

Summary
The photoreceptor rhodopsin (Rho) becomes active when a tethered inverse agonist ligand (11CR) is photoconverted to an agonist (ATR). The ligand-binding pocket of inactive rhodopsin is completely enclosed, whereas active rhodopsin displays pores accessible from the lipid bilayer. Stabilization of active rhodopsin impedes 11CR binding and photoreceptor dark adaptation. Here, we used genetic code expansion and bioorthogonal labeling to engineer Rho mutants that serve as FRET sensors for measuring 11CR binding kinetics and energetics. We found that mutations that alter a channel between transmembrane helices 5 and 6 (TM5/6) dramatically affect 11CR binding kinetics but not agonist release kinetics. Our data provide direct experimental evidence for 11CR entry between TM5/6 in Rho that involves dynamic allosteric control of the ligand entry channel. Our findings provide a conceptual framework for understanding the function of G protein-coupled receptors with hydrophobic ligands that are hypothesized to enter their binding pockets through transmembrane pores.
Subject areas: Cellular neuroscience, Cell biology, Biophysics
Graphical abstract
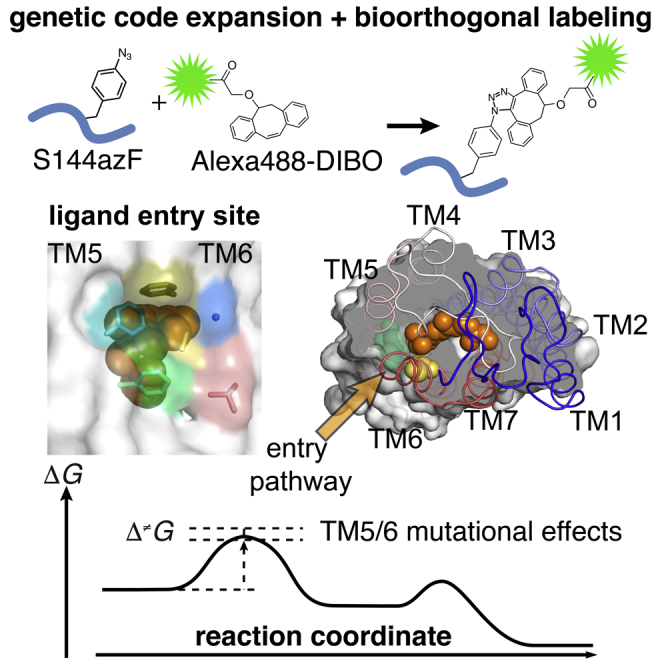
Highlights
-
•
Site-specific bioorthogonal labeling enables a FRET-based sensor for ligand binding
-
•
11-cis-retinal enters the ligand-binding channel of opsin through the lipid bilayer
-
•
The retinal entry site is located between TM5 and TM6
-
•
Retinal entry involves dynamic allosteric control of the ligand entry channel
Cellular neuroscience; Cell biology; Biophysics
Introduction
The visual photoreceptor rhodopsin (Rho) is G protein-coupled receptor (GPCR) that consists of an apoprotein opsin and its native ligand 11-cis-retinal (11CR). 11CR is covalently linked to opsin via a protonated Schiff base (PSB) bond (Palczewski and Kiser, 2020; Wald, 1968). 11CR is a potent inverse agonist that stabilizes Rho in its inactive state with extremely low basal activity. Upon photoactivation, 11CR absorbs a photon to isomerize to all-trans-retinal (ATR), triggering a series of conformational changes to produce the active Meta-II Rho state capable of activating transducin. The Schiff base linkage then hydrolyzes, allowing ATR to dissociate from the ligand-binding site. In a process known as “regeneration,” 11CR enters the empty ligand-binding channel of opsin to form a transient opsin-11CR complex. The formation of a PSB completes the regeneration of Rho.
Many high-resolution structures for GPCRs that bind diffusible ligands show openings connecting the extracellular surface to their orthosteric ligand-binding site. However, the crystal structures of opsin do not reveal a direct channel from the extracellular environment to the 11CR binding site. Instead, high-resolution structures of Meta-II opsin (Choe et al., 2011; Standfuss et al., 2011) and ligand-free opsin (Scheerer et al., 2008) reveal openings in the transmembrane (TM) helical bundle at both ends of the ligand-binding pocket specific to the active conformation, suggesting that ATR exits and 11CR enters the pocket using these two openings in the TM region (Hildebrand et al., 2009). Moreover, the high partition coefficient of retinal into the lipid bilayer indicates that retinal predominantly exists in the lipid-solubilized form (Frederiksen et al., 2012). An earlier molecular dynamics simulation also suggested an intramembranous pathway (Wang and Duan, 2007). However, the “active conformation” model is incongruent with certain experimental observations (Schafer and Farrens, 2015). On the other hand, recent structures of opsin bound with non-retinal small molecules reveal a channel open toward the extracellular side when the receptor was stabilized by a bulky moiety protruding between the extracellular end of TM5 and TM6 (Mattle et al., 2018). Nonetheless, it is unclear whether 11CR can induce the formation of a similar channel to open toward the extracellular surface.
Here, we employ an engineered opsin that serves as a sensitive fluorescence sensor (Tian et al., 2014) for 11CR binding kinetics to identify the 11CR entry pathway. We examine how mutations designed to alter the ligand-binding channel affect the 11CR entry rate and ATR exit rate. Mutations in the channel nearest to the β-ionone end of 11CR (TM5/6) caused a ∼100-fold variation in the 11CR entry kinetics but only a 3-fold variation in the ATR release kinetics. We combined one of three TM5 mutations with I189P, which is situated in the middle of the channel and defines a key difference between rod and cone photoreceptors. The energetic additivity of the double mutant suggests that the mutations in the TM5/6 channel exert localized effects on 11CR entry. We contend that our data and several other lines of evidence fit best with a previously proposed model (Schafer and Farrens, 2015) in which the opsin conformer permissive for 11CR binding (Ops∗∗) resembles that of the inactive state, and the ligand binding channel needs to be dynamically adjusted to the incoming ligand. Our data provide direct experimental evidence for 11CR entry through a pore between TM5/6 and suggest an “allosteric” mechanism in ligand binding in other GPCRs as well.
Results
FRET-based assays for retinal entry kinetics
Based on the dark-state Rho and Meta-II structures (Figure 1), we hypothesized that the residues close to the β-ionone ring end of the bound retinal might play a role in 11CR entry or ATR release. To avoid severely disrupting the overall folding of the TM bundle, we chose to mutate residues (F208, F212, L266, A269, G270, and F273) whose side chains do not protrude into the TM interface. We heterologously expressed these mutants in HEK cells, immunopurified the receptors, reconstituted them into lipidic bicelles, and measured the 11CR entry kinetics as well as the ATR release kinetics using the fluorescence resonance energy transfer (FRET) assays as described previously (Figure 2A) (Farrens and Khorana, 1995; Schafer et al., 2016; Tian et al., 2017a, 2017b). Briefly, 11CR entry kinetics was measured by monitoring the quenching of either the intrinsic tryptophan (Trp) fluorescence (Figures 2B–2E) or an extrinsic fluorescent reporter, Alexa488 (Figures 2B–2F). ATR release kinetics was measured using a process that was essentially the inverse of Trp fluorescence quenching (Farrens and Khorana, 1995). For the Alexa488-based assays, we first substituted a residue in the second intracellular loop of Rho (S144) with an unnatural amino acid, p-azido-L-phenylalanine (azF); then we attached Alexa488 specifically to this site with a bioorthogonal chemistry (Figures 2C and S1) (Tian et al., 2014, 2017a). These two FRET-assays are functionally complementary: the Trp-based assay reports formation and dissociation of the opsin-retinal complex, whereas the Alexa488-based assay reports the formation of a mature pigment with the PSB characteristic for dark-state Rho (Figure 2D). Previously, we have shown that S144-Alexa488 Rho exhibits nearly identical retinal binding kinetics and energetics as wt Rho (Tian et al., 2017a), and that the regenerated Rho can be repeatedly photoactivated, confirming the functional integrity of this mutant (Tian et al., 2014). Although the Alexa488-based assay requires an additional modification step, Alexa488 fluorescence falls into a clean window of retinal absorbance; therefore, it is not limited by the inner filter effect as the Trp fluorescence (Tian et al., 2017a). The Alexa488-based assay is more suitable for measuring the retinal entry kinetics for “slow” mutants, for which much higher retinal concentrations are required to drive regeneration to completion. In this study, the Alexa488-based assay enabled us to analyze the binding kinetics of slow mutants at lower temperatures.
Figure 1.

Comparing the transmembrane surfaces and ligand-binding channel of the inactive Rho and active Meta-II Rho
(A) The side-view of 11CR-bound, inactive Rho (PDB: 1U19) and ATR-bound, and active Meta-II Rho (PDB: 3PXO). Retinal is shown as spheres in orange and the stick and the surface representations of the mutated residues are highlighted in colors.
(B) Photoactivation causes the cis-trans isomerization of retinal leading to serial conformational changes, including the emergence of two openings in Meta-II Rho. The TM5/6 opening is designated as Opening A and TM1/7 opening as Opening B (retinal, orange; F208, yellow; F212, lime green).
Figure 2.

Engineering FRET sensors for measuring retinal entry and release kinetics
(A) The assay for measuring retinal entry and release kinetics. The purified Rho is reconstituted in lipidic bicelles.
(B) Design of FRET sensors utilizing the intrinsic Trp fluorescence and extrinsic fluorophore reporter. The location of five Trp residues (blue) and S144 (green) relative to the 11-cis-retinylidene group (orange) in Rho (PDB: 1U19).
(C) Bioorthogonal, site-specific labeling of S144 with strain-promoted alkyne-azide cycloaddition (SpAAC).
(D) The energy transfer scheme for Trp-based and Alexa 488-based assay. Note that energy transfer occurs between Trp and dark-state as well as Meta-II Rho, but is specific to dark-state Rho in the case of Alexa 488.
(E) Representative trace of Trp-based FRET assay.
(F) Representative trace of Alexa 488-based FRET assay.
TM5/6 mutations locally alter retinal entry kinetics
We tested a panel of 24 mutants (Figures 3A and 3B, Table S1). For Rho carrying additional mutations, the 11CR entry kinetics measurements obtained from the Trp-based and Alexa488-based assays were comparable (Figures 3A and 3B, Table S1). We found that mutations at the 11CR putative entry site caused a ∼100-fold variation in the 11CR entry kinetics (G270L, 4.2 ± 0.4; F212W: 0.043 ± 0.022, data measured from Trp-based assay and normalized to the wt Rho), but only a modest 3-fold variation in ATR exit kinetics (F212A 2.82 ± 1.0; A269F: 0.91 ± 0.07). This 3-fold variation in ATR release kinetics resulting from TM5/6 mutations is comparable to an earlier report on zebrafish Rho (Morrow and Chang, 2015). The observation that the 11CR entry rate is much more affected than the ATR release rate suggested that these TM5/6 sites might be an integral part of the 11CR entry site.
Figure 3.

TM5/6 mutations primarily alter 11CR entry kinetics but not ATR release kinetics
The 11CR entry kinetics is measured with Trp-based (kon, Trp) and/or Alexa 488-based (kon, Alexa 488) FRET assays, and ATR release kinetics (koff, Trp) measured with Trp-based FRET assay.
(A) Data are grouped based on the site of mutations (mean ± STD).
(B) Data from (A) shown in descending order based on kon, Trp.
Next, we asked whether the observed mutational effects were because of short-range or potential longer-range disruptions to the ligand-binding channel. A straightforward test was performed to examine whether or not the mutations of residues on the opposite end of the ligand-binding channel at the pore between TM1 and TM7 behave in the mirror direction (i.e., whether mutating these sites cause a greater variation in the ATR release rate than in the 11CR binding rate). We tested T94I (Figure 4A) and found a moderate decrease in the 11CR entry kinetics (0.80 ± 0.04) and a significant decrease on the ATR release kinetics (0.17 ± 0.02) (Table S1). However, a caveat here is that these TM1/7 residues being relatively juxtaposed to the SB bond with retinal, may simultaneously affect the Schiff base chemistry and the folding of the putative ligand-binding channel (Janz and Farrens, 2004). Alternatively, the hydrophobic substitution T94I could lead to a reduction of the local concentration of water molecules available for the SB hydrolysis reaction, and it would be difficult to dissect their respective contributions to the overall mutational effect.
Figure 4.

Double mutant analysis reveals localized mutational effects
(A) Two TM5 sites (F208, teal; F212, lime green), I189 (magenta), and T94I (forest green) highlighted in Rho (PDB: 1U19).
(B) Mutational effect at site 189. I189P simultaneously increases the 11CR entry and ATR release kinetics (mean ± STD).
(C) The Eyring plot for I189P and I189A mutants. The kinetics (mean ± STD) were measured using the Alexa 488-based assay.
(D) Double logarithmic plot of kon (mean ± STD), Trp of TM5 single mutant versus TM5/I189P double mutants. I189P reduces the activation free energy (Δ‡G) by 1.6 ± 0.3 kcal mol−1. The fitting equation is ln(kon(I189P)) = (0.997 ± 0.093)×ln(kon(I189)) + (2.69 ± 0.55) (R2 = 0.98).
(E) Logarithmic plot of koff (mean ± STD), Trp in TM5 single mutant and TM5/I189P double mutants. I189P reduces the activation free energy (Δ‡G) by 1.6 ± 0.8 kcal mol−1. The fitting equation is ln(kon(I189P)) = (1.09 ± 0.23)×ln(kon(I189)) + (2.6 ± 1.4) (R2 = 0.92).
(F) Summation of ΔΔ‡G in single mutants vs. ΔΔ‡G in the corresponding double mutants.
We reasoned that a better approach would be to test a site that alters the middle segment of the ligand-binding channel. We therefore chose to examine I189, a conserved molecular determinant that distinguishes between rod and cone opsins. In rod receptors, site 189 is invariably an Ile or Val, whereas in cone receptors, a Pro is very highly conserved (Ebrey and Koutalos, 2001). One exception is lamprey rhodopsin, which is an evolutionary intermediate between cones and rods and carries P189 (Yanagawa et al., 2015). In Rho, I189 is located at the end of a β-hairpin formed by the second extracellular loop that functions as the “cap” of the ligand-binding channel (Figure 4A). The I189P mutation in Rho has been reported to increase the Meta-II decay rate (Kuwayama et al., 2002), thereby increasing the regeneration rate (Imai et al., 2005) and elevating the thermal isomerization rate of 11CR to ATR (Yanagawa et al., 2015).
Among the I189 mutants tested here (Figure 4B, Tables S1 and S2), we found that the reduced size of the aliphatic side chain (I189A, I189V) only slightly accelerated 11CR entry kinetics and reduced the ATR release rate. Eyring plots of the kinetics data showed that I189A only made a slight difference to the transition energy (Figure 4C, Table S2). We also observed a decreased thermal stability for I189A at higher temperatures (unpublished data). Substitution of an aliphatic Ile with a polar Thr moderately slowed down 11CR entry and accelerated ATR release. Overall, the effects of the side chain size and polarity are modest. In contrast, we observed that the I189P mutation dramatically increased both 11CR entry and ATR release rate (Figure 4B), which is consistent with previous reports (Imai et al., 2005; Kuwayama et al., 2002). Back-converting the Pro in a chicken green cone opsin to Ile significantly slowed down 11CR binding and ATR release. Eyring analysis indicated that the I189P substitution accelerated 11CR entry by increasing the entropic factor. These data suggest that I189P significantly eases the structural constraints on retinal movement within the channel, possibly through proline’s ability to introduce kinks into the peptide backbone.
It was well established that the summation of the energetic effects of single mutations is nearly equal to the free energy change measured in the corresponding multiple mutants (Dill, 1997; Wells, 1990). Deviation from this simple additivity rule is a strong indicator that 1) the mutated residues are interacting with each other directly or indirectly or 2) the mutation causes a change in mechanism or rate-limiting step. Therefore, we measured the 11CR entry and ATR release rates for three double mutants consisting of a TM5 mutation and I189P. We found that the I189P mutation and TM5/6 mutations on 11CR entry rate were highly energetically additive (Figures 4D–4F). In the natural logarithmic plot of the 11CR entry and ATR release kinetics, the slope was fitted to 1.00 ± 0.09 (R2 = 0.98) and 1.1 ± 0.2 (R2 = 0.92), respectively (Figures 4D and 4E).
We calculated the change of activation free energy (ΔΔ‡G) for these mutants. We found that the summation of ΔΔ‡G for single mutants is close to the ΔΔ‡G of corresponding double mutants (Figure 4F). I189P reduced Δ‡Gon by 1.6 ± 0.3 kcal mol−1 and Δ‡Goff by 1.6 ± 0.8 kcal mol−1. This additivity shows that I189 and the TM5/6 site are noninteracting structural determinants for the retinal-binding channel. Intriguingly, the changes of Δ‡Gon and Δ‡Goff are statistically the same, suggesting that I189P reduces constriction for the retinal ligands to traverse through the ligand-binding pathway in either direction. The lack of synergistic effects between I189P and TM5 mutations suggest that the observed mutational effects at the TM5/6 channel are localized within a short-range. Thus, the most parsimonious explanation for the observed large variation in 11CR entry kinetics is that the TM5/6 channel constitutes the 11CR entry site.
The mutational effects at TM5/6 are dependent on the side-chain polarity
We further analyzed mutations at three TM5/6 sites based on their side chain properties (Figure 5, Tables S3–S5). We tested polar mutations at three sites (F208, F212, and A269). Ser and Tyr differ only from Ala and Phe in having additional hydroxyl groups, and the comparison of Ser/Ala and Tyr/Phe pairs are particularly meaningful as these substitutions involve minimal steric changes apart from the hydroxyl group. We also included Thr, an analogue of Ser. We found that all the polar mutations, except for F208Y, reduced the 11CR entry kinetics (Figure 5A). We could not measure the retinal entry kinetics for A269Y because this mutant simply could not be regenerated even after prolonged incubation with a high concentration of 11CR (15 μm, over 48 h) (i.e., 11CR entry for A269Y is either indefinitely slow or the mutation irreversibly denatures the receptor).
Figure 5.

Comparison of the effects of polar and hydrophobic mutations on 11CR entry kinetics
(A) The effect of polar mutations on 11CR entry kinetics (mean ± STD).
(B) The effect of hydrophobic substitutions on 11CR entry kinetics (mean ± STD). The A269W mutant only expressed at very low level and failed to regenerate.
(C–E) The Eyring plots of the 11CR entry kinetics (mean ± STD) for the following mutants: F208 A/W, F212 A/W, and F273 A/W. The kinetics were measured using the Alexa 488-based assay.
The data for hydrophobic mutations revealed a more complex pattern (Figure 5B). We varied the steric hindrance at these sites by installing a series of hydrophobic substitutions (Ala, Phe, Leu, and Trp). For F212 and A269, 11CR entry kinetics is negatively correlated with the size of the side chain. The F273 mutations are the opposite of that of F212 and A269; therefore, decreasing the side-chain size led to slower retinal binding. For mutations at F208, increasing the side-chain size (F208W) or decreasing it (F208A) both resulted in slower 11CR entry kinetics.
We further measured the temperature dependence of 11CR entry kinetics for six mutants, F208 A/W, F212 A/W, and F273 A/W using the Alexa 488-based 11CR entry assay (Figures 5D–5F). All of the Eyring plots for the wt and the mutants showed a high degree of linearity (R2 = 0.92–0.97). Two mutations, F208W and F212A, caused significant change in the activation enthalpy. F208W increased the activation enthalpy (Δ‡H) by 4.8 ± 3.1 kcal mol−1, whereas F212A reduced it by 3.7 ± 1.6 kcal mol−1. The thermodynamic effects of F273A/W are less pronounced compared with F208A/W and F212A/W.
Discussion
FRET-based assays have been widely used to study intramolecular and intermolecular dynamics. When it comes to studying ligand binding, a challenge is to find the appropriate energy transfer partner with the ligand. In the case of Rho, the intrinsic Trp fluorescence serves as a good reporter for retinal entry, except for the slower mutants. However, in most cases, Trp fluorescence has insufficient spectral overlapping with the ligand. Our bioorthogonal labeling strategy represents an attractive alternative: because the labeling is modular, site-specific, and the fluorophore can be chosen with flexibility, RET-assays can be developed to probe ligand binding in a variety of GPCR-ligand pairs. Moreover, genetic code expansion eliminates the need to generate and validate single-accessible Cys mutant that is often required by the classic site-specific protein labeling methods.
An earlier study on retinal entry kinetics reported nonlocalized mutational effects and did not pinpoint a discrete entry site (Piechnick et al., 2012). The design of the preceding study was limited in three important ways. First, if the side chain of a residue protrudes into the TM domain, mutating this site can disrupt the overall receptor folding. Second, for ATR release, it is well established that hydroxylamine, which cleaves the SB bond, accelerates Meta-II decay in both wt Rho and mutants by about two-orders of magnitude, suggesting that SB hydrolysis could be a rate-limiting step of ATR release. Third, this study used DDM-solubilized Rho carrying a N2C/D282C stabilizing mutation, which appeared to be necessary for Rho regeneration in micelles. We addressed each of these issues in the design of the present study. We chose to focus on residues whose side chains faced outward from the TM region. A mutation affecting SB chemistry is likely to cause a greater change in the apparent ATR release rate than a mutation affecting the channel property. Because residues at TM1/7 are prone to affect both SB chemistry and folding of the channel, the mutational effects at these sites are difficult to interpret. Thus, we focused on the TM5/6 end of the ligand-binding channel and only mutated residues whose side chains face outward. Our bicelle system also provides a more native-like environment to obviate the requirement for an extra disulfide bond. A potential caveat of this lipid-based system is that dissociated lipids may compete with retinal for ligand entry, as suggested by a structural study (Blankenship et al., 2015).
Earlier studies also suggested that 11CR binding with opsin requires an “active” opsin conformation; however, several new experimental observations contradict this “active conformation” model (Hildebrand et al., 2009; Piechnick et al., 2012). First, we have shown that the opsin reconstituted in the lipidic bilayer of bicelles robustly recombines with 11CR 1 h after photoactivation, whereas opsin in DDM failed to bind with 11CR in a temperature-dependent manner (Tian et al., 2017a). An EPR (electron paramagnetic resonance) DEER spectroscopy study also found that the cytoplasmic end of TM6 reset spontaneously within 30 min in nanodiscs but failed to do so in DDM micelles (Van Eps et al., 2017). Second, a constitutively activating mutation M257Y and transducin-mimic peptide fused to the C-terminal tail of Rho, both shifting the equilibrium of the receptor population toward the active state (Deupi et al., 2012; Han et al., 1998; Tsukamoto and Farrens, 2013), selects for ATR rather than 11CR (Schafer and Farrens, 2015). These independent lines of experimental evidence together predict that the permissive opsin conformer for 11CR entry (Ops∗∗) is characterized by TM6 resetting, rather than outward movement of TM6, i.e., it shows greater resemblance to the inactive conformation than to the active conformation.
Our data show that 1) mutating the TM5/6 end of the ligand-binding channel causes a much greater change to the 11CR entry kinetics than to the ATR release kinetics, and 2) these mutational effects on the TM5/6 pore are localized. In contrast, mutations on TM4, which is not part of the ligand binding channel, barely perturbed the retinal entry kinetics (Tian et al., 2015). These observations strongly support an entry site between TM5/6 to allow 11CR to access the ligand-binding channel, and that this entry site roughly co-localizes with the TM5/6 opening observed in the Meta-II Rho.
We initially made the prediction that mutational effects at the putative entry site would be dominated by steric factors (i.e., larger side chains should slow down 11CR entry, whereas smaller side chains should accelerate it). We indeed found two sites, F212 and A269, which conformed to this predication. The consistent trend observed for the polar mutations at F212 and A269 further suggests that these two sites act in concert. On the other hand, we also observed residues F208, F273, and G270L deviate from the predicted pattern. F208A, F208W, and F273A slowed down 11CR entry, whereas F273A and G270L accelerated it. In fact, similar observations were reported for F208A and F273A (Schafer and Farrens, 2015). Experiments designed to modify the size of opening are implicitly based on the assumption that a conformational change occurs before the binding event, and that the ligand stabilizes the permissive, typically high-energy, conformation for binding. The role of “conformational selection” is widely appreciated in Rho (Schafer and Farrens, 2015; Schafer et al., 2016) as well as a wide-range of GPCRs (Latorraca et al., 2017). The “conformational selection” model takes a static view of the ligand binding channel. In the case of Rho regeneration, the underlying assumptions are: 1) the size of the 11CR entry opening is sufficiently large to accommodate the cross section of 11CR, including the β-ionone ring; 2) the size of the opening remains roughly constant over the course of ligand entry; 3) the entry opening must promptly close right after ligand entry to protect the PSB.
Here, we propose an “allosteric opening” model for 11CR entry (Figure 6). The pore opening does not have to be sufficiently large in the very beginning. Rather, this opening is dynamically reformed as 11CR traverses through the channel, possibly accompanied by additional conformational change. Without the inverse agonist 11CR stabilizing the inactive state, the apoprotein opsin alone is likely to be more flexible and dynamic than Rho. Single-molecule fluorescence experiments (Maeda et al., 2018) and electrophysiological recordings (Sato et al., 2019) have confirmed the existence of multiple conformers for opsin (Oprian et al., 1987). This flexibility of the opsin backbone makes it conceivable for 11CR to force open TM5/6. In fact, the notion of dynamically-adjusted ligand-binding channels has been suggested by molecular dynamic simulations on β2 adrenergic receptors (Dror et al., 2011) and human protease-activated receptor 1 (Zhang et al., 2012).
Figure 6.

A model for 11CR entry in opsin
The energetic cost for reforming the opening might underlie the notably high activation barrier (Δ‡Gon = 13.5 kcal mol−1) for 11CR binding with opsin (Tian et al., 2017a). By comparison, Δ‡Gon for high affinity ligands for β2 adrenergic receptors (Rasmussen et al., 2011; Rosenbaum et al., 2007) is lower by 3 kcal mol−1. The extraordinarily high energy barrier may arise from the additional conformational change of the protein backbone required for 11CR entry and the energetic cost for Schiff base formation.
The general trend for polar mutations supports the hypothesis that TM5/6 is the 11CR entry site. In such a scenario, the aldehyde group should precede the polyene chain and the β-ionone ring into the channel. A plausible mechanism is that the carbonyl group of retinal may interact with the substituted hydroxyl group through dipole-dipole interaction or hydrogen bonding to form a transiently stabilized intermediate, resulting in slower binding kinetics. Alternately, these additional polar groups in the low dielectric membrane environment may be forced to hydrogen-bond with nearby polar residues and thus limit the flexibility of the entry site to undergo the necessary conformational change. Importantly, our model rationalizes the counterintuitive mutational effects of F208 A/W, F273 A/W, and G270L. Although steric hindrance is necessarily localized, localized effect is not limited to steric hindrance. A mutation not only changes the packing density in situ but also affects any interaction with nearby residues; for example, W265, a pivotal residue for opsin conformational switch (Standfuss et al., 2011) and the incoming 11CR. As a result, steric hindrance alone may not dominate the network effect of a mutation on 11CR binding kinetics; the altered energetic cost of opening expansion must be taken into account. Additional molecular dynamics simulation may be needed to explore how these mutations alter the energetic landscape of ligand channels.
The “allosteric” model and the “conformational selection” model are not strictly mutually exclusive. To dissect their respective contributions, it is important to elucidate the initial state of the opening for 11CR immediately before the ligand binding process. An entry pore that requires weakening of electron density between TM5 and 6 may arise from the following plausible mechanisms: 1) the opening entirely emerge as a result of the interaction between the incoming 11CR and opsin; 2) a small population of Ops∗∗ preexists in equilibrium (Schafer and Farrens, 2015); 3) the real structure of inactive opsin is in fact different from the apoprotein in the Meta-II structure and the inactive opsin itself contains such an entry site and qualifies as Ops∗∗. Our data cannot formally distinguish among these possibilities. In future studies, NMR should be utilized to elucidate the conformational dynamics of opsin in the course of 11CR entry. In particular, the sites of interest identified in this study could be replaced with NMR-active unnatural amino acids to probe the conformational dynamics (Jones et al., 2010).
Conclusion
Accumulating structural studies of GPCRs reveal a lateral binding pocket that is inaccessible from the extracellular milieu (Szlenk et al., 2019). Although intermembranous ligand binding pathways for hydrophobic ligands appears plausible, current understanding of binding pathways in GPCRs is primarily derived from computational simulations (Alfonso-Prieto et al., 2019; Elisi et al., 2021; Ibrahim and Clark, 2019; Marino and Filzola, 2018; Stanley et al., 2016; Wang and Duan, 2007). Direct experimental evidence on intermembranous ligand entry sites is rare (Bokoch et al., 2018; Piechnick et al., 2012). Here we addressed the question of native ligand (11CR) entry in the visual photoreceptor Rho, a prototypical GPCR. Through the engineering of a sensitive FRET sensor, improved mutagenesis design, and extensive energetic analysis, we were able to show that amino acid residues close to a putative pore between TM5 and TM6 display the expected characteristics of a ligand entry site. Mutating these sites caused a much greater variation in the 11CR entry kinetics compared with the ATR release kinetics. The effects of the side chain substitutions lining the pore were localized. Based on our own data and the existing literature, we propose that 11CR entry involves an allosteric opening of a pore in a permissive opsin conformation, which dynamically adjusts to the incoming 11CR. Our findings provide experimental evidence on one of the entry pathways proposed in computational studies (Wang and Duan, 2007) and highlight the importance of understanding how conformational dynamics regulates ligand binding in a GPCR. Moreover, our approach of engineering a site-specific FRET reporter and double-mutant energetic analysis can be extended to study ligand binding in a wide range of intermolecular interactions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. Coli TOP10 cells | ThermoFisher Scientific | |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa 488-DIBO | ThermoFisher Scientific | |
| p-azido-L-phenylalanine | Chem Impex | Cat# 06162 |
| 1D4 monoclonal antibody | Hybridoma culture in house | (Molday and MacKenzie, 1983) |
| n-dodecyl-β-D-maltoside (DDM) | Anatrace | Cat# D310 |
| 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) |
Anatrace | Cat# C316 |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | Cat# 850427 |
| Experimental models: Cell lines | ||
| FreeStyle™ 293-F Cells | ThermoFisher Scientific | Cat# R79007 |
| Recombinant DNA | ||
| PMT4.Rho | (Franke et al., 1988) | |
| pcDNA.RS | (Ye et al., 2009) | |
| pSVB.Yam | (Ye et al., 2008) | |
| Software and algorithms | ||
| Prism | GraphPad | |
| Origin | OriginLab | |
| Others | ||
| SPEX Fluorolog tau-311 spectrofluorometer | Horiba | N/A |
| Lambda800 Spectrophotometer | Perkin Elmer | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thomas P. Sakmar (sakmar@rockefeller.edu).
Materials availability
DNA constructs generated in the study are available upon request from the Lead Contact with a material transfer agreement.
Experimental model and subject details
Cell culture
The HEK293F suspension cell line was obtained from ThermoFisher Scientific and maintained according to the manufacturer’s protocol.
Method details
Molecular biology
E. Coli TOP10 cells (Invitrogen) were used for plasmid propagation and isolation. Mutations were introduced into bovine rhodopsin using the QuikChange site-directed mutagenesis kit (Agilent) according to the manufacturer’s protocol.
Heterologous expression of wt and mutant Rho in mammalian cell culture
The wt and azF-tagged Rho were expressed in HEK293F suspension cell culture. The HEK293F suspension cells were cultured in serum-free FreeStyle 293 expression medium in a 125-mL disposable, sterile, polycarbonate Erlenmeyer flask (Corning) at 37°C in 5% CO2 atmosphere and constantly shaked at 125 rpm. Transfection of suspension cells was done using FreeStyleMax according to the manufacturer’s protocol. For a 30-mL culture, 38.6 μg of plasmid DNA (for amber codon suppression 18.4 μg of pMT4.Rho containing the amber codon, 18.4 μg of pSVB.Yam, and 1.84 μg pcDNA.RS-azF were mixed together) was used. For wt and Rho mutants without unnatural amino acids, cells were harvested 48 h post-transfection. For azF-tagged Rho mutants, FreeStyle 293 expression medium was supplemented with 1 mm azF and the cells were harvested 96 h post-transfection. The total cell number upon harvesting normally ranged from 6×107 to 8×107. The harvested cells were resuspended in DPBS (Gibco, supplemented with leupeptin and aprotinin, Sigma) at a density of 107 cells/mL in a 15-mL conical, polypropylene tube (Falcon). In the dark room, 11-cis-retinal (1.4 mm ethanol solution) was added into the cell suspension to a final concentration of 5 μm (Oprian et al., 1987; Starace and Knox, 1998). The suspension was nutated at 4 °C at least overnight. Excess 11-cis-retinal was removed by spinning down the cells and discarding the supernatant fraction. The regenerated cells can be immediately used, or may be stored at −20°C for several months.
Immunopurification and bioorthogonal labeling
1D4-Sepharose 2B was prepared from 1D4 mAb and CNBr-activated Sepharose 2B (2 mg IgG per mL packed beads) as described (Knepp et al., 2011; Oprian et al., 1987). The 11-cis-retinal regenerated cells expressing Rho mutants were lysed with the solubilization buffer (1 mL per 107 cells, 1% (w/v) DDM, 50 mm HEPES or Tris-HCl, pH 6.8, 100 mm NaCl, 1 mm CaCl2 with Complete EDTA-free Protease Inhibitor Cocktail, Roche) for at least 1 h at 4°C. The lysate was cleared by centrifugation at 100,000×g for 30 min and incubated overnight at 4°C with 1D4-mAb-sepharose 2B (100 μL). The resin was transferred into a 1.5-mL Eppendorf tube and washed three times for 30 min each with 0.5 mL reaction buffer (0.1% DDM in DPBS, pH 7.2). AzF-tagged mutants was labeled before the elution step. Non-azF-tagged mutants was directly eluted with a competitive peptide. Alexa 488-DIBO was obtained from Molecular Probes/Thermo Fisher Scientific as a dry powder, dissolved in DMSO (5 mm), and stored at −20°C. Then the reaction buffer (200 μL) was mixed with the resin (100 μL) to give 300-μL slurry. The Alexa 488-DIBO stock solution (5 mm in DMSO) was directly added to into the reaction mixture to give the appropriate final concentration. The reaction was agitated with a thermomixer at 25°C. For labeling S144azF, we used 50 μm of Alexa 488-DIBO and allowed the reaction to proceed for 18 h. The reaction was stopped by centrifugation and removal of the supernatant fraction. The resin was then transferred into a microporous centrifugal filtering unit (Microcon-MC pore size 0.48 μm, Millipore). The resin was first washed with the reaction/wash buffer for three times (30 min incubation each time) to deplete the unreacted dyes, and then with a low-salt buffer (0.1% (w/v) DDM, 2 mm sodium phosphate buffer, pH 6.0). The receptor was eluted with elution buffer (100 μL, no less than the volume of the resin; 0.33 mg/mL C9 peptide in 0.1% (w/v) DDM, 2 mm sodium phosphate buffer, pH 6.0). The resin was incubated with the elution buffer on ice for at least 1 h. The purified receptor was collected in a clean 1.5-mL Eppendorf tube. The elution was repeated a second time. The dark−light difference spectrum at 500 nm was used to calculate the concentration of rhodopsin using an extinction coefficient of 40,600 m−1cm−1. The Alexa 488 concentration was calculated from 495-nm absorbance in the light spectrum using the extinction coefficient 73,000 m−1cm−1. The combined elutions were supplemented with 150 mm NaCl and characterized by UV-Vis spectroscopy. Purified samples were stored at −80°C. The yield from 107 HEK293F cells was typically 0.5–1 μg Alexa488-labeled azF-Rho (Tian et al., 2014).
Reconstitution of purified Rho mutants into POPC/CHAPS bicelles
The POPC lipids were dissolved in 20% (w/v) CHAPS solution at 1:1 POPC-to-CHAPS ratio (w/w) using repeated freeze-thaw cycles and diluted with water to make a 10% (w/v) stock bicelle solution. The 10% stock solution was aliquoted, kept at −20°C and thawed prior to use. The final working solution (1% (w/v) POPC, 1% (w/v) CHAPS, 125 mM KCl, 25 mM MES, 25 mM HEPES, 12.5 mM KOH, pH 6.0) was prepared freshly by diluting 10% POPC/CHAPS stock solution with the corresponding concentrated buffer stocks. At the beginning of the retinal binding assay, the frozen purified Rho samples (30 μL) were thawed on ice and diluted into the freshly prepared bicelle buffer (450 μL).
Measurement of 11CR entry kinetics or ATR release kinetics based on quenching of Trp or Alexa 488 fluorescence
All of the 11CR and ATR release assays were performed in 1% POPC/CHAPS bicelle solutions (10 mg/mL POPC and 10 mg/mL CHAPS). Fluorescence measurement was done on an SPEX Fluorolog tau-311 spectrofluorometer (Horiba) in photon-counting mode. The temperature was controlled by circulating pre-heated or pre-chilled water through the recording chamber. 30 μL of purified Rho was added into 450-μL of POPC/CHAPS buffer. In the Trp-based fluorescence experiment, the excitation wavelength was 295 nm with a 0.6-nm band-pass, and the emission was measured at 330 nm with a 15-nm band-pass. The concentration of Rho was typically 0.25–0.30 μm. In the Alexa488-based experiment, the sample was excited at 488 nm with a 0.2-nm band-pass, and the emission was measured at 520 nm with a 15-nm band-pass. The concentrations of Alexa488-Rho in these experiments were lower, normally in the range of 5–50 nm. The sample was constantly stirred to facilitate mixing. The fluorescence signal was integrated for 2 s in 30-s intervals. For each measurement, 11CR ethanolic stock solution was freshly diluted in POPC/CHAPS bicelle buffer and used within 12 h. The actual concentration of 11CR was determined by UV-vis spectroscopy using the extinction coefficient of 11CR in POPC/CHAPS bicelles (ε378 nm = 25,600 m−1 cm−1) (Tian et al., 2017a). 20 μL of 11CR working dilution was added to the cuvette to give a final concentration in the range of 0.5–30 μm, adjusted based on the first measurement of 11CR entry kinetics. The decrease of Trp or Alexa488 fluorescence was fitted with a pseudo first-order exponential decay model to derive the apparent regeneration rate (kobs). The second-order rate constant (k2) for the recombination reaction between opsin and retinal was calculated as k2 = kobs/[retinal]. The full details of preparing POPC/CHAPS buffer and retinal concentration measurement have been reported in an earlier publication (Tian et al., 2017a).
Quantification and statistical analysis
To calculate the kinetics of retinal entry and release, the fluorescence traces was fitted with a single exponential model using Origin. Data analysis was performed using GraphPad Prism. Data points with ≥3 trials in the figures and Tables S1–S5 are shown as mean ± STD. For a few mutants that were difficult to regenerate (n = 1 trial), STD was omitted.
Acknowledgments
We acknowledge the generous support from the Crowley Family Fund and the Danica Foundation. This work has also been generously supported by an International Research Alliance with Professor Thue W. Schwartz at the Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) through an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen. We also acknowledge the support from NIH R01EY012049 (T.P.S. and T.H.) as well as the Tri-Institutional Training Program in Chemical Biology (H.T.). We thank Drs. Belinda S.W. Chang and Elsa C.Y. Yan for advice and assistance in the initial phase of the study.
Author contributions
H.T., T.P.S., and T.H. designed the study, analyzed the results, and wrote the manuscript . H.T., T.H., K.M.G., and M.K. conducted the experiments.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
This research was carried out at an institution that is strongly committed to furthering the cause of diversity, equity, and inclusion at our organization and the broader research community through its recruitment, retention, and mentorship practices. As such, we note that the majority of this paper’s authors identify as female.
Published: April 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104060.
Contributor Information
Thomas P. Sakmar, Email: sakmar@rockefeller.edu.
Thomas Huber, Email: hubert@rockefeller.edu.
Supplemental information
Data and code availability
The original fluorescence traces used to calculate 11CR binding and ATR unbinding kinetics are available upon reasonable request to T.P.S. and T.H.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
References
- Alfonso-Prieto M., Navarini L., Carloni P. Understanding ligand binding to G-protein coupled receptors using multiscale simulations. Front. Mol. Biosci. 2019;6:29. doi: 10.3389/fmolb.2019.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship E., Vahedi-Faridi A., Lodowski D.T. The high-resolution structure of activated opsin reveals a conserved solvent network in the transmembrane region essential for activation. Structure. 2015;23:2358–2364. doi: 10.1016/j.str.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch M.P., Jo H., Valcourt J.R., Srinivasan Y., Pan A.C., Capponi S., Grabe M., Dror R.O., Shaw D.E., DeGrado W.F., et al. Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry. 2018;57:5748–5758. doi: 10.1021/acs.biochem.8b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H.W., Kim Y.J., Park J.H., Morizumi T., Pai E.F., Krauss N., Hofmann K.P., Scheerer P., Ernst O.P. Crystal structure of metarhodopsin ii. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- Deupi X., Edwards P., Singhal A., Nickle B., Oprian D., Schertler G., Standfuss J. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-ii. Proc. Natl. Acad. Sci. U S A. 2012;109:119–124. doi: 10.1073/pnas.1114089108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill K.A. Additivity principles in biochemistry. J. Biol. Chem. 1997;272:701–704. doi: 10.1074/jbc.272.2.701. [DOI] [PubMed] [Google Scholar]
- Dror R.O., Pan A.C., Arlow D.H., Borhani D.W., Maragakis P., Shan Y., Xu H., Shaw D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrey T., Koutalos Y. Vertebrate photoreceptors. Prog. Retin. Eye Res. 2001;20:49–94. doi: 10.1016/s1350-9462(00)00014-8. [DOI] [PubMed] [Google Scholar]
- Elisi G.M., Scalvini L., Lodola A., Mor M., Rivara S. Free-energy simulations support a lipophilic binding route for melatonin receptors. J. Chem. Inf. Model. 2021;62:210–222. doi: 10.1021/acs.jcim.1c01183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrens D.L., Khorana H.G. Structure and function in rhodopsin. Measurement of the rate of metarhodopsin ii decay by fluorescence spectroscopy. J. Biol. Chem. 1995;270:5073–5076. doi: 10.1074/jbc.270.10.5073. [DOI] [PubMed] [Google Scholar]
- Franke R.R., Sakmar T.P., Oprian D.D., Khorana H.G. A single amino acid substitution in rhodopsin (lysine 248-leucine) prevents activation of transducin. J. Biol. Chem. 1988;263:2119–2122. [PubMed] [Google Scholar]
- Frederiksen R., Boyer N.P., Nickle B., Chakrabarti K.S., Koutalos Y., Crouch R.K., Oprian D., Cornwall M.C. Low aqueous solubility of 11-cis-retinal limits the rate of pigment formation and dark adaptation in salamander rods. J. Gen. Physiol. 2012;139:493–505. doi: 10.1085/jgp.201110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M., Smith S.O., Sakmar T.P. Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry. 1998;37:8253–8261. doi: 10.1021/bi980147r. [DOI] [PubMed] [Google Scholar]
- Hildebrand P.W., Scheerer P., Park J.H., Choe H.W., Piechnick R., Ernst O.P., Hofmann K.P., Heck M. A ligand channel through the G protein coupled receptor opsin. PLoS One. 2009;4:e4382. doi: 10.1371/journal.pone.0004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim P., Clark T. Metadynamics simulations of ligand binding to Gpcrs. Curr. Opin. Struct. Biol. 2019;55:129–137. doi: 10.1016/j.sbi.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Imai H., Kuwayama S., Onishi A., Morizumi T., Chisaka O., Shichida Y. Molecular properties of rod and cone visual pigments from purified chicken cone pigments to mouse rhodopsin in situ. Photochem. Photobiol. Sci. 2005;4:667–674. doi: 10.1039/b416731g. [DOI] [PubMed] [Google Scholar]
- Janz J.M., Farrens D.L. Role of the retinal hydrogen bond network in rhodopsin Schiff base stability and hydrolysis. J. Biol. Chem. 2004;279:55886–55894. doi: 10.1074/jbc.M408766200. [DOI] [PubMed] [Google Scholar]
- Jones D.H., Cellitti S.E., Hao X., Zhang Q., Jahnz M., Summerer D., Schultz P.G., Uno T., Geierstanger B.H. Site-specific labeling of proteins with NMR-active unnatural amino acids. J. Biomol. NMR. 2010;46:89–100. doi: 10.1007/s10858-009-9365-4. [DOI] [PubMed] [Google Scholar]
- Knepp A.M., Grunbeck A., Banerjee S., Sakmar T.P., Huber T. Direct measurement of thermal stability of expressed Ccr5 and stabilization by small molecule ligands. Biochemistry. 2011;50:502–511. doi: 10.1021/bi101059w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwayama S., Imai H., Hirano T., Terakita A., Shichida Y. Conserved proline residue at position 189 in cone visual pigments as a determinant of molecular properties different from rhodopsins. Biochemistry. 2002;41:15245–15252. doi: 10.1021/bi026444k. [DOI] [PubMed] [Google Scholar]
- Latorraca N.R., Venkatakrishnan A.J., Dror R.O. Gpcr dynamics: structures in motion. Chem. Rev. 2017;117:139–155. doi: 10.1021/acs.chemrev.6b00177. [DOI] [PubMed] [Google Scholar]
- Maeda R., Hiroshima M., Yamashita T., Wada A., Sako Y., Shichida Y., Imamoto Y. Shift in conformational equilibrium induces constitutive activity of G-protein-coupled receptor, rhodopsin. J. Phys. Chem. B. 2018;122:4838–4843. doi: 10.1021/acs.jpcb.8b02819. [DOI] [PubMed] [Google Scholar]
- Marino K.A., Filizola M. Investigating small-molecule ligand binding to G protein-coupled receptors with biased or unbiased molecular dynamics simulations. Methods Mol. Biol. 2018;1705:351–364. doi: 10.1007/978-1-4939-7465-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattle D., Kuhn B., Aebi J., Bedoucha M., Kekilli D., Grozinger N., Alker A., Rudolph M.G., Schmid G., Schertler G.F.X., et al. ligand channel in pharmacologically stabilized rhodopsin. Proc. Natl. Acad. Sci. U S A. 2018;115:3640–3645. doi: 10.1073/pnas.1718084115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday R.S., MacKenzie D. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry. 1983;22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- Morrow J.M., Chang B.S. Comparative mutagenesis studies of retinal release in light-activated zebrafish rhodopsin using fluorescence spectroscopy. Biochemistry. 2015;54:4507–4518. doi: 10.1021/bi501377b. [DOI] [PubMed] [Google Scholar]
- Oprian D.D., Molday R.S., Kaufman R.J., Khorana H.G. Expression of a synthetic bovine rhodopsin Gene in monkey kidney cells. Proc. Natl. Acad. Sci. U S A. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K., Kiser P.D. Shedding new light on the generation of the visual chromophore. Proc. Natl. Acad. Sci. U S A. 2020;117:19629–19638. doi: 10.1073/pnas.2008211117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechnick R., Ritter E., Hildebrand P.W., Ernst O.P., Scheerer P., Hofmann K.P., Heck M. Effect of channel mutations on the uptake and release of the retinal ligand in opsin. Proc. Natl. Acad. Sci. U S A. 2012;109:5247–5252. doi: 10.1073/pnas.1117268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S.G., Choi H.J., Fung J.J., Pardon E., Casarosa P., Chae P.S., Devree B.T., Rosenbaum D.M., Thian F.S., Kobilka T.S., et al. Structure of a nanobody-stabilized active state of the Β2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D.M., Cherezov V., Hanson M.A., Rasmussen S.G., Thian F.S., Kobilka T.S., Choi H.J., Yao X.J., Weis W.I., Stevens R.C., et al. Gpcr engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Sato S., Jastrzebska B., Engel A., Palczewski K., Kefalov V.J. Apo-opsin exists in equilibrium between a predominant inactive and a rare highly active state. J. Neurosci. 2019;39:212–223. doi: 10.1523/JNEUROSCI.1980-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer C.T., Farrens D.L. Conformational selection and equilibrium governs the ability of retinals to bind opsin. J. Biol. Chem. 2015;290:4304–4318. doi: 10.1074/jbc.M114.603134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer C.T., Fay J.F., Janz J.M., Farrens D.L. Decay of an active Gpcr: conformational dynamics Govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc. Natl. Acad. Sci. U S A. 2016;113:11961–11966. doi: 10.1073/pnas.1606347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P., Park J.H., Hildebrand P.W., Kim Y.J., Krauss N., Choe H.W., Hofmann K.P., Ernst O.P. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Standfuss J., Edwards P.C., D'Antona A., Fransen M., Xie G., Oprian D.D., Schertler G.F. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley N., Pardo L., Fabritiis G.D. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016;6:22639. doi: 10.1038/srep22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace D.M., Knox B.E. Cloning and expression of a Xenopus short wavelength cone. Pigment. Exp. Eye Res. 1998;67:209–220. doi: 10.1006/exer.1998.0507. [DOI] [PubMed] [Google Scholar]
- Szlenk C.T., Gc J.B., Natesan S. Does the lipid bilayer orchestrate access and binding of ligands to transmembrane orthosteric/allosteric sites of G protein-coupled receptors? Mol. Pharmacol. 2019;96:527–541. doi: 10.1124/mol.118.115113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Naganathan S., Kazmi M.A., Schwartz T.W., Sakmar T.P., Huber T. Bioorthogonal fluorescent labeling of functional G-protein-coupled receptors. Chem. Biochem. 2014;15:1820–1829. doi: 10.1002/cbic.201402193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Sakmar T.P., Huber T. Micelle-enhanced bioorthogonal labeling of genetically encoded azido groups on the lipid-embedded surface of a Gpcr. Chem. Biochem. 2015;16:1314–1322. doi: 10.1002/cbic.201500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Sakmar T.P., Huber T. The energetics of chromophore binding in the visual photoreceptor rhodopsin. Biophys. J. 2017;113:60–72. doi: 10.1016/j.bpj.2017.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H., Sakmar T.P., Huber T. Measurement of slow spontaneous release of 11-cis-retinal from rhodopsin. Biophys. J. 2017;112:153–161. doi: 10.1016/j.bpj.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H., Farrens D.L. A constitutively activating mutation alters the dynamics and energetics of a key conformational change in a ligand-free G protein-coupled receptor. J. Biol. Chem. 2013;288:28207–28216. doi: 10.1074/jbc.M113.472464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N., Caro L.N., Morizumi T., Kusnetzow A.K., Szczepek M., Hofmann K.P., Bayburt T.H., Sligar S.G., Ernst O.P., Hubbell W.L. Conformational equilibria of light-activated rhodopsin in nanodiscs. Proc. Natl. Acad. Sci. U S A. 2017;114:E3268–E3275. doi: 10.1073/pnas.1620405114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald G. Molecular basis of visual excitation. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- Wang T., Duan Y. Chromophore channeling in the G-protein coupled receptor rhodopsin. J. Am. Chem. Soc. 2007;129:6970–6971. doi: 10.1021/ja0691977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J.A. Additivity of mutational effects in proteins. Biochemistry. 1990;29:8509–8517. doi: 10.1021/bi00489a001. [DOI] [PubMed] [Google Scholar]
- Yanagawa M., Kojima K., Yamashita T., Imamoto Y., Matsuyama T., Nakanishi K., Yamano Y., Wada A., Sako Y., Shichida Y. Origin of the low thermal isomerization rate of rhodopsin chromophore. Sci. Rep. 2015;5:11081. doi: 10.1038/srep11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S., Huber T., Vogel R., Sakmar T.P. FTIR analysis of GPCR activation using azido probes. Nat. Chem. Biol. 2009;5:397–399. doi: 10.1038/nchembio.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S., Kohrer C., Huber T., Kazmi M., Sachdev P., Yan E.C.Y., Bhagat A., RajBhandary U.L., Sakmar T.P. Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis. J. Biol. Chem. 2008;283:1525–1533. doi: 10.1074/jbc.M707355200. [DOI] [PubMed] [Google Scholar]
- Zhang C., Srinivasan Y., Arlow D.H., Fung J.J., Palmer D., Zheng Y., Green H.F., Pandey A., Dror R.O., Shaw D.E., et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original fluorescence traces used to calculate 11CR binding and ATR unbinding kinetics are available upon reasonable request to T.P.S. and T.H.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.