

Abstract
Herein are described substrate oxidations with H2O2 catalyzed by [FeII(IndH)(CH3CN)3](ClO4)2 [IndH = 1,3-bis(2′-pyridylimino)isoindoline], involving a spectroscopically characterized (μ-oxo)(μ-1,2-peroxo)diiron(III) intermediate (2) that is capable of olefin epoxidation and alkane hydroxylation including cyclohexane. Species 2 also converts ketones to lactones with a decay rate dependent on [ketone], suggesting direct nucleophilic attack of the substrate carbonyl group by the peroxo species. In contrast, peroxo decay is unaffected by the addition of olefins or alkanes, but the label from H2 18O is incorporated into the the epoxide and alcohol products, implicating a high-valent iron–oxo oxidant that derives from O–O bond cleavage of the peroxo intermediate. These results demonstrate an ambiphilic diferric–peroxo intermediate that mimics the range of oxidative reactivities associated with O2-activating nonheme diiron enzymes.
Nonheme diiron enzymes are involved in many oxidative metabolic pathways.1 This class includes those that hydroxylate strong C–H bonds like soluble methane monooxygenase (sMMO)2 and deoxyhypusine hydroxylase (hDOHH), which helps to regulate cell proliferation in humans.3 These enzymes activate O2 at diiron(II) active sites that form (μ-1,2-peroxo)diiron(III) centers called P.4a,5 For sMMO, P converts into a diiron(IV) intermediate Q that is directly responsible for methane oxidation.4b An analogous O–O bond cleavage step is proposed for hDOHH,5b but the putative high-valent oxidant has not been trapped. Yet another diiron enzyme, cyanobacterial aldehyde deformylating oxygenase (cADO), converts fatty aldehydes into alkanes, a reaction initiated by nucleophilic attack of the corresponding peroxo intermediate on the aldehyde functionality of the substrate.6,7 These examples illustrate the mechanistic diversity of the peroxodiiron(III) unit in this family of enzymes.
In recent years, synthetic peroxodiiron(III) complexes have served to model such nonheme diiron enzyme intermediates.1 Kodera et al. have described a (μ-oxo)(μ-1,2-peroxo)diiron-(III) complex supported by a bis-TPA [TPA = tris(pyridyl-2-methyl)amine] ligand that undergoes O–O bond cleavage to oxidize the benzylic C–H bonds of toluene.8 More recently, Kaizer et al. have reported a peroxodiferric species supported by 2-(4-thiazolyl)benzimidazole ligands that exhibits ambiphilic reactivity in aldehyde deformylation, oxidation of the O–H bonds of phenols, and oxidative demethylation of DMA.9 In this study, we focus on another (μ-oxo)(μ-1,2-peroxo)diiron(III) intermediate that exhibits even greater oxidative versatility.
Previously, [FeII(IndH)(CH3CN)3](ClO4)2 [1; IndH = 1,3-bis(2′-pyridylimino)isoindoline] has been found to react with H2O2 at 25 °C in acetonitrile (MeCN) to form a transient green species 2 (Figure 1 left). This species has been identified as [FeIII2(μ-O)(μ-1,2-O2)(IndH)2(solv)2]2+ and shown to oxidize thioanisoles and benzyl alcohols.10a Remarkably, we herein demonstrate 2 to be an even more versatile and powerful oxidant that converts cyclic ketones to lactones, epoxidizes olefins, and even oxidizes cyclohexane.
Figure 1.
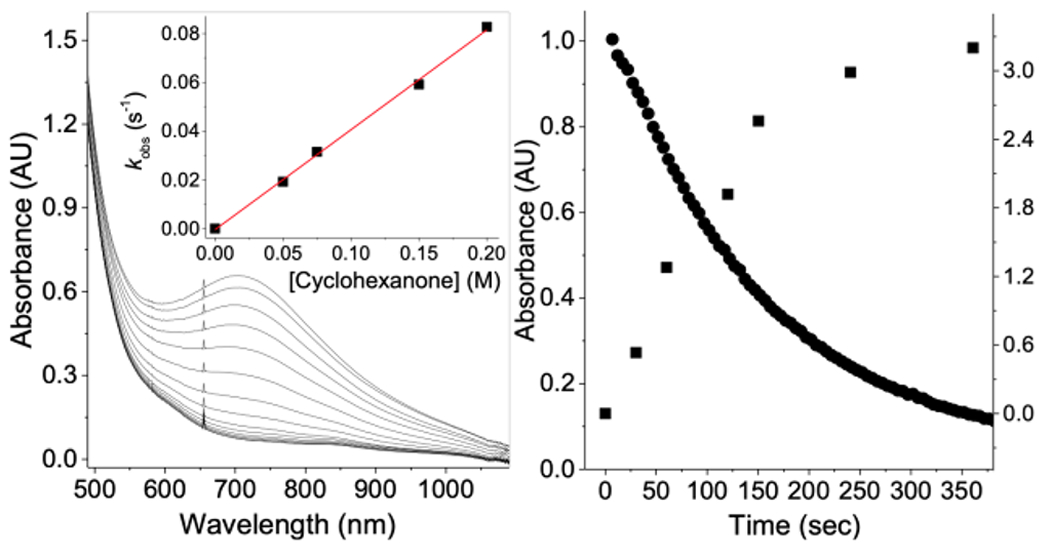
Left: Spectral changes upon cyclohexanone oxidation by 2 in MeCN at 5 °C with [1] = 0.5 × 10−3 M and [H2O2] = 2.0 mM. Inset: Plot of the rate constants for 2 decay versus [cyclohexanone]. Right: Oxidation of cyclohexane (0.1 M) by 1 (1 mM) and H2O2 (0.1 M) at 25 °C, monitoring the change in [2] (●) at 680 nm and the concentration of cyclohexanone formed (mM, ■) versus time.
In a previous publication, we reported that 2 decayed over 10 min at 5 °C with kdecay = 1.16(5) × 10−3 s−1 (Figure S1) and with ΔH‡ = 81(5) kJ mol−1 and ΔS‡ = −10(10) J mol−1 K−1 (Figure S2a), reflecting a unimolecular decay process.10a,b In our current study, we find that the addition of cyclohexanone accelerates its decay at rates that depend linearly on [cyclohexanone] (Figure 1, left), indicating a direct reaction between 2 and cyclohexanone with k2 = 0.4 M−1 s−1. Caprolactone is formed with up to 12.5 TON (relative to 1; see Table 1), demonstrating catalytic conversion of cyclohexanone to caprolactone. Under these conditions, the decay of 2 exhibits ΔH‡ = 22(1) kJ mol−1 and ΔS‡ = −170(10) J mol−1 K−1 (Figure S2a), parameters similar to those for PhCHO oxidation by 2.10b When the experiment is carried out in the presence of H2 18O, no 18O is incorporated into the lactone product (Figure S2b), analogous to Baeyer–Villiger oxidations.11 Complex 2 thus acts as a nucleophilic oxidant (Scheme 1, top left) and represents a rare example of a diferric–peroxo complex that performs the role proposed for the corresponding intermediate in the cADO mechanism.7
Table 1.
Substrate Oxidation Rates at 25 °C, Products, and Turnover Numbers for 1-Catalyzed Reactionsa
| substrate [DC–H in kcal mol−1] | k2 (M−1 s−1) | product [% 18O]b | TON |
|---|---|---|---|
| cyclohexanone | 0.4 | caprolactone | 13 |
| PhSMe | 10.5 | PhS(O)Me [43]b | 56 |
| cyclooctene | 4.2 | epoxide [44]b | 36 |
| 1-octene | 1.2 | epoxide | 20 |
| Ph3CH [81] | 0.072 | Ph3COH [50]b | 10 |
| PhCH(CH3)2 [84] | 0.054 | cumyl alcohol | 7 |
| PhCH2CH3 [87] | 0.019 | PhC(O)Me | 12 |
| PhCH3 [90] | 0.008 | PhCHO (KIE 37; Hammett Hρ = −0.42) | 5 |
| c-C8H16 [96] | 0.0062 | C8H14O | 7 |
| c-C6H12 [99] | 0.0045 | C6H10O | 3 |
0.1 mmol of substrate, 0.25 mmol of H2O2, 1 μmol of 1, and 10 mL of CH3CN.
% 18O label incorporated into oxidation products. Reaction conditions: 4 mM 1, 4 mM H2O2, 250 mM substrate, 25 °C.
Scheme 1.
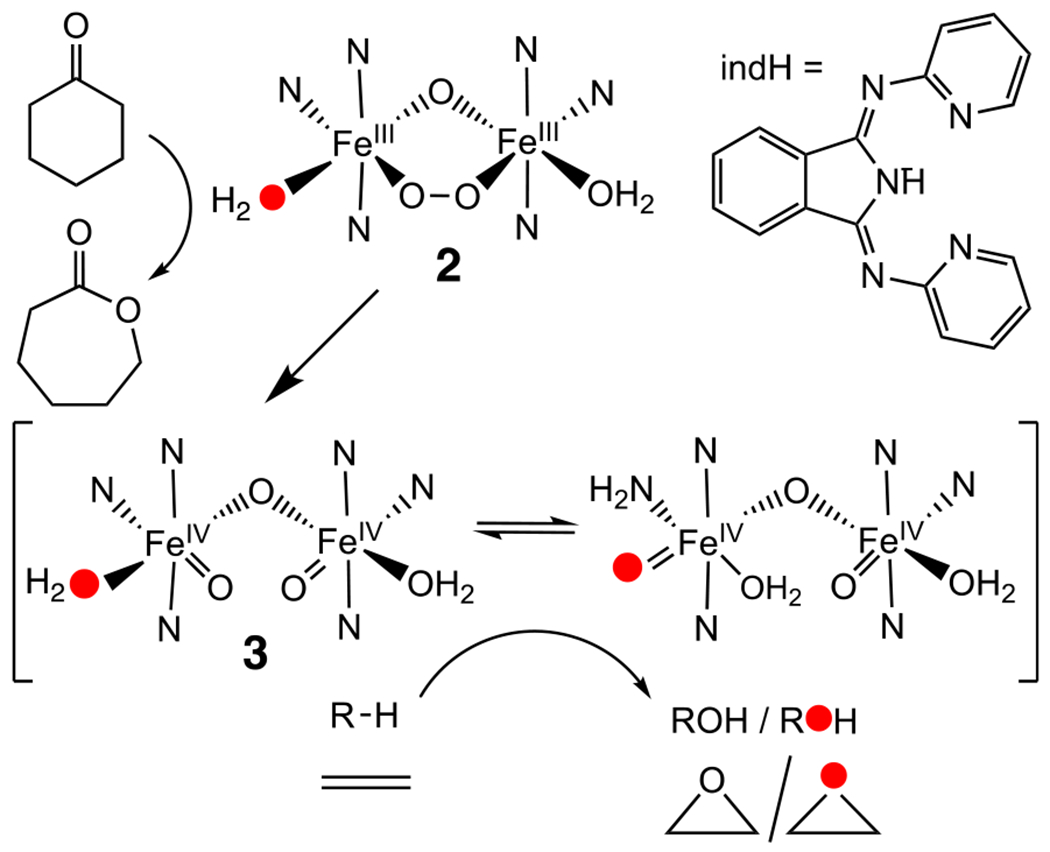
Proposed Oxidation Mechanism via 2
More significant than acting as a nucleophilic oxidant, 2 also carries out catalytic electrophilic hydrocarbon oxidation (Table 1). Unlike its reaction with cyclohexanone, 2 decays at a rate of ~0.02 s−1 at 25 °C, which is independent of the nature of the hydrocarbon and its concentration (Figure S3). These results show that 2 itself cannot be the actual oxidant but must evolve into a more powerful species to generate the oxidized products (Figure 1, right). Interestingly, the second-order rate constants associated with hydrocarbon oxidation show a linear dependence on [substrate] (Figure 2, left) and span over a 1000-fold range, with O-atom transfer to a C═C bond being much faster than C–H bond cleavage (Table 1). These observations indicate that the rate-determining step for these catalytic reactions must involve substrate oxidation. Kinetic measurements of toluene oxidation (see the Table S2 data with the gray background and Figure S4) show a first-order (not half-order) dependence on [2], excluding the possibility that 2 dissociates into two mononuclear FeIV═O units.
Figure 2.
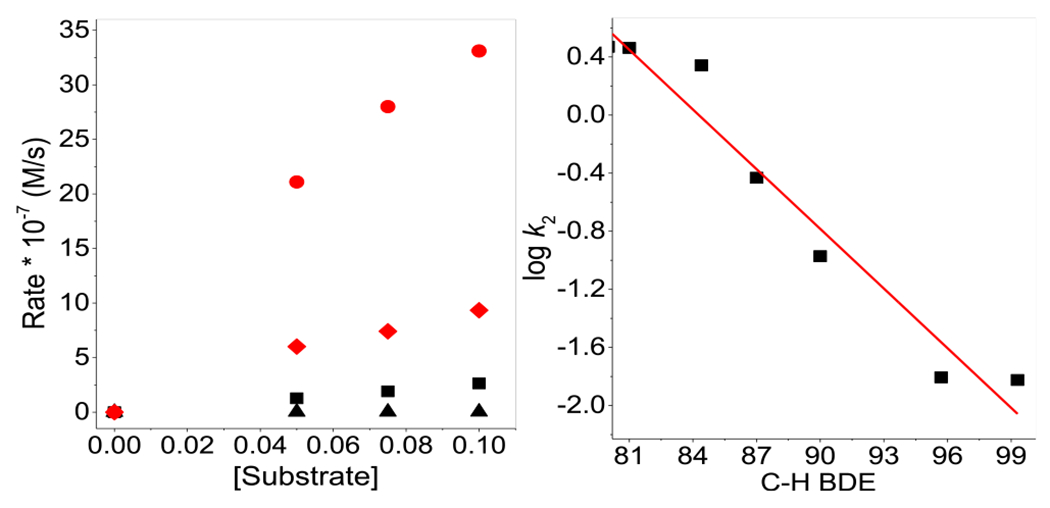
Left: Plot of the product formation rates versus [substrate] in reactions of cyclooctene (red ●), 1-octene (red ◆), cumene (black ■), and cyclohexane (black ▲) with 0.05 mM 1 and 250 mM H2O2 in CH3CN at 25 °C. Right: Linear correlation between log kox per substrate H atom and the corresponding C–H BDEs.
The oxidative reactivity of 2 resembles that of nonheme FeIV═O complexes.12 As shown in Figure 2 (right), there is a linear correlation between the logarithms of the product formation rate constants normalized per equivalent target H atom for substrates that undergo C–H bond attack versus C–H bond dissociation enthalpies (BDEs), showing that C–H bond cleavage is an important component of the product formation step. In addition, a nonclassical H/D kinetic isotope effect (KIE) of 37 for toluene is observed (Figure S4), concurring with Kodera’s observations for a related diiron catalyst supported by a dinucleating ligand.8b Such large nonclassical values are associated with nonheme iron(IV) oxo complexes12 and are much larger than the classical values for catalytic alkane hydroxylations by mononuclear Fe(L)/H2O2 systems involving FeV═O oxidants.13
Further insight into the nature of the oxidant has been obtained from H2 18O-labeling experiments, which show significant label incorporation into the oxidized products. As much as 40–50% 18O is incorporated into the PhS(O)Me, cyclooctene oxide, and Ph3COH products, which represent three different types of reactions (Table 1). Because labeled oxygen from H2 18O cannot exchange into a peroxide moiety without prior O–O bond cleavage, the results show that the actual oxidant in these electrophilic reactions must involve a species formed subsequent to O–O bond cleavage of 2 and capable of undergoing label exchange with H2 18O. When examined as a function of the H2 18O concentration, the % 18O incorporated into cyclooctene oxide increases linearly with [H2 18O] but plateaus above 0.6 M H2 18O (Figure 3, left). This saturation behavior implicates a reversible H2O binding step prior to O-atom transfer to the substrate. Indeed, the high degree of 18O-label incorporation observed indicates reaction conditions that approach complete equilibration within the putative FeIV(O)(OH2) unit. Moreover, 18O-label incorporation into cyclooctene oxide is found to be independent of the cyclooctene concentration (10–300 equiv; Figure S6), suggesting that 18O-label exchange is much more facile than substrate oxidation.
Figure 3.
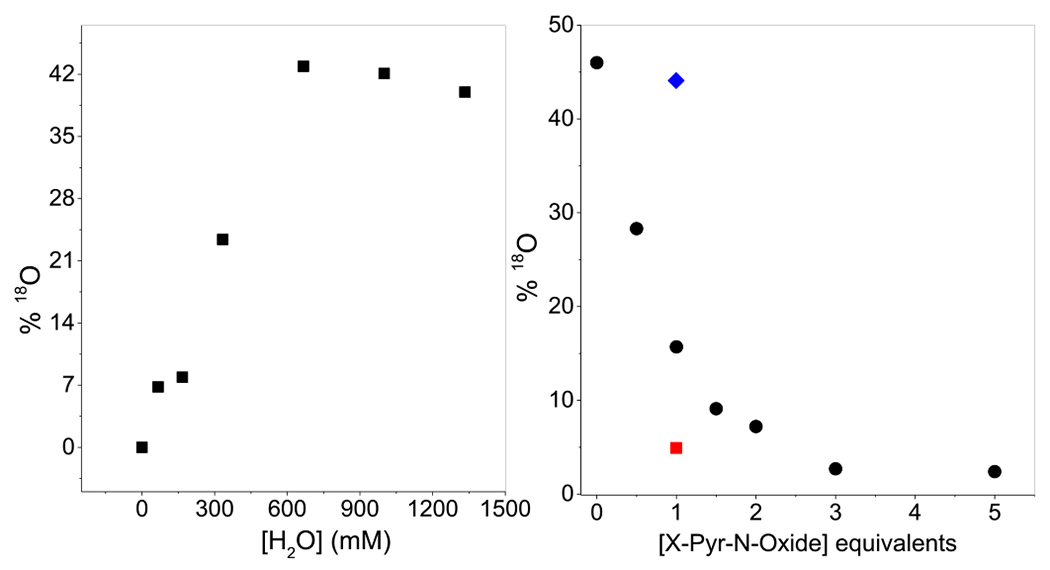
Left: Plot of % 18O incorporation into product versus [H2 18O] in cyclooctene oxidation (300 equiv) with 1 and H2O2 (5 equiv) in CH3CN at 25 °C. Right: % 18O labeling of the epoxide product of cyclooctene (200 equiv) by 1 (4 mM), H2O2 (2 equiv), and H2 18O (250 equiv), with added PyO (black ●), 4-MeO-PyO (red ■), or 4-NO2-PyO (blue ◆).
The observed saturation behavior for 18O incorporation in cyclooctene oxidation (Figure 3, left) resembles that reported for Fe(TPA)-catalyzed hydrocarbon oxidations with H2O2 as the oxidant.14 For the latter, the percentage of incorporation of 18O from H2 18O into cyclohexanol and cyclooctane-1,2-diol products increased linearly with [H2 18O] and maximized above 0.3 M with added H2O. These results were rationalized by H2 18O exchange into the site cis to the hydroperoxo unit on the [(TPA)FeIII(OOH)(solv)]2+ intermediate. Subsequent O–O bond heterolysis formed a putative cis-FeV(O)(18OH) oxidant, which, in turn, underwent oxo–hydroxo tautomerization to introduce 18O to the high-valent Fe═O unit, thereby accounting for the observed partial 18O labeling of the products. An analogous mechanism can be formulated for 2, which has an available site on each Fe center for label exchange with H2 18O to allow 18O incorporation into the putative high-valent O═FeIV–O–FeIV═O oxidant 3 (Scheme 1). Oxidant 3 is analogous to the oxidant that Kodera postulated for his diferric–peroxo intermediate, which is supported by an octadentate 6,6′-(ethylene-bridged)-bis(TPA) ligand,8 but the Kodera oxidant does not have solvent-exchangeable sites that allow exchange with H2 18O to afford the labeled products.
This proposed mechanism is further supported by the drop in % 18O incorporation found for the cyclooctene oxide product with increasing equivalents of pyridine-N-oxide (PyO) added into the reaction mixture, reflecting the competition between PyO and H2O for the labile sixth site on each Fe (Figure 3, right). Furthermore, introducing 1 equiv of the more basic 4-MeO-PyO instead of PyO results in a lower % 18O incorporation, while adding 1 equiv of the less basic 4-NO2-PyO has very little effect. These results show that PyO competes with H2 18O for binding to 2 in Scheme 1. As a control, PyO added to the reaction mixture in place of H2O2 produces no epoxide, showing that PyO does not act as an oxidant in this reaction.
Given that 2 can carry out both nucleophilic and electrophilic transformations, we have also conducted competitive oxidations between cyclohexanone and 1-octene or toluene. Consistent with our mechanistic hypothesis, electrophilic oxidation products decrease in yield with higher [cyclohexanone], raising the yield of caprolactone (Figure 4). These results show that nucleophilic oxidation of cyclohexanone competes with the O–O bond cleavage step required to generate the electrophilic oxidant responsible for 1-octene or toluene oxidation.
Figure 4.
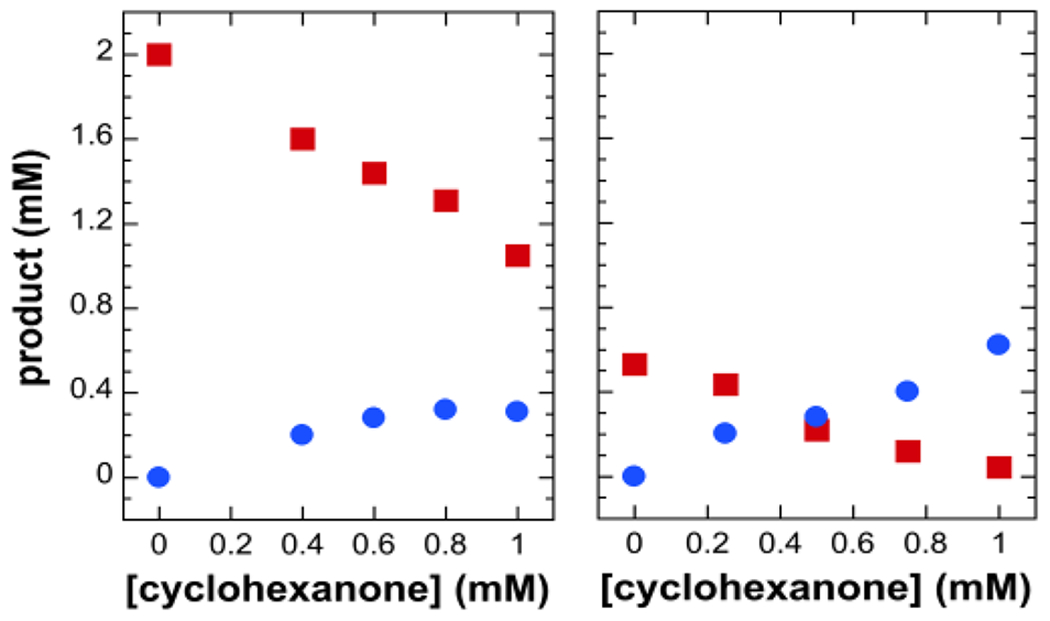
Yields of 1-octene oxide (red ■, left) and PhCHO (red ■, right) from the oxidations of 1-octene (left) and toluene (right) by 1/H2O2 versus added cyclohexanone. Caprolactone, the cyclohexanone oxidation product, is represented by blue ● in both panels. Reaction conditions: 0.1 mM 1, 0.25 M H2O2, 0.1 M substrate in CH3CN, 25 °C.
Among the many diferric–peroxo complexes characterized thus far as models for peroxo intermediates in nonheme diiron enzymes,1 2 stands out as the only synthetic diferric–peroxo species reported to date found to oxidize cyclohexane and the only one that carries out both nucleophilic and electrophilic oxidations under catalytic conditions. Specifically, 2 catalytically converts cyclohexanone to caprolactone in competition with the epoxidation of C═C bonds and the hydroxylation of aliphatic C–H bonds as strong as those in cyclohexane (Table 1). In fact, caprolactone formation is favored over epoxidation and hydroxylation (Figure 4). Whereas the rate of 2 decay is dependent on the cyclohexanone concentration, it is unaffected by the addition of either olefins or alkanes. These results show that 2 reacts directly with cyclohexanone to initiate its lactonization but must evolve into a more powerful oxidant that oxidizes the latter substrates. In support, 18O from H2 18O is incorporated into the epoxide and alcohol products (Table 1), indicating prior cleavage of the O–O bond of 2 to form the high-valent iron–oxo species that oxidizes the hydrocarbons. Diferric–peroxo intermediate 2 is thus quite a versatile reagent, with its diverse reactivity supporting the notion that nonheme diiron enzymes share a common diferric–peroxo intermediate that can be tuned to perform the electrophilic functions of hydroxylases like sMMO and the nucleophilic functions of deformylating enzymes like cADO.1
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to the memory of Professor Elena Rybak-Akimova, an enthusiastic investigator of iron–oxygen chemistry. This work was supported by the Hungarian Research Fund (Grants K108489, TKP2020-IKA-07, and GINOP-2.3.2-15-2016-00049 to J.K.) and the U.S. National Institutes of Health (Grants R01 GM-38767 and R35 GM-131721 to L.Q.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c03468.
Additional data and experimental details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.inorgchem.1c03468
The authors declare no competing financial interest.
Contributor Information
Williamson N. Oloo, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Miklós Szávuly, Research Group of Bioorganic and Biocoordination Chemistry, University of Pannonia, 8200 Veszprém, Hungary.
József Kaizer, Research Group of Bioorganic and Biocoordination Chemistry, University of Pannonia, 8200 Veszprém, Hungary.
Lawrence Que, Jr., Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Jasniewski AJ; Que L Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Banerjee R; Jones JC; Lipscomb JD Soluble Methane Monooxygenase. Annu. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- (3).Wolff EC; Kang KR; Kim YS; Park MH Posttranslational Synthesis of Hypusine: Evolutionary Progresion and Specificity of the Hypusine Modifocation. Amino Acids 2007, 33, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Liu KE; Valentine AM; Wang D; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ Characterization of a Diiron(III)-Peroxo Intermediate in the Reaction Cycle of Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc 1995, 117, 10174–10185. [Google Scholar]; (b) Lee S-K; Fox BG; Froland WA; Lipscomb JD; Münck E A Transient Intermediate of the Methane Monooxygenase Catalytic Cycle Containing an FeIVFeIV Cluster. J. Am. Chem. Soc 1993, 115, 6450–6451. [Google Scholar]
- (5).(a)Vu VV; Emerson JP; Martinho M; Kim YS; Münck E; Park MH; Que L Jr. Human Deoxyhypusine Hydroxylase, an Enzyme Involved in Regulating Cell Growth, Activates O2 with a Nonheme Diiron Center. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14814–14819. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jasniewski AJ; Engstrom LM; Vu VV; Park MH; Que L X-ray Absorption Spectroscopic Characterization of the Diferric-peroxo Intermediate of Human Deoxyhypusine Hydroxylase in the Presence of its Substrate eIF5a. JBIC, J. Biol. Inorg. Chem 2016, 21, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schirmer A; Rude MA; Li X; Popova E; del Cardayre SB Microbial Biosynthesis of Alkanes. Science 2010, 329, 559–562. [DOI] [PubMed] [Google Scholar]
- (7).Pandelia ME; Li N; Nørgaard H; Warui DM; Rajakovich LJ; Chang W.-c.; Booker SJ; Krebs C; Bollinger JM Jr. Substrate-Triggered Addition of Dioxygen to the Diferrous Cofactor of Aldehyde-Deformylating Oxygenase to Form a Diferric-peroxo Intermediate. J. Am. Chem. Soc 2013, 135, 15801–15812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Kodera M; Kawahara Y; Hitomi Y; Nomura T; Ogura T; Kobayashi Y Reversible O-O Bond Scission of Peroxo-diiron(III) to High-spin Oxodiiron(IV) in Oxygen Activation of a Diiron Center with a Bistpa Dinucleating Ligand as a Soluble Methane Monoxygenase Model. J. Am. Chem. Soc 2012, 134, 13236–13239. [DOI] [PubMed] [Google Scholar]; (b) Kodera M; Ishiga S; Tsuji T; Sakurai K; Hitomi Y; Shiota Y; Sajith PK; Yoshizawa K; Mieda K; Ogura T Formation and High Reactivity of the anti-Dioxo Form of High-Spin μ-Oxodioxodiiron(IV) as the Active Species That Cleaves Strong C–H Bonds. Chem. - Eur. J 2016, 22, 5924–5936. [DOI] [PubMed] [Google Scholar]; (c) Kodera M; Itoh M; Kano K; Funabiki T; Reglier M A Diiron Center Stabilized by a Bis-TPA Ligand as a Model of Soluble Methane Monooxygenase: Predominant Alkene Epoxidation with H2O2. Angew. Chem., Int. Ed 2005, 44, 7104–7106. [DOI] [PubMed] [Google Scholar]
- (9).Török P; Unjaroen D; Viktória Csendes F; Giorgi M; Browne WR; Kaizer J A Nonheme Peroxodiiron(III) Complex Exhibiting Both Nucleophilic and Electrophilic Oxidation of Organic Substrates. Dalton Trans. 2021, 50, 7181–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Pap JS; Cranswick MA; Balogh-Hergovich É; Baráth G; Giorgi M; Rohde GT; Kaizer J; Speier G; Que L An Iron(II)[1,3-bis(2′-pyridylimino)isoindoline] Complex As a Catalyst for Substrate Oxidation with H2O2: Evidence for a Transient Peroxodiiron(III) Species. Eur. J. Inorg. Chem 2013, 2013, 3858–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kripli B; Szávuly M; Csendes FV; Kaizer J Functional Models of Nonheme Diiron Enzymes: Reactivity of the μ-Oxo-μ-1,2-peroxo-diiron(III) Intermediate in Electrophilic and Nucleophilic Reactions. Dalton Trans. 2020, 49, 1742–1746. [DOI] [PubMed] [Google Scholar]
- (11).(a) Cho J; Jeon S; Wilson SA; Liu LV; Kang EA; Braymer JJ; Lim MH; Hedman B; Hodgson KO; Valentine JS; Solomon EI; Nam W Structure and Reactivity of a Mononuclear Non-haem Iron(III)-Peroxo Complex. Nature 2011, 478, 502–505. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McDonald AR; Van Heuvelen KM; Guo Y; Li F; Bominaar EL; Münck E; Que L Jr. Characterization of a Thiolate Iron(III) Peroxy Dianion Complex. Angew. Chem., Int. Ed 2012, 51, 9132–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shokri A; Que L Jr. Conversion of Aldehyde to Alkane by a Peroxoiron(III) Complex, A Functional Model for the Cyanobacterial Aldehyde Deformylating Oxygenase. J. Am. Chem. Soc 2015, 137, 7686–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Kaizer J; Klinker EJ; Oh NY; Rohde J-U; Song WJ; Stubna A; Kim J; Münck E; Nam W; Que L Jr. Nonheme FeIVO Complexes That Can Oxidize the C–H Bonds of Cyclohexane at Room Temperature. J. Am. Chem. Soc 2004, 126, 472–473. [DOI] [PubMed] [Google Scholar]; (b) Biswas AN; Puri M; Meier KK; Oloo WN; Rohde GT; Bominaar EL; Münck E; Que L Jr. Modeling TauD-J: A High-Spin Nonheme Oxoiron(IV) Complex with High Reactivity Towards C–H Bonds. J. Am. Chem. Soc 2015, 137, 2428–2431. [DOI] [PubMed] [Google Scholar]; (c) Mitra M; Nimir H; Demeshko S; Bhat SS; Malinkin SO; Haukka M; Lloret-Fillol J; Lisensky GC; Meyer F; Shteinman AA; Browne WR; Hrovat DA; Richmond MG; Costas M; Nordlander E Nonheme Iron(IV)-Oxo Complexes of Two New Pentadentate Ligands and Their Hydrogen-Atom and Oxygen-Atom Transfer Reactions. Inorg. Chem 2015, 54, 7152–7164. [DOI] [PubMed] [Google Scholar]; (d) Monte Pérez I; Engelmann X; Lee Y-M; Yoo M; Kumaran E; Farquhar ER; Bill E; England J; Nam W; Swart M; Ray K A Highly Reactive Oxoiron(IV) Complex Supported by a Bioinspired N3O Macrocyclic Ligand. Angew. Chem., Int. Ed 2017, 56, 14384–14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Chen K; Que L Jr. Stereospecific Alkane Hydroxylation by Nonheme Iron Catalysts: Mechanistic Evidence for an FeV═O Active Species. J. Am. Chem. Soc 2001, 123, 6327–6337. [DOI] [PubMed] [Google Scholar]; (b) Chen K; Costas M; Kim J; Tipton AK; Que L Jr. Olefin cis-Dihydroxylation versus Epoxidation by Nonheme Iron Catalysts: Two Faces of an FeIII–OOH Coin. J. Am. Chem. Soc 2002, 124, 3026–3035. [DOI] [PubMed] [Google Scholar]
- (14).(a) Bernadou J; Meunier B ‘Oxo-hydroxo Tautomerism’ as a Useful Mechanistic Tool in Oxygenation Reactions Catalysed by Water-soluble Metalloporphyrins. Chem. Commun 1998, 2167–2173. [Google Scholar]; (b) Puri M; Company A; Sabenya G; Costas M; Que L Jr. Oxygen-Atom Exchange Between H2O and Nonheme Oxoiron(IV) Complexes: Ligand Dependence and Mechanism. Inorg. Chem 2016, 55, 5818–5827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.