

Abstract
Dysfunction and degeneration of CNS cholinergic systems is a significant component of multi-system pathology in Parkinson’s disease (PD). We review the basic architecture of human CNS cholinergic systems and the tools available for studying changes in human cholinergic systems. Earlier post-mortem studies implicated abnormalities of basal forebrain corticopetal cholinergic (BFCC) and pedunculopontine-laterodorsal tegmental (PPN-LDT) cholinergic projections in cognitive deficits and gait-balance deficits, respectively. Recent application of imaging methods, particularly molecular imaging, allowed more sophisticated correlation of clinical features with regional cholinergic deficits. BFCC projection deficits correlate with general and domain specific cognitive deficits, particularly for attentional and executive functions. Detailed analyses suggest that cholinergic deficits within the salience and cingulo-opercular task control networks, including both neocortical, thalamic, and striatal nodes, are a significant component of cognitive deficits in non-demented PD subjects. Both BFCC and PPN-LDT cholinergic projection systems, and striatal cholinergic interneuron (SChI), abnormalities are implicated in PD gait-balance disorders. In the context of experimental studies, these results indicate that disrupted attentional functions of BFCC and PPN-LDT cholinergic systems underlie impaired gait-balance functions. SChI dysfunction likely impairs intra-striatal integration of attentional and motor information. Thalamic and entorhinal cortex cholinergic deficits may impair multi-sensory integration. Overt degeneration of CNS systems may be preceded by increased activity of cholinergic neurons compensating for nigrostriatal dopaminergic deficits. Subsequent dysfunction and degeneration of cholinergic systems unmasks and exacerbates functional deficits secondary to dopaminergic denervation. Research on CNS cholinergic systems dysfunctions in PD requires a systems-level approach to understanding PD pathophysiology.
Keywords: Acetylcholine, Attention, Basal forebrain, Pedunculopontine nucleus, Striatum, Mild cognitive impairment, Cognition
1. Introduction
Parkinson’s disease (PD) is now recognized widely as a multi-system neurodegenerative syndrome with multiple clinical manifestations. The multi-system nature of PD explains the diversity of PD clinical manifestations, but involvement of multiple brain systems is a challenge for identifying the pathophysiologic underpinnings of important clinical features. Unlike the cardinal motor deficits of bradykinesia, rigidity, and tremor, associated with deficient nigrostriatal dopaminergic signaling, the highly varied cognitive and behavioral deficits of PD likely reflect combined effects of varying degrees of pathology in numerous CNS systems with both cortical and subcortical components. Of particular importance may be consequences of pathologies within subcortical cholinergic neurons, Basal Forebrain Cholinergic Corticopetal (BFCC) and Pedunculopontine-Laterodorsal Tegmental (PPN-LDT) projection systems, and striatal cholinergic interneurons (SChIs). An interesting convergence of clinical research and expanding understanding of the normal functions of these systems implicates dysfunction and/or degeneration of these cholinergic systems in important clinical features of PD. These results also emphasize the importance of interpreting clinical features of PD and underlying pathologies though the lens of modern systems level neuroscience concepts (see also chapter “Cognitive control and Parkinson’s disease” by Cavanagh et al. in this volume). Anti-muscarinic cholinergic agents were historically used to ameliorate PD tremor but largely abandoned because of cognitive side-effects. Acetylcholinesterase inhibitors are presently used to treat cognitive deficits but with modest benefits (Seppi et al., 2019). Improved understanding of the effects of cholinergic systems deficits in PD may provide avenues for improved symptomatic therapies.
2. Cholinergic systems organization and functions
Acetylcholine (ACh) is the primary (small molecule) neurotransmitter of several brain projection systems and one major population of brain interneurons (see below). It is important to bear in mind that many cholinergic neurons also express peptide neurotransmitters-neuromodulators and some cholinergic neurons co-express other small molecule neurotransmitters. Cholinergic neuron populations not discussed in this chapter are motor neurons, preganglionic autonomic system neurons, and spinal cholinergic interneurons. Cholinergic neurons and terminals are identified by the expression and activity of several proteins associated uniquely with ACh synthesis and turnover. Important markers include the cytosolic enzyme Choline Acetyltransferase (ChAT; CHAT), responsible for ACh synthesis from Choline and Acetyl-CoA; the Vesicular Acetylcholine Transporter (VChT; SLC18A3), the synaptic vesicle protein that pumps ACh from the cytosol into synaptic vesicles; and the High Affinity Choline Transporter (HCT; SLC5A7), the plasmalemmal protein scavenging choline from synaptic clefts. Strongly, but not uniquely, associated with cholinergic neurons and synapses is acetylcholinesterase (AChase; ACHE), the protein responsible for terminating cholinergic neurotransmission by hydrolysis of intrasynaptic ACh. Some non-cholinergic neurons, notably dopaminergic nigrostriatal neurons, express AChase, and significant AChase expression is found in some regions with modest cholinergic innervation (Heiland and Greenfield, 1999). Much of the cerebellar cortex is a notable example of a mismatch with significant AChase expression but minimal density of cholinergic terminals (Zhang et al., 2016).
Cholinergic neurotransmission is mediated by both metabotropic (G-protein coupled; muscarinic; mAChR) and ionotropic (nicotinic; nAChR) receptors. There are five metabotropic (m1–m5) receptors with varying signal transduction mechanisms. Nicotinic receptors are cation permeated pentamers composed of different types of subunits with complex stoichiometry. There are 10 α subunit genes, four β subunit genes, and genes for δ, ε, and γ subunits (Changeux and Taly, 2008). Varying subunit stoichiometry leads to varying functional properties with a dizzying array of potential subunit composition permutations. Brain nAChR populations, however, appear to be dominated by a more restricted number of subunit combinations, particularly α4β2* (*potential inclusion of other subunits) heteromeric receptors and α7 homomeric receptors (Paterson and Nordberg, 2000). Further multiplying the complexity of cholinergic neurotransmission is the varying synaptic location of both mAChRs and nAChRs. Both mAChRs and nAChRs are expressed as post-synaptic receptors and as pre-synaptic receptors with localization on cholinergic (pre-synaptic homoreceptors) and non-cholinergic (pre-synaptic heteroreceptors) terminals.
The organization of CNS cholinergic systems is conserved across vertebrate phyla with modest deviations from the general pattern. Some species may have cholinergic cortical interneurons thought to be absent in humans. Recent PET imaging data indicates that cholinergic systems organization in humans largely conforms to the mammalian pattern (Fig. 1; Albin et al., 2018b)
FIG. 1.

[18F]FEOBV binding in human brain: Dorsal to ventral survey of averaged transaxial images for 29 normal subjects. Images scaled to peak SUVR of 3.0. Regions with notable [18F]FEOBV binding include the striatal complex, thalamus, amygdala, hippocampal formation, neocortical mantle, mesopontine junction, and portions of the cerebellum.
Reprinted with permission from Albin, R.L., Bohnen, N.I., Muller, M.L.T.M., et al., 2018a. Regional vesicular acetylcholine transporter distribution in human brain: A [(18)F]fluoroethoxybenzovesamicol positron emission tomography study. J. Comp. Neurol. 526, 2884–2897.
BFCC:
The Basal Forebrain Cholinergic Corticopetal (BFCC) complex is the most extensively studied and best characterized CNS cholinergic projection system (Ballinger et al., 2016; Schmitz and Zaborszky, 2021; Záborszky et al., 2018). The BF lies rostral to the hypothalamus and ventral to the striato-pallidal complex and is composed of different types of neurons projecting to the neocortical mantle and related structures. Cholinergic neurons are only one component of the BF, which includes glutamatergic and GABAergic neurons. With conventional histology methods, BFCC neurons are relatively large (magnocellular) and relatively intensely stained (hyperchromic) cells. While a continuous column of neurons, the BF is conventionally divided into four nuclei—the medial septal nucleus (mSN), the vertical and horizontal limbs of the diagonal band of Broca, and the nucleus basalis of Meynert—Substantia Innominata (nBM-SI). BFCC neurons exhibit a parallel organization but are not uniformly distributed within the BF—they tend to form clusters within the conventionally defined BF nuclei. Based partly on non-human primate data, Mesulam et al. (1983) defined four groups of BFCC neurons: Ch1 in the mSN; Ch2 in the vertical limb of the diagonal band; Ch3 in the horizontal limb of the diagonal band; and Ch4 in the nBM-SI. Zaborszky et al. (2008) recently used a careful analysis of human post-mortem material to develop a probabilistic map of BFCC components for human imaging studies. In this analysis, there are three major clusters of BFCC neurons: a septal cluster (merged Ch1–Ch2), a horizontal limb of the diagonal band cluster (Ch3), and a nBM-SI cluster (Ch4). This parcellation corresponds with BFCC connections. Ch1–Ch2 project primarily to the hippocampal formation, Ch3 to the olfactory bulb and piriform cortex, and Ch4 to the neocortical mantle and amygdala.
The BFCC was historically conceived as a highly diffuse, non-specific projection system with individual BFCC neurons innervating many cortical regions, though with roughly topographic organization of nBM-SI (Ch4) projections to the neocortex (Liu et al., 2015). Recent connectional studies demonstrated considerable specificity of BFCC neuron termination patterns (Gielow and Zaborszky, 2017; Zaborszky et al., 2015). The BFCC appears to consistent of sub-clusters of cholinergic neurons with similar termination patterns. Individual BFCC neurons project to several cortical fields with the apparent governing rule that inter-connected cortical fields receive cholinergic afferents from sub-clusters of BFCC neurons. Recent evidence suggests that BFCC sub-clusters function independently (Gombkoto et al., 2021). BF cholinergic neurons also target parts of the thalamus, though the relationship between the pattern of thalamic projections and corticopetal projections is unknown.
BFCC axons ramify widely in target regions. Wu et al. (2014) used a genetic labeling strategy to illuminate the morphology of individual murine BFCC neuron axonal arbors. These neurons produce large terminal arbors, whose intracortical spatial extent is on the order of cubic millimeters. The post-mortem human studies of Mesulam et al. (1992) indicate regionally specific variations in the density of and in the laminar distribution of cholinergic terminals of different human neocortical fields. Recent PET study of regional cholinergic terminal density confirms inhomogeneous distribution of BFCC terminals with higher density of cholinergic terminals in anterior cingulate and perirolandic cortices and an overall anterior to posterior gradient with mildly higher cholinergic terminal density in frontal cortices (Albin et al., 2018b).
BFCC pathways are implicated in learning, memory, and attentional functions (Ballinger et al., 2016; Záborszky et al., 2018). Modulation of attention is probably the best defined functional role of BFCC neurons (Sarter and Lustig, 2019, 2020; Sarter et al., 2021). BFCC neuron activity is critical for two complementary aspects of attention—“bottom-up” and “top-down” attentional functions. Bottom-up attentional function is typically described as “stimulus-driven”; highly salient stimuli and events attract attention largely without effort or volition. In contrast, top-down attention is “goal driven,” conscious, and effortful, and can also be used to describe cognitive control or executive functions such as maintenance of attention on tasks, resisting or recovering from distraction from salient but irrelevant stimuli, and control of task switching. Although bottom-up and top-down attentional functions are conventionally classified as cognitive functions, normal attentional function is a critical element of normal motor functions (see Section 6).
PPN-LDT:
Cholinergic neurons at the mesopontine junction are conventionally classified as components of the Pedunculopontine Nucleus-Laterodorsal Tegmental (PPN-LDT) complex (Martinez-Gonzalez et al., 2011; Mena-Segovia and Bolam, 2017; Pienaar et al., 2017). The distribution of these mesopontine cholinergic neurons is often used to define the extent of the PPN-LDT complex (PPN = Ch5 and LDT = Ch6 in the Mesulam et al., 1983 nomenclature). The PPN-LDT complex is an ambiguous concept. As pointed out by Mesulam et al. (1989), there are no obvious fiber tracts or other anatomic features delineating the boundaries of the PPN-LDT complex. As with the BF, cholinergic neurons are only one component of the PPN-LDT, which also contains heterogeneous populations of glutamatergic, GABAergic, and glycinergic neurons. In contrast to BFCC neurons, cholinergic PPN-LDT neurons appear to be a diffusely projecting system. Single-cell tract tracing studies indicate that individual cholinergic PPN-LDT cholinergic neurons give rise to widely ramifying ascending and descending projections innervating a variety of target nuclei. Targets of cholinergic PPN-LDT neurons include all components of the basal ganglia, the BF, multiple thalamic nuclei, the superior colliculus, and a variety of brainstem targets. Ascending projections to the thalamus are particularly robust.
The functional roles of PPN-LDT cholinergic neurons are unclear. Conventional definitions of the PPN-LDT overlap with the physiologically defined Mesencephalic Locomotor Region (MLR), an area where low intensity electrical stimulation produces locomotion in decerebrate preparations. This led to conflation of the PPN-LDT and MLR and an impression that descending projections of cholinergic mesopontine neurons were critical drivers of locomotion (reviewed in Albin et al., 2018a). This does not appear to be correct and this conception of PPN-LDT cholinergic neuron function tended to obscure the importance of the substantial ascending projections to midbrain and forebrain structures. Interconnections with basal ganglia nuclei are so extensive that some authors discuss the PPN-LDT as a component or partner of the basal ganglia. Some data suggests important behavioral functions of PPN-LDT cholinergic neurons, perhaps as a relatively rapid “alerting” channel for identification of significant stimuli, placing cholinergic PPN-LDT neurons within the general category of subserving attentional functions (Gut and Mena-Segovia, 2019; Gut and Winn, 2016). The PPN-LDT, broadly considered (not just cholinergic neurons), receives considerable polymodal sensory input and may constitute a channel for rapid transmission of sensory information to PPN-LDT target regions. Some experiments suggest PPN-LDT involvement in forms of learning, sensorimotor gating, and evaluation of action outcomes. Several of these proposed functions overlap with well characterized functions of the basal ganglia, with which PPN-LDT neurons are intimately connected, notably forms of reinforcement learning and action-outcome evaluation. The concept of PPN-LDT cholinergic neurons subserving some type of attentional function is consistent with the high density of cholinergic PPN-LDT afferents to the superior colliculus and multiple thalamic nuclei. A principal function of the superior colliculus is orientation toward meaningful stimuli. Considerable recent data indicates that thalamic nuclei are coordinators of cortical function and particularly important modulators of attention (Halassa and Kastner, 2017).
Striatal Cholinergic Interneurons (SChIs):
SChIs are the only cholinergic interneuron population of the human brain. These interneurons are a small fraction, 2–3%, of neurons within the striatum, but give rise to unusually luxuriant terminals (Gonzales and Smith, 2015). The density of presynaptic cholinergic terminals and synaptic markers—ChAT activity, VChT expression, HAT expression, and AChase expression/activity—is easily the highest in the CNS. It is likely that SChIs are mediators of multiple aspects of striatal function. SChIs receive cortical, thalamic, and dopaminergic nigrostriatal afferents, and innervate multiple other neuronal elements of the striatum, including medium spiny projection neurons and dopaminergic afferent terminals. SChIs, for example, may regulate nigrostriatal terminal dopamine release (Brimblecombe et al., 2018). A basic feature of striatal-basal ganglia architecture is the existence of roughly parallel and relatively functionally specialized cortico-striato-pallidothalamo-cortical circuits. With their abundant and diffuse terminals, SChIs are positioned to transmit information across cortico-striato-pallidothalamo-cortical circuits. SChIs and their terminals, however, exhibit inhomogeneous distribution with the striatum. Recent stereological studies of human post-mortem specimens indicate modestly higher SChI density in the caudate than putamen (Bernacer et al., 2007). Striatal SChI terminals/synapses exhibit a striking form of inhomogeneity. ChAT immunohistochemical and AChase histochemical studies of numerous mammalian species, including human post-mortem specimens, indicate the presence of striosome-matrix architecture with a rostral-caudal gradient. Delineated by a number of neurochemical-histochemical-synaptic markers, striosomes are tubular domains tunneling through the surrounding matrix (Gerfen, 1992). Striosomes exhibit relatively lower density of cholinergic terminals-synapses. Striosome-matrix differentiation is most obvious in the caudate with more even distribution of cholinergic synapses-terminals in the putamen. Based on connectional studies, striosomal medium spiny striatal projection neurons are suggested to play particular roles in “limbic” functions such as motivation (Gerfen, 1992).
It is likely that SChIs are involved in many aspects of striatal function. Interesting behavioral experiments, for example, suggest that SChIs are critical for maintaining behavioral flexibility (Matamales et al., 2016). SChIs likely integrate and transmit information from corticostriate glutamatergic and nigrostriatal dopaminergic afferents to medium spiny neurons. Recent experiments indicate that SChIs are critical for complex movement control, consistent with the view that these interneurons integrate cortico-striatal transfer of movement-related, attended cues with striatal motor sequencing (Avila et al., 2020; see Section 6).
Other Cholinergic Projection Systems:
The functions of other CNS cholinergic projection systems are less well understood and not explored in the context of PD. Cholinergic medial vestibular neurons (MVNs) innervate portions of the cerebellar cortex, notably the uvula, the flocculi, and the nodulus (Barmack et al., 1992). The latter two are the phylogenetically ancient vestibulocerebellum, intimately connected to vestibular nuclei, and involved in maintenance of posture, balance, oculomotor control, and oculomotor adaptation. In what may be a uniquely human feature, human PET imaging studies indicate significant cholinergic innervation of the entire cerebellar vermis (Fig. 1). The origin of these vermal cholinergic terminals is unknown, though expansion of MVN afferents is a plausible candidate.
Other CNS cholinergic projections include the medial habenula to interpeduncular nucleus projection (Ch7), the parabigeminal nucleus to superior colliculus projection (Ch8), and a recently described cholinergic projection from the prepositus hypoglossi nuclei to the cerebellar cortex (Sugimura and Saito, 2021; only described in rodents), likely important for eye movement control. The basal interstitial nucleus is a group of neurons embedded in the white matter underlying the vestibulocerebellum, innervating the floccular cortex. In primates, some of these neurons may be cholinergic (Jaarsma et al., 2018).
3. Tools to study cholinergic systems in humans
Identification of cholinergic deficits in and correlation with clinical features of PD is a direct function of the availability of methods that can be deployed in human studies. Until relatively recently, studies of cholinergic system changes in PD were limited to analysis of post-mortem tissues. These studies used either biochemical methods measuring the expression of cholinergic terminal markers, mainly regional ChAT activity, or conventional histopathologic, histochemical, or immunohistochemical methods examining the integrity of cholinergic neurons and terminals. These early studies demonstrated heterogeneous loss of BFCC and PPN-LDT cholinergic neurons in PD. As these were post-mortem studies, correlation with clinical features was limited to qualitative interpretations (see below). In addition to limited correlation with clinical features, post-mortem studies depending on cholinergic-synaptic markers may be limited by disease-related modulation of cholinergic marker expression. There is evidence, for example, of up-regulation of ChAT activity in Mild Cognitive Impairment (MCI) and early Alzheimer’s disease (AD) subjects (DeKosky et al., 2002). Similarly, older biochemical studies of AD comparing nBM-SI cholinergic neuron density to ChAT activity indicated disproportionate loss of ChAT activity, suggesting down-regulation of ChAT expression or activity of BFCC neurons in AD (Candy et al., 1986).
The application of modern imaging methods, single photon emission computed tomography (SPECT), positron emission tomography (PET), and MRI, made systematic clinical correlation studies feasible. These methods are analogues of the methods used for post-mortem studies. SPECT and PET methods use tracers targeting markers of cholinergic terminals/synapses with MRI morphometric and connectional methods examining structural changes in nuclei and pathways containing cholinergic neurons and projections. MRI methods are more widely available but as these nuclei and pathways do not exclusively contain cholinergic neurons/projections, must be interpreted cautiously. Recent human MRI morphometric and connectional studies of the BFCC, summarized concisely and thoughtfully by Schmitz and Zaborszky (2021), provided considerable novel information about connections and possible functional roles of BFCC neurons in humans. These data are useful for interpretating changes in MRI morphometric and connectional measures found in studies of neurodegenerative disorders.
The pioneering molecular imaging method was [123I]iodobenzovesamicol (IBVM) SPECT, a vesamicol derivative targeting VChTs (Kuhl et al., 1996). As with all SPECT methods, this method has significant limitations in terms of anatomic resolution and quantification. The initial PET methods targeted AChase activity, based on the concept that AChase is strongly associated with cholinergic synapse density. These methods used tracers—[11C]methyl-4-piperidinyl propionate (PMP) and N-[11C]-methyl-4-piperidyl acetate (MP4A)—AChase substrates with polar metabolites that remain trapped in brain, allowing quantification of regional AChase activity as a proxy for cholinergic synapse density. Potential drawbacks of AChase PET are that AChase enzyme activity might be modulated by physiologic or disease-related processes, weakening the relationship between regional AChase activity and cholinergic synaptic density. As mentioned above, in some regions (e.g., cerebellar cortex), AChase expression is not tightly linked to cholinergic synapse density. Another defect is that some regions, striatum and cerebellum, have high levels of regional AChase activity with tracer metabolite retention as a function of tracer delivery (regional cerebral blood flow) rather than regional AChase activity. Finally, PMP and MP4A are also butyrylcholinesterase substrates, though to a much lesser extent than they are AChase substrates. Changes in butyrylcholinesterase expression or activity might influence the results of PET studies with these tracers.
More recently developed PET tracers target the VChT. These include [18F] fluoroethoxybenzovesamicol (FEOBV) and [18F](−)-(1-(−8-(2-fluoroethoxy)-3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(4-fluorophenyl)-methanone (VAT), providing more specific measures of regional cholinergic terminal density with excellent anatomic resolution. Proteins associated with presynaptic vesicles—VChT in cholinergic terminals, the Type 2 vesicular monoamine transporter (VMAT2) in CNS monoaminergic terminals, SV2A for all synaptic vesicles—are favored targets for PET tracers aimed at quantifying synaptic terminal density. Expression or activity of these proteins is suggested to be less likely to undergo significant physiologic or disease-related modulation that could confound measurement of regional terminal density. This is a reasonable assumption but may not always be correct. Basic studies indicate that VMAT2 expression and activity are actively regulated in synaptic terminals (Mulvihill, 2019). Experience with the VMAT2 ligand dihydrotetrabenazine indicates that marked changes in intracellular dopamine concentrations can affect VMAT2 ligand binding and recent studies suggest that striatal VChT tracer binding is modulated by dopamine D2 receptor manipulations (Kilbourn et al., 2008; Liu et al., 2020; Schildt et al., 2020; Tong et al., 2008). Recent experience with FEOBV PET in PD suggests disease related modulation of tracer binding (see Section 7).
4. Cholinergic system changes in PD: Post-mortem studies
Documentation of BF neurodegeneration in PD has a venerable history. Lewy initially described the eponymous Lewy body (LB) and neuronal loss in magnocellular nBM-SI cells, now known to be BFCC neurons, as well as in dorsal motor nucleus of the vagus neurons. The discovery of LBs in and substantia nigra neuronal loss came later. Lewy’s findings were subsequently confirmed by Hassler, who later suggested that nBM-SI pathology in PD was related to cognitive impairment (summarized in Liu et al., 2015). Following the discovery (or perhaps re-discovery, as there were prior neuropathologic reports) of BF pathology in AD and correlated cortical cholinergic marker deficits, there were reports of substantial cholinergic BFCC neuron loss in PD. These pioneering studies utilized conventional histological and immunohistological analysis of BFCC neuronal perikaryal, AChase histochemistry, and neurochemical measurements of cholinergic markers, particularly regional ChAT activity, in BF nuclei and BFCC target regions (Agid et al., 1989; Candy et al., 1986; Liu et al., 2015). These studies demonstrated reduced cholinergic marker expression in both the hippocampal formation and neocortical regions with complementary results found in evaluations of BF cholinergic markers and assessments of BFCC perikaryal densities. These studies not only indicated BFCC neuron degeneration in PD but also considerable variation in the magnitude of BFCC degeneration and potential correlation with the magnitude of cognitive impairments. While limited due to relatively sparse pre-mortem clinical characterizations, demented PD subjects (PDD) were reported to exhibit greater depletion of cortical cholinergic markers and loss of BFCC neurons than cognitively intact PD subjects. Data from post-mortem studies also indicated that the magnitude of forebrain cholinergic deficits secondary to BF neurodegeneration is greater in PD than AD and that demented PD subjects exhibited the greatest BFCC deficits. This latter result was subsequently replicated in initial AChase PET imaging studies (see below). Another interesting result emerging from these early studies was that control subjects exhibited age-related declines in BFCC perikaryal density and cortical cholinergic marker expression, the latter indicating age-related loss of cortical cholinergic afferents, another finding replicated in imaging studies.
Hall et al. (2014) more recently reported a careful, combined morphometric and biochemical post-mortem study comparing PD subjects with intact cognition (N = 5–8), PDD (N = 6–8), and controls (N = 5–8). nBM-SI (Ch4) cholinergic neurons were depleted in PDD subjects and there was a trend toward nBM cholinergic neuron depletion in PD subjects with intact cognition. Mean numbers of mSN-vertical limb of the diagonal band (Ch1–2) cholinergic neurons were equivalent across all three groups, but considerably more variation in PDD subjects and PD subjects with intact cognition. Measurements of ChAT activity in frontal cortex and hippocampus were consistent with morphometry results. Mean frontal cortex ChAT activity was reduced in PDD subjects and in PD subjects with intact cognition but with considerably greater variation in PD subjects with intact cognition. A similar profile was found with hippocampal ChAT activity measurements. PDD subjects exhibited markedly reduced hippocampal ChAT activity and PD subjects with intact cognition exhibited hippocampal ChAT activities intermediate between PDD and control subjects. Hall et al. also showed a higher density of Lewy pathology in PDD subject BFs than in the BFs of PD subjects with intact cognition. This gradient of Lewy pathology was also seen in the hippocampus. Overall, this data suggested that nBM-SI (Ch4) cholinergic neurons are initially more affected than mSN-vertical limb of the diagonal band (Ch1–2) cholinergic neurons in PD.
PPN-LDT cholinergic neuron degeneration in in PD was reported initially in by Hirsch et al. (1987), who documented an approximately 50% loss of cholinergic PPN-LDT neurons in some post-mortem specimens from PD subjects. Even greater losses of cholinergic PPN-LDT neurons were noted in Progressive Supranuclear Palsy (PSP) post-mortem specimens. Similar results in PSP and PD were reported also by Zweig et al. (1987, 1989). Cholinergic neurons are not the only PPN-LDT neuronal population affected. Both GABAergic and glycinergic PPN-LDT neurons are diminished in PD, though to a significantly lesser extent (15–20% decreases) than cholinergic neurons (Pienaar et al., 2013).
In their original proposal for staging PD pathology, Braak et al. (2003) include the PPN-LDT region as a site of accumulating Lewy neurite and LB pathology, approximately paralleling the accumulation of Lewy pathology in the substantia nigra. In their earlier study, Zweig et al. (1989) suggested a reasonably close correlation between the magnitude of PPN-LDT cholinergic neuron loss and the magnitude of SNc dopaminergic neuron loss. As post-mortem studies are typically based on advanced disease, they usually say little about disease natural history, heterogeneity, or specific relationships between pathologic features and clinical features. The discovery of PPN-LDT cholinergic neuron loss in PD and PSP, in the context of these neurons as possible MLR components, suggested that degeneration of this group of cholinergic neurons might be related to gait and balance difficulties. Both Hirsch et al. (1987) and subsequent reports describe significant variation in the magnitude of PPN-LDT cholinergic neuron loss in PD with some specimens exhibiting normal or near normal cholinergic neuron numbers. PSP specimens, however, consistently showed marked depletion of cholinergic PPN-LDT neurons. Hirsch et al. (1987) also reported PPN-LDT ChAT activity measurements in control, PD, and PSP specimens. Paralleling the anatomic results, the ChAT activity measurements revealed a broad range in PD specimens, overlapping well into the normal range of the controls while PSP specimen ChAT activities were consistently depressed. In a subsequent study, Karachi et al. (2010) assessed PPN-LDT neuron integrity in post-mortem specimens from PD subjects classified as fallers (n = 6) or non-fallers (n = 6) on the basis of UPDRSIII evaluations. PPN-LDT cholinergic neuron counts from non-fallers exhibited a very modest decrease (~10%) but faller PD subjects had an approximately 40% decrease in PPN-LDT cholinergic neurons.
The aggregate post-mortem data indicated degeneration of BFCC and PPN-LDT projection systems in PD and linked these changes to cognitive deficits and treatment-refractory motor deficits.
5. Cholinergic system changes in PD: Imaging studies and cognitive deficits
The initial IBVM SPECT study of Kuhl et al. (1996) contrasted a group of younger (N = 9; mean age = 59) non-demented PD subjects and older (N = 6; mean age = 77) demented PD (PDD) subjects. The former exhibited modest decreases,~20%, in occipital cortical IBVM binding while the demented subjects exhibited more marked, ~40%, reductions in IBVM binding throughout the neocortical mantle. The first AChase PET studies were reported by Shinotoh et al. (1999), using MP4A PET to compare PD and PSP subjects with controls. Their PD subjects exhibited moderate, ~20%, reductions in cortical tracer uptake PD subjects with non-significant reductions in cortex of PSP subjects. Results similar to those of Kuhl et al. were subsequently reported by Hilker et al. (2005) with MP4A PET. This group documented modest parieto-occipital cortex decreases in tracer uptake in non-demented PD subjects with significantly diminished tracer uptake throughout the cortical mantle in PDD subjects. Hilker et al. also used [18F]fluorodopa PET to assess striatal dopaminergic innervation, and found no difference in striatal dopaminergic deficits between non-demented PD and PDD subjects. In a prior study, Bohnen et al. (2003) used PMP PET to study differences between non-demented PD, PDD, Dementia with Lewy body (DLB), and Alzheimer’s disease (AD) subjects, the latter three approximately matched for dementia severity. Whole neocortical tracer uptake was most reduced in PDD subjects, least reduced in AD subjects, and with intermediate reductions in non-demented PD subjects. Shimada et al. (2009) used MP4A PET to evaluate cortical cholinergic innervation in early PD, including de novo subjects, more advanced PD without dementia, PDD, and DLB subjects. Reductions in tracer uptake were found in all groups with significant, pan-cortical reductions in PDD and DLB. Notable additional findings were that some regional deficits, notably in occipital cortices, were present even in de novo PD subjects, and that there was considerable variation in the degree of cortical cholinergic deficits with some more advanced PD subjects exhibiting only modest posterior cortical cholinergic deficits.
These imaging studies confirmed the association of diffuse neocortical cholinergic deficits with marked cognitive deficits in PD and suggested other features of cholinergic systems deficits in PD. These include significant variation in the degree of cholinergic deficits among PD subjects and a suggestion that neocortical cholinergic deficits initially manifest in posterior cortices—occipital and parietal—with subsequent posterior to anterior progression. This work formed some of the background for the Dual Syndrome model of PD cognitive deficits of Kehagia et al. (2013). In their model, earlier cognitive deficits reflect frontostriatal loop dysfunctions driven by striatal dopaminergic deficits with later cognitive deficits reflecting more posterior cortical—temporal, parietal, occipital—dysfunctions driven heavily by BFCC deficits. In terms of progression of PD cognitive deficits, this model captures important aspects of PD natural history (Williams-Gray et al., 2009). This model, however, may be misleading as to the role of BFCC deficits (see below) and doesn’t incorporate the increasing burden of cortical α-synucleinopathy.
Complementary data exploring the relationship between BFCC dysfunction-degeneration and cognitive impairments in PD was obtained in several correlative MRI morphometry studies. Choi et al. (2012) studied PD subjects with normal cognition (PD-NC), PD-MCI, and PDD subjects. PD-MCI and PDD, but not PD-NC subjects, exhibited diminished nBM-SI (Ch4) volumes with a correlation between nBM-SI volume and a measure of general cognitive performance. Choi et al. also described correlations between nBM-SI volumes and domain specific measures of cognitive performance. Several studies used MRI data from the Parkinson’s Progression Markers Initiative (PPMI), which enrolled de novo PD subjects, to assess relationships between cognitive declines and BF volumes. Ray et al. (2018) found that smaller nBM-SI volumes at enrollment predicted cognitive decline with a higher probability of developing PD-MCI. Schulz et al. (2018) compared baseline data from PPMI subjects with and without cognitive impairments, finding that those with cognitive impairments had lower nBM-SI volume. Similar to the results of Ray et al., Schulz et al. found that lower baseline nBM-SI volume in cognitively intact PPMI subjects predicted subsequent cognitive decline. Barrett et al. (2019) reported results from both the PPMI dataset and a cohort of more advanced PD subjects. In both datasets, nBM-SI volume correlated with global measures of cognitive performance and some domain specific measures. Similarly, in an analysis of non-demented PD subjects enrolled in the COPPADIS study, Grothe et al. (2021) found that nBM-SI volume correlated with a general measure of cognitive function.
More detailed correlation of PD clinical features and cholinergic systems changes was pioneered by Bohnen et al. (2006, 2012) using PMP PET. In an initial study, mean neocortical PMP uptake was correlated with selected cognitive domain measures. Tracer uptake deficits correlated best with measures of attentional and executive function, a result consistent with well characterized functions of BFCC projections. In a subsequent, larger study of moderately advanced, non-demented PD subjects (N = 101), Bohnen et al. (2012) evaluated both neocortical and thalamic AChase activity. Approximately one-third of PD subjects exhibited reduced mean neocortical AChase activity, defined as below the fifth percentile of the control distribution. Subjects with diminished neocortical AChase activity had somewhat more advanced PD. Approximately one-fifth of the PD subjects had diminished thalamic tracer uptake. PD subjects with diminished thalamic AChase activity generally, though not invariably, had diminished neocortical AChase activity. Those with diminished neocortical AChase activity were impaired on global measures of cognition, and exhibited impairments in the domains of verbal learning, executive function, and attention, but not visuospatial function. Parallel evaluation of striatal dopaminergic terminals with [11C]DTBZ PET revealed evidence of independent striatal dopaminergic deficit impacts on cognitive measures.
PET studies with the more anatomically resolute VChT ligand [18F]FEOBV allowed detailed exploration of correlations between changes in regional cholinergic terminal densities and cognitive deficits in PD. van der Zee et al. (2021) studied 86 moderately advanced, non-demented PD subjects. The most marked FEOBV binding deficits were found in occipital, parietal, and temporal cortices (Fig. 2).
FIG. 2.

VChT deficits in Parkinson’s disease brain: Statistical parametric voxel-based analysis (false discovery rate corrected P < 0.05) showing significantly diminished [18F]FEOBV binding in moderately advanced, non-demented Parkinson’s disease subjects (N = 89) compared with healthy controls. Extensive depletion of tracer binding in posterior cortices.
Reprinted with permission from van der Zee, S., Müller, M.L.T.M., Kanel, P., van Laar, T., Bohnen, N.I., 2021. Cholinergic denervation patterns across cognitive domains in Parkinson’s disease. Mov. Disord. 36, 642–650
Consistent with results from the PMP PET study of Bohnen et al. (2012), global neocortical FEOBV binding was correlated with a global measure of cognition, measures of memory function, attentional function, and executive function but not language or visuospatial functions. In voxel-based analyses, FEOBV binding was correlated with domain specific cognitive measures of memory, attention, and executive functions. Widespread regional correlations were found for these domain specific measures, with overlapping correlated regions including the cingulate cortex, opercular and insular regions, the visual thalamus (notably the lateral geniculate nucleus), and the hippocampal formation. These results implicate degeneration of regionally specific BFCC projections from the septal complex (Ch1–Ch2) and nBM-SI (Ch4), and from the PPN-LDT in cognitive deficits in early to moderately advanced PD.
For both the PD and control groups, seven main principal components (PCs) were identified but the spatial patterns of PD and normal PCs had overlapping but significantly different distributions. In PD subjects, multivariate regression including PC scores, l-dopa equivalent dose, disease duration, and MDS-UPDRSIII scores demonstrated that three PC values and MDS-UPDRSIII scores predicted performance on a global measure of cognitive function. One PC included the cingulate cortex, the second the cerebellum, and the third, bilateral frontal pole and left orbitofrontal regions. Subsequent multivariate regressions for individual cognitive domains indicated that different combinations of these three PCs predicted memory, attentional, executive, and language performance. Cingulate cortex PC values and MDS-UPDRSIII scores predicted memory, executive function, attention, and language function.
Together with the prior voxel-based analyses, the PC analyses suggests that degeneration of BFCC afferents to specific frontal regions is a significant mediator of cognitive dysfunction in mild to moderate PD. This is despite the fact that cholinergic terminal deficits appear most marked in occipital and parietal cortices. Some of the implicated frontal regions—anterior cingulate cortex, insular cortex, and opercular cortex, plus portions of subcortical structures such as the striatum and specific thalamic nuclei—are components of cognitive control networks such as the salience and cingulo-opercular task control (COTC) networks. These cognitive control networks oversee and coordinate cognitive domain specific networks (see chapter “Cognitive control and Parkinson’s disease” by Cavanagh et al. in this volume) and dysfunction of these networks is predicted to have downstream ramifications with degraded performance of domain specific cognitive functions. This view of the role of cholinergic deficits in PD cognitive dysfunction modifies the Dual Syndrome model. Rather than driving posterior cortical dysfunction and associated mainly with PDD, cholinergic projection systems dysfunction may be a driver of cognitive dysfunction in early to moderate PD secondary to dysfunction of cognitive control network frontal cortex nodes.
6. Cholinergic system changes in PD: Imaging studies and gait-balance deficits
Parallel data pointed to cholinergic deficits contributing to another morbid aspect of PD; gait and balance problems (Bohnen et al., 2009, 2013). PD subjects with cortical AChase deficits had slower gaits, independent of striatal dopaminergic denervation, indeed, PD subjects without cortical AChase deficits had normal gait speed under these test conditions (Bohnen et al., 2013). In a complementary, recent MRI morphometry study of more advanced PD subjects, Dalrymple et al. (2021) found that reduced nBM-SI volume was associated with slower gait speed and impaired performance on a dual task gait measure. Studies using other measures of cortical cholinergic function also indicated that cortical cholinergic deficits are associated with slower gait in PD (Rochester et al., 2012). PD subjects with thalamic AChase deficits were more likely to have a history of falls (Bohnen et al., 2009). Similarly, in their earlier MP4A PET study, Shinotoh et al. (1999) found that PSP subjects, a notoriously fall-prone group, had approximately 40% reductions in thalamic tracer uptake.
Abundant data indicates that normal gait and balance performance depend on integration of what are conventionally regarded as cognitive functions (Sarter et al., 2014). Attentional functions are essential for normal gait performance and maintenance of balance. Numerous studies in both normal elderly and PD subjects indicate that diminished attentional capacity and reduced ability to deal with distractors results in gait abnormalities and increased fall risk. Morbid gait and balance impairments in PD include both chronic impairments (usually termed postural instability and gait disorder [PIGD]), and the episodic impairment described as freezing of gait (FoG). Both greatly increase fall risk. PIGD is largely unresponsive to conventional dopamine replacement treatments (DRT), and while some FoG is DRT responsive, most is not. Absent DRT responsiveness suggested that both chronic and episodic gait-balance impairments reflect degeneration of CNS systems extrinsic to the nigrostriatal dopaminergic projection. In conjunction with the known role of attentional deficits in PD-related gait-balance disorders and falls, and AChase PET data, dysfunction and/or degeneration of the BFCC and PPN-LDT cholinergic projection systems were strong candidates as substrates of PD-related chronic and episodic gait balance disorders.
Using a challenging rat gait-balance task, Kucinski et al. (2013) evaluated the interactions of toxin-induced nigrostriatal dopaminergic deficits and BFCC deficits. Rats with targeted lesions of the dopaminergic innervation of the dorsomedial striatum and of BFCC afferents to cortex innervating the dorsomedial striatum (dual lesion [DL]) exhibited marked increases in fall frequency compared with sham lesion or single lesion (either nigrostriatal dopaminergic or BFCC lesion) controls. Because of the restricted extent of their nigrostriatal lesions, DL animals do not exhibit the marked motor deficits of rats with major striatal dopaminergic denervation. DL animals performed well on tests of skilled forelimb use but also exhibited gait deficits. Fall frequency correlated with performance deficits on a Sustained Attention Test (SAT). Rodent SAT performance is a measure of BFCC attentional function with a homologous human SAT also linked to BFCC functions. In a particularly interesting analysis, Kucinski et al. (2013) demonstrated that fall rates correlated with the extent of striatal dopaminergic lesions, suggesting that BFCC neuron activity compensated partially for effects of nigrostriatal dopaminergic denervation. These results are consistent with a model in which BFCC neuron activity can partially compensate for the consequences of nigrostriatal dopaminergic denervation with DRT-refractory clinical deficits emerging as these compensating cholinergic systems degenerate (see below).
Correlative studies using PMP PET imaging directly implicate abnormalities of BFCC and PPN-LDT cholinergic projections in impaired attentional functions in PD subjects. Using behavioral measures relatively specific for “top down” and “bottom-up” attentional functions, Kim et al. (2017, 2019a) found that neocortical PMP uptake predicted “top-down” attentional performance and that thalamic PMP uptake predicted “bottom-up” attentional performance. As mentioned above, PD subjects with thalamic deficits generally also have neocortical deficits. In a complementary study, Müller et al. (2013) used PMP PET to examine the relationship between BFCC and PPN-LDT cholinergic projection intregrity and postural control in PD. Thalamic PMP uptake, but not neocortical uptake, predicted postural sensory integration. Kim et al. (2019b) also examined an aspect of executive control, conflict processing, with results indicating that both nigrostriatal dopaminergic projections and BFCC projections mediate conflict processing and suggesting that BFCC function may compensate for failing nigrostriatal dopaminergic functions in conflict processing.
Application of FEOBV PET allowed more resolute correlative analyses. Bohnen et al. (2019) studied 94 moderately advanced PD subjects, 40% with a fall history and 15% with FoG. Volume-of-interest based correlations indicated that thalamic FEOBV binding deficits were associated with fall history and that caudate and limbic archicortex FEOBV binding deficits were associated with FoG. Voxel-based analyses were pursued for more detailed correlations, suggesting that right visual thalamus cholinergic deficits, particularly LGN deficits, right caudate cholinergic deficits, and bilateral frontal cholinergic deficits were correlated with fall history. FoG was correlated with FEOBV binding reductions in the bilateral striatum, temporal, and mesiofrontal limbic regions. Bohnen et al. (2021) subsequently obtained complementary results using a data-driven approach, step-wise regression with forward selection, and volume-of-interest analysis of this dataset to assess relationships between regional FEOBV binding changes and PD motor features, including PIGD. Overall PIGD severity, measured by UPDRSIII scores, was predicted by medial geniculate, lateral geniculate, and entorhinal cortex FEOBV binding deficits. UPDRSIII scores specifically reflecting non-episodic aspects of PIGD were predicted by medial geniculate, entorhinal cortex, and internal segment of the globus pallidus FEOBV binding deficits. Analogous results were obtained with voxel-based analyses. Combined with the prior analyses, these results implicate BFCC, PPN-LDT, and SChI deficits in both episodic (falls, FoG), and non-episodic (chronic PIGD) features of PD.
MRI-based studies implicate PPN-LDT abnormalities in the pathophysiology of PD gait-balance disorders (Fling et al., 2013; Schweder et al., 2010; Snijders et al., 2011; Youn et al., 2012; Zwergal et al., 2013). While these methods are not specific to cholinergic neurons-projections, these results are consistent with a major role for dysfunction-degeneration of PPN-LDT cholinergic neurons in PD gait-balance disorders. Using measures of PPN-LDT structural integrity and connectivity, these studies implicate PPN-LDT dysfunction and degeneration in the pathophysiology of FoG in PD. In recent PPMI data analysis, Craig et al. (2020) demonstrated that baseline abnormalities of PPN-LDT microstructure, presumably reflecting neurodegeneration, predicted the emergence of gait-balance features.
The FEOBV data indicating involvement of striatal cholinergic deficits in DRT-refractory gait-balance dysfunctions points to degeneration or dysfunction of SChIs in this aspect of PD pathophysiology. The connectional anatomy of SChIs suggested that these interneurons integrate attentional information from the cortex with movement related information encoded by nigrostriatal dopaminergic signaling (Sarter et al., 2021). Avila et al. tested this hypothesis using the rat DL lesion model and the complex gait task developed by Kucinski et al. (2013), plus a novel task based on cued turning. Using chemogenetic methods to manipulate SChI activity, Avila et al. (2020) found that SChI inhibition in normal rats duplicated the gait deficits, increased fall propensity, and turning deficits of DL rats. Chemogenetic SChI excitation in DL rats ameliorated their deficits. These results indicate the integrative role of SChIs and underscore the artificiality of conventional cognitive vs motor function distinctions.
7. Cholinergic system changes in PD: Early compensation: Upregulation?
As discussed above, work with the rat DL model is consistent with a scenario in which BFCC neuron activity compensates for impaired nigrostriatal dopaminergic signaling. When BFCC function fails, this unmasks DRT-refractory deficits. A possible mechanism of compensation for dopaminergic deficits is upregulation of cholinergic neurotransmission. Some PET imaging study data is consistent with cholinergic systems upregulation in response to nigrostriatal dopaminergic deficits. van der Zee et al. (2022) studied 57 recently diagnosed, untreated PD subjects with FEOBV PET. Approximately 30% of these subjects met criteria for PD-MCI, a result consistent with several studies of cognition in early PD. In a voxel-based analysis, both PD-NC and PD-MCI subjects exhibited regional FEOBV binding deficits, notably in posterior cortices. These results were notable for evidence of increased FEOBV binding in numerous cortical and subcortical regions, including the cerebellum, cingulate cortex, putamen, gyrus rectus, hippocampal formation, and amygdala. van der Zee also found that regional FEOBV binding increases were more pronounced in PD-NC subjects than in PD-MCI subjects. Legault-Denis et al. (2021) reported analogous results in a comparison of PD-NC, PD-MCI, and control subjects. In this study, PD-NC subjects exhibited supra-normal hippocampal FEOBV binding with PD-MCI subjects exhibiting hippocampal FEOBV binding comparable to normal subjects. The results of these two studies are consistent with a model in which increased cholinergic neuron function in several systems compensates for nigrostriatal deficits with emergence of clinically manifest cognitive deficits as cholinergic system upregulation declines with disease progression.
Bedard et al. (2019) previously reported increased FEOBV binding in numerous regions in subjects with isolated REM sleep behavior disorder, a precursor of PD and closely related synucleinopathies. Analogous results were reported by Liu et al. (2018), who applied PMP PET to a cohort of LRRK2 mutation carriers, both with and without PD. Both groups of LRRK2 mutation carriers exhibited increased PMP uptake in cortical and subcortical regions. The concept of transient, early compensatory upregulation of cholinergic neuron activity followed by subsequent declining function and degeneration is consistent with data from other systems. Sossi et al. (2010) described significantly increased striatal dopamine turnover in pre-manifest LRRKK2 mutant carriers with modest losses of striatal dopamine terminals.
8. Conclusions
The confluence of earlier post-mortem and recent imaging data indicates that dysfunction-degeneration of CNS cholinergic systems is an important contributor to morbid features of PD, particularly the DRT-refractory features of cognitive impairment and gait-balance deficits. Recent imaging data, in conjunction with expanding knowledge of the normal structure and functions of brain cholinergic systems, points to specific roles of several cholinergic system dysfunctions in these important PD clinical features. The best documented and understood aspect of CNS cholinergic system dysfunction in PD is the contribution of these dysfunctions to DRT-refractory gait and balance problems. The exploration of the relationship of cholinergic deficits to PD gait-balance problems has instructive features. First is the utility of combining good imaging tools with careful clinical characterization. A second instructive feature is the considerable value of well-targeted animal model experiments evaluating specific hypotheses derived from clinical research. In the case of PD gait-balance dysfunction, illuminating experiments were feasible because of a large body of prior basic research investigating the functional roles of the BFCC system. Third is the importance of abandoning conventional functional divisions of motor and cognitive functions in favor of neurobiologically grounded integrative concepts of functional performance. A corollary is the equivalent necessity of discarding simplistic single system concepts of pathophysiology and assessing the interactions of systems disrupted in PD (see also chapter “Cognition and serotonin in Parkinson’s disease” by Frouni et al. in this volume).
The accumulated clinical and experimental data indicate that brain cholinergic systems are critical to the integration of several aspects of normal gait performance—BFCC projections as important mediators of attentional functions with SChIs as integrators of attentional and motor information within the striatum. PPN-LDT cholinergic neuron are clearly important in gait and balance control, but as there is less basic understanding of the functional roles of PPN-LDT cholinergic neurons, it is difficult to be specific about their functional roles. The Müller et al. (2013) data indicating that thalamic cholinergic deficits are associated with impaired postural sensory integration is consistent with the suggestion that PPN-LDT neurons are a channel for rapid integration of polymodal sensory inputs. A significant body of recent data indicates that visual thalamus nuclei such as the pulvinar and lateral geniculate nuclei play important roles in modulating attention and coordinating the interactions of cortical fields (Halassa and Kastner, 2017). Loss of thalamic cholinergic afferents is predicted to disrupt these functions, as found in the Kim et al. (2017) PMP PET study of “bottom-up” attentional performance in PD subjects. Additional nodes targeted by BFCC or PPN-LDT afferents and with cholinergic deficits might impede other integrative functions, such as those needed for multi-sensory processing and spatial navigation, include the medial and lateral geniculate nuclei, and entorhinal cortex.
Correlations between cortical cholinergic deficits with both global and domain specific measures of cognitive impairments in PD also point to the importance of examining the impacts of CNS cholinergic system deficits via systems level perspectives. These data suggest that cognitive deficits in mild to moderate PD are related to dysfunction of higher order cognitive control networks coordinating the function of domain specific networks. In the context of recent data indicating that the BFCC is constituted by clusters of neurons innervating interconnected cortical fields, it is plausible that early to moderate PD is characterized by preferential degeneration and/or dysfunction of sub-clusters of BFCC neurons innervating the frontal cortical nodes of the salience and COTC cognitive control networks.
It is plausible also that during progression of PD, declining cholinergic denervation of posterior cortices becomes more impactful, resulting in additional cognitive deficits. Declining visuospatial dysfunction with deficient spatial navigation, common in advancing PD, is a plausible candidate for an important clinical feature driven by progressing neocortical cholinergic denervation. Retrosplenial cortex (RSC) is a neocortical hub critical for spatial memory and navigation. Experimental data indicates that cholinergic neurotransmission within the RSC is critical for normal spatial navigation (Anzalone et al., 2009). RSC integrates spatial information from the hippocampal formation and anterior thalamus, and receives a unique pattern of BFCC afferents (Brennan et al., 2020; Solari and Hangya, 2018). Disrupted BFCC signaling is likely to impede integration of spatial information in RSC and could contribute to impaired spatial navigation in PD. Functions centered on the RSC would also be disrupted by impaired anterior thalamic function secondary to loss of PPN-LDT cholinergic afferents.
The examples of disrupted attentional-motor integration related to PD-gait balance problems, the potential role of neocortical cholinergic denervation in disrupting cognitive control networks, and more speculatively, the roles of degenerating cholinergic projections in RSC-related functions, all point to the importance of investigating the consequences of CNS cholinergic systems dysfunction-degeneration in the context of our growing circuit-systems level understanding of brain functions (Fig. 3). This perspective is necessary to properly understand the nature of deficits secondary to cholinergic systems changes in PD and likely necessary to select targets for potential interventions. Cholinergic deficits within different nodes of specific circuits, entorhinal cortex and RSC, for example, might have synergistic effects. It is also possible that cholinergic deficits of other systems are involved in major PD features. Initial analyses implicate cerebellar cortical cholinergic deficits, presumably reflecting MVN pathology, in the pathophysiology of tremor in PD (Bohnen et al., 2021; Seidel et al., 2015). This result is consistent with a substantial body of data pointing to cerebellar dysfunction in PD tremor (Madelein van der Stouwe et al., 2020).
FIG. 3.
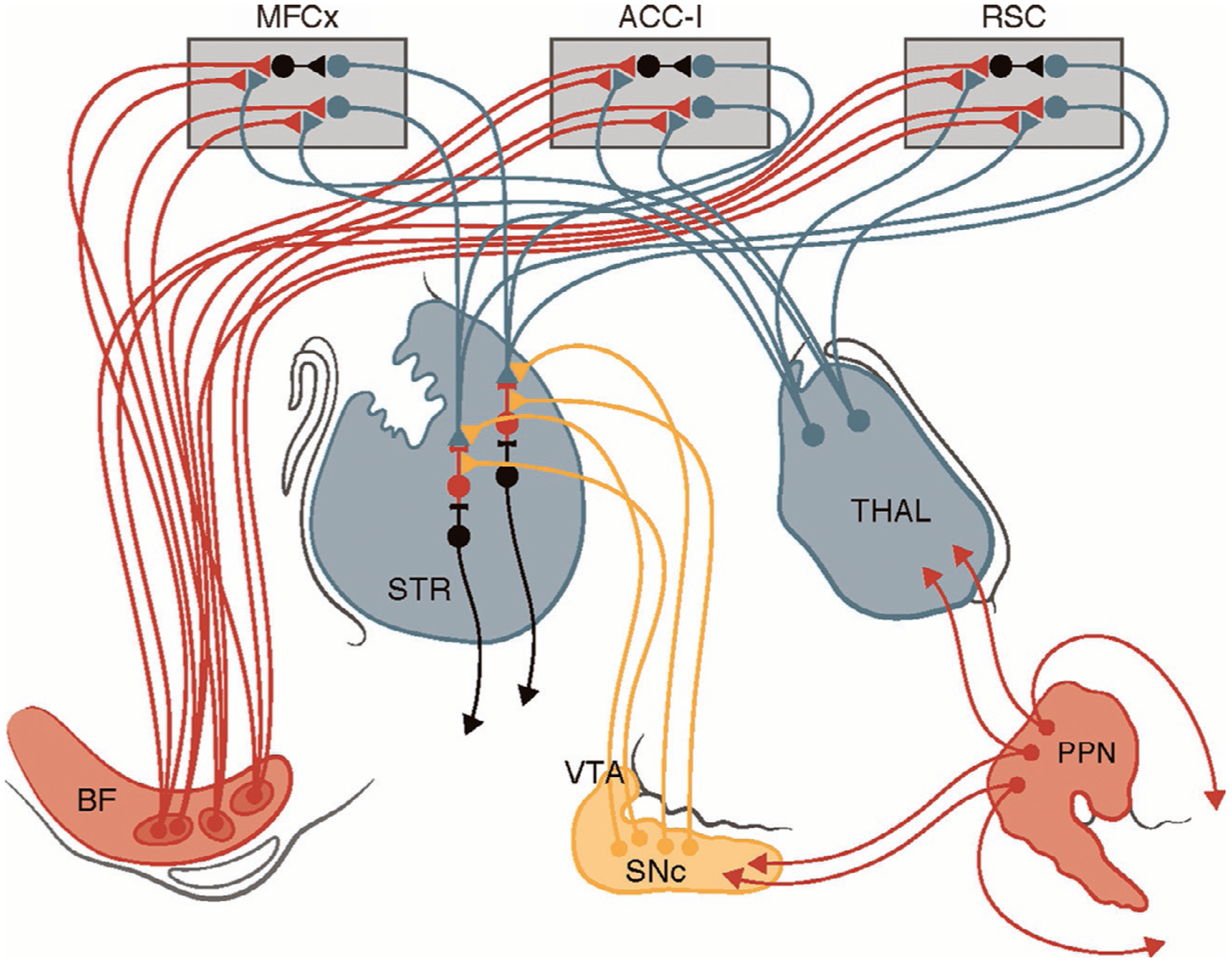
Attentional-motor interfaces: Schematic of major brain cholinergic projection systems, BFCC and PPN-LDT, and their interactions with important cortical and subcortical targets. Convergence-integration of attentional and motor information at SChIs. Important cholinergic inputs to attentional and cognitive control circuit nodes in MFCx, ACC-I, RSC, and THAL. BF = basal forebrain, Str = striatum, MFCx = medial frontal cortex, ACC-I = anterior cingulate cortex–insular cortex, RSC = retrosplenial cortex, THAL = thalamus, VTA = ventral tegmental area, SN = substantia nigra, PPN = pedunculopontine-laterodorsotegmental nucleus.
The FEOBV PET study of early PD by van der Zee et al. (2022) suggests that early PD is characterized by upregulation of cholinergic systems activity. This result is consistent with FEOBV and PMP PET data suggesting that upregulation occurs during the PD prodrome, raising the possibility that cholinergic system compensation for deficits caused by impaired nigrostriatal dopaminergic signaling is an important feature of prodromal to early PD. This concept is supported by the experimental data indicating that compensation, and its subsequent decline with loss of BFCC neurons, plays an important role in PD gait-balance problems (Kucinski et al., 2013). These findings suggest a model in which transition from normal cognition to MCI parallels and might be driven by relative normalization and subsequent decline of cholinergic systems activity. This scenario considerably modifies and extends the Dual Syndrome model. In a modified model, PD patients, and likely some individuals in the PD prodrome, experience some cognitive deficits secondary to striatal dopaminergic deficits causing dysfunction of frontostriatal loops. These deficits are partially remediated by increased activity of cholinergic systems but as these systems fail, the full consequences of nigrostriatal dopamine deficiency are unmasked. Likely superimposed on, and interacting with, the unmasked nigrostriatal dopaminergic deficits are additional impairments related primarily to cholinergic systems degeneration causing dysfunctions of cognitive control networks. With disease progression, and as suggested by the original Dual Syndrome model, posterior neocortical cholinergic deficits add to burden of cognitive dysfunctions in PD. In many PD subjects, the combination of nigrostriatal dopaminergic deficits and cholinergic system deficits may underly MCI and mild dementia, which often overlaps with MCI. In later disease, the superimposed effects of diffusely distributed synucleinopathy drives further cognitive declines. Given heterogeneity of cholinergic systems deficits in PD, it plausible that varying cholinergic systems deficits are major contributors to the considerable heterogeneity found in the clinical manifestations, particularly cognitive and gait-balance impairments, and progression of PD.
The important roles of cholinergic system deficits in important clinical features of PD suggests avenues for novel pharmacologic interventions. The wide variety of ACh receptors, both metabotropic and ionotropic, are potential targets for interventions to palliate important DRT-refractory features of PD. The available agents for augmenting cholinergic neurotransmission, AChase inhibitors, are non-specific and in some cases, have only modest effects on CNS AChase activity (Bohnen et al., 2005). These factors likely account for their relatively modest effects on cognitive impairments and little effect on gait-balance impairments (Mancini et al., 2019; Seppi et al., 2019). Both preclinical and clinical experiments suggests that subtype specific agonists are worth pursuing for treatment of these DRT-refractory features of PD (Albin et al., 2021; Koshy Cherian et al., 2019; Kucinski and Sarter, 2021).
Acknowledgments
The authors acknowledge support from P50NS123067, the Parkinson’s Foundation, the Michael J. Fox Foundation, and the W. Garfield Weston Foundations’ Weston Brain Institute. We thank our research participants in the United States and the Netherlands.
References
- Agid Y, Cervera P, Hirsch E, et al. , 1989. Biochemistry of Parkinson’s disease 28 years later: a critical review. Mov. Disord 4, S126–S144. [DOI] [PubMed] [Google Scholar]
- Albin RL, Bohnen NI, Muller MLTM, et al. , 2018b. Regional vesicular acetylcholine transporter distribution in human brain: a [(18)F]fluoroethoxybenzovesamicol positron emission tomography study. J. Comp. Neurol 526, 2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Müller MLTM, Bohnen NI, et al. , 2021. α4β2* nicotinic cholinergic receptor target engagement in Parkinson disease gait-balance disorders. Ann. Neurol 90, 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Surmeier DJ, Tubert C, et al. , 2018a. Targeting the pedunculopontine nucleus in Parkinson’s disease: time to go back to the drawing board. Mov. Disord 33, 1871–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone S, Roland J, Vogt B, Savage L, 2009. Acetylcholine efflux from retrosplenial areas and hippocampal sectors during maze exploration. Behav. Brain Res 201, 272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila C, Kucinski A, Sarter M, 2020. Complex movement control in a rat model of parkinsonian falls: bidirectional control by striatal cholinergic interneurons. J. Neurosci 40, 6049–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger EC, Ananth M, Talmage DA, Role LW, 2016. Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91, 1199–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmack NH, Baughman RW, Eckenstein FP, 1992. Cholinergic innervation of the cerebellum of rat, rabbit, cat, and monkey as revealed by choline acetyltransferase activity and immunohistochemistry. J. Comp. Neurol 317, 233–249. [DOI] [PubMed] [Google Scholar]
- Barrett MJ, Sperling SA, Blair JC, et al. , 2019. Lower volume, more impairment: reduced cholinergic basal forebrain grey matter density is associated with impaired cognition in Parkinson disease. J. Neurol. Neurosurg. Psychiatr 90, 1251–1256. [DOI] [PubMed] [Google Scholar]
- Bedard MA, Aghourian M, Legault-Denis C, et al. , 2019. Brain cholinergic alterations in idiopathic REM sleep behaviour disorder: a PET imaging study with 18F-FEOBV. Sleep Med. 58, 35–41. [DOI] [PubMed] [Google Scholar]
- Bernacer J, Prensa L, Gimenez-Amaya JM, 2007. Cholinergic interneurons are differentially distributed in the human striatum. PLoS One 2, e1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Frey KA, Studenski S, et al. , 2013. Gait speed in Parkinson disease correlates with cholinergic degeneration. Neurology 81, 1611–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kanel P, Koeppe RA, et al. , 2021. Regional cerebral cholinergic nerve terminal integrity and cardinal motor features in Parkinson’s disease. Brain Commun. 3, fcab109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kanel P, Zhou Z, et al. , 2019. Cholinergic system changes of falls and freezing of gait in Parkinson’s disease. Ann. Neurol 85, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Hendrickson R, et al. , 2005. Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatr 76, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Hendrickson R, et al. , 2006. Cognitive correlates of cortical cholinergic denervation in Parkinson’s disease and parkinsonian dementia. J. Neurol 253, 242–247. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Kaufer DI, Ivanco LS, Lopresti B, et al. , 2003. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch. Neurol 60, 45–1748. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Müller ML, Koeppe RA, et al. , 2009. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 73, 1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Müller ML, Kotagal V, et al. , 2012. Heterogeneity of cholinergic denervation in Parkinson’s disease without dementia. J. Cereb. Blood. Flow. Metab 32, 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, et al. , 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Brennan EKW, Sudhakar SK, Jedrasiak-Cape I, John TT, Ahmed OJ, 2020. Hyperexcitable neurons enable precise and persistent information encoding in the superficial retrosplenial cortex. Cell Rep. 30, 1598–1612.e8. [DOI] [PubMed] [Google Scholar]
- Brimblecombe KR, Threlfell S, Dautan D, et al. , 2018. Targeted activation of cholinergic interneurons accounts for the modulation of dopamine by striatal nicotinic receptors. eNeuro 5. ENEURO.0397–17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candy JM, Perry EK, Perry RH, et al. , 1986. The current status of the cortical cholinergic system in Alzheimer’s disease and Parkinson’s disease. Prog. Brain Res 70, 105–132. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Taly A, 2008. Nicotinic receptors, allosteric proteins and medicine. Trends Mol. Med 14, 93–102. [DOI] [PubMed] [Google Scholar]
- Choi SH, Jung TM, Lee JE, et al. , 2012. Volumetric analysis of the substantia innominata in patients with Parkinson’s disease according to cognitive status. Neurobiol. Aging 33, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Craig CE, Jenkinson NJ, Brittain JS, et al. , 2020. Pedunculopontine nucleus microstructure predicts postural and gait symptoms in Parkinson’s disease. Mov. Disord 35, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Dalrymple WA, Huss DS, Blair J, et al. , 2021. Cholinergic nucleus 4 atrophy and gait impairment in Parkinson’s disease. J. Neurol 268, 95–101. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, et al. , 2002. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol 51, 145–155. [DOI] [PubMed] [Google Scholar]
- Fling BW, Cohen RG, Mancini M, et al. , 2013. Asymmetric pedunculopontine network connectivity in parkinsonian patients with freezing of gait. Brain 136, 2405–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, 1992. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15, 133–139. [DOI] [PubMed] [Google Scholar]
- Gielow MR, Zaborszky L, 2017. The input-output relationship of the cholinergic basal forebrain. Cell Rep. 18, 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombkoto P, Gielow M, Varsanyi P, Chavez C, Zaborszky L, et al. , 2021. Contribution of the basal forebrain to corticocortical network interactions. Brain Struct. Funct 226, 1803–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales KK, Smith Y, 2015. Cholinergic interneurons in the dorsal and ventral striatum: anatomical and functional considerations in normal and diseased conditions. Ann. N. Y. Acad. Sci 1349, 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Labrador-Espinosa MA, Jesús S, et al. , 2021. In vivo cholinergic basal forebrain degeneration and cognition in Parkinson’s disease: imaging results from the COPPADIS study. Parkinsonism Relat. Disord 88, 68–75. [DOI] [PubMed] [Google Scholar]
- Gut NK, Mena-Segovia J, 2019. Dichotomy between motor and cognitive functions of midbrain cholinergic neurons. Neurobiol. Dis 128, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut NK, Winn P, 2016. The pedunculopontine tegmental nucleus-A functional hypothesis from the comparative literature. Mov. Disord 3, 615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Kastner S, 2017. Thalamic functions in distributed cognitive control. Nat. Neurosci 20, 1669–1679. [DOI] [PubMed] [Google Scholar]
- Hall H, Reyes S, Landeck N, et al. , 2014. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 137, 2493–2508. [DOI] [PubMed] [Google Scholar]
- Heiland B, Greenfield SA, 1999. Rat locomotion and release of acetylcholinesterase. Pharmacol. Biochem. Behav 62, 81–87. [DOI] [PubMed] [Google Scholar]
- Hilker R, Thomas AV, Klein JC, et al. , 2005. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 65, 1716–1722. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F, 1987. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc. Natl. Acad. Sci. U. S. A 84, 5976–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Blot FGC, Wu B, et al. , 2018. The basal interstitial nucleus (BIN) of the cerebellum provides diffuse ascending inhibitory input to the floccular granule cell layer. J. Comp. Neurol 526, 2231–2256. [DOI] [PubMed] [Google Scholar]
- Karachi C, Grabli D, Bernard FA, et al. , 2010. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J. Clin. Invest 120, 2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW, 2013. Cognitive impairment in Parkinson’s disease: the dual syndrome hypothesis. Neurodegener. Dis 11, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbourn MR, Butch ER, Desmond T, et al. , 2008. In vivo [11C]dihydrotetrabenazine binding in rat striatum: sensitivity to dopamine concentrations. Nucl. Med. Biol 37, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Bohnen NI, Müller MLTM, Lustig C, 2019b. Compensatory dopaminergic-cholinergic interactions in conflict processing: evidence from patients with Parkinson’s disease. Neuroimage 190, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Müller MLTM, Bohnen NI, Sarter M, Lustig C, 2017. Thalamic cholinergic innervation makes a specific bottom-up contribution to signal detection: evidence from Parkinson’s disease patients with defined cholinergic losses. Neuroimage 149, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Müller MLTM, Bohnen NI, Sarter M, Lustig C, 2019a. The cortical cholinergic system contributes to the top-down control of distraction: evidence from patients with Parkinson’s disease. Neuroimage 190, 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy Cherian A, Kucinski A, Wu R, de Jong IEM, Sarter M, 2019. Co-treatment with rivastigmine and idalopirdine reduces the propensity for falls in a rat model of falls in Parkinson’s disease. Psychopharmacology (Berl) 236, 1701–1715. [DOI] [PubMed] [Google Scholar]
- Kucinski A, Paolone G, Bradshaw M, Albin RL, Sarter M, 2013. Modeling fall propensity in Parkinson’s disease: deficits in the attentional control of complex movements in rats with cortical-cholinergic and striatal-dopaminergic deafferentation. J. Neurosci 33, 16522–16539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucinski A, Sarter M, 2021. Reduction of falls in a rat model of PD falls by the M1 PAM TAK-071. Psychopharmacology (Berl) 238, 1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl DE, Minoshima S, Fessler JA, et al. , 1996. In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann. Neurol 40, 399–410. [DOI] [PubMed] [Google Scholar]
- Legault-Denis C, Aghourian M, Rosa-Neto P, et al. , 2021. Normal cognition in Parkinson’s disease may involve hippocampal cholinergic compensation: a PET imaging study with [18F]-FEOBV. Parkinsonism Relat. Disord 91, 162–166. [DOI] [PubMed] [Google Scholar]
- Liu AK, Chang RC, Pearce RK, Gentleman SM, 2015. Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer’s and Parkinson’s disease. Acta Neuropathol. 129, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Luo Z, Gu J, et al. , 2020. The impact of dopamine D(2)-like agonist/antagonist on [(18)F]VAT PET measurement of VAChT in the brain of nonhuman primates. Eur. J. Pharm. Sci 143, 105152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Wile DJ, Fu JF, Valerio J, et al. , 2018. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: a cross-sectional PET study. Lancet Neurol. 17, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madelein van der Stouwe AM, Nieuwhof F, Helmich RC, 2020. Tremor pathophysiology: lessons from neuroimaging. Curr. Opin. Neurol 2020 (33), 474–481. [DOI] [PubMed] [Google Scholar]
- Mancini M, Chung K, Zajack A, et al. , 2019. Effects of augmenting cholinergic neurotransmission on balance in Parkinson’s disease. Parkinsonism Rel. Disord 69, 40–47. [DOI] [PubMed] [Google Scholar]
- Martinez-Gonzalez C, Bolam JP, Mena-Segovia J, 2011. Topographical organization of the pedunculopontine nucleus. Front. Neuroanat 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamales M, Skrbis Z, Hatch RJ, et al. , 2016. Aging-related dysfunction of striatal cholinergic interneurons produces conflict in action selection. Neuron 90, 362–373. [DOI] [PubMed] [Google Scholar]
- Mena-Segovia J, Bolam JP, 2017. Rethinking the pedunculopontine nucleus: from cellular organization to function. Neuron 94, 7–18. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Geula C, Bothwell MA, Hersh LB, 1989. Human reticular formation: cholinergic neurons of the pedunculopontine and laterodorsal tegmental nuclei and some cytochemical comparisons to forebrain cholinergic neurons. J. Comp. Neurol 283, 611–633. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Hersh LB, Mash DC, Geula C, 1992. Differential cholinergic innervation within functional subdivisions of the human cerebral cortex: a choline acetyltransferase study. J. Comp. Neurol 318, 316–328. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH, 1983. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol 214, 170–197. [DOI] [PubMed] [Google Scholar]
- Müller ML, Albin RL, Kotagal V, et al. , 2013. Thalamic cholinergic innervation and postural sensory integration function in Parkinson’s disease. Brain 136, 3282–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill KG, 2019. Presynaptic regulation of dopamine release: role of the DAT and VMAT2 transporters. Neurochem. Int 122, 94–105. [DOI] [PubMed] [Google Scholar]
- Paterson D, Nordberg A, 2000. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol 61, 75–111. [DOI] [PubMed] [Google Scholar]
- Pienaar IS, Elson JL, Racca C, et al. , 2013. Mitochondrial abnormality associates with type-specific neuronal loss and cell morphology changes in the pedunculopontine nucleus in Parkinson disease. Am. J. Pathol 183, 1826–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienaar IS, Vernon A, Winn P, 2017. The cellular diversity of the pedunculopontine nucleus: relevance to behavior in health and aspects of Parkinson’s disease. Neuroscientist 23, 415–431. [DOI] [PubMed] [Google Scholar]
- Ray NJ, Bradburn S, Murgatroyd C, et al. , 2018. In vivo cholinergic basal forebrain atrophy predicts cognitive decline in de novo Parkinson’s disease. Brain 141, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochester L, Yarnall AJ, Baker MR, et al. , 2012. Cholinergic dysfunction contributes to gait disturbance in early Parkinson’s disease. Brain 135, 2779–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Albin RL, Kucinski A, Lustig C, 2014. Where attention falls: increased risk of falls from the converging impact of cortical cholinergic and midbrain dopamine loss on striatal function. Exp. Neurol 25, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Avila C, Kucinski A, Donovan E, 2021. Make a left turn: cortico-striatal circuitry mediating the attentional control of complex movements. Mov. Disord 36, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C, 2019. Cholinergic double duty: cue detection and attentional control. Curr. Opin. Psychol 29, 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C, 2020. Forebrain cholinergic signaling: wired and phasic, not tonic, and causing behavior. J. Neurosci 40, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildt A, de Vries EJF, Willemsen ATM, et al. , 2020. Effect of dopamine D2 receptor antagonists on [18F]-FEOBV binding. Mol. Pharm 17, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, Zaborszky L, 2021. Spatial topography of the basal forebrain cholinergic projections: organization and vulnerability to degeneration. Handb. Clin. Neurol 179, 159–173. [DOI] [PubMed] [Google Scholar]
- Schulz J, Pagano G, Fernandez Bonfante JG, et al. , 2018. Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson’s disease. Brain 141, 1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweder PM, Hansen PC, Green AL, et al. , 2010. Connectivity of the pedunculopontine nucleus in parkinsonian freezing of gait. Neuroreport 21, 914–916. [DOI] [PubMed] [Google Scholar]
- Seidel K, Mahlke J, Siswanto S, et al. , 2015. The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol. 25, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppi K, Ray Chaudhuri K, Coelho M, et al. , 2019. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov. Disord 34, 180–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Hirano S, Shinotoh H, et al. , 2009. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73, 273–278. [DOI] [PubMed] [Google Scholar]
- Shinotoh H, Namba H, Yamaguchi M, et al. , 1999. Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson’s disease and progressive supranuclear palsy. Ann. Neurol 46, 62–69. [PubMed] [Google Scholar]
- Snijders AH, Leunissen I, Bakker M, Overeem S, Helmich RC, Bloem BR, et al. , 2011. Gait-related cerebral alterations in patients with Parkinson’s disease with freezing of gait. Brain 134, 59–72. [DOI] [PubMed] [Google Scholar]
- Solari N, Hangya B, 2018. Cholinergic modulation of spatial learning, memory and navigation. Eur. J. Neurosci 48, 2199–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernández R, Nandhagopal R, et al. , 2010. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov. Disord 25, 2717–2723. [DOI] [PubMed] [Google Scholar]
- Sugimura T, Saito Y, 2021. Distinct proportions of cholinergic neurons in the rat prepositus hypoglossi nucleus according to their cerebellar projection targets. J. Comp. Neurol 529, 1541–1552. [DOI] [PubMed] [Google Scholar]
- Tong J, Wilson AA, Boileau I, Houle S, Kish SJ, 2008. Dopamine modulating drugs influence striatal (+)-[11C]DTBZ binding in rats: VMAT2 binding is sensitive to changes in vesicular dopamine concentration. Synapse 62, 873–876. [DOI] [PubMed] [Google Scholar]
- van der Zee S, Müller MLTM, Kanel P, van Laar T, Bohnen NI, 2021. Cholinergic denervation patterns across cognitive domains in Parkinson’s disease. Mov. Disord 36, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee S, Kanel P, Gerritsen MJJ, et al. , 2022. Altered brain cholinergic innervation in newly diagnosed Parkinson’s disease patients with and without cognitive impairment. Mov. Disord 10.1002/mds.28913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Gray CH, Evans JR, Goris A, et al. , 2009. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 132, 2958–2969. [DOI] [PubMed] [Google Scholar]
- Wu H, Williams J, Nathans J, 2014. Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife 3, e02444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J, Cho JW, Lee WY, et al. , 2012. Diffusion tensor imaging of freezing of gait in patients with white matter changes. Mov. Disord 27, 760–764. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Csordas A, Mosca K, et al. , 2015. Neurons in the basal forebrain project to the cortex in a complex topographic organization that reflects corticocortical connectivity patterns: an experimental study based on retrograde tracing and 3D reconstruction. Cereb. Cortex 25, 118–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Záborszky L, Gombkoto P, Varsanyi P, et al. , 2018. Specific basal forebrain-cortical cholinergic circuits coordinate cognitive operations. J. Neurosci 38, 9446–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Hoemke L, Mohlberg H, et al. , 2008. Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. Neuroimage 42, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhou P, Yuan T, 2016. The cholinergic system in the cerebellum: from structure to function. Rev. Neurosci 27, 769–776. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Jankel WR, Hedreen JC, et al. , 1989. The pedunculopontine nucleus in Parkinson’s disease. Ann. Neurol 26, 41–46. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Whitehouse PJ, Casanova MF, et al. , 1987. Loss of pedunculopontine neurons in progressive supranuclear palsy. Ann. Neurol 22, 18–25. [DOI] [PubMed] [Google Scholar]
- Zwergal A, La Fougère C, Lorenzl S, et al. , 2013. Functional disturbance of the locomotor network in progressive supranuclear palsy. Neurology 80, 634–641. [DOI] [PubMed] [Google Scholar]