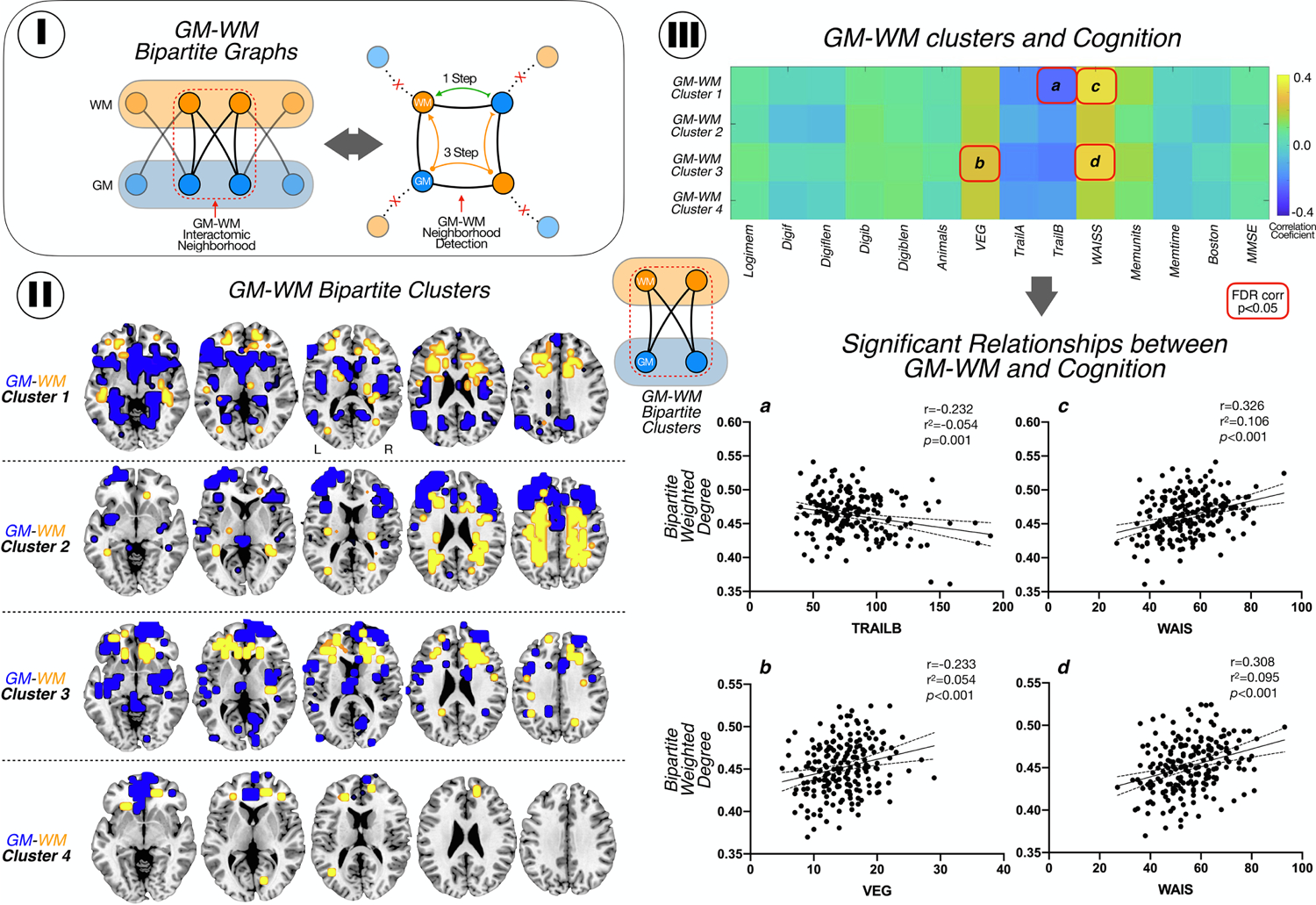
GM-WM Bipartite Interactions and Modularity. I. GM-WM bipartite graphs: we computed the network distances between all bipartite graph nodes (left). Then, we identified quadrangle motifs by selecting nodes that connects to other nodes directly (1-Step connectivity), and indirectly in 3-steps (right). II. Location of modules of GM (blue) and WM (yellow) interconnected. III. Pearson correlation matrix between modules and cognitive test. We found a negative correlation with trail B and module 1 and positive correlations between module 1 and 3 and WAIS-R; and between VEG and module 1. The correlations are shown with 95% confidence interval. Logimem: Logical memory immediate; Digitf: digit spam forward total number of trial correct; Digitflen: Digit spam forward length; Digitb: digit spam backward total number of trial correct; Digitblen: Digit spam backward length; Animals: Category fluency test animals; VEG: Category fluency test vegetables; Trail A: Trail making test part A; Trail B: Trail making test part B; WAIS: WAIS-R Digit Symbol; Memunits: Logical memory delayed, units recall; Memtime: time between logical memory test; Boston: Boston naming test; MMSE: MiniMental State Exam.
Longitudinal Neuroimaging Connectivity Analysis
We investigated the temporal and spatial relationship, simultaneously, between GM and WM lesions using network science tools, as previously done (Sepulcre et al., 2018). We developed a longitudinal weighted-degree analysis (also known as hubs) to investigate the temporal relationships between the GM and WM lesions. We used the longitudinal data of Dataset 1 for this analysis. We calculated the GM and WM lesions maps in time 1 and time 2 and computed the association matrices between all voxels of GM signal in time 1 and WM lesions in time 2 and GM signal in time 2 and WM lesions in time 1; as well as WM lesions in time 1 and GM in time 2 and WM lesions in time 2 and GM in time 1. The association matrices were controlled by age, brain size, the time between the first and the second MRI, whole brain GM mean density and WM lesions mean density using partial correlation. We subtracted the correlation between time 1 and time 2 (Figure 4-I). Links surviving a false discovery rate (FDR) at q-level < 0.05 were used. We obtained weighted degree maps to visualize GM and WM hubs in both relationships, namely, GM1-to-WM2 and WM1-to-GM2 temporal interactions (Figure 4-I/II/III).
Figure 4.
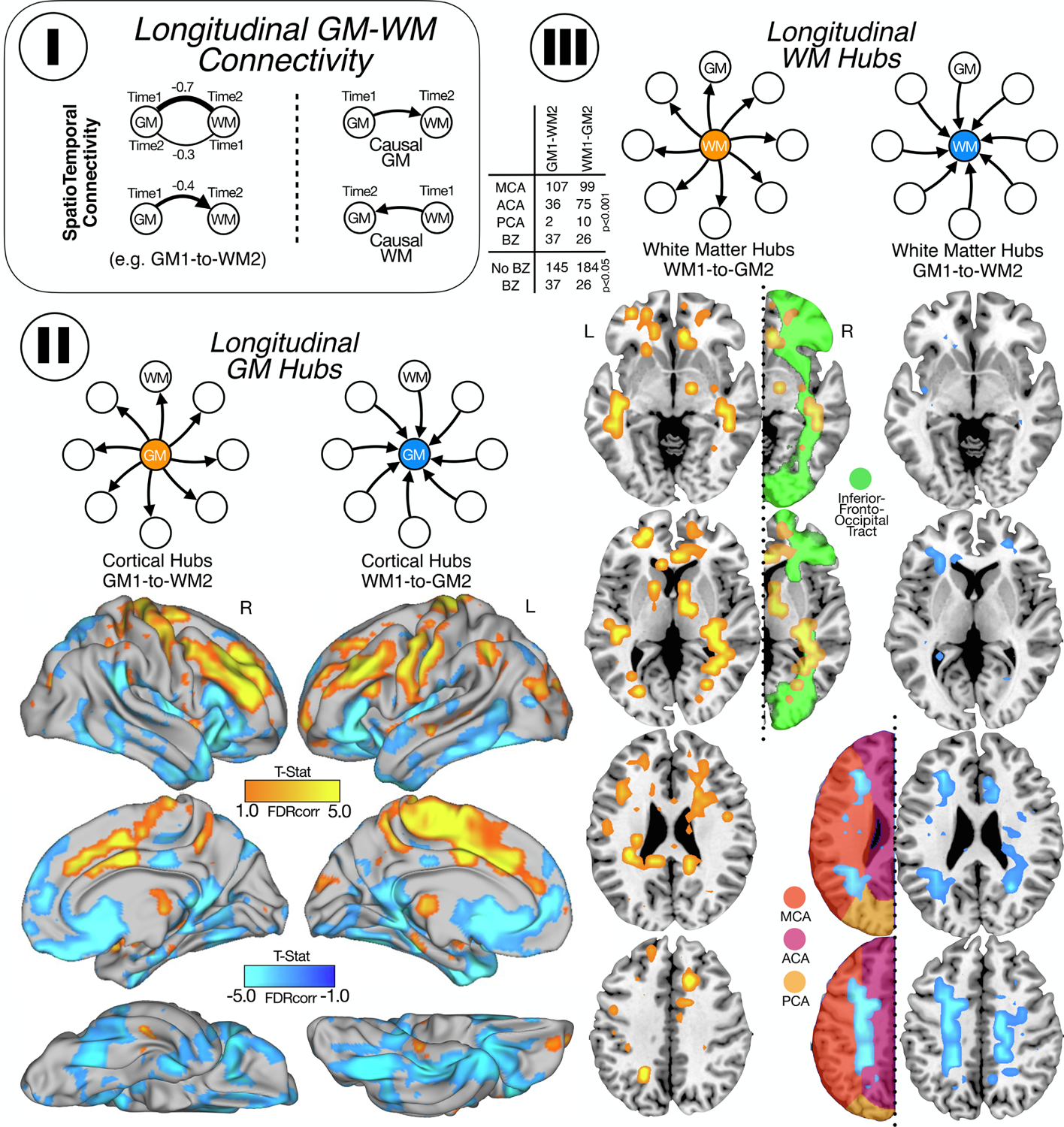
Longitudinal GM-WM connectivity. I. First, we calculate the weighted degree for GM and WM in time 1 and in time 2. We subtracted the correlations (time 1 minus time 2) and obtained maps for GM and WM. Those regions with positive correlations mean that the causal relationships go from time 1 to time 2 and negative correlations mean the causal relationship go from time 2 to time 1. II. Cortical maps for GM hubs: in orange (positive correlations) are shown the cortical areas that lead to secondary degeneration of the WM and in blue (negative correlation) are shown the cortical areas secondary degenerated due to the presence of WM lesions. III. WM maps for WM hubs: in orange (positive) are shown the WM zones that cause degeneration in the GM, mostly located in the inferior front occipital tract and in blue (negative) are shown the WM areas affected by primary vascular damage from GM, mostly border zone between MCA and ACA. For GM and WM maps, we show the statistically significant weighted-degree t-statistic maps.
Neuroimaging-Genetics Analysis
To gain biological insights about brain connectomic findings, we investigated the spatial cerebral similarities between our GM1-to-WM2 and WM1-to-GM2 cortical maps and protein-coding genetic profiles using the AHBA. The AHBA is a unique publicly available database that provides whole-brain, high- resolution genome-wide expression values (Shen et al., 2012). The complete transcriptome dataset consists of 58,692 measurements of gene expression of 20,737 genes in 3,702 brain samples obtained from 6 individuals (French and Paus, 2015). We used a surface anatomical transformation (French and Paus, 2015) of the AHBA data based on the 68 cortical regions of the Desikan–Killiany atlas (Desikan et al., 2006), which covers the entire cortex and uses individual vectors of the median cortical expression of protein-coding genes across 68 cortical regions. To find gene expression profiles putatively associated to vascular damage vulnerability, we performed a spatial similarity analysis between the discovered connectomic phenotypes from the longitudinal analysis (GM maps for GM1 to WM2 and WM1 to GM2 and AHBA data by means of MATLAB in-house–developed code based on a linear regression approach (MATLAB R2019b; The MathWorks Inc.) (Bueichekú et al., 2020; Diez and Sepulcre, 2018). Later, we focused our investigations on genes that showed high spatial similarity with our longitudinal GM-WMH connectomic map (statistically significant R-values were retained using a 95% threshold for the confidence interval (±2 SD), assigning positive R-values to the GM1-WM2 map and negative R-values to the WM1-GM2 map). The neurogenetics analysis led to two sets of genes (one on the positive and another one in the negative tail of the histogram; Figure 5-I) that expressed which protein-coding genes have a biological relevance in the GM-WM relationships. From the obtained gene set, we used four specific approaches to characterize its biological meaning: 1) we searched for previously reported genes (a priori genes) related to vascular functionality or SVD (bibliographic review); 2) we performed fold enrichment analyses for Gene Ontology biological processes (Ashburner et al., 2000; The gene Onthology Consortium, 2021) and Reactome pathways; 3) we explored potential single cell gene expression patterns (of pericytes, smooth muscle cells, vascular fibroblast-like cells or microglia) of a priori genes related with the risk of SVD obtained in the spatial similarity analysis using a mouse molecular atlas of brain vasculature (He et al., 2018; Vanlandewijck et al., 2018); and 4) we used Genemania (Mostafavi et al., 2008) gene-gene network interactions from co-expressions, colocalizations, genetic interactions, pathways, predicted physical interactions and shared proteins domains and Cytoscape (Lopes et al., 2011) software to perform an interactome analysis, including centrality assessments (degree and closeness centrality), to confirm which genes obtained in the spatial similarity analysis (in both left and right tail of the histogram) play a central role in the GM-WM lesions connectomic-genetic interactions.
Figure 5.
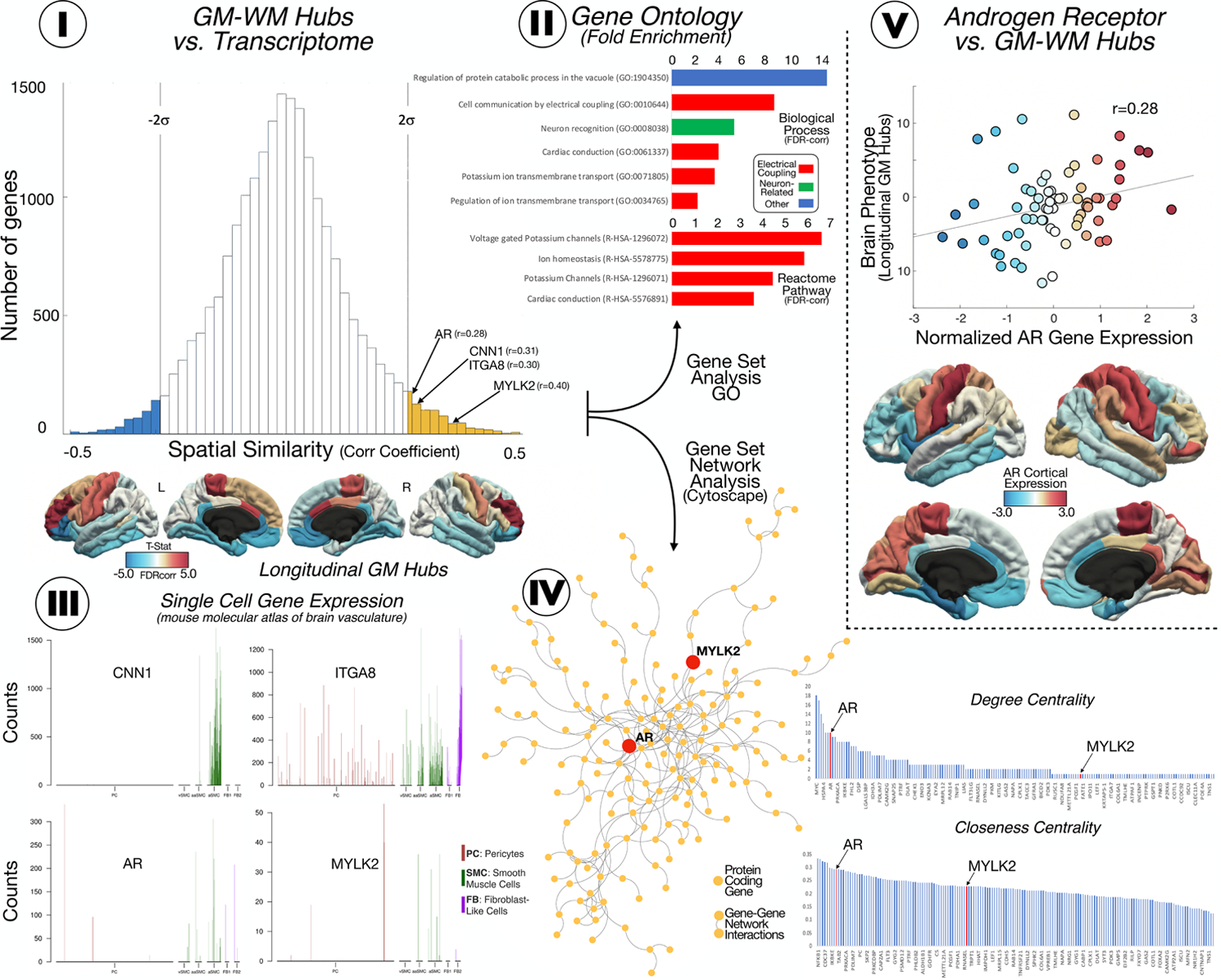
I. Spatial similarity analysis between the discovered connectomic phenotypes (longitudinal GM maps) and AHBA data. In the histogram are represented the 20,737 genes identified in the AHBA (Y-axis) in our cortical map (X-axis). The genes founded beyond +/− 2SD were considered significantly correlated with our map. Interestingly, we found in the positive tail of the histogram genes that was previously associated with the SVD. II. Fold enrichment analysis after FDR correction for the genes significantly associated with the risk of SVD (>2SD); that shows the implication of these genes in cell communication, muscle conduction and ion channels and transports. III. Single gene expression for the genes related with the risk of SVD, with high expression in smooth muscle cells and pericytes. IV. Interactomic analysis of the a priori set of SVD genes, showing that AR had a top centrality (both degree centrality and closeness centrality). V. Spatial similarity analysis for the AR gene and the longitudinal cortical map; showing its most expression in motor and sensitive primary areas and motor supplementary area in both hemisphere, regions identified in the longitudinal cortical map as causative of WM lesions.
Results
Connectivity Interactions Between Grey Matter and White Matter Lesions
We found that GM and WM lesions showed significant connectivity relationships across the human brain (Figure 2). For example, there are GM regions in orbitofrontal cortex, dorsolateral frontal cortex, insula and anterior cingulate cortex -in both, Dataset 1 and Dataset 2 cohorts-, where volumetric signal in GM is heavily associated by the occurrence of WM lesions in the supratentorial space (Figure 2-II; upper row). As for WM areas, there are leukoaraiotic regions in supratentorial WM, in middle cerebral artery (MCA) and anterior cerebral artery (ACA) watershed territories -in both cohorts- that are negatively related to GM volume across the cortical mantle, meaning that the more lesions in WM, the less volume of GM (Figure 2-II; bottom row). Our seed-based analysis confirmed local regional specificity between WM lesions and GM interactions, such as frontal WM and GM, as well as more widespread WM lesions and GM relationships, such as fronto-parietal WM lesions and GM volume changes mostly in frontal lobes (Figure 2-III). For the Dataset 1, the voxel with highest weighted degree in WM is located in MCA-ACA left border-zone region; and is highly connected with left dorsolateral frontal cortex. The voxel with highest weighted degree in GM is in the cingulate cortex; and is highly connected with spread voxels of WM, mostly located in MCA-ACA watershed zones. For the Dataset 2 cohort, the results were similar for the projections from WM to GM (adding insula as cortex target) but the highest weighted degree region of GM was the supramarginal gyrus, also highly connected with supratentorial watershed WM. Finally, we observed the adjustment by the diagnosis of hypertension did not significantly change the GM and WM lesions weighted degree maps (Supplementary Figure 1).
Cognition and Connectivity Neighborhoods of Grey Matter and White Matter Lesions
Using a bipartite modularity analysis, we observed 4 main modules of highly stable voxels of GM volumetric changes and WM lesions (Figure 3-II): 1) module one, in which the GM of medial temporal lobes and fronto-basal regions interacts with prefrontal WM tracts; 2) module 2 in which prefrontal and frontal lateral GM interacts with supratentorial WM; 3) module 3, with GM located in insula and prefrontal cortex interacts with WM tracts of prefrontal lobes and 4) module 4, with medial frontal cortex interacting with tiny regions of WM in frontal lobes.
We observed that GM-WM lesions modules, that is combined systems integrating GM and WM at the same time, explain the cognitive performance of the sample, particularly in the WAIS-R, VEG and Trail B scores (correlations and scatterplots in Figure 3-III; FDR-corrected) for modules 1 and 3 (involving the GM-WM connections in frontal and temporal lobes) without significant association with modules 2 and 4 (involving mostly the connections of supratentorial WM with the prefrontal and frontal lateral cortex). We did not find any significant FDR-corrected correlation with cognitive test when GM or WM modules are taken independently.
Temporal Interactions from Grey Matter to White Matter Lesions (and vice versa)
Our longitudinal analysis not only corroborated areas of high GM-WM lesions connectivity degree found in the cross-sectional study but also discovered that specific regions in the brain display predominant vulnerability for GM1-to-WM2 or WM1-to-GM2 degeneration (Figure 4). We observed two distinctive progression patterns: 1) one in which GM volume changes, in areas such as the dorsolateral frontal cortex, pre and post Rolandic gyrus, supplementary motor area and some cingulate areas, that lead to the eventual appearance of secondary WM lesions located in supratentorial WM, in watershed regions of MCA and ACA (Figure 4, yellow cortical areas in II, and right column in III), and 2) another one in which WM lesions, particularly in the inferior fronto-occipital fasciculus, produce subsequent cortical degeneration on areas such as temporal pole, anterior cingulate cortex, insula, orbitofrontal cortex and inferior temporal ACA (Figure 4, blue cortical areas in II, and left column in III). Finally, we observed the adjustment by the diagnosis of hypertension did not significantly change the GM1-to-WM2 and WM1-to-GM2 maps (Supplementary Figure 1).
Neurogenetic Signatures of Grey Matter and White Matter Network Degeneration
The biological mechanism that drives GM and WM alterations in SVD are likely to be interdependent or even causally related. Our cross-sectional and longitudinal findings described specific connectomic phenotypes that support that view. Consequently, we investigated whether these connectomic phenotypes display biological potential at the SVD mechanistic level. Thus, we performed a spatial cerebral similarity analysis with the AHBA. Interestingly, the GM1-to-WM2 phenotype identified a gene set of 683 protein-coding genes, in which several a priori genes have been associated to SVD and vascular smooth muscle cells, namely AR, CNN1, CSF3, ITGA8 and MYLK2 genes (Figure 5-I). Moreover, we analyzed this entire set of genes from the GM1-to-WM2 phenotype (or GM1-to-WM2 related genes) and we observed significant fold enrichments in ionic transmembrane transport, cardiac conduction and electrical cell communication functionalities (Figure 5-I/II), genetic domains that are known to be related not just to cardiac cells but also to vessels cells (e.g. smooth muscle cells).
The WM1-to-GM2 phenotype showed significantly cortical similarity with 476 genes in which we identified IL1-b, IL-6 and TRIBE3 as potentially relevant for SVD pathology. However, the entire set of genes associated to this type of connectivity pattern did not reach any fold enrichment significance. The complete lists of genes (for GM1-to-WM2 and for WM1-to-GM2) with their description are included in Supplementary Material.
Next, we used a single-cell database to verify whether AR, CNN1, ITGA8 and MYLK 2 are specifically expressed in brain vascular cells (pericytes, smooth muscle cells and vascular fibroblast-like cells). Among these four genes, only CNN1 and AR display high cellular specificity for arteriolar smooth muscle cells (Figure 5-III). Finally, we used the entire gene set of 683 genes obtained from the GM1-to-WM2 connectivity map and an interactome analysis to investigate whether any of them play central roles from independent data of gene-gene network interactions. We found that only AR displayed a central interactomic role in this genetic network (both degree centrality and closeness centrality; Figure 5-IV) suggesting a core role in the overall GM-WM lesions connectomic-genetics. Figure 5-V shows the cortical spatial similarity between AR gene (i.e., brain regions linked to the expression of this gene) and GM1-to-WM2 connectomic phenotype (Figure 5-V).
Discussion
The brain pathology of SVD beyond the accumulation of WM lesions is poorly understood. Diffuse arteriopathy of the GM and WM perforating vessels due to dysfunctional cerebral blood flow regulation has been postulated as a key factor in the development of SVD (Promjunyakul et al., 2018). The current study aimed to close the gap in understanding the complex relationship of GM and WM pathological changes from the viewpoint of cerebral connectomics. We hypothesize that alterations in GM and WM do not occur in isolation but in modular coordination to arise final SVD phenotypes. Particularly, we speculate that WM lesions and GM neurodegeneration both share the ability to be the primary source of vascular-molecular dysfunctions, in which secondary degeneration might occur in distant areas of WM or GM, depending on the primary source. This multilayer interdigitation of primary and secondary damage makes graph theory an ideal framework to disentangle them.
Previous studies have shown that WMH could cause structural connectivity alterations related to SVD. Habes et al. show that normal aging and WMH cause different affectation patterns of GM: WMH could primarily affect the orbitofrontal cortex and temporal pole (Habes et al., 2016). Liu et al., using a measure of density fiber, found less density fiber in frontal, temporal, and parietal regions related to SVD in cognitively normal subjects (Liu et al., 2020). Using novel cross-sectional and longitudinal graph theory metrics, we find converging evidence similar to those structural connectivity descriptions. Critically, we provide new data about the bidirectional nature of GM and WM interactions. Our results show that GM changes can lead to secondary WM degeneration in vascular border zones areas (irrigated by pial arteries of MCA and ACA), whereas WM lesions can lead to secondary GM degeneration in cortical projecting areas. Moreover, we found this GM-WM communication is organized in close and stable connectivity neighborhoods; a network organization feature that seems critical for an adequate cognitive performance. In fact, we observed that the GM-WM connectivity neighborhoods are related with cognition, particularly with the executive function, which affectation is the main feature of vascular cognitive decline (O’Brien and Thomas, 2015). Previous studies of structural and functional connectivity in SVD have shown that the global brain network is altered, and this alteration mediates the results in cognitive scores (Lawrence et al., 2014). Here we go one step further and saw that connectivity modules between GM and WM (and not isolated GM or WM regions) are key in this SVD-cognitive relationship.
The pathophysiology interpretation of SVD has evolved over the years, from the chronic hypoperfusion as the central hypothesis of brain damage to the endothelium and BBB disruption as critical points of the disease (Wardlaw et al., 2019). Recent studies have shown that vascular reactivity is impaired in the cerebral vasculature before developing the disease (Sam et al., 2016; Shi et al., 2020), supporting the hypothesis of chronic vascular impairment with global brain hypoperfusion. Some other studies have pointed out that WMH might be just the consequence of altered cerebral blood flow somewhere else (Shi et al., 2016). Moreover, others have suggested that BBB alterations in WMH could be the primary cause of cerebral damage (Muñoz Maniega et al., 2017; Wardlaw et al., 2017). However, studies developed in spontaneously hypertensive stroke-prone rats models show credible evidence that vascular wall damage with narrowing of the arterioles (Jandke et al., 2018) and an alteration of the vascular reactivity seems to be an early causal phenomenon (in which an initial vasodilatation and increase of oxygen exchange eventually compromises and evolves into vasoconstriction and reduction of oxygen exchange). Interestingly, wall damage activates the inflammatory response that leads to the BBB break (Mencl et al., 2013). Moreover, neuropathological studies have found that these primary local effects produce changes in distant areas of the brain. For instance, it has been observed glial apoptosis in cortical-subcortical WM junctions remotely to focal ischemic areas in the CADASIL brain (Viswanathan et al., 2006). This suggests that secondary degeneration from local lesions either GM or WM can potentially affect distant regions, creating a complex network of interacting cerebral degeneration. Our connectomic findings correspond with these observations. On one hand, we found that primary GM changes can lead to WM lesions in border vascular zones, meaning that the primary affectation of the pial arteries in the GM might induce vascular and molecular dysfunctionality in distal capillaries within vascular-vulnerable zones of the human brain. On the other hand, we also found that the primary WM lesions in association fasciculus, such as the fronto-occipital fasciculus, can lead to secondary GM degeneration in projecting areas at the frontal lobe, with little effect of hypertension. Thus, our results suggest that there are more pathological factors behind the sporadic SVD than the chronic hypoperfusion theory behind the appearance of WM lesions. In light of the GM-WM bidirectional causal damage interactions found in this study, we believe that sporadic SVD is the result not only of primary WM lesions with secondary degeneration of cortical areas, but also the occurrence of primary mis-regulation of cerebral blood flow in GM pial arteries that infringe damage toward downstream and distal artery territories.
The diversity of underlying pathologies and vascular risk factors related to SVD make its genetic background a complex scenario which remains largely unclear (Iadecola, 2013). In this study, the integration of neuroimaging connectivity SVD phenotypes and transcriptomic data from the AHBA has helped us to identify several genetic signatures that might shed light for future studies. Particularly, we found that AR, CNN1, MYLK2 and ITGA8 gene expression levels are highly similar to the SVD-related map which primary longitudinal degeneration of in situ GM led to secondary vascular damage in distant WM. We also observed that the entire genetic profile was highly enriched on genetic functionalities that are putatively related to smooth muscle cells and their biological processing domains (ion transmembrane transport and electrical conduction). These findings point to plausible causative roles of cortical alterations in the SVD pathological mechanisms. Interestingly, among the 4 candidate genes, AR (gene coding for the androgen receptor protein) stood out as the most central interactomic role in a genetic network. AR has been identified previously in GWAS studies of vascular dementia, with almost 4-times higher risk of vascular dementia when present (Schrijvers et al., 2012). There is an increasing number of studies that demonstrate the role of AR in the vascular damage (Son et al., 2010), and vascular risk factors as hypertension (Huang et al., 2016). The AR gene is highly expressed in vascular smooth muscle cells (Son et al., 2010) and the activity of androgen/AR signaling were pointed as risk factors of hypertension and stroke (Huang et al., 2016). The results of our study complement the existing evidence, as the AR gene was significantly expressed in those cortical regions that lead to degeneration in the WM and confirm our hypothesis of primary vascular damage in GM that might cause WM lesions. Regarding the reverse pattern -primary vascular damage in WM leading to secondary GM degeneration- we identified several brain damage and inflammatory related genes associated to (IL6, IL1b and TRIBE3 genes). This proinflammatory genetic profile did not reach any significant fold enrichment and therefore, they seem less conclusive compare to our findings with the AR centered gene set, however, it deserves future investigations. For instance, IL6 and IL1b are both implicated in the inflammatory cascade, codifying proinflammatory cytokines and both are well-known ischemic damage biomarkers; both are related to the outcome of ischemic stroke (Martínez-Sánchez et al., 2014; Ramiro et al., 2018). Recently, Staszewski et al. found that IL-6 relates to the radiological progression of small vessel disease (Staszewski et al., 2018)). Moreover, in brain hypoperfusion models, animals with lower levels of Il1b and Il6 had lower necrotic cells in the hippocampal cells and better performance in the cognitive test (Ishida et al., 2020). These findings support the idea that IL6 and IL1b genes in cortical areas can explain in part some of the secondary degeneration process occurring in SVD.
In conclusion, using graph theory-based analysis and connectomic-genetic approaches, this study has led us to interpret SVD as a bidirectional degeneration process between GM and WM lesions. This framework opens new venues to understand the underlying interactomic mechanism in which the SVD develops as well as to investigate key pathophysiology features associated to its molecular background. Importantly, our findings support the central role of AR for the brain lesion vulnerability and network breakdown in the context of SVD.
Supplementary Material
Highlights.
We unveiled the bidirectional nature of neurodegeneration (ND) and vascular damage (VD) in the cerebral small vessel disease (SVD).
We described that ND-VD bidirectional connectomics are key to understand cognitive performance.
The ND-VD connectomics led us to characterize cortical expression levels of candidate genes linked to SVD.
We discovered that the Androgen Receptor (AR) protein coding gene is a central gene that confer vulnerability for brain lesion development of SVD.
This study advances our understanding of the primary and secondary damage, biological mechanisms, and genetic signatures of SVD.
Acknowledgment:
This research was supported by grants from the National Institutes of Health (NIH; R01AG061811, and R01AG061445 to J.S.) and from the Institute of Health Carlos III (CM18/00175 to RGZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interest:
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Al-Janabi OM, Bauer CE, Goldstein LB, Murphy RR, Bahrani AA, Smith CD, Wilcock DM, Gold BT, Jicha GA, 2019. White matter hyperintensity regression: Comparison of brain atrophy and cognitive profiles with progression and stable groups. Brain Sci. 9. 10.3390/brainsci9070170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson JLR, Jenkinson M, Smith S, 2007. Non-linear registration aka spatial normalisation, FMRIB Technical Report TRO7JA2. [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, 2000. Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bueichekú E, Aznárez-Sanado M, Diez I, d’Oleire Uquillas F, Ortiz-Terán L, Qureshi AY, Suñol M, Basaia S, Ortiz-Terán E, Pastor MA, Sepulcre J, 2020. Central neurogenetic signatures of the visuomotor integration system. Proc. Natl. Acad. Sci 117, 6836–6843. 10.1073/pnas.1912429117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craggs LJL, Yamamoto Y, Deramecourt V, Kalaria RN, 2014. Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol. 24, 495–509. 10.1111/bpa.12177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ, 2006. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–980. 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- Diez I, Larson AG, Nakhate V, Dunn EC, Fricchione GL, Nicholson TR, Sepulcre J, Perez DL, 2020. Early-life trauma endophenotypes and brain circuit–gene expression relationships in functional neurological (conversion) disorder. Mol. Psychiatry 10.1038/s41380-020-0665-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez I, Sepulcre J, 2021. Unveiling the neuroimaging-genetic intersections in the human brain. Curr. Opin. Neurol 34, 480–487. 10.1097/WCO.0000000000000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez I, Sepulcre J, 2018. Neurogenetic profiles delineate large-scale connectivity dynamics of the human brain. Nat. Commun 9, 1–10. 10.1038/s41467-018-06346-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud G, Smith S, Jenkinson M, Behrens T, Johansen-Berg H, Vickers J, James S, Voets N, Watkins K, Matthews PM, James A, 2007. Anatomically related grey and white matter abnormalities in adolescent-onset schizophrenia. Brain 130, 2375–2386. 10.1093/brain/awm184 [DOI] [PubMed] [Google Scholar]
- Flores CO, Poisot T, Valverde S, Weitz JS, 2016. BiMat: A MATLAB package to facilitate the analysis of bipartite networks. Methods Ecol. Evol 7, 127–132. 10.1111/2041-210X.12458 [DOI] [Google Scholar]
- Fornito A, Arnatkevičiūtė A, Fulcher BD, 2019. Bridging the Gap between Connectome and Transcriptome. Trends Cogn. Sci 23, 34–50. 10.1016/j.tics.2018.10.005 [DOI] [PubMed] [Google Scholar]
- French L, Paus T, 2015. A FreeSurfer view of the cortical transcriptome generated from the Allen Human Brain Atlas. Front. Neurosci 9, 1–5. 10.3389/fnins.2015.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CD, Johnsrude IS, Ashburner J, Henson RNA, Friston KJ, Frackowiak RSJ, 2001. A Voxel-Based Morphometric Study of Ageing in 465 Normal Adult Human Brains. Neuroimage 14, 21–36. 10.1006/nimg.2001.0786 [DOI] [PubMed] [Google Scholar]
- Habes M, Erus G, Toledo JB, Zhang T, Bryan N, Launer LJ, Rosseel Y, Janowitz D, Doshi J, Van Der Auwera S, Von Sarnowski B, Hegenscheid K, Hosten N, Homuth G, Völzke H, Schminke U, Hoffmann W, Grabe HJ, Davatzikos C, 2016. White matter hyperintensities and imaging patterns of brain ageing in the general population. Brain 139, 1164–1179. 10.1093/brain/aww008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habes M, Sotiras A, Erus G, Toledo JB, Janowitz D, Wolk DA, Shou H, Bryan NR, Doshi J, Völzke H, Schminke U, Hoffmann W, Resnick SM, Grabe HJ, Davatzikos C, 2018. White matter lesions spatial heterogeneity, links to risk factors, cognition, genetics, and atrophy. Neurology 91, E964–E975. 10.1212/WNL.0000000000006116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Vanlandewijck M, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Segerstolpe Å, Liu J, Gustafsson S, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C, 2018. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 5, 180160. 10.1038/sdata.2018.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Cheng L, Sepulcre J, Johnson KA, Fakhri GE, Lu YM, Li Q, 2015. A spectral graph regression model for learning brain connectivity of Alzheimer’s disease. PLoS One 10, 1–24. 10.1371/journal.pone.0128136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-K, Lee SO, Chang E, Pang H, Chang C, 2016. Androgen receptor (AR) in cardiovascular diseases. J. Endocrinol 229, R1–R16. 10.1530/JOE-15-0518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, 2013. The Pathobiology of Vascular Dementia. Neuron 80, 844–866. 10.1016/j.neuron.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Saito M, Sato S, Koepsell H, Taira E, Hirose M, 2020. SGLT1 participates in the development of vascular cognitive impairment in a mouse model of small vessel disease. Neurosci. Lett 727, 134929. 10.1016/j.neulet.2020.134929 [DOI] [PubMed] [Google Scholar]
- Jandke S, Garz C, Schwanke D, Sendtner M, Heinze HJ, Carare RO, Schreiber S, 2018. The association between hypertensive arteriopathy and cerebral amyloid angiopathy in spontaneously hypertensive stroke-prone rats. Brain Pathol. 28, 844–859. 10.1111/bpa.12629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C, Benjamin P, Zeestraten E, Lawrence AJ, Barrick TR, Markus HS, 2016. Longitudinal patterns of leukoaraiosis and brain atrophy in symptomatic small vessel disease. Brain 139, 1136–1151. 10.1093/brain/aww009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMontage P, Benzinger T, JC M, Keefe S, Hornbeck R, Xiong C, Grant E, Hassenstab J, Krista V, Andrei G, Raichle M, Cruchaga C, Marcus D, 2019. OASIS-3: Longitudinal Neuroimaging, CLinica, and Cognitive Dataset for Normal Aging and Alzheimer Disease. MedXriv. 10.1101/2019.12.13.19014902 [DOI] [Google Scholar]
- Lawrence AJ, Chung AW, Morris RG, Markus HS, Barrick TR, 2014. Structural network efficiency is associated with cognitive impairment in small-vessel disease. Neurology 83, 304–311. 10.1212/WNL.0000000000000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yang Y, Reis C, Tao T, Li W, Li X, Zhang JH, 2018. Cerebral Small Vessel Disease. Cell Transplant. 27, 1711–1722. 10.1177/0963689718795148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Shi L, Zhu Wenhao, Yang S, Sun P, Qin Y, Tang X, Zhang S, Yao Y, Wang Z, Zhu Wenzhen, Wang D, 2020. Fiber Connectivity Density in Cerebral Small-Vessel Disease Patients With Mild Cognitive Impairment and Cerebral Small-Vessel Disease Patients With Normal Cognition. Front. Neurosci 14, 1–11. 10.3389/fnins.2020.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD, Dopazo J, 2011. Cytoscape Web: An interactive web-based network browser. Bioinformatics 27, 2347–2348. 10.1093/bioinformatics/btq430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus HS, 2008. Genes, endothelial function and cerebral small vessel disease in man. Exp. Physiol 93, 121–127. 10.1113/expphysiol.2007.038752 [DOI] [PubMed] [Google Scholar]
- Martínez-Sánchez P, Gutiérrez-Fernández M, Fuentes B, Masjuán J, de L.Cases MA, Novillo-López ME, Díez-Tejedor E, 2014. Biochemical and inflammatory biomarkers in ischemic stroke: translational study between humans and two experimental rat models. J. Transl. Med 12, 220. 10.1186/s12967-014-0220-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencl S, Garz C, Niklass S, Braun H, Göb E, Homola G, Heinze HJ, Reymann KG, Kleinschnitz C, Schreiber S, 2013. Early microvascular dysfunction in cerebral small vessel disease is not detectable on 3.0 Tesla magnetic resonance imaging: A longitudinal study in spontaneously hypertensive stroke-prone rats. Exp. Transl. Stroke Med 5, 5–9. 10.1186/2040-7378-5-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q, 2008. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 9, S4. 10.1186/gb-2008-9-s1-s4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz Maniega S, Chappell FM, Valdés Hernández MC, Armitage PA, Makin SD, Heye AK, Thrippleton MJ, Sakka E, Shuler K, Dennis MS, Wardlaw JM, 2017. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J. Cereb. Blood Flow Metab 37, 644–656. 10.1177/0271678X16635657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JT, Thomas A, 2015. Vascular dementia. Lancet 386, 1698–1706. 10.1016/S0140-6736(15)00463-8 [DOI] [PubMed] [Google Scholar]
- Ortiz-Terán L, Diez I, Ortiz T, Perez DL, Aragón JI, Costumero V, Pascual-Leone A, El Fakhri G, Sepulcre J, 2017. Brain circuit-gene expression relationships and neuroplasticity of multisensory cortices in blind children. Proc. Natl. Acad. Sci. U. S. A 114, 6830–6835. 10.1073/pnas.1619121114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster L, Chen W, Sudlow CLM, 2009. Genetic determinants of white matter hyperintensities on brain scans: A systematic assessment of 19 candidate gene polymorphisms in 46 studies in 19 000 subjects. Stroke 40, 2020–2026. 10.1161/STROKEAHA.108.542050 [DOI] [PubMed] [Google Scholar]
- Promjunyakul NO, Dodge HH, Lahna D, Boespflug EL, Kaye JA, Rooney WD, Silbert LC, 2018. Baseline NAWM structural integrity and CBF predict periventricular WMH expansion over time. Neurology 90, e2107–e2118. 10.1212/WNL.0000000000005684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro L, Simats A, García-Berrocoso T, Montaner J, 2018. Inflammatory molecules might become both biomarkers and therapeutic targets for stroke management. Ther. Adv. Neurol. Disord 11, 175628641878934. 10.1177/1756286418789340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi B, Narkhede A, Last BS, Budge M, Tosto G, Manly JJ, Schupf N, Mayeux R, Brickman AM, 2018. The effect of white matter hyperintensities on cognition is mediated by cortical atrophy. Neurobiol. Aging 64, 25–32. 10.1016/j.neurobiolaging.2017.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam K, Crawley AP, Conklin J, Poublanc J, Sobczyk O, Mandell DM, Venkatraghavan L, Duffin J, Fisher JA, Black SE, Mikulis DJ, 2016. Development of White Matter Hyperintensity Is Preceded by Reduced Cerebrovascular Reactivity. Ann. Neurol 80, 277–285. 10.1002/ana.24712 [DOI] [PubMed] [Google Scholar]
- Schrijvers EMC, Schürmann B, Koudstaal PJ, Van Den Bussche H, Van Duijn CM, Hentschel F, Heun R, Hofman A, Jessen F, Kölsch H, Kornhuber J, Peters O, Rivadeneira F, Rüther E, Uitterlinden AG, Riedel-Heller S, Dichgans M, Wiltfang J, Maier W, Breteler MMB, Ikram MA, 2012. Genome-wide association study of vascular dementia. Stroke 43, 315–319. 10.1161/STROKEAHA.111.628768 [DOI] [PubMed] [Google Scholar]
- Sepulcre J, Grothe MJ, d’Oleire Uquillas F, Ortiz-Terán L, Diez I, Yang H-SS, Jacobs HILL, Hanseeuw BJ, Li Q, El-Fakhri G, Sperling RA, Johnson KA, 2018. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nat. Med 24, 1910–1918. 10.1038/s41591-018-0206-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafto MA, Tyler LK, Dixon M, Taylor JR, Rowe JB, Cusack R, Calder AJ, Marslen-Wilson WD, Duncan J, Dalgleish T, Henson RN, Brayne C, Cam-CAN, Matthews FE, 2014. The Cambridge Centre for Ageing and Neuroscience (Cam-CAN) study protocol: A cross-sectional, lifespan, multidisciplinary examination of healthy cognitive ageing. BMC Neurol. 14, 1–25. 10.1186/s12883-014-0204-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen EH, Overly CC, Jones AR, 2012. The Allen Human Brain Atlas. Trends Neurosci. 35, 711–714. 10.1016/j.tins.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Shi Y, Thrippleton MJ, Blair GW, Dickie DA, Marshall I, Hamilton I, Doubal FN, Chappell F, Wardlaw JM, 2020. Small vessel disease is associated with altered cerebrovascular pulsatility but not resting cerebral blood flow. J. Cereb. Blood Flow Metab 40, 85–99. 10.1177/0271678X18803956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Thrippleton MJ, Makin SD, Marshall I, Geerlings MI, De Craen AJM, Van Buchem MA, Wardlaw JM, 2016. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab 36, 1653–1667. 10.1177/0271678X16662891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM, 2004. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23, S208–S219. 10.1016/j.neuroimage.2004.07.051 [DOI] [PubMed] [Google Scholar]
- Son BK, Akishita M, Iijima K, Ogawa S, Maemura K, Yu J, Takeyama K, Kato S, Eto M, Ouchi Y, 2010. Androgen receptor-dependent transactivation of growth arrest-specific gene 6 mediates inhibitory effects of testosterone on vascular calcification. J. Biol. Chem 285, 7537–7544. 10.1074/jbc.M109.055087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staszewski J, Piusińska-Macoch R, Brodacki B, Skrobowska E, Stepien A, 2018. IL-6, PF-4, sCD40 L, and homocysteine are associated with the radiological progression of cerebral small-vessel disease: a 2-year follow-up study. Clin. Interv. Aging Volume 13, 1135–1141. 10.2147/CIA.S166773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan R, Traylor M, Rutten-Jacobs L, Markus H, 2017. New insights into mechanisms of small vessel disease stroke from genetics. Clin. Sci 131, 515–531. 10.1042/CS20160825 [DOI] [PubMed] [Google Scholar]
- Ter Telgte A, Wiegertjes K, Gesierich B, Baskaran BS, Marques JP, Kuijf HJ, Norris DG, Tuladhar AM, Duering M, De Leeuw FE, 2020. Temporal Dynamics of Cortical Microinfarcts in Cerebral Small Vessel Disease. JAMA Neurol. 77, 643–647. 10.1001/jamaneurol.2019.5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The gene Onthology Consortium, 2021. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. 10.1093/nar/gkaa1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T, Cotlarciuc I, Yadav S, Hasan N, Bentley P, Levi C, Worrall BB, Meschia JF, Rost N, Sharma P, 2016. Candidate-gene analysis of white matter hyperintensities on neuroimaging. J. Neurol. Neurosurg. Psychiatry 87, 260–266. 10.1136/jnnp-2014-309685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C, 2018. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. 10.1038/nature25739 [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Gray F, Bousser MG, Baudrimont M, Chabriat H, 2006. Cortical neuronal apoptosis in CADASIL. Stroke 37, 2690–2695. 10.1161/01.STR.0000245091.28429.6a [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Makin SJ, Valdés Hernández MC, Armitage PA, Heye AK, Chappell FM, Muñoz-Maniega S, Sakka E, Shuler K, Dennis MS, Thrippleton MJ, 2017. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: evidence from a cohort study. Alzheimer’s Dement. 13, 634–643. 10.1016/j.jalz.2016.09.006 [DOI] [Google Scholar]
- Wardlaw JM, Smith C, Dichgans M, 2019. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 18, 684–696. 10.1016/S1474-422(19)30079-1 [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, Lindley RI, O’Brien JT, Barkhof F, Benavente OR, Black SE, Brayne C, Breteler M, Chabriat H, DeCarli C, de Leeuw FE, Doubal F, Duering M, Fox NC, Greenberg S, Hachinski V, Kilimann I, Mok V, van Oostenbrugge R, Pantoni L, Speck O, Stephan BCM, Teipel S, Viswanathan A, Werring D, Chen C, Smith C, van Buchem M, Norrving B, Gorelick PB, Dichgans M, 2013. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838. 10.1016/S1474-4422(13)70124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Q, Ortiz-Terán L, Diez I, Perez DL, Ginsburg J, El Fakhri G, Sepulcre J, 2018. Sequence Alterations of Cortical Genes Linked to Individual Connectivity of the Human Brain. Cereb. Cortex 29, 1–8. 10.1093/cercor/bhy262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Stamova B, Jickling G, Tian Y, Zhan X, Ander BP, Liu D, Turner R, Rosand J, Goldstein LB, Furie KL, Verro P, Johnston SC, Sharp FR, Decarli CS, 2010. Distinctive RNA expression profiles in blood associated with white matter hyperintensities in brain. Stroke 41, 2744–2749. 10.1161/STROKEAHA.110.591875 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.